

Abstract
Objectives:
Multiple organ failure in critically ill patients is associated with poor prognosis but biomarkers contributory to pathogenesis are unknown. Previous studies support a role for Fas-mediated apoptosis in organ dysfunction. Our objectives were to test for associations between soluble Fas (sFas) and multiple organ failure, identify protein quantitative trait loci, and determine associations between genetic variants and multiple organ failure.
Design:
Retrospective observational cohort study
Setting:
Four academic ICUs at U.S. hospitals
Patients:
Genetic analyses were completed in a discovery (n=1589) and validation set (n=863). Fas gene expression and flow cytometry studies were completed in outpatient research participants (n=250).
Interventions:
None
Measurements and Main Results:
In discovery and validation sets of critically ill patients, we tested for associations between enrollment plasma sFas concentrations and sequential organ failure assessment (SOFA) score on day 3. We conducted a genome-wide association study of plasma sFas (discovery n=1042) and carried forward a single nucleotide variant (SNV) in the FAS gene, rs982764, for validation (n=863). We further tested whether the SNV in FAS (rs982764) was associated with SOFA score, FAS transcriptional isoforms, and Fas cell surface expression. Higher plasma sFas was associated with higher day 3 SOFA scores in both the discovery (β = 4.07, p<0.001) and validation (β =6.96, p<0.001) sets. A SNV in FAS (rs982764G) was associated with lower plasma sFas concentrations and lower day 3 SOFA score in meta-analysis (−0.21, p=0.02). SNV rs982764G was also associated with a lower relative expression of the transcript for soluble as opposed to transmembrane Fas and higher cell surface expression of Fas on CD4+ T cells.
Conclusions:
We found that SNV rs982764G was associated with lower plasma sFas concentrations in a discovery and validation population and SNV rs982764G was also associated with lower organ dysfunction on day 3. These findings support further study of the Fas pathway as a potential mediator of organ dysfunction in critically ill patients.
Keywords: CD95, soluble Fas, Organ Dysfunction Scores, sepsis, single nucleotide polymorphism, quantitative trait loci, single nucleotide variant
Introduction
Four million patients are admitted annually to the intensive care unit (ICU) and mortality is conservatively estimated at 12%(1). Often systemic organ dysfunction precedes death in the ICU, with mortality rates greater than 60% when three or more organs simultaneously fail(2, 3). Dysregulated cell death due to excessive apoptosis, or programmed cell death, has been implicated in the development of organ failure in the ICU, including acute respiratory distress syndrome (ARDS), acute kidney injury (AKI), sepsis, and multi-organ dysfunction syndrome(4–7).
Fas is a type 1 membrane receptor, which mediates apoptosis through binding Fas ligand (8). Soluble Fas (sFas) is a truncated form of Fas believed to result from alternative mRNA splicing through skipping of exon 6 of FAS that encodes the transmembrane domain(9–12). The Fas pathway is thought to affect both immune modulation via apoptosis of leukocytes and end organ damage via apoptosis of epithelial cells(13–16). Observational studies have shown that higher levels of sFas are associated with poor outcomes in ARDS(4, 17, 18). Studies in critically ill cohorts have shown that common genetic polymorphisms in the Fas/FasL related genes are associated with ARDS susceptibility and development of AKI in ICU populations(19, 20). Thus, previous association studies show a link between the Fas/FasL system and organ dysfunction. However, protein quantitative trait loci (pQTL) for sFas have not been identified. If these loci are discovered, associations between them and multiple organ dysfunction could be utilized for future causal analyses between sFas and organ dysfunction.
In this study, we conducted a genome wide association study in a multi-center discovery set to determine single nucleotide variants (SNV) associated with sFas levels. Then, using the strongest candidate SNV within the FAS gene, we tested the hypothesis that lower sFas concentrations are associated with organ dysfunction in the ICU, as measured by Sequential Organ Failure Assessment (SOFA) score on day 3(21). We then replicated our findings in an external validation set.
Materials and Methods
Detailed methods are available in the Supplemental Digital Content – Supplemental Methods.
Discovery set (iSPAAR consortium). Subjects (n=1589) were all critically ill with genome wide genotyping data and clinical variables that are publicly available(22). The parent study included only Caucasian subjects to reduce confounding due to population stratification. For a subset of subjects (n=1042) plasma was obtained within 48 hours of enrollment. For a partially overlapping subset of patients (n=1072), an adjusted SOFA score(21) excluding the Glasgow-coma scale (GCS) component was available (Supplemental Digital Content - Figure S1).
Validation set (HMC SIRS). Critically ill patients (n=863) were prospectively enrolled after meeting criteria for the systemic inflammatory response syndrome (SIRS) (23). Subjects were Caucasian and blood was obtained within 24 hours of ICU admission. All subjects had a SOFA score and the GCS component was excluded to match the discovery set.
