

Abstract
Several pharmacogenetics studies have identified an association between a greater metformin-dependent reduction in HbA1c levels and the minor A allele at rs2289669 in intron 10 of SLC47A1, encoding multidrug and toxin extrusion 1 (MATE1), a presumed metformin transporter. It is currently unknown if the rs2289669 locus is a cis-eQTL, which would validate its role as predictor of metformin efficacy. We looked at association between common genetic variants in the SLC47A1 gene region and HbA1c reduction after metformin treatment using locus-wise meta-analysis from the MetGen consortium. CRISPR-Cas9 was applied to perform allele editing of, or genomic deletion around, rs2289669 and of the closely linked rs8065082 in HepG2 cells. The genome-edited cells were evaluated for SLC47A1 expression and splicing. None of the common variants including rs2289669 showed significant association with metformin response. Genomic editing of either rs2289669 or rs8065082 did not alter SLC47A1 expression or splicing. Experimental and in silico analyses show that the rs2289669-containing haploblock does not appear to carry genetic variants that could explain its previously reported association with metformin efficacy.
Introduction
Glycaemic response to metformin varies widely between type 2 diabetes patients (1), motivating the need for an optimized, precision medicine-type approach. Accordingly, previous studies have explored the association of genetic variants in metformin-transport genes with metformin response (2,3); some of this work has focused on SLC47A1 (4). This gene encodes a transmembrane cationic transporter MATE1 (multidrug and toxin extrusion 1), which modulates metformin efflux from the probable effector organs—liver and kidney (5–10); concordantly, several pharmacogenetics studies have reported an enhanced HbA1c-lowering effect associated with carriers of the A-allele at the intronic SNP rs2289669 (4,11–14). In the Diabetes Prevention Program, a randomized controlled trial of progression to diabetes in people at high risk of the disease, metformin-treated carriers of the T-allele at rs8065082 (in tight linkage disequilibrium with rs2289669) progressed more slowly than the major (C) allele homozygotes at rs8065082 (15). Despite these study-specific findings, the metformin HbA1c-lowering effect associated with rs2289669 was not supported by analyses within the MetGen consortium (16). Thus, there remains uncertainty about whether rs2289669 (or SNPs in high LD) affects metformin response; also no experimental studies have been performed to determine potential biological function of rs2289669 or rs8065082.
It is conceivable that the intronic genetic variants at rs2289669, or closely linked variants, could affect SLC47A1 gene expression or splicing, ultimately determining metformin efflux rate from liver and kidney. To investigate this, we combined in silico analysis of SLC47A1 variants with CRISPR-Cas9-mediated genome engineering of hepatocyte-like HepG2 cells to study the in vitro effects of metformin, conditional on SLC47A1 variation at rs2289669 or rs8065082.
Results
In silico functional annotation of SNPs in linkage disequilibrium with the tag SNP rs2289669
To find putative causal SNPs linked to rs2289669 we analyzed all SNPs in LD (linkage disequilibrium) r2 > 0.8 using the PredictSNP2 algorithm (17), which collates analyses of potential functional relevance of genetic variants using several external analysis tools (CADD, DANN, FATHMM, FunSeq2, GWAVA). The algorithm predicted only neutral mutations, i.e. did not return any putative causal SNPs, even after expanding the genomic region to include SNPs in LD r2 > 0.2. We also used WashU EpiGenome Browser (v52.5.2) to manually search the 5.7 kb rs2289669 haploblock for liver ChIP-Seq peaks, including histone modifications and liver-specific transcription factor binding sites (TFBS), proximal to any SNPs in tight LD with rs2289669. We found rs8065082 to be located close to a predicted enhancer region (large H3K27ac peak) and close to binding sites for JUND1, HNF4G, FOXA1 (Fig. 1). Based on these functional annotations, we considered rs8065082 to be the most probable functional variant. Additionally, we found the rs2289669 SNP to be proximal to the EGR1 binding site, therefore possibly functional. Four other common genetic variants in tight LD with rs2289669 are parts of Alu repeats and they do not overlap with the analyzed chromatin feature peaks.
Figure 1.
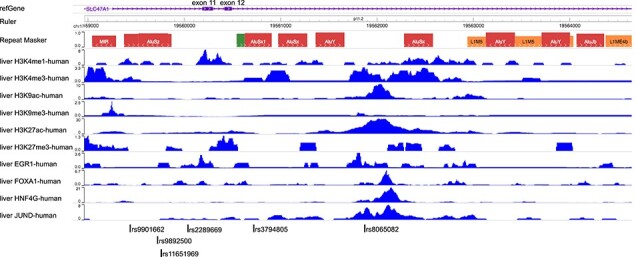
Functional annotation and identification of probable causal SNPs in SLC47A1 (MATE1) that may influence hepatic response to metformin. A screenshot from WashU Epigenome Browser showing human liver ChIP-seq peaks on different chromatin features mapped to the rs2289669 haploblock. Other common genetic variants identified in the haploblock are marked below the image. Alu repeats are denoted in the RepeatMasker track.
