

Abstract
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability and the leading monogenic cause of autism. The condition stems from loss of fragile X mental retardation protein (FMRP), which regulates a wide range of ion channels via translational control, protein-protein interactions, and second messenger pathways. Rapidly increasing evidence demonstrates that loss of FMRP leads to numerous ion channel dysfunctions, i.e., channelopathies, which in turn contribute significantly to FXS pathophysiology. Consistent with this, pharmacological or genetic interventions that target dysregulated ion channels effectively restore neuronal excitability, synaptic function, and behavioral phenotypes in FXS animal models. Recent studies further support a role for direct and rapid FMRP-channel interactions in regulating ion channel function. This review lays out the current state of knowledge in the field regarding channelopathies and the pathogenesis of FXS, including promising therapeutic implications.
Fragile X syndrome (FXS) is the leading monogenic cause of intellectual disability and autism. Affecting both males (~1/4000) and females (~1/5000-8000), FXS is typically associated with cognitive dysfunction (language delay, intellectual disabilities, learning deficits), social and behavioral problems (anxiety, autism spectrum disorders), neurological deficits (seizures, abnormal sleep patterns), and morphological abnormalities (dysmorphic faces and macroorchidism)1. The syndrome is caused by loss of fragile X mental retardation protein (FMRP), which most commonly results from the expansion of an unstable triplet CGG-repeat motif (>200) located within the 5’-untranslated region of the FMR1 gene on the X chromosome. This expansion causes gene hypermethylation and transcriptional silencing. Rare mutations in FMR1 that result in ‘loss-of-function FMRP’ have also been linked to FXS2.
Increasing evidence suggests that the symptoms of FXS stem from disruptions in neuronal activity3. Indeed, FMRP, with its multiple RNA-binding and protein-protein interaction domains (BOX 1), is highly expressed throughout the nervous system4. It is involved in a broad range of physiological functions, including genome stabilization, RNA-editing, pre-RNA splicing, regulation of cell differentiation, and modulation of ion channel function, neuronal excitability, and synaptic plasticity. Specific to ion channel function, FMRP works through multiple direct and/or indirect mechanisms to control their expression and activity (TABLE 1)3,5,6. Indeed, many types of ion channels are dysfunctional in Fmr1 knockout (KO) models, including voltage-dependent ion channels (Na+ channels, K+ channels, Ca2+ channels, and HCN channels), voltage-independent ion channels (SK channels), and ligand-gated ion channels (ionotropic-glutamatergic and GABAergic receptors). While these abnormalities are not caused by genetic mutations in the ion channel genes themselves, we broadly refer here to these ion channel dysfunctions as channelopathies. At the functional level, these channelopathies contribute to neuronal and network hyperexcitability, cognitive dysfunction, and behavioral abnormalities, all of which are part of FXS pathophysiology (FIG. 1). In this review, we first summarize evidence of hyperexcitability at the behavioral, circuit, cellular, and synaptic level in FXS. We then present findings in support of channelopathies underlying these excitability-related defects, including key molecular mechanisms. Finally, we discuss implications for future therapeutic strategies targeting ion channels in FXS.
Box 1|. FMRP functional domains.
The multi-domain structure of FMRP enables it to modulate the functions of ion channels through multiple mechanisms including regulation of channel translation, surface expression, and/or gating dynamics, or by indirectly regulating channel activity through second-messenger signaling pathways. FMRP is a protein of 632 amino acids with a molecular weight of ~80 kDa. The N-terminal domain (aa1-202) contains two Tudor (Agenet) domains (AG1 aa3-49 and AG2 aa63-113, both of which are involved in protein-protein interactions), one nuclear localization signal (NLS, aa115-154) and one K Homology (KH) domain (KH0, aa126-202). The central domain (aa203-404) comprises the KH1 (aa216-280) and KH2 (aa281-404) motifs that mediate RNA binding. The C-terminal domain contains the RGG box (aa527-552), a high-affinity RNA-binding domain which interacts with G-quadruplex RNAs and is essential for FMRP’s association with polyribosomes. The C-terminus also harbors a low complexity domain (LCD, aa466-632), which can promote dynamic interactions with proteins and RNAs. The LCD is implicated in the formation of ribonucleoprotein particles. Schematic representation shows FMRP functional domains159. Three major domains are color-coded. Numbers indicate position within the amino acid sequence.

Table 1 |.
Mechanisms of FMRP regulation of ion channels
| Channel/modulation | Direction of modulation by FMRP | Refs |
|---|---|---|
| Translational control | ||
| Kv1.2 | ⇧ or ⇩ | 58, 65, 95 |
| Kv3.1 | ⇩ | 67, 68 |
| Kv4.2 | ⇧ or ⇩ | 95–97 |
| Slack | ⇄ or ⇩ | 72, 95 |
| BK | ⇧ | 158 |
| Cav1.3 | ⇧ or ⇩ | 88, 99 |
| Cav2.1 | ⇧ | 89, 95 |
| Cav2.3 | ⇩ | 90, 95 |
| HCN | ⇧ or ⇩ | 56, 57, 95, 102 |
| AMPAR | ⇧ or ⇩ | 59, 83, 104, 105 |
| NMDAR | ⇧ or ⇩ | 59, 111–113, 116 |
| GABAAR | ⇩ | 64, 119–125 |
| Ion channel activity via FMRP-channel interactions | ||
| Slack | ⇧ | 72 |
| BK | ⇧ | 57, 62, 80 |
| SK | ⇧ or ⇩ | 51, 86 |
| Kv1.2 | ⇧ | 65 |
| Kv4 | ⇩ | 91 |
| Cav3 | ⇩ | 91 |
| Ion channel surface expression via FMRP-channel interactions | ||
| Kv1.2 | ⇧ | 65 |
| Cav2.1 | ⇧ | 60, 89 |
| Cav2.2 | ⇩ | 60 |
| HCN | ⇧ or ⇩ | 102 |
| AMPAR | ⇧ or ⇩ | 59, 108 |
| Second messenger pathways | ||
| INaP | ⇩ | 52 |
| Unidentified | ||
| NKCC1 | ⇩ | 145, 146 |
⇧ or ⇩, FMRP increases or decreases channel function, respectively;
⇄, no significant change in channel function;
Figure 1 |. Overview of channelopathies in FXS.
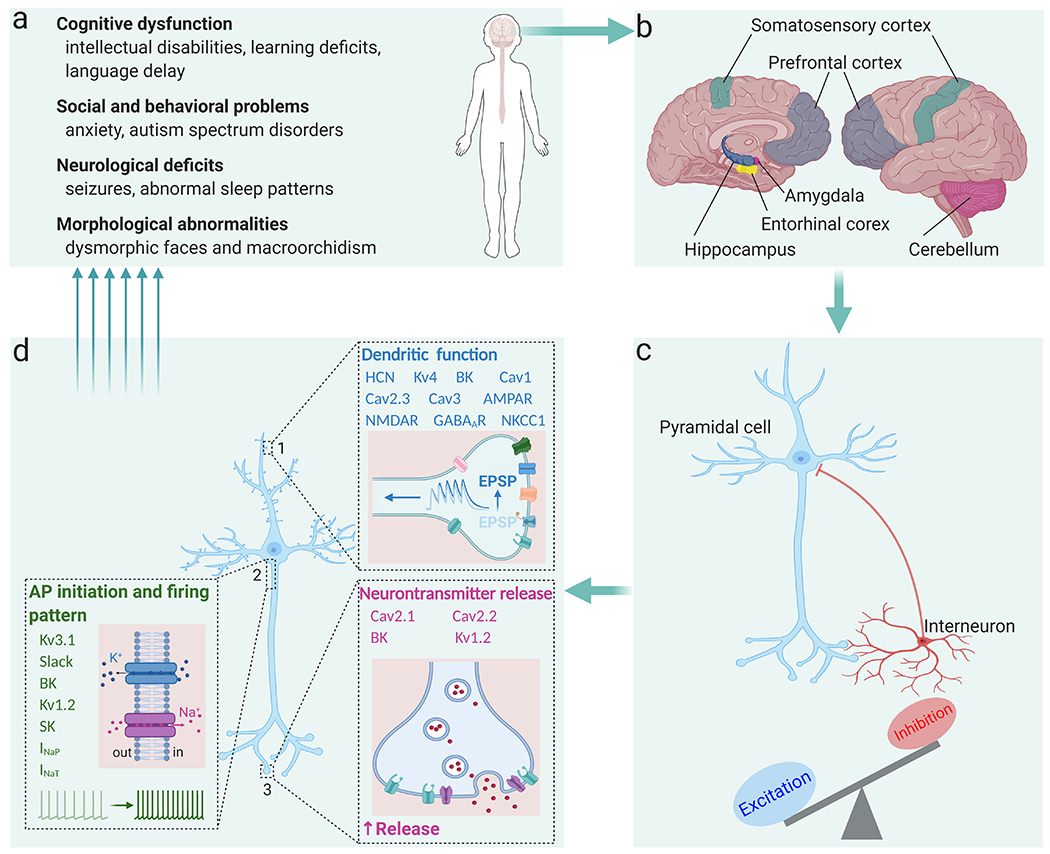
a | Though FXS clinical manifestations are complex and vary considerably among individuals, most FXS patients exhibit cognitive dysfunction (intellectual disabilities, learning deficits, language delay), social and behavioral problems (anxiety, autism spectrum disorders), neurological deficits (seizures, abnormal sleep patterns), indicative of cellular and network hyperexcitability. b | Brain regions that are affected in FXS and are discussed in this review (somatosensory cortex, prefrontal cortex, hippocampus, entorhinal cortex, amygdala, cerebellum) are shown in a medial view of the brain (left panel, brain stem and cerebellum removed) and in a lateral view of the brain (right panel). c | A cartoon representation of the basic unitary neural circuit showing increased circuit excitability and excitation/inhibition imbalance, a key feature of the circuit changes observed in FXS. d | Hyperexcitability defects observed in FXS can be attributed to channelopathy-caused functional abnormalities in (1) AP initiation and firing; (2) neurotransmitter release; and/or (3) dendritic function, which may underlie various FXS phenotypes shown in a (multiple arrows). Ion channels contributing to these excitability defects are listed in corresponding inserts 1, 2 or 3. Insert 1, The FXS-related hyperexcitability is apparent in elevated neuronal firing frequency. These changes involve functional alterations in both potassium channels and sodium channels, including Kv1.2, Kv3.1, Slack, BK, SK, INaP and INaT. AP traces in light-green and green represent normal and FXS conditions, respectively, indicating that loss of FMPR increases AP firing frequency. Insert 2, The transduction of neuronal firing into neurotransmitter release at presynaptic terminals is mediated by Ca2+ influx through the voltage-gated calcium channels (VGCCs). Cav2.1 and Cav2.2 channels are the predominant VGCCs supporting neurotransmitter release in central neurons. FMRP can also indirectly regulate neurotransmitter release by controlling AP peak and duration, which in turn determine the amplitude and duration of presynaptic calcium influx through VGCCs. This mechanism is mediated by a subset of voltage-gated K+ channels, including Kv1.2 and BK channels. Insert 3, Dendritic excitability is governed, in large part, by ion channels, a number of which are dysregulated in the absence of FMRP, including HCN, Kv4, BK, Cav1, Cav2.3, Cav3, AMPAR, NMDAR, GABAAR and NKCC1. EPSC waveforms in light-blue and blue denote normal and FXS conditions, respectively, indicating that loss of FMRP enhances EPSC integration and thus increases dendritic excitability.
Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BK, large-conductance calcium-activated potassium channel; Cav1, voltage-gated calcium channel L-type; Cav2.1, voltage-gated calcium channel P/Q type; Cav2.2, voltage-gated calcium channel N type; Cav2.3, voltage-gated calcium channel R type; Cav3, voltage-gated calcium channel T type; GABAAR, γ-aminobutyric acid type A receptor; HCN, hyperpolarization-activated, cyclic nucleotide-gated ion channel; INaP, persistent sodium current carried by voltage-gated sodium channel; INaT, transient sodium current carried by voltage-gated sodium channel; Kv1.2, voltage-gated potassium channel subfamily A member 2; Kv3, voltage-gated potassium channel subfamily C; Kv4, voltage-gated potassium channel subfamily D; NKCC1, Na+–K+–Cl− cotransporter; NMDAR, N-methyl-D-aspartate receptor; NT, neurotransmitter; SK, small-conductance calcium-activated potassium channel; Slack, sodium-activated potassium channel.
Evidence of hyperexcitability in FXS
The clinical phenotypes of FXS are quite complex and vary considerably across individuals. However, most exhibit neurological symptoms indicative of network hyperexcitability, including seizures, hyperarousal, hyperactivity, and hypersensitivity to sensory stimuli7. Neuronal hyperexcitability is also consistently observed in FXS animal models, data from which indicate intrinsic hyperexcitability and/or excitation/inhibition (E/I) imbalance in neuronal circuits3,8,9. We summarize the findings from both human patients and animal models below.
Behavioral abnormalities/symptoms
Epilepsy—an outward expression of circuit-level hyperexcitability—occurs in 10–20% of individuals with FXS. Seizure patterns on electroencephalography (EEG) typically present as benign focal epilepsy of childhood10, though many affected individuals continue to have seizures after the age of 2011. FXS patients are also hyper-responsive to sensory stimulation and display tactile defensiveness12,13. Sensory hyperreactivity can cause a hyperaroused state characterized by disruptions in circadian rhythms, including frequent awakenings from sleep14,15. The seizures and hyperresponsivity are recapitulated in FXS models16; Fmr1 KO mice have a lower threshold for audiogenic seizures17–23, hypersensitivity to sensory stimuli24–26, and hyperactivity27–29.
Circuit and network dysfunctions
Increased excitability at the network level has been measured in patients using various non-invasive techniques. For example, EEG abnormalities are commonly observed in FXS individuals11,15,30, including centrotemporal spikes, slowed background rhythm, and multifocal epileptiform discharges. Sensory hypersensitivity is also apparent in the elevated amplitude of evoked potentials in response to auditory and/or visual stimuli31, including heightened event-related brain potentials within auditory cortex of children with FXS32. Looking into the mechanisms of cortical network hyperexcitability in FXS individuals, Morin-Parent et al.9 used transcranial magnetic stimulation to show that patients with FXS have significantly reduced short-interval intracortical inhibition (mediated by GABAA receptors), increased long-interval intracortical inhibition (mediated by GABAB receptors), and increased intracortical facilitation (mediated by glutamatergic receptors) compared to healthy controls. Overall, the data support that loss of FMRP leads to network hyperexcitability, hypersensitivity to sensory stimuli, and E/I imbalance.
Some of the specific neurological symptoms associated with FXS are also indicative of circuit dysfunction in particular brain regions. For example, tactile defensiveness, learning and memory deficits, fear and anxiety—all of which are common in FXS—can be linked to circuit/network abnormalities in the somatosensory cortex, hippocampus, or amygdala, respectively. Data from FXS animal models confirm the presence of circuit abnormalities and marked E/I imbalance in these regions. In the somatosensory cortex of Fmr1 KO mice, circuit hyperexcitability manifests as prolonged UP states33–35. Studies also find elevated cortical network synchrony in layer 2/3 of barrel cortex36, whereas network inhibition during the UP states is less synchronous33. Also in this model, Domanski and colleagues observe diminished feed-forward inhibition (FFI) and increased E/I ratio during trains of activity in layer 4 barrel cortex neurons37. Similar network E/I imbalance is also observed in primary visual cortex38 and auditory brain stem39–41 of FXS animal models. In the hippocampal circuits of Fmr1 KO mice, hyperexcitability manifests in elevated epileptiform activity42,43 as well as abnormally high spiking probability and reduced spiking precision in hippocampal FFI circuits44,45. These defects are associated with a marked shift in E/I circuit balance, primarily a result of reduced activity of FFI hippocampal interneurons. In the amygdala of Fmr1 KO mice, though the overall E/I balance appears to be maintained, the temporal window of peak excitation to peak inhibition is significantly narrower as a result of reduced tonic GABAAR-mediated inhibition46,47. This narrowing leads to circuit hyperexcitability and abnormal amygdala functions46,48–50.
Collectively, these data support the presences of hyperexcitable circuits and networks in FXS individuals and animal models, some of which may underlie characteristic FXS symptoms.
Synaptic and cellular defects
Some of the behavioral and network-level hallmarks of FXS are driven by cellular and synaptic mechanisms. There is now extensive evidence of changes in synaptic transmission and plasticity, as well as dysregulated cellular excitability in FXS. For example, in animal models neurons across multiple brain regions exhibit an abnormally high action potential (AP) firing rate, including in hippocampus, somatosensory cortex, entorhinal cortex, and the auditory brainstem33,36,37,51–54.
In terms of dendritic function and synaptic plasticity, Fmr1 KO mice have a higher threshold for spike-timing-dependent plasticity in the prefrontal cortex55, and changes in the input resistance of dendritic compartments, which can significantly impact dendritic summation and thus cellular excitability37,56–58. In line with elevated dendritic excitability, higher single spine glutamate currents and elevated dendritic gain (resulting in higher AP output) are observed in somatosensory cortex layer 4 stellate cells of Fmr1 KO mice59. In addition, there are major FMRP-related abnormalities in neurotransmitter release across several types of neurons. For instance, glutamate release is elevated in the axon terminals of dorsal root ganglion neurons60,61 and at the hippocampal CA3-CA1 synapse during spike trains43,62,63.
Several studies have also observed synaptic deficits in inhibitory interneurons, including reduced GABA release in the hippocampus of Fmr1 KO mice44,64, attributable in part to excessive presynaptic GABAB receptor signaling in inhibitory presynaptic terminals44. In contrast, Yang and colleagues report increased GABA release from basket cells in the cerebellum of Fmr1 KO mice, resulting in reduced firing frequency among postsynaptic Purkinje neurons65. Though contradictory at first glance, Purkinje neurons are inhibitory, and thus the circuit-level effect of these changes are similar—both reduce circuit inhibition and promote hyperexcitability. Similarly, in the amygdala, loss of FMRP causes a marked reduction in the frequency and amplitude of inhibitory postsynaptic currents (IPSCs), reduced tonic inhibition, and decreased GABA availability48. In the subiculum, loss of FMRP leads to selective reduction of tonic inhibition, but not phasic IPSCs, suggesting that specific subtypes of GABAA receptors mediating tonic inhibitory currents are downregulated66.
The Roles of Channelopathies in FXS
Overview
Most of the pathological changes in excitability associated with FXS can be attributed to ion channel dysfunction that occurs as a result of the loss of FMRP, which regulates multiple types of channels across various neuronal subcompartments (FIG 1. and TABLE 1). The specific functional consequences of FMRP loss include changes in AP initiation and firing pattern (involving various K+, and Na+ channels), neurotransmitter release (involving Ca2+ and K+ channels), and dendritic function (involving K+, Ca2+, and HCN channels, as well as AMPAR, NMDAR and GABAAR) (BOX 2, TABLE 2). In turn, these functional effects are likely part of the mechanisms underlying cognitive and behavioral hallmarks of FXS. In the following sections we discuss recent advances in our understanding of channelopathy-based mechanisms of FXS pathophysiology, and potential new therapeutic strategies stemming from our growing knowledge of FXS-related ion channel defects. Where applicable, we also review efforts to investigate potential therapeutic interventions that target particular channelopathies (TABLE 3).
Box 2 |. Mechanisms underlying excitability defects in FXS models.
Loss of FMRP dysregulates a large number of ion channels. This in turn affects multiple aspects of neuronal excitability, including changes in AP initiation and firing rate, presynaptic vesicle release, dendritic properties, and synaptic plasticity (TABLE 2).
The combined activity of voltage-gated Na+ and K+ channels determine AP shape, firing rate and pattern. For example, channels that active around threshold potentials, such as SK channels and Na+ channels carrying INaP, contribute to AP initiation. Na+ channels carrying INaT determine AP rising speed and amplitude. Several families of voltage-gated K+ channels act in concert to determine AP repolarization dynamics, and thus AP duration.
AP firing patterns are transduced into synaptic vesicle release at synaptic terminals via activation of VGCCs, among which P/Q- and N-type Ca2+ channels are the major source of Ca2+ for synaptic vesicle release in central neurons.
Multiple channels act together to control dendritic excitability, input resistance, and thus spatial and temporal summation, including K+ channels, VGCCs, and HCN channels. Many of these channels are also involved in the initiation and maintenance of various forms of synaptic plasticity. Ligand-gated ion channels/ionotropic receptors (AMPA, NMDA and GABAA receptors) function to directly evoke excitatory or inhibitory synaptic currents.
Many channels are expressed in multiple cellular compartments and be activated by different factors, in cell-type and brain-region specific manners.
Table 2 |.
