

Abstract
Gammaherpesviruses are ubiquitous pathogens that establish lifelong infections in the vast majority of adults worldwide. Importantly, these viruses are associated with numerous malignancies and are responsible for significant human cancer burden. These virus-associated cancers are due, in part, to the ability of gammaherpesviruses to successfully evade the innate immune response throughout the course of infection. In this review we will summarize the current understanding of how gammaherpesviruses are detected by innate immune sensors, how these viruses evade recognition by host cells, and how this knowledge can inform novel therapeutic approaches for these viruses and their associated diseases.
Keywords: KSHV, EBV, MHV68, gammaherpesvirus, innate immunity, IFN, NF-κB
Graphical Abstract
Introduction
Herpesviruses are a family of ancient viruses that have co-evolved with their hosts over millions of years. While there are three subfamilies of Herpesviridae (alpha-, beta-, and gammaherpesvirinae) comprising over 100 identified viral species, all of these viruses share numerous features, including a large double-stranded DNA (dsDNA) genome, icosahedral capsid, tegument, host cell-derived lipid envelope, and a unique biphasic lifecycle. Here we will highlight the interactions between the host innate immune response and gammaherpesviruses, with specific focus on the two identified human gammaherpesviruses, Epstein-Barr virus (EBV, human gammaherpesvirus 4) and Kaposi’s sarcoma-associated herpesvirus (KSHV, human gammaherpesvirus 8), as well as the murine model of gammaherpesvirus infection, murine gammaherpesvirus 68 (MHV68, murid gammaherpesvirus 4).
Unique among herpesviruses, gammaherpesviruses are bona fide oncogenic agents, and are responsible for considerable cancer burden. Gammaherpesviruses can infect numerous cell types, however they are generally lymphotropic, with EBV, KSHV, and MHV68 showing a predilection for B cells. As such, many of the malignancies associated with gammaherpesviruses are B cell lymphomas, although other cancers are also driven by these viruses. For example, EBV is associated with numerous B, T, and NK cell lymphomas and lymphoproliferative diseases, nasopharyngeal carcinoma (NPC), gastric carcinoma, and other cancers (reviewed in [1]). KSHV is the etiologic agent of its namesake, Kaposi’s sarcoma (KS), as well as primary effusion lymphoma (PEL) and the plasmablastic form of multicentric Castleman’s disease (MCD) (reviewed in [2]). KSHV has recently also been linked to osteosarcoma [3]. The precise mechanisms underpinning gammaherpesviruses-driven oncogenesis have been an active area of research since the discovery of EBV over 50 years ago and are still being defined. However, immunodeficiencies due to genetic mutation, irradiation, HIV infection, or other means are the greatest risk factor for developing gammaherpesvirus-driven cancers. Thus, while more than 95% of adults are infected with at least one gammaherpesvirus, the ability of a competent immune system to detect and control virally infected cells is sufficient to prevent oncogenesis in most individuals.
Latency.
Like all other described herpesviruses, gammaherpesviruses utilize two distinct life cycles termed lytic replication and latency. During latency, the virus exists as a circular episome of chromatinized DNA in the nucleus of the infected host cell. The virus persists by relying on cellular division and the host DNA replication machinery to passively replicate the viral genome. In fact, EBV, KSHV, and MHV68 ensure faithful genome replication and segregation into daughter cells by tethering their episomal DNA to a host chromosome through their multifunctional proteins EBV nuclear antigen 1 (EBNA1), latency-associated nuclear antigen (LANA), or mLANA, respectively. Depending on the virus, the cell type infected, and the immunocompetency of the host, the virus may express a range of latency genes and non-coding RNAs, or in some instances only non-coding RNAs. The genes expressed in latency typically have roles in circumventing normal proliferative and apoptotic checkpoints, as well as subverting detection by the immune system. In the case of EBV, distinct latent gene expression programs have been defined, and are transcriptionally regulated via the use of distinct latent promoters (reviewed in [4]). However, it is important to note that exceptions to these programs are frequently observed, particularly in the context of cancer. Notably, the number of genes expressed during latency is negatively correlated with the immunocompetency of the host, and the essential latency genes are generally poorly immunogenic. KSHV encodes a latency locus, comprising two promoters that facilitate the transcription of six viral genes believed to be expressed in all latently infected cells (reviewed in [5]). Interestingly, additional genes have been shown to be expressed at low levels during latency [6].
Lytic replication.
Latency represents the default pathway for EBV and KSHV upon de novo infection. However, in order to transmit from cell to cell, or individual to individual, these viruses must also undergo reactivation from latency into the lytic cycle. Lytic replication is typically associated with expression of nearly all viral genes and the production of infectious progeny virions. Triggers that can induce reactivation from latency to lytic replication are diverse and include factors such as B cell receptor stimulation [7–9], toll-like receptor (TLR) signaling [10, 11], hypoxia [12, 13], cellular differentiation [14–17], and stress hormones [18]. Importantly, as lytic replication involves the expression of many more viral gene products compared to latency, and typically resolves with the destruction of the infected cell, this process is vastly more immunostimulatory than latency, which can prove nearly undetectable to the immune system. Therefore, while subversion of cellular innate immune sensors and antiviral pathways is a broadly utilized strategy amongst gammaherpesviruses and is critical for numerous aspects of their biology, the very nature of their biphasic life cycle allows gammaherpesviruses to avoid immune detection and persist for the lifetime of the infected individual.
Antiviral signaling pathways.
With respect to innate immune pathways involved in the recognition of gammaherpesvirus infection, which will be thoroughly discussed in the next section, nearly all converge on the interferon regulatory factor (IRF)-mediated production of type I interferons (IFNs) and/or nuclear factor-κB (NF-κB)-mediated production of inflammatory cytokines. Type I IFN production is initiated following activation of multiple innate immune sensors. These pathways converge on the activation of tank binding kinase 1 (TBK1) and/or IκB kinase ε (IKKε) to phosphorylate IRF3 and IRF7. IRF3 and IRF7 can function each as homodimers, or as an IRF3/IRF7 heterodimer, to induce the transcription of IFN-α and IFN-β. Canonically, IRF3 homodimers induce an initial short burst of IFN-α and IFN-β which stimulate the transcription of IRF7, resulting in a positive feedback loop in which IRF7 facilitates the transcription of more IFNs. The type I IFNs are then released from the cell where they will signal through the IFN-α/β receptor (IFNAR) in an autocrine and paracrine fashion to induce the robust expression of interferon stimulated genes (ISGs) (Figure 1).
Fig. 1. Recognition of gammaherpesvirus infection results in the production of type I IFNs and inflammatory cytokines.