MetGen analysis of SNPs in SLC47A1 and association with metformin glycaemia-lowering effect
We explored the association between all common variants in the SLC47A1 gene and glycaemic response to metformin in more than 10 000 MetGen participants of European ancestry. Locus-wise meta-analysis is presented in Fig. 2. None of the variants showed significant association with glycaemic response to metformin after correction for multiple testing (P > 2.73 × 10−4). Table 2 features the top associations from the locus-wise meta-GWAS. The A allele at rs2289669 also showed no significant association (−0.007 ± 0.013, P = 0.58).
Figure 2.
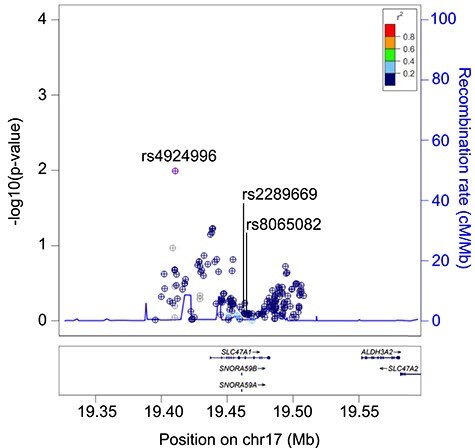
Regional association plots around the SLC47A1 locus. The plot shows locus-wise meta-analysis association between the SLC47A1 common variants and glycaemic response to metformin in the MetGen consortium dataset.
Table 2.
SNPs in the SLC47A1 gene nominally associated with glycaemic response to metformin
| CHR | BP | EAF | SNP | EA | NEA | beta | SE | P | n_samples |
|---|---|---|---|---|---|---|---|---|---|
| 17 | 19 507 233 | 0.85 | rs4924996 | A | T | 0.047 | 0.018 | 0.010 | 10 519 |
| 17 | 19 533 874 | 0.45 | rs2252281 | C | T | 0.022 | 0.012 | 0.071 | 12 582 |
| 17 | 19 535 008 | 0.45 | rs2453580 | C | T | 0.023 | 0.012 | 0.067 | 12 582 |
| 17 | 19 535 247 | 0.44 | rs2453581 | T | C | 0.023 | 0.012 | 0.060 | 12 580 |
| 17 | 19 535 753 | 0.44 | rs2453582 | T | C | 0.024 | 0.012 | 0.059 | 12 575 |
CHR is chromosome number, BP is chromosome base pair (coordinate), EAF is effect allele frequency, EA is effect allele, NEA is non-effect allele, SE is standard error of the mean, P is P-value, n_samples is number of individuals.
SLC47A1 expression in HepG2 cells allele-edited at rs2289669 and rs8065082
So far, in silico analyses and the MetGen meta-analysis provided little evidence for any functional significance of the rs2289669 locus in metformin HbA1c-lowering efficacy. Nevertheless, we also experimentally evaluated if the two SNPs proximal to TFBS (i.e. rs2289669 and rs8065082) modulate SLC47A1 gene expression. Here, we used CRISPR-Cas9 to perform allele substitution at the rs2289669 and rs8065082 loci in hepatocyte-like HepG2 cells. These cells are heterozygous for both SNPs; thus, we performed allele substitutions G-to-A or A-to-G at rs2289669, and C-to-T or T-to-C at rs8065082 (Fig. 3a and b). To obtain homozygous populations for each of the four genotypes we generated at least three single cell-derived clonal populations per genotype. Finally, we analyzed these cells for SLC47A1 gene expression using primer/probe sets that cover different exons of the gene. In rs2289669-edited cells, G/G and A/A genotypes had similar SLC47A1 gene expression (P > 0.05), albeit with one of the qPCR probe sets the P-value was 0.03 (Fig. 3c). Also, in rs8065082-edited cells, C/C and T/T genotypes had similar SLC47A1 gene expression (P > 0.05) (Fig. 3d). We also did not observe any apparent alternative splicing of the SLC47A1 transcript around exons 5–17 in the allele-edited cells (the SNPs are proximal to exons 11 and 12) (Fig. 3e).
Figure 3.
