

Abstract
Protein-protein interfaces have been attracting great attention due to their critical roles in protein-protein interactions and the fact that human disease-related mutations are generally enriched in them. Recently, substantial research progress has been made in this field, which have significantly promoted the understanding and treatment of various human diseases. For example, many studies have discovered the properties of disease-related mutations. Besides, as more large-scale experimental data become available, various computational approaches have been proposed to advance our understanding of disease mutations from the data. Here, we overview recent advances in characteristics of disease-related mutations at protein-protein interfaces, mutation effects on protein interactions, and investigation of mutations on specific diseases.
Keywords: Disease-related mutations, Protein-protein interfaces, Binding affinity, Deep learning, Pathogenicity
Introduction
The protein-protein interface makes crucial contributions to the specificity and strength of protein interactions, which are essential to most biological processes, and determines the mechanism by which proteins fulfill their functions [1]. The loss or alteration of protein functions caused by amino acid mutations can result in diseases and these mutations are hence referred to as ‘disease-related mutations’ [2]. Disease-related mutations have been reported to be enriched at protein-protein interfaces [3, 4, 5, 6], and are more evolutionarily conserved than other surface residues [7, 8, 9]. Therefore, studies of disease-related mutations involved in protein-protein interfaces provide important insights for deciphering disease mechanisms and potential treatments. For example, ECLAIR, a unified machine learning framework of Interactome INSIDER [3], computationally predicted protein-protein interfaces of 185,957 binary interactions with previously unresolved interfaces in human and seven other model organisms on a large scale, which extensively makes the downstream studies possible. Cheng et al. [10] used the predicted protein-protein interfaces in Interactome INSIDER to demonstrate that network-predicted oncoPPIs, which is a protein interaction with a significant enrichment in interface mutations across individuals, are closely related to patient survival and drug resistance/sensitivity either in human cancer cell lines or in patient-derived xenografts. This finding provides promising prognostic markers and pharmacogenomic biomarkers for potential clinical guidance.
Large-scale studies have reported that disease-related mutations tend to cause large geometrical and physicochemical changes of mutation sites, which in turn affects the stability of protein interactions by changing their binding affinity [6, 7, 11]. In the past few years, many computational methods have been proposed to predict mutation effects on protein interactions, especially their effects on the binding affinity of protein interactions [12]. Although there is often a trade-off between accuracy of experimental methods and efficiency of computational methods, the computational methods have achieved remarkable successes given that experimental methods are generally laborious, costly, and time-consuming. In the following sections, we focus on recent studies on the characteristics of disease-related mutations at protein-protein interfaces, advances in the identification of mutational effects on protein interactions, and investigations of mutations on specific human diseases.
Mutations at protein-protein interfaces on human diseases
It has been widely demonstrated that disease-related mutations preferentially localize to protein-protein interaction interfaces [3, 4, 5, 13, 14]. Figure 1 shows that a disease-related mutation located at an interface disrupts the interaction and implicates the corresponding disease [9]. Moreover, mutations on the same protein can cause distinct clinical diseases by disrupting its interactions with different partners [3, 4, 5]. David et al. [14] examined the frequencies of 2,420 disease-related mutations in three different regions of proteins (protein core, interface, and surface noninterface) and compared the quantified preferences of disease-related mutations at protein interfaces with that of the other two regions. Their results showed that disease-related mutations preferentially occur at interfaces than surface noninterface with an odds ratio (OR) of 1.59. Additionally, a similar observation (OR=1.44) has been reported by another study [2].
Figure 1. A disease-related interface mutation G352R on SMAD4.