Type I IFNs bind and activate the type I IFN receptor. IFNAR, resulting in the phosphorylation of STAT1 and STAT2, by JAK1 and Tyk2, respectively. Phosphorylated STAT1 and STAT2 dimerize, complex with IRF9, and translocate to the nucleus to induce the transcription of ISGs. Canonical NF-κB signaling is initiated by various stimuli, including the ligation of most TLRs. The activated IKK complex (IKKα, IKKβ, and IKKγ) mediates the phosphorylation of the inhibitory IκB subunit of the NF-κB complex. The resulting degradation of IκB allows the active NF-κB complex (p50 and p65/RelA) to translocate the nucleus and mediate the induction of inflammatory genes. IFN, interferon; IFNAR, interferon-a /b receptor; STAT, signal transducer and activator of transcription; JAK, Janus kinase; Tyk, tyrosine kinase; IRF, interferon regulatory factor; ISGs, interferon stimulated genes; NF-κB, nuclear factor kappa B; IKK, IκB kinase; IκB, inhibitor of NF-κB. Adapted from “Interferon Pathway” and “NF-KB Signaling Pathway” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
NF-κB signaling proceeds through two related pathways, termed the canonical and non-canonical pathways (reviewed in [19]). The canonical NF-κB signaling pathway is stimulated by activation of pattern recognition receptors (PRRs) and, as such, is the pathway most relevant to the innate immune response to viral infection. In this pathway, a heterodimer of NF-κB proteins, namely p50 and p65/RelA, are retained in the cytosol by the regulatory protein, IκB (Figure 1). Upon PRR stimulation, the protein IκB kinase (IKK) is activated and phosphorylates a phosphodegron on IκB, leading to its proteasomal degradation. This releases the NF-κB heterodimer, which translocates to the nucleus and mediates the transcription of inflammatory genes (Figure 1).
The above cellular responses to gammaherpesvirus infection require initial detection by PRRs. The PRRs that have been implicated in the biology of gammaherpesvirus infection can be divided into four categories: TLRs, RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and intracellular DNA sensors. Hypothetically, these receptors should be capable of detecting viral intruders at nearly every stage of the gammaherpesvirus lifecycle. However, as will be exemplified below, gammaherpesviruses employ a diverse arsenal of genes to limit detection and prevent subsequent antiviral responses. In this review we will summarize i) the current knowledge of how the gammaherpesviruses EBV, KSHV, and MHV68 are recognized via innate immune pathways, ii) the various mechanisms by which these viruses subvert these responses, and iii) how our knowledge surrounding innate immune recognition of, and subversion by, gammaherpesviruses can be employed to develop novel therapeutics for their associated malignancies.
Innate immune sensing of gammaherpesvirus infection
The innate immune system plays a critical role in the biology of gammaherpesvirus infection. Many different innate immune sensors have been described to recognize virus-associated molecules, such as DNA, RNA, double-stranded RNA (dsRNA), structural and non-structural viral proteins, and lipids. Furthermore, virus-mediated perturbations in certain host processes, such as cellular metabolism, or virus-induced cellular damage, can also induce an antiviral state. As such, numerous innate immune sensors have been implicated in the detection of latent and lytic viral processes, resulting in the rapid induction of type I IFNs and the restriction of viral replication. Here we will review the sensors relevant to gammaherpesvirus infection and discuss some of the unanswered questions and scientific challenges in this area.
Toll-like receptors.
TLRs are a family of transmembrane receptors present in mammals and insects with well-described roles in innate immunity. As PRRs, TLRs recognize an array of conserved motifs associated with microbial pathogens. There are 10 TLRs encoded by the human genome (TLR1-TLR10) and 13 in mice (TLR1-TLR13); however, murine TLR10 is non-functional due to insertion of a retroviral element [20]. TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the cell surface and recognize extracellular constituents of pathogens, such as lipids and proteins, while TLR3, TLR7, TLR8, and TLR9 are found within endosomes and are activated by nucleic acids [21–28] (reviewed in [29, 30]). All TLRs, except for TLR3, recruit the adaptor protein myeloid differentiation primary response 88 (MyD88) [21]. MyD88 then recruits members of the IRAK family, which in turn recruit the E3 ubiquitin ligase TRAF6, which activates TAK1 [31, 32]. Activated TAK1 subsequently activates NF-κB and MAPK signaling pathways, resulting in the expression of inflammatory cytokines [32]. TLR3 does not recruit MyD88, but instead recruits TRIF to drive the TRIF-RIP1-TAK1 signaling pathway, similarly resulting in the activation of NF-κB and MAPK signaling [33–35]. In pDCs, TLR7 and TLR9 activation results in similar MyD88-dependent activation of NF-κB and MAPK signaling. However, MyD88 can also associate with IRAK family members and TRAF3 to activate IRF7, resulting in the robust production of type I IFNs [36–40]. TLR4 is unique in its ability to promote MyD88- and TRIF-dependent activation of NF-κB and MAPK signaling pathways [33, 34]. Furthermore, TLR3 and TLR4 activation can recruit TRIF to promote the activation of TBK1, resulting in the activation of IRF3 and the production of type I IFNs [33–35].
Multiple TLRs have been demonstrated to be involved in the detection of gammaherpesviruses (Figure 2). TLR2, which recognizes a diverse array of pathogen-associated molecular patterns (PAMPs), was shown to be activated by EBV [41]. Stimulation of HEK293 cells transfected with a construct encoding human TLR2 with infectious or UV-inactivated EBV resulted in significant increase in NF-κB activity. Furthermore, expression of the inflammation-associated chemokine MCP-1 was significantly increased in primary human monocytes stimulated with EBV, and this increase in expression was primarily dependent on TLR2 expression. In addition to EBV, MHV68 has also been demonstrated to activate TLR2 [42]. As with EBV, stimulation of TLR2 expressing HEK293 cells with MHV68 resulted in elevated NF-κB activity. Additionally, infection of TLR2−/− mice with MHV68 resulted in sub-optimal production of the inflammatory cytokines, IL-6, IFN-α, and IFN-β at early time points post infection, in an anatomic site-specific manner. Interestingly, while stimulation of TLR2−/− embryonic fibroblasts resulted in an approximate 2-fold decrease in IL-6 and IFN-α secretion, the absence of MyD88 completely abolished production of these cytokines, foreshadowing a role for other MyD88-dependent TLRs in the response to MHV68. Notably, TLR2 has previously been reported to be involved in the detection of alpha- and betaherpesviruses, suggesting that TLR2 may recognize features conserved across the herpesvirus family [43–45].
Fig. 2. Gammaherpesvirus infection can be sensed by multiple TLRs including TLRs 2, 3, 4, 7, and 9.

