

Abstract
Genome-wide association studies (GWAS) are highly effective at identifying common risk variants for schizophrenia. Rare risk variants are also important contributors to schizophrenia aetiology but, with the exception of large copy number variants (CNVs), are difficult to detect with GWAS. Exome and genome sequencing, which have accelerated the study of rare variants, are expensive so alternative methods are needed to aid detection of rare variants. Here we re-analyse an Irish schizophrenia GWAS dataset (n=3,473) by performing identity-by-descent (IBD) mapping followed by exome sequencing of individuals identified as sharing risk haplotypes to search for rare risk variants in coding regions. We identified 45 rare haplotypes (>1cM) that were significantly more common in cases than controls. By exome sequencing 105 haplotype carriers, we investigated these haplotypes for functional coding variants that could be tested for association in independent GWAS samples. We identified one rare missense variant in PCNT but did not find statistical support for an association with schizophrenia in a replication analysis. However, IBD mapping can prioritize both individual samples and genomic regions for follow-up analysis but genome rather than exome sequencing may be more effective at detecting risk variants on rare haplotypes.
Keywords: IBD mapping, GWAS, rare variants
Introduction
Schizophrenia [MIM 181500] is a heritable mental disorder with a lifetime risk of ~1%. The emerging genetic architecture points to a spectrum of risk variation from common variants, likely to have subtle effects on gene expression, to functionally deleterious variants. Analysis of common risk variants has identified 108 independent schizophrenia associated risk loci (median odds ratio (OR) = 1.08) (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014), which collectively explain a small fraction of genetic risk (Loh et al., 2015). Analysis of rare copy number variants (CNVs) has identified a global enrichment of CNV burden in schizophrenia patients and eight individual risk loci reaching genome-wide significance (4 < OR < 68) (Marshall et al., 2017). The risk CNVs are rare, even in schizophrenia cases, and were identified by large case-control studies or meta-analyses of array-based data (reviewed in Marshall et al., 2017). Such rare variants, having greater penetrance and a potentially clearer relationship to disease risk, may be most informative for functional follow-up studies (Karayiorgou, Flint, Gogos, Malenka, & Genetic and Neural Complexity in Psychiatry Working, 2012).
Next-generation sequencing (NGS) has allowed the search for rare risk variants to extend to the exome and genome using both case-control and family-based designs. To date, genome sequencing has been limited to exploratory studies of multiplex families (Homann et al., 2016). A number of reasonably large case-control exome sequencing studies have now been reported and have identified gene-disruptive and putatively protein-damaging ultra-rare variants to be more abundant in cases than controls (Genovese et al., 2016). Initially, some small gene sets were thought highly enriched for rare disruptive variants (S. M. Purcell et al., 2014) but subsequent analyses suggest the enrichment is within a broader set of potentially synaptic genes. Results from the largest study to date, an analysis of 4,877 cases and 6,203 controls did not succeed in implicating any individual gene or risk variant after correction for multiple testing (Genovese et al., 2016).
The association of schizophrenia with decreased fecundity has prompted the study of trios samples to detect de novo variants. Data from CNV studies support de novo variation as a contributing factor to the genetic aetiology of schizophrenia (Rees, Moskvina, Owen, O’Donovan, & Kirov, 2011). This has been confirmed by exome sequencing in trios implicating genes involved in synaptic function and chromatin remodelling, but also suggesting shared pathophysiology with other neurodevelopmental disorders such as autism and intellectual disability (Fromer et al., 2014; McCarthy et al., 2014). But like the case-control studies, the trios studies did not specifically identify a new risk gene after multiple test correction. It took the novel combined analysis of case-control and trios exome data to identify SETD1A, a gene involved in chromatin remodelling, as a site of rare loss-of-function (LoF) variants increasing risk of schizophrenia after genome-wide correction (Singh et al., 2016).
With limited numbers of trios available for analysis and case-control sequencing studies remaining very expensive, the study of identity-by-descent (IBD) segments in genome-wide association study (GWAS) data is an alternative method for finding rare risk variants for schizophrenia. IBD segments represent chromosomal regions that are inherited without recombination from a common ancestor, and a signature of their presence is strong linkage disequilibrium across the segment that can be detected in GWAS data (Hou et al., 2013). IBD methods have been successfully applied in founder populations to identify genes for both monogenic (Brooks et al., 2009) and complex disorders (Morrow et al., 2008). In outbred populations, IBD methods have been successfully applied to map genes for neurodevelopmental disorders using array-based SNP data with follow-up Sanger sequencing (Ercan-Sencicek et al., 2010) and using exome sequencing data (Krawitz et al., 2010) but these studies used family-based samples. IBD methods have been applied to population-based GWAS data for multiple sclerosis but with limited success (Lin et al., 2013; Westerlind et al., 2015).
Here, we detail a novel approach to rare risk variant detection in schizophrenia that combines IBD analysis of GWAS data and exome sequencing. We used IBD analysis of GWAS data from a homogeneous sample of Irish schizophrenia patients and controls (Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2, 2012) to search for shared haplotype segments in distantly related individuals and tested for haplotype segments that were more frequent in cases than controls. This approach identified a subset of key individuals to be sequenced and reduced the search space for mid to high effect rare variants that may have arisen de novo in more recent generations but have not been removed from the population by purifying selection (Lupski, Belmont, Boerwinkle, & Gibbs, 2011). After exome sequencing haplotype carriers, we investigated these segments to identify rare and potentially functional coding variants and then tested these variants for association in both our Irish and large international case-control samples.