Mechanisms underlying excitability defects in FXS models
| Channel | Changes | Functional abnormality | Refs |
|---|---|---|---|
| AP initiation and firing pattern | |||
| Kv3.1 | ↑ | ↑AP repolarization speed and firing rate | 53, 68 |
| Slack | ↓ | ↑ AP duration and firing rate | 73 |
| BK | ↓ | ↑ AP duration, ↓ fAHP | 62 |
| Kv1.2 | ↓ | ↑ AP duration | 65 |
| SK | ↓ ↑ |
↓ AP threshold and mAHP, ↑ AP firing rate ↑ mAHP, ↓AP firing rate |
51
86 |
| INaP | ↑ | ↓ AP threshold, ↑ AP firing rate | 52 |
| INaT | ↑ | ↑ AP rising speed and amplitude ↑ AP firing rate, ↓ AP duration |
54 |
| Neurotransmitter release | |||
| Cav2.1 | ↓ | May be a compensation for Cav2.2 | 89 |
| Cav2.2 | ↑ | ↑ Ca2+ influx and vesicle release | 60, 61 |
| BK | ↓ | ↑ Ca2+ influx and vesicle release | 62 |
| Kv1.2 | ↓ | ↑ Ca2+ influx and vesicle release | 65 |
| Dendritic function | |||
| Kv4 | ↑ | ↑ threshold for LTP, Lack of TBS-induced LTP | 91, 97 |
| Cav3 | ↑ | ↑ Ca2+ influx | 88 |
| BK | ↓ | ↑ dendritic excitability, ↑ sensory sensitivity, ↑ dendritic spines | 57, 77 |
| Cav1 | ↓ | ↑ threshold for tLTP | 99,55 |
| ↑ | ↑ progenitor differentiation to glutamate-responsive cells | 88 | |
| Cav2.3 | ↑ | ↑ Ca2+ influx | 90 |
| HCN | ↓ | ↑ input resistance and summation | 56, 102 |
| ↑ | ↓ input resistance and summation | 57,59,102 | |
| AMPAR | ↓ | ↓ LTP and ↑ LTD | 104, 105 |
| ↑ | ↓ LTP, ↑ AMPAR current ↑ silent dendritic spines |
59, 83 | |
| NMDAR | ↓ | ↓ NMDAR currents | 113, 116 |
| ↑ | ↑ NMDAR mEPSC frequency | 59 | |
| GABAAR | ↓ | ↓ inhibition | 64,120,121 |
| NKCC1 | ↑ | delayed GABAAR polarity switch delayed maturation of glutamatergic synapses |
145, 146 |
↑, increase; ↓, decrease; AP, action potential; fAHP, fast afterhyperpolarization; LTD, long-term depression; LTP, long-term potentiation; mEPSC, miniature excitatory postsynaptic current; MF, mossy fiber; RMP, resting membrane potential; TBS, theta burst stimulation; tLTP, spike-timing-dependent long-term potentiation.
Table 3 |.
Manipulations that rescue cellular, circuit and behavioral phenotypes in FXS models
| Target | Rescue strategy | Behavioral phenotypes normalized | Refs |
|---|---|---|---|
| Kv1.2 | Oral administration of Kv1.2 agonist DHA | Acoustic startle reflex and social interaction | 65 |
|
| |||
| Kv3.1 | Kv3.1 positive modulator AUT2 | MNTB neurons firing rate Auditory brain response |
53 |
|
| |||
| Kv4 | Kv4 blocker heteropodatoxin | TBS-induced LTP in hippocampus | 97 |
|
| |||
| Slack | FMRP1-298 | Slack open probability. | 72, 73 |
|
|
|||
| BK | Intracellular application of FMRP1-298 Genetic upregulation of BK activity by ablation of BKβ4 subunit |
AP duration, BK open probability, glutamate release Short-term plasticity Epileptiform activity |
43, 62, 76 |
| Intraperitoneal injection of BK channel opener BMS-204352 | Acoustic startle reflex, locomotor activity, nest building, grooming, social interactions, anxiety and spatial memory | 57, 77, 84 | |
|
| |||
| SK | Intracellular administration of FMRP1-298, Genetic reintroduction of FMRP1-234 or SK channel opener 1-EBIO or NS309 |
AP threshold, mAHP and AP firing rate MF-CA3 input-output transmission. |
51 |
|
| |||
| NMDAR | Bath application of NMDAR coagnoists glycine or D-serine | NMDAR-dependent LTP in dentate gyrus | 113 |
|
| |||
| GABAAR | Intraperitoneal injection of GABAAR agonist ganaxolone, acamprosate or metadoxine. | UP states, locomotor activity, anxiety, marble burying behavior and sensory hyperresponsiveness. | 127,131 |
|
| |||
| NKCC1 | Intraperitoneal injection of NKCC1 inhibitor bumetanide | GABAAR reversal potential of L4 somatosensory neurons Excitatory synapse development LTP in somatosensory cortex Whisker-evoked responses in the cortex |
146 |
|
| |||
| FMRP loss | Intravenous injection of FMRP297-tat | Cav3 and Kv4 currents Cerebellar MF-granule cell LTP Protein levels (APP, αCaMKII and PSD95), locomotor activity |
91 |
αCaMKII, alpha-Ca2+/calmodulin-dependent protein kinase II; AUT2, ((4-(53oxy)-2-(1-methylethyl) benzonitrile; CP, critical period; DHA, docosahexaenoic acid; mAHP, medium afterhyperpolarization; MF, mossy fiber; NS309, 3-Oxime-6,7-dichloro-1H-indole-2,3-dione; MNTB, medial nucleus of the trapezoid body; PSD, postsynaptic density.
AP Initiation and Firing Patterns
The FXS-related hyperexcitability noted at the behavioral and circuit level is apparent in lowered AP threshold and elevated firing frequency characteristic of Fmr1 KO animal models. These changes involve functional alterations to both potassium (K+) channels and sodium (Na+) channels.
The role of K+ channels
Potassium channels crucially contribute to establishing resting membrane potential, and setting AP dynamics and firing patterns. FMRP regulates the translation of a number of K+ channels, as well as their activity and surface expression (FIG. 2).
Figure 2 |. FMRP controls ion channel functions through multiple mechanisms.

There are four major mechanisms by which FMRP regulates channel functions. a | FMRP regulates ion channel’s gating. By interacting directly with the pore-forming subunit (1) and/or with the regulatory subunit (2) of ion channels, FMRP modulates the function of the pore-forming subunit and thus regulates neuronal excitability and neurotransmitter release. Ion channels regulated by FMRP through this mechanism include Slack, BK, SK, Kv1.2, Kv4.3 and Cav3. b | By binding to mRNA of ion channels, FMRP regulates ion channel translation and thus modulates neural excitability. Ion channels regulated by FMRP through this mechanism include Slack, BK, Kv1.2, Kv3.1, Kv4.2, Cav1.3, Cav2.1, Cav2.3, HCN, AMPAR, NMDAR and GABAAR. c | By binding to ion channels, FMRP modulates ion channel trafficking and surface expression. Ion channels regulated by FMRP through this mechanism include Kv1.2, Cav2.1, Cav2.2, HCN and AMPAR. d | FMRP controls ion channel activity via second messenger pathways. By modulating mGluR5-PKC-PLC pathway, FMRP modulates persistent sodium current (INaP) and thus regulates AP threshold and excitability. Note that the same channel can be modulated via multiple mechanisms. Also, the direction of changes, extent and the mechanism of channel modulation by FMRP is often brain-region, cell-type and even subcellular-compartment specific
Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BK, large-conductance calcium-activated potassium channel; Cav1.3, voltage-gated calcium channel with α1D subunit L-type; Cav2.1, voltage-gated calcium channel P/Q type; Cav2.2, voltage-gated calcium channel N type; Cav2.3, voltage-gated calcium channel R type; Cav3, voltage-gated calcium channel T type; GABAAR, γ-aminobutyric acid type A receptor; HCN, hyperpolarization-activated, cyclic nucleotide-gated ion channel; INaP, persistent sodium current carried by voltage-gated sodium channel; Kv1.2, voltage-gated potassium channel subfamily A member 2; Kv3.1, voltage-gated potassium channel subfamily C member 1; Kv4.2, voltage-gated potassium channel subfamily D member 2; Kv4.3, voltage-gated potassium channel subfamily D member 3; mGluR5, metabotropic glutamate receptor subtype 5; NMDAR, N-methyl-D-aspartate receptor; PKC, protein kinase C; PLC, phosphoinositide phospholipase C; SK, small-conductance calcium-activated potassium channel; Slack, sodium-activated potassium channel.
Kv3.1 channels.
FMRP directly regulates the expression of delayed rectifier K+ channel (Kv3.1) mRNA67,68. The rapid voltage-dependent activation/deactivation characteristics of Kv3.1 facilitate high firing rates in cells that express these channels. For example, there is normally a gradient of Kv3.1 expression within the sound localization circuitry of the brainstem (the medial nucleus of the trapezoid body, MNTB). However, when FMRP is absent, the normal gradient distribution of Kv3.1 is flattened and the levels of Kv3.1b protein are higher overall69. Consequently, the high-threshold (Kv3.1-dependent) current is increased in the MNTB of Fmr1 KO mice, leading to faster AP repolarization and higher firing rates (i.e., hyperexcitability)53.
Therapeutic implications.
Since increased Kv3.1 channel expression contributes to impaired encoding and processing of auditory information in Fmr1 KO mice68, these channels are promising therapeutic targets. Indeed, the Kv3.1 channel modulator AUT2 normalizes AP firing rate in auditory brainstem neurons and restores the auditory brainstem response in vivo in Fmr1 KO mice53. Targeting Kv3.1 channels may have an even broader therapeutic potential to reduce sensory hypersensitivity across multiple modalities in FXS, as Kv3.1 channels are widely expressed in rapidly firing neurons throughout the nervous system70. However, it remains to be determined if loss of FMRP also increases excitability among these rapidly firing neurons in various brain regions in a Kv3.1-dependent manner.
Slack channels.
Slack channels are widely expressed in the brain, including in brainstem, cerebellum, prefrontal cortex, and hippocampus. As a major component of the delayed outward current, Slack channels are critical in regulating AP firing patterns and temporal precision71. FMRP directly modulates Slack channel activity72. In a seminal study, Kaczmarek and colleagues show that the N-terminal domain of FMRP interacts with the Slack channel C-terminus to increase the frequency of the channel opening (i.e. gating) in a rapid and reversible manner72,73. Through this protein-protein interaction, FMRP dynamically and precisely tunes Slack channel activity and thus neuronal firing. In the absence of FMRP, Slack currents are significantly reduced, thus increasing neuronal excitability and reducing temporal precision of spiking, as observed in the auditory brainstem of Fmr1 KO mice72. Together with the aforementioned effects on Kv3.1 channels, the FMRP-related decrease in Slack currents likely contributes to auditory hypersensitivity in FXS.
Interestingly, an additional “nonconducting” function for Slack channels in regulating neuronal firing has been reported in Aplysia bag cell neurons in which activation of Slack channels is linked to protein synthesis-dependent, prolonged changes in neuronal firing patterns73. This function raises the possibility that Slack-FMRP interactions may link changes in neuronal firing to changes in protein translation, known to be disrupted in FXS.
BK channels.