Our primary outcome measurement was day 3 SOFA score without the GCS component in subjects who survived to day 3. We chose this outcome because we hypothesized, given prior findings, that sFas might participate in altering risk for organ dysfunction such as respiratory and renal failure. We chose a time point that occurred after plasma sample acquisition but relatively proximal to the sampling to minimize noise from secondary events and interventions.
Genotyping
Discovery subjects were genotyped using the Illumina 660Quad BeadChip (San Diego, CA). We removed SNVs with a minor allele frequency (MAF) <0.03, an overall call rate<90%, and for deviation from Hardy-Weinberg Equilibrium (HWE) (p< 0.001)(24). This resulted in 488,966 SNVs. Imputation was performed via the Michigan Imputation Server with 1000 Genomes Project phase 3 data as the reference panel (25, 26). We additionally analyzed imputed SNVs within 10kB of the 5’ and 3’ end of the FAS gene. Validation subjects were genotyped for the SNV rs982764 by Taqman™ PCR.
Plasma sFas measurements
We measured plasma sFas protein concentrations using an antibody-based assay (Meso Scale Discovery, Gaithersburg, MD).
Quantitative RT-PCR
Healthy Subject mRNA cohort: We recruited healthy subjects and genotyping was performed on the Illumina Human 1M beadchip array (San Diego, CA)(27). RNA was purified from unstimulated whole blood. We selectively performed SYBR green quantitative RT-PCR in all subjects homozygous for the minor allele rs982764G (n=17), heterozygotes (n=20), and major allele homozygotes (n=25). Two distinct forward primers were used for the isoforms containing exon 6 (membrane Fas) and for skipping exon 6 (soluble Fas). The same reverse primer was used for both isoforms. The relative expression of sFas to Fas was calculated as 2− (ΔCt sFas-Fas).
Flow Cytometry
Benaroya Research Institute (BRI) cohort: Subjects (n=188) were genotyped on the Illumina ImmunoChip (San Diego, CA). Subjects were healthy or had diabetes. Peripheral blood mononuclear cells were stained with fluorophore-conjugated monoclonal antibodies against CD3, CD8, CD4, CD45RA, CD19, CD62L, and Fas (CD95)(28–30). Hierarchical gating schemes are shown in Supplemental Digital Content - Figure S2.
Data Analysis
In the discovery and validation sets, we tested for associations between log10-transformed sFas concentration and day 3 SOFA without GCS in subjects who survived to day 3 by multivariate linear regression adjusting for age, gender, and sepsis (sepsis-2 definition).
In the discovery genome-wide association study (GWAS), we used linear regression to test for associations with each variant and log10-transformed plasma sFas concentration, using an additive model adjusting for age, gender, sepsis and the first three principal components of ancestry (PCAs). In the validation phase, we tested for associations between rs982764 and log10-transformed plasma sFas concentration adjusting for age, gender and sepsis. We did not adjust for PCAs as no genome-wide data is available for this set. Genetic association testing was performed using Golden Helix Software (Bozeman, MT).
We further tested for associations between rs982764 genotype and sFas Ct/Fas Ct Ratio in the healthy subject mRNA cohort and median florescent intensity of Fas on memory CD4+ T cells, B cells, or CD8+ long-term effector memory cells in the Benaroya Research Cohort. Linear regression analyses using an additive genetic model were performed using Stata 14 (College Station, TX). Except for the GWAS (significant p< 1 × 10−8), a p value <0.05 was considered significant.
All studies were approved by human subject committees at the respective institutions (UW IRB# 1389, 3181, 37361, and BRI IRB #07109).
Results
Subjects in both the discovery and validation were predominantly male (58 and 63% respectively) and middle aged (mean age 58±17 and 55±16; Table 1). Sepsis was the predominant form of critical illness (76% and 71% respectively). Subjects in the discovery set were on average more severely ill on admission than the validation set (mean APACHE III score of 78 and 65, respectively). Subjects in the discovery set had a higher proportion of ARDS (64%) and higher persistent systemic organ dysfunction (mean day 3 SOFA of 5). Clinical characteristics of subjects within each area of the Venn diagram shown in Supplemental Digital Content - Figure S1 (i.e. genotyped, plasma sFas only, SOFA only) were similar (Supplemental Digital Content - Table S1). The mean (±SD) plasma sFas concentrations were 11082 (±7265) pg/mL and 13419 (±7670) pg/mL in the discovery and validation set respectively.
Table 1.