Materials and Methods
Discovery Sample
The discovery sample included cases and controls from the Irish Schizophrenia Genomics Consortium/WTCCC2 GWAS study, which has previously been described (Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2, 2012). Cases were recruited through community mental health services and inpatient units in the Republic of Ireland and Northern Ireland following similar research protocols and with local ethics approval. All participants were interviewed using a structured clinical interview (Structured Clinical Interview for DSM-III-R, Structured Clinical Interview for DSM-IV (First, Spitzer, Robert, Gibbon, & Williams, 2002); Schedule for Affective Disorders and Schizophrenia [Lifetime Version] (Endicott & Spitzer, 1978); or Schedule for Clinical Assessment in Neuropsychiatry (World Health Organization, 1992)). Diagnosis of a major psychotic disorder was made by the consensus lifetime best estimate method using DSM-III-R or DSM-IV criteria with all available information (interview, family or staff report, and chart review). All cases were over 18 years of age, were of Irish origin (being of Irish parents and having all four grandparents born in Ireland or the United Kingdom), and had been screened to exclude substance-induced psychotic disorder or psychosis due to a general medical condition. The final analysis included samples from 1,635 participants whose SNP data passed quality control filters, described below, and met DSM-IV criteria for schizophrenia (or a related disorder).
Control individuals were ascertained with written informed consent from the Trinity Biobank and represented blood donors from the Irish Blood Transfusion Service recruited in the Republic of Ireland. They met the same ethnicity criteria as cases but were not specifically screened for psychiatric illness. Individuals taking regularly prescribed medication are excluded from blood donation in Ireland and donors are not financially remunerated. As the lifetime prevalence of schizophrenia is relatively low (<1%) and there was no obvious reason for individuals with schizophrenia to be overrepresented in the control subjects, the fraction of putative case individuals in the control collection was expected to be small. Following QC, the control sample included 1,838 participants.
Replication Samples
GWAS data from 41 schizophrenia studies was accessed through the Psychiatric Genomics Consortium (PGC2 data) and a description of the individual datasets (which are based on individuals from Europe, North America, Australia and Asia, and genotyped on a variety of arrays) can be found in the 2014 PGC2 publication (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Where possible, variants of interest were imputed in these datasets (maximum possible sample: 28,918 cases and 35,910 controls). A further independent replication dataset from the UK comprised 5,585 cases from two collections, Cardiff COGS and CLOZUK, and 8,103 controls from the UK Blood Service donors and the 1958 British Birth Cohort. Samples were genotyped using the Illumina HumanExome and HumanOmniExpressExome BeadChip arrays, as described by Richards et al. (Richards et al., 2016)
Discovery GWAS: Genotype Calling
All samples were genotyped using the Affymetrix 6.0 platform totalling 893,634 autosomal SNPs, either at the Affymetrix (Santa Clara, California) or Broad Institute (Cambridge, Massachusetts) laboratories. For all samples passing laboratory quality control, raw intensities (from the .CEL files) were renormalized within collections using CelQuantileNorm (http://outmodedbonsai.sourceforge.net/). These normalized intensities were used to call genotypes with an updated version of the Chiamo software (https://mathgen.stats.ox.ac.uk/genetics_software/chiamo/chiamo.html), adapted for Affymetrix 6.0 SNP data.
Discovery GWAS: Quality Control
The primary GWAS has previously been reported and details of the original quality control methods were described therein (Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2, 2012). For this analysis, SNP data was subjected to additional quality filters (variants with minor allele frequency <0.001, SNP missingness ≥0.02, Hardy-Weinberg equilibrium p≤10−6 were removed). SNP positions were updated to hg19. As the aim of this study was to identify shared regions of IBD between distantly related individuals, the previous stringent control on cryptic relatedness was relaxed such that the proportion of genome sharing between any possible pair of individuals was <0.1 (i.e. more distantly related than first cousins), rather than <0.05 as in the original study. In order to ensure a homogeneous sample, 4 individuals were removed that were identified as outliers in a multi-dimensional scaling (MDS) analysis of pairwise genome-wide IBS distances (performed in PLINK (S. Purcell et al., 2007)).
Discovery GWAS: Identification of IBD segments
IBD segments shared between pairs of individuals were detected using the Refined IBD algorithm (B. L. Browning & Browning, 2013) within BEAGLE 4.0 (r1230) (S. R. Browning & Browning, 2007). Candidate segments were required to have a minimum length of 1cM and the likelihood ratio of an IBD model (one haplotype shared IBD) compared to a non-IBD model (no haplotypes shared IBD), i.e. the LOD score, was required to be >3. These are the default values as implemented in (B. L. Browning & Browning, 2013).
Discovery GWAS: IBD mapping
The efficient multiple-IBD (EMI) algorithm (Qian, Browning, & Browning, 2014) was used to identify clusters of individuals sharing a haplotype IBD. EMI was run using a window size of 200kb and a cluster density of 0.5 (as in Qian, Browning, & Browning, 2014). IBD segments with LOD scores between 3 and 40 were weighted 0.8, IBD segments with LOD scores greater than 40 were weighted as 1. This resulted in a set of haplotype cluster files for each chromosome, detailing the genomic coordinates of each identified IBD segment (haplotype), and the individuals sharing each haplotype. Cluster files were converted to plink format for association testing. The association analysis was performed in PLINK (S. Purcell et al., 2007) and compared individual haplotype frequencies in cases versus controls. Haplotypes occurring significantly more frequently in cases than controls at p≤0.01 were taken forward for further analysis. A second strategy employed locus-based tests as opposed to the single haplotype tests performed previously. Each of the genomic regions containing multiple different IBD segments was considered a single locus, and the individual haplotypes within them were tested en masse using SKAT-O (Lee et al., 2012). Of the three significant loci detected at a family-wise error rate of 0.05, any individual haplotype within these loci more frequent in cases than controls at p≤0.05 was taken forward for further analysis. For each of the 45 haplotype segments selected from the two strategies, and where DNA was available for at least two individuals sharing that haplotype, exome sequencing was performed on each individual. A total of 118 individuals were sequenced, representing 32 haplotype segments.