Large conductance voltage-and Ca2+-activated K+ (BK) channels are widely expressed throughout the brain, and in nearly every cellular compartment of neurons75. Being both voltage- and calcium-activated, these channels play critical roles in regulating neuronal excitability, AP duration, firing, and neurotransmitter release75. FMRP targets BK channels, regulating translation and function43,57,62,76–80. Specifically, the N-terminal domain of FMRP interacts with the channel’s regulatory β4 subunit to increase BK channel’s Ca2+ sensitivity and thus open probability43,62. FMRP can also directly bind to the BK channel’s pore-forming α subunit76,80 to modulate channel gating and increase BK currents80. Indeed, studies of both Fmr1 KO mice and stem cell-derived neurons from FXS patients report that the loss of FMRP reduces BK channel activity, which in turn leads to a wide range of functional consequences. For example, in hippocampal and cortical neurons these effects include increased firing, excessive AP broadening, elevated presynaptic Ca2+ influx, and increased glutamate release43,62,76,81.
More recent studies support the idea that the loss of FMRP-BK channel interactions contribute to FXS pathophysiology. Particularly interesting is the discovery that a rare FMRP missense mutation (R138Q), which is associated with a partial clinical FXS phenotype (intellectual disability and seizures) in at least one human patient, impairs the protein’s translation-independent presynaptic functions, including its ability to interact with BK channels to regulate AP duration and control glutamate release (i.e., excitability)76. In drosophila, this mutation impairs the presynaptic-specific function of FMRP to control axonal elaboration in the neuromuscular junction76. And in mouse cortical neurons, while the R138Q mutation preserves FMRP’s canonical mRNA binding and translational regulation functions—generally associated with (but not limited to) postsynaptic compartments—it impairs interactions with BK channels that ultimately control glutamate release76. These results suggest a model in which the effects of R138Q mutation are caused by disruption of FMRP’s protein-protein interactions, including FMRP-BK channel interactions, which are important in regulating neuronal excitability and glutamate release, while retaining translational regulation functions of FMRP.
Despite the compelling alignment of both clinical and preclinical data linking these specific FMRP functions to a specific subset of FXS phenotypes, the story is likely more complicated. Two other individuals with FXS who harbor the same mutation present with different clinical phenotypes: one (male) presents with a more extensive set of FXS features, including intellectual disability, ADHD, autism, seizures, craniofacial changes, and hyperextensibility2; the other (unrelated female) presents with mild intellectual disability and attention deficits82. Data from a rodent model of FXS specific to the R138Q mutation indicate that at least some postsynaptic functions of FMRP are also impacted by this mutation; surface expression of AMPA receptors is increased, as is dendritic spine density, hippocampal LTP is impaired, and there are socio-cognitive deficits at the behavioral level83. These data suggest that the R138Q mutation may disrupt FMRP interactions with other channels as well, resulting in both pre- and postsynaptic defects.
Therapeutic implications.
Upregulating BK channel activity may be a useful approach to normalize a range of excitability-associated phenotypes in FXS. Indeed, genetically upregulating BK channel activity normalizes multiple synaptic and circuit defects in an FXS mouse model, including AP broadening, elevated glutamate release, and epileptiform activity in the hippocampal circuit43. Moreover, the BK channel opener BMS-204352 corrects a number of behavioral phenotypes in Fmr1 KO mice, including hyperactivity, hypersensitivity to sensory stimuli, social defects, anxiety, deficient spatial memory, impaired nest building, and excessive grooming57,77,84. Together, these studies suggest that targeting BK channels may have therapeutic potential in alleviating a number of excitability-associated FXS phenotypes.
Kv1.2 channels.
Kv1.2 channels are widely distributed across various cellular compartments of neurons, and play important roles in regulating intrinsic excitability and AP initiation85. In Fmr1 KO mice, the Kv1-mediated current is downregulated in one population of layer 5 pyramidal cells (PCs) of the medial prefrontal cortex (called long tract-projecting neurons), leading to increased excitability of these neurons58. Interestingly, this defect appears to be cell-type specific, as it is not observed in the neighboring intratelencephalic projecting PCs of the same brain region. Mechanistically, FMRP binds Kv1.2 channel mRNA, as well as interacts directly with the channel through protein-protein interactions to regulate channel activity and surface expression. Specifically, recent studies indicate that the N-terminus of FMRP directly binds to a phosphorylated serine motif on the C-terminus of Kv1.2 to form a dimer-dimer configuration in which two C-terminal strands of the same Kv1.2 channel bind to one FMRP dimer65. This interaction enhances axonal trafficking, membrane expression, and gating dynamics of Kv1.2 in axonal terminals of cerebellar basket cells, ultimately impacting presynaptic excitability and Ca2+ influx. Consequently, loss of FMRP-Kv1.2 interaction in Fmr1 KO basket cells increases the duration and amplitude of APs, leading to enhanced presynaptic Ca2+ influx and excessive GABA release onto Purkinje neurons65. Since Purkinje neurons are inhibitory, the overall effect of these changes is to promote circuit hyperexcitability.
Therapeutic implications.
As Purkinje neurons are the sole output cells from cerebellar cortex, the Kv1.2 channelopathy is likely to lead to abnormal cerebellar functions. Targeting Kv1.2 may thus help normalize cerebellar circuit hyperexcitability and cerebellar-related abnormalities in FXS. Indeed, the therapeutic potential of one Kv1.2 agonist, docosahexaenoic acid, was recently tested in an FXS mouse model, where it was found to rectify the dysregulated cerebellar inhibition in vitro and to rescue acoustic startle reflex and social interaction phenotypes in vivo65.
SK channels.
Voltage-independent K+ channels also play key roles in regulating neuronal excitability by modulating the resting membrane potential, AP threshold, and afterpotentials85. Within the context of Fmr1 KO mice, there is evidence of hypofunction of small conductance Ca2+-activated K+ (SK) channel, which is linked to neuronal hyperexcitability (lower AP threshold, smaller medium AHP, increased firing rate, and exaggerated synaptic gain) in CA3 PCs51. The SK-current defect stems from the loss of FMRP interactions with the SK2 channel isoform, though channel expression is unaffected. This idea is further supported by the finding that all observed excitability defects in CA3 PCs of Fmr1 KO mice are normalized by acute intracellular reintroduction of the N-terminal FMRP fragment (aa1-298), or the genetic reintroduction of an even shorter N-terminal FMRP fragment (aa1-234), both of which lack the KH2 domain and are thus incapable of translational regulation51. Interestingly, similar to other FXS-related channelopathies, the SK defects are cell-type specific. Within a single Fmr1 KO animal, changes to the SK current in CA1 PCs mirror those apparent in CA3: SK current goes up, as does spike rate variability and medium AHP, but AP rate goes down86. Similar cell-type specific changes are also reported for Kv158, and HCN channels56–58 (TABLE 1).
The role of Na+ channels
In most neurons, AP initiation and rising phase are governed by voltage-gated Na+ channel availability and activation/inactivation dynamics. Na+ channels can produce two distinct currents: the fast transient Na+ current (INaT) and the non-inactivating persistent Na+ current (INaP). INaP is active at subthreshold voltages and thus is critical for AP initiation, while INaT underlies the AP rising phase85. Thus, disruption to either current is likely to impact neuronal excitability.
INaP within the context of FXS.
In Fmr1 KO mice, INaP is abnormally increased in the entorhinal cortex layer 3 PCs, i.e., neurons that project directly to CA1 region. This upregulation of INaP leads to profound excitability defects, including lower AP threshold and increased AP firing52. Unlike many K+ channel defects, which are mediated by FMRP-channel interactions within individual neurons, INaP-mediated defects are not apparent in neurons isolated from their circuit. This indicates that the defects are not cell-autonomous. Indeed, the enhanced INaP is caused by exaggerated activation of mGluR5 signaling, which in turn acts on Na+ channels through phospholipase C (PLC) and protein kinase C (PKC)52. This signaling mechanism requires circuit activity and is distinct from the well-established mGluR5 signaling cascade affecting local translation in FXS animal models. Thus, Na+ channel dysregulation is likely a major contributor to neuronal hyperexcitability in the FXS mouse model, at least in this subpopulation of cortical neurons.
INaT within the context of FXS.
While the above study did not include direct measures of INaT, observation of increased AP rising speed, an INaT-dependent parameter, suggests that INaT is also increased in Fmr1 KO neurons52. Indeed, a direct evidence of increased INaT resulting in larger and faster rising APs and a higher gain of somatic excitability is reported in layer 2/3 prefrontal cortex neurons of Fmr1 KO mice54. This study also finds that the observed excitability changes are accompanied by a large depolarizing shift in the activation of A-type K+ channels. This shift is expected to decrease AP threshold, as is the case when INaP is higher. However, no such changes were observed54, indicating that some form of compensatory changes may be involved.
Taken together, the data discussed here reflect a complex interplay of celltype-specific Na+ and K+ channelopathies caused by loss of FMRP. In nearly all cases, the deficits contribute to overall neuronal and circuit hyperexcitability. To elucidate further mechanistic detail will require careful consideration of celltype-specific effects, as well as the impact of compensatory changes.
Neurotransmitter Release
The transduction of neuronal firing into neurotransmitter release at presynaptic terminals is mediated by Ca2+ influx through the voltage-gated calcium channels (VGCC)87. Interestingly, in FXS models all five types of mammalian VGCCs [P/Q (Cav2.1), N (Cav2.2), R (Cav2.3), L (Cav1), and T (Cav3)] are affected (FIG. 2, TABLE 2)6,60,61,88–91. N- and P/Q-type channels are the predominant VGCCs supporting neurotransmitter release at most central synapses and are discussed here; those VGCC types that contribute to dendritic excitability changes in FXS are discussed in the next section.
N-type and P/Q-type VGCCs
The N-type VGCC (Cav2.2) plays a prominent role in neurotransmitter release at presynaptic terminals. It was also the first Ca2+ channel found to be directly modulated by FMRP through protein-protein interactions60. The mechanism of this interaction is distinct from the known FMRP interactions with K+ channels; in the case of N-type VGCC, it is FMRP’s C-terminal domain that interacts with N-type channel’s synaptic targeting domain60,61 to suppress the channels’ presynaptic localization and surface expression. Consequently, loss of FMRP increases N-type VGCC surface expression and glutamate release at presynaptic terminals of sensory neurons in dorsal root ganglia60,61.
The P/Q-type (Cav2.1) channel is also critical for AP-evoked neurotransmitter release in many types of neurons. It is involved in various forms of synaptic plasticity as well as spatial learning and memory in mice92. In cortical neurons, overall levels of P/Q-type channels are unaltered by FMRP loss. However, we know that FMRP can bind to these channels to increase their surface expression60,89; this is opposite of the effect of FMRP interactions with N-type channels. Thus, loss of FMRP results in reduced surface expression of P/Q-type channels, while surface expression of N-type channels is simultaneously increased89. Interestingly, it has been suggested that N- and P/Q-type channels have channel-type preferred slots in hippocampal synapses, which can be occupied by the competing channel type upon certain disease conditions93. Thus, the loss of FMRP may contribute to synaptic transmission deficits by creating an imbalance between the surface levels of N- and P/Q-type channels, which differentially contribute to the efficiency of neurotransmitter release and synaptic plasticity93.