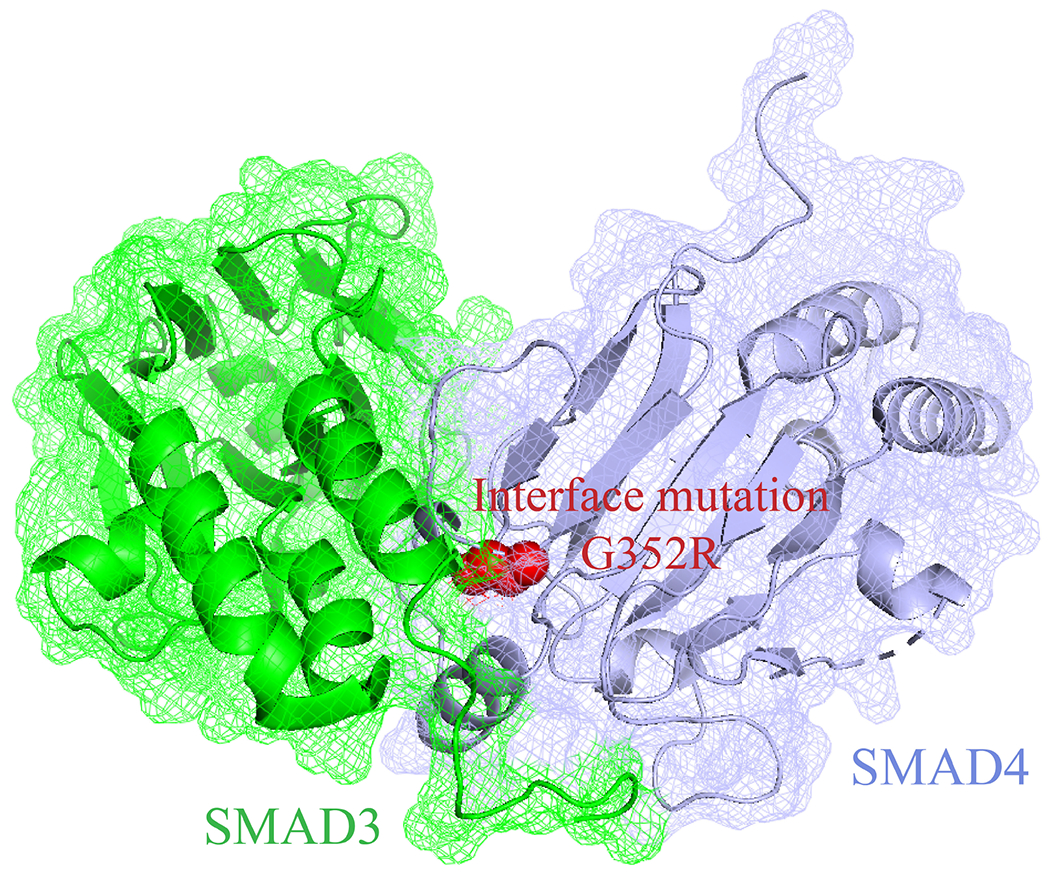
This mutation disrupts the SMAD4 interaction with SMAD3, and implicates the TGFβ/SMAD signaling pathway in the formation of juvenile polyposis.
Interface residues, which are located over a large surface area, can be distinguished into ‘core residues’ and ‘rim residues’ based on their solvent accessibility in the bound state of two interacting proteins [15]. Interface rim residues are partially solvent-accessible and surround the interface core region at which the residues are completely buried as a result of the protein-protein interaction. It has been shown that residues on the interface core region generally make more contributions than those on the rim to protein interactions [2, 13]. Specifically, Navío et al. [2] showed this tendency by examining 2,062 residues in the interface core and rim regions. The authors concluded that interface residues that make an energetic contribution to the protein complex stability or related to human diseases are more likely to locate at the interface core region rather than rim region, and quantified this preference using OR (OR = 2.11). However, disease-related mutations did not show any preference to be located at the interface rim regions than the non-interface surfaces [13]. Moreover, the significance of preference for the neutral mutations, which do not cause any genetic disorders, for interface core vs rim has not been established [2].
The binding affinity change (ΔΔG) is an important factor to discriminate disease-related mutations from neutral mutations. While random amino acid substitutions including neutral and disease-related mutations tend to decrease the binding free energe [6, 13, 16], disease-related mutations are more likely to cause a decrease in their binding affinity [6, 13]. Jemimah et al. [6] performed a statistical analysis to explore the change in binding affinity caused by neutral and disease-related mutations. They found that most of the disease-related mutations decrease the binding affinity and showed that there exists a significant difference in the effect on protein stability between neutral and disease-related mutations.
Mutations may introduce various types of effects, which include reduction in hydrophobic region, overpacking, decrease in electrostatic interactions etc., and cause protein instability and, eventually, loss of protein interactions [14, 17]. For example, an amino acid substitution from a smaller residue to a larger residue could lead to steric clashes. On the other hand, a mutation from a larger amino acid to a smaller amino acid could create a spatial gap. Either of the cases is likely to impair protein stability [17]. Besides, the impact is more significant if the mutation is carried out by energetic hotspots or residues in the protein core [13]. The change of hydrophobicity of residue caused by mutations in the interface region also brings about loss of protein interactions. As interface residues are more hydrophobic than surface noninterface residues [18], their substitution to charged or polar residues could disrupt protein interactions [14]. While destabilization is a more common effect, disease-related mutations could also stabilize the proteins and complexes [19, 20]. This suggests that, although the decrease in binding affinity and protein instability could be the dominant effects of disease-related mutations, they could bring other types of effects, or even opposite effects, to protein structures and protein interactions, which requires more comprehensive analysis to understand their mechanisms.