TLRs 2 and 4 are present on the plasma membrane and recognize viral lipids and proteins, while TLRs 3, 7, and 9 are intracellular sensors embedded within endosomal compartments and recognize viral DNA and RNA. Signaling through TLRs is mediated through the adaptor proteins TRIF (TLRs 3 and 4) or MyD88 (TLRs 2, 4, 7, and 9). TRIF recruits downstream proteins which lead to the activation and nuclear translocation of various IRFs, which transcribe type I IFNs. MyD88 recruits downstream proteins which lead to the activation and nuclear translocation of NF-κB, which transcribes proinflammatory and antiviral cytokines. TLRs 7 and 9 have been demonstrated to sense KSHV, EBV, and MHV68 infection, while TLRs 3 and 4 have been shown to be involved in KSHV detection and TLR2 has been shown to be involved in both EBV and MHV68 detection. TLR, Toll-like receptor; MyD88, myeloid differentiation primary response 88; TRIF, TIR domain-containing adaptor inducing IFN-β; NF-κB, nuclear factor kappa B; IRF, interferon regulatory factor. Adapted from “TLR Signaling Pathway” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
In addition to TLR2, TLR7 and TLR9 have been found to contribute to the detection of gammaherpesvirus infection (Figure 2). In contrast to TLR2, TLR7 and TLR9 are predominantly endosomal, expressed most robustly by B cells, macrophages, and dendritic cells (DCs), and recognize single-stranded RNA (ssRNA) and CpG DNA, respectively [23, 25, 46]. Interferon production by DCs, particularly plasmacytoid DCs (pDCs), is a crucial aspect of the innate immune response to gammaherpesvirus infection [47]. Interestingly, TLR7 and TLR9 are the only TLRs expressed at substantial levels in human pDCs [48], supporting the established role of these cells in the detection of viral pathogens. Indeed, TLR9 has been demonstrated to be involved in DC-mediated detection of MHV68 and is important for the early control of lytic replication; however, TLR9 does not impact long term latency [49]. In another model, TLR9 expression was involved in the protection from gammaherpesvirus-induced lung fibrosis, and required for robust IFN production in the lungs of MHV68 infected animals [50]. Furthermore, maximal IFN production by pDCs stimulated with MHV68 was found to require TLR9 expression, whereas TLR7 expression was dispensable [51]. Surprisingly, deficiency in both TLR7 and TLR9 resulted in a greater abrogation of IFN production than the loss of TLR9 alone, suggesting that TLR7 may contribute to gammaherpesvirus detection in some contexts. Another study expanded on these data, reporting that, while MyD88 expression is important for the optimal control of MHV68 in the peritoneal cavity, MyD88 expression supports latency and reactivation in the spleen [52]. Fascinatingly, by using chimeric animals, the authors found that MHV68 latency is preferentially established in MyD88-sufficient splenocytes, suggesting dual roles for the MyD88/IRAK/TRAF6 signaling pathway in the context of gammaherpesvirus infection. Finally, the TLR7/8 pathway was found to control KSHV reactivation from latency [10].
Consistent with the above findings in mice, TLR9 activity is required for robust IFN production by pDCs following stimulation with EBV and KSHV [53–55]. Chemical inhibition of TLR9 with inhibitory oligodeoxyribonucleotides dramatically reduced IFN production by pDCs following stimulation with EBV and KSHV [54, 55], suggesting that TLR9 is the primary TLR involved in the pDC detection of gammaherpesviruses. Interestingly, inhibition of TLR7 with the compound IRF 661 modestly reduced activation of pDCs by EBV [55], but showed no effect when combined with TLR9 inhibition, further suggesting that TLR9 is the primary innate sensor triggered in pDCs during gammaherpesvirus infection. Monocytes, which can differentiate into macrophages and a subset of DCs, were found to utilize both TLR9 and TLR2 in the detection of EBV [55]. Finally, TLR3, which is activated by dsRNA, is triggered during KSHV infection of THP-1 monocytes and is required for optimal expression of inflammatory cytokines and chemokines [56].
RIG-I-like receptors.
The RLRs are a family of PRRs capable of detecting foreign RNAs in the cytosol. Importantly, foreign RNAs often exhibit characteristics that allow for the differentiation of these RNAs from host. For example, the RLR melanoma differentiation-associated protein 5 (MDA5) recognizes long dsRNA with high strand complementarity, while the RLR RIG-I binds short, 5’ di- and triphosphorylated RNAs with base-paired ends, as well as dsRNA [57–61] (Figure 3). Fascinatingly, while gammaherpesviruses are dsDNA viruses that replicate in the nucleus, numerous reports have highlighted the relevance of these RNA sensors in the detection of gammaherpesvirus infection.
Fig. 3. Intracellular detection of viral nucleic acids can occur via RNA sensing by RIG-I and MDA5, or DNA sensing by cGAS.

RIG-I generally recognizes short, di- and triphosphorylated RNAs, as well as dsRNA, while MDA5 is activated by long dsRNA. Ligation of RIG-I or MDA5 by their respective ligands results in the activation of MAVS, which is associated with the mitochondria. The sensor cGAS is activated by foreign dsDNA and produces the second messenger cGMP to induce the activation of STING. Activated MAVS and STING can induce both ISG and inflammatory gene expression via activation of the TBK1/IRF3 signaling axis and the IKK/NF-κB signaling axis, respectively. RIG-I has been demonstrated to be activated upon infection with EBV, KSHV, and MHV68, while MDA5 is activated during KSHV reactivation. The cGAS/STING signaling pathway has been shown to be involved in the detection of KSHV and MHV68. RIG-I, retinoic acid-inducible gene I; MDA5, melanoma differentiation-associated protein 5; cGAS, cyclic GFP-AMP synthase; Cgmp, cyclic guanosine monophosphate; STING, stimulator of interferon genes; ISG, interferon stimulated gene; TBK, tank-binding kinase; IRF, interferon regulatory factor; NF-kB, nuclear factor kappa B; IKK, IκB kinase. Figure created using BioRender.com.
Early reports of RLR involvement in the sensing of gammaherpesviruses showed that overexpression of RIG-I resulted in elevated type I IFN expression in EBV+, but not EBV−, Burkitt lymphoma cell lines [62]. Similarly, knockdown of RIG-I significantly reduced inflammatory cytokine production following EBV reactivation from a gastric carcinoma cell line [63], demonstrating the bona fide relevance of RLRs in multiple EBV+ cell types. One mechanism proposed for the detection of EBV infection through RIG-I involves cellular sensing of EBV-encoded small RNAs (EBERS), the viral non-coding RNAs robustly expressed during all latency programs [62, 64]. Furthermore, RNA polymerase III, which transcribes non-coding RNAs such as tRNAs and 5s rRNA, also transcribes AT-rich DNA into dsRNA with a 5’ triphosphate capable of being detecting by RIG-I [65]. Importantly, EBERs are transcribed by RNA polymerase III, and this mechanism of transcription has been proposed to facilitate RIG-I mediated detection of EBV [65, 66]. Notably, EBV employs multiple mechanisms to subvert RLR activation, as discussed below, further highlighting the relevance of RNA sensors during infection with this DNA virus.