Subject Characteristics
| Characteristic | Discovery | Validation |
|---|---|---|
| Genotyped (n=1589) | Genotyped (n=863) | |
| Patient Age, mean ± SD | 58 (±17) | 55 (±16) |
| Male patients, n (%) | 928 (58) | 543 (63) |
| Caucasian, n (%) | 1589 (100) | 863 (100) |
| Source of critical illness, n (%) | ||
| Sepsis | 1201 (76) | 612 (71) |
| Trauma | 110 (7) | 0 |
| Other | 278 (17) | 251 (29) |
| APACHE III score, mean ± SD | 78 (±27) | 65 (±25) |
| Comorbidities, n (%) | ||
| Diabetes | 381 (24) | 228 (26) |
| Cirrhosis | 74 (5) | 90 (10) |
| End Stage Renal Disease | 73 (5) | 22 (3) |
| ARDS, n (%) | 1022 (64) | 231 (27) |
| SOFA, mean ± SD | 5.2 (±2.8) | 2.7 (±2.6) |
| Plasma sFas (pg/mL), mean ± SD | 11082 (±7265) | 13419 (±7670) |
| Mortality, n (%) | 281 (18) | 120 (14) |
Plasma sFas and Organ Dysfunction.
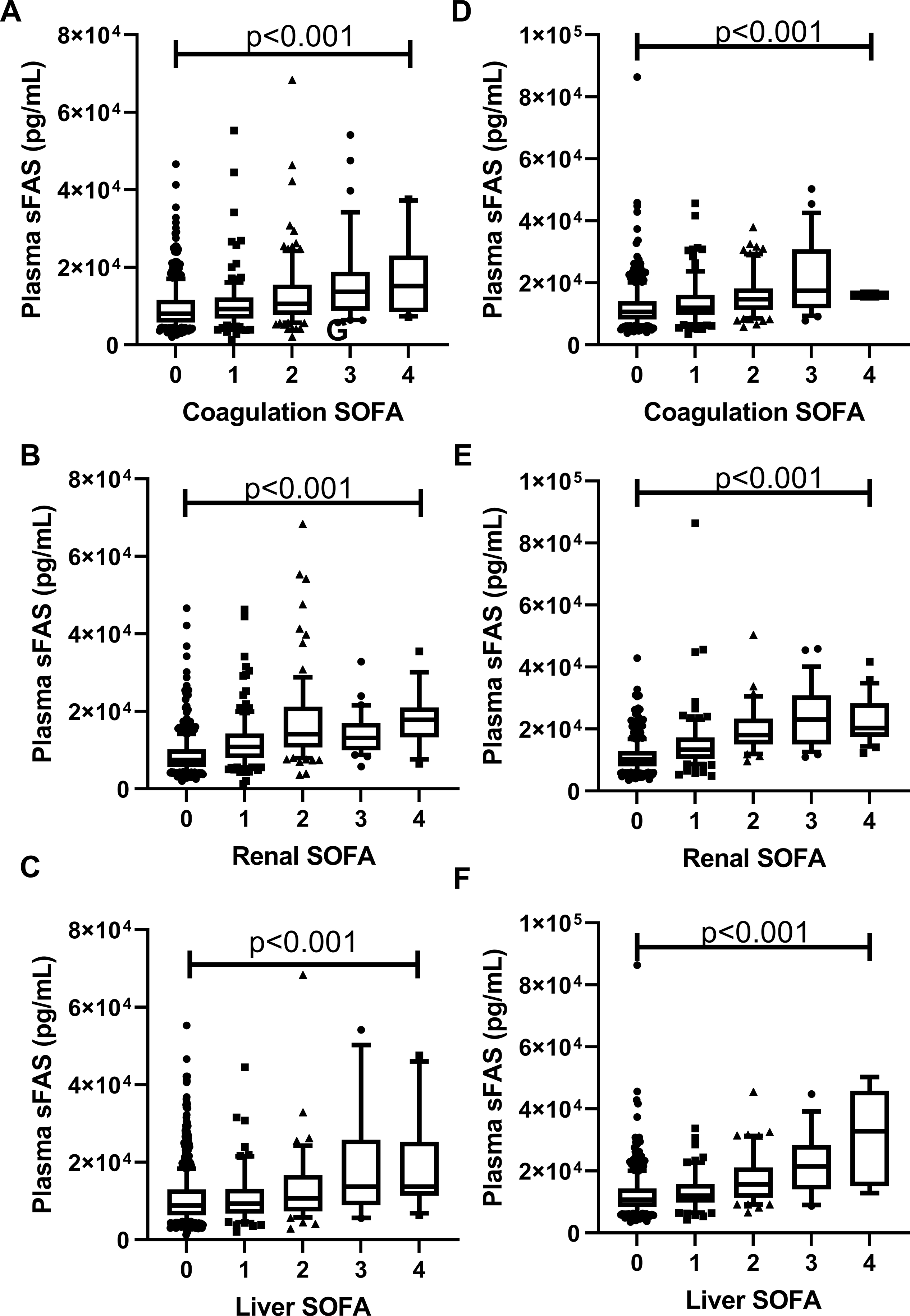
In the discovery set, we found that a one log10 unit increase in plasma sFas concentration was associated with a 4.40 point (95% CI 3.53 to 5.28, p<0.001) higher day 3 SOFA score (Table 2). This finding was also seen in the validation set, with one log10 unit change in sFas concentrations associated with a 6.96 point (95% CI 5.95 to 7.97, p<0.001; Table 2) higher day 3 SOFA score. Plasma sFas was broadly associated with higher organ dysfunction including coagulation (Figure 1A), renal (Figure 1B), and liver (Figure 1C), cardiovascular (Supplemental Digital Content – Figure S3), in both the discovery and validation sets (Figure 1D–F and Supplemental Digital Content Figure S3); all p<0.001). Respiratory organ dysfunction was not associated with higher sFas level in the discovery set (Supplemental Digital Content – Figure S3, p=0.06), possibly due to the fact the degree of respiratory dysfunction was skewed towards higher values in this group. These results demonstrate robust associations between higher plasma sFas and higher systemic organ dysfunction in critically ill patients.