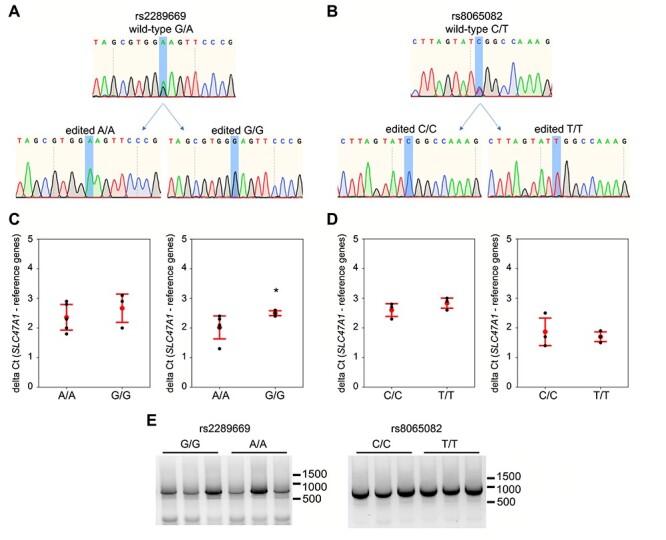
Two putative causal genetic variants in SLC47A1 were assessed for cis-eQTL and effect on splicing in HepG2 cells. (a, b) Representative Sanger sequencing traces of HepG2 genome at rs2289669 locus (a) and at rs8065082 locus (b), before (wild-type) and after CRISPR-Cas9-mediated HDR editing and single-cell cloning to obtain the respective homozygous genotype. (c, d) qPCR for SLC47A1 on cDNA from rs2289669-edited (c) or rs8065082-edited (d) HepG2 cells. Probes against two different exon boundaries of the SLC47A1 transcript were used (exons 1–2 on the left, and exons 14–15 on the right in each subfigure). Graphs show mean delta Ct (red dot) and standard deviation (red error bars), and also individual biological replicates’ mean Ct values obtained from three technical replicates (black dots). Mann–Whitney U test was used to calculate P-values, with asterisk denoting P < 0.05. (e) Qualitative assessment of splicing of SLC47A1 transcripts covering the range of exons 5–17. RNA from three biological replicates each of allele-edited cell populations was used to run one-step end-stage RT-PCR.
SLC47A1 expression in HepG2 cells carrying small deletions around rs2289669 and rs8065082
To further validate that the rs2289669 and rs8065082 are not parts of any enhancer or repressor elements, we used CRISPR-Cas9 to create small deletions around the two SNPs, in introns 10 and 12, respectively. Here, we obtained very high, >85%, editing efficiency, allowing us to circumvent the single-cell cloning procedure that might confound the experimental outcome. We deleted 44 bp (hg38 chr17:19559999 to 19 560 043) around the rs2289669 locus and 36–37 bp (hg38 chr17:19561851–19 561 887) around the rs8065082 locus, in both cases also deleting the SNPs. Here, we generated three biological replicate cell populations and three mock negative control populations using non-specific gRNA (three separate transfections each) (Fig. 4a and b). Neither deletion mutation affected SLC47A1 gene expression when assayed by qPCR (P > 0.05) (Fig. 4c and d). Also, the deletions did not appear to influence the splicing of the SLC47A1 in the exon range 5–17 (Fig. 4e).
Figure 4.
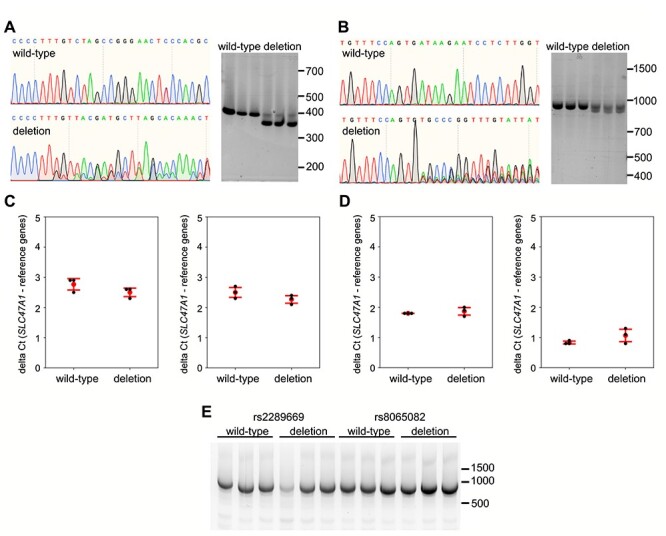
Assessment of effect on splicing and SLC47A1 expression after deletion of genomic DNA around rs2289669 and rs8065082 in HepG2 cells. (a, b) Representative Sanger sequencing traces of HepG2 genome at rs2289669 locus (a) and at rs8065082 locus (b), before (wild-type) and after CRISPR-Cas9-mediated deletion (deletion). Right panels in each subfigure show agarose-gel images of PCR amplicons of the respective loci before (wild-type) and after deletion (deletion), generated from three biological replicates. (c, d) qPCR for SLC47A1 on cDNA from wild-type cells versus cells with rs2289669 deletion (c) or rs8065082 deletion (d). Probes against two different exon boundaries of the SLC47A1 transcript were used (exons 1–2 on the left, and exons 14–15 on the right in each subfigure). Graphs show mean delta Ct (red dot) and standard deviation (red error bars), and also individual biological replicates’ mean Ct values obtained from three technical replicates (black dots). Mann–Whitney U test was used to calculate P-values, with no statistical significance obtained in all tests. e) Qualitative assessment of splicing of SLC47A1 transcripts covering the range of exons 5–17. RNA from three biological replicates each of wild-type versus deletion-carrying cell populations was used to run one-step end-stage RT-PCR.