Exome Capture and Sequencing
Exome capture was performed using the Solution Phase Exome Capture method, which is an optimization of the NimbleGen SeqCap EZ Library protocol and the BioO Scientific NEXTflex™ (Illumina Compatible) Sequencing Kit. (See Technology Note: Targeted Sequencing with NimbleGen SeqCap EZ Libraries and Illumina TruSeq DNA Sample Preparation kit). Briefly, sonicated genomic DNA ranging from 1–5μg was used to create Illumina Barcoded libraries. Approximately 1μg of the pre-capture library was hybridized with the NimbleGen SeqCap EZ Human Exome Library v3.0 probes for 72 hours at 47°C. (For further details see the NimbleGen SeqCap EZ Exome User’s Guide and TechNote for paired-end libraries). Following dual capture, PCR enrichment and QC evaluation, samples with Bioanalyzer traces resulting in broad peaks ranging from 250bp–850bp and producing the highest peak around 400bp (DNA insert plus adaptors) were pooled (4–6 captures per pool). Libraries were sequenced on 1–2 lanes on a HiSeq 2000 with a Paired End 101 run including a 9 base pair index read for the barcode detection.
Data Processing and Variant Calling
Sequence reads from the Illumina HiSeq 2000 runs were de-multiplexed using the Illumina Casava v1.8 pipeline, aligned to hg19 using the BWA aligner (Li & Durbin, 2009), allowing 2 mismatches in the 30-base seed. Alignments were then paired, imported to binary (bam) format, sorted and indexed using SAMtools (Li et al., 2009). Picard was then used to fix any mate pair information altered by the sorting. Bamtools (Barnett, Garrison, Quinlan, Strömberg, & Marth, 2011) was used to filter alignments to retain only properly paired reads (reads aligned with appropriate insert size and orientation). PCR duplicates were removed using Picard. Bamtools was then used to select alignments with a minimum mapping quality score of 20. Target coverage for each NimbleGen exome capture was assessed using Picard’s HSmetrics utility, and both depth and breadth of coverage were reviewed for each sample. The Genome Analysis Toolkit (GATK v3.3) (DePristo et al., 2011) was used for local read realignment around indels, and for base quality score recalibration using corrections for base position within the Illumina read, for sequence context, and for platform-reported quality. Variants were called using the GATK Haplotype Caller in joint calling mode, followed by Variant Quality Score Recalibration (VQSR). Twelve samples were excluded due to low coverage.
Variant Filtering and Phasing
The VCF file was filtered to retain variants passing variant quality recalibration and to ensure ≥10X depth at the individual level (using vcftools (Danecek et al., 2011)) and quality by depth (QD) ≥5 at the variant level (using GATK (DePristo et al., 2011)). Samples were checked for concordance with GWAS data; all samples but one were >99% concordant for overlapping SNPs. The remaining discordant sample was determined to be a result of sample mix-up at the GWAS stage. Thus, sequence data was available for 105 individuals, representing 32 individual haplotype segments. As the analysis is focused on variants shared IBD by at least two individuals, all singleton and private doubleton variants were excluded. Allele counts were updated and non-polymorphic variants (post-filtering) removed. Variant IDs were updated to dbSNP142 rs# IDs where available (using SnpSift (Cingolani, Patel, et al., 2012)). Variants were annotated with SnpEff (Cingolani, Platts, et al., 2012) and the Variant Effect Predictor (VEP) (McLaren et al., 2010). The data was phased using Beagle 4.0 (r1230) and the haplotypes shared IBD between individuals, detected through the IBD mapping step, were identified.
Sanger Sequencing
Carriers of the PCNT variant rs143796569 (NC_000021.8:g.47817316G>A) identified through the exome study were Sanger sequenced (primer sequences are available on request) and all individuals were confirmed as heterozygotes. Selected individuals that were predicted to be carriers following imputation (see below) were also verified by Sanger sequencing.
Imputation
The exome sequencing data was merged with existing GWAS data for the 105 sequenced individuals and the resulting dataset was used to create a reference panel using SHAPEIT v2 (Delaneau, Marchini, & Zagury, 2012). This Irish reference panel was merged with the 1000 Genomes Phase I integrated panel (Dec 2013 release; http://www.internationalgenome.org/) and used to impute identified variants of interest into (1) the full Irish GWAS dataset and (2) individual PGC2 GWAS datasets. Association of variants with schizophrenia was tested using a score test implemented in SNPTEST v2 (Wellcome Trust Case Control Consortium, 2007).
Results
Figure 1 is a flow chart that summarizes the analytical methods used in this study. Seeking to identify relatively recent variants impacting on schizophrenia risk in a homogenous population, we performed IBD mapping using data generated from a previously published GWAS of Irish schizophrenia patients and controls (Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2, 2012). We identified segments of the genome (>1cM) predicted to be shared IBD between pairs of individuals using the Refined IBD algorithm within the Beagle software (B. L. Browning & Browning, 2013). We subsequently performed clustering of the identified segments using the efficient multiple-IBD (EMI) algorithm (Qian et al., 2014) to identify haplotype segments shared by at least three individuals. As a result, 11,442 haplotype segments (each shared IBD between at least 3 people) were identified.