Indirect modulation of calcium influx
In addition to controlling presynaptic surface expression of VGCCs, FMRP can also indirectly regulate neurotransmitter release by controlling AP duration (which in turn determines the duration of presynaptic calcium influx through VGCCs). This mechanism is mediated by a subset of voltage-gated K+ channels that regulate AP duration, and has major implications for glutamate and GABA release, both of which are significantly impacted in Fmr1 KO mice. As discussed above, reduced BK channel activity in hippocampal and cortical excitatory neurons of Fmr1 KO mice leads to extensive AP broadening. This in turn elevates presynaptic calcium influx and glutamate release62. Similarly, the increased AP duration apparent in cerebellar basket cells (secondary to reduced Kv1.2 channel activity) leads to excessive GABA release65. VGCC-K+ channel activity interdependence is often reciprocal; for example, VGCCs are the main source of Ca2+ to activate BK channels, whereas BK channels can physically couple to pore-forming α-subunits of L, P/Q, and N-type VCGGs94. This reciprocal interdependence is present to various extents across different types of central neurons. Thus, it likely contributes to neuronal hyperexcitability within the context of FXS.
Dendritic function
There are numerous reports of disrupted dendritic function in FXS models, including changes in excitability, gain, integration, and multiple forms of long-term synaptic plasticity localized to the dendritic compartment37,55–59. These functions are governed, in large part, by ion channels, a number of which are dysregulated in the absence of FMRP (BOX 2, TABLE 2).
K+ channels
Kv4 channels.
The Kv4.2 channel regulates hippocampal and neocortical dendritic excitability and synaptic plasticity. FMRP can bind Kv4.2 mRNA and regulate its translation95–97. FMRP can also directly interact with and regulate Kv4 activity. Specifically, reduced A-type K+ currents (carried by Kv4 channels) in hippocampal PCs of Fmr1 KO mice are associated with hyperpolarizing shifts in channel’s activation and inactivation kinetics98, suggesting changes to channel gating properties. Moreover, FMRP constitutively binds to the complex of Kv4 and Cav3 channels in cerebellar granule cells to modulate activity of both channels91. In both cases, FMRP decreases channel currents primarily through a strong hyperpolarizing shift in inactivation kinetics, while channel density is unchanged91. The Cav3-Kv4-FMRP interaction has important functional consequences within the context of FXS. In Fmr1 KO mice, the theta burst-induced form of long-term potentiation (LTP) is absent in mossy fiber-granule cell synapses of cerebellar cortex, a change that is attributed to increased Kv4 current91. Significantly, acute intracellular administration of the N-terminal FMRP fragment (aa1–297) is sufficient to restore LTP defects; it directly modulates Cav3-Kv4 channel complex activity in the absence of translational changes91.
Therapeutic implications.
The fact that FMRP directly modulates Cav3-Kv4 complex activity raises the possibility that this interaction can be targeted as a therapeutic strategy. When the N-terminal fragment of FMRP is modified to facilitate blood-brain barrier crossing (conjugation to tat; FMRP297-tat) and infused into Fmr1 KO mice, it rapidly improves multiple functions at the cellular and behavioral levels, including restoration of Cav3-Kv4 complex activity, rescue of cerebellar mossy fiber LTP, and normalization of hyperactivity in the open field test91. Interestingly, FMRP297-tat also restores levels of several selected proteins (PSD95, CamKII) for at least 24 hours. It remains unclear whether the observed changes in synaptic protein levels are a cause or a consequence of normalized LTP, since the FMRP297 fragment lacks the ability to associate with polyribosomes and regulate translation. Additionally, there are still no data directly linking improvements in hyperactivity in vivo with restored Cav3-Kv4 complex function or the normalization of mossy fiber LTP. Understanding the mechanisms underlying FMRP297-tat treatment is important because FMRP297-tat is likely taken up by a wide range of excitatory and inhibitory neurons throughout the brain, including cerebellum but also cortex, hippocampus, and other brain areas91. However, regardless of the precise mechanisms, a tat-conjugate approach is a promising strategy through which to reintroduce a combination of different FMRP segments containing various functional domains, thus wielding broad power to address FXS channelopathies.
BK channels.
In additional to their prominent presynaptic roles in controlling AP duration and glutamate release, BK channels are also localized to dendrites and regulate dendritic excitability. In Fmr1 KO mice, BK channel activity is reduced in dendrites of neocortical neurons, leading to exaggerated Ca2+ influx accompanying backpropagating APs and reduced dendritic spike threshold57. The BK channel opener BMS-204352 rescues a wide range of behavioral phenotypes in these mice, which likely reflects the effect of normalizing both pre- and post-synaptic functions57,77,84.
Calcium channels
L-type VGCCs.
In addition to the Cav3 (T-type) VGCC discussed above, a large proportion of depolarization-induced Ca2+influx in dendrites is mediated by L-type (Cav1) VGCCs. The L-type channels regulate dendritic excitability, gene expression, synaptic plasticity, and neural differentiation and migration87. Cav1.3 mRNA is FMRP target; channel expression is downregulated in the frontal cortex and cerebellum of Fmr1 KO mice99. In the dendritic spines of prefrontal cortex layer 2/3 neurons from Fmr1 KO mice, the reduced expression of L-type channels is linked with defects in spike timing-dependent plasticity55. Interestingly, in both FXS human patients and mouse models, Ca2+ influx through L-type VGCCs is increased in neural progenitors derived from stem cells88. Specifically, the ratio of L-type to T-type channels increases, leading to enhanced progenitor differentiation to glutamate-responsive cells. These findings suggest that there are significant implications of upregulated L-type channel function on differentiation, migration, and fate determination in neural progenitor cells within the context of FXS88.
R-type VGCCs.
The R-type calcium current is carried by Cav2.3, whose mRNA is also targeted by FMRP95. This channel regulates burst-firing and afterdepolarization in dendrites, and its inhibition disrupts a form of mGluR-dependent long-term depression (LTD)87. Consistent with profound alterations in dendritic excitability and calcium spiking in neurons lacking FMRP, Cav2.3 expression and R-type Ca2+ current are increased in brain synaptosomes and hippocampal cultured neurons from Fmr1 KO mice90. Moreover, loss of FMRP occludes mGluR-dependent Cav2.3 upregulation90. We do not yet completely understand how these defects contribute to altered dendritic excitability in FXS models, as other factors are also involved in modulating excitability following mGluR stimulation, including the downregulation of several K+ channels100.
HCN channels
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are active at rest and therefore play a crucial role in controlling the resting membrane potential, input resistance, and thus dendritic excitability101. They are prominently expressed in the soma and dendrites of excitatory neurons, following an expression gradient that increases toward more distal regions to regulate the integration of synaptic events occurring at different dendritic locations. Numerous cell type-specific alterations in HCN channels are observed in FXS models. For example, in CA1 PCs of Fmr1 KO mice, HCN1 subunit expression and dendritic HCN currents are elevated. This in turn decreases input resistance, reduces temporal summation, and increases membrane resonance frequency56. In contrast, in both layer 5 PCs of the somatosensory cortex and layer 4 stellate cells in these mice, HCN1 subunit expression and HCN current are decreased, leading to opposite changes in excitability, i.e., increased dendritic gain57,59. Mechanistically, FMRP bidirectionally regulates HCN channels via a protein-protein interaction mechanism in dendrites of hippocampal CA1 PCs and prefrontal cortex neurons102. FMRP associates with the HCN-TRIP8b complex to down-regulate (in hippocampus) or up-regulate (in prefrontal cortex) the number of functional dendritic HCN channels. This previously unrecognized mechanism expands the repertoire of modulatory mechanisms by which FMRP regulates ion channel functions and excitability (FIG. 2), in opposite directions depending upon the cellular milieu. If this type of regulation is observed more broadly in future studies, it may explain many cell-type specific changes in channel expression/activity in neurons lacking FMRP.
Ligand-gated ion channels
The ligand-gated ion channels are a group of postsynaptic neurotransmitter receptors, also known as ionotropic receptors. There are three major ionotropic receptors involved in FXS: AMPA-, NMDA-, and GABAA receptors (Rs). The AMPARs and NMDARs are nonselective cation channels, while GABAAR is a Cl− channel. In addition to these channels, we discuss the impact of FMRP loss on the chloride transporters that maintain intracellular Cl− ion homeostasis, as these transporters determine the GABAAR polarity switch early in development involved in synaptogenesis and synapse maturation.
AMPARs.
AMPARs mediate fast excitatory synaptic transmission in the brain. Altering the number of postsynaptic AMPARs is a fundamental mechanism of activity-dependent plasticity at excitatory synapses, including some forms of LTP and LTD. Local protein synthesis in dendrites is required for stable expression of LTP and LTD, and there is now evidence that these newly expressed proteins are regulated by specific RNA-binding proteins, including FMRP103–108. Thus, FMRP may indirectly regulate synaptic plasticity through AMPAR-dependent functions59,83,103–108. In Fmr1 KO mice, expression of the AMPAR subunit GluA1 is decreased in cortex (but not in the hippocampus or cerebellum104), as is LTP104,105. AMPARs also show brain region/cell-type specific defects in FXS models. Specifically, in the hippocampus, FMRP promotes GluA1 surface expression in adult-born granule cells of dentate gyrus, but has no effect on total GluA1 protein levels107,108. Consequently, loss of FMRP decreases surface expression of GluA1, reduces GluA1-mediated AMPA currents, and delays dendritic maturation. In contrast, layer 4 stellate cells of primary somatosensory cortex in Fmr1 KO mice show elevated single-spine excitation with normal spine morphology, in part due to increased AMPAR currents59. Consistent with this, studies of a new FXS mouse model (point missense mutation R138Q in FMRP) reveal an increase in both total and surface expression of AMPAR subunits GluA1 and GluA2, decreased thickness of postsynaptic density, and reduced LTP83.
Therapeutic implications.
The evidence presented above raise the possibility that positive allosteric modulators of AMPARs could be useful for restoring some of the synaptic deficits associated with FXS. However, a double-blind clinical trial of one such compound, CX516, did not show significant improvements in cognition, language, or attention/executive function in individuals with FXS109. AMPARs also show brain region-/cell type-specific defects in FXS models. Thus, globally targeting AMPARs is unlikely to be effective.
NMDARs.