Table 2.
Multivariate analysis of sFas association with SOFA score
| sFas (log10(pg/mL)) | n | Unadjusted β (95% CI) | p | Adjusted β1 (95%CI) | p |
|---|---|---|---|---|---|
|
| |||||
| Discovery | 525 | 4.40 (3.53, 5.28) | <0.001 | 4.07 (3.13, 5.01) | <0.001 |
| Validation | 579 | 7.11 (6.10, 8.12) | <0.001 | 6.96 (5.95, 7.97) | <0.001 |
Adjusted for age, gender, sepsis
Figure 1. Plasma sFas Concentrations and associations with SOFA organ dysfunction.
Plasma sFas concentrations by SOFA score for the discovery set for coagulation (A), Renal (B), and Liver (C) components. Validation set plasma sFAS concentrations by coagulation (F), Renal (G), and Liver (H) components. P values shown are from linear regression. Whiskers extend from 10th to 90th percent value and box ranged from the 25th to 75th percentile with line at the median.
SNVs and plasma sFas.
No SNV reached genome-wide significance in the GWAS of plasma sFas levels in the discovery set (Supplemental Digital Content - Table S2). The quantile-quantile plot did not suggest residual confounding (Supplemental Digital Content - Figure S4). While not genome-wide significant, one of the strongest associations was observed with an SNV located within an intron of FAS (rs982764; p=1.81×10−5)(Supplemental Digital Content - Figure S5). The minor allele for rs982764 was associated with lower sFas levels (additive model, β=−0.05)(Figure 2A). The SNV with the strongest association with sFas levels was rs11663956 in Laminin Subunit Alpha 1(LAMA1), encoding Laminin α1 (Supplemental Digital Content - Table S2). The third most highly associated SNV (rs820371; p=5.6×10−6) was located within an intron of Myosin Light Chain Kinase (MYLK). We foucused on the variants within the FAS gene given that these variants could represent cis-pQTL, and and, thus, have high biologic plausibility. We sought to more finely map the peak association within FAS using densely imputed SNVs but the peak of association remained at rs982764 (Supplemental Digital Content - Figure S6). An imputed, intronic SNV was equivalently associated (rs7911226) and was in perfect linkage disequilibrium (R2=1.0, D’=1.0) with rs982764. In a sensitivity analysis limiting to subjects with sepsis (n=681), rs982764 remained associated with lower plasma concentrations (log10-transformed) of sFas (β=−0.05, 95% CI: −0.03, −0.07, p=2.47×10−4). Similarly, in the validation set, each copy of the minor allele was associated with lower plasma sFas (β=−0.04, p=0.009)(Figure 2B).
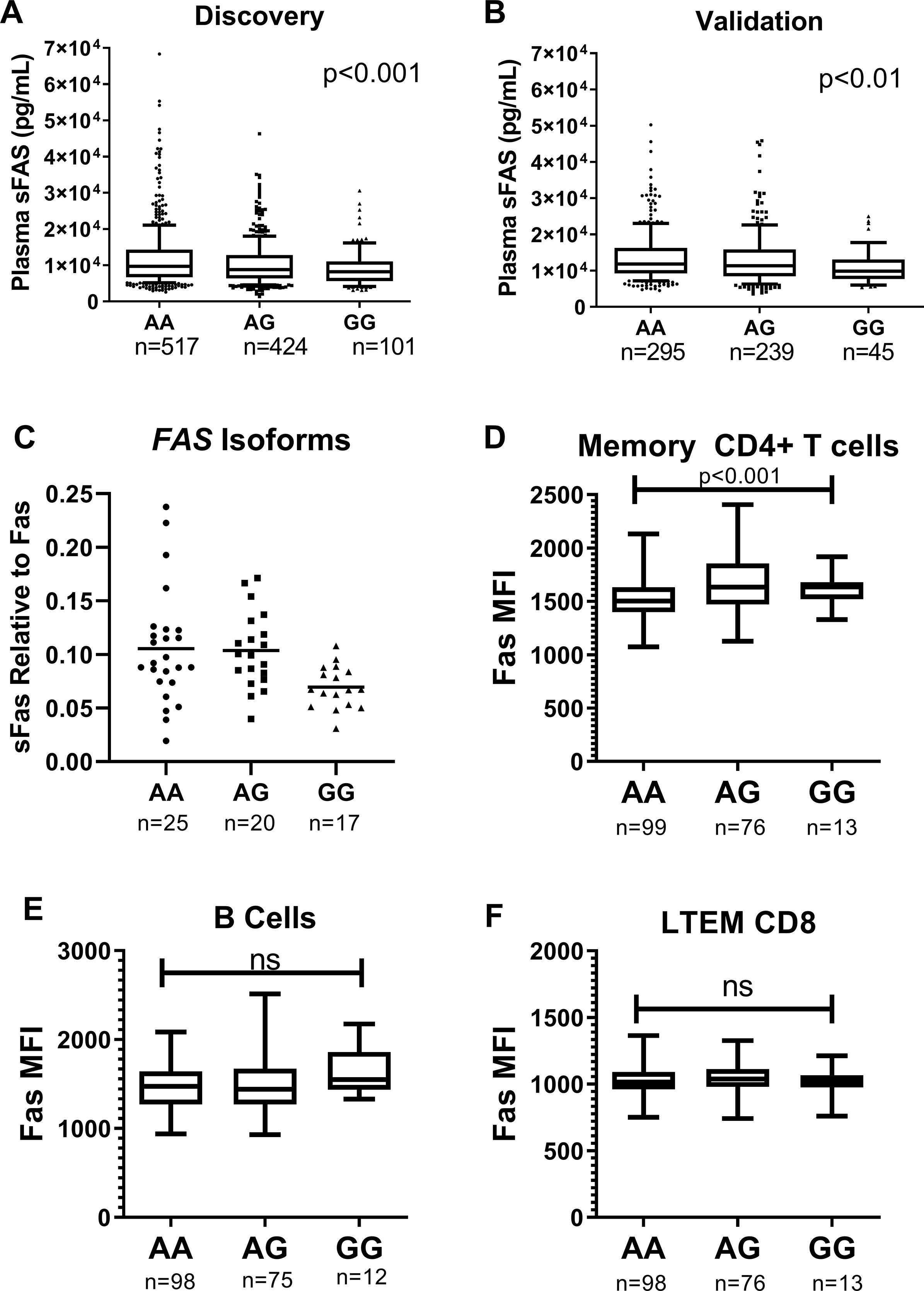
Figure 2. rs982764 and Relationship to Fas.
