

ABSTRACT
The RASopathies are a group of disorders caused by a germline mutation in one of the genes encoding a component of the RAS/MAPK pathway. These disorders, including neurofibromatosis type 1, Noonan syndrome, cardiofaciocutaneous syndrome, Costello syndrome and Legius syndrome, among others, have overlapping clinical features due to RAS/MAPK dysfunction. Although several of the RASopathies are very rare, collectively, these disorders are relatively common. In this Review, we discuss the pathogenesis of the RASopathy-associated genetic variants and the knowledge gained about RAS/MAPK signaling that resulted from studying RASopathies. We also describe the cell and animal models of the RASopathies and explore emerging RASopathy genes. Preclinical and clinical experiences with targeted agents as therapeutics for RASopathies are also discussed. Finally, we review how the recently developed drugs targeting RAS/MAPK-driven malignancies, such as inhibitors of RAS activation, direct RAS inhibitors and RAS/MAPK pathway inhibitors, might be leveraged for patients with RASopathies.
KEY WORDS: Cardiofaciocutaneous syndrome, Costello syndrome, Legius syndrome, Noonan syndrome, RAS, RASopathies
Summary: RASopathies are developmental disorders caused by germline pathogenic variants in RAS/MAPK pathway genes. Here, we review the preclinical studies that provide the basis for future interventional clinical trials for RASopathy patients.
Introduction
The RASopathies are a clinically defined group of disorders caused by a germline mutation in one of the genes encoding components of the RAS/MAPK pathway (Rauen, 2013; Tidyman and Rauen, 2009), typically resulting in increased signaling through this pathway. The RAS/MAPK pathway is a signal transduction cascade that is central to such normal cellular processes as proliferation, survival, differentiation and metabolism. This pathway is essential for normal development, with knockout mouse models of several components resulting in embryonic lethality due to, in many cases, abnormal vascularization of the embryo and/or placenta (Table S1). The pathway is composed of the RAS proteins, RAS guanine nucleotide exchange factors (GEFs), RAS GTPase-activating proteins (GAPs), RAS effector proteins and their targets, and other pathway modulators.
The specific classification of RASopathies is a matter of some debate among experts (Gross et al., 2020a). However, the RASopathies are generally accepted to include neurofibromatosis type 1 (NF1), Noonan syndrome (NS), Noonan syndrome with multiple lentigines (NS-ML), cardiofaciocutaneous syndrome (CFC), Costello syndrome (CS), Legius syndrome (LS), central conducting lymphatic anomalies syndrome (CCLA), SYNGAP1 syndrome, and capillary malformation arteriovenous malformation syndrome (CM-AVM). Many of the RASopathies have overlapping clinical features; however, each syndrome also has its unique characteristics. The overlapping features can include short stature, dysmorphic facial features, congenital heart disease, lymphatic dysfunction and intellectual disability. RASopathies are a common cause of non-immune hydrops fetalis (Sparks et al., 2020), as well as other features that can be observed on prenatal ultrasound such as increased nuchal translucency, cystic hygroma, congenital heart disease or pleural effusions (Scott et al., 2021). Each of the RASopathies are single-gene inheritance disorders. NS, CFC, CCLA and CM-AVM can be caused by alterations in one of several genes, while NF1, NS-ML, CS, LS and SYNGAP1 syndrome are each linked to alterations in one specific gene.
In this Review, we discuss the pathogenesis of the RASopathy-associated genetic variants (Table 1; Fig. S1) and the knowledge gained about RAS/MAPK signaling that resulted from studying RASopathies. We also describe the cell and animal models of the RASopathies (Fig. 1) and explore emerging RASopathy genes. Preclinical and clinical experiences with targeted agents as therapeutics for RASopathies are also discussed, together with the potential of therapeutics developed for RAS/MAPK-driven malignancies for the prevention or treatment of RASopathies. This article serves as an update to the excellent and comprehensive reviews on the non-NF1 RASopathies (Hernandez-Porras and Guerra, 2017; Jindal et al., 2015; Schuhmacher et al., 2017). Animal models of NF1 have recently been reviewed (Williams and Largaespada, 2020) and will thus not be discussed here.
Table 1.
Common RASopathy-associated mutations

Fig. 1.

Modeling the RASopathies. (A) Biochemical assays with purified proteins. These assays can compare the impacts of RASopathy-associated mutations on the enzymatic activities of the chosen proteins. Some examples include kinase, phosphatase, GEF-stimulated exchange, GEF-independent exchange, GAP-stimulated hydrolysis and GAP-independent hydrolysis activities. In addition, purified protein can be used to determine the impact of RASopathy variants on protein–protein interactions. Bmax, maximum specific binding; Kd, dissociation constant. (B) Transfection of RASopathy variants into mammalian cells. These assays assess the ability of the variants to stimulate the RAS/MAPK pathway, as well as define the subcellular localization of the variants in comparison to the wild-type protein. (C) Expression of RASopathy variants in model organisms. These models facilitate the study of the impact of RAS/MAPK pathway activation at the organismal level. In Drosophila melanogaster, aberrant RAS/MAPK signaling in the eye leads to small, rough eyes, while aberrant signaling in the wing leads to ectopic wing development. Similarly, aberrant RAS/MAPK signaling in the zebrafish (Danio rerio) embryo leads to elongated embryos with an increased major:minor axis ratio, and aberrant RAS/MAPK signaling in developing Caenorhabditis elegans embryos leads to various vulvar phenotypes. These phenotypes can be quantified, elucidating the genotype/phenotype correlations when comparing RASopathy-associated variants in these systems. The D. melanogaster, D. rerio, C. elegans and Mus musculus orthologs of each of the human RASopathy-associated genes are listed in Table S2. (D) Patient-derived cell lines. Fibroblast lines, lymphoblastoid lines and induced pluripotent stem cells can be used in a variety of cell signaling and phenotypic assays, provided an appropriate control line is available for comparison. Model organisms and patient-derived cell lines can both be used for preclinical drug screening. (E) Knock-in/knockout mouse models. These can be used to determine the developmental stage at which a variant can trigger the RASopathy phenotype, as well as identify the cell of origin for the RASopathy phenotypes. Germline expression of RASopathy variants in mouse models can identify genetic modifiers of the phenotype through backcrossing onto different genetic backgrounds. Moreover, these mouse models can be used in preclinical studies of potential therapeutics. Knockout models of RASopathy genes are summarized in Table S1, while knock-in models are described in Tables S3-S7, which are organized by RASopathy.
NS
NS is a relatively common disorder with a prevalence of 1:1000 to 1:2500 live births (Nora et al., 1974). The most common forms of NS are inherited in an autosomal-dominant fashion; however, rare autosomal recessive forms have been described (Johnston et al., 2018; van Der Burgt and Brunner, 2000). NS is caused by germline pathogenic variants in one of several RAS/MAPK pathway genes. In particular, 50% of NS cases are caused by a gain-of-function mutation in PTPN11, 10% by mutation in SOS1, 10% by mutation in RIT1 and 5% by mutation in RAF1, and an additional 5% of cases are caused by mutations in KRAS. The remaining 20% of cases are caused by rarer mutations in NRAS, RRAS, LZTR1 and others (Tartaglia and Gelb, 2005).
Most patients with NS have one or more of the following clinical features: (1) facial dysmorphic features; (2) congenital heart disease; and/or (3) postnatal proportional short stature. Additional features include endocrine abnormalities, such as low body mass index, delayed puberty onset, cryptorchidism or Sertoli cell dysfunction; skeletal abnormalities or decreased bone mineral density; neurocognitive abnormalities; pulmonary lymphangiectasias; and easy bruising (Romano et al., 2010). Patients with NS are at increased risk of developing malignancies such as neuroblastoma, juvenile myelomonocytic leukemia (JMML), low-grade glioma, giant cell lesions and rhabdomyosarcoma (Romano et al., 2010). The severity of the clinical features of NS varies by genotype (Table 1). The pathogeneses of the genetic subtypes of NS are discussed below:
PTPN11
Protein tyrosine phosphatase, non-receptor 11 (PTPN11; also known as SHP2) is a tyrosine phosphatase and molecular scaffold. Its phosphatase activity is required for full activation of the RAS/MAPK pathway. Active PTPN11 dephosphorylates many substrates (Hill et al., 2019). The cancer- and NS-associated mutations in PTPN11 occur at the interface between the N-terminal SH2 domain at residues D62, Y63 or Q79, and in the PTP domain at residue N308, and relieve auto-inhibitory intramolecular interactions (Table 1; Fig. S1). NS-associated PTPN11 mutants thus have increased phosphatase activity (Keilhack et al., 2005; Martinelli et al., 2008; Tartaglia et al., 2006).
NS-associated PTPN11 mutations have been studied in several model systems. Transfecting mutant PTPN11 transgenes into mammalian cells in vitro increased signaling through the RAS/MAPK pathway (Fragale et al., 2004; Kontaridis et al., 2006). Zebrafish embryos expressing NS-associated ptpn11 (also known as ptpn11a) mutants display increased major:minor axis ratios (Fig. 1) (Jopling et al., 2007), and adult mutants have cardiac defects that can be rescued by treatment with the MEK inhibitor CI-1040 (Bonetti et al., 2014). Drosophila expressing NS-associated PTPN11 mutants display ectopic wing veins (Fig. 1) (Oishi et al., 2006) and have impaired long-term memory, a phenotype that is rescued by treatment with the pre-clinical PTPN11 inhibitor NSC-87877 (Pagani et al., 2009). Moreover, induced pluripotent stem cells (iPSCs) derived from NS patients with PTPN11N308D and PTPN11D61N displayed impaired early neuroectodermal development due to enhanced gliogenesis and shortened neurites. This phenotype was rescued by treatment with the preclinical compound PHPS1, a cell-permeable phospho-tyrosine mimetic that inhibits PTPN11 phosphatase activity (Ju et al., 2020). Knock-in mouse models of Ptpn11N308D/+ and Ptpn11D61G/+ (Table S3) display small body size, facial dysmorphia, splenomegaly, and deficits in spatial learning and memory (Araki et al., 2009, 2004). The Ptpn11D61G/+ mice display a variety of cardiac phenotypes, with penetrance depending on the genetic background. Intriguingly, postnatal treatment of Ptpn11D61G/+ mice with the MEK inhibitor SL327 or the HMG-CoA reductase inhibitor lovastatin rescued the observed spatial learning deficits (Lee et al., 2014), whereas postnatal treatment of Ptpn11D61Y/+ mice with the tyrosine kinase inhibitor dasatinib reversed their cardiac dysfunction (Yi et al., 2016). Studies with conditional knock-in mice have elucidated the cell of origin and developmental stage responsible for NS phenotypes. For example, tissue-specific transgenic expression of Ptpn11Q79R in neural crest-derived tissues leads to craniofacial dysmorphia, which can be prevented with prenatal treatment with the tool compound U0126, a copper-chelating MEK inhibitor (Nakamura et al., 2009). Mice engineered to express Ptpn11Q79R in the fetal cardiomyocytes develop ventricular septal defects and heart failure in an ERK1/2 (also known as MAPK3/1)-dependent manner. Interestingly, mice expressing the same Ptpn11Q79R transgene in postnatal cardiomyocytes have no cardiac phenotype, underscoring that both the cell type and the developmental stage of the cell are important for this phenotype (Nakamura et al., 2007). Testing of candidate MEK, PTPN11 and SOS1 inhibitors in these models of NS could inform which NS phenotypes are reversible, which genotypes are most amenable to targeted therapy and which drugs are best tolerated.
SOS1
Son of sevenless homolog 1 (SOS1) is a member of the RAS GEF family that activates the RAS proteins by catalyzing the exchange of bound GDP for GTP. In the basal state, the RHO GEF and PH domains of SOS1 occlude the allosteric site, preventing its full activation. A further level of auto-inhibition is achieved by the N-terminal histone-like domain interacting with the linker between the PH and REM domains (Fig. S1), locking the RHO GEF/PH domains into position (Gureasko et al., 2010; Sondermann et al., 2004). NS-associated SOS1 mutations cluster either in the region of the RHO GEF domain that interacts with the allosteric site (M269) or in the region of the allosteric site that interacts with the histone-like domain (R552; Table 1). Both relieve auto-inhibitory interactions, increasing SOS1 GEF activity on RAS and thus signaling through the MAPK pathway (Lepri et al., 2011; Nakamura et al., 2017; Roberts et al., 2007; Tartaglia et al., 2007; Zenker et al., 2007).
Mouse models of M269- or R552-altered SOS1 have not yet been reported. However, mice expressing Sos1E846K, an activating mutation in the GEF domain, display cardiac hypertrophy and splenomegaly (Table S3). The viability of these mice depends on the genetic background. Importantly, prenatal treatment with the MEK inhibitor PD-0325901 rescued the perinatal lethality of mice homozygous for Sos1E846K (Chen et al., 2010). Clinical whole-exome sequencing (WES) of NS patients also identified mutations in SOS2, which shares 65% sequence identity with SOS1. These SOS2 variants cluster in a region of the RHO GEF domain that interacts with the allosteric site and lead to increased signaling through the MAPK pathway in these patients (Cordeddu et al., 2015; Lissewski et al., 2021; Yamamoto et al., 2015).
RAF1
RAF1 (also known as CRAF) is a member of the RAF family of serine/threonine kinases and a RAS effector. In the basal state, the N-terminal autoinhibitory region of RAF1, composed of the RAS-binding and cysteine-rich domains (Fig. S1), occludes the kinase domain (Fig. 2). This autoinhibitory conformation is stabilized by the binding of a 14-3-3 dimer, an interaction mediated by phosphorylation of RAF1 at S259. Activated RAS recruits RAF to the plasma membrane, disrupts the auto-inhibitory binding between the N-terminal regulatory domain and C-terminal kinase domain, and induces RAF dimerization, all of which activates its kinase activity (Terrell and Morrison, 2019). Active RAF phosphorylates and activates MEK1 (also known as MAP2K1) and MEK2 (also known as MAP2K2), which in turn phosphorylate and activate ERK1 and ERK2. Both cancer- and NS-associated RAF1 mutations cluster around S259 (Table 1). These variants (e.g. S257L) are constitutively dephosphorylated at the inhibitory S259 phospho-site, leading to constitutive activation (Molzan et al., 2010; Pandit et al., 2007; Razzaque et al., 2007).
Fig. 2.

Pathogenesis of Noonan syndrome with loose anagen hair (NS-LAH) and related disorders. (A) In the basal state, MRAS and HRAS/KRAS/NRAS (depicted as RAS) are GDP bound, and RAF1 is held in the autoinhibited confirmation by 14-3-3 binding to phospho-serines 259 and 621. (B) The NS-LAH-associated SHOC2S2G causes aberrant myristylation and membrane localization of SHOC2. Activation of MRAS, which can occur via guanine nucleotide exchange factor activity or via mutation, myristylation of SHOC2, or mutation of the PPP1CB phosphatase, facilitates the formation of a complex between MRAS-GTP and PPP1CB, SHOC2 and SCRIB. RAF1 membrane localization via binding to RAS-GTP puts RAF1 in proximity to the MRAS/SHOC2/PPP1CB complex, which allows PPP1CB to dephosphorylate S259 on RAF1. (C) In the active state, S259-dephosphorylated RAF1 is now able to homo- or heterodimerize with other RAF proteins, activating its kinase activity and triggering the signaling through MEK to ERK. 14-3-3 binding to S621 stabilizes the dimerization.
The biological effects of NS-associated RAF1 mutations have been studied in several models. Cardiomyocytes derived from RAF1S257L iPSCs (iPSC-CMs) display increased surface area and myofibrillar disarray. Importantly, treatment of these myocytes with the MEK inhibitors PD98059 or trametinib resolved the myofibrillar disarray but did not reduce the cell surface area (Jaffre et al., 2019), while treatment with U0126 reduced both the cell surface area and the myofibrillar disarray. Mouse models of S257-altered RAF1 have not yet been reported; however, a knock-in Raf1L613V mouse model displays a variety of NS-like phenotypes, including cardiac hypertrophy and an indolent myeloproliferative neoplasm (Table S3). Importantly, treatment with the MEK inhibitor PD-0325901 (mirdametinib) at weaning reversed the observed cardiac defects in this model (Holter et al., 2019).
RAS superfamily
HRAS, KRAS and NRAS are the ‘founding’ members of a larger superfamily of at least 35 related human proteins, which also includes MRAS, RRAS and RIT1 (Colicelli, 2004). RAS proteins function as binary, guanine nucleotide-regulated, on-off switches. In normal quiescent cells, RAS is predominantly cytosolic, GDP bound and inactive. Growth factors activate RAS GEFs to promote nucleotide exchange and form active RAS-GTP at the plasma membrane. Membrane localization is required for the activation of RAS proteins. This localization is mediated by a combination of C-terminal farnesylation, palmitoylation and electrostatic interactions (Cox et al., 2015). Once in the active, GTP-bound conformation, membrane-bound RAS can bind to a variety of effectors, including the RAF and PI3 kinases, to transmit downstream signals (Pantsar, 2020).
The RAS proteins were first identified in transforming retroviruses and are mutated in 30% of human cancers. In sporadic cancer, RAS proteins are typically mutated at one of three main hotspots, G12, G13 or Q61. These mutations render RAS variably GAP insensitive, thus persistently GTP bound and active (Cox and Der, 2010). By contrast, RAS mutation at A146 increases the fraction of GTP-bound activated RAS by enabling it to spontaneously exchange GDP for GTP in the absence of a RAS GEF (Janakiraman et al., 2010).