Similarly, cells lacking functional RIG-I were found to be more permissive to KSHV and MHV68 infection than cells expressing wild-type RIG-I [67]. RIG-I expression also attenuated KSHV lytic gene expression following chemical induction of viral reactivation. The adaptor protein mitochondrial antiviral signaling protein (MAVS), on which MDA5 and RIG-I converge to induce IFN and NF-κB signaling (Figure 3), was also found to be required for control of KSHV infection and reactivation [68, 69]. Fascinatingly, dsRNA was also shown to accumulate in the cytoplasm of cells reactivating KSHV [68], suggesting that RLRs are indeed activated by RNA ligands rather than through unconventional mechanisms. Unlike the EBV transcripts reported to be detected by RIG-I, one study reported that RNA polymerase III was dispensable for RIG-I-dependent sensing of KSHV [70]. Instead, it was found that certain regions of the KSHV genome, including protein coding regions, give rise to RNAs detected by RIG-I independent of RNA polymerase III activity. Interestingly, it has also been reported that RNA polymerase III-dependent transcription of host non-coding RNAs, such as vault RNAs, NOP14, and GINS1, can be sensed by RIG-I and MDA5 to activate MAVS and downstream interferon and NF-κB signaling pathways [69, 71]. Furthermore, infection with MHV68 and KSHV was found to result in an accumulation of these stimulatory host RNAs, providing another mechanism for the RLR-mediated sensing of gammaherpesviruses. ADAR1 expression was found to attenuate IFN production during KSHV reactivation by preventing RLR activation, and knockdown of ADAR1 correlated with an accumulation of GINS1 and NOP14 RNAs when reactivation was induced [72]. Taken together these data point to an unexpected mechanism by which cells sense gammaherpesvirus infection through the recognition of stimulatory viral and host RNAs.
DNA sensors.
Intracellular DNA can be recognized by several different sensors, resulting in the activation of the adaptor protein stimulator of interferon genes (STING) and subsequent transcription of IFNs and inflammatory cytokines (Figure 3). Two DNA sensors in particular, cyclic GMP-AMP synthase (cGAS) and interferon gamma inducible protein 16 (IFI16), have been implicated in the detection of gammaherpesviruses. The protein cGAS is a DNA sensor that binds dsDNA in a length-dependent manner, with longer DNA molecules driving more robust activation [73, 74]. Originally identified as a cytosolic DNA sensor, a plethora of recent studies have clearly demonstrated that cGAS also resides in the nucleus and shuttles back and forth between the nucleus and cytoplasm [75–78].
As dsDNA viruses, gammaherpesviruses would logically need to combat recognition by DNA sensors such as cGAS. Indeed, three independent laboratories demonstrated that the cGAS/STING pathway can sense KSHV [79–81]. Knockdown of cGAS or STING significantly augmented KSHV reactivation, suggesting a critical role for this DNA sensing pathway in the maintenance of KSHV latency.
STING activity is tightly regulated through posttranslational modifications, and chronic STING activation is associated with several autoinflammatory diseases. One such regulatory mechanism involves protein phosphatase 6 catalytic subunit (PPP6C), which interacts with STING and removes activating phosphate groups. As expected, knockdown of PPP6C resulted in greater induction of type I IFNs, inhibition of herpes simplex virus 1 (HSV-1) and vesicular stomatitis virus (VSV) replication, and reduced KSHV reactivation [82].
Extracellular vesicles contain cellular constituents such as proteins, lipids, and nucleic acids and are released from healthy and cancerous cells to facilitate intercellular communication. Extracellular vesicles isolated from KSHV-infected endothelial cells were reported to induce a robust IFN signature when added to uninfected endothelial cells through a mechanism requiring cGAS and STING [83]. Fascinatingly, extracellular vesicles purified from KSHV+ PEL cell lines and patient samples were shown to induce dramatic cellular reprogramming of endothelial cells but had no effect on IFN-β expression or STING activation [84]. Thus, it is possible that cell type-specific differences in extracellular vesicle cargo may impact innate immune pathway activation during gammaherpesvirus infection.
The cGAS/STING pathway was also found to be relevant to in vivo infection with MHV68. Viral titers were modestly increased in the spleens and lungs of cGAS-deficient mice infected with MHV68, confirming the importance of cGAS/STING signaling in the context of gammaherpesvirus infection of an intact, otherwise immunocompetent host [85]. Furthermore, mesenchymal stem cells were demonstrated to be permissive to MHV68 infection in vitro and in vivo, and were capable of activating cGAS/STING-dependent signaling following stimulation with MHV68 DNA [86]. While de novo MHV68 infection of mesenchymal stem cells did not induce a measurable increase in IFN signaling, possibly due to viral antagonism of host sensing, chemical activation of the cGAS/STING pathway significantly attenuated MHV68 infection.
Notably, research into the role of DNAs sensors, such as cGAS, in the context of EBV infection is scarce. While studies of KSHV often make use of endothelial cells (the cell type that gives rise to KS), fibroblasts, myeloid cells, and B cells, most studies of EBV infection and reactivation have been restricted to B cells. Fascinatingly, uninfected B cells were found to lack detectable STING expression [87]. Furthermore, while EBV-infected B cells do express cGAS and STING, stimulation of a STING-expressing lymphoblastoid cell line (LCL) with dsDNA failed to induce IFN production. Additionally, reconstitution of STING in EBV-negative B cell lines was unable to rescue IFN production downstream of dsDNA stimulation. This is in contrast to murine B cells, which do produce IFNs in response to STING agonists [88]. Hence, while EBV-infected B cells may express cGAS and STING, it is possible that the virus somehow inhibits activation of this pathway in B cells. Importantly, the role of cGAS/STING signaling in the context of EBV infection of other cell types has not been fully characterized.
As mentioned above, human and murine cells express multiple sensors of DNA. In addition to cGAS, the DNA binding protein IFI16 has been implicated in the detection of gammaherpesvirus DNA. Predominantly a nuclear protein, IFI16 is thought to play a role in the sensing of nuclear herpesvirus genomes but can shuttle between the nucleus and cytosol. Considerable crosstalk is believed to occur between IFI16 and cGAS [89–91], but IFI16 has also been reported to interact with STING directly to drive the production of type I IFNs [92, 93]. Furthermore, IFI16 is capable of activating inflammasome complexes, leading to the production of IL-1β and IL-18 [94, 95].