Genotype rs982763G is associated with lower plasma sFas in the discovery (A) and validation (B) sets. Number of subjects of each genotype is shown on the x-axis. Whiskers extend from 10th to 90th percent value and box ranged from the 25th to 75th percentile with line at the median. P value represents multiple linear regression adjusted for age, gender, and sepsis using an additive genetic model. C) RNA was collected from unstimulated whole blood samples from healthy controls of differing genotype. Quantitative RT-PCR RNA was performed for transcripts containing exon 6 (membrane bound “Fas”) and skipping exon 6 (soluble Fas “sFas”). The relative expression of sFas to Fas was calculated as 2− (ΔCt sFas-Fas)). Median florescent intensity (MFI) of cell surface FAS on D) memory CD4+ T cells, E) B cells, and F) long term effector memory CD8+ T cells. P values are for linear regression using an additive genetic model.
SNV and Organ Dysfunction.
We then tested for associations between rs982764 and organ dysfunction. We found that in an additive model that the minor allele (G) of rs982764 trended towards lower day 3 SOFA score in both the discovery (β=−0.25, 95% CI: −0.49, 0.00, p=0.056) and validation (β=−0.18, 95% CI: −0.45, 0.10) sets (Table 3). In a pooled meta-analysis, the minor allele of rs982764 was associated with lower day 3 SOFA scores (β=−0.21, 95% CI: −0.40, −0.03, p=0.02; Table 3).
Table 3.
Association of rs982764 with SOFA score
| Subject Set | n | β (95% CI)1 | p | Adjusted β (95% CI)2 | p |
|---|---|---|---|---|---|
|
| |||||
| Discovery | 1072 | −0.24 (−0.48, 0.02) | 0.07 | −0.25 (−0.49, 0.00) | 0.06 |
| Validation | 861 | −0.18 (−0.45, 0.10) | 0.21 | −0.18 (−0.45, 0.10) | 0.20 |
| Meta-analysis | −0.21 (−0.39, −0.02) | 0.03 | −0.21 (−0.40, −0.03) | 0.02 | |
Effect size and direction for change in mean SOFA for each copy of minor allele of rs982764 (A>G)
Adjusted for age, gender, sepsis
SNV, Alternative Splicing, and Fas cell surface expression
We wanted to test splice variation as a potential mechanism for the association between rs982764 and plasma sFAS and utilized two additional cohorts with mRNA and flow cytometry data available. First, we tested whether rs982764 was associated with differences in FAS mRNA isoform proportions. Differential expression of FAS mRNA splice variants due to rs982764 could lead to differences in sFas production. We used PCR assays to detect FAS mRNA splice variants with exon 6 (exon 5–6-7) or that skip exon 6 (exon 5–7, skip 6). Exon 6 is the region encoding the transmembrane domain and exclusion of exon 6 is thought to produce the soluble form of Fas (sFas)(9, 10). We compared the relative expression of the two isoforms by rs982764 genotype in mRNA from whole blood obtained from a cohort of healthy subjects. We found that the minor allele of rs982764 was associated with lower relative expression of the isoform that excludes exon 6 (p<0.05; Figure 2C) which supports the hypothesis that the G allele of rs982764 reduces sFas production through altered RNA splicing.