Discussion
The outcome of hyperglycaemia-lowering metformin treatment can vary between patients (18,19), conceivably because of the patient’s genotype at metformin-regulating loci (20), among other explanations. One of the candidate genes steering metformin efflux from the effector organs (liver, kidney) is SLC47A1, in which the A allele of the intronic SNP rs2289669 or the T allele of the closely linked rs8065082 have been associated with improved metformin response in several studies: first in a smaller cohort (4), then in the Diabetes Prevention Program study (15), and in Iranian (21), Chinese (22) and European (14) populations. In contrast, the MetGen consortium showed no association of rs2289669 with metformin efficacy (16), as is the case with at least two other studies (23,24). Here, we explored the MetGen consortium data further: in data collected from approximately 10 000 individuals, we explored the effect of multiple SLC47A1 variants, but found no association with the HbA1c-lowering effects of metformin. This is in line with our previous report in the MetGen with smaller sample size (16). Intriguingly, in this dataset, the variants rs2289669 and rs8065082 had P-values of 0.58 and 0.83, respectively.
In our experimental studies, allele editing or genomic deletion of rs2289669 or rs8065082 did not appear to markedly alter SLC47A1 gene expression or splicing. Except in one experiment, SLC47A1 expression assayed by qPCR probes against different parts of the SLC47A1 transcript did not appear to be altered in single cell-derived clonal populations of rs2289669 A/A versus G/G, or rs8065082 C/C versus T/T cells. In these experiments we often observed a rather large variance in SLC47A1 expression between clones of the same genotype, which is probably due to unknown confounding factors originating during the single-cell cloning process. This process, unfortunately, was unavoidable in order to generate pure homozygous cell populations. To account for this, however, we also validated the absence of any gene-regulatory elements in the examined loci by generating small deletions around both rs2289669 and rs8065082; this editing, unlike allele editing, was highly efficient, deeming single-cell cloning unnecessary. In effect, we obtained low variance in SLC47A1 expression within cell populations of the same genotype, but also here we did not observe any deletion-mediated altered SLC47A1 expression. Altogether, we observed rather homogenous null effects on gene expression following the genomic editing of rs2289669 and rs8065082 loci. Hence, the in silico and the experimental studies on the previously reported metformin efficacy-associated rs2289669 locus suggest that the gene variation in this locus probably does not impact on SLC47A1 expression or splicing.
Throughout the past few decades, the ‘candidate gene’ approach has often been used to identify potential loss/gain-of-function mutations in genes suspected to be pertinent to specific phenotypes. However, relatively few discoveries made using this approach have been replicated in GWAS studies, which are widely viewed as being free from the types of bias that candidate gene studies are prone to. Nevertheless, where the candidacy of a gene is founded on robust evidence, and the subsequent association analyses are performed properly, some results from candidate gene studies have proven to be robust to the interrogation method (e.g. PPARG and KCNJ11 variants in type 2 diabetes). The role of SLC47A1 in metformin response is well established. Nevertheless, the net effect of metformin in glucose regulation is likely to be multifaceted, not only resulting from the cellular metformin flux rate, but also from the abundance of its target proteins, differential organ distribution, individual organ deterioration, possibly even blood pressure and cardiovascular status. Several studies also suggest the involvement of gut cells and gut microbiota as regulators of metformin release (25–28). Other liver or kidney metformin transporters, such as OCT1 (29), or proteins, such as GLUT2 (3), may also affect metformin efficacy. While the present study does not completely rule out a role for SLC47A1 in this complex regulatory network controlling metformin response, it is unlikely that the SLC47A1 variants reported to date play a functional role in liver-mediated metformin action.
Limitations
In our experimental approach, we focused on deletion mutants, as they can be quickly produced and data obtained from them are relatively homogeneous. Nevertheless, that these experiments yielded null results leaves open the possibility that effects exist that could not be detected under the current experimental conditions (i.e. our findings are false negative). In some cases, generating more independent replicates (single-cell clones) of allele-edited cells can improve the signal-to-noise ratio, making it easier to detect small effects on gene expression. However, because the deletion mutant cell populations yielded relatively precise data, we elected not to proceed with additional single-cell cloning, as we concluded that to do so would utilize precious resources with little hope that the results would change.
In this study we used one hepatocyte-like cell line for genomic editing—possibly editing other cell lines, even from other target organs, would generate different results, but the apparent absence of an SLC47A1-expression-modulating effect of rs2289669 or rs8065082 makes this unlikely. Four other common SNPs in tight LD with rs2289669 were not examined here, but are unlikely to be causal given the in silico predictions described above, mainly because of their location within repetitive DNA elements and that they are distal to gene expression-associated chromatin features. Furthermore, GTEx data on SLC47A1 gene expression in liver show an absence of rs2289669-proximal eQTLs. At this point, we cannot rule out the possibility that the signal reported in other studies might come from a rare coding variant in tight linkage disequilibrium with rs2289669.
In summary, based on our functional genomics experiments and the new meta-analysis of epidemiological data, it appears highly unlikely that the SLC47A1 SNP rs2289669 (or variants in LD) or SLC47A1 transcriptional regulation are involved in liver-mediated metformin action.