KRAS
The RAS protein that is most commonly altered in NS is KRAS. NS-associated KRAS mutations are not found at the oncogenic hotspots; rather, they commonly occur at V14, P34 or T58I (Table 1; Fig. S1). In addition to these single-nucleotide variants, copy-number gain of the KRAS locus is also associated with NS-like phenotypes (Gilbert-Dussardier et al., 2016). Like the cancer-associated mutations, NS-associated mutations result in accumulation of GTP-bound KRAS through enhanced nucleotide exchange or impaired GTP hydrolysis, albeit to a lesser extent than the cancer-associated variants (Gremer et al., 2011; Schubbert et al., 2007).
Several mouse models of KRAS-associated NS have been developed (Table S3). KrasV14I knock-in mice display a variety of NS-like phenotypes, including cardiac hypertrophy and an indolent myeloproliferative neoplasm, with the phenotypes altered by the genetic background of the model. Importantly, prenatal treatment of KrasV14I mice with the MEK inhibitor PD-0325901 (mirdametinib) prevented perinatal lethality and cardiac defects, but postnatal treatment did not reverse these phenotypes (Hernandez-Porras et al., 2014, 2015, 2016), indicating that modulating the MAPK pathway at the right developmental time is essential. KrasP34R and KrasT58I mouse models of NS were recently described. Interestingly, the KrasP34R mice have perinatal lethality due to a lung sacculation defect, while the KrasT58I mice display cardiac hypertrophy (Wong et al., 2020). Treatment studies in these mouse models have not yet been reported.
NRAS
Because KRAS alterations are found in NS, and HRAS alterations are typically found in CS, as discussed below, investigators performed targeted sequencing for the third ‘classical’ RAS isoform, NRAS, in patients with clinically diagnosed NS without previously known mutations. Two recurrent NRAS alterations were identified, T50I and G60E (Table 1; Fig. S1). Both are expected to affect the ability of NRAS to interact with effectors, GEFs or GAPs. Although NRASG60E is a rare somatic variant in JMML, in large part, the NS-associated NRAS variants are different from those identified in NRAS-altered malignancies (Denayer et al., 2012).
NS-associated NRAS mutants have been studied in vitro and in vivo. Overexpression studies in mammalian cells showed that each of these alterations increase signaling through the MAPK pathway. Biochemical studies showed that NRASG60E is insensitive to GAP-stimulated hydrolysis, and molecular dynamics simulations suggest that NRAST50I occupies a more signaling-competent orientation without affecting intrinsic or GAP-stimulated GTP hydrolysis (Cirstea et al., 2010). NRASI24N, which was identified in a Dutch NS cohort, also increased MAPK signaling when overexpressed in mammalian cells. Importantly, the authors of this study also showed that nrasI24N and nrasQ60E, but not nrasT50I, cause the early developmental defect of an oval-shaped yolk sac when introduced as transgenes into zebrafish embryos, and that this could be prevented by MEK inhibition (Runtuwene et al., 2011). No mouse models of NRAS-mutated NS have been developed to date.
RRAS
Protein interaction network analyses suggested that alterations in RAS-related protein (RRAS), a small GTPase that shares 48% amino acid sequence identity to HRAS, might be associated with the RASopathies. Targeted sequencing in a panel of NS patients without previously known mutations identified two RRAS mutations, V55M and G39dup (Flex et al., 2014). Somatic mutations in RRAS were also identified in the circulating leukocytes of JMML patients, including RRASG39dup and RRASQ87L (Flex et al., 2014), a mutation analogous to Q61L in the classical RAS isoforms. These mutations are all expected to affect the guanine-binding ability of RRAS.
In vitro biochemical studies showed that both RRASV55M and RRASG39dup displayed increased GEF-stimulated nucleotide exchange compared to the wild-type protein, and both RRASQ87L and RRASG39dup displayed decreased intrinsic and GAP-stimulated GTP hydrolysis (Flex et al., 2014; Suzuki et al., 1997). When overexpressed in mammalian cells, RRASV55M and RRASG39dup increased RAS/MAPK signaling, and transgenic Caenorhabditis elegans embryos expressing ras-1G27dup, the ortholog of RRASG39dup (Table S2), displayed abnormal vulval morphogenesis (Flex et al., 2014). Recurrent variants in the closely related RRAS2 (also known as TC21) were recently identified in patients with NS. One of these, RRAS2A70T, displays increased GEF-catalyzed guanine nucleotide exchange and impaired intrinsic and GAP-stimulated GTP hydrolysis in vitro, and stimulates the RAS/MAPK pathway in mammalian cells (Capri et al., 2019). NS-associated RRAS and RRAS2 variants have yet to be modeled in mice.
RIT1
In NS patients who did not carry known NS-associated gene mutations, WES revealed recurrent mutations in the small GTPase Ras-like without CAAX 1 (RIT1; Table 1) (Aoki et al., 2013; Bertola et al., 2014; Chen et al., 2014). RIT1 is ubiquitously expressed and plays a role in neuronal survival, proliferation and differentiation. The NS-associated mutations in RIT1 are clustered in the switch II region (Fig. S1). Their expression in Drosophila or zebrafish models results in ectopic wing veins and major:minor axis dysmorphia, respectively (Aoki et al., 2013). Knock-in mouse models of Rit1M90I and Rit1A57G display facial dysmorphia, cardiac hypertrophy and splenomegaly (Castel et al., 2019; Takahara et al., 2019) (Table S3). Importantly, two infants with RIT1-associated NS and heart failure were treated with the MEK inhibitor trametinib and showed clinically significant improvement (Andelfinger et al., 2019), suggesting that drugs targeting the RAS/MAPK pathway might be meaningful treatment options for these patients.
ERK2
ERK2 and its homolog ERK1 are phosphorylated downstream of MEK1/2 in the RAS/MAPK pathway. Recently, ERK2 variants have been identified in patients with clinical features of NS (Motta et al., 2020a). Expression of these variants in mammalian cell cultures increased ERK2 translocation to the nucleus and increased the phosphorylation of ERK targets in a growth factor-dependent manner, whereas their expression in C. elegans caused the multi-vulva phenotype consistent with aberrant RAS/MAPK pathway activity (Motta et al., 2020a). Interestingly, the aberrant MAPK signaling observed in ERK2 variant-expressing mammalian cells was inhibited by the MEK inhibitor trametinib (Motta et al., 2020a), indicating that MEK inhibition may be a therapeutic option for patients with ERK2-associated NS, despite ERK2 being downstream of MEK in the RAS/MAPK pathway.
LZTR1
Several WES studies identified likely pathogenic variants in leucine zipper-like transcriptional regulator 1 (LZTR1) in NS patients without mutations in known NS-associated genes (Johnston et al., 2018; Yamamoto et al., 2015). LZTR1, which is an adaptor for the E3 ubiquitin ligase CUL3, is recurrently altered in schwannomatosis (Piotrowski et al., 2014) and was identified as a tumor suppressor in glioblastoma multiforme (Frattini et al., 2013), and to associate with both autosomal-dominant and autosomal-recessive forms of NS. The mechanism by which LZTR1 affects the RAS/MAPK pathway was not immediately clear; however, overexpression of LZTR1 mutants such as G248R or genetic depletion of endogenous LZTR1 in mammalian cells activated MAPK (Bigenzahn et al., 2018; Motta et al., 2019). Several groups independently discovered that LZTR1 binds to RAS family GTPases, including HRAS, KRAS, NRAS, MRAS and RIT1, facilitating their ubiquitination by CUL3 and subsequent proteasomal degradation (Fig. 3). Decreased LZTR1 expression increases RAS stability and, thus, persistent MAPK signaling (Bigenzahn et al., 2018; Castel et al., 2019; Steklov et al., 2018). The NS-associated mutants in the Kelch domains of LZTR1 disrupt the interaction with RAS, preventing ubiquitination and degradation (Steklov et al., 2018). Deletion of lztr1 in zebrafish causes cardiac hypertrophy (Nakagama et al., 2020), while deletion of Lztr1 in Drosophila causes ectopic wing veins (Bigenzahn et al., 2018), consistent with increased MAPK signaling. In mouse models, homozygous knockout of Lztr1 is embryonic lethal, and Lztr1+/− mice display craniofacial, pulmonary and cardiac abnormalities (Steklov et al., 2018) (Table S3). Prenatal treatment with the MEK inhibitor pimasertib only partially reversed the embryonic lethality of Lztr1−/− and had no effect on the pulmonary phenotype (Sewduth et al., 2020). Additionally, iPSC-CMs from NS patients with LZTR1 alterations are larger than iPSC-CMs from unaffected individuals, suggesting that the cardiac hypertrophy observed in LZTR1-associated NS is linked to increased cardiomyocyte size. Treatment of LZTR1-mutant iPSC-CMs with the MEK inhibitor U0126 only slightly decreased their size (Hanses et al., 2020). Therefore, MEK inhibition might not be effective as a monotherapy in patients with LZTR1-associated NS.
Fig. 3.
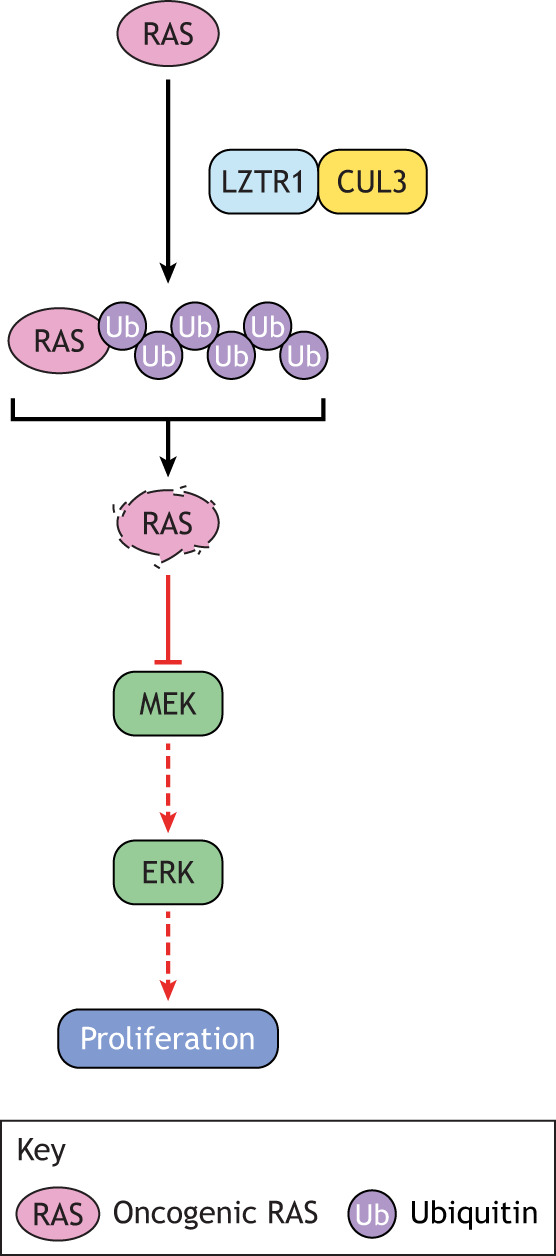
Pathogenesis of LZTR1-associated Noonan syndrome (NS). LZTR1 facilitates the CUL3-mediated polyubiquitination of several RAS isoforms, which leads to their degradation. Loss-of-function alterations in LZTR1, then, increase RAS stability and signaling through the RAS/MAPK pathway, driving NS phenotype.
SHOC2
Alterations in the molecular scaffold SHOC2 are associated with development of the NS spectrum disorder, Noonan syndrome with loose anagen hair [NS-LAH; Online Mendelian Inheritance in Man (OMIM) 607721]. NS-LAH is most commonly caused by the S2G germline pathogenic mutation in SHOC2 (Fig. S1). Patients with NS-LAH have easily pluckable hair, as well as NS-like facial features, growth failure, mild cognitive impairment, and cardiac defects including mitral valve and septal defects. SHOC2 forms a ternary complex with MRAS and the PPP1CB phosphatase that dephosphorylates the RAF kinases to facilitate full activation of the RAS/MAPK pathway (Young et al., 2018) (Fig. 2). The NS-LAH-associated S2G mutation causes SHOC2 myristylation and its mislocalization to the plasma membrane (Cordeddu et al., 2009). Additionally, activating mutations in MRAS (T68I, G23R/V), which augment its binding to SHOC2 and PPP1CB, have also been identified in patients with NS and hypertrophic cardiomyopathy (Higgins et al., 2017; Motta et al., 2020b), indicating a likely genotype–phenotype continuum between NS and NS-LAH.
CBL
Alterations in the E3 ubiquitin ligase CBL are associated with development of a NS spectrum disorder (NS-like disorder with or without JMML; OMIM 613563). CBL mutations most commonly occur at Y371H. Patients with pathogenic germline CBL variants have subtle clinical features, like craniofacial abnormalities, café-au-lait macules, cryptorchidism, and growth and developmental delay, but a striking predisposition for JMML. The loss of the wild-type CBL allele via uniparental disomy is commonly observed in the JMML clone. CBL mutations associated with this NS-like disorder impair its ubiquitin ligase function and consequently impair the ability of CBL to induce the degradation of cell surface receptors upon ligand stimulation, ultimately resulting in prolonged activity of RAS/MAPK signaling (Martinelli et al., 2010; Niemeyer et al., 2010; Perez et al., 2010).
In summary, NS is a disorder caused by alterations in one of an expanding list of RAS/MAPK pathway genes. The severity of symptoms and the potential treatment options vary by genotype, suggesting that genetic testing is informative for prognosis and for personalized treatment strategies for NS spectrum disorder patients.
NS-ML
NS-ML (formerly LEOPARD syndrome; OMIM 151100) is a rare autosomal-dominant syndrome with a poorly characterized prevalence (Sarkozy et al., 2008). Patients have many of the same clinical characteristics as NS patients, with the addition of sensorineural hearing loss, multiple pigmented skin lesions (lentigines) and ocular hypertelorism. Whereas pulmonary valve stenosis is the most common congenital heart defect in NS, hypertrophic cardiomyopathy (HCM) and electrocardiogram abnormalities are more common in NS-ML. In addition, short stature is less common in NS-ML than in NS. Patients with NS-ML are predisposed to neuroblastoma and acute myelogenous leukemia (Gelb and Tartaglia, 1993). NS-ML is caused by germline pathogenic variants in PTPN11, most commonly Y279C, T468M and Q506P (Table 1). The NS-ML PTPN11 variants are therefore different from those that cause NS. NS-ML PTPN11 variants are confined to the protein phosphatase domain and decrease its activity. Importantly, nonsense and frameshift alterations in PTPN11 are associated with metachondromatosis (OMIM 156250), an autosomal-dominant syndrome in which affected patients manifest multiple cartilage tumors (Bowen et al., 2011).
Ptpn11 knockout in the murine germline is embryonic lethal (Yang et al., 2006) (Table S1); however, knocking out Ptpn11 in mesenchymal progenitors results in a metachondromatosis-like phenotype rather than an NS-ML one (Yang et al., 2013), which suggests that some residual activity of PTPN11 is responsible for the NS-ML phenotype. Several other PTPN11-based model systems recapitulate the clinical phenotypes of NS-ML. Indeed, NS-ML-associated PTPN11 variants activate multiple signaling pathways, including RAS/MAPK, PI3K/AKT, FAK and MEK5/ERK5 (Kontaridis et al., 2006; Yu et al., 2014), when overexpressed in mammalian cells, through mechanisms that are both dependent and independent of residual phosphatase activity. Introduction of NS-ML-associated ptpn11 variants in zebrafish increases the major:minor axis ratio and cardiac edema in embryos (Jopling et al., 2007), and causes craniofacial dysmorphia, increased melanophore formation (Bonetti et al., 2014) and cardiac asymmetry defects (Stewart et al., 2010) in adult fish. In addition, expression of NS-ML-associated PTPN11 variants in Drosophila results in ectopic wing vein and rough eye phenotypes (Oishi et al., 2009). PTPN11T468M iPSC-CMs are larger than wild-type ones and have increased MEK phosphorylation, correlating with a potential hypertrophic state (Carvajal-Vergara et al., 2010). Knock-in mouse models of Ptpn11T468M and Ptpn11Y279C have also been developed (Table S4) and display small size, cardiomegaly and facial dysmorphia. The Ptpn11T468M mice have splenomegaly, and the Ptpn11Y279C mice have pectus abnormalities (Marin et al., 2011; Tajan et al., 2014). Postnatal treatment of Ptpn11Y279C mice with the mTOR inhibitor rapamycin, the AKT inhibitor ARQ 092 or the tyrosine kinase inhibitor dasatinib reverses the hypertrophic cardiomyopathy, suggesting that these agents might represent therapeutic options for NS-ML patients (Marin et al., 2011; Wang et al., 2017; Yi et al., 2016). Consistent with this idea, a patient with NS-ML-associated malignant HCM caused by PTPN11Q510E was treated with the mTOR inhibitor everolimus as a bridge to cardiac transplant and experienced improvement in heart failure severity (Hahn et al., 2015). Some patients with NS-ML may therefore require different therapies than those used to treat patients with classical NS.
CS
Unlike NS, CS is quite rare, with prevalence estimates ranging from 1:380,000 in the UK (Giannoulatou et al., 2013) to 1:1,290,000 in Japan (Abe et al., 2012). CS is an autosomal-dominant disorder caused exclusively by pathogenic germline variants in HRAS (Rauen, 2007). Somatic alterations in HRAS have been identified in transitional cell carcinoma of the bladder, thyroid cancers, head and neck squamous cell carcinoma (Moore et al., 2020), and pediatric cancers such as embryonal rhabdomyosarcoma (Vaseva and Yohe, 2020). Unlike the NS-associated variants in genes encoding various RAS proteins, the CS-associated HRAS mutations overlap with cancer-associated ones (Fig. S1). In most cases of CS (80%), the causative HRAS variant is G12S (Table 1) (Gripp et al., 2006). Several rare G12 variants, including G12V, G12D, G12C and G12E, are associated with the development of a more-severe form of CS that is lethal in infancy (Weaver et al., 2014). In addition to single-nucleotide variants, alterations that affect the splicing of HRAS (Guil et al., 2003) have been reported in patients with CS (Pantaleoni et al., 2017). Almost all reported cases of CS are de novo (Rauen, 2007).