NMDARs are involved in synaptic transmission, plasticity, and experience-dependent synaptic refinement during development. FMRP regulates transport and stabilization of NMDAR subunits GluN1, GluN2B110, and GluN2A111. In the dentate gyrus of Fmr1 KO mice, NMDAR currents are decreased and LTP is reduced112. The reduction in NMDAR-dependent synaptic plasticity in this region is accompanied by lower levels of GluN1, GluN2A, and GluN2B subunits. Indeed, the NMDAR co-agonists glycine and D-serine can restore NMDAR-dependent LTP in these mice to WT levels113,114. At the behavioral level, NMDAR hypofunction is also linked to impaired context discrimination in adult Fmr1 KO mice115. Most recently, Yau and colleagues found that lower NMDAR currents in the dentate gyrus of Fmr1 KO mice is associated with a significant decrease in dendritic complexity (i.e., reduced dendritic length and number of intersections)116. Yet again, evidence points to cell-type specific changes in NMDAR levels and functions as a result of FMRP loss. NMDAR signaling is elevated in stellate cells of somatosensory cortex layer 4, and dendritic morphology is normal59. In these cells, the increase in dendritic gain and enhanced summation appear to be caused by a 3-fold increase in the number of poly-synaptic spines and an increase in intrinsic excitability. Interestingly, while the loss of FMRP causes abnormal synaptogenesis, there is little correlation between spine structure and function, at least at the age of animals used in this study59.
GABAARs.
Fast inhibitory synaptic neurotransmission in the brain is mostly mediated by GABA and its ionotropic target, GABAARs117. GABAAR is a pentameric ion channel that is selectively permeable to Cl− ions. Animal models of FXS show abnormalities in GABAAR expression and function, as do human FXS patients (for review see ref 118). Kooy and colleagues initially found decreased expression of GABAAR δ subunit mRNA in Fmr1 KO mice119. Further studies from multiple groups found that the protein levels of more than half of GABAAR subunits (including α1–α4, β1-β3, γ1, γ2 and δ) are lower in FXS mouse models48,64,120–125, and throughout the brain of FXS patients126. In addition, the amount of GABA itself is also lower across multiple brain regions in FXS mouse models127,128. Such hypofunction of the GABAergic system is likely to cause profound circuit hyperexcitability. Indeed, Morin-Parent and colleagues demonstrate that reduced GABAAR function causes impaired short-interval intracortical inhibition, which contributes to cortical hyperexcitability in individuals with FXS9.
Therapeutic implications.
Given the critical role of the GABAergic system in network excitability, GABAAR agonists have the potential to ameliorate excitability-associated deficits in FXS129,130. The GABAAR agonists acamprosate, ganaxolone, and metadoxine can improve multiple defects in Fmr1 KO models, including neuromuscular junction overgrowth and locomotor defects131, prolonged cortical UP state and anxiety132, and marble burying behavior and sensory hyperresponsiveness127. However, when these compounds were tested in clinical trials of individuals with FXS, there were no significant improvements in outcome measures133–136. Another strategy is to target metabotropic GABABR receptors to alleviate FXS deficits by normalizing the GABAergic system. However, again, although the GABABR agonist baclofen restores cognitive and behavioral deficits in animal models137–140, it did not impact primary outcome measures in clinical trials141,142. These results highlight the limitations of targeting a single aspect of excitability defects in FXS. Ultimately, the most effective treatment strategies are likely to be multifactorial.
Chloride transporters
In addition to wielding fine control over circuit excitability, GABAergic transmission plays a critical trophic role in cortical development through its early depolarizing action. During the first few postnatal weeks (critical period) in rodents, activating GABAAR depolarizes neurons, increasing their excitability and promoting Ca2+ entry143. This in turn regulates cell migration, proliferation, and synaptogenesis. The early depolarizing effect of GABA results from a transient high intracellular Cl− concentration that is only present early in development and is determined by the expression pattern of two Cl− cotransporters: Na+–K+–Cl− cotransporter (NKCC1) and K+–Cl− cotransporter (KCC2). The NKCC1, which transports Cl− into neurons, is expressed at high levels early after birth, while the levels of KCC2, which extrudes Cl− from neurons, gradually increases over the course of development144. In the FXS mouse model, NKCC1 expression stays high through the end of the critical period, thus delaying the GABAAR polarity switch and disrupting the normal trophic function of GABA145,146,147.
Therapeutic implications.
Systemic administration of bumetanide to inhibit NKCC1 during the critical period rectifies the intracellular Cl− imbalance in layer 4 somatosensory cortex neurons and corrects the development of thalamocortical excitatory synapses in Fmr1 KO mice146. Inhibiting NKCC1 during early development also leads to broad remodeling of the proteome in the barrel cortex, and restores adult tactile response maps in Fmr1 KO mice146. These findings provide rationale for considering NKCC1 as a novel therapeutic target in FXS.
Caveats of targeting channelopathies
As we have presented above, channelopathies play wide and varied roles in FXS pathophysiology. This makes ion channel-targeting therapies of particular interest. Indeed, multiple ion channel modulators show promise based on their ability to normalize cellular-, circuit-, and behavior-level abnormalities in FXS animal models (TABLE 3). We note above many cases in which specific ion channel-targeting therapies have already been tested in both animals and human patients. However, in most cases clinical trials fail to show efficacy. In fact, recent studies emphasize that loss of FMRP can have diverse effects on the same ion channel depending on the brain region, cell-type, or even subcellular compartment56,58,98. This will make targeting any given ion channel for therapeutic intervention in FXS particularly challenging. Recent modeling studies also indicate that many cellular and synaptic pathologies in Fmr1 KO mice are antagonistic on the circuit level, and hence may be compensatory to the primary pathology37. Thus, while there has been major progress towards identifying the molecular dysfunctions at synaptic and cellular levels, we need to better understand the overall effects of various channelopathies on circuit functions to design more effective therapeutic strategies. Moreover, because no single channelopathy can account for all, or even most of defects in FXS, a combinatory approach is almost certainly necessary. Alternatively, a strategy that targets the primary causes of ion channel dysfunctions (such as loss of interactions with, or translational regulation by, FMRP) might prove more efficient. For example, a tat-conjugate approach to reintroduce N-terminal FMRP fragment via intravenous injection restores ion channel and synaptic functions in at least part of the brain, as well as locomotion activity at the behavioral level in Frm1 KO mice91. Finally, a role for astrocytes in modulating neuronal excitability and plasticity in FXS is beginning to emerge148–150. This may represent another potential target through which to normalize excitability deficits in FXS.
Implications for ASD
FXS is one of the many causes of autism spectrum disorders (ASD) and it has comorbidities with other genetic causes and also with idiopathic ASD. FXS and FMRP-loss-independent ASD, while genetically distinct, share significant clinical phenotypes. Indeed, their similarities suggest some level of common pathophysiology, including similar ion channel-based deficits151. There are multiple types of ion channels and transporters that are affected, in similar ways, in both FXS and FMRP-loss-independent ASD152, including Kv4.2153,154, BK channels155, Na+ channels156, Ca2+ (L, R and T-type) channels, HCN1157, GABAAR, and NKCC1152. Moreover, channelopathies responsible for hyperexcitability of somatosensory neurons are part of the core developmental pathology in ASD models, leading to region-specific brain abnormalities during the critical period157. Peripherally-restricted GABAAR agonists, which reduce hyperexcitability within peripheral sensory circuits, improve a number of ASD-associated behaviors in mouse models, including tactile over-reactivity, anxiety-like behaviors, and certain social impairments157. Together, these findings indicate that what we learn about the role of channelopathies in FXS pathogenesis is likely to have important implications for ASD stemming from other causes.
Concluding remarks
We now understand that FMRP wields control over neuronal and circuit excitability by regulating ion channels (FIG. 1, TABLES 1,2). Indeed, FMRP modulates the physiological functions of many different types of ion channels, each of which contributes uniquely to controlling intrinsic excitability, AP properties, firing patterns, synaptic transmission, synaptic plasticity, and dendritic integration. FMRP can regulate ion channel function via protein-protein interactions with pore-forming or regulatory subunits, or channel expression, and some channels are regulated via both mechanisms (FIG. 2). FMRP can also indirectly modulate channel activity through second messenger pathways. The various mechanisms through which FMRP regulates channel function may be brain region-, cell type-, or even subcellular compartment-specific. Given that direct FMRP-channel interactions are rapid and reversible, this mechanism means that ion channels can be precisely and dynamically tuned, which is critical for normal excitability, synaptic plasticity, and information processing, and, ultimately, normal behavioral and cognitive functions. Consequently, loss of FMRP, as occurs in FXS, triggers a series of channelopathies that contribute to circuit hyperexcitability-related abnormalities which in turn underlie core symptoms of FXS. Focusing on the role of channelopathies in such neuronal and circuit-level defects will be crucial in understanding the mechanistic basis of FXS pathophysiology and in designing novel therapeutic strategies.
Acknowledgements
This work was supported in part by an R35 grant NS111596 to VAK from NINDS. We would like to apologize to the colleagues whose work could not be cited in this review due to space limitations. Figures 1 and 2 were created with BioRender.com.