Next, we tested whether the minor allele of rs982764 was associated with cell surface expression of Fas in a second cohort of subjects with available flow cytometry data for circulating leukocytes. We found that surface expression of Fas measured by mean florescent intensity (MFI) was higher on CD4+ T cells for each copy of the minor allele of rs982764 (Figure 2D). These findings provide further support for the hypothesis that the minor allele of rs982764 leads to lower circulating sFas through altered isoform ratios favoring production of transmembrane Fas. We did not observe a significant difference in cell surface Fas expression on B cells and long-term effector memory CD8+ T cells by rs982764 genotype (Figures 2E and 2F, respectively).
Discussion
Our studies extend prior work demonstrating a strong association between higher circulating sFas and more severe organ dysfunction in critically ill patients and provides the first evidence of a quantitative trait loci (QTL) for plasma levels of sFas in acute critical illness. We showed that this QTL is marked by rs982764, a SNV located in the intron 4–5 in FAS. It is associated with both decreased levels of plasma sFas and lower levels of organ failure in meta-analyses in these cohorts. These findings provide evidence supporting associations between sFas and organ failure in critically ill patients.
Our findings that higher plasma sFas is associated with increased organ dysfunction as measured by SOFA score is consistent with previous studies of AKI, ARDS, and MODS(18–20, 31). We have previously shown that sFas concentrations are associated with a distinct form of non-resolving AKI in studies that employed the set used here for validation (32). While we observed robust associations between sFas and organ dysfunction in both the discovery and validation sets we did not find an association between sFas and respiratory failure in the discovery set. This exception is notable given our own work showing that other genetic variants in FAS associate with risk for ARDS(19). These previously published variants in FAS were not associated with lung injury in this discovery cohort. We postulate that our finding in the discovery set could be due to the very different overall proportion of subjects with severe respiratory failure at the time of enrollment and, thus, potential differences in timing of sample acquisition relative to onset of organ failure. Future studies will need to clarify the temporal relationship between the onset of the cause of critical illness (e.g. pneumonia, trauma) and sFas levels and, in turn, organ failure onset.
Our finding that the variants within FAS are strongly associated with circulating sFas levels in two critically ill cohorts provides very strong evidence that this locus is a true QTL for sFas production. Prior evidence from patients with coronary artery disease suggests that rs982764 may be a QTL for sFas in the population more generally (33) though this is the first report that suggests a link between genetic modulation of sFas levels and risk for a clinical outcome. We also provide new evidence that this QTL may influence sFas levels through altered levels of FAS mRNA isoforms. There is insufficient evidence to explain how sFas levels might affect organ dysfunction or whether the effect might be more directly related to altered CD4+ T cell expression of Fas and modulation of immune cell apoptosis. Our findings with this variant are supported by strong biologic plausibility due to the “cis” location and the effect on relevant mRNA isoforms, however it only explains 1.6% of the variance in plasma sFas levels. Future work will need to clarify the other major factors (e.g. demographic, environmental) influencing sFas levels.
While no SNV reached genome-wide significance in our discovery phase, variants in genes other than FAS also showed highly suggestive associations with sFas levels including variants within myosin light chain kinase (MYLK), a gene previously associated with risk for ARDS(34, 35). Notably, Myosin light chain kinase enzyme activity is also linked to cellular apoptosis(36). The SNV most strongly associated with sFas levels was located in LAMA1, a gene implicated in organ development and shown to modulate the pulmonary response to lung injury and fibrosis, biologic processes relevant to acute organ dysfunction(37). Future work will need to clarify whether these loci might be true “trans” QTL for sFas levels and whether these loci affect clinical outcomes in which Fas-mediated cell death is pathophysiologically relevant.