Materials and Methods
Functional annotation
The publicly available database HaploReg v4.1 (30) was used to detect all SNPs in tight linkage disequilibrium (LD) of r2 ≥ 0.8 with the reported, metformin efficacy-associated, tag SNP rs2289669 (4). The location of the SNPs relative to specific histone modifications and potential TFBS was further analyzed using the WashU EpiGenome Browser and publicly available ENCODE ChIP-seq data for liver (31,32). Further analyses for finding putative causal SNPs in LD with the tag SNP rs2289669 were performed using PredictSNP2 (17); the parameters are listed in Supplemental Appendix 2.
The SLC47A1 locus and glycaemic response to metformin
We looked at association between common genetic variants around SLC47A1 (± 115 kb) and glycaemic response to metformin using a meta-GWAS data consisting of ~10 519 metformin-treated individuals of European origin from the MetGen consortium (PMID: 27500523). Analysis was performed using glycaemic response as defined by an absolute HbA1c reduction within the first 18 months of therapy as an outcome variable and individual variants as independent variable using the additive genetic model adjusted for baseline HbA1c. Statistical significance was considered after Bonferroni correction (0.05/number of SNPs).
Cell culture
HepG2 cells, originally isolated from a human male donor, were purchased from Sigma-Aldrich (Sweden) and cultured in the growth medium (DMEM with 5.56 mM glucose, 1 mM pyruvate, 2 mM L-glutamine, ThermoFisher Scientific, Sweden) supplemented with 10% FBS (HyClone, Cytiva, Sweden) and 1% (100 U/ml) penicillin/streptomycin (Merck, Sweden). Prior to CRISPR editing, genomic DNA was isolated (DNeasy Blood and Tissue kit, Qiagen) and regions containing rs2289669 and rs8065082 were PCR-amplified and Sanger-sequenced to determine the SNP genotypes of the HepG2 cells. The cells were checked routinely for mycoplasma contamination.
CRISPR-Cas9 genome editing design
All reagents used in CRISPR-Cas9-mediated genome editing were purchased from IDT DNA, unless stated otherwise. Control mock CRISPR-Cas9 gRNA was also from IDT DNA. Genome editing of HepG2 cells was performed using ribonucleoprotein (Alt-R® S.p. Cas9 nuclease V3 + synthetic sgRNA) delivery through nucleofection, with or without ssDNA donor templates. In all reactions, 125 pmol Cas9 protein was incubated with 150 pmol sgRNA for 20 min, before being mixed with 1 × 106 cells suspended in 100 μl nucleofector reagent V (Lonza, Sweden), to which 300 pmol ssDNA donor template (where appropriate) and 4 μM electroporation enhancer were added. Nucleofection was performed using program T-028 on Nucleofector 2b device (Lonza, Sweden). Transfected cells were immediately transferred from cuvettes to a 6-well plate containing growth medium (DMEM, 10% FBS) supplemented with 30 μM HDR enhancer where single-base editing was performed. The medium was replaced with fresh growth medium after 24 h. Cells were then expanded for at least 1 week, until they resumed normal growth rate. The single nucleotide-edited cells were seeded at around 500 cells per 10 cm dish to obtain single-cell clones with homozygous SNP genotypes without random indels. Cells were allowed to grow over approximately 2–3 weeks before individual colonies were picked and seeded into 96-well plates. Genomic DNA was extracted from all expanded clones using QuickExtract DNA Extraction Solution (Lucigen) for genotyping by PCR, as described below. In contrast, cells with deletion editing were not cloned, but expanded for 2 weeks for downstream experiments.
The sgRNA spacer sequences and ssDNA donor template sequences for the respective genome editing experiment are listed in Table 1.
Table 1.
Cas9 sgRNA spacer sequences and ssDNA donor template sequences used for genomic editing of HepG2 cells
| rs2289669 G-to-A substitution | |
|---|---|
| Cas9 sgRNA spacer sequence | 5’-CGGGAACTCCCACGCTACTG-3’ |
| ssDNA donor template sequence | 5’-AGTTTGTGCTAAGCATCGTAACCTGGGGCTCAGTTTCCACAGTAGCGTGGAAGTTCCCGGCTAGACAAAGGGGATGTTGCAAATCAGTCTTTTCAAAACT-3’ |
| rs2289669 A-to-G substitution | |
| Cas9 sgRNA spacer sequence | 5’-CGGGAACTTCCACGCTACTG-3’ |
| ssDNA donor template sequence | 5’-AGTTTGTGCTAAGCATCGTAACCTGGGGCTCAGTTTCCACAGTAGCGTGGGAGTTCCCGGCTAGACAAAGGGGATGTTGCAAATCAGTCTTTTCAAAACT-3’ |
| rs8065082 C-to-T substitution | |
| Cas9 sgRNA spacer sequence | 5’-TTAGTATCGGCCAAAGTGCC-3’ |
| ssDNA donor template sequence | 5’-TATTTTGTTTCCAGTGATAAGAATCCTCTTGGTTTCTTAGTATTGGCCAAAGTGCCCGGTTTGTAGTAGTTGCATAATATGTTT-3’ |
| rs8065082 T-to-C substitution | |
| Cas9 sgRNA spacer sequence | 5’-TTAGTATTGGCCAAAGTGCC-3’ |