KSHV infection of endothelial cells was shown to interact with apoptosis-associated speck-like protein containing a CARD (ASC) to promote the activation of caspase-1 and the resulting maturation of pro-IL-1β to IL-1β [95]. IFI16 colocalized with both ASC and the KSHV nuclear episomes, and knockdown of IFI16 attenuated inflammasome activation upon KSHV infection. Upon recognition of KSHV DNA, IFI16 was postulated to associate with ASC and procaspase-1 and translocate to the cytosol where the inflammasome complex becomes activated and forms perinuclear aggregates. Furthermore, endothelial cells and B cells latently infected with KSHV were found to exhibit constitutive IFI16-dependent inflammasome activation [96]. Alphaherpervirus infection has been shown to initiate IFI16-dependent activation of STING [97], and while de novo KSHV infection induces a lower degree of IFN signaling, this mechanism of IFI16-dependent IFN induction has also been purported to occur during KSHV infection [98, 99]. Further characterization of IFI16-mediated detection of KSHV demonstrated that p300-dependent acetylation of IFI16 occurs in the nucleus of KSHV-infected cells, and that acetylation is required for proper cytoplasmic translocation of the IFI16-containing inflammasome complex and for activation of STING by IFI16 [98]. The tumor suppressor BRCA1 has been reported to participate in the IFI16 inflammasome, promote p300-mediated acetylation of IFI16, and augment IFI16-driven antiviral responses [99]. Furthermore, interaction of histone H2B with the IFI16-BRCA1 complex has been demonstrated to skew the consequence of IFI16-mediated recognition of viral DNA towards IFN production, rather than inflammasome activation, via cytoplasmic interactions with cGAS and STING [100].
As observed with KSHV, constitutive inflammasome activation was also reported in B cells and epithelial cells latently infected with EBV [101]. Notably this activity was observed in cell lines comprising each of the three predominant EBV latency states. Furthermore, while this study did less to characterize the mechanisms of EBV detection, the authors demonstrated co-localization of IFI16 with the EBV genome in latently infected B cells. Further studies demonstrated that knockdown of IFI16 resulted in a significant increase in EBV gene expression and replication in latently infected B cells [102]. Thus, based on a limited number of published studies, detection of EBV through DNA sensors appears to operate similarly to the detection of other herpesviruses.
Current challenges.
Our cumulative understanding of how gammaherpesviruses are detected and restricted by the innate immune system has advanced significantly over the last 10–15 years. The recent discoveries of numerous PRRs relevant to gammaherpesvirus infection has paved the way for novel approaches to target EBV− and KSHV-driven diseases, and new tools such as CRISPR editing and humanized mouse models have allowed for cleaner and more thorough characterization of innate immune system components in the context of gammaherpesvirus infection. Despite these advances, further research is needed to fill gaps in the dogma of how these viruses are controlled by innate immune sensors and cells. For example, despite the breadth of knowledge that has been garnered regarding DNA sensor activation during KSHV infection, relatively little is known about how EBV may be sensed by receptors such as cGAS and IFI16. Additionally, the activation of RNA sensors such as TLR3 and RLRs during dsDNA virus infection is an interesting observation that deserves further characterization, particularly in the context of an intact host. Furthermore, while EBV and KSHV are generally considered lymphotropic, numerous cell types are permissive to infection with these viruses. EBV infection of other cell types is biologically relevant and EBV+ non-B cell cancers are a growing problem in some countries, including the United States [103, 104]. As such, cell type-specific responses to infection need to be more fully characterized.
Advanced in vitro tools allow for the careful analysis of viral recognition during gammaherpesvirus reactivation and have been used to describe the cellular and molecular mechanisms by which reactivation is sensed and restricted. However, these approaches are often limited to particular cell lines, may utilize cell lines irrelevant to gammaherpesvirus biology, or may be performed in cell lines with dysfunctional innate immune pathways. While de novo infection of primary cells provides a more biologically relevant model in which to study innate immune recognition, it can be technically challenging and is not always amenable to the advanced molecular techniques that can be performed in immortalized or transformed cell lines. Tools such as humanized mice and the MHV68 model of gammaherpesvirus infection offer unique and tractable approaches to examine the relative importance of innate immune constituents in the context of an intact host. Advances in humanized mouse models of gammaherpesvirus infection have allowed for elegant studies of a human immune response to EBV and KSHV. The data generated from such studies highlights the clear importance of NK cells in the control of EBV lytic replication [105], further characterizing what has been postulated based on studies of human tonsils and PBMCs [106–109]. The importance of DCs has been similarly highlighted from these studies [47]. Furthermore, the MHV68 system enables the use of murine genetics to study cell type-specific responses to infection and allows for the most delicate in vivo experiments of gammaherpesvirus biology. Thus, through more thorough characterization of cell type-specific responses, the development of biologically relevant in vitro systems, and complimentary approaches using humanized and/or biologically related mouse models, a more complete picture of the innate immune recognition of gammaherpesvirus infection can be described.
Evasion of innate immunity by gammaherpesviruses
Gammaherpesviruses, like many viruses, have evolved multiple mechanisms by which to evade the various host innate immune sensors discussed above. KSHV, EBV, and MHV68 each encode multiple viral proteins that counteract the ability of these sensors and their downstream targets to mount an effective response against infection, thus helping to ensure successful viral propagation (summarized in Table 1). Some of these viral proteins are homologues of cellular innate immune proteins such as IRFs and cytokines, and can interfere with cellular functions. Additionally, these viruses can hijack host cell proteins such as phosphatases to dampen innate immune signaling, thus reducing the ability of an infected cell to establish an antiviral state and subsequently limiting viral detection and clearance. In the following section, we discuss strategies each of these gammaherpesviruses use to limit the innate immune response to viral infection.
Table 1.