Our study has several limitations. First, a significant proportion of the subjects in the discovery set were enrolled through interventional clinical trials and all patients were recruited at academic medical centers which may limit the generalizability of our results. Second, we limited this study to Caucasian subjects only in an effort to reduce the potential confounding effects of population stratification. Future work will need to determine whether our findings are relevant to more diverse populations. Third, testing associations with SOFA scores on days other than day 3 would have been informative, but some missing data and timing differences between the datasets prevented us performing these analyses. Fourth, our findings of an association between rs982764 and organ failure was marginal when tested in the discovery and validation sets individually while showing a significant association in the meta-analysis. Our estimates from this study suggest that sample sizes of greater than 3000 subjects would be necessary to fully power future studies. While this finding will need to be further replicated, the fact that the direction of effect was consistent in the two independent cohorts provides some reassurance as to its validity. Finally, we did not have access to any critically ill study populations that had simultaneous measures of genotype, gene expression, plasma protein measurements, and clinical outcomes. However, the consistency of the direction of effects observed with the G allele of rs982764 G in the different study groups (i.e. reduced exon 6-negative mRNA isoform, increased cell surface Fas, reduced circulating sFas) supports a testable model linking the G allele to sFas and, in turn, organ failure in the critically ill. Because of some missingness in these cohorts, we were not able to perform a mendelian randomization or mediation analysis to draw more direct causation. However, the associations resulted here provide support for this type of structured analysis of the Fas pathway in critical illness.
Conclusions
We have found that SNV rs982764-G located in intron 4–5 of FAS is associated with lower plasma sFas in two large critically ill cohorts. This SNV is also associated with decreased persistent systemic organ dysfunction. Finally, the G allele is associated with decreased relative expression of an mRNA isoform likely to code for sFas while also associated with increased cell surface expression of Fas on T-cells. These findings provide evidence for a potential role of the Fas pathway in the development of organ failure in critical illness.
Supplementary Material
Acknowledgments
Funding Support: This work was supported by the NIH: NCATS UL1 TR002319, NHLBI R01HL060710 and NHLBI RC2 HL101779, NIA U19AG023122, NIAID AI101990, AI083455, NIDDK DK097672. The funding sources had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Copyright Form Disclosure: Dr. Mikacenic’s institution received funding from the National Center for Advancing Translational Sciences (UL1 TR002319), the National Heart, Lung, and Blood Institute (NHLBI) (R01HL060710, RC2 HL101779), the National Institute on Aging (U19AG023122), the National Institute of allergy and Infectious Diseases (AI101990, AI083455), and the National Institute of Diabetes and Digestive and Kidney Issues (DK097672). Drs. Mikacenic, Bhatraju, Robinson-Cohen, Long, Cerosaletti, Calfee, Matthay, Christie, Meyer, Christiani, and Wurfel received support for article research from the National Institutes of Health (NIH). Drs. Cerosaletti, Calfee, and Meyer’s institutions received funding from the NIH. Dr. Cerosaletti’s institution received funding from the Department of Defense, the American Diabetes Association, and the Juvenile Diabetes Research Foundation. Drs. Calfee and Matthay’s institutions received funding from Gentech/Roche. Dr. Calfee’s institution received funding from Bayer; she received funding from Quark, Vasomune, Gen1e Life Sciences, and Prometic. Dr. Matthay received funding from Novartis and Citius Pharmaceuticals. Dr. Russell received funding from Asahi Kesai Pharmaceuticals of America, IB Therapeutics LLC, and Ferring Pharmaceuticals; he received funding from Grifols; he disclosed that he is the inventor of two patents owned by the University of British Colombia and Ferring, that he is a founder, director and shareholder in Cyon Therapetutics Inc and a shareholder in Molecular You Corp, that he is a member of the Data Safety Monitoring Board of an NIH-sponsored trial of plasma in COVID-19 (PASS-IT-ON). Dr. Meyer’s institution received funding from the NHLBI (HL137006, HL137915), Quantum Leap Healthcare Collaborative, Biomarck, Inc, and Athersys Inc The Marcus Foundation. Dr. Wurfel’s institution received funding from the NHLBI. The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Young MP, Birkmeyer JD: Potential reduction in mortality rates using an intensivist model to manage intensive care units. Eff Clin Pract 2000; 3:284–289 [PubMed] [Google Scholar]
- 2.Vincent JL, de Mendonça A, Cantraine F, et al. : Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: results of a multicenter, prospective study. Working group on “sepsis-related problems” of the European Society of Intensive Care Medicine. Crit Care Med 1998; 26:1793–1800 [DOI] [PubMed] [Google Scholar]
- 3.Nfor TK, Walsh TS, Prescott RJ: The impact of organ failures and their relationship with outcome in intensive care: analysis of a prospective multicentre database of adult admissions. Anaesthesia 2006; 61:731–738 [DOI] [PubMed] [Google Scholar]
- 4.Imai Y, Parodo J, Kajikawa O, et al. : Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA 2003; 289:2104–2112 [DOI] [PubMed] [Google Scholar]