| ssDNA donor template sequence | 5’-TATTTTGTTTCCAGTGATAAGAATCCTCTTGGTTTCTTAGTATCGGCCAAAGTGCCCGGTTTGTAGTAGTTGCATAATATGTTT-3’ |
| rs2289669 deletion | |
| Cas9 sgRNA spacer sequences | 5’-GTGCTAAGCATCGTAACCTG-3’ |
| 5’-GAGTTCCCGGCTAGACAAAG-3’ | |
| rs8065082 deletion | |
| Cas9 sgRNA spacer sequences | 5’-ACCAAGAGGATTCTTATCAC-3’ |
| 5’-TTAGTATCGGCCAAAGTGCC-3’ | |
| 5’-TTAGTATTGGCCAAAGTGCC-3’ | |
Genotyping of rs2289669 and rs8065082 loci in HepG2 cells
For genotyping, PCR reaction was used followed by Sanger sequencing. For the PCR, 1 μl of extracted DNA was mixed with 12 μl nuclease-free water, 15 μl 2x PCR master mix AmpliTaq Gold 360 (ThermoFisher) and 1 μl each of 10 mM primer. The primer sequences for amplifying rs2289669 locus were 5’-AGGAACATGGTTGTGCAG-3′ and 5’-CAAGAGCCTCGGGTAAAG-3′, and for the rs8065082 locus: 5’-AGGCCACCTATGGTTGTT-3′ and 5’-TTAGTTCTGTCCGCTCCA-3′. The reaction was run on a BioRad C1000 Touch Thermal Cycler using AmpliTaq Gold 360-recommended conditions, with annealing temperature 58°C. PCR products were purified using GeneJet PCR Purification Kit (ThermoFisher) and sequenced using sequencing primers: for rs2289669, 5’-GGGTCTCAGCACCTGTAATC-3′ and for rs8065082, 5’-AAAGATCAGAGGCTCCACTG-3′. Where applicable, PCR products were resolved on a 1% agarose gel with GelRed (ThermoFisher) and visualized on a UV board. CRISPR-Cas9-mediated deletion efficiency was analyzed using ICE Synthego online tool (https://ice.synthego.com) and the Sanger sequencing data.
Gene expression analysis
RNA was extracted from HepG2 cells using SV Total RNA Isolation System (Promega). RNA purity and concentration were measured using Nanodrop (Nanodrop). A total of 500 ng RNA was converted to cDNA using RevertAid first strand cDNA synthesis kit (ThermoFisher). For qPCR, three technical replicates per expression assay and biological replicate were run. Expression assays were for SLC47A1 exon 1–2 and exon 14–15 (ThermoFisher or IDT), and for reference genes ACTB, PPIA, TBP (all from IDT). qPCR was run in QuantStudio 7 Flex Real-Time PCR System (ThermoFisher) using PrimeTime Gene Expression Master Mix (IDT) according to manufacturer’s instructions. The qPCR gene expression analysis was performed by subtracting the geometric mean of the three reference genes’ Ct from the Ct of SLC47A1. Mann–Whitney U test was used to test the null hypothesis that two compared genotypes do not differ in SLC47A1 gene expression. At least three biological replicates of each genotype were used in each test, and the delta Ct data were expressed as means and standard deviation.
Assessment of alternative splicing
A total of 200 ng RNA extracted from rs2289669- and rs8065082-edited cells, three biological replicates each, was used to detect presence of any alternatively spliced isoforms of SLC47A1. Primers against exon 5 (5’-ACGATCTTCATTCCAGCTCTTC-3′) and against exon 17 (5’-TTCCGGCAAAGGTTCTTCTT-3′) were used together with SuperScript III™ One-Step RT-PCR System (ThermoFisher) to amplify the SLC47A1 cDNA spanning exons 5 through 17. The resulting PCR products were run on a 1% agarose gel with GelRed (ThermoFisher) and visualized using UV board. The bands were cut out, the DNA purified, and Sanger-sequenced to confirm the SLC47A1 sequence identity.
Supplementary Material
Acknowledgements
We acknowledge the use of the dataset from MetGen Plus Consortium. Link to the website: http://pgrn2016.weebly.com/metgen.html.
All authors contributed to conception and design of the study, interpretation of data, drafting or revising of the article, and approved the final version of the manuscript. Acquisition of data was performed by SK, MH, JD-R, MK, MetGen Plus Consortium.
All data are available on request from the authors.
Conflict of Interest Statement. All authors declare no conflict of interest.
Funding
European Research Council (ERC-2015-CoG-681742 NASCENT).
Full author list as Supplemental Appendix 1.
Contributor Information
Sebastian Kalamajski, Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Lund University, Malmö 20502, Sweden.
Mi Huang, Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Lund University, Malmö 20502, Sweden.
Jonathan Dalla-Riva, Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Lund University, Malmö 20502, Sweden.
Maria Keller, Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Lund University, Malmö 20502, Sweden; IFB Adiposity Diseases, University of Leipzig, Leipzig 04103, Germany.
Adem Y Dawed, Division of Population Health and Genomics, Ninewells Hospital and School of Medicine, University of Dundee, Dundee DD2 1UB, Scotland, UK.