Figure 1:
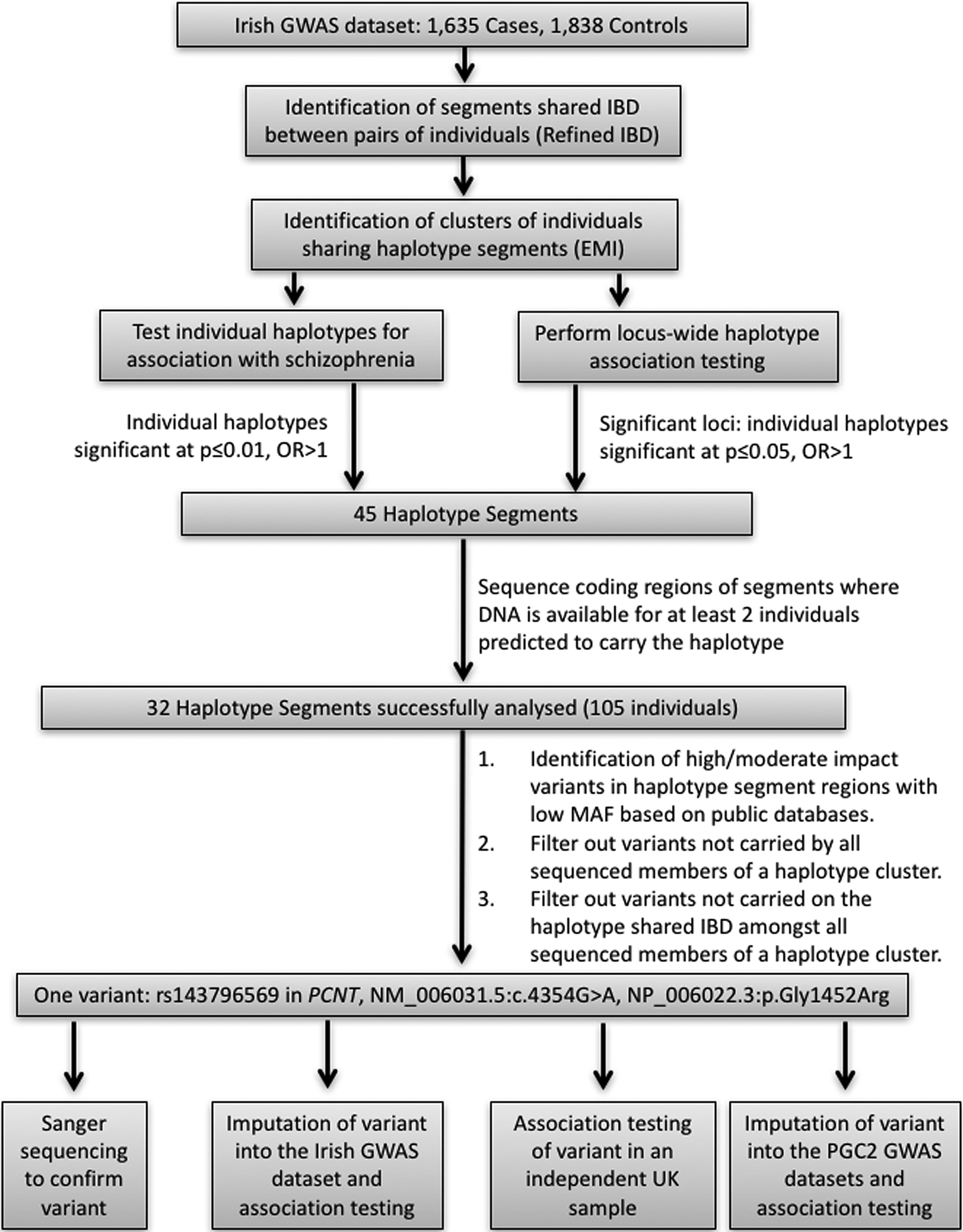
Flow chart highlighting the methods used in our IBD analysis of an Irish GWAS dataset. This study identified rs143796569 in PCNT as a putative rare risk variant for schizophrenia but subsequent analyses in independent case-control samples did not support association with schizophrenia.
For each of the 11,442 segments, the proportion of haplotype carriers in the patient group was compared with the proportion of haplotype carriers in the control group. Following correction for multiple testing, no individual haplotype segment was significantly associated with schizophrenia. However, as this was considered an exploratory analysis, twenty-eight haplotype segments that were more frequent in cases than controls at uncorrected p≤0.01 (listed in Supplementary Table 1) were taken forward for further analysis. To capture the potential impact of multiple risk variants at a locus, multiple different IBD segments at a locus were collapsed and the individual haplotypes within them were tested en masse using SKAT-O (Lee et al., 2012). Forty loci were tested and three were significant after controlling for a family-wise error rate of 0.05 by resampling. Within these loci, 17 individual haplotype segments were observed more frequently in cases than controls at p≤0.05 (listed in Supplementary Table 1) and were taken forward for further analysis. In total, 45 haplotype segments were selected for further analysis.
Hypothesising that rare haplotypes more frequently observed in cases than controls may harbour susceptibility variants of moderate to large effect, we focused on the coding sequence of the prioritised segments. Where DNA was available, we performed exome sequencing in carriers of the prioritised haplotypes (118 individuals). One hundred and five individuals passed quality control, allowing us to analyse 32 of the selected 45 haplotype segments (each with at least 2 carriers successfully sequenced).
Of the identified sequence variants, those lying within haplotype segment regions were extracted and annotated with SnpEff and VEP. Initially high impact variants (e.g. frameshift, nonsense, splice site variants) that were either novel or with a minor allele frequency ≤0.01 in public databases (1000 Genomes European population, the NHLBI GO Exome Sequencing Project (ESP; https://esp.gs.washington.edu/drupal/) European Ancestry population, the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) European population) were assessed. However, of the 30 variants identified, none were carried by all sequenced members of a haplotype cluster. We then assessed moderate impact variants (e.g. missense variants, inframe indels) that were either novel or with a minor allele frequency ≤0.01 in public databases. Of the 159 moderate impact variants identified, three variants were carried by all sequenced members of a relevant haplotype cluster (Table 1). One of these variants was also carried by additional sequenced cases not identified in the original IBD analysis (see Table 1). Each variant was examined to determine if it was carried on the shared IBD haplotype identified in the original analysis. This was only the case for one of the variants; a rare, non-synonymous variant in the PCNT gene, rs143796569 (chr21:47817316 G>A; hg19; NP_006022.3:p.Gly1452Arg), that was confirmed by Sanger sequencing. The variant causes a glycine to arginine change in the protein and is predicted to be “probably damaging” by PolyPhen (Adzhubei et al., 2010) but “tolerated” by SIFT (Kumar, Henikoff, & Ng, 2009). Of the 11 cases and 1 control that were identified as sharing a haplotype at this locus, DNA was available for 5 individuals (all cases), which were sequenced; all 5 carried the alternate allele of the PCNT variant on the shared haplotype. The variant had been previously identified in other studies, with a minor allele frequency of 0.0025 in 33,330 non-Finnish Europeans (ExAC), but was not observed in the 1000 Genomes data.