Infants with CS frequently have polyhydramnios in utero and are born prematurely but large for their gestational age. Children with CS have coarser facial features than those with NS, as well as macrocephaly, prominent foreheads, epicanthal folds, short noses and low-set, posteriorly rotated ears (Goodwin et al., 2014). Patients fail to thrive in the perinatal period due to swallowing difficulties and increased energy expenditure, and the gastrointestinal involvement progresses to include gastrointestinal reflux, oral aversion and constipation (Leoni et al., 2016). CS patients have unique dermatological abnormalities including cutaneous papillomas (Siegel et al., 2012), as well as a variety of musculoskeletal abnormalities, such as hypotonia, elbow contractures and laxity of the small joints (Leoni et al., 2021). Like NS, CS also causes congenital heart defects, including HCM, septal defects and arrhythmias (Lin et al., 2011). Patients with CS have profound cognitive deficits (Axelrad et al., 2011), as well as anatomic abnormalities of the central nervous system (Gripp et al., 2010). Importantly, they have the highest risk of developing a malignancy of any of the RASopathies patients, particularly rhabdomyosarcoma, neuroblastoma and transitional cell carcinoma of the bladder (Rauen, 2007). Although the average age at diagnosis of this bladder carcinoma in the general population is 73 years, it has been diagnosed in individuals with CS who are 20 years old or younger (Gripp, 2005). CS is therefore a classical cancer predisposition syndrome.
Several models of CS have been developed. Neuronal progenitors from HRASG12S iPSCs display an extended progenitor phase, increased soma size and decreased neurite outgrowth, which may contribute to the neurodevelopmental disorders associated with CS (Rooney et al., 2016). Transgenic zebrafish expressing hrasbG12V in the germline display phenotypes consistent with CS: shortened body length, craniofacial dysmorphia, precocious ossification, myocardial hypertrophy, increased incidence of cancer, and oncogene-induced senescence in the brain and heart (Santoriello et al., 2009). The initial CS mouse model (Table S5), HrasG12V−IRES-β-geo, has a phenotype of hypertension and myocardial and kidney fibrosis, but does not develop tumors (Schuhmacher et al., 2008). A subsequent model, CC-FR-HrasG12V, has a high perinatal mortality rate and CS-like craniofacial defects, and develops benign skin papillomas and malignant angiosarcoma (Chen et al., 2009). Importantly, these mice also have a skeletal muscle myopathy, which can be reversed by postnatal treatment with the MEK inhibitor PD-0325901 (Tidyman et al., 2021). A HrasG12S mouse model has also been established. These mice have craniofacial abnormalities, cardiomegaly, poor weight gain and impaired metabolism, consistent with CS patients (Oba et al., 2018). These mice did not develop papillomas or angiosarcomas.
In summary, the available mouse models of CS demonstrate some, but not all, of the clinical features of CS. Treatment studies in CS models suggest that patients with CS may benefit from RAS/MAPK-specific therapies, as discussed in more detail below.
CFC
Like CS, CFC is a rare RASopathy. Its worldwide prevalence is not known, but is estimated at 1:810,000 in Japan (Abe et al., 2012). CFC is inherited in an autosomal-dominant manner and is caused by germline pathogenic variants in BRAF (75%), MAP2K1 (which encodes MEK1), MAP2K2 (which encodes MEK2) or, rarely, KRAS (Niihori et al., 2006; Rodriguez-Viciana et al., 2006). CFC patients have craniofacial features reminiscent of NS, congenital heart disease, thin, curly and friable hair with sparse eyebrows and eyelashes, hyperkeratotic skin and frequent hemangiomas. These patients also have short stature and fail to thrive, most often due to gastrointestinal reflux and oral aversion. Neurological involvement is common in CFC and manifests as cognitive impairment, learning disabilities, hypotonia, motor and speech delay, and epilepsy. Patients with CFC may also have an increased risk of malignancy (Pierpont et al., 2014; Rauen, 2016).
BRAF
In cancers such as melanoma, BRAF is typically mutated at the V600 codon (Fig. S1), allowing for the BRAF kinase activation in the absence of both dimerization and an active RAS (Freeman et al., 2013). By contrast, the most common CFC-associated mutations in BRAF occur at Q257 (Table 1). This residue is predicted to be a part of both the 14-3-3 interface (Park et al., 2019) and the plasma membrane interface (Travers et al., 2018), but the precise mechanism by which Q257 mutations activate BRAF are currently unknown. BRAFQ257R has increased in vitro kinase activity compared to wild type. Similarly, transfection of BRAFQ257R into mammalian cells yields greater increases in MEK and ERK phosphorylation than transfection of wild-type BRAF (Rodriguez-Viciana et al., 2006).
The biological consequences of CFC-associated BRAF mutations have been studied in several models. BRAFT599R and BRAFQ257R iPSC-CMs are hypertrophic and have altered calcium homeostasis. Treatment with U0126 reversed the hypertrophic phenotype (Josowitz et al., 2016). Expression of BRAFQ257R in zebrafish embryos increased the major:minor axis ratio, a phenotype that was prevented by treatment with the MEK inhibitor CI-1040 (Anastasaki et al., 2009). The first murine model of BRAF-associated CFC (Table S6) was based on a knock-in of the oncogenic BrafV600E mutation. Although this variant is not associated with CFC, the model has been used to study how Braf activation leads to the CFC phenotype in mice. In particular, the Braf +/LSLV600E mice have decreased viability, splenomegaly, facial dysmorphia, small size, hyperactivity and epilepsy (Urosevic et al., 2011). Importantly, these mice also develop spontaneous lung adenomas and melanoma (Urosevic et al., 2011). The second model also involves an alteration in the kinase domain, LSL-Braf L597V and recapitulates the phenotype of Braf+/LSLV600E animals; however, instead of epilepsy, the mice can have spontaneous cerebrovascular accidents. They also develop skin papillomas and intestinal polyps, but no malignant tumors. These mice have a skeletal myopathy, which is due to failed myoblast differentiation (Andreadi et al., 2012). Treating myoblasts derived from the LSL-Braf L597V mice with the MEK inhibitor PD-0325901 rescued myogenic differentiation (Maeda et al., 2021). A Braf Q241R/+ mouse model has also been developed, which corresponds to BRAF Q257R in CFC patients. The phenotypes of these mice vary across genetic backgrounds, but in general present as decreased viability, small size, cardiomegaly, lymphangiectasia, impairments in contextual fear conditioning and decreased esophageal motility. Prenatal exposure of Braf Q241R/+ mice to PD-0325901 prevented perinatal lethality, and prenatal exposure to the MEK inhibitor MEK162 prevented the esophageal dysmotility (Inoue et al., 2014; Moriya et al., 2015).
MEK1/2
MEK1 and MEK2, the products of MAP2K1 and MAP2K2, respectively, are dual-specificity kinases that phosphorylate ERK1 and ERK2 in the RAS/MAPK signaling pathway. MEK1/2 are the only phosphorylation targets of active RAF kinases (Roskoski, 2012). The majority of the cancer- and CFC-associated variants in MEK1/2 occur at the interface of the auto-inhibitory region, e.g. MEK2K57N, and the kinase domain, e.g. MEK1Y130C and MEK2E203K (Table 1; Fig. S1). These mutations increase the in vitro kinase activity (Ordan et al., 2018), and their expression in mammalian cells increases signaling through the RAS/MAPK pathway, which can be blocked by U0126 (Rodriguez-Viciana et al., 2006; Senawong et al., 2008). However, some of the CFC-associated MEK mutations confer resistance to the MEK inhibitor trametinib (Emery et al., 2017), indicating that MEK inhibitors may not always be appropriate therapeutics for patients with MEK-associated CFC, despite the preclinical finding that elongated zebrafish embryos, a phenotype caused by expression of map2k1Y130C, can be reversed by the MEK inhibitors CI-1040 and PD-0325901 (Anastasaki et al., 2009). Expression of Dsor1Y130C, the MAP2K1 ortholog in Drosophila (Table S2), leads to larval cuticle deficits and ectopic wing veins (Jindal et al., 2017). In addition, a knock-in mouse model of Map2k1Y130C has been developed (Table S6). The Map2k1Y130C mice have cardiac and craniofacial abnormalities, increased activated astrocytes in the sensory cortex and hippocampus, as well as increased oligodendrocyte precursors in the sensory cortex (Aoidi et al., 2018). These neuropathological changes could be important for the pathogenesis of the neurological phenotypes of CFC; however, neurobehavioral testing has not yet been performed with this mouse model. No treatment studies in these mice have been reported to date.
In summary, CFC is a rare RASopathy that is caused by alterations in one of several components of the RAS/MAPK pathway. The clinical features of CFC, as well as the associated genotypes, overlap with those of NS.
LS
LS (OMIM 611431) is a rare, autosomal-dominant disorder caused by germline pathogenic variants in SPRED1 (Brems et al., 2007). The prevalence of LS is currently not well characterized (Brems et al., 2012), but variants in SPRED1 are commonly identified in patients with a clinical diagnosis of NF1 who do not have detectable alterations in NF1 (Messiaen et al., 2009). LS patients have skin manifestations, such as café-au-lait macules similar to those of NF1 patients; however, neurofibromas, which are common in NF1, are not a feature of LS. Additionally, some LS patients have dysmorphic facial features similar to those of NS and musculoskeletal features, including pectus excavatum or carinatum and unilateral postaxial polydactyly. Neurological features are also common and include learning disabilities, attention deficit hyperactivity disorder and developmental delay. Lipomas are common and malignancies such as acute myelogenous leukemia have been reported (Brems et al., 2012).
SPRED1 is a molecular scaffold that binds to the NF1 protein and recruits it to the plasma membrane, where NF1 exerts its RAS GAP function. The LS-associated SPRED1 variants, including nonsense, frameshift and missense mutations, prevent NF1 membrane localization (Yan et al., 2020). Expression of LS-associated SPRED1 variants in mammalian cells increased signaling through the RAS/MAPK pathway (Brems et al., 2007).
Spred1 knockout mice are viable but display airway hyper-responsiveness and facial dysmorphia (Denayer et al., 2008; Inoue et al., 2005). Neurobehavioral testing demonstrates that Spred1 knockout mice have impaired spatial learning and memory (Borrie et al., 2021a), as well as altered social and communicative behavior (Borrie et al., 2021b) (Table S7). Importantly, postnatal MEK inhibition with PD-0325901 does not affect the spatial learning and memory phenotypes but does rescue the altered social behavior phenotypes (Borrie et al., 2021a,b). Knockout of the related Spred2 results in an obsessive-compulsive behavioral phenotype (Ullrich et al., 2018), as well as impaired growth, cardiac hypertrophy, splenomegaly and craniofacial malformation (Ullrich et al., 2019). Interestingly, a recent report described a case series of patients with an autosomal-recessive NS-like phenotype resulting from loss of SPRED2 function (Motta et al., 2021), further validating SPRED2 as a RASopathy gene.
LS, therefore, is a RASopathy with clinical features similar to NF1. The similarity between these disorders is likely to be due to the dependence of NF1 on SPRED1 for GAP activity (Stowe et al., 2012). Treatment studies in LS animal models indicate that some, but not all, of the features of LS may be ameliorated by MEK inhibition.
CCLA
Germline alterations in several RAS/MAPK genes have been identified in patients with lymphatic anomalies. In some cases, the lymphatic anomaly is the only RASopathy-associated clinical manifestation (Sevick-Muraca and King, 2014). Recently, a recurrent alteration in ARAF, S214P, was identified in patients with CCLA (Li et al., 2019). This ARAF residue is analogous to S259 in RAF1, which is phosphorylated and bound by 14-3-3 in the auto-inhibited state (Fig. 2). Somatic ARAF alterations at S214 are also found in lung adenocarcinoma (Imielinski et al., 2014). Expressing ARAFS214P in mammalian cells decreases the association with 14-3-3 and increases ERK phosphorylation (Li et al., 2019). Additionally, human primary dermal lymphatic endothelial cells expressing ARAFS214P have increased lymphangiogenic capacity compared to wild type, a phenotype that is reversed by treatment with the MEK inhibitor trametinib (Li et al., 2019). Expression of arafS214P in zebrafish leads to dilated lymphatic vessels, which can be reversed by treatment with the MEK inhibitor cobimetinib (Li et al., 2019). Moreover, a patient with CCLA associated with ARAFS214P was treated with trametinib on a compassionate-use basis, which improved pulmonary function and decreased oxygen requirement, indicating meaningful clinical improvement (Li et al., 2019).
CM-AVM
CM-AVM is an autosomal-dominant disorder affecting 1:100,000 people of northern European origin (Revencu et al., 2008). Approximately 50% of CM-AVM cases are caused by pathogenic germline variants in RASA1, which encodes the p120-RAS GAP (Eerola et al., 2003; Revencu et al., 2008), with the remaining caused by other RAS/MAPK pathway genes such as EPHB4 (Amyere et al., 2017). The defining characteristic of CM-AVM syndrome is the presence of multifocal cutaneous capillary malformations, with or without concurrent arteriovenous malformations in the skin, muscle, bone or internal organs, although some patients also have cardiac defects and may also have an increased risk of cancer (Boon et al., 2005). Morpholinos targeting both zebrafish RASA1 alleles, rasa1 (also known as rasa1a) and rasa2, cause an embryonic vascular defect marked by an increase in mTOR signaling, which was rescued by treatment with the PI3K/mTOR inhibitor NVP-BEZ235 (Kawasaki et al., 2014). In mice, homozygous knockout of the RAS GAP domain exons of Rasa1 either in the germline or exclusively in the blood vessel endothelial compartment is lethal at mid-gestation due to the resulting vascular defects (Lapinski et al., 2007, 2012; Lubeck et al., 2014). Treatment studies have not yet been reported in these mouse models.
SYNGAP1 syndrome
SYNGAP1 syndrome is an autosomal-dominant disorder caused by germline loss-of-function alterations in SYNGAP1, which encodes a RAS GAP that is part of a subfamily that also includes DAB2IP, RASAL2 and RASAL3. The exact prevalence of the disorder is unknown, but germline SYNGAP1 alterations are found in 0.5-1% of children with neurodevelopmental disorders. The clinical features of SYNGAP1 syndrome include global developmental delay, hypotonia, seizures, skeletal abnormalities, strabismus, constipation, failure to thrive and autism spectrum disorders (Parker et al., 2015). Homozygous knockout of Syngap1 in mice is embryonic lethal; heterozygous mice display profound neurobehavioral changes (Nakajima et al., 2019). Both increased RAS/MAPK signaling and long-term potentiation impairment were observed in the hippocampal slices from heterozygous mice, but treatment with the MEK inhibitor PD-0325901 did not improve the long-term potentiation impairment (Kopanitsa et al., 2018).
Emerging RASopathy genes
Next-generation sequencing has identified several genes that were altered in patients with RASopathies; however, the functional validation of these as RASopathy-associated genes is not yet complete. These include RASA2, SPRY1, and MAP3K8 (Chen et al., 2014). Here, we discuss RASopathy genes that have been identified or validated since this topic was originally defined (Tidyman and Rauen, 2016a,b).
KAT6B (also known as MYST4) is a histone H3 lysine 23 (H3K23ac) acetyltransferase. The epigenetic implications of H3K23 acetylation are not well studied. A balanced translocation predicted to cause loss of KAT6B function was identified in a patient with NS. In addition to decreased H3K23 acetylation, cells with decreased expression of KAT6B show increased signaling through the RAS/MAPK pathway. Mice that are homozygous for a hypomorphic mutation in Kat6b show features reminiscent of NS: failure to thrive, craniofacial dysmorphia and brain abnormalities (Kraft et al., 2011). However, alterations in KAT6B are also observed in several other developmental disorders with features similar to those of the RASopathies, including Say-Barber-Biesecker-Young-Simpson syndrome (OMIM 603736) (Campeau et al., 2012), so further validation is needed.
Individuals with 6p-interstitial deletion syndrome (OMIM 612582) have NS-like clinical features, such as short stature, intellectual disability, craniofacial dysmorphia and cardiac abnormalities. The RAS-responsive element-binding protein 1 (RREB1) gene is deleted in individuals with this syndrome. RREB1 is a transcription factor that binds to RAS-responsive elements in the promoters of genes encoding components of the MAPK pathway, such as HRAS, MAP2K2 and FGFR4. Promoter-bound RREB1 represses the expression of these MAPK pathway genes in part through its recruitment of the CtBP1 transcriptional co-repressor complex. Rreb+/− mice are viable and display short stature, craniofacial dysmorphia and cardiac hypertrophy. Increased Hras expression and increased ERK phosphorylation are observed in cardiac tissue from these mice. These results suggest that RREB1, like KAT6B, may function as a RASopathy gene through a transcriptional regulation mechanism (Kent et al., 2020).
Recurrent alterations in the RHO family GTPase CDC42 have recently been identified in patients with clinical features of NS. These alterations cause vulvar phenotypes consistent with aberrant RAS/MAPK activity when expressed in C. elegans (Fig. 1), although their direct impact on ERK phosphorylation has not yet been assessed (Martinelli et al., 2018). This observation opens up the possibility of RASopathy-causing variants in other subfamilies of GTPases. Importantly, variants in another RHO family GTPase, RAC1, are associated with developmental delay (Reijnders et al., 2017).