References
- 1.Penagarikano O, Mulle JG & Warren ST The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet 8, 109–29 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Sitzmann AF, Hagelstrom RT, Tassone F, Hagerman RJ & Butler MG Rare FMR1 gene mutations causing fragile X syndrome: A review. Am J Med Genet A 176, 11–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Contractor A, Klyachko VA & Portera-Cailliau C Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 87, 699–715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devys D, Lutz Y, Rouyer N, Bellocq JP & Mandel JL The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 4, 335–40 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Brager DH & Johnston D Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res Bull 103, 11–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferron L Fragile X mental retardation protein controls ion channel expression and activity. J Physiol 594, 5861–5867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salcedo-Arellano MJ, Hagerman RJ & Martinez-Cerdeno V Fragile X syndrome: clinical presentation, pathology and treatment. Gac Med Mex 156, 60–66 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Nelson SB & Valakh V Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 87, 684–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morin-Parent F, Champigny C, Lacroix A, Corbin F & Lepage JF Hyperexcitability and impaired intracortical inhibition in patients with fragile-X syndrome. Transl Psychiatry 9, 312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berry-Kravis E Epilepsy in fragile X syndrome. Dev Med Child Neurol 44, 724–8 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Sabaratnam M, Vroegop PG & Gangadharan SK Epilepsy and EEG findings in 18 males with fragile X syndrome. Seizure 10, 60–3 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Miller LJ et al. Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet 83, 268–79 (1999). [PubMed] [Google Scholar]
- 13.Merenstein SA et al. Molecular-clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet 64, 388–94 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Kronk R et al. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep 33, 679–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carotenuto M et al. Polysomnographic Findings in Fragile X Syndrome Children with EEG Abnormalities. Behav Neurol 2019, 5202808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scharf SS, Gasparini F, Spooren W & Lindemann L in Fragile X Syndrome: From Genetics to Targeted Treatment (eds. Willemsen R & Kooy RF) 363–399 (Elsevier, 2017). [Google Scholar]
- 17.Chen L & Toth M Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–50 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Musumeci SA et al. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41, 19–23 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Yan QJ, Rammal M, Tranfaglia M & Bauchwitz RP Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–66 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Osterweil EK et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curia G, Gualtieri F, Bartolomeo R, Vezzali R & Biagini G Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front Cell Neurosci 7, 46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross C et al. Selective role of the catalytic PI3K subunit p110beta in impaired higher order cognition in fragile X syndrome. Cell Rep 11, 681–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Busquets-Garcia A et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19, 603–7 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Rotschafer S & Razak K Altered auditory processing in a mouse model of fragile X syndrome. Brain Res 1506, 12–24 (2013). [DOI] [PubMed] [Google Scholar]
- 25.He CX et al. Tactile Defensiveness and Impaired Adaptation of Neuronal Activity in the Fmr1 Knock-Out Mouse Model of Autism. J Neurosci 37, 6475–6487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frankland PW et al. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 9, 417–25 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Dansie LE et al. Long-lasting effects of minocycline on behavior in young but not adult Fragile X mice. Neuroscience 246, 186–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oddi D et al. Early social enrichment rescues adult behavioral and brain abnormalities in a mouse model of fragile X syndrome. Neuropsychopharmacology 40, 1113–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heulens I, D’Hulst C, Van Dam D, De Deyn PP & Kooy RF Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res 229, 244–9 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Heard TT et al. EEG abnormalities and seizures in genetically diagnosed Fragile X syndrome. Int J Dev Neurosci 38, 155–60 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Knoth IS, Vannasing P, Major P, Michaud JL & Lippe S Alterations of visual and auditory evoked potentials in fragile X syndrome. Int J Dev Neurosci 36, 90–7 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Castren M, Paakkonen A, Tarkka IM, Ryynanen M & Partanen J Augmentation of auditory N1 in children with fragile X syndrome. Brain Topogr 15, 165–71 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Gibson JR, Bartley AF, Hays SA & Huber KM Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol 100, 2615–26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hays SA, Huber KM & Gibson JR Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci 31, 14223–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ronesi JA et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 15, 431–40, S1 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goncalves JT, Anstey JE, Golshani P & Portera-Cailliau C Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci 16, 903–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domanski APF, Booker SA, Wyllie DJA, Isaac JTR & Kind PC Cellular and synaptic phenotypes lead to disrupted information processing in Fmr1-KO mouse layer 4 barrel cortex. Nat Commun 10, 4814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goel A et al. Impaired perceptual learning in a mouse model of Fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat Neurosci 21, 1404–1411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Pino E, Gessele N & Koch U Enhanced Excitatory Connectivity and Disturbed Sound Processing in the Auditory Brainstem of Fragile X Mice. J Neurosci 37, 7403–7419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCullagh EA, Salcedo E, Huntsman MM & Klug A Tonotopic alterations in inhibitory input to the medial nucleus of the trapezoid body in a mouse model of Fragile X syndrome. J Comp Neurol 525, 3543–3562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rotschafer SE, Marshak S & Cramer KS Deletion of Fmr1 alters function and synaptic inputs in the auditory brainstem. PLoS One 10, e0117266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chuang SC et al. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci 25, 8048–55 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng PY & Klyachko VA Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J Physiol 594, 83–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wahlstrom-Helgren S & Klyachko VA GABAB receptor-mediated feed-forward circuit dysfunction in the mouse model of fragile X syndrome. J Physiol 593, 5009–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wahlstrom-Helgren S & Klyachko VA Dynamic Balance of Excitation and Inhibition Rapidly Modulates Spike Probability and Precision in Feed-Forward Hippocampal Circuits. J Neurophysiol, 116(6): 2564–2575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin BS, Corbin JG & Huntsman MM Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J Neurophysiol 112, 890–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin BS et al. Rescue of deficient amygdala tonic gamma-aminobutyric acidergic currents in the Fmr−/y mouse model of fragile X syndrome by a novel gamma-aminobutyric acid type A receptor-positive allosteric modulator. J Neurosci Res 94, 568–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olmos-Serrano JL et al. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci 30, 9929–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals inhibitory synapse abnormalities in amygdala of an FXS mouse model, and provides evidence that pharmacological approaches targeting the GABAergic system may be a viable therapeutic approach to correct amygdala-dependent symptoms in FXS.
- 49.Vislay RL et al. Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J Neurosci 33, 7548–58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suvrathan A, Hoeffer CA, Wong H, Klann E & Chattarji S Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A 107, 11591–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng PY et al. Voltage-Independent SK-Channel Dysfunction Causes Neuronal Hyperexcitability in the Hippocampus of Fmr1 Knock-Out Mice. J Neurosci 39, 28–43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng PY & Klyachko VA Increased Persistent Sodium Current Causes Neuronal Hyperexcitability in the Entorhinal Cortex of Fmr1 Knockout Mice. Cell Rep 16, 3157–3166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work identifies Na+ channel dysregulation as a major cause of neuronal hyperexcitability in cortical FXS neurons and uncovers a mechanism via which increased mGluR5 signaling causes neuronal hyperexcitability in FXS mouse model.
- 53.El-Hassar L et al. Modulators of Kv3 Potassium Channels Rescue the Auditory Function of Fragile X Mice. J Neurosci 39, 4797–4813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Routh BN et al. Increased transient Na(+) conductance and action potential output in layer 2/3 prefrontal cortex neurons of the fmr1(-/y) mouse. J Physiol 595, 4431–4448 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meredith RM, Holmgren CD, Weidum M, Burnashev N & Mansvelder HD Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–38 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Brager DH, Akhavan AR & Johnston D Impaired dendritic expression and plasticity of h-channels in the fmr1(−/y) mouse model of fragile X syndrome. Cell Rep 1, 225–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports elevated dendritic expression of HCN channels coupled with impaired h-channel plasticity, which may underlie some of the cognitive impairments associated with FXS.
- 57.Zhang Y et al. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(−/y) mice. Nat Neurosci 17, 1701–9 (2014). [DOI] [PubMed] [Google Scholar]; This work reveals dysfunctions of HCN and BK channels within the dendritic compartment, and provides evidence that BK channel openers have potential to address the sensory hypersensitivity aspects of FXS.
- 58.Kalmbach BE, Johnston D & Brager DH Cell-Type Specific Channelopathies in the Prefrontal Cortex of the fmr1−/y Mouse Model of Fragile X Syndrome. eNeuro 2, ENEURO.0114–ENEURO.0115. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows celltype-specific alterations in A-type K+ channels and HCN channels in two types of neighboring neurons in the prefontal cortex of Fmr1 KO mice, which highlights the importance of understanding celltype-specific changes in neuronal excitability within the context of FXS.
- 59.Booker SA et al. Altered dendritic spine function and integration in a mouse model of fragile X syndrome. Nat Commun 10, 4813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferron L, Nieto-Rostro M, Cassidy JS & Dolphin AC Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat Commun 5, 3628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demostrates that FMRP regulates surface expression of N-type VGCCs, and thus neurotransmitter release, in peripheral sensory neurons via protein-protein interactions.
- 61.Ferron L et al. FMRP regulates presynaptic localization of neuronal voltage gated calcium channels. Neurobiol Dis 138, 104779 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deng PY et al. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work presents initial evidence that FMRP directly interacts with BK channels to regulate neurotransmitter release in central neurons, which provides the rationale for investigating the BK channel as a promising therapeutic target to treat a number of excitability-related deficits in FXS.
- 63.Deng PY, Sojka D & Klyachko VA Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci 31, 10971–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang N et al. Decreased surface expression of the delta subunit of the GABAA receptor contributes to reduced tonic inhibition in dentate granule cells in a mouse model of fragile X syndrome. Exp Neurol 297, 168–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang YM et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol Psychiatry (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curia G, Papouin T, Seguela P & Avoli M Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex 19, 1515–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darnell JC et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–99 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Strumbos JG, Brown MR, Kronengold J, Polley DB & Kaczmarek LK Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci 30, 10263–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strumbos JG, Polley DB & Kaczmarek LK Specific and rapid effects of acoustic stimulation on the tonotopic distribution of Kv3.1b potassium channels in the adult rat. Neuroscience 167, 567–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rudy B & McBain CJ Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci 24, 517–26 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Kim GE & Kaczmarek LK Emerging role of the KCNT1 Slack channel in intellectual disability. Front Cell Neurosci 8, 209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown MR et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci 13, 819–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This seminal study provides the first evidence that FMRP directly regulates ion channel activity through protein-protein interactions, which opened a new field investigating FMRP’s physiological role in controlling ion channel function.
- 73.Zhang Y et al. Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J Neurosci 32, 15318–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doll CA & Broadie K Neuron class-specific requirements for Fragile X Mental Retardation Protein in critical period development of calcium signaling in learning and memory circuitry. Neurobiol Dis 89, 76–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salkoff L, Butler A, Ferreira G, Santi C & Wei A High-conductance potassium channels of the SLO family. Nat Rev Neurosci 7, 921–31 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Myrick LK et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci U S A 112, 949–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that loss of the translation-independent functions of FMRP is linked with a subset of FXS clinical features, suggesting that domain-specific functions of FMRP in pre- and postsynaptic compartments may contribute to different aspects of FXS pathophysiology.
- 77.Hebert B et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by a BKCa channel opener molecule. Orphanet J Rare Dis 9, 124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Short B FMRP differentially regulates BK channels. J Gen Physiol 152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Contractor A Broadening roles for FMRP: big news for big potassium (BK) channels. Neuron 77, 601–3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kshatri A et al. Differential regulation of BK channels by fragile X mental retardation protein. J Gen Physiol 152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Telias M, Kuznitsov-Yanovsky L, Segal M & Ben-Yosef D Functional Deficiencies in Fragile X Neurons Derived from Human Embryonic Stem Cells. J Neurosci 35, 15295–306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents an extensive functional analysis of FXS neurons derived in vitro from embrionic stem cells of FXS individuals that provides a useful tool for studying molecular mechanisms underlying the impaired neuronal functions in FXS.
- 82.Diaz J, Scheiner C & Leon E Presentation of a recurrent FMR1 missense mutation (R138Q) in an affected female. Translational Science of Rare Diseases 139–144 (2018). [Google Scholar]
- 83.Prieto M et al. Abnormal AMPAR-mediated synaptic plasticity, cognitive and autistic-like behaviors in a missense Fmr1 mutant mouse model of Fragile X syndrome. BioRxiv, 10.1101/2020.04.19.048819 (2020). [DOI] [Google Scholar]
- 84.Carreno-Munoz MI et al. Potential Involvement of Impaired BKCa Channel Function in Sensory Defensiveness and Some Behavioral Disturbances Induced by Unfamiliar Environment in a Mouse Model of Fragile X Syndrome. Neuropsychopharmacology 43, 492–502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bean BP The action potential in mammalian central neurons. Nat Rev Neurosci 8, 451–65 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Dwivedi D, Chattarji S & Bhalla US Impaired Reliability and Precision of Spiking in Adults But Not Juveniles in a Mouse Model of Fragile X Syndrome. eNeuro 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Catterall WA Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3, a003947 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Danesi C et al. Increased Calcium Influx through L-type Calcium Channels in Human and Mouse Neural Progenitors Lacking Fragile X Mental Retardation Protein. Stem Cell Reports 11, 1449–1461 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castagnola S et al. New Insights Into the Role of Cav2 Protein Family in Calcium Flux Deregulation in Fmr1-KO Neurons. Front Mol Neurosci 11, 342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gray EE et al. Disruption of GpI mGluR-Dependent Cav2.3 Translation in a Mouse Model of Fragile X Syndrome. J Neurosci 39, 7453–7464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhan X et al. FMRP(1-297)-tat restores ion channel and synaptic function in a model of Fragile X syndrome. Nat Commun 11, 2755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that FMRP is part of a Cav3-Kv4 ion channel complex in cerebellar neurons, and describes a promising tat-conjugate approach to reintroduce an FMRP fragment to improve function in the cerebellum on multiple levels (channels, synapses, behavior) in an FXS mouse model.