Table 1:
Moderate impact variants carried by all sequenced members of a relevant haplotype cluster
| Variant | Location (hg19) | Gene | Impact | Number of haplotype cluster individuals sequenced | Additional carriers in sequenced non-cluster individuals | Alternative allele on IBD haplotype? | IBD analysis: Haplotype Frequency (Cases) | IBD analysis: Haplotype Frequency (Controls) | Imputation: Variant MAF in full Irish GWAS (Cases) | Imputation: Variant MAF in full Irish GWAS (Controls) |
|---|---|---|---|---|---|---|---|---|---|---|
| rs115796168a | chr1:247737493 | GCSAML | Asn/His | 2 | 0 | No | 0.0031 | 0 | N.P. | N.P. |
| rs140065392b | chr13:114624033 | LINC00452 | Arg/Cys | 2 | 3 | No | 0.0018 | 0 | N.P. | N.P. |
| rs147006683c | chr19:58579679 | ZNF135 | His/Gln | 3 | 3 | No | 0.0024 | 0 | N.P. | N.P. |
| rs143796569d | chr21:47817316 | PCNT | Gly/Arg | 5 | 0 | Yes | 0.0034 | 0.0003 | 0.0091 | 0.0054 |
N.P. = Not Performed; further analysis was not performed for this variant as it was not carried on the shared IBD haplotype identified in the original analysis.
Variants mapped to the following loci identified in the IBD analysis (see Supplementary Table 1):
1_clst698,
13_clst239,
19_clst121,
21_clst23.
We used the exome and GWAS data of the 105 sequenced individuals, in conjunction with 1000 Genomes data, to generate a merged reference panel in order to impute unobserved genotypes at the PCNT locus. The rs143796569 variant was successfully imputed in the original Irish GWAS data and predicted to be carried by all individuals sharing the haplotype segment; however, it was also predicted to be carried by several additional cases and controls. When tested for association with schizophrenia in the full GWAS sample, the minor allele of the imputed variant was present at a higher frequency in cases compared with controls, although this was not statistically significant (p=0.057, odds ratio=1.77, 95% confidence interval (C.I.)= 0.98–3.17).
The haplotype segment originally identified comprised 205 GWAS SNPs, covering almost 1 Mb on chromosome 21 (chr21:47,013,876–47,964,259; hg19). We investigated the haplotypes of the additional carriers of the rs143796569 rare allele identified through imputation and determined that these individuals all share a shorter, ~500kb haplotype that is identical to the 3’ end of the 1Mb haplotype (chr21:47,503,311–47,964,259; hg19). Thus, while the rs143796569 rare allele is carried on the 1Mb rare haplotype, it is not exclusive to it.
We nevertheless sought to examine the rs143796569 variant in independent datasets. Using the 1000 Genomes/Irish merged reference panel, we attempted to impute the variant in 41 GWAS datasets from the PGC2 schizophrenia study; the variant was successfully imputed (info>0.5) and analysed in 7 datasets (see Table 2), comprising 6,889 cases and 8,043 controls. Random effects meta-analysis of all 7 datasets was not significant (p=0.58, odds ratio=1.32, 95% C.I.= 0.49–3.59). We also assessed an independent dataset from the UK, in which the variant had been directly genotyped. Again, there was no evidence for association with schizophrenia (p=0.32).
Table 2:
Association analysis of rs143796569 in 7 datasets from the PGC2 schizophrenia GWAS
| PGC2 dataset | No. Cases | No. Controls | MAF in Cases | MAF in Controls | P-value | Odds Ratio |
|---|---|---|---|---|---|---|
| scz_aber_eur | 719 | 697 | 0.0051 | 0.0014 | 0.071 | 3.510 |
| scz_caws_eur | 396 | 284 | 0.0016 | 0.0009 | 0.601 | 2.791 |
| scz_edin_eur | 367 | 284 | 0.0068 | 0 | 0.054 | 5.771 |
| scz_mgs2_eur | 2638 | 2482 | 0.0029 | 0.0038 | 0.544 | 0.805 |
| scz_pewb_eur | 574 | 1812 | 0.0007 | 0.0032 | 0.051 | 0.274 |
| scz_s234_eur | 1980 | 2274 | 0.0011 | 0.0005 | 0.146 | 4.790 |
| scz_swe1_eur | 215 | 210 | 0 | 0.0039 | 0.172 | 0.098 |
Discussion
We performed IBD mapping in an Irish schizophrenia GWAS dataset and selected 45 genomic regions where a rare haplotype, >1cM in length, was more frequent in cases compared to controls. For 32 of these regions, we were able to perform exome sequencing on at least two individuals that shared a rare haplotype. Using functional annotation tools and data on allele frequencies from a number of large-scale exome and genome sequencing projects, we examined these shared haplotypes for rare protein-altering variants that could increase risk of schizophrenia. We succeeded in identifying a single missense variant in PCNT that was present on a rare haplotype and more frequent in sequenced cases compared to controls. Using 1000 Genomes and our Irish exome sequencing data as a merged reference panel, we were able to impute this variant into our full Irish GWAS dataset; although the rare allele was more frequent in cases than controls, the test for association was not statistically significant (p=0.057). Subsequent efforts in larger independent replication samples did not provide further support for association between this variant and schizophrenia.
We consider two possible explanations for our findings in this region: 1. Our detection of the rare 1Mb haplotype in 11 cases versus 1 control in our GWAS sample was a chance result and does not reflect a rare risk variant at this locus. This spurious association led us to the PCNT variant, which is not associated with schizophrenia. 2. This 1Mb haplotype does carry a rare risk variant but it is not the PCNT variant and we could not find it using our approach here. When we imputed the PCNT variant into our GWAS sample, we found that it is not exclusively present on the 1Mb haplotype. As such, if there is a rare risk variant on the 1Mb haplotype that is responsible for the initial association signal after IBD analysis, it is not in complete linkage disequilibrium with the PCNT variant. Frustratingly, we could not find any variant, regardless of functional impact on coding sequence, in our exome data that was exclusive to and would effectively tag the 1Mb haplotype. Thus, we could test the PCNT variant in replication samples, but in the absence of a tagging variant we were not able to specifically test the 1Mb haplotype itself for independent evidence of association with schizophrenia.