Approaches to identifying mutation effects on protein interactions
The change in binding affinity of protein interactions caused by mutations can further affect the stability of protein interactions and the function of proteins involved and, eventually, cause diseases. Effects of mutations on protein-protein binding sites can be assessed by the change in binding free energy, which is one of the most significant factors contributing to pathogenicity [11, 21]. The binding affinity can be quantitatively measured through various experimental methods, including Isothermal Titration Calorimetry (ITC), Förster Resonance Energy Transfer (FRET), Surface Plasmon Resonance (SPR), and Fluorescence Polarization (FP) [11, 22]. These methods provide accurate measures of binding affinity change, however, they are laborious, expensive and time-consuming and, more importantly, not feasible to large-scale datasets in practice. These drawbacks of experimental methods motivated the development of fast and reliable computational approaches to predicting protein-protein ΔΔG in large-scale studies. In the past decades, as shown in Table 1, many computational approaches have been proposed to meet the needs, which can be broadly categorized into classical energy-based methods and machine learning-based methods [12, 23].
Table 1.
Summary of representative computational approaches for ΔΔG prediction.
| Categories | Names | Webservers/Softwares | Unique features/advantages | References |
|---|---|---|---|---|
| Energy-based | FoldX | http://foldxsuite.crg.eu | Uses a rotamer library | [24, 25] |
| Rosetta | - | Uses a rotamer library, focusing on alanine mutations | [26] | |
| CC/PBSA | - | Considers structural flexibility | [27] | |
| ZEMu | https://simtk.org/projects/rnatoolbox | Uses a multiscale method which models flexibility of mutation region | [28] | |
| Flex ddG | https://github.com/Kortemme-Lab/flex_ddG_tutorial | Samples conformational diversity using “backrub” to generate an ensemble of models | [29] | |
| BeAtMuSiC | http://babylone.ulb.ac.be/beatmusic | Uses the coarse-grained representation of protein structures | [30] | |
| Contact potentials-based model | - | Uses atomic and residue contact potentials | [31] | |
| BindProfX | https://zhanglab.dcmb.med.umich.edu/BindProfX | Calculates ΔΔG as the logarithm of relative probability of mutant residues over wild-type ones | [32] | |
| SSIPe | https://zhanglab.ccmb.med.umich.edu/SSIPe https://github.com/tommyhuangthu/SSIPe | Combines interface profiles derived from structural and sequence homology searches with a physics-based energy | [33] | |
| Machine learning-based | mCSM-PPI2 | http://biosig.unimelb.edu.au/mcsm_ppi2 | Integrates mCSM graph-based signatures, evolutionary information, inter-residue non-covalent interaction networks analysis and energetic terms with ETs | [36] |
| iSEE | https://github.com/haddocking/iSee | Combines structural, evolutionary, and energetic features with RF | [37] | |
| TopNetTree | https://codeocean.com/capsule/2202829/tree/v1 | Integrates topological describtors with CNN-assisted GBDT | [38] | |
| MutaBind2 | https://lilab.jysw.suda.edu.cn/research/mutabind2 | Combines a set of scoring functions with RF | [39] | |
| ELASPIC2 | http://elaspic.kimlab.org https://gitlab.com/elaspic/elaspic2 | Incoraporates features generated using pre-trained TNN and GNN, and employs GBDT with a ranking object function | [40] | |
| ProAffiMuSeq | https://web.iitm.ac.in/bioinfo2/proaffimuseq | Considers the functional classes | [41] | |
| MuPIPR | https://github.com/guangyu-zhou/MuPIPR | An end-to-end deep learning framework using Bi-LSTM, RCNN and MLP without the need of hand-crafted features | [23] | |
| SAAMBE-SEQ | http://compbio.clemson.edu/saambe_webserver/indexSEQ.php#started | Employs GBDT on a set of features, and doesn’t require the knowledge of interfacial residue | [42] |
Classical energy-based methods typically rely on physical/empirical energies and/or statistical potentials to find the optimal models. Some of these methods, such as FoldX [24, 25], Rosetta [26], CC/PBSA [27], ZEMu [28], Flex ddG [29], etc. use physical energies. The physical energies mainly come from van der Waals, solvation, hydrogen bonds, water bridges, electrostatic, entropy, Lennard Jones interactions. Furthermore, the statistical potentials are also used to predict ΔΔG, such methods include BeAtMuSiC [30], contact potentials-based model [31], etc. These statistical potentials describe the correlations between numbers of pairwise inter-residue contacts, pairwise inter-residue distances, amino acid types, backbone torsion angles and solvent accessibilities. The most recently published energy-based methods including BindProfX [32] and SSIPe [33] combined such energies with other information (such as protein interface profiles) to predict ΔΔG and have achieved better performance than other energy-based models. However, these methods require the structures of mutated complexes as an input, which in turn limits their applicability drastically.