- 5.Havasi A, Borkan SC: Apoptosis and acute kidney injury. Kidney Int 2011; 80:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotchkiss RS, Strasser A, McDunn JE, et al. : Cell death. N Engl J Med 2009; 361:1570–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linkermann A, Chen G, Dong G, et al. : Regulated cell death in AKI. J Am Soc Nephrol 2014; 25:2689–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wajant H: The Fas signaling pathway: more than a paradigm. Science 2002; 296:1635–1636 [DOI] [PubMed] [Google Scholar]
- 9.Cheng J, Zhou T, Liu C, et al. : Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 1994; 263:1759–1762 [DOI] [PubMed] [Google Scholar]
- 10.Cascino I, Fiucci G, Papoff G, et al. : Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol 1995; 154:2706–2713 [PubMed] [Google Scholar]
- 11.Liu C, Cheng J, Mountz JD: Differential expression of human Fas mRNA species upon peripheral blood mononuclear cell activation. Biochem J 1995; 310 ( Pt 3):957–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papoff G, Cascino I, Eramo A, et al. : An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J Immunol 1996; 156:4622–4630 [PubMed] [Google Scholar]
- 13.Liles WC, Kiener PA, Ledbetter JA, et al. : Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med 1996; 184:429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alderson MR, Tough TW, Davis-Smith T, et al. : Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med 1995; 181:71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrero R, Tanino M, Smith LS, et al. : The Fas/FasL pathway impairs the alveolar fluid clearance in mouse lungs. Am J Physiol Lung Cell Mol Physiol 2013; 305:L377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ortiz-Arduan A, Danoff TM, Kalluri R, et al. : Regulation of Fas and Fas ligand expression in cultured murine renal cells and in the kidney during endotoxemia. Am J Physiol 1996; 271:F1193–1201 [DOI] [PubMed] [Google Scholar]
- 17.Matute-Bello G, Liles WC, Steinberg KP, et al. : Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 1999; 163:2217–2225 [PubMed] [Google Scholar]
- 18.Albertine KH, Soulier MF, Wang Z, et al. : Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 2002; 161:1783–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glavan BJ, Holden TD, Goss CH, et al. : Genetic variation in the FAS gene and associations with acute lung injury. Am J Respir Crit Care Med 2011; 183:356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhatraju P, Hsu C, Mukherjee P, et al. : Associations between single nucleotide polymorphisms in the FAS pathway and acute kidney injury. Crit Care 2015; 19:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vincent JL, Moreno R, Takala J, et al. : The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996; 22:707–710 [DOI] [PubMed] [Google Scholar]
- 22.dbGaP | phs000631.v1.p1 | ARDSnet and the iSPAAR Consortium: Genomic Basis of Susceptibility and Outcomes in Patients with the Acute Respiratory Distress Syndrome (ARDS) [Internet]. [cited 2014 Jul 11] Available from: http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000631.v1.p1
- 23.Ko DC, Shukla KP, Fong C, et al. : A genome-wide in vitro bacterial-infection screen reveals human variation in the host response associated with inflammatory disease. Am J Hum Genet 2009; 85:214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson CA, Pettersson FH, Clarke GM, et al. : Data quality control in genetic case-control association studies. Nat Protoc 2010; 5:1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The 1000 Genomes Browsers | 1000 Genomes [Internet]. [cited 2017 Apr 25] Available from: http://www.internationalgenome.org/1000-genomes-browsers
- 26.Das S, Forer L, Schönherr S, et al. : Next-generation genotype imputation service and methods. Nat Genet 2016; 48:1284–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wurfel MM, Gordon AC, Holden TD, et al. : Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am J Respir Crit Care Med 2008; 178:710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwedhelm K, Thorpe J, Murray SA, et al. : Attenuated IL-2R signaling in CD4 memory T cells of T1D subjects is intrinsic and dependent on activation state. Clin Immunol 2017; 181:67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lord JD, Long SA, Shows DM, et al. : Circulating integrin alpha4/beta7+ lymphocytes targeted by vedolizumab have a pro-inflammatory phenotype. Clin Immunol 2018; 193:24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hundhausen C, Roth A, Whalen E, et al. : Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci Transl Med 2016; 8:356ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papathanassoglou ED, Moynihan JA, McDermott MP, et al. : Expression of Fas (CD95) and Fas ligand on peripheral blood mononuclear cells in critical illness and association with multiorgan dysfunction severity and survival. Crit Care Med 2001; 29:709–718 [DOI] [PubMed] [Google Scholar]
- 32.Bhatraju PK, Robinson-Cohen C, Mikacenic C, et al. : Circulating levels of soluble Fas (sCD95) are associated with risk for development of a nonresolving acute kidney injury subphenotype. Crit Care 2017; 21:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Folkersen L, Fauman E, Sabater-Lleal M, et al. : Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet 2017; 13:e1006706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao L, Grant A, Halder I, et al. : Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol 2006; 34:487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christie JD, Ma S-F, Aplenc R, et al. : Variation in the myosin light chain kinase gene is associated with development of acute lung injury after major trauma. Crit Care Med 2008; 36:2794–2800 [DOI] [PubMed] [Google Scholar]
- 36.Fazal F, Gu L, Ihnatovych I, et al. : Inhibiting myosin light chain kinase induces apoptosis in vitro and in vivo. Mol Cell Biol 2005; 25:6259–6266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee C-M, Cho SJ, Cho W-K, et al. : Laminin α1 is a genetic modifier of TGF-β1-stimulated pulmonary fibrosis. JCI Insight 2018; 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.