Ola Hansson, Department of Clinical Sciences, Genomics, Diabetes and Endocrinology, Lund University, Malmö 20502, Sweden; Finnish Institute for Molecular Medicine, Helsinki University, Helsinki 00014, Finland.
Ewan R Pearson, Division of Population Health and Genomics, Ninewells Hospital and School of Medicine, University of Dundee, Dundee DD2 1UB, Scotland, UK.
Hindrik Mulder, Department of Clinical Sciences, Unit of Molecular Metabolism, Lund University, Malmö 20502, Sweden.
Paul W Franks, Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Lund University, Malmö 20502, Sweden; Department of Nutrition, Harvard T.H. Chan School of Public Health, Boston, MA 02115, USA.
References
- 1. Zolk, O. (2012) Disposition of metformin: variability due to polymorphisms of organic cation transporters. Ann. Med., 44, 119–129. [DOI] [PubMed] [Google Scholar]
- 2. Dawed, A.Y., Zhou, K., vanLeeuwen, N., Mahajan, A., Robertson, N., Koivula, R., Elders, P.J.M., Rauh, S.P., Jones, A.G., Holl, R.W.et al. (2019) Variation in the plasma membrane monoamine transporter (PMAT) (encoded by SLC29A4) and organic cation transporter 1 (OCT1) (encoded by SLC22A1) and gastrointestinal intolerance to metformin in type 2 diabetes: an IMI DIRECT study. Diabetes Care, 42, 1027–1033. [DOI] [PubMed] [Google Scholar]
- 3. Zhou, K., Yee, S.W., Seiser, E.L., vanLeeuwen, N., Tavendale, R., Bennett, A.J., Groves, C.J., Coleman, R.L., van derHeijden, A.A., Beulens, J.W.et al. (2016) Variation in the glucose transporter gene SLC2A2 is associated with glycemic response to metformin. Nat. Genet., 48, 1055–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Becker, M.L., Visser, L.E., vanSchaik, R.H., Hofman, A., Uitterlinden, A.G. and Stricker, B.H. (2009) Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: a preliminary study. Diabetes, 58, 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hibma, J.E., Zur, A.A., Castro, R.A., Wittwer, M.B., Keizer, R.J., Yee, S.W., Goswami, S., Stocker, S.L., Zhang, X., Huang, Y.et al. (2016) The effect of famotidine, a MATE1-selective inhibitor, on the pharmacokinetics and pharmacodynamics of metformin. Clin. Pharmacokinet., 55, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kusuhara, H., Ito, S., Kumagai, Y., Jiang, M., Shiroshita, T., Moriyama, Y., Inoue, K., Yuasa, H. and Sugiyama, Y. (2011) Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther., 89, 837–844. [DOI] [PubMed] [Google Scholar]
- 7. Masuda, S., Terada, T., Yonezawa, A., Tanihara, Y., Kishimoto, K., Katsura, T., Ogawa, O. and Inui, K. (2006) Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol., 17, 2127–2135. [DOI] [PubMed] [Google Scholar]
- 8. Otsuka, M., Matsumoto, T., Morimoto, R., Arioka, S., Omote, H. and Moriyama, Y. (2005) A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. U. S. A., 102, 17923–17928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toyama, K., Yonezawa, A., Masuda, S., Osawa, R., Hosokawa, M., Fujimoto, S., Inagaki, N., Inui, K. and Katsura, T. (2012) Loss of multidrug and toxin extrusion 1 (MATE1) is associated with metformin-induced lactic acidosis. Br. J. Pharmacol., 166, 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsuda, M., Terada, T., Mizuno, T., Katsura, T., Shimakura, J. and Inui, K. (2009) Targeted disruption of the multidrug and toxin extrusion 1 (mate1) gene in mice reduces renal secretion of metformin. Mol. Pharmacol., 75, 1280–1286. [DOI] [PubMed] [Google Scholar]
- 11. Nies, A.T., Damme, K., Kruck, S., Schaeffeler, E. and Schwab, M. (2016) Structure and function of multidrug and toxin extrusion proteins (MATEs) and their relevance to drug therapy and personalized medicine. Arch. Toxicol., 90, 1555–1584. [DOI] [PubMed] [Google Scholar]
- 12. Stocker, S.L., Morrissey, K.M., Yee, S.W., Castro, R.A., Xu, L., Dahlin, A., Ramirez, A.H., Roden, D.M., Wilke, R.A., McCarty, C.A.et al. (2013) The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin. Pharmacol. Ther., 93, 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Becker, M.L., Visser, L.E., vanSchaik, R.H., Hofman, A., Uitterlinden, A.G. and Stricker, B.H. (2010) Interaction between polymorphisms in the OCT1 and MATE1 transporter and metformin response. Pharmacogenet. Genomics, 20, 38–44. [DOI] [PubMed] [Google Scholar]
- 14. Tkac, I., Klimcakova, L., Javorsky, M., Fabianova, M., Schroner, Z., Hermanova, H., Babjakova, E. and Tkacova, R. (2013) Pharmacogenomic association between a variant in SLC47A1 gene and therapeutic response to metformin in type 2 diabetes. Diabetes Obes. Metab., 15, 189–191. [DOI] [PubMed] [Google Scholar]