Machine learning methods capture the relationship between the ΔΔG and a set of generally important features extracted from the protein structure, sequence, energy, evolution, etc.. Due to the ever-increasing mutation data availability in public databases such as SKEMPI 2.0 [34] and PROXiMATE [35], a high number of machine learning-based methods have been proposed in recent years. Some of them, such as mCSM-PPI2 [36], iSEE [37], TopNetTree [38], MutaBind2 [39], ELASPIC2 [40], etc., have been developed with structure-based features using extra trees (ETs), random forest (RF), gradient boosting decision tree (GBDT), convolutional neural network (CNN), transformer neural network (TNN), graph neural network (GNN) and so on. Several machine learning-based methods, which give better representations of structures of mutated complexes, typically include the aforementioned energies as features. However, these methods, like the energy-based methods, suffer in terms of their applicability due to their reliance on the input structures, even though the structures of a small portion of mutated complexes can be approximated by modeling 3D structures from sequences using homology modeling. Therefore, some sequence-based methods, such as ProAffiMuSeq [41], MuPIPR [23], SAAMBE-SEQ [42], etc., have been proposed using bidirectional long short-term memory (Bi-LSTM), recurrent convolutional neural network (RCNN), multi-layer perceptron (MLP), GBDT and so on. Over the past few years, deep learning has shown remarkable success and the explosive growth in its application to various fields including bioinformatics [43, 44]. For studying mutational effects on protein interactions, several deep learning models such as TopNetTree, MuPIPR, have been developed and achieves satisfactory performance. Especially, MuPIPR achieves success in an end-to-end manner without the need of hand-crafted features, which indicates the strong representation power and practical advantages of deep learning. Along with further accumulation of data and advances in computational technology, deep learning will lead to enormous opportunities for machine learning-based methods to learn the complicated patterns between various protein features and the ΔΔG.
Feature design also plays a crucial role in computational approaches. The knowledge of feature importance would greatly contribute to the understanding of mechanisms of mutation impact on ΔΔG and the development of novel computational approaches [12]. Several methods have reported the relative importance of features they used. Specifically, iSEE reveals that the Position Specific Scoring Matrix (PSSM) value of the wildtype amino acid and the difference of PSSM values between mutant and wildtype residues are the most important features. The PSSM captures the evolutionary conservation information of a specific amino acid. In SAAMBE-SEQ, the PSSM-based evolution and conservation scores were also found to be the two most important features. It is worth noting such evolution and conservation information can be obtained through protein sequence without the need for any structural information.
The pathogenicity of mutations affecting protein interaction on human diseases
Mutations that affect protein interaction interfaces influence the formation of protein complexes. They lead to phenotypic changes and have been demonstrated to play “driver” roles in human cancers as well as other genetic diseases. A vast number of studies have discovered that pathogenic mutations cause different diseases by disrupting protein interactions or changing the binding affinity of specific protein complexes [45, 46, 47, 48, 49, 50, 51]. Kato et al. [48] reported that the cancer-derived substitution mutation is likely to occur at highly conserved amino acids of the ubiquitously transcribed tetratricopeptide repeat on the X chromosome (UTX). The authors also observed that several mutations in the tetratricopeptide repeat (TPR) alter the interactions of UTX with core components of mixed-lineage leukemia complexes (MLL3/4 complexes), the interactions with crucial importance in the tumor suppressor function. Yan et al. [50] investigated the structural basis of the pathogenicity of Legius syndrome mutations and discovered that these mutations reduce the binding affinity between SPRED1 and NF1. This finding supports the hypothesis proposed in [51] that mutations in SPRED1 could cause Legius syndrome due to an inability to form the NF1-SPRED1 complex. Chen et al. [52] showed that missense mutations disrupting interactions of autism spectrum disorder (ASD) proteins are enriched in individuals with ASD and further constructed and analyzed an “ASD disrupted network” by connecting all disrupted pairs of proteins to explore the risk genes of ASD.