While we have shown that this approach can identify rare risk haplotypes in GWAS data, the challenge in subsequent analysis of these haplotypes is that they are large, making the task of tagging them and pinpointing potential risk variants difficult. For the vast majority of the IBD haplotypes we found to be more frequent in cases compared to controls, we did not find a suitable variant to bring forward for association analysis. We concentrated on variants that we detected in coding sequences of genes via exome sequencing. The advantages to this approach were that we were better able to assess the functional impact of variants on protein coding genes and we could use large datasets from exome sequencing projects to measure the frequency of coding variants detected in European populations. It is possible that we missed functional variants in these haplotypes because they exist outside of coding regions or because they are difficult to detect using exome sequencing, e.g CNVs (Tan et al., 2014). Full analysis of the haplotypes using complete genome data would better capture all variants. Interpretation of these data will remain challenging until knowledge to support functional annotation of non-coding regions increases. Importantly, genome sequencing would provide a better opportunity to at least identify variants that exclusively tag rare risk haplotypes and can be used in replication association studies, even if the task of finding actual causal variants persists.
As with any approach using GWAS data, larger sample sizes will provide more power to detect associations. An accurate IBD method for rare variant analysis of GWAS data is very appealing, as there are vast quantities of GWAS data available for re-analysis. The challenge is that these data were generated on numerous cohorts drawn from many diverse populations. IBD mapping is more effective in relatively homogeneous samples such as the Irish data used here. Increasing sample size means analysis of more heterogeneous samples, which can cause false-positive IBD mapping signals (S. R. Browning & Thompson, 2012).
The availability of whole genome sequence data for a case-control sample would make IBD mapping irrelevant because the utility of identifying IBD segments is in their capacity to infer untyped variants, which would be present in sequence data (S. R. Browning & Thompson, 2012). At present however, the cost of genome sequencing has not dropped sufficiently to make this feasible. In the meantime, the application of IBD mapping to GWAS data warrants investigation. Our study was able to find large rare haplotypes that were significantly more frequent in cases compared to controls but full sequencing of the prioritised genomic segments rather than exome sequencing may have been more effective for mapping or tagging risk variants for replication analysis.
Supplementary Material
Acknowledgements
We wish to thank all patients and their support staff, and all healthy volunteers for participating in the data collection on which this manuscript is based. Recruitment, genotyping and analysis was supported by Science Foundation Ireland grants (12/IP/1670, 12/IP/1359 and 08/IN.1/B1916), US National Institutes of Health grants (R01-MH083094, R01-MH041953) and the Wellcome Trust Case Control Consortium 2 project grant (085475/B/08/Z). We thank EMI author Yu Qian for providing a script to convert EMI cluster files to plink format. CSHL funding was from a generous gift of The Stanley Family. We acknowledge the support of the Trinity Biobank in providing control samples for this analysis.
Conflict of Interest
Dr McCombie has participated in Illumina sponsored meetings over the past four years and received travel reimbursement and an honorarium for presenting at these events. Illumina had no role in decisions relating to the study/work to be published, data collection and analysis of data and the decision to publish. Dr McCombie has also participated in Pacific Biosciences sponsored meetings over the past three years and received travel reimbursement for presenting at these events. Dr McCombie is a founder and shareholder of Orion Genomics, which focuses on plant genomics and cancer genetics. Other authors declare no conflict of interest.
Author list for Wellcome Trust Case Control Consortium 2:
Peter Donnelly, Lesley Bates, Ines Barroso, Jenefer M. Blackwell, Elvira Bramon, Matthew A. Brown, Juan P. Casas, Aiden Corvin, Panos Deloukas, Audrey Duncanson, Janusz Jankowski, Hugh S. Markus, Christopher G. Mathew, Colin N. A. Palmer, Robert Plomin, Anna Rautanen, Stephen J. Sawcer, Richard C. Trembath, Ananth C. Viswanathan, Nicholas W.Wood, Chris C. A. Spencer, Gavin Band, Céline Bellenguez, Colin Freeman, Garrett Hellenthal, Eleni Giannoulatou, Lucinda Hopkins, Matti Pirinen, Richard Pearson, Amy Strange, Zhan Su, Damjan Vukcevic, Cordelia Langford, Sarah E. Hunt, Sarah Edkins, Rhian Gwilliam, Hannah Blackburn, Suzannah J. Bumpstead, Serge Dronov, Matthew Gillman, Emma Gray, Naomi Hammond, Alagurevathi Jayakumar, Owen T.McCann, Jennifer Liddle, Simon C. Potter, Radhi Ravindrarajah, Michelle Ricketts, Matthew Waller, Paul Weston, Sara Widaa, and Pamela Whittaker.