Gammaherpesvirus innate immune evasion proteins and microRNAs
| Protein | Description | Evasion function |
|---|---|---|
|
| ||
| KSHV | ||
| ORF45 | Immediate-early tegument protein | Inhibits IRF7; negatively regulates cGAS-STING |
| K-bZIP (K8) | Late nuclear protein | Binds to IFN-β promoter and inhibits IRF3 activity |
| RIF (ORF10) | Late protein | Inhibits type I IFN production |
| ORF64 | Large tegument protein | Decreases RIG-I ubiquitination |
| LANA (ORF73) | Latency regulator | Decreases IFN-β production; negatively regulates cGAS-STING |
| RTA (ORF50) | Lytic transactivator | Lowers MyD88 expression; inhibits IRF7 expression; decreases TRIF stability and TLR3 signaling |
| ORF52 | Late tegument protein | Inhibits cGAS; inhibits IRF3 activation |
| ORF54 | Viral dUTPase | Inhibits type I IFN activity |
| vIRF1 | Homologous to cellular IRFs | Decreases IRF3 activity; inhibits MAVS; inhibits TLR3 signaling; negatively regulates cGAS-STING |
| vIRF2 | Homologous to cellular IRFs | Inhibits IRF1 and IRF3; inhibits TLR3 signaling |
| vIRF3 | Homologous to cellular IRFs | Inhibits IRF7; inhibits TLR3 signaling |
| vIRF4 | Homologous to cellular IRFs | Prevents IRF7 dimerization |
| ORF63 | Homologous to cellular NLRP1 | Inhibits inflammasome activity |
| vIL-6 | Homologous to cellular IL-6 | Blocks anti-proliferative effects of IFN signaling |
| ORF36 | Viral protein kinase | Negatively regulates cGAS-STING; inhibits IFN-β activity |
| miR-K9 | Viral microRNA | Inhibits IRAK1 and MyD88 |
| miR-K5 | Viral microRNA | Inhibits MyD88 |
| EBV | ||
| BGLF5 | Exonuclease | Downregulates TLR9 |
| BARF1 | Oncoprotein | Reduces IFN-α production |
| EBNA1 | Latent and lytic protein involved in viral genomic maintenance | Inhibits NF-κB signaling |
| BGLF2 | Tegument protein | Decreases phosphorylation of NF-κB subunits |
| BPLF1 | Large tegument protein; deubiquitinase | Downregulates TLR signaling; deubiquitinates TRAF6 |
| BZLF1 (ZTA) | Lytic transactivator | Inhibits IRF7; activates SOCS proteins |
| BRLF1 (RTA) | Lytic transactivator | Downregulates IRF3 and IRF7; decreases IFN-β levels |
| BILF4 (LF2) | Tegument protein | Prevents IRF7 dimerization |
| BFRF1 | Early protein | Inhibits IRF3 and production of IFN-β |
| LMP-1 | Latent membrane oncoprotein | Downregulates TLR9; reduces Tyk2 and STAT2 phosphorylation |
| LMP-2 | Latent membrane oncoprotein | Decreases STAT3 activation; reduces STAT1, JAK, and Tyk2 phosphorylation; degrades intracellular IFNAR |
| BHRF1 | Anti-apoptotic protein | Induces fission of mitochondria and degradation of MAVS |
| BGLF4 | Viral protein kinase | Inhibits IRF3; reduces STAT1 phosphorylation; decreases NF-κB signaling |
| miR-BART15 | Viral microRNA | Decreases NLRP3 expression |
| miR-BART16 | Viral microRNA | Inhibits IFN-α signaling and ISG production |
| miR-BART6-3p | Viral microRNA | Downregulates RIG-I and IFN-β |
| MHV68 | ||
| ORF36 | Viral protein kinase | Inhibits transcription of IFN-β |
| ORF64 | Large tegument protein | Inhibits IFN signaling |
| ORF54 | Viral dUTPase | Inhibits type I IFN activity; decreases expression of IFNAR |
| RTA (ORF50) | Lytic transactivator | Ubiquitinates NF-κB subunits, resulting in their degradation |
| ORF11 | Late protein | Prevents the interaction of TBK1 and IRF3 |
KSHV.
As mentioned above, KSHV encodes proteins that have been shown to directly interact with innate immune signaling components and thus dampen the host antiviral response. One of these proteins, ORF45, interacts with and inhibits IRF7 activation which, in turn, prevents IRF7 translocation to the nucleus and the transcription of IFN-α and IFN-β [110]. Later experimentation by the same group suggested that ORF45 achieves this IRF7 inhibition by binding IRF7 and inducing a conformational change that renders IRF7 inactive [111]. Another KSHV protein, K-bZIP, binds to the IFN-β promoter and induces low levels of IFN-β production. Although counterintuitive, the results of this K-bZIP/IFN-β interaction were shown to decrease IRF3 function and impair downstream IRF3 chemokines such as CXCL11 and RANTES [112]. Additionally, the KSHV protein RIF (ORF10) binds to IFN signaling components and inhibits the type I IFN response [113], while the large tegument protein of KSHV, ORF64, inhibits RIG-I-mediated IFN production by decreasing ubiquitination of RIG-I [67]. Finally, LANA, a KSHV protein responsible for latency maintenance, was shown to significantly reduce IFN-β production at both the mRNA and protein level, and this was mediated, at least in part, by preventing IRF3 binding to the IFN-β promoter region [114].
The KSHV latent-to-lytic switch protein, RTA (ORF50), also possesses immunomodulatory properties and suppresses expression of several host innate immune RNAs and proteins. RTA decreases MyD88 transcripts in vitro by slowing down MyD88 RNA synthesis [115] and decreases MyD88 protein levels in cells by targeting MyD88 for proteasomal degradation. This reduction in MyD88 protein is achieved by an inherent E3 ubiquitin ligase activity of RTA, which polyubiquitinates MyD88 for proteasomal trafficking. Furthermore, this RTA-mediated degradation of MyD88 leads to downregulated TLR4 signaling and decreased activation of type I IFN and NF-κB signaling [116]. These data echo the findings of previous studies, which demonstrated that KSHV downregulated TLR4 and several components of the TLR4 signaling pathway, including MyD88, IRAK1, and downstream pro-inflammatory cytokines such as IL-6 and IL-8 [117, 118]. Additionally, endothelial cells were more permissible to KSHV infection in the absence of TLR4 and, conversely, were protected from KSHV infection when TLR4 was exogenously stimulated [118]. In a similar fashion to the above ubiquitination of MyD88, RTA also targets IRF7 for proteasomal degradation through its E3 ubiquitin ligase activity, resulting in decreased levels of IFN-α and IFN-β transcripts [119]. Lastly, RTA targets the adaptor molecule TRIF. RTA modulates both the expression and stability of TRIF, leading to decreased TLR3 signaling. The mechanism was shown to involve proteasomal degradation of TRIF, but whether RTA itself or another cellular E3 ligase mediated the ubiquitination of TRIF was unclear [120].
Complementing the above arsenal of immunomodulatory proteins are the viral IRFs (vIRFs). vIRFs are homologous to cellular IRFs and act to suppress the functions of host IRFs. vIRF1 interrupts IRF3 signaling by binding to the transcriptional cofactors CBP/p300 and preventing IRF3-mediated transcription, thus disrupting downstream antiviral protein production [121]. vIRF1 also traffics to the mitochondria, where it interacts with MAVS and hinders MAVS-dependent immune responses such as IFN-β production and apoptotic activity [122]. vIRF2 inhibits both IRF1 and IRF3 functions, suppressing IFN signaling and downstream IFN targets [123]. Interestingly, the mechanism underlying this observed vIRF2-mediated IRF3 inhibition was shown to involve caspase-3. Caspase-3 helps to regulate IRF3 levels within the cell and can mediate IRF3 degradation. vIRF2 can enhance this caspase-3-mediated IRF3 degradation process, facilitating more rapid turnover of IRF3 and a subsequent dampening of the cellular innate immune response [124]. vIRF3 binds to IRF7 and decreases IFN-α transcription by disrupting the ability of IRF7 to associate with DNA [125], while vIRF4 prevents dimerization of IRF7 [126]. Interestingly, vIRF1–3 can inhibit TLR3 signaling, but only vIRF1 and vIRF2 inhibit TLR3-mediated IFN-β and CXCL10 production. Furthermore, vIRF1 and vIRF2 appear to achieve this IFN-β downregulation via differing mechanisms, as only vIRF1 was shown to modulate IRF3 phosphorylation and nuclear accumulation [127]. A subsequent report revealed that vIRF1 associates with the cellular ISG15 E3 ligase, HERC5, which led to decreased ISG15 activity and a corresponding loss in IRF3 protein [128]. Thus, vIRFs collectively act to suppress type I IFN signaling and ISG activity.