- 15. Jablonski, K.A., McAteer, J.B., deBakker, P.I., Franks, P.W., Pollin, T.I., Hanson, R.L., Saxena, R., Fowler, S., Shuldiner, A.R., Knowler, W.C.et al. (2010) Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes, 59, 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dujic, T., Zhou, K., Yee, S.W., vanLeeuwen, N., deKeyser, C.E., Javorsky, M., Goswami, S., Zaharenko, L., Hougaard Christensen, M.M., Out, M.et al. (2017) Variants in pharmacokinetic transporters and Glycemic response to metformin: a Metgen meta-analysis. Clin. Pharmacol. Ther., 101, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bendl, J., Musil, M., Stourac, J., Zendulka, J., Damborsky, J. and Brezovsky, J. (2016) PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput. Biol., 12, e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kahn, S.E., Haffner, S.M., Heise, M.A., Herman, W.H., Holman, R.R., Jones, N.P., Kravitz, B.G., Lachin, J.M., O'Neill, M.C., Zinman, B.et al. (2006) Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med., 355, 2427–2443. [DOI] [PubMed] [Google Scholar]
- 19. Turner, R.C., Cull, C.A., Frighi, V. and Holman, R.R. (1999) Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) group. JAMA, 281, 2005–2012. [DOI] [PubMed] [Google Scholar]
- 20. Nasykhova, Y.A., Tonyan, Z.N., Mikhailova, A.A., Danilova, M.M. and Glotov, A.S. (2020) Pharmacogenetics of type 2 diabetes-progress and prospects. Int. J. Mol. Sci., 21, 6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mousavi, S., Kohan, L., Yavarian, M. and Habib, A. (2017) Pharmacogenetic variation of SLC47A1 gene and metformin response in type2 diabetes patients. Mol. Biol. Res. Commun., 6, 91–94. [PMC free article] [PubMed] [Google Scholar]
- 22. He, R., Zhang, D., Lu, W., Zheng, T., Wan, L., Liu, F. and Jia, W. (2015) SLC47A1 gene rs2289669 G>a variants enhance the glucose-lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res. Clin. Pract., 109, 57–63. [DOI] [PubMed] [Google Scholar]
- 23. Raj, G.M., Mathaiyan, J., Wyawahare, M. and Priyadarshini, R. (2018) Lack of effect of the SLC47A1 and SLC47A2 gene polymorphisms on the glycemic response to metformin in type 2 diabetes mellitus patients. Drug Metab. Pers. Ther., 33, 175–185. [DOI] [PubMed] [Google Scholar]
- 24. Christensen, M.M., Brasch-Andersen, C., Green, H., Nielsen, F., Damkier, P., Beck-Nielsen, H. and Brosen, K. (2011) The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet. Genomics, 21, 837–850. [DOI] [PubMed] [Google Scholar]
- 25. Duca, F.A., Cote, C.D., Rasmussen, B.A., Zadeh-Tahmasebi, M., Rutter, G.A., Filippi, B.M. and Lam, T.K. (2015) Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med., 21, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Napolitano, A., Miller, S., Nicholls, A.W., Baker, D., Van Horn, S., Thomas, E., Rajpal, D., Spivak, A., Brown, J.R. and Nunez, D.J. (2014) Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS One, 9, e100778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buse, J.B., DeFronzo, R.A., Rosenstock, J., Kim, T., Burns, C., Skare, S., Baron, A. and Fineman, M. (2016) The primary glucose-lowering effect of metformin resides in the gut, not the circulation: results from short-term pharmacokinetic and 12-week dose-ranging studies. Diabetes Care, 39, 198–205. [DOI] [PubMed] [Google Scholar]
- 28. Forslund, K., Hildebrand, F., Nielsen, T., Falony, G., Le Chatelier, E., Sunagawa, S., Prifti, E., Vieira-Silva, S., Gudmundsdottir, V., Pedersen, H.K.et al. (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature, 528, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graham, G.G., Punt, J., Arora, M., Day, R.O., Doogue, M.P., Duong, J.K., Furlong, T.J., Greenfield, J.R., Greenup, L.C., Kirkpatrick, C.M.et al. (2011) Clinical pharmacokinetics of metformin. Clin. Pharmacokinet., 50, 81–98. [DOI] [PubMed] [Google Scholar]
- 30. Ward, L.D. and Kellis, M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou, X., Li, D., Zhang, B., Lowdon, R.F., Rockweiler, N.B., Sears, R.L., Madden, P.A., Smirnov, I., Costello, J.F. and Wang, T. (2015) Epigenomic annotation of genetic variants using the roadmap Epigenome browser. Nat. Biotechnol., 33, 345–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou, X., Maricque, B., Xie, M., Li, D., Sundaram, V., Martin, E.A., Koebbe, B.C., Nielsen, C., Hirst, M., Farnham, P.et al. (2011) The human Epigenome browser at Washington University. Nat. Methods, 8, 989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.