The 14-3-3 family of proteins play a role both in RAF auto-inhibition maintenance and in their dimerization (Fig. 2). Recently, a germline variant in 14-3-3zeta (YWHAZ) was identified in a patient with a clinical diagnosis of CFC. Expression of this variant in Xenopus tropicalis increased BRAF and RAF1 binding, ERK phosphorylation and decreased body length, consistent with YWHAZ functioning as a RASopathy gene (Popov et al., 2019).
The genes discussed here have been identified as altered in small subsets of patients. In addition to these, several genes encoding components of the RAS/MAPK pathway also merit further study, even though they have not yet been identified as RASopathy genes. As new candidate RASopathy genes continue to emerge, existing RASopathy clinical sequencing panels may need to be expanded.
Targeting aberrant RAS signaling in the RASopathies
Recently, the U.S. Food and Drug Administration (FDA) approved the MEK inhibitor selumetinib for the treatment of some patients with NF1, but no treatments are currently available for patients with non-NF1 RASopathies. In several of the mouse or other model system studies described above, a MEK inhibitor was used as a prenatal preventative therapy or postnatal treatment, raising the possibility that MEK inhibitors may be effective in the treatment of non-NF1 RASopathies. For example, in the Sos1E846K and KrasV14I mouse models of NS (Chen et al., 2010; Hernandez-Porras et al., 2014) and the BrafQ241R mouse model of CFC (Inoue et al., 2014), MEK inhibitors given prenatally effectively prevented the manifestations of the modeled RASopathy. MEK inhibition also reversed RASopathy phenotypes in PTPN11- and NRAS-associated zebrafish models of NS (Bonetti et al., 2014; Runtuwene et al., 2011), BRAF- and MAP2K1-associated zebrafish models of CFC (Anastasaki et al., 2009), and ARAF-associated zebrafish models of CCLA (Li et al., 2019). Excitingly, MEK inhibitors were effective in treating some, if not all, of the other RASopathy manifestations, in the Ptpn11D61G and Raf1L613V knock-in mouse models of NS (Lee et al., 2014; Wu et al., 2011).
However, there are some murine models in which MEK inhibition had no effect on the RASopathy phenotype. MEK inhibitors were not effective when given prenatally to the Lztr1 knockout model of NS (Sewduth et al., 2020) and the Syngap1 heterozygous knockout mouse model of SYNGAP1 syndrome (Kopanitsa et al., 2018). In addition, despite the fact that prenatal administration prevented RASopathy phenotypes, postnatal administration did not ameliorate the phenotypes of KrasV14I knock-in NS mice (Hernandez-Porras et al., 2014), indicating how important the timing of the intervention may be. Responses to MEK inhibition in Spred1 knock-out mouse models of LS underline the importance of careful preclinical studies, because although the social behavior phenotypes of these mice were affected by MEK inhibition, the spatial learning and memory phenotypes were not (Borrie et al., 2021a,b). Most excitingly, however, patients with RIT1-associated NS and HCM had clinical improvement in their heart failure symptoms when treated with trametinib (Andelfinger et al., 2019), providing the strongest evidence to date that MEK inhibition may be an effective treatment strategy for patients with RASopathies.
Other drug classes are also effective in models of RASopathies, particularly NS-ML. As discussed above, postnatal treatment of NS-ML mouse models with inhibitors of the PI3K/mTOR/AKT pathway or with the tyrosine kinase inhibitor dasatinib reversed cardiomyopathy phenotypes (Marin et al., 2011; Wang et al., 2017; Yi et al., 2016), and treatment of a NS-ML patient with the mTOR inhibitor everolimus provided clinical benefit (Hahn et al., 2015). Treatment of a PTPN11-associated NS mouse model with dasatinib also reversed cardiomyopathy (Yi et al., 2016). PI3K/mTOR inhibitors are also effective in a RASA1-associated CM-AVM zebrafish model (Emery et al., 2017). Inhibition of other pathways, then, may be more beneficial than inhibition of the RAS/MAPK pathway in some RASopathies, although this hypothesis has yet to be formally tested.
Recent work by the National Cancer Institute's RAS initiative, industry and academic laboratories has yielded a large number of novel agents for the treatment of cancers driven by RAS/MAPK pathway alterations (Moore et al., 2020). These agents could represent treatment options for patients with RASopathies and fall into three classes: inhibitors of RAS activation, direct RAS inhibitors and inhibitors of the MAPK pathway (Fig. 4).
Fig. 4.
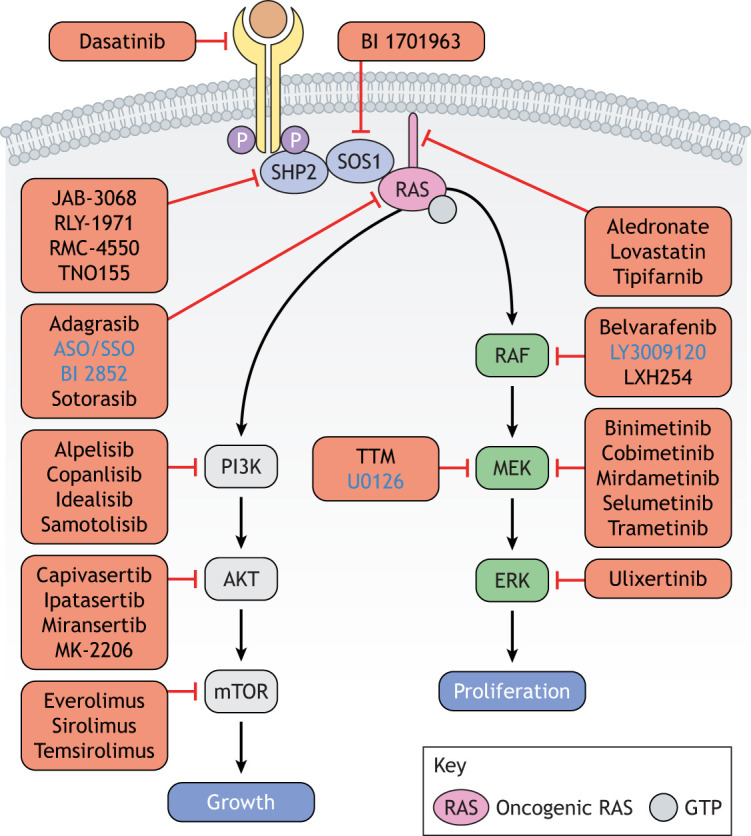
Treating RASopathies. Researchers have developed several classes of drugs that target RAS activation and components of the MAPK or the PI3K/AKT pathway. Drugs that inhibit RAS activation include those that target RAS directly (e.g. sotorasib), inhibit RAS membrane localization (e.g. tipifarnib), prevent the activity of SHP2 (e.g. RMC-4550), inhibit the interaction of RAS with its exchange factor SOS1 (e.g. BI 1701963), or block the kinase activity of receptor tyrosine kinases (e.g. dasatinib). Please see the text for more details. Blue font indicates preclinical tool compounds; black font indicates a drug in clinical development. Several have been FDA approved for the treatment of cancer, including sotorasib, trametinib and copanlisib, among others, and could potentially be used for the treatment of RASopathies. TTM, tetrathiomolybate.
Inhibitors of RAS activation
Inhibitors of RAS activation include compounds that inhibit RAS prenylation or its interactions with proteins necessary for its activation, such as SHP2 and SOS1. As discussed above, C-terminal prenylation is required for RAS membrane localization and, thus, activation. Both the statins, e.g. atorvastatin and lovastatin, and the N-bisphosphonates, e.g. alendronate and pamidronate, are metabolic inhibitors that prevent the synthesis of prenylation substrates. Either statins or N-bisphosphonates, then, might prevent RAS prenylation and, thus, its activation in patients with RASopathies (Baranyi et al., 2020). These drugs are well studied and relatively well tolerated for the treatment of chronic conditions such as hypercholesterolemia and osteoporosis, respectively, and, thus, represent potential long-term treatment options for adult and pediatric patients with RASopathies. Importantly, lovastatin reverses learning defects in the Ptpn11D61G/+ mouse model of NS (Lee et al., 2014), but it is not clear if this response is associated with a decrease in RAS activation. However, based on these results, a phase 3 clinical trial of a statin for treatment of growth and bone abnormalities in children with NS is currently underway (NCT02713945). The statins have not been studied in other RASopathy models. By contrast, the farnesyl transferase inhibitors (FTIs), such as tipifarnib, directly inhibit the enzyme responsible for farnesylation, a type of prenylation. NRAS and KRAS bypass the requirement for farnesylation by undergoing geranyl geranylation, an alternative post-translational modification that also allows for membrane localization. However, HRAS, the RAS isoform affected in CS, cannot be alternatively modified, and, thus, its membrane localization and cellular function are suppressed by FTIs (Cox et al., 2015). FTIs may therefore represent a potential therapeutic approach for CS patients specifically.
Additional targeted agents that prevent RAS activation have recently been identified. BI 1701963, which inhibits the interaction between RAS and SOS1 to inhibit RAS activation, is in clinical development for solid tumors harboring a KRAS mutation, but it also represents a potential RASopathy therapeutic (Hillig et al., 2019), although it has not yet been tested in RASopathy models. Allosteric inhibitors of wild-type PTPN11 that hold the phosphatase in the auto-inhibited confirmation and therefore limit signaling through the RAS/MAPK pathway are currently in clinical trials for cancer (Yuan et al., 2020). These drugs, which include RMC-4630, have not yet been tested in RASopathy models, but are promising potential therapeutics for all RASopathies with aberrant MAPK activity except for PTPN11-associated NS, because these inhibitors have decreased binding affinity for variant PTPN11.
Direct RAS inhibitors
Direct inhibition of RAS proteins was considered impossible until the development of several direct covalent inhibitors of KRASG12C, including sotorasib (AMG 510) (Hong et al., 2020), which has recently been FDA approved for non-small cell lung cancer harboring KRASG12C. These drugs have not yet been tested in children and are likely to have limited utility in individuals with RASopathies, mainly because KRASG12C is uncommon in the RASopathies (Sebastian et al., 2021). Conversely, BI 2825, a preclinical tool compound that inhibits KRAS regardless of a particular mutation, represents an exciting potential therapeutic for RASopathy patients (Kessler et al., 2019). Agents that modify RAS expression have also been developed. These include cell-permeable antisense oligonucleotides that decrease expression of KRAS (Ross et al., 2017) and splice-switching oligonucleotides that induce the skipping of exon 2 in the maturation of HRAS mRNA, yielding an N-terminally truncated protein unable to bind guanine nucleotides (Hartung et al., 2016). These oligonucleotides have not yet been tested clinically or in RASopathy models.
RAS/MAPK pathway inhibitors
The MEK inhibitor selumetinib is the first FDA-approved treatment for patients with a RASopathy, in particular, pediatric patients with NF1 who have symptomatic, inoperable plexiform neurofibromas (Gross et al., 2020b). The clinical benefit of selumetinib in NF1 has inspired the preclinical testing of MEK inhibitors in non-NF1 RASopathies, as discussed above. However, in addition to MEK inhibitors, other agents inhibit signaling through the RAS/MAPK pathway. For example, the MEK1/2 kinases require copper as a cofactor for their kinase activity. Copper chelation with ammonium tetrathiomolybdate, which is used clinically for individuals with Wilson disease, might represent a novel method by which to decrease MAPK signaling in individuals with RASopathies (Brady et al., 2017). Indeed, the tool compound U0126, which inhibits MEK in part due to its ability to chelate copper, is effective in reversing RASopathy phenotypes in iPSCs derived from patients with RAF1-associated NS and in BRAF- and MEK-associated CFC, as discussed above.
Several pan-RAF inhibitors are in clinical development, including LXH254 (Monaco et al., 2021) and belvarafenib (Yen et al., 2021). These drugs inhibit MAPK signaling in the absence of BRAFV600E, while the BRAFV600E-specific inhibitors, such as vemurafenib, paradoxically activate the MAPK pathway if administered in the absence of this mutation (Peng et al., 2015). ERK inhibitors, such as ulixertinib, are also in clinical development in pediatrics and may benefit individuals with RASopathies (Sullivan et al., 2018). The pan-RAF and ERK inhibitors also merit testing in RASopathy models.
Taken together, the preclinical evaluation of targeted agents specific for the RAS/MAPK pathway in models of RASopathies has revealed that MEK inhibitors show therapeutic promise. The key remaining challenges to be investigated in RASopathy model systems include careful characterization of which particular features of which RASopathies might be reversed with MEK inhibition. Thorough preclinical testing of novel RAS/MAPK targeted agents could also yield treatment options for patients.
Conclusions
In the past several years, next-generation sequencing has enabled the identification of several new RASopathy genes, limiting the number of RASopathy cases without a genetic diagnosis. However, the expansion of patient genotyping has introduced some complexity into the discussion of what defines a RASopathy. For example, mosaic alterations affecting the RAS/MAPK pathway have been identified in congenital melanocytic, keratinocytic epidermal and sebacceous nevi. This discovery led to these birthmarks being termed ‘cutaneous mosaic RASopathies’ (Hafner and Groesser, 2013). Although these disorders are not established RASopathies like those discussed above, patients with cutaneous mosaic RASopathies are at risk of developing RAS-associated malignancies, and, furthermore, established RASopathies may occur in mosaicism. Patients with cutaneous mosaic RASopathies may therefore benefit from being included in RASopathy treatment protocols.
Defining RASopathy genes is similarly challenging. The generally accepted definition of a RASopathy gene is a gene involved in the RAS/MAPK pathway that is altered in the germline of a patient diagnosed with a RASopathy. Typically, in model systems, the expression of the RASopathy-associated variant leads to aberrant RAS/MAPK signaling (Fig. 1). However, some recently identified RASopathy genes have challenged this definition. For example, A2ML1, a gene altered in a patient with a clinical diagnosis of NS (Chen et al., 2014), encodes a protease inhibitor and component of the complement cascade of the immune system. This protein has no known link to the RAS/MAPK pathway and functional validation of the variants have yielded conflicting results, such that its role as a RASopathy gene has been questioned (Brinkmann et al., 2021). In addition, WES in a family affected by hereditary pancreatic cancer revealed a nonsense mutation in the gene encoding the small GTPase Rab-like protein 3 (RABL3), resulting in a truncated protein. Pancreatic cancer is not a RASopathy-associated malignancy; however, zebrafish models of hereditary pancreatic cancer with homozygous truncating mutations in rabl3 displayed stunted growth, craniofacial dysmorphia, abnormal swimming behavior and decreased bone mineralization consistent with a RASopathy-like phenotype (Nissim et al., 2019), raising the possibility that RABL3 is a RASopathy gene. Rigorous and systematic functional validation of candidate RASopathy genes is therefore essential for the development of accurate RASopathy diagnostic sequencing panels.
Currently, many patients receive a RASopathy diagnosis from prenatal genetic testing. In the majority of cases, the identification of a germline pathogenic variant in a RASopathy gene reveals not only the patient's genotype, but also the clinical diagnosis. For PTPN11, the specific variant can distinguish between a NS and a NS-ML diagnosis. However, both NS and CFC are associated with the same KRAS variants. The clinical diagnosis, then, depends upon the patient's clinical course. Future work will be needed to identify cooperating genetic variants or environmental conditions to ensure the accuracy of prenatal genetic diagnosis.
Recently, there has been a dramatic increase in the number of drugs that inhibit the RAS/MAPK pathway (Fig. 4). These were originally designed for the treatment of malignancies but may provide clinical benefit for RASopathy patients. The barriers to the initiation of clinical trials for patients with RASopathies include disease rarity, inter-patient phenotypic variability, lacking knowledge of natural history without treatment, incomplete knowledge of which clinical manifestations might be treated by postnatal administration of a RAS/MAPK-targeted drug, and the need for long-term drug tolerability. The evaluation of novel therapeutics in RASopathy models is thus critical for the development of interventional clinical trials for the non-NF1 RASopathies.
The available preclinical data suggest that MEK inhibition may be an effective treatment strategy in some, but not all, RASopathies. NS-ML models are sensitive to inhibition of the PI3K/mTOR pathway (Marin et al., 2011; Wang et al., 2017), and responses to MEK inhibition were limited in some models (Borrie et al., 2021a; Hernandez-Porras et al., 2014; Kopanitsa et al., 2018; Sewduth et al., 2020). In addition, some MEK inhibitors may not be effective in MEK-associated CFC because the single-nucleotide variant associated with the RASopathy could prevent inhibitor binding (Emery et al., 2017).
There are several important questions related to the use of MEK inhibitors for patients with RASopathies that can be answered using RASopathy models, beyond predicting efficacy. For example, the various MEK inhibitors in clinical development, such as selumetinib, trametinib, binimetinib (MEK162), cobimetinib and mirdametinib (PD-0325901) (Fig. 4), have not been compared in the same model to determine which may be most effective in a particular disease context. Additionally, for postnatal dosing, a drug is usually given by gavage at weaning. An alternative, ‘MEKi-in-milk’ protocol, in which the drug is given to lactating mothers, has been employed effectively in mouse models of NF1 (Jecrois et al., 2021), and may represent a strategy to deliver MEK inhibitors at an earlier developmental stage in non-NF1 RASopathy models.
The choice of drug dose is also important. For NF1, clinical efficacy is observed at doses lower than the recommended adult cancer dose, which also do not completely abrogate ERK phosphorylation (Jousma et al., 2015). Lower doses of MEK inhibitors may therefore be effective and have greater long-term tolerability in models of non-NF1 RASopathies. The use of the novel agents discussed above (Fig. 4), which can be selected specifically for the patient's genotype, may also improve tolerability.