The pathogenicity of disease mutations can also be derived from the alteration of binding properties of proteins and their interactome [4, 19, 53, 54, 55], of which little research has been conducted as the perturbations of interactome is much more difficult to analyze than the alterations in single disease proteins. Mehnert et al. [53] analyzed cancer mutations in the Dyrk2 protein kinase and found that these mutations significantly change the Dyrk2 interaction network. For more information, readers may refer to Wanker et al. [55], which reviewed the studies about the perturbed interactors of the huntingtin (HTT) protein with mutants and their pathobiological roles in the disease.
Moreover, mutations of SARS-Cov-2 proteins that affect specific protein interactions have significant impacts on its infectivity. It is well known that the coronavirus disease 2019 (COVID-19) has stronger infectivity than the SARS coronavirus 2003 [56]. The key factor causing this difference discovered by Wang et al. [57] is the mutation of a hydrophobic residue in the SARS-CoV sequence to Lys417 in SARS-CoV-2. It enhances protein interactions between SARS-CoV-2 and the host receptor ACE2 which serves as a major receptor for SARS-CoV-2 in human cells. Besides, several SARS-CoV-2 mutations (e.g., D614G [58, 59], N501Y [60]) were also observed to enhance the binding affinity between SARS-CoV-2 and ACE2. These mutations might either increase the infection rates or be related to a higher case fatality rate from the strains of SARS-CoV-2 found in different countries. Rawat et al. [61] performed a mutational analysis for the interactions of three strains of NL63, SARS-CoV and SARS-CoV-2 with ACE2, respectively. They found the mutation of the conserved Gly residue in all three strains (Gly537 in NL63, Gly488 in SARS-CoV and Gly502 in SARS-CoV-2) significantly reduced the binding affinity, which revealed its importance for both stabilizing and interacting with ACE2. Further, the SARS-CoV-2 mutations could also change intraviral protein interactions and might influence the transmission of SARS-CoV-2 and the treatment of COVID-19 [62].
Conclusion
The significance of protein-protein interfaces has prompted rigorous research and resulted in great insights that enhance our understanding of molecular mechanism of protein interactions. Many studies have demonstrated that disease-related mutations at interfaces may lead to various effects such as destabilizing protein interactions, decreasing binding affinity, etc. Although experimental methods allow us to accurately measure such effects, many computational approaches have been proposed to efficiently study protein functions with large-scale datasets. Especially, deep learning has been given considerable attention due to the accumulation of large data and powerful computational resources, and its strong feature representation ability. The deep exploration of disease-related mutations will enable the novel and rational drug design.
Highlights.
Disease-related mutations preferentially localize to protein-protein interfaces.
Mutations, in general, destabilize protein-protein interactions and protein structures.
Deep learning shows great advantages and potential in mutation effect prediction.
Pathogenic mutations may lead to different diseases by disrupting or changing the binding affinity.
Acknowledgements
We appreciate the valuable advices from Dr. Yugandhar Kumar for the manuscript writing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflict of interest.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Cukuroglu E, Engin HB, Gursoy A, Keskin O: Hot spots in protein–protein interfaces: Towards drug discovery. Prog Biophys Mol Biol 2014, 116:165–173. [DOI] [PubMed] [Google Scholar]
- 2**.Navío D, Rosell M, Aguirre J, de la Cruz X, Fernández-Recio J: Structural and Computational Characterization of Disease-Related Mutations Involved in Protein-Protein Interfaces. Int J Mol Sci 2019, 20:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]; Considering the structural location of amino acid residues (Buried, Surface, Interface rim, and Interface core), the authors studied the effect of disease-related mutations on protein-protein interactions. They comprehensively discussed the structural characterization of protein interactions, the distribution of disease-related and neutral mutations across the different interface regions, and the substitution susceptibility of distinct amino acids.
- 3.Meyer MJ, Beltrán JF, Liang S, Fragoza R, Rumack A, Liang J, Wei X, Yu H: Interactome INSIDER: a structural interactome browser for genomic studies. Nat Methods 2018, 15:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahni N, Yi S, Taipale M, Fuxman Bass Juan I, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras Georgios I, Wang Y, et al. : Widespread Macromolecular Interaction Perturbations in Human Genetic Disorders. Cell 2015, 161:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Wei X, Thijssen B, Das J, Lipkin SM, Yu H: Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol 2012, 30:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6*.Jemimah S, Gromiha MM: Insights into changes in binding affinity caused by disease mutations in protein-protein complexes. Comput Biol Med 2020, 123:103829. [DOI] [PubMed] [Google Scholar]; This paper performs a statistical study to show the relationship between ΔΔG and disease-related/neutral mutations. The authors observed a large proportion of disease-causing mutations cause the decrease of the binding affinity. The results are given according to the disease classes. Additionally, the authors provide other factors potentially affecting the disease development.