KSHV also encodes proteins other than vIRFs that are homologous to cellular immune factors. KSHV ORF63 was the first described protein from a human virus with homology to NLRs. ORF63, homologous to NLRP1, blocks both NLRP1- and NLRP3-mediated inflammasome activity and inhibits IL-1β production as well as activation of caspase-1 [129]. Additionally, expression of vIL-6, a KSHV-secreted cytokine similar to cellular IL-6, is upregulated following viral exposure to IFN-α. vIL-6 blocks the anti-proliferative effects of IFN signaling, allowing for survival and persistence of virally-infected cells despite the presence of IFN-α in the cellular environment [130]. Thus, gammaherpesviruses have evolved to evade host detection by exploiting host cell protein sequences for their own advantage.
As mentioned in the previous section, the cGAS-STING pathway is another mechanism by which host cells detect gammaherpesvirus infection. Accordingly, KSHV encodes several proteins that modulate cGAS-mediated sensing of viral DNA. KSHV ORF52 binds to cGAS and blocks its enzymatic activity, subsequently inhibiting downstream IRF3 activation [80]. Interestingly, other gammaherpesvirus homologues of ORF52, including those of EBV and MHV68, were shown to possess this same anti-cGAS/IRF3 functionality. Additionally, after identifying cGAS-STING as a KSHV sensor, Ma et al. performed a screen to identify KSHV proteins that might function to counteract cGAS-STING signaling. Results of the screen identified several hits, including ORF36 (the KSHV viral protein kinase which functions similarly to cellular serine/threonine kinases), ORF45, ORF55, ORF57, ORF73, and vIRF1, as negative regulators of the cGAS-STING pathway. All six proteins inhibited cGAS-STING-mediated IFN-β production, and vIRF1 was further shown to block phosphorylation of STING by TBK1 [79]. Thus, these data collectively reveal a multi-pronged approach employed by KSHV to circumvent sensing of foreign DNA by the host cell.
As exemplified above, virally-encoded proteins are widely used by KSHV to counteract immune sensing. Interestingly, however, cellular proteins can also be utilized by KSHV as a defense mechanism against host innate immunity. Two recent studies by Ni et al. and Yu et al. elucidated a novel mechanism by which KSHV utilizes cellular phosphatases to evade the innate immune response [82, 131]. The cGAS-STING regulator screen performed by Ma et al. also uncovered ORF48 to be involved in cGAS-STING signaling inhibition. A successive study found ORF48 to interact with the host protein phosphatase PPP6C. Further experimentation revealed PPP6C to inhibit IRF3, but not NF-κB, activation and to reduce IFN-β production by dephosphorylation of STING [82]. However, the exact role ORF48 plays in this mechanism beyond interaction with PPP6C remains to be elucidated. Similarly, the KSHV protein ORF33 was shown to interact with PPM1G, a host cell phosphatase capable of dephosphorylating STING and MAVS. This ORF33-PPM1G-mediated suppression of STING and MAVS activation resulted in decreased production of IFN-β, inhibition of IRF3 activation, and reduced transcription of downstream target genes such as ISG56 [131]. The host NLR, NLRX1, was likewise shown to suppress JAK/STAT signaling and IFN-β transcription, leading to increased KSHV replication, potentially through association with a yet-unidentified KSHV protein [132]. Finally, upon infection of endothelial cells, KSHV upregulates the host protein heme oxygenase-1, which produces the TLR4 signaling inhibitor carbon monoxide as a catalytic byproduct [133]. Together, these data demonstrate innovative mechanisms by which host cell machinery can be successfully hijacked by KSHV to impair immune responses.
EBV.
Like KSHV, EBV also possesses a wide array of viral proteins used to subvert innate immunity. Several of these proteins are conserved across gammaherpesviruses, and the KSHV and MHV68 homologues for these EBV proteins can be found in Table 2. The EBV exonuclease BGLF5 targets host immune signaling by depleting TLR9 mRNA levels, leading to a subsequent reduction in TLR9 protein expression in the cell [134]. Another EBV protein, LMP-1, which will be discussed more below, also targets TLR9 at the transcriptional level through an NF-κB-dependent mechanism, leading to reduced TLR9 expression [135]. Additionally, the EBV oncoprotein BARF1 can reduce IFN-α production in human monocytes [136], and EBV EBNA1 decreases NF-κB signaling in cancer cells by preventing phosphorylation and nuclear accumulation of multiple key proteins involved in perpetuating the NF-κB signaling cascade [137]. Similarly, the EBV tegument protein BGLF2 blocks phosphorylation of the p65 NF-κB subunit, leading to decreased NF-κB signaling [138]. Finally, EBV can inhibit TLR signaling through the function of the viral large tegument protein BPLF1, which possess deubiquitinase (DUB) activity. Interestingly, BPLF1 can negatively regulate TLR signaling through both the MyD88 and TRIF adaptor proteins and, mechanistically, this observed TLR downregulation was linked to DUB activity of BPLF1 through mutation studies [139]. Furthermore, BPLF1 targets TRAF6 for deubiquitination, leading to suppression of the NF-κB pathway and enhanced viral replication [140]. As protein ubiquitination serves to perpetuate the signaling cascade in TLRs as well as other antiviral receptor signaling pathways, viral deubiquitinases can therefore serve as innate immune defense modulators.
Table 2.