The ultimate goal of all of these efforts – improved genetic diagnosis, effective preclinical models and accurate drug development – is to provide meaningful therapeutic options for RASopathy patients.
Supplementary Material
Acknowledgments
This article is part of a collection ‘The RAS Pathway: Diseases, Therapeutics and Beyond’, which was launched in a dedicated Special Issue guest edited by Donita Brady and Arvin Dar. See related articles in this collection at https://journals.biologists.com/dmm/collection/5089/The-RAS-Pathway.
Acknowledgements
The authors are grateful to L. Schoyer, L. Schill, B. Stronach, P. A. Randazzo, D. K. Morrison, R. L. Kortum, A. Gross, G. Ney, D. R. Stewart and K. L. Rossman for helpful discussions and review of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
This work was supported by the Intramural Research Program of the National Cancer Institute.
References
- Abe, Y., Aoki, Y., Kuriyama, S., Kawame, H., Okamoto, N., Kurosawa, K., Ohashi, H., Mizuno, S., Ogata, T., Kure, S.et al. (2012). Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: findings from a nationwide epidemiological survey. Am. J. Med. Genet. A 158A, 1083-1094. 10.1002/ajmg.a.35292 [DOI] [PubMed] [Google Scholar]
- Amyere, M., Revencu, N., Helaers, R., Pairet, E., Baselga, E., Cordisco, M., Chung, W., Dubois, J., Lacour, J.-P., Martorell, L.et al. (2017). Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation 136, 1037-1048. 10.1161/CIRCULATIONAHA.116.026886 [DOI] [PubMed] [Google Scholar]
- Anastasaki, C., Estep, A. L., Marais, R., Rauen, K. A. and Patton, E. E. (2009). Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum. Mol. Genet. 18, 2543-2554. 10.1093/hmg/ddp186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andelfinger, G., Marquis, C., Raboisson, M.-J., Théoret, Y., Waldmüller, S., Wiegand, G., Gelb, B. D., Zenker, M., Delrue, M.-A. and Hofbeck, M. (2019). Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK-inhibition. J. Am. Coll. Cardiol. 73, 2237-2239. 10.1016/j.jacc.2019.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadi, C., Cheung, L.-K., Giblett, S., Patel, B., Jin, H., Mercer, K., Kamata, T., Lee, P., Williams, A., McMahon, M.et al. (2012). The intermediate-activity L597VBRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 26, 1945-1958. 10.1101/gad.193458.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoidi, R., Houde, N., Landry-Truchon, K., Holter, M., Jacquet, K., Charron, L., Krishnaswami, S. R., Yu, B. D., Rauen, K. A., Bisson, N.et al. (2018). Mek1Y130C mice recapitulate aspects of human cardio-facio-cutaneous syndrome. Dis. Model. Mech. 11, dmm031278. 10.1242/dmm.031278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki, Y., Niihori, T., Banjo, T., Okamoto, N., Mizuno, S., Kurosawa, K., Ogata, T., Takada, F., Yano, M., Ando, T.et al. (2013). Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am. J. Hum. Genet. 93, 173-180. 10.1016/j.ajhg.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, T., Mohi, M. G., Ismat, F. A., Bronson, R. T., Williams, I. R., Kutok, J. L., Yang, W., Pao, L. I., Gilliland, D. G., Epstein, J. A.et al. (2004). Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10, 849-857. 10.1038/nm1084 [DOI] [PubMed] [Google Scholar]
- Araki, T., Chan, G., Newbigging, S., Morikawa, L., Bronson, R. T. and Neel, B. G. (2009). Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc. Natl. Acad. Sci. USA 106, 4736-4741. 10.1073/pnas.0810053106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrad, M. E., Schwartz, D. D., Katzenstein, J. M., Hopkins, E. and Gripp, K. W. (2011). Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am. J. Med. Genet. C Semin. Med. Genet. 157C, 115-122. 10.1002/ajmg.c.30299 [DOI] [PubMed] [Google Scholar]
- Baranyi, M., Buday, L. and Hegedus, B. (2020). K-Ras prenylation as a potential anticancer target. Cancer Metastasis Rev. 39, 1127-1141. 10.1007/s10555-020-09902-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger, L.-F., Roy, S., Tremblay, M., Brott, B., Steff, A.-M., Mourad, W., Hugo, P., Erikson, R. and Charron, J. (2003). Mek2 is dispensable for mouse growth and development. Mol. Cell. Biol. 23, 4778-4787. 10.1128/MCB.23.14.4778-4787.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola, D. R., Yamamoto, G. L., Almeida, T. F., Buscarilli, M., Jorge, A. A. L., Malaquias, A. C., Kim, C. A., Takahashi, V. N. V., Passos-Bueno, M. R. and Pereira, A. C. (2014). Further evidence of the importance of RIT1 in Noonan syndrome. Am. J. Med. Genet. A 164A, 2952-2957. 10.1002/ajmg.a.36722 [DOI] [PubMed] [Google Scholar]
- Bigenzahn, J. W., Collu, G. M., Kartnig, F., Pieraks, M., Vladimer, G. I., Heinz, L. X., Sedlyarov, V., Schischlik, F., Fauster, A., Rebsamen, M.et al. (2018). LZTR1 is a regulator of RAS ubiquitination and signaling. Science 362, 1171-1177. 10.1126/science.aap8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti, M., Paardekooper Overman, J., Tessadori, F., Noël, E., Bakkers, J. and den Hertog, J. (2014). Noonan and LEOPARD syndrome Shp2 variants induce heart displacement defects in zebrafish. Development 141, 1961-1970. 10.1242/dev.106310 [DOI] [PubMed] [Google Scholar]
- Boon, L. M., Mulliken, J. B. and Vikkula, M. (2005). RASA1: variable phenotype with capillary and arteriovenous malformations. Curr. Opin. Genet. Dev. 15, 265-269. 10.1016/j.gde.2005.03.004 [DOI] [PubMed] [Google Scholar]
- Borrie, S. C., Horner, A. E., Yoshimura, A., Legius, E., Kopanitsa, M. V. and Brems, H. (2021a). Impaired instrumental learning in Spred1−/− mice, a model for a rare RASopathy. Genes Brain Behav. 20, e12727. 10.1111/gbb.12727 [DOI] [PubMed] [Google Scholar]
- Borrie, S. C., Plasschaert, E., Callaerts-Vegh, Z., Yoshimura, A., D'Hooge, R., Elgersma, Y., Kushner, S. A., Legius, E. and Brems, H. (2021b). MEK inhibition ameliorates social behavior phenotypes in a Spred1 knockout mouse model for RASopathy disorders. Mol. Autism 12, 53. 10.1186/s13229-021-00458-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen, M. E., Boyden, E. D., Holm, I. A., Campos-Xavier, B., Bonafé, L., Superti-Furga, A., Ikegawa, S., Cormier-Daire, V., Bovee, J. V., Pansuriya, T. C.et al. (2011). Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 7, e1002050. 10.1371/journal.pgen.1002050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady, D. C., Crowe, M. S., Greenberg, D. N. and Counter, C. M. (2017). Copper chelation inhibits BRAFV600E-driven melanomagenesis and counters resistance to BRAFV600E and MEK1/2 inhibitors. Cancer Res. 77, 6240-6252. 10.1158/0008-5472.CAN-16-1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brems, H., Chmara, M., Sahbatou, M., Denayer, E., Taniguchi, K., Kato, R., Somers, R., Messiaen, L., De Schepper, S., Fryns, J.-P.et al. (2007). Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 39, 1120-1126. 10.1038/ng2113 [DOI] [PubMed] [Google Scholar]
- Brems, H., Pasmant, E., Van Minkelen, R., Wimmer, K., Upadhyaya, M., Legius, E. and Messiaen, L. (2012). Review and update of SPRED1 mutations causing Legius syndrome. Hum. Mutat. 33, 1538-1546. 10.1002/humu.22152 [DOI] [PubMed] [Google Scholar]
- Brinkmann, J., Lissewski, C., Pinna, V., Vial, Y., Pantaleoni, F., Lepri, F., Daniele, P., Burnyte, B., Cuturilo, G., Fauth, C.et al. (2021). The clinical significance of A2ML1 variants in Noonan syndrome has to be reconsidered. Eur. J. Hum. Genet. 29, 524-527. 10.1038/s41431-020-00743-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, W., Rudolph, J. L., Harrison, S. M. W., Jin, L., Frantz, A. L., Harrison, D. A. and Andres, D. A. (2011). An evolutionarily conserved Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress resistance. Mol. Biol. Cell 22, 3231-3241. 10.1091/mbc.e11-05-0400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau, P. M., Lu, J. T., Dawson, B. C., Fokkema, I. F. A. C., Robertson, S. P., Gibbs, R. A. and Lee, B. H. (2012). The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum. Mutat. 33, 1520-1525. 10.1002/humu.22141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capri, Y., Flex, E., Krumbach, O. H. F., Carpentieri, G., Cecchetti, S., Lissewski, C., Rezaei Adariani, S., Schanze, D., Brinkmann, J., Piard, J.et al. (2019). Activating mutations of RRAS2 are a rare cause of Noonan syndrome. Am. J. Hum. Genet. 104, 1223-1232. 10.1016/j.ajhg.2019.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vergara, X., Sevilla, A., D'Souza, S. L., Ang, Y.-S., Schaniel, C., Lee, D.-F., Yang, L., Kaplan, A. D., Adler, E. D., Rozov, R.et al. (2010). Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465, 808-812. 10.1038/nature09005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel, P., Cheng, A., Cuevas-Navarro, A., Everman, D. B., Papageorge, A. G., Simanshu, D. K., Tankka, A., Galeas, J., Urisman, A. and McCormick, F. (2019). RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science 363, 1226-1230. 10.1126/science.aav1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., Mitsutake, N., LaPerle, K., Akeno, N., Zanzonico, P., Longo, V. A., Mitsutake, S., Kimura, E. T., Geiger, H., Santos, E.et al. (2009). Endogenous expression of HrasG12V induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc. Natl. Acad. Sci. USA 106, 7979-7984. 10.1073/pnas.0900343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P.-C., Wakimoto, H., Conner, D., Araki, T., Yuan, T., Roberts, A., Seidman, C., Bronson, R., Neel, B., Seidman, J. G.et al. (2010). Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J. Clin. Invest. 120, 4353-4365. 10.1172/JCI43910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P.-C., Yin, J., Yu, H.-W., Yuan, T., Fernandez, M., Yung, C. K., Trinh, Q. M., Peltekova, V. D., Reid, J. G., Tworog-Dube, E.et al. (2014). Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc. Natl. Acad. Sci. USA 111, 11473-11478. 10.1073/pnas.1324128111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirstea, I. C., Kutsche, K., Dvorsky, R., Gremer, L., Carta, C., Horn, D., Roberts, A. E., Lepri, F., Merbitz-Zahradnik, T., Konig, R.et al. (2010). A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat. Genet. 42, 27-29. 10.1038/ng.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli, J. (2004). Human RAS superfamily proteins and related GTPases. Science‘s STKE 2004, re13. 10.1126/stke.2502004re13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeddu, V., Di Schiavi, E., Pennacchio, L. A., Ma'ayan, A., Sarkozy, A., Fodale, V., Cecchetti, S., Cardinale, A., Martin, J., Schackwitz, W.et al. (2009). Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet. 41, 1022-1026. 10.1038/ng.425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeddu, V., Yin, J. C., Gunnarsson, C., Virtanen, C., Drunat, S., Lepri, F., De Luca, A., Rossi, C., Ciolfi, A., Pugh, T. J.et al. (2015). Activating mutations affecting the Dbl homology domain of SOS2 cause Noonan syndrome. Hum. Mutat. 36, 1080-1087. 10.1002/humu.22834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, A. D. and Der, C. J. (2010). Ras history: the saga continues. Small GTPases 1, 2-27. 10.4161/sgtp.1.1.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, A. D., Der, C. J. and Philips, M. R. (2015). Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin. Cancer Res. 21, 1819-1827. 10.1158/1078-0432.CCR-14-3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado, P., Cubelos, B., Calleja, E., Martinez-Martin, N., Cipres, A., Merida, I., Bellas, C., Bustelo, X. R. and Alarcon, B. (2009). Essential function for the GTPase TC21 in homeostatic antigen receptor signaling. Nat. Immunol. 10, 880-888. 10.1038/ni.1749 [DOI] [PubMed] [Google Scholar]
- Denayer, E., Ahmed, T., Brems, H., Van Woerden, G., Borgesius, N. Z., Callaerts-Vegh, Z., Yoshimura, A., Hartmann, D., Elgersma, Y., D'Hooge, R.et al. (2008). Spred1 is required for synaptic plasticity and hippocampus-dependent learning. J. Neurosci. 28, 14443-14449. 10.1523/JNEUROSCI.4698-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denayer, E., Peeters, H., Sevenants, L., Derbent, M., Fryns, J. P. and Legius, E. (2012). NRAS mutations in Noonan Syndrome. Mol. Syndromol. 3, 34-38. 10.1159/000338467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eerola, I., Boon, L. M., Mulliken, J. B., Burrows, P. E., Dompmartin, A., Watanabe, S., Vanwijck, R. and Vikkula, M. (2003). Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 73, 1240-1249. 10.1086/379793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery, C. M., Monaco, K.-A., Wang, P., Balak, M., Freeman, A., Meltzer, J., Delach, S. M., Rakiec, D., Ruddy, D. A., Korn, J. M.et al. (2017). BRAF-inhibitor associated MEK mutations increase RAF-dependent and -independent enzymatic activity. Mol. Cancer Res. 15, 1431-1444. 10.1158/1541-7786.MCR-17-0211 [DOI] [PubMed] [Google Scholar]
- Esteban, L. M., Fernandez-Medarde, A., Lopez, E., Yienger, K., Guerrero, C., Ward, J. M., Tessarollo, L. and Santos, E. (2000). Ras-guanine nucleotide exchange factor sos2 is dispensable for mouse growth and development. Mol. Cell. Biol. 20, 6410-6413. 10.1128/MCB.20.17.6410-6413.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban, L. M., Vicario-Abejon, C., Fernandez-Salguero, P., Fernandez-Medarde, A., Swaminathan, N., Yienger, K., Lopez, E., Malumbres, M., McKay, R., Ward, J. M.et al. (2001). Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell. Biol. 21, 1444-1452. 10.1128/MCB.21.5.1444-1452.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flex, E., Jaiswal, M., Pantaleoni, F., Martinelli, S., Strullu, M., Fansa, E. K., Caye, A., De Luca, A., Lepri, F., Dvorsky, R.et al. (2014). Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum. Mol. Genet. 23, 4315-4327. 10.1093/hmg/ddu148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragale, A., Tartaglia, M., Wu, J. and Gelb, B. D. (2004). Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum. Mutat. 23, 267-277. 10.1002/humu.20005 [DOI] [PubMed] [Google Scholar]
- Frattini, V., Trifonov, V., Chan, J. M., Castano, A., Lia, M., Abate, F., Keir, S. T., Ji, A. X., Zoppoli, P., Niola, F.et al. (2013). The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 45, 1141-1149. 10.1038/ng.2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, A. K., Ritt, D. A. and Morrison, D. K. (2013). Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751-758. 10.1016/j.molcel.2012.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb, B. D. and Tartaglia, M. (1993). Noonan syndrome with multiple lentigines. In GeneReviews® [Internet] (ed. Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K. and Amemiya A.). Seattle: University of Washington. [PubMed] [Google Scholar]
- Giannoulatou, E., McVean, G., Taylor, I. B., McGowan, S. J., Maher, G. J., Iqbal, Z., Pfeifer, S. P., Turner, I., Burkitt Wright, E. M. M., Shorto, J.et al. (2013). Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc. Natl Acad. Sci. USA 110, 20152-20157. 10.1073/pnas.1311381110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert-Dussardier, B., Briand-Suleau, A., Laurendeau, I., Bilan, F., Cave, H., Verloes, A., Vidaud, M., Vidaud, D. and Pasmant, E. (2016). Copy number variants and rasopathies: germline KRAS duplication in a patient with syndrome including pigmentation abnormalities. Orphanet J. Rare Dis. 11, 101. 10.1186/s13023-016-0479-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux, S., Tremblay, M., Bernard, D., Cardin-Girard, J.-F., Aubry, S., Larouche, L., Rousseau, S., Huot, J., Landry, J., Jeannotte, L.et al. (1999). Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 9, 369-372. 10.1016/S0960-9822(99)80164-X [DOI] [PubMed] [Google Scholar]
- Goodwin, A. F., Oberoi, S., Landan, M., Charles, C., Massie, J. C., Fairley, C., Rauen, K. A. and Klein, O. D. (2014). Craniofacial and dental development in Costello syndrome. Am. J. Med. Genet. A 164A, 1425-1430. 10.1002/ajmg.a.36475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer, L., Merbitz-Zahradnik, T., Dvorsky, R., Cirstea, I. C., Kratz, C. P., Zenker, M., Wittinghofer, A. and Ahmadian, M. R. (2011). Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum. Mutat. 32, 33-43. 10.1002/humu.21377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp, K. W. (2005). Tumor predisposition in Costello syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 137C, 72-77. 10.1002/ajmg.c.30065 [DOI] [PubMed] [Google Scholar]