Author list for Schizophrenia Working Group of the Psychiatric Genomics Consortium:
Stephan Ripke, Benjamin M. Neale, Aiden Corvin, James T. R. Walters, Kai-How Farh, Peter A Holmans, Phil Lee, Brendan Bulik-Sullivan, David A. Collier, Hailiang Huang, Tune H. Pers, Ingrid Agartz, Esben Agerbo, Margot Albus, Madeline Alexander, Farooq Amin, Silviu A. Bacanu, Martin Begemann, Richard A. Belliveau, Judit Bene, Sarah E. Bergen, Elizabeth Bevilacqua, Tim B. Bigdeli, Donald W. Black, Richard Bruggeman, Nancy G. Buccola, Randy L. Buckner, William Byerley, Wiepke Cahn, Guiqing Cai, Dominique Campion, Rita M. Cantor, Vaughan J. Carr, Noa Carrera, Stanley V. Catts, Kimberley D. Chambert, Raymond C. K. Chan, Ronald Y. L. Chan, Eric Y. H. Chen, Wei Cheng, Eric F. C. Cheung, Siow Ann Chong, C Robert Cloninger, David Cohen, Nadine Cohen, Paul Cormican, Nick Craddock, James J. Crowley, David Curtis, Michael Davidson, Kenneth L. Davis, Franziska Degenhardt, Jurgen Del Favero, Ditte Demontis, Dimitris Dikeos, Timothy Dinan, Srdjan Djurovic, Gary Donohoe, Elodie Drapeau, Jubao Duan, Frank Dudbridge, Naser Durmishi, Peter Eichhammer, Johan Eriksson, Valentina Escott-Price, Laurent Essioux, Ayman H. Fanous, Martilias S. Farrell, Josef Frank, Lude Franke, Robert Freedman, Nelson B. Freimer, Marion Friedl, Joseph I. Friedman, Menachem Fromer, Giulio Genovese, Lyudmila Georgieva, Ina Giegling, Paola Giusti-Rodríguez, Stephanie Godard, Jacqueline I. Goldstein, Vera Golimbet, Srihari Gopal, Jacob Gratten, Lieuwe de Haan, Christian Hammer, Marian L. Hamshere, Mark Hansen, Thomas Hansen, Vahram Haroutunian, Annette M. Hartmann, Frans A. Henskens, Stefan Herms, Joel N. Hirschhorn, Per Hoffmann, Andrea Hofman, Mads V. Hollegaard, David M. Hougaard, Masashi Ikeda, Inge Joa, Antonio Julià, Luba Kalaydjieva, Sena Karachanak-Yankova, Juha Karjalainen, David Kavanagh, Matthew C. Keller, James L. Kennedy, Andrey Khrunin, Yunjung Kim, Janis Klovins, James A. Knowles, Bettina Konte, Vaidutis Kucinskas, Zita Ausrele Kucinskiene, Hana Kuzelova-Ptackova, Anna K. Kähler, Claudine Laurent, Jimmy Lee, S Hong Lee, Sophie E. Legge, Bernard Lerer, Miaoxin Li, Tao Li, Kung-Yee Liang, Jeffrey Lieberman, Svetlana Limborska, Carmel M. Loughland, Jan Lubinski, Jouko Lönnqvist, Milan Macek, Patrik K. E. Magnusson, Brion S. Maher, Wolfgang Maier, Jacques Mallet, Sara Marsal, Manuel Mattheisen, Morten Mattingsdal, Robert W. McCarley, Colm McDonald, Andrew M. McIntosh, Sandra Meier, Carin J. Meijer, Bela Melegh, Ingrid Melle, Raquelle I. Mesholam-Gately, Andres Metspalu, Patricia T. Michie, Lili Milani, Vihra Milanova, Younes Mokrab, Derek W. Morris, Ole Mors, Kieran C. Murphy, Robin M. Murray, Inez Myin-Germeys, Bertram Müller-Myhsok, Mari Nelis, Igor Nenadic, Deborah A. Nertney, Gerald Nestadt, Kristin K. Nicodemus, Liene Nikitina-Zake, Laura Nisenbaum, Annelie Nordin, Eadbhard O’Callaghan, Colm O’Dushlaine, F Anthony O’Neill, Sang-Yun Oh, Ann Olincy, Line Olsen, Jim Van Os, Christos Pantelis, George N. Papadimitriou, Sergi Papiol, Elena Parkhomenko, Michele T. Pato, Tiina Paunio, Milica Pejovic-Milovancevic, Diana O. Perkins, Olli Pietiläinen, Jonathan Pimm, Andrew J. Pocklington, Alkes Price, Ann E. Pulver, Shaun M. Purcell, Digby Quested, Henrik B. Rasmussen, Abraham Reichenberg, Mark A. Reimers, Alexander L. Richards, Joshua L. Roffman, Panos Roussos, Douglas M. Ruderfer, Veikko Salomaa, Alan R. Sanders, Ulrich Schall, Christian R. Schubert, Thomas G. Schulze, Sibylle G. Schwab, Edward M. Scolnick, Rodney J. Scott, Larry J. Seidman, Jianxin Shi, Engilbert Sigurdsson, Teimuraz Silagadze, Jeremy M. Silverman, Kang Sim, Petr Slominsky, Jordan W. Smoller, Hon-Cheong So, Chris C. A. Spencer, Eli A. Stahl, Hreinn Stefansson, Stacy Steinberg, Elisabeth Stogmann, Richard E. Straub, Eric Strengman, Jana Strohmaier, T Scott Stroup, Mythily Subramaniam, Jaana Suvisaari, Dragan M. Svrakic, Jin P. Szatkiewicz, Erik Söderman, Srinivas Thirumalai, Draga Toncheva, Sarah Tosato, Juha Veijola, John Waddington, Dermot Walsh, Dai Wang, Qiang Wang, Bradley T. Webb, Mark Weiser, Dieter B. Wildenauer, Nigel M. Williams, Stephanie Williams, Stephanie H. Witt, Aaron R. Wolen, Emily H. M. Wong, Brandon K. Wormley, Hualin Simon Xi, Clement C. Zai, Xuebin Zheng, Fritz Zimprich, Naomi R. Wray, Kari Stefansson, Peter M. Visscher, Rolf Adolfsson, Ole A. Andreassen, Douglas H. R. Blackwood, Elvira Bramon, Joseph D. Buxbaum, Anders D. Børglum, Ariel Darvasi, Enrico Domenici, Hannelore Ehrenreich, Tõnu Esko, Pablo V. Gejman, Michael Gill, Hugh Gurling, Christina M. Hultman, Nakao Iwata, Assen V. Jablensky, Erik G. Jönsson, Kenneth S. Kendler, George Kirov, Jo Knight, Todd Lencz, Douglas F. Levinson, Qingqin S. Li, Jianjun Liu, Anil K. Malhotra, Steven A. McCarroll, Andrew McQuillin, Jennifer L. Moran, Preben B. Mortensen, Bryan J. Mowry, Michael J. Owen, Aarno Palotie, Carlos N. Pato, Tracey L. Petryshen, Danielle Posthuma, Brien P. Riley, Dan Rujescu, Pak C. Sham, Pamela Sklar, David St Clair, Daniel R. Weinberger, Jens R. Wendland, Thomas Werge, Mark J. Daly, Patrick F. Sullivan & Michael C. O’Donovan
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7(4), 248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, & Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12), 1691–1692. doi: 10.1093/bioinformatics/btr174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P, Marcaillou C, Vanpeene M, Saraiva JP, Stockholm D, Francke S, … Philippi A (2009). Robust physical methods that enrich genomic regions identical by descent for linkage studies: confirmation of a locus for osteogenesis imperfecta. BMC Genet, 10, 16. doi: 10.1186/1471-2156-10-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, & Browning SR (2013). Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics, 194(2), 459–471. doi: 10.1534/genetics.113.150029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, & Browning BL (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet, 81(5), 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, & Thompson EA (2012). Detecting rare variant associations by identity-by-descent mapping in case-control studies. Genetics, 190(4), 1521–1531. doi: 10.1534/genetics.111.136937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, & Lu X (2012). Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front Genet, 3, 35. doi: 10.3389/fgene.2012.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang l. L., Coon M, Nguyen T, Wang L, … Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 6(2), 80–92. doi: 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, … Group GPA (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. doi: 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, & Zagury JF (2012). A linear complexity phasing method for thousands of genomes. Nat Methods, 9(2), 179–181. doi: 10.1038/nmeth.1785 [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, … Daly MJ (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet, 43(5), 491–498. doi: 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J, & Spitzer RL (1978). A diagnostic interview: the schedule for affective disorders and schizophrenia. Arch Gen Psychiatry, 35(7), 837–844. [DOI] [PubMed] [Google Scholar]