- 7.Jubb HC, Pandurangan AP, Turner MA, Ochoa-Montaño B, Blundell TL, Ascher DB: Mutations at protein-protein interfaces: Small changes over big surfaces have large impacts on human health. Prog Biophys Mol Biol 2017, 128:3–13. [DOI] [PubMed] [Google Scholar]
- 8.Teppa E, Zea DJ, Marino-Buslje C: Protein–protein interactions leave evolutionary footprints: High molecular coevolution at the core of interfaces. Protein Sci 2017, 26:2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fragoza R, Das J, Wierbowski SD, Liang J, Tran TN, Liang S, Beltran JF, Rivera-Erick CA, Ye K, Wang T-Y, et al. : Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat Commun 2019, 10:4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Cheng F, Zhao J, Wang Y, Lu W, Liu Z, Zhou Y, Martin WR, Wang R, Huang J, Hao T, et al. : Comprehensive characterization of protein–protein interactions perturbed by disease mutations. Nat Genet 2021, 53:342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors showed disease-associated germline mutations are significantly enriched in sequences encoding protein-protein interfaces. Somatic missense mutations are also significantly enriched in protein-protein interfaces. They also developed a human structurally resolved macromolecular interactome framework for comprehensive identification of protein interaction-perturbing alleles in human disease, and showed widespread protein interaction network perturbations caused by both disease-associated germline and somatic mutations.
- 11.Kucukkal TG, Petukh M, Li L, Alexov E: Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr Opin Struct Biol 2015, 32:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Geng C, Xue LC, Roel-Touris J, Bonvin AMJJ: Finding the ΔΔG spot: Are predictors of binding affinity changes upon mutations in protein–protein interactions ready for it? WIREs Comput Mol Sci 2019, 9:e1410. [Google Scholar]; In this review, the authors summarized various aspects of the prediction of the mutation impact on the binding affinity. They focused on predictors that consider 3D structure information, excluding the more rigorous free-energy perturbation methods. Emphasis was given to training and evaluation of models, ΔΔG databases, data selection, and existing ΔΔG predictors.
- 13.David A, Sternberg MJE: The Contribution of Missense Mutations in Core and Rim Residues of Protein–Protein Interfaces to Human Disease. J Mol Biol 2015, 427:2886–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.David A, Razali R, Wass MN, Sternberg MJE: Protein–protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum Mutat 2012, 33:359–363. [DOI] [PubMed] [Google Scholar]
- 15.Bogan AA, Thorn KS: Anatomy of hot spots in protein interfaces. J Mol Biol 1998, 280:1–9. [DOI] [PubMed] [Google Scholar]
- 16.Petukh M, Kucukkal TG, Alexov E: On Human Disease-Causing Amino Acid Variants: Statistical Study of Sequence and Structural Patterns. Hum Mutat 2015, 36:524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue P, Li Z, Moult J: Loss of Protein Structure Stability as a Major Causative Factor in Monogenic Disease. J Mol Biol 2005, 353:459–473. [DOI] [PubMed] [Google Scholar]
- 18.Jones S, Thornton JM: Principles of protein-protein interactions. Proc Natl Acad Sci USA 1996, 93:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishi H, Tyagi M, Teng S, Shoemaker BA, Hashimoto K, Alexov E, Wuchty S, Panchenko AR: Cancer Missense Mutations Alter Binding Properties of Proteins and Their Interaction Networks. PLOS ONE 2013, 8:e66273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Norris J, Schwartz C, Alexov E: In Silico and In Vitro Investigations of the Mutability of Disease-Causing Missense Mutation Sites in Spermine Synthase. PLOS ONE 2011, 6:e20373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng Y, Alexov E: Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abbasi WA, Asif A, Ben-Hur A, Minhas FuAA: Learning protein binding affinity using privileged information. BMC Bioinformatics 2018, 19:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23**.Zhou G, Chen M, Ju CJT, Wang Z, Jiang J-Y, Wang W: Mutation effect estimation on protein–protein interactions using deep contextualized representation learning. NAR Genom Bioinform 2020, 2:lqaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents MuPIPR, an end-to-end deep learning framework with only sequence information and without the need of hand-crafted features to estimate the mutation effects on protein interactions. The architecture of MuPIPR includes three components: a contextualized amino acid representation mechanism, a protein sequence level Siamese encoder by leveraging the RCNN, and multiple-layer perceptron regressors for estimating quantifiable property changes in protein interactions upon mutations.