- Gripp, K. W., Lin, A. E., Stabley, D. L., Nicholson, L., Scott, C. I., Jr., Doyle, D., Aoki, Y., Matsubara, Y., Zackai, E. H., Lapunzina, P.et al. (2006). HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am. J. Med. Genet. A 140, 1-7. 10.1002/ajmg.a.31047 [DOI] [PubMed] [Google Scholar]
- Gripp, K. W., Hopkins, E., Doyle, D. and Dobyns, W. B. (2010). High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am. J. Med. Genet. A 152A, 1161-1168. 10.1002/ajmg.a.33391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A. M., Frone, M., Gripp, K. W., Gelb, B. D., Schoyer, L., Schill, L., Stronach, B., Biesecker, L. G., Esposito, D., Hernandez, E. R.et al. (2020a). Advancing RAS/RASopathy therapies: An NCI-sponsored intramural and extramural collaboration for the study of RASopathies. Am. J. Med. Genet. A 182, 866-876. 10.1002/ajmg.a.61485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A. M., Wolters, P. L., Dombi, E., Baldwin, A., Whitcomb, P., Fisher, M. J., Weiss, B., Kim, A. R., Bornhorst, M., Shah, A. C.et al. (2020b). Selumetinib in children with inoperable plexiform neurofibromas. N. Engl. J. Med. 382, 1430-1442. 10.1056/NEJMoa1912735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guil, S., de La Iglesia, N., Fernandez-Larrea, J., Cifuentes, D., Ferrer, J. C., Guinovart, J. J. and Bach-Elias, M. (2003). Alternative splicing of the human proto-oncogene c-H-ras renders a new Ras family protein that trafficks to cytoplasm and nucleus. Cancer Res. 63, 5178-5187. [PubMed] [Google Scholar]
- Gureasko, J., Kuchment, O., Makino, D. L., Sondermann, H., Bar-Sagi, D. and Kuriyan, J. (2010). Role of the histone domain in the autoinhibition and activation of the Ras activator Son of Sevenless. Proc. Natl. Acad. Sci. USA 107, 3430-3435. 10.1073/pnas.0913915107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner, C. and Groesser, L. (2013). Mosaic RASopathies. Cell Cycle 12, 43-50. 10.4161/cc.23108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, A., Lauriol, J., Thul, J., Behnke-Hall, K., Logeswaran, T., Schanzer, A., Bogurcu, N., Garvalov, B. K., Zenker, M., Gelb, B. D.et al. (2015). Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: palliative treatment with a rapamycin analog. Am. J. Med. Genet. A 167A, 744-751. 10.1002/ajmg.a.36982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanses, U., Kleinsorge, M., Roos, L., Yigit, G., Li, Y., Barbarics, B., El-Battrawy, I., Lan, H., Tiburcy, M., Hindmarsh, R.et al. (2020). Intronic CRISPR repair in a preclinical model of Noonan syndrome-associated cardiomyopathy. Circulation 142, 1059-1076. 10.1161/CIRCULATIONAHA.119.044794 [DOI] [PubMed] [Google Scholar]
- Hartung, A.-M., Swensen, J., Uriz, I. E., Lapin, M., Kristjansdottir, K., Petersen, U. S. S., Bang, J. M. V., Guerra, B., Andersen, H. S., Dobrowolski, S. F.et al. (2016). The splicing efficiency of activating HRAS mutations can determine Costello syndrome phenotype and frequency in cancer. PLoS Genet. 12, e1006039. 10.1371/journal.pgen.1006039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano, N., Mori, Y., Oh-hora, M., Kosugi, A., Fujikawa, T., Nakai, N., Niwa, H., Miyazaki, J.-I., Hamaoka, T. and Ogata, M. (2003). Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8, 847-856. 10.1046/j.1365-2443.2003.00680.x [DOI] [PubMed] [Google Scholar]
- Henkemeyer, M., Rossi, D. J., Holmyard, D. P., Puri, M. C., Mbamalu, G., Harpal, K., Shih, T. S., Jacks, T. and Pawson, T. (1995). Vascular system defects and neuronal apoptosis in mice lacking Ras GTPase-activating protein. Nature 377, 695-701. 10.1038/377695a0 [DOI] [PubMed] [Google Scholar]
- Hernandez-Porras, I. and Guerra, C. (2017). Modeling RASopathies with genetically modified mouse models. Methods Mol. Biol. 1487, 379-408. 10.1007/978-1-4939-6424-6_28 [DOI] [PubMed] [Google Scholar]
- Hernandez-Porras, I., Fabbiano, S., Schuhmacher, A. J., Aicher, A., Canamero, M., Camara, J. A., Cusso, L., Desco, M., Heeschen, C., Mulero, F.et al. (2014). K-RasV14I recapitulates Noonan syndrome in mice. Proc. Natl. Acad. Sci. USA 111, 16395-16400. 10.1073/pnas.1418126111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Porras, I., Jimenez-Catalan, B., Schuhmacher, A. J. and Guerra, C. (2015). The impact of the genetic background in the Noonan syndrome phenotype induced by K-RasV14I. Rare Dis. 3, e1045169. 10.1080/21675511.2015.1045169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Porras, I., Schuhmacher, A. J., Garcia-Medina, R., Jimenez, B., Canamero, M., de Martino, A. and Guerra, C. (2016). K-RasV14I-induced Noonan syndrome predisposes to tumour development in mice. J. Pathol. 239, 206-217. 10.1002/path.4719 [DOI] [PubMed] [Google Scholar]
- Higgins, E. M., Bos, J. M., Mason-Suares, H., Tester, D. J., Ackerman, J. P., MacRae, C. A., Sol-Church, K., Gripp, K. W., Urrutia, R. and Ackerman, M. J. (2017). Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2, e91225. 10.1172/jci.insight.91225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, K. S., Roberts, E. R., Wang, X., Marin, E., Park, T. D., Son, S., Ren, Y., Fang, B., Yoder, S., Kim, S.et al. (2019). PTPN11 plays oncogenic roles and is a therapeutic target for BRAF wild-type melanomas. Mol. Cancer Res. 17, 583-593. 10.1158/1541-7786.MCR-18-0777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillig, R. C., Sautier, B., Schroeder, J., Moosmayer, D., Hilpmann, A., Stegmann, C. M., Werbeck, N. D., Briem, H., Boemer, U., Weiske, J.et al. (2019). Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc. Natl. Acad. Sci. USA 116, 2551-2560. 10.1073/pnas.1812963116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holter, M. C., Hewitt, L. T., Koebele, S. V., Judd, J. M., Xing, L., Bimonte-Nelson, H. A., Conrad, C. D., Araki, T., Neel, B. G., Snider, W. D.et al. (2019). The Noonan Syndrome-linked Raf1L613V mutation drives increased glial number in the mouse cortex and enhanced learning. PLoS Genet. 15, e1008108. 10.1371/journal.pgen.1008108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, D. S., Fakih, M. G., Strickler, J. H., Desai, J., Durm, G. A., Shapiro, G. I., Falchook, G. S., Price, T. J., Sacher, A., Denlinger, C. S.et al. (2020). KRASG12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207-1217. 10.1056/NEJMoa1917239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser, M., Luckett, J., Chiloeches, A., Mercer, K., Iwobi, M., Giblett, S., Sun, X. M., Brown, J., Marais, R. and Pritchard, C. (2001). MEK kinase activity is not necessary for Raf-1 function. EMBO J. 20, 1940-1951. 10.1093/emboj/20.8.1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski, M., Greulich, H., Kaplan, B., Araujo, L., Amann, J., Horn, L., Schiller, J., Villalona-Calero, M. A., Meyerson, M. and Carbone, D. P. (2014). Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J. Clin. Invest. 124, 1582-1586. 10.1172/JCI72763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, H., Kato, R., Fukuyama, S., Nonami, A., Taniguchi, K., Matsumoto, K., Nakano, T., Tsuda, M., Matsumura, M., Kubo, M.et al. (2005). Spred-1 negatively regulates allergen-induced airway eosinophilia and hyperresponsiveness. J. Exp. Med. 201, 73-82. 10.1084/jem.20040616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, S.-I., Moriya, M., Watanabe, Y., Miyagawa-Tomita, S., Niihori, T., Oba, D., Ono, M., Kure, S., Ogura, T., Matsubara, Y.et al. (2014). New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum. Mol. Genet. 23, 6553-6566. 10.1093/hmg/ddu376 [DOI] [PubMed] [Google Scholar]
- Jaffre, F., Miller, C. L., Schanzer, A., Evans, T., Roberts, A. E., Hahn, A. and Kontaridis, M. I. (2019). Inducible pluripotent stem cell-derived cardiomyocytes reveal aberrant extracellular regulated kinase 5 and mitogen-activated protein kinase kinase 1/2 signaling concomitantly promote hypertrophic cardiomyopathy in RAF1-associated Noonan syndrome. Circulation 140, 207-224. 10.1161/CIRCULATIONAHA.118.037227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janakiraman, M., Vakiani, E., Zeng, Z., Pratilas, C. A., Taylor, B. S., Chitale, D., Halilovic, E., Wilson, M., Huberman, K., Ricarte Filho, J. C.et al. (2010). Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 70, 5901-5911. 10.1158/0008-5472.CAN-10-0192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jecrois, E. S., Zheng, W., Bornhorst, M., Li, Y., Treisman, D. M., Muguyo, D., Huynh, S., Andrew, S. F., Wang, Y., Jiang, J.et al. (2021). Treatment during a developmental window prevents NF1-associated optic pathway gliomas by targeting Erk-dependent migrating glial progenitors. Dev. Cell 56, 2871-2885.e6. 10.1016/j.devcel.2021.08.004 [DOI] [PubMed] [Google Scholar]
- Jindal, G. A., Goyal, Y., Burdine, R. D., Rauen, K. A. and Shvartsman, S. Y. (2015). RASopathies: unraveling mechanisms with animal models. Dis. Model. Mech. 8, 1167. 10.1242/dmm.022442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal, G. A., Goyal, Y., Yamaya, K., Futran, A. S., Kountouridis, I., Balgobin, C. A., Schupbach, T., Burdine, R. D. and Shvartsman, S. Y. (2017). In vivo severity ranking of Ras pathway mutations associated with developmental disorders. Proc. Natl. Acad. Sci. USA 114, 510-515. 10.1073/pnas.1615651114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, L., Greenbaum, D., Cichowski, K., Mercer, K., Murphy, E., Schmitt, E., Bronson, R. T., Umanoff, H., Edelmann, W., Kucherlapati, R.et al. (1997). K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 11, 2468-2481. 10.1101/gad.11.19.2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J., van der Smagt, J. J., Rosenfeld, J. A., Pagnamenta, A. T., Alswaid, A., Baker, E. H., Blair, E., Borck, G., Brinkmann, J., Craigen, W.et al. (2018). Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 20, 1175-1185. 10.1038/gim.2017.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling, C., van Geemen, D. and den Hertog, J. (2007). Shp2 knockdown and Noonan/LEOPARD mutant Shp2-induced gastrulation defects. PLoS Genet. 3, e225. 10.1371/journal.pgen.0030225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josowitz, R., Mulero-Navarro, S., Rodriguez, N. A., Falce, C., Cohen, N., Ullian, E. M., Weiss, L. A., Rauen, K. A., Sobie, E. A. and Gelb, B. D. (2016). Autonomous and non-autonomous defects underlie hypertrophic cardiomyopathy in BRAF-mutant hiPSC-derived cardiomyocytes. Stem Cell Reports 7, 355-369. 10.1016/j.stemcr.2016.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousma, E., Rizvi, T. A., Wu, J., Janhofer, D., Dombi, E., Dunn, R. S., Kim, M.-O., Masters, A. R., Jones, D. R., Cripe, T. P.et al. (2015). Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of neurofibromatosis type 1. Pediatr. Blood Cancer 62, 1709-1716. 10.1002/pbc.25546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju, Y., Park, J. S., Kim, D., Kim, B., Lee, J. H., Nam, Y., Yoo, H.-W., Lee, B. H. and Han, Y.-M. (2020). SHP2 mutations induce precocious gliogenesis of Noonan syndrome-derived iPSCs during neural development in vitro. Stem Cell Res. Ther. 11, 209. 10.1186/s13287-020-01709-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katata, Y., Inoue, S.-I., Asao, A., Kobayashi, S., Terui, H., Inoue-Shibui, A., Abe, T., Niihori, T., Aiba, S., Ishii, N.et al. (2020). Costello syndrome model mice with a Hras G12S mutation are susceptible to develop house dust mite-induced atopic dermatitis. Cell Death Dis. 11, 617. 10.1038/s41419-020-02845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki, J., Aegerter, S., Fevurly, R. D., Mammoto, A., Mammoto, T., Sahin, M., Mably, J. D., Fishman, S. J. and Chan, J. (2014). RASA1 functions in EPHB4 signaling pathway to suppress endothelial mTORC1 activity. J. Clin. Invest. 124, 2774-2784. 10.1172/JCI67084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhack, H., David, F. S., McGregor, M., Cantley, L. C. and Neel, B. G. (2005). Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J. Biol. Chem. 280, 30984-30993. 10.1074/jbc.M504699200 [DOI] [PubMed] [Google Scholar]
- Kent, O. A., Saha, M., Coyaud, E., Burston, H. E., Law, N., Dadson, K., Chen, S., Laurent, E. M., St-Germain, J., Sun, R. X.et al. (2020). Haploinsufficiency of RREB1 causes a Noonan-like RASopathy via epigenetic reprogramming of RAS-MAPK pathway genes. Nat. Commun. 11, 4673. 10.1038/s41467-020-18483-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler, D., Gmachl, M., Mantoulidis, A., Martin, L. J., Zoephel, A., Mayer, M., Gollner, A., Covini, D., Fischer, S., Gerstberger, T.et al. (2019). Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 116, 15823-15829. 10.1073/pnas.1904529116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H., Lee, H.-K., Takamiya, K. and Huganir, R. L. (2003). The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J. Neurosci. 23, 1119-1124. 10.1523/JNEUROSCI.23-04-01119.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu, M. and Ruoslahti, E. (2005). R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 11, 1346-1350. 10.1038/nm1324 [DOI] [PubMed] [Google Scholar]
- Kontaridis, M. I., Swanson, K. D., David, F. S., Barford, D. and Neel, B. G. (2006). PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 281, 6785-6792. 10.1074/jbc.M513068200 [DOI] [PubMed] [Google Scholar]
- Kopanitsa, M. V., Gou, G., Afinowi, N. O., Bayes, A., Grant, S. G. N. and Komiyama, N. H. (2018). Chronic treatment with a MEK inhibitor reverses enhanced excitatory field potentials in Syngap1+/− mice. Pharmacol. Rep. 70, 777-783. 10.1016/j.pharep.2018.02.021 [DOI] [PubMed] [Google Scholar]
- Kraft, M., Cirstea, I. C., Voss, A. K., Thomas, T., Goehring, I., Sheikh, B. N., Gordon, L., Scott, H., Smyth, G. K., Ahmadian, M. R.et al. (2011). Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J. Clin. Invest. 121, 3479-3491. 10.1172/JCI43428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapinski, P. E., Bauler, T. J., Brown, E. J., Hughes, E. D., Saunders, T. L. and King, P. D. (2007). Generation of mice with a conditional allele of the p120 Ras GTPase-activating protein. Genesis 45, 762-767. 10.1002/dvg.20354 [DOI] [PubMed] [Google Scholar]
- Lapinski, P. E., Kwon, S., Lubeck, B. A., Wilkinson, J. E., Srinivasan, R. S., Sevick-Muraca, E. and King, P. D. (2012). RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J. Clin. Invest. 122, 733-747. 10.1172/JCI46116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y.-S., Ehninger, D., Zhou, M., Oh, J.-Y., Kang, M., Kwak, C., Ryu, H.-H., Butz, D., Araki, T., Cai, Y.et al. (2014). Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat. Neurosci. 17, 1736-1743. 10.1038/nn.3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni, C., Onesimo, R., Giorgio, V., Diamanti, A., Giorgio, D., Martini, L., Rossodivita, A., Tartaglia, M. and Zampino, G. (2016). Understanding growth failure in Costello syndrome: increased resting energy expenditure. J. Pediatr. 170, 322-324. 10.1016/j.jpeds.2015.11.076 [DOI] [PubMed] [Google Scholar]
- Leoni, C., Romeo, D. M., Pelliccioni, M., Di Gia, M., Onesimo, R., Giorgio, V., Flex, E., Tedesco, M., Tartaglia, M., Rigante, D.et al. (2021). Musculo-skeletal phenotype of Costello syndrome and cardio-facio-cutaneous syndrome: insights on the functional assessment status. Orphanet J. Rare Dis. 16, 43. 10.1186/s13023-021-01674-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepri, F., De Luca, A., Stella, L., Rossi, C., Baldassarre, G., Pantaleoni, F., Cordeddu, V., Williams, B. J., Dentici, M. L., Caputo, V.et al. (2011). SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum. Mutat. 32, 760-772. 10.1002/humu.21492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D., March, M. E., Gutierrez-Uzquiza, A., Kao, C., Seiler, C., Pinto, E., Matsuoka, L. S., Battig, M. R., Bhoj, E. J., Wenger, T. L.et al. (2019). ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat. Med. 25, 1116-1122. 10.1038/s41591-019-0479-2 [DOI] [PubMed] [Google Scholar]
- Lin, A. E., Alexander, M. E., Colan, S. D., Kerr, B., Rauen, K. A., Noonan, J., Baffa, J., Hopkins, E., Sol-Church, K., Limongelli, G.et al. (2011). Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/MAPK pathway syndrome. Am. J. Med. Genet. A 155A, 486-507. 10.1002/ajmg.a.33857 [DOI] [PubMed] [Google Scholar]