- Ercan-Sencicek AG, Stillman AA, Ghosh AK, Bilguvar K, O’Roak BJ, Mason CE, … State MW (2010). L-histidine decarboxylase and Tourette’s syndrome. N Engl J Med, 362(20), 1901–1908. doi: 10.1056/NEJMoa0907006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- First M, Spitzer R, Robert L, Gibbon M, & Williams J (2002). Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition. In. New York: Biometrics Research, New York State Psychiatric Institute. [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, … O’Donovan MC (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature, 506(7487), 179–184. doi: 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landen M, … McCarroll SA (2016). Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci, 19(11), 1433–1441. doi: 10.1038/nn.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann OR, Misura K, Lamas E, Sandrock RW, Nelson P, McDonough SI, & DeLisi LE (2016). Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol Psychiatry, 21(12), 1690–1695. doi: 10.1038/mp.2016.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Faraci G, Chen DT, Kassem L, Schulze TG, Shugart YY, & McMahon FJ (2013). Amish revisited: next-generation sequencing studies of psychiatric disorders among the Plain people. Trends Genet, 29(7), 412–418. doi: 10.1016/j.tig.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2. (2012). Genome-wide association study implicates HLA-C*01:02 as a risk factor at the major histocompatibility complex locus in schizophrenia. Biol Psychiatry, 72(8), 620–628. doi: 10.1016/j.biopsych.2012.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiorgou M, Flint J, Gogos JA, Malenka RC, & Genetic and Neural Complexity in Psychiatry Working, G. (2012). The best of times, the worst of times for psychiatric disease. In Nat Neurosci (Vol. 15, pp. 811–812). United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, … Robinson PN (2010). Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet, 42(10), 827–829. doi: 10.1038/ng.653 [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, & Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc, 4(7), 1073–1081. doi: 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, … Team, N. G. E. S. P. E. L. P. (2012). Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet, 91(2), 224–237. doi: 10.1016/j.ajhg.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14), 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, … Subgroup, G. P. D. P. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Charlesworth J, Stankovich J, Perreau VM, Brown MA, & Taylor BV (2013). Identity-by-descent mapping to detect rare variants conferring susceptibility to multiple sclerosis. PLoS One, 8(3), e56379. doi: 10.1371/journal.pone.0056379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Bhatia G, Gusev A, Finucane HK, Bulik-Sullivan BK, Pollack SJ, … Price AL (2015). Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat Genet, 47(12), 1385–1392. doi: 10.1038/ng.3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, & Gibbs RA (2011). Clan genomics and the complex architecture of human disease. Cell, 147(1), 32–43. doi: 10.1016/j.cell.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, … Sebat J (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet, 49(1), 27–35. doi: 10.1038/ng.3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, … Corvin A (2014). De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry, 19(6), 652–658. doi: 10.1038/mp.2014.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, & Cunningham F (2010). Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics, 26(16), 2069–2070. doi: 10.1093/bioinformatics/btq330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, … Walsh CA (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science, 321(5886), 218–223. doi: 10.1126/science.1157657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, … Sham PC (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 81(3), 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, … Sklar P (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature, 506(7487), 185–190. doi: 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Browning BL, & Browning SR (2014). Efficient clustering of identity-by-descent between multiple individuals. Bioinformatics, 30(7), 915–922. doi: 10.1093/bioinformatics/btt734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees E, Moskvina V, Owen MJ, O’Donovan MC, & Kirov G (2011). De novo rates and selection of schizophrenia-associated copy number variants. Biol Psychiatry, 70(12), 1109–1114. doi: 10.1016/j.biopsych.2011.07.011 [DOI] [PubMed] [Google Scholar]
- Richards AL, Leonenko G, Walters JT, Kavanagh DH, Rees EG, Evans A, … O’Donovan MC (2016). Exome arrays capture polygenic rare variant contributions to schizophrenia. Hum Mol Genet, 25(5), 1001–1007. doi: 10.1093/hmg/ddv620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511(7510), 421–427. doi: 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, … Barrett JC (2016). Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci, 19(4), 571–577. doi: 10.1038/nn.4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R, Wang Y, Kleinstein SE, Liu Y, Zhu X, Guo H, … Zhu M (2014). An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum Mutat, 35(7), 899–907. doi: 10.1002/humu.22537 [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 447(7145), 661–678. doi: 10.1038/nature05911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlind H, Imrell K, Ramanujam R, Myhr KM, Celius EG, Harbo HF, … Hillert J (2015). Identity-by-descent mapping in a Scandinavian multiple sclerosis cohort. Eur J Hum Genet, 23(5), 688–692. doi: 10.1038/ejhg.2014.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. (1992). Schedules for Clinical Assessment in Neuropsychiatry (Version 1.0). In. Geneva: WHO. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.