- 92.Nanou E, Scheuer T & Catterall WA Calcium sensor regulation of the CaV2.1 Ca2+ channel contributes to long-term potentiation and spatial learning. Proc Natl Acad Sci U S A 113, 13209–13214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cao YQ et al. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 43, 387–400 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Contet C, Goulding SP, Kuljis DA & Barth AL BK Channels in the Central Nervous System. Int Rev Neurobiol 128, 281–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Darnell JC et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gross C, Yao X, Pong DL, Jeromin A & Bassell GJ Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 31, 5693–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study and Ref. 97 together show that FMRP regulates Kv4.2 channel expression with implications for altered dendritic excitability and plasticity in FXS mouse model.
- 97.Lee HY et al. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; see Ref. 96.
- 98.Routh BN, Johnston D & Brager DH Loss of functional A-type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J Neurosci 33, 19442–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen L, Yun SW, Seto J, Liu W & Toth M The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience 120, 1005–17 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Gereau R.W.t. & Conn PJ Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci 15, 6879–89 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shah MM Cortical HCN channels: function, trafficking and plasticity. J Physiol 592, 2711–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brandalise F et al. Fragile X Mental Retardation Protein bidirectionally controls dendritic Ih in a cell-type specific manner between mouse hippocampus and prefrontal cortex. J Neurosci (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that FMRP regulates HCN channels via a cell-autonomous, protein-protein interactions and regulates its targets in opposite directions depending upon the cellular milieu.
- 103.Huber KM, Gallagher SM, Warren ST & Bear MF Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99, 7746–50 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li J, Pelletier MR, Perez Velazquez JL & Carlen PL Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci 19, 138–51 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Larson J, Jessen RE, Kim D, Fine AK & du Hoffmann J Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci 25, 9460–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hu H et al. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci 28, 7847–62 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lim CS et al. Pharmacological rescue of Ras signaling, GluA1-dependent synaptic plasticity, and learning deficits in a fragile X model. Genes Dev 28, 273–89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo W et al. Fragile X Proteins FMRP and FXR2P Control Synaptic GluA1 Expression and Neuronal Maturation via Distinct Mechanisms. Cell Rep 11, 1651–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Berry-Kravis E et al. Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J Child Adolesc Psychopharmacol 16, 525–40 (2006). [DOI] [PubMed] [Google Scholar]
- 110.Schutt J, Falley K, Richter D, Kreienkamp HJ & Kindler S Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem 284, 25479–87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Edbauer D et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65, 373–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yun SH & Trommer BL Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res 89, 176–82 (2011). [DOI] [PubMed] [Google Scholar]
- 113.Bostrom CA et al. Rescue of NMDAR-dependent synaptic plasticity in Fmr1 knock-out mice. Cereb Cortex 25, 271–9 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Bostrom C et al. Hippocampal dysfunction and cognitive impairment in Fragile-X Syndrome. Neurosci Biobehav Rev 68, 563–574 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Eadie BD, Cushman J, Kannangara TS, Fanselow MS & Christie BR NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 22, 241–54 (2012). [DOI] [PubMed] [Google Scholar]
- 116.Yau SY, Bettio L, Chiu J, Chiu C & Christie BR Fragile-X Syndrome Is Associated With NMDA Receptor Hypofunction and Reduced Dendritic Complexity in Mature Dentate Granule Cells. Front Mol Neurosci 11, 495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smart TG & Stephenson FA A half century of gamma-aminobutyric acid. Brain Neurosci Adv 3, 2398212819858249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van der Aa N & Kooy RF GABAergic abnormalities in the fragile X syndrome. Eur J Paediatr Neurol 24, 100–104 (2020). [DOI] [PubMed] [Google Scholar]
- 119.Gantois I et al. Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis 21, 346–57 (2006). [DOI] [PubMed] [Google Scholar]; This study and Ref. 120 report hypofunction of GABAA receptors in an FXS mouse model, which points to the GABAA receptor as a promising target for treatment of the behavioral and epileptic phenotypes associated with FXS.
- 120.D’Hulst C et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res 1121, 238–45 (2006). [DOI] [PubMed] [Google Scholar]; See Ref 119.
- 121.Sabanov V et al. Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice. Neuropharmacology 116, 71–81 (2017). [DOI] [PubMed] [Google Scholar]
- 122.Vien TN et al. Compromising the phosphodependent regulation of the GABAAR beta3 subunit reproduces the core phenotypes of autism spectrum disorders. Proc Natl Acad Sci U S A 112, 14805–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.D’Hulst C, Atack JR & Kooy RF The complexity of the GABAA receptor shapes unique pharmacological profiles. Drug Discov Today 14, 866–75 (2009). [DOI] [PubMed] [Google Scholar]
- 124.Adusei DC, Pacey LK, Chen D & Hampson DR Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology 59, 167–71 (2010). [DOI] [PubMed] [Google Scholar]
- 125.El Idrissi A et al. Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett 377, 141–6 (2005). [DOI] [PubMed] [Google Scholar]
- 126.D’Hulst C et al. Positron Emission Tomography (PET) Quantification of GABAA Receptors in the Brain of Fragile X Patients. PLoS One 10, e0131486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Braat S et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 14, 2985–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Davidovic L et al. A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res 21, 2190–202 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huntsman MM & Kooy RF in Fragile X Syndrome From Genetics to Targeted Treatment (eds. Willemsen R & Kooy RF) 205–215 (2017). [Google Scholar]
- 130.Braat S & Kooy RF Insights into GABAAergic system deficits in fragile X syndrome lead to clinical trials. Neuropharmacology 88, 48–54 (2014). [DOI] [PubMed] [Google Scholar]
- 131.Hutson RL, Thompson RL, Bantel AP & Tessier CR Acamprosate rescues neuronal defects in the Drosophila model of Fragile X Syndrome. Life Sci 195, 65–70 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Schaefer TL et al. Acamprosate in a mouse model of fragile X syndrome: modulation of spontaneous cortical activity, ERK1/2 activation, locomotor behavior, and anxiety. J Neurodev Disord 9, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Erickson CA, Mullett JE & McDougle CJ Brief report: acamprosate in fragile X syndrome. J Autism Dev Disord 40, 1412–6 (2010). [DOI] [PubMed] [Google Scholar]
- 134.Erickson CA et al. Impact of acamprosate on plasma amyloid-beta precursor protein in youth: a pilot analysis in fragile X syndrome-associated and idiopathic autism spectrum disorder suggests a pharmacodynamic protein marker. J Psychiatr Res 59, 220–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ligsay A et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J Neurodev Disord 9, 26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Di Miceli M & Gronier B Pharmacology, Systematic Review and Recent Clinical Trials of Metadoxine. Rev Recent Clin Trials 13, 114–125 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Silverman JL et al. GABAB Receptor Agonist R-Baclofen Reverses Social Deficits and Reduces Repetitive Behavior in Two Mouse Models of Autism. Neuropsychopharmacology 40, 2228–39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sinclair D et al. GABA-B Agonist Baclofen Normalizes Auditory-Evoked Neural Oscillations and Behavioral Deficits in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. eNeuro 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stoppel LJ et al. R-Baclofen Reverses Cognitive Deficits and Improves Social Interactions in Two Lines of 16p11.2 Deletion Mice. Neuropsychopharmacology 43, 513–524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Henderson C et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med 4, 152ra128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Berry-Kravis EM et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med 4, 152ra127 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Erickson CA et al. STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J Autism Dev Disord 44, 958–64 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Represa A & Ben-Ari Y Trophic actions of GABA on neuronal development. Trends Neurosci 28, 278–83 (2005). [DOI] [PubMed] [Google Scholar]
- 144.Liu R, Wang J, Liang S, Zhang G & Yang X Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol 10, 1407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.He Q, Nomura T, Xu J & Contractor A The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci 34, 446–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study and the following one (Ref.146) provide strong evidence that targeting the Cl− trasporter NKCC1 during the critical period restores synapse development in cortical neurons and produces a long-lasting correction of somatosensory circuit function in FXS mice.
- 146.He Q et al. Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol Psychiatry 24, 1732–1747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; see Ref 145.
- 147.Tyzio R et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–9 (2014). [DOI] [PubMed] [Google Scholar]
- 148.Hodges JL et al. Astrocytic Contributions to Synaptic and Learning Abnormalities in a Mouse Model of Fragile X Syndrome. Biol Psychiatry 82, 139–149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cheng C, Lau SK & Doering LC Astrocyte-secreted thrombospondin-1 modulates synapse and spine defects in the fragile X mouse model. Mol Brain 9, 74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Higashimori H et al. Selective Deletion of Astroglial FMRP Dysregulates Glutamate Transporter GLT1 and Contributes to Fragile X Syndrome Phenotypes In Vivo. J Neurosci 36, 7079–94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Guglielmi L et al. Update on the implication of potassium channels in autism: K(+) channelautism spectrum disorder. Front Cell Neurosci 9, 34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schmunk G & Gargus JJ Channelopathy pathogenesis in autism spectrum disorders. Front Genet 4, 222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee H, Lin MC, Kornblum HI, Papazian DM & Nelson SF Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 23, 3481–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lin MA, Cannon SC & Papazian DM Kv4.2 autism and epilepsy mutation enhances inactivation of closed channels but impairs access to inactivated state after opening. Proc Natl Acad Sci U S A 115, E3559–E3568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Laumonnier F et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry 163, 1622–9 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Kruth KA, Grisolano TM, Ahern CA & Williams AJ SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: a role for pluripotent stem cells? Mol Autism 11, 23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Orefice LL et al. Targeting Peripheral Somatosensory Neurons to Improve Tactile-Related Phenotypes in ASD Models. Cell 178, 867–886 e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals a potential therapeutic strategy targeting E/I imbalance in peripheral mechanosensory circuits to treat tactile over-reactivity and select ASD-related behaviors.
- 158.Liao L, Park SK, Xu T, Vanderklish P & Yates JR 3rd. Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc Natl Acad Sci U S A 105, 15281–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Myrick LK, Hashimoto H, Cheng X & Warren ST Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum Mol Genet 24, 1733–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]