- Lissewski, C., Chune, V., Pantaleoni, F., De Luca, A., Capri, Y., Brinkmann, J., Lepri, F., Daniele, P., Leenders, E., Mazzanti, L.et al. (2021). Variants of SOS2 are a rare cause of Noonan syndrome with particular predisposition for lymphatic complications. Eur. J. Hum. Genet. 29, 51-60. 10.1038/s41431-020-00708-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck, B. A., Lapinski, P. E., Bauler, T. J., Oliver, J. A., Hughes, E. D., Saunders, T. L. and King, P. D. (2014). Blood vascular abnormalities in Rasa1R780Q knockin mice: implications for the pathogenesis of capillary malformation-arteriovenous malformation. Am. J. Pathol. 184, 3163-3169. 10.1016/j.ajpath.2014.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, Y., Tidyman, W. E., Ander, B. P., Pritchard, C. A. and Rauen, K. A. (2021). Ras/MAPK dysregulation in development causes a skeletal myopathy in an activating BrafL597V mouse model for cardio-facio-cutaneous syndrome. Dev. Dyn. 250, 1074-1095. 10.1002/dvdy.309 [DOI] [PubMed] [Google Scholar]
- Marin, T. M., Keith, K., Davies, B., Conner, D. A., Guha, P., Kalaitzidis, D., Wu, X., Lauriol, J., Wang, B., Bauer, M.et al. (2011). Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Invest. 121, 1026-1043. 10.1172/JCI44972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli, S., Torreri, P., Tinti, M., Stella, L., Bocchinfuso, G., Flex, E., Grottesi, A., Ceccarini, M., Palleschi, A., Cesareni, G.et al. (2008). Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes. Hum. Mol. Genet. 17, 2018-2029. 10.1093/hmg/ddn099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli, S., De Luca, A., Stellacci, E., Rossi, C., Checquolo, S., Lepri, F., Caputo, V., Silvano, M., Buscherini, F., Consoli, F.et al. (2010). Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 87, 250-257. 10.1016/j.ajhg.2010.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli, S., Krumbach, O. H. F., Pantaleoni, F., Coppola, S., Amin, E., Pannone, L., Nouri, K., Farina, L., Dvorsky, R., Lepri, F.et al. (2018). Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am. J. Hum. Genet. 102, 309-320. 10.1016/j.ajhg.2017.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan, T. F., Conte, N., West, D. B., Jacobsen, J. O., Mason, J., Warren, J., Chen, C.-K., Tudose, I., Relac, M., Matthews, P.et al. (2017). Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet. 49, 1231-1238. 10.1038/ng.3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer, K., Giblett, S., Green, S., Lloyd, D., DaRocha Dias, S., Plumb, M., Marais, R. and Pritchard, C. (2005a). Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 65, 11493-11500. 10.1158/0008-5472.CAN-05-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer, K., Giblett, S., Oakden, A., Brown, J., Marais, R. and Pritchard, C. (2005b). A-Raf and Raf-1 work together to influence transient ERK phosphorylation and Gl/S cell cycle progression. Oncogene 24, 5207-5217. 10.1038/sj.onc.1208707 [DOI] [PubMed] [Google Scholar]
- Messiaen, L., Yao, S., Brems, H., Callens, T., Sathienkijkanchai, A., Denayer, E., Spencer, E., Arn, P., Babovic-Vuksanovic, D., Bay, C.et al. (2009). Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA 302, 2111-2118. 10.1001/jama.2009.1663 [DOI] [PubMed] [Google Scholar]
- Molzan, M., Schumacher, B., Ottmann, C., Baljuls, A., Polzien, L., Weyand, M., Thiel, P., Rose, R., Rose, M., Kuhenne, P.et al. (2010). Impaired binding of 14-3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased Ras signaling. Mol. Cell. Biol. 30, 4698-4711. 10.1128/MCB.01636-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco, K.-A., Delach, S., Yuan, J., Mishina, Y., Fordjour, P., Labrot, E., McKay, D., Guo, R., Higgins, S., Wang, H. Q.et al. (2021). LXH254, a potent and selective ARAF-sparing inhibitor of BRAF and CRAF for the treatment of MAPK-driven tumors. Clin. Cancer Res. 27, 2061-2073. 10.1158/1078-0432.CCR-20-2563 [DOI] [PubMed] [Google Scholar]
- Moore, A. R., Rosenberg, S. C., McCormick, F. and Malek, S. (2020). RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19, 533-552. 10.1038/s41573-020-0068-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya, M., Inoue, S.-I., Miyagawa-Tomita, S., Nakashima, Y., Oba, D., Niihori, T., Hashi, M., Ohnishi, H., Kure, S., Matsubara, Y.et al. (2015). Adult mice expressing a Braf Q241R mutation on an ICR/CD-1 background exhibit a cardio-facio-cutaneous syndrome phenotype. Hum. Mol. Genet. 24, 7349-7360. 10.1093/hmg/ddv435 [DOI] [PubMed] [Google Scholar]
- Motta, M., Fidan, M., Bellacchio, E., Pantaleoni, F., Schneider-Heieck, K., Coppola, S., Borck, G., Salviati, L., Zenker, M., Cirstea, I. C.et al. (2019). Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum. Mol. Genet. 28, 1007-1022. 10.1093/hmg/ddy412 [DOI] [PubMed] [Google Scholar]
- Motta, M., Pannone, L., Pantaleoni, F., Bocchinfuso, G., Radio, F. C., Cecchetti, S., Ciolfi, A., Di Rocco, M., Elting, M. W., Brilstra, E. H.et al. (2020a). Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am. J. Hum. Genet. 107, 499-513. 10.1016/j.ajhg.2020.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta, M., Sagi-Dain, L., Krumbach, O. H. F., Hahn, A., Peleg, A., German, A., Lissewski, C., Coppola, S., Pantaleoni, F., Kocherscheid, L.et al. (2020b). Activating MRAS mutations cause Noonan syndrome associated with hypertrophic cardiomyopathy. Hum. Mol. Genet. 29, 1772-1783. 10.1093/hmg/ddz108 [DOI] [PubMed] [Google Scholar]
- Motta, M., Fasano, G., Gredy, S., Brinkmann, J., Bonnard, A. A., Simsek-Kiper, P. O., Gulec, E. Y., Essaddam, L., Utine, G. E., Guarnetti Prandi, I.et al. (2021). SPRED2 loss-of-function causes a recessive Noonan syndrome-like phenotype. Am. J. Hum. Genet. 108, 2112-2129. 10.1016/j.ajhg.2021.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagama, Y., Takeda, N., Ogawa, S., Takeda, H., Furutani, Y., Nakanishi, T., Sato, T., Hirata, Y., Oka, A. and Inuzuka, R. (2020). Noonan syndrome-associated biallelic LZTR1 mutations cause cardiac hypertrophy and vascular malformations in zebrafish. Mol. Genet. Genomic Med. 8, e1107. 10.1002/mgg3.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima, R., Takao, K., Hattori, S., Shoji, H., Komiyama, N. H., Grant, S. G. N. and Miyakawa, T. (2019). Comprehensive behavioral analysis of heterozygous Syngap1 knockout mice. Neuropsychopharmacol. Rep. 39, 223-237. 10.1002/npr2.12073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T., Colbert, M., Krenz, M., Molkentin, J. D., Hahn, H. S., Dorn, G. W., II and Robbins, J. (2007). Mediating ERK1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J. Clin. Invest. 117, 2123-2132. 10.1172/JCI30756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T., Gulick, J., Pratt, R. and Robbins, J. (2009). Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc. Natl. Acad. Sci. USA 106, 15436-15441. 10.1073/pnas.0903302106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, Y., Umeki, N., Abe, M. and Sako, Y. (2017). Mutation-specific mechanisms of hyperactivation of Noonan syndrome SOS molecules detected with single-molecule imaging in living cells. Sci. Rep. 7, 14153. 10.1038/s41598-017-14190-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer, C. M., Kang, M. W., Shin, D. H., Furlan, I., Erlacher, M., Bunin, N. J., Bunda, S., Finklestein, J. Z., Gorr, T. A., Mehta, P.et al. (2010). Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 42, 794-800. 10.1038/ng.641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niihori, T., Aoki, Y., Narumi, Y., Neri, G., Cave, H., Verloes, A., Okamoto, N., Hennekam, R. C., Gillessen-Kaesbach, G., Wieczorek, D.et al. (2006). Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 38, 294-296. 10.1038/ng1749 [DOI] [PubMed] [Google Scholar]
- Nissim, S., Leshchiner, I., Mancias, J. D., Greenblatt, M. B., Maertens, O., Cassa, C. A., Rosenfeld, J. A., Cox, A. G., Hedgepeth, J., Wucherpfennig, J. I.et al. (2019). Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat. Genet. 51, 1308-1314. 10.1038/s41588-019-0475-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora, J. J., Nora, A. H., Sinha, A. K., Spangler, R. D. and Lubs, H. A. (1974). The Ullrich-Noonan syndrome (Turner phenotype). Am. J. Dis. Child. 127, 48-55. 10.1001/archpedi.1974.02110200050007 [DOI] [PubMed] [Google Scholar]
- Nunez Rodriguez, N., Lee, I. N., Banno, A., Qiao, H. F., Qiao, R. F., Yao, Z., Hoang, T., Kimmelman, A. C. and Chan, A. M.-L. (2006). Characterization of R-Ras3/M-Ras null mice reveals a potential role in trophic factor signaling. Mol. Cell. Biol. 26, 7145-7154. 10.1128/MCB.00476-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba, D., Inoue, S.-I., Miyagawa-Tomita, S., Nakashima, Y., Niihori, T., Yamaguchi, S., Matsubara, Y. and Aoki, Y. (2018). Mice with an oncogenic HRAS mutation are resistant to high-fat diet-induced obesity and exhibit impaired hepatic energy homeostasis. EBioMedicine 27, 138-150. 10.1016/j.ebiom.2017.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi, K., Gaengel, K., Krishnamoorthy, S., Kamiya, K., Kim, I.-K., Ying, H., Weber, U., Perkins, L. A., Tartaglia, M., Mlodzik, M.et al. (2006). Transgenic Drosophila models of Noonan syndrome causing PTPN11 gain-of-function mutations. Hum. Mol. Genet. 15, 543-553. 10.1093/hmg/ddi471 [DOI] [PubMed] [Google Scholar]
- Oishi, K., Zhang, H., Gault, W. J., Wang, C. J., Tan, C. C., Kim, I.-K., Ying, H., Rahman, T., Pica, N., Tartaglia, M.et al. (2009). Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum. Mol. Genet. 18, 193-201. 10.1093/hmg/ddn336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordan, M., Pallara, C., Maik-Rachline, G., Hanoch, T., Gervasio, F. L., Glaser, F., Fernandez-Recio, J. and Seger, R. (2018). Intrinsically active MEK variants are differentially regulated by proteinases and phosphatases. Sci. Rep. 8, 11830. 10.1038/s41598-018-30202-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani, M. R., Oishi, K., Gelb, B. D. and Zhong, Y. (2009). The phosphatase SHP2 regulates the spacing effect for long-term memory induction. Cell 139, 186-198. 10.1016/j.cell.2009.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages, G., Guerin, S., Grall, D., Bonino, F., Smith, A., Anjuere, F., Auberger, P. and Pouyssegur, J. (1999). Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286, 1374-1377. 10.1126/science.286.5443.1374 [DOI] [PubMed] [Google Scholar]
- Pandit, B., Sarkozy, A., Pennacchio, L. A., Carta, C., Oishi, K., Martinelli, S., Pogna, E. A., Schackwitz, W., Ustaszewska, A., Landstrom, A.et al. (2007). Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 39, 1007-1012. 10.1038/ng2073 [DOI] [PubMed] [Google Scholar]
- Pantaleoni, F., Lev, D., Cirstea, I. C., Motta, M., Lepri, F. R., Bottero, L., Cecchetti, S., Linger, I., Paolacci, S., Flex, E.et al. (2017). Aberrant HRAS transcript processing underlies a distinctive phenotype within the RASopathy clinical spectrum. Hum. Mutat. 38, 798-804. 10.1002/humu.23224 [DOI] [PubMed] [Google Scholar]
- Pantsar, T. (2020). The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 18, 189-198. 10.1016/j.csbj.2019.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, E., Rawson, S., Li, K., Kim, B.-W., Ficarro, S. B., Pino, G. G.-D., Sharif, H., Marto, J. A., Jeon, H. and Eck, M. J. (2019). Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 575, 545-550. 10.1038/s41586-019-1660-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, M. J., Fryer, A. E., Shears, D. J., Lachlan, K. L., McKee, S. A., Magee, A. C., Mohammed, S., Vasudevan, P. C., Park, S.-M., Benoit, V.et al. (2015). De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am. J. Med. Genet. A 167A, 2231-2237. 10.1002/ajmg.a.37189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, S.-B., Henry, J. R., Kaufman, M. D., Lu, W.-P., Smith, B. D., Vogeti, S., Rutkoski, T. J., Wise, S., Chun, L., Zhang, Y.et al. (2015). Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28, 384-398. 10.1016/j.ccell.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Perez, B., Mechinaud, F., Galambrun, C., Ben Romdhane, N., Isidor, B., Philip, N., Derain-Court, J., Cassinat, B., Lachenaud, J., Kaltenbach, S.et al. (2010). Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J. Med. Genet. 47, 686-691. 10.1136/jmg.2010.076836 [DOI] [PubMed] [Google Scholar]
- Pierpont, M. E. M., Magoulas, P. L., Adi, S., Kavamura, M. I., Neri, G., Noonan, J., Pierpont, E. I., Reinker, K., Roberts, A. E., Shankar, S.et al. (2014). Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 134, e1149-e1162. 10.1542/peds.2013-3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski, A., Xie, J., Liu, Y. F., Poplawski, A. B., Gomes, A. R., Madanecki, P., Fu, C., Crowley, M. R., Crossman, D. K., Armstrong, L.et al. (2014). Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 46, 182-187. 10.1038/ng.2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov, I. K., Hiatt, S. M., Whalen, S., Keren, B., Ruivenkamp, C., van Haeringen, A., Chen, M.-J., Cooper, G. M., Korf, B. R. and Chang, C. (2019). A YWHAZ variant associated with cardiofaciocutaneous syndrome activates the RAF-ERK pathway. Front. Physiol. 10, 388. 10.3389/fphys.2019.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, C. A., Bolin, L., Slattery, R., Murray, R. and McMahon, M. (1996). Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr. Biol. 6, 614-617. 10.1016/S0960-9822(02)00548-1 [DOI] [PubMed] [Google Scholar]
- Qian, X., Esteban, L., Vass, W. C., Upadhyaya, C., Papageorge, A. G., Yienger, K., Ward, J. M., Lowy, D. R. and Santos, E. (2000). The Sos1 and Sos2 Ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J. 19, 642-654. 10.1093/emboj/19.4.642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen, K. A. (2007). HRAS and the Costello syndrome. Clin. Genet. 71, 101-108. 10.1111/j.1399-0004.2007.00743.x [DOI] [PubMed] [Google Scholar]
- Rauen, K. A. (2013). The RASopathies. Annu. Rev. Genomics Hum. Genet. 14, 355-369. 10.1146/annurev-genom-091212-153523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen, K. A. (2016). Cardiofaciocutaneous Syndrome. In GeneReviews® [Internet] (ed. Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K. and Amemiya A.). Seattle: University of Washingto; n. [Google Scholar]
- Razzaque, M. A., Nishizawa, T., Komoike, Y., Yagi, H., Furutani, M., Amo, R., Kamisago, M., Momma, K., Katayama, H., Nakagawa, M.et al. (2007). Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 39, 1013-1017. 10.1038/ng2078 [DOI] [PubMed] [Google Scholar]
- Reijnders, M. R. F., Ansor, N. M., Kousi, M., Yue, W. W., Tan, P. L., Clarkson, K., Clayton-Smith, J., Corning, K., Jones, J. R., Lam, W. W. K.et al. (2017). RAC1 missense mutations in developmental disorders with diverse phenotypes. Am. J. Hum. Genet. 101, 466-477. 10.1016/j.ajhg.2017.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revencu, N., Boon, L. M., Mulliken, J. B., Enjolras, O., Cordisco, M. R., Burrows, P. E., Clapuyt, P., Hammer, F., Dubois, J., Baselga, E.et al. (2008). Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum. Mutat. 29, 959-965. 10.1002/humu.20746 [DOI] [PubMed] [Google Scholar]
- Roberts, A. E., Araki, T., Swanson, K. D., Montgomery, K. T., Schiripo, T. A., Joshi, V. A., Li, L., Yassin, Y., Tamburino, A. M., Neel, B. G.et al. (2007). Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 39, 70-74. 10.1038/ng1926 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., Tetsu, O., Tidyman, W. E., Estep, A. L., Conger, B. A., Cruz, M. S., McCormick, F. and Rauen, K. A. (2006). Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311, 1287-1290. 10.1126/science.1124642 [DOI] [PubMed] [Google Scholar]
- Romano, A. A., Allanson, J. E., Dahlgren, J., Gelb, B. D., Hall, B., Pierpont, M. E., Roberts, A. E., Robinson, W., Takemoto, C. M. and Noonan, J. A. (2010). Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126, 746-759. 10.1542/peds.2009-3207 [DOI] [PubMed] [Google Scholar]
- Rooney, G. E., Goodwin, A. F., Depeille, P., Sharir, A., Schofield, C. M., Yeh, E., Roose, J. P., Klein, O. D., Rauen, K. A., Weiss, L. A.et al. (2016). Human iPS cell-derived neurons uncover the impact of increased Ras signaling in Costello syndrome. J. Neurosci. 36, 142-152. 10.1523/JNEUROSCI.1547-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski, R.Jr. (2012). MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 417, 5-10. 10.1016/j.bbrc.2011.11.145 [DOI] [PubMed] [Google Scholar]
- Ross, S. J., Revenko, A. S., Hanson, L. L., Ellston, R., Staniszewska, A., Whalley, N., Pandey, S. K., Revill, M., Rooney, C., Buckett, L. K.et al. (2017). Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 9, eaal5253. 10.1126/scitranslmed.aal5253 [DOI] [PubMed] [Google Scholar]