- 24.Guerois R, Nielsen JE, Serrano L: Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J Mol Biol 2002, 320:369–387. [DOI] [PubMed] [Google Scholar]
- 25.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L: The FoldX web server: an online force field. Nucleic Acids Res 2005, 33:W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kortemme T, Baker D: A simple physical model for binding energy hot spots in protein–protein complexes. Proc Natl Acad Sci USA 2002, 99:14116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benedix A, Becker CM, de Groot BL, Caflisch A, Böckmann RA: Predicting free energy changes using structural ensembles. Nat Methods 2009, 6:3–4. [DOI] [PubMed] [Google Scholar]
- 28.Dourado DFAR, Flores SC: A multiscale approach to predicting affinity changes in protein–protein interfaces. Proteins 2014, 82:2681–2690. [DOI] [PubMed] [Google Scholar]
- 29.Barlow KA, Ó Conchúir S, Thompson S, Suresh P, Lucas JE, Heinonen M, Kortemme T: Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein–Protein Binding Affinity upon Mutation. J Phys Chem B 2018, 122:5389–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dehouck Y, Kwasigroch JM, Rooman M, Gilis D: BeAtMuSiC: prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res 2013, 41:W333–W339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moal IH, Fernandez-Recio J: Intermolecular Contact Potentials for Protein–Protein Interactions Extracted from Binding Free Energy Changes upon Mutation. J Chem Theory Comput 2013, 9:3715–3727. [DOI] [PubMed] [Google Scholar]
- 32.Xiong P, Zhang C, Zheng W, Zhang Y: BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. J Mol Biol 2017, 429:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Huang X, Zheng W, Pearce R, Zhang Y: SSIPe: accurately estimating protein–protein binding affinity change upon mutations using evolutionary profiles in combination with an optimized physical energy function. Bioinformatics 2020, 36:2429–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents SSIPe that combines protein interface profiles with a physics-based energy function for accurate ΔΔG prediction. To offset the statistical limits of the PPI structure and sequence databases, amino acid-specific pseudocounts were introduced to enhance the profile accuracy.
- 34.Jankauskaitė J, Jiménez-García B, Dapkūnas J, Fernández-Recio J, Moal IH: SKEMPI 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019, 35:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jemimah S, Yugandhar K, Michael Gromiha M: PROXiMATE: a database of mutant protein–protein complex thermodynamics and kinetics. Bioinformatics 2017, 33:2787–2788. [DOI] [PubMed] [Google Scholar]
- 36.Rodrigues CHM, Myung Y, Pires DEV, Ascher DB: mCSM-PPI2: predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res 2019, 47:W338–W344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geng C, Vangone A, Folkers GE, Xue LC, Bonvin AMJJ: iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins 2019, 87:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38*.Wang M, Cang Z, Wei G-W: A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat Mach Intell 2020, 2:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors introduced site-specific persistent homology that is tailored for protein interaction analysis and explored the utility of site-specific persistent homology and machine learning algorithm for characterizing protein interactions that are associated with site-specific mutations. They further proposed TopNetTree which integrates topological descriptor with CNN-assisted GBDT to predict ΔΔG of protein interactions.