- Runtuwene, V., van Eekelen, M., Overvoorde, J., Rehmann, H., Yntema, H. G., Nillesen, W. M., van Haeringen, A., van der Burgt, I., Burgering, B. and den Hertog, J. (2011). Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis. Model. Mech. 4, 393-399. 10.1242/dmm.007112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoriello, C., Deflorian, G., Pezzimenti, F., Kawakami, K., Lanfrancone, L., d'Adda di Fagagna, F. and Mione, M. (2009). Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis. Model. Mech. 2, 56-67. 10.1242/dmm.001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozy, A., Digilio, M. C. and Dallapiccola, B. (2008). Leopard syndrome. Orphanet J. Rare Dis. 3, 13. 10.1186/1750-1172-3-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber, J., Grimbergen, L.-A., Overwater, I., Vaart, T. V., Stedehouder, J., Schuhmacher, A. J., Guerra, C., Kushner, S. A., Jaarsma, D. and Elgersma, Y. (2017). Mechanisms underlying cognitive deficits in a mouse model for Costello Syndrome are distinct from other RASopathy mouse models. Sci. Rep. 7, 1256. 10.1038/s41598-017-01218-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert, S., Bollag, G., Lyubynska, N., Nguyen, H., Kratz, C. P., Zenker, M., Niemeyer, C. M., Molven, A. and Shannon, K. (2007). Biochemical and functional characterization of germ line KRAS mutations. Mol. Cell. Biol. 27, 7765-7770. 10.1128/MCB.00965-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher, A. J., Guerra, C., Sauzeau, V., Canamero, M., Bustelo, X. R. and Barbacid, M. (2008). A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J. Clin. Invest. 118, 2169-2179. 10.1172/JCI34385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher, A. J., Hernandez-Porras, I., Garcia-Medina, R. and Guerra, C. (2017). Noonan syndrome: lessons learned from genetically modified mouse models. Expert Rev. Endocrinol. Metab. 12, 367-378. 10.1080/17446651.2017.1361821 [DOI] [PubMed] [Google Scholar]
- Scott, A., Di Giosaffatte, N., Pinna, V., Daniele, P., Corno, S., D'Ambrosio, V., Andreucci, E., Marozza, A., Sirchia, F., Tortora, G.et al. (2021). When to test fetuses for RASopathies? Proposition from a systematic analysis of 352 multicenter cases and a postnatal cohort. Genet. Med. 23, 1116-1124. 10.1038/s41436-020-01093-7 [DOI] [PubMed] [Google Scholar]
- Sebastian, M., Eberhardt, W. E. E., Hoffknecht, P., Metzenmacher, M., Wehler, T., Kokowski, K., Alt, J., Schütte, W., Büttner, R., Heukamp, L. C.et al. (2021). KRAS G12C-mutated advanced non-small cell lung cancer: A real-world cohort from the German prospective, observational, nation-wide CRISP Registry (AIO-TRK-0315). Lung Cancer 154, 51-61. 10.1016/j.lungcan.2021.02.005 [DOI] [PubMed] [Google Scholar]
- Senawong, T., Phuchareon, J., Ohara, O., McCormick, F., Rauen, K. A. and Tetsu, O. (2008). Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and RAF inhibition: implications for therapeutic options. Hum. Mol. Genet. 17, 419-430. 10.1093/hmg/ddm319 [DOI] [PubMed] [Google Scholar]
- Sevick-Muraca, E. M. and King, P. D. (2014). Lymphatic vessel abnormalities arising from disorders of Ras signal transduction. Trends Cardiovasc. Med. 24, 121-127. 10.1016/j.tcm.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewduth, R. N., Pandolfi, S., Steklov, M., Sheryazdanova, A., Zhao, P., Criem, N., Baietti, M. F., Lechat, B., Quarck, R., Impens, F.et al. (2020). The Noonan syndrome gene Lztr1 controls cardiovascular function by regulating vesicular trafficking. Circ. Res. 126, 1379-1393. 10.1161/CIRCRESAHA.119.315730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, D. H., Mann, J. A., Krol, A. L. and Rauen, K. A. (2012). Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br. J. Dermatol. 166, 601-607. 10.1111/j.1365-2133.2011.10744.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondermann, H., Soisson, S. M., Boykevisch, S., Yang, S.-S., Bar-Sagi, D. and Kuriyan, J. (2004). Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell 119, 393-405. 10.1016/j.cell.2004.10.005 [DOI] [PubMed] [Google Scholar]
- Sparks, T. N., Lianoglou, B. R., Adami, R. R., Pluym, I. D., Holliman, K., Duffy, J., Downum, S. L., Patel, S., Faubel, A., Boe, N. M.et al. (2020). Exome sequencing for prenatal diagnosis in nonimmune hydrops fetalis. N. Engl. J. Med. 383, 1746-1756. 10.1056/NEJMoa2023643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steklov, M., Pandolfi, S., Baietti, M. F., Batiuk, A., Carai, P., Najm, P., Zhang, M., Jang, H., Renzi, F., Cai, Y.et al. (2018). Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362, 1177-1182. 10.1126/science.aap7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, R. A., Sanda, T., Widlund, H. R., Zhu, S., Swanson, K. D., Hurley, A. D., Bentires-Alj, M., Fisher, D. E., Kontaridis, M. I., Look, A. T.et al. (2010). Phosphatase-dependent and -independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Dev. Cell 18, 750-762. 10.1016/j.devcel.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowe, I. B., Mercado, E. L., Stowe, T. R., Bell, E. L., Oses-Prieto, J. A., Hernández, H., Burlingame, A. L. and McCormick, F. (2012). A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 26, 1421-1426. 10.1101/gad.190876.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, R. J., Infante, J. R., Janku, F., Wong, D. J. L., Sosman, J. A., Keedy, V., Patel, M. R., Shapiro, G. I., Mier, J. W., Tolcher, A. W.et al. (2018). First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer Discov. 8, 184-195. 10.1158/2159-8290.CD-17-1119 [DOI] [PubMed] [Google Scholar]
- Suzuki, J., Kaziro, Y. and Koide, H. (1997). An activated mutant of R-Ras inhibits cell death caused by cytokine deprivation in BaF3 cells in the presence of IGF-I. Oncogene 15, 1689-1697. 10.1038/sj.onc.1201333 [DOI] [PubMed] [Google Scholar]
- Tajan, M., Batut, A., Cadoudal, T., Deleruyelle, S., Le Gonidec, S., Saint Laurent, C., Vomscheid, M., Wanecq, E., Treguer, K., De Rocca Serra-Nedelec, A.et al. (2014). LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from diet-induced obesity. Proc. Natl. Acad. Sci. USA 111, E4494-E4503. 10.1073/pnas.1406107111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara, S., Inoue, S.-I., Miyagawa-Tomita, S., Matsuura, K., Nakashima, Y., Niihori, T., Matsubara, Y., Saiki, Y. and Aoki, Y. (2019). New Noonan syndrome model mice with RIT1 mutation exhibit cardiac hypertrophy and susceptibility to β-adrenergic stimulation-induced cardiac fibrosis. EBioMedicine 42, 43-53. 10.1016/j.ebiom.2019.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi, K., Kohno, R.-I., Ayada, T., Kato, R., Ichiyama, K., Morisada, T., Oike, Y., Yonemitsu, Y., Maehara, Y. and Yoshimura, A. (2007). Spreds are essential for embryonic lymphangiogenesis by regulating vascular endothelial growth factor receptor 3 signaling. Mol. Cell. Biol. 27, 4541-4550. 10.1128/MCB.01600-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia, M. and Gelb, B. D. (2005). Noonan syndrome and related disorders: genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 6, 45-68. 10.1146/annurev.genom.6.080604.162305 [DOI] [PubMed] [Google Scholar]
- Tartaglia, M., Martinelli, S., Stella, L., Bocchinfuso, G., Flex, E., Cordeddu, V., Zampino, G., Burgt, I., Palleschi, A., Petrucci, T. C.et al. (2006). Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet. 78, 279-290. 10.1086/499925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia, M., Pennacchio, L. A., Zhao, C., Yadav, K. K., Fodale, V., Sarkozy, A., Pandit, B., Oishi, K., Martinelli, S., Schackwitz, W.et al. (2007). Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 39, 75-79. 10.1038/ng1939 [DOI] [PubMed] [Google Scholar]
- Terrell, E. M. and Morrison, D. K. (2019). Ras-mediated activation of the Raf family kinases. Cold Spring Harb. Perspect. Med. 9, a033746. 10.1101/cshperspect.a033746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman, W. E. and Rauen, K. A. (2009). The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230-236. 10.1016/j.gde.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman, W. E. and Rauen, K. A. (2016a). Expansion of the RASopathies. Curr. Genet. Med. Rep. 4, 57-64. 10.1007/s40142-016-0100-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman, W. E. and Rauen, K. A. (2016b). Pathogenetics of the RASopathies. Hum. Mol. Genet. 25, R123-R132. 10.1093/hmg/ddw191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman, W. E., Goodwin, A. F., Maeda, Y., Klein, O. D. and Rauen, K. A. (2021). MEK-inhibitor-mediated rescue of skeletal myopathy caused by activating Hras mutation in a Costello syndrome mouse model. Dis. Model. Mech. 15, dmm049166. 10.1242/dmm.049166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers, T., Lopez, C. A., Van, Q. N., Neale, C., Tonelli, M., Stephen, A. G. and Gnanakaran, S. (2018). Molecular recognition of RAS/RAF complex at the membrane: role of RAF cysteine-rich domain. Sci. Rep. 8, 8461. 10.1038/s41598-018-26832-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich, M., Weber, M., Post, A. M., Popp, S., Grein, J., Zechner, M., Guerrero Gonzalez, H., Kreis, A., Schmitt, A. G., Uceyler, N.et al. (2018). OCD-like behavior is caused by dysfunction of thalamo-amygdala circuits and upregulated TrkB/ERK-MAPK signaling as a result of SPRED2 deficiency. Mol. Psychiatry 23, 444-458. 10.1038/mp.2016.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich, M., Aßmus, B., Augustin, A. M., Häbich, H., Abeßer, M., Martin Machado, J., Werner, F., Erkens, R., Arias-Loza, A.-P., Umbenhauer, S.et al. (2019). SPRED2 deficiency elicits cardiac arrhythmias and premature death via impaired autophagy. J. Mol. Cell. Cardiol. 129, 13-26. 10.1016/j.yjmcc.2019.01.023 [DOI] [PubMed] [Google Scholar]
- Umanoff, H., Edelmann, W., Pellicer, A. and Kucherlapati, R. (1995). The murine N-ras gene is not essential for growth and development. Proc. Natl. Acad. Sci. USA 92, 1709-1713. 10.1073/pnas.92.5.1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urosevic, J., Sauzeau, V., Soto-Montenegro, M. L., Reig, S., Desco, M., Wright, E. M. B., Canamero, M., Mulero, F., Ortega, S., Bustelo, X. R.et al. (2011). Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc. Natl. Acad. Sci. USA 108, 5015-5020. 10.1073/pnas.1016933108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Der Burgt, I. and Brunner, H. (2000). Genetic heterogeneity in Noonan syndrome: evidence for an autosomal recessive form. Am. J. Med. Genet. 94, 46-51. 10.1002/1096-8628(20000904)94:1<46::AID-AJMG10>3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- Vaseva, A. V. and Yohe, M. E. (2020). Targeting RAS in pediatric cancer: is it becoming a reality? Curr. Opin. Pediatr. 32, 48-56. 10.1097/MOP.0000000000000856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viosca, J., Schuhmacher, A. J., Guerra, C. and Barco, A. (2009). Germline expression of H-RasG12V causes neurological deficits associated to Costello syndrome. Genes Brain Behav. 8, 60-71. 10.1111/j.1601-183X.2008.00443.x [DOI] [PubMed] [Google Scholar]
- Wang, J., Chandrasekhar, V., Abbadessa, G., Yu, Y., Schwartz, B. and Kontaridis, M. I. (2017). In vivo efficacy of the AKT inhibitor ARQ 092 in Noonan Syndrome with multiple lentigines-associated hypertrophic cardiomyopathy. PLoS ONE 12, e0178905. 10.1371/journal.pone.0178905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, K. N., Wang, D., Cnota, J., Gardner, N., Stabley, D., Sol-Church, K., Gripp, K. W., Witte, D. P., Bove, K. E. and Hopkin, R. J. (2014). Early-lethal Costello syndrome due to rare HRAS tandem base substitution (c.35_36GC>AA; p.G12E)-associated pulmonary vascular disease. Pediatr. Dev. Pathol. 17, 421-430. 10.2350/14-05-1488-OA.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K. B. and Largaespada, D. A. (2020). New model systems and the development of targeted therapies for the treatment of neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Genes (Basel) 11, 477. 10.3390/genes11050477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnowski, L., Zimmer, A. M., Beck, T. W., Hahn, H., Bernal, R., Rapp, U. R. and Zimmer, A. (1997). Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 16, 293-297. 10.1038/ng0797-293 [DOI] [PubMed] [Google Scholar]
- Wong, J. C., Perez-Mancera, P. A., Huang, T. Q., Kim, J., Grego-Bessa, J., Del Pilar Alzamora, M., Kogan, S. C., Sharir, A., Keefe, S. H., Morales, C. E.et al. (2020). KrasP34R and KrasT58I mutations induce distinct RASopathy phenotypes in mice. JCI Insight 5, e140495. 10.1172/jci.insight.140495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., Simpson, J., Hong, J. H., Kim, K.-H., Thavarajah, N. K., Backx, P. H., Neel, B. G. and Araki, T. (2011). MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1L613V mutation. J. Clin. Invest. 121, 1009-1025. 10.1172/JCI44929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., Yin, J., Simpson, J., Kim, K.-H., Gu, S., Hong, J. H., Bayliss, P., Backx, P. H., Neel, B. G. and Araki, T. (2012). Increased BRAF heterodimerization is the common pathogenic mechanism for Noonan syndrome-associated RAF1 mutants. Mol. Cell. Biol. 32, 3872-3890. 10.1128/MCB.00751-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, G. L., Aguena, M., Gos, M., Hung, C., Pilch, J., Fahiminiya, S., Abramowicz, A., Cristian, I., Buscarilli, M., Naslavsky, M. S.et al. (2015). Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J. Med. Genet. 52, 413-421. 10.1136/jmedgenet-2015-103018 [DOI] [PubMed] [Google Scholar]
- Yan, W., Markegard, E., Dharmaiah, S., Urisman, A., Drew, M., Esposito, D., Scheffzek, K., Nissley, D. V., McCormick, F. and Simanshu, D. K. (2020). Structural insights into the SPRED1-neurofibromin-KRAS complex and disruption of SPRED1-neurofibromin interaction by oncogenic EGFR. Cell Rep. 32, 107909. 10.1016/j.celrep.2020.107909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W., Klaman, L. D., Chen, B., Araki, T., Harada, H., Thomas, S. M., George, E. L. and Neel, B. G. (2006). An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev. Cell 10, 317-327. 10.1016/j.devcel.2006.01.002 [DOI] [PubMed] [Google Scholar]
- Yang, W., Wang, J., Moore, D. C., Liang, H., Dooner, M., Wu, Q., Terek, R., Chen, Q., Ehrlich, M. G., Quesenberry, P. J.et al. (2013). Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491-495. 10.1038/nature12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen, I., Shanahan, F., Lee, J., Hong, Y. S., Shin, S. J., Moore, A. R., Sudhamsu, J., Chang, M. T., Bae, I., Dela Cruz, D.et al. (2021). ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 594, 418-423. 10.1038/s41586-021-03515-1 [DOI] [PubMed] [Google Scholar]
- Yi, J., Chen, M., Wu, X., Yang, X., Xu, T., Zhuang, Y., Han, M. and Xu, R. (2010). Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev. Dyn. 239, 2005-2013. 10.1002/dvdy.22343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, J.-S., Huang, Y., Kwaczala, A. T., Kuo, I. Y., Ehrlich, B. E., Campbell, S. G., Giordano, F. J. and Bennett, A. M. (2016). Low-dose dasatinib rescues cardiac function in Noonan syndrome. JCI Insight 1, e90220. 10.1172/jci.insight.90220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, L. C., Hartig, N., Boned Del Rio, I., Sari, S., Ringham-Terry, B., Wainwright, J. R., Jones, G. G., McCormick, F. and Rodriguez-Viciana, P. (2018). SHOC2-MRAS-PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis. Proc. Natl. Acad. Sci. USA 115, E10576-E10585. 10.1073/pnas.1720352115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Z.-H., Zhang, R.-Y., Walls, C. D., Chen, L., Zhang, S., Wu, L., Liu, S. and Zhang, Z.-Y. (2014). Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry 53, 4136-4151. 10.1021/bi5002695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, X., Bu, H., Zhou, J., Yang, C.-Y. and Zhang, H. (2020). Recent advances of SHP2 inhibitors in cancer therapy: current development and clinical application. J. Med. Chem. 63, 11368-11396. 10.1021/acs.jmedchem.0c00249 [DOI] [PubMed] [Google Scholar]
- Zenker, M., Horn, D., Wieczorek, D., Allanson, J., Pauli, S., van der Burgt, I., Doerr, H.-G., Gaspar, H., Hofbeck, M., Gillessen-Kaesbach, G.et al. (2007). SOS1 is the second most common Noonan gene but plays no major role in cardio-facio-cutaneous syndrome. J. Med. Genet. 44, 651-656. 10.1136/jmg.2007.051276 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.