- 39.Zhang N, Chen Y, Lu H, Zhao F, Alvarez RV, Goncearenco A, Panchenko AR, Li M: MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience 2020, 23:100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strokach A, Lu TY, Kim PM: ELASPIC2 (EL2): Combining Contextualized Language Models and Graph Neural Networks to Predict Effects of Mutations. J Mol Biol 2021, 433:166810. [DOI] [PubMed] [Google Scholar]
- 41.Jemimah S, Sekijima M, Gromiha MM: ProAffiMuSeq: sequence-based method to predict the binding free energy change of protein–protein complexes upon mutation using functional classification. Bioinformatics 2020, 36:1725–1730. [DOI] [PubMed] [Google Scholar]
- 42.Li G, Pahari S, Murthy AK, Liang S, Fragoza R, Yu H, Alexov E: SAAMBE-SEQ: a sequence-based method for predicting mutation effect on protein–protein binding affinity. Bioinformatics 2021, 37:992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Tian S, Li Y, Fang Q, Tan R, Pan Y, Huang C, Xu Y, Gao X: Modern deep learning in bioinformatics. J Mol Cell Biol 2020, 12:823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Huang C, Ding L, Li Z, Pan Y, Gao X: Deep learning in bioinformatics: Introduction, application, and perspective in the big data era. Methods 2019, 166:4–21. [DOI] [PubMed] [Google Scholar]
- 45.Muda K, Bertinetti D, Gesellchen F, Hermann JS, von Zweydorf F, Geerlof A, Jacob A, Ueffing M, Gloeckner CJ, Herberg FW: Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc Natl Acad Sci USA 2014, 111:E34–E43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin Guan N, Corominas R, Lemmens I, Yang X, Tavernier J, Hill David E, Vidal M, Sebat J, Iakoucheva Lilia M: Spatiotemporal 16p11.2 Protein Network Implicates Cortical Late Mid-Fetal Brain Development and KCTD13-Cul3-RhoA Pathway in Psychiatric Diseases. Neuron 2015, 85:742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, et al. : Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kato H, Asamitsu K, Sun W, Kitajima S, Yoshizawa-Sugata N, Okamoto T, Masai H, Poellinger L: Cancer-derived UTX TPR mutations G137V and D336G impair interaction with MLL3/4 complexes and affect UTX subcellular localization. Oncogene 2020, 39:3322–3335. [DOI] [PubMed] [Google Scholar]
- 49.Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. : Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 2015, 47:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan W, Markegard E, Dharmaiah S, Urisman A, Drew M, Esposito D, Scheffzek K, Nissley DV, McCormick F, Simanshu DK: Structural Insights into the SPRED1-Neurofibromin-KRAS Complex and Disruption of SPRED1-Neurofibromin Interaction by Oncogenic EGFR. Cell Rep 2020, 32:107909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernández H, Burlingame AL, McCormick F: A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Gems Dev 2012, 26:1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52*.Chen S, Wang J, Cicek E, Roeder K, Yu H, Devlin B: De novo missense variants disrupting protein–protein interactions affect risk for autism through gene co-expression and protein networks in neuronal cell types. Mol Autism 2020, 11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors showed that de novo missense variants that disrupt protein interactions are enriched in individuals with ASD. Genes encoding disrupted complementary interactors are likely to be risk genes, and an interaction network built from these proteins is enriched for ASD proteins. Genes identifed by disrupted protein interactions are expressed early in development and in excitatory and inhibitory neuronal lineages.
- 53.Mehnert M, Ciuffa R, Frommelt F, Uliana F, van Drogen A, Ruminski K, Gstaiger M, Aebersold R: Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat Commun 2020, 11:3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jafri M, Wake NC, Ascher DB, Pires DEV, Gentle D, Morris MR, Rattenberry E, Simpson MA, Trembath RC, Weber A, et al. : Germline Mutations in the CDKN2B Tumor Suppressor Gene Predispose to Renal Cell Carcinoma. Cancer Discov 2015, 5:723–729. [DOI] [PubMed] [Google Scholar]
- 55.Wanker EE, Ast A, Schindler F, Trepte P, Schnoegl S: The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J Neurochem 2019, 151:507–519. [DOI] [PubMed] [Google Scholar]
- 56.Kuiken T, Fouchier RAM, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, Laman JD, de Jong T, van Doornum G, Lim W, et al. : Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003, 362:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Wang Y, Liu M, Gao J: Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc Natl Acad Sci USA 2020, 117:13967–13974. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors found a key mutation from a hydrophobic residue in the SARS-CoV sequence to Lys417 in SARS-CoV-2 results in greater electrostatic complementarity. Both electrostatic effects and enhanced hydrophobic packing lead to conformation shift toward a more tilted binding groove in the complex in comparison with the SARS-CoV complex. Hydrophobic contacts in the complex of the SARS-CoV–neutralizing antibody 80R are disrupted in the SARS-CoV-2 homology complex model.
- 58.Ozono S, Zhang Y, Ode H, Sano K, Tan TS, Imai K, Miyoshi K, Kishigami S, Ueno T, Iwatani Y, et al. : SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat Commun 2021, 12:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Becerra-Flores M, Cardozo T: SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract 2020, 74:e13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ali F, Kasry A, Amin M: The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med Drug Discov 2021, 10:100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rawat P, Jemimah S, Ponnuswamy PK, Gromiha MM: Why are ACE2 binding coronavirus strains SARS-CoV/SARS-CoV-2 wild and NL63 mild? Proteins 2021, 89:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu S, Tian C, Liu P, Guo D, Zheng W, Huang X, Zhang Y, Liu L: Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J Med Virol 2021, 93:2132–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]