KSHV and MHV68 homologues of EBV innate immune evasion proteins
| EBV | KSHV | MHV68 |
|---|---|---|
|
| ||
| BGLF5 | ORF37/SOX | muSOX |
| BARF1 | ORF60 | ORF60 |
| EBNA1 | LANA | mLANA |
| BGLF2 | ORF33 | ORF33 |
| BPLF1 | ORF64 | ORF64 |
| BRLF1 (RTA) | ORF50 | ORF50 |
| BFRF1 | ORF67 | ORF67 |
| BHRF1 | ORF16/vBcl-2 | vBcl-2 |
| BGLF4 | ORF36 | ORF36 |
In addition to the above TLR and NF-κB modulation, EBV has also been shown to modulate host IRFs through various mechanisms. BZLF1 (also known as ZTA), one of the first viral proteins expressed upon EBV infection, inhibits the antiviral effects of IRF7. Studies to date suggest that this IRF7 modulation by BZLF1 can be either direct or indirect. One study demonstrated that, although BZLF1 and IRF7 did not directly interact, IRF7 activation was indirectly modulated by BZLF1 through activation of suppressor of cytokine signaling (SOCS) proteins. Specifically, BZLF1 upregulated the expression of SOCS3 which, in turn, led to inhibition of IFN-α production [141]. This decrease in IFN-α can then reduce further activation of IFNAR and its downstream signaling, leading to an inhibition in IRF7 activation. However, another study showed that BZLF1 directly binds IRF7 in vitro and not only represses IRF7 activation by RNA substrates, but also decreases IRF7-mediated type I IFN and downstream ISG production [142]. This work was expanded upon when another EBV immediate-early protein, BRLF1, was shown to downregulate IRF3 and IRF7 protein levels in infected cells, which correlated with an observed decrease in IRF3 and IRF7 mRNA levels in transfected cells [143]. Additionally, BRLF1 decreased IFN-β production as well as IFN-β promoter activity in transfected cells, echoing the repressed type I IFN response mediated by BZLF1. Similarly, the EBV tegument protein BILF4 (LF2) was identified as an IRF7 regulator, binding to IRF7 and preventing its dimerization and subsequent induction of IFN-α [144]. Finally, the EBV early protein BFRF1 was recently shown to negatively regulate IRF3 activation and IFN-β transcription [145]. Altogether, these data demonstrate how EBV has evolved to evade the antiviral effects of IRF signaling through the actions of multiple viral proteins.
The EBV latent membrane oncoproteins, LMP-1 and LMP-2, promote oncogenesis in part by downregulating the innate immune response in host cells. LMP-1 directly associates with the IFN signaling protein Tyk2, reducing phosphorylation of not only Tyk2 but also STAT2 and dampening the signaling propagation of IFN-α [146]. LMP-2A modulates the NF-κB pathway, decreasing activation of STAT3 and the production of the pro-inflammatory cytokine, IL-6 [147]. Follow-up experimentation revealed that both LMP-2A and LMP-2B reduced phosphorylation of STAT1, JAK, and Tyk2, and lessened transcription of ISGs. Interestingly, this observed IFN pathway modulation was shown to be mediated by LMP-2A/2B facilitating the degradation of intracellular, but not cell surface, IFNAR [148]. Thus, LMP-1 and LMP-2 interfere with type I IFN signaling which can aid in the establishment of EBV latency and persistence in host cells.
EBV can also utilize tripartite motif (TRIM) proteins, which are E3 ubiquitin ligases, for immune escape and host cell persistence. Xing et al. showed that TRIM29 expression was elevated in EBV-positive NPC tissue compared to healthy tissue, and that knockdown of TRIM29 in the context of EBV infection resulted in increased type I IFN production and decreased EBV replication. Furthermore, TRIM29 interacts directly with STING, mediating the degradation of STING via ubiquitination [149]. Conversely, stifling of TRIM protein activity can also potentiate viral activity. EBV BPLF1 negatively regulates TRIM25, preventing its activating interaction with RIG-I and resulting in suppressed IFN signaling. Mechanistically, BPLF1 was shown to modulate the ubiquitination of TRIM25 and cause TRIM25 protein aggregation, resulting in ligase inhibition [150].
A recent study on the viral anti-apoptotic protein BHRF1 revealed an intriguing mechanism by which EBV circumvents RIG-I- and MDA5-mediated signaling through the adaptor protein MAVS. Instead of targeting RIG-I or MAVS for degradation or negatively interacting with their downstream signaling proteins, BHRF1 targets the mitochondrial organelles themselves. BHRF1 expression was shown to induce fission of mitochondria which eventually led to their degradation and, along with it, the degradation of MAVS. This sequentially resulted in a block in IFN-β signaling and IRF3 activity [151]. These data provide interesting insight into how viruses can evolve to escape immune recognition by attacking physical structures of host cells that are important for mediating antiviral responses.
The EBV viral protein kinase, BGLF4, has been shown to counteract the innate immune response through several distinct mechanisms. First, BGLF4 inhibits IRF3 signaling through its kinase activity. Phosphorylation of IRF3 at certain sites can lead to protein degradation rather than activation, and BGLF4-mediated phosphorylation of IRF3 at Ser123/173 and Thr180 results in a decrease in IRF3-specific IFN-β promoter binding [152]. Additionally, BGLF4 reduces phosphorylation of STAT1 at Tyr701 and suppresses IFN-β promoter activity. Finally, BGLF4 targets NF-κB signaling by phosphorylating the host protein ubiquitously expressed transcript (UXT), which is involved in the nuclear functions of NF-κB. BGLF4-mediated phosphorylation of UXT at Thr3 was shown to reduce UXT’s interaction with NF-κB, resulting in decreased NF-κB effector functions and an enhancement of the lytic cycle of EBV [153]. These studies underscore the utility of a viral protein kinase in the context of innate immune evasion and, altogether, these above reports echo the immunomodulatory properties described above for KSHV-encoded proteins.
In addition to viral proteins, viral microRNAs (miRNAs) can also be used to circumvent host innate immunity. The EBV miRNA miR-BART15 binds to the 3’ end of NLRP3, decreasing NLRP3 protein expression as well as IL-1β production [154]. Furthermore, the EBV miRNA miR-BART16 inhibits IFN-α signaling and the expression of downstream proteins such as IFIT1 and ISG15 [155]. Mechanistically, miR-BART16 was shown in the same study to bind the transcriptional cofactor CBP, negatively regulating CBP expression and resulting in increased cellular proliferation in the context of suppressed IFN signaling. Additionally, EBV miR-BART6–3p downregulates RIG-I and IFN-β as well as multiple immunomodulatory proteins that perpetuate these signaling pathways and their downstream targets [156]. KSHV similarly utilizes miRNAs to target STAT signaling and the production of ISGs [157], as well as to mediate the downregulation of several TLR4 signaling components as mentioned earlier. Specifically, KSHV miR-K9 targets IRAK1, leading to diminished NF-κB signaling, while KSHV miR-K9 and miR-K5 jointly target MyD88, leading to a reduction in pro-inflammatory cytokine levels [117]. Overall, as exemplified above, human gammaherpesviruses have evolved multiple mechanisms involving host proteins, viral proteins, and viral miRNAs to avoid immune surveillance and persist long-term in the host. Accordingly, these proteins and miRNAs can represent therapeutic targets for human gammaherpesvirus infection and their associated malignancies, and this possibility will be discussed in a later section.
MHV68.
As a model for gammaherpesvirus infection and pathogenesis, the findings of MHV68 immune evasion can potentially be applied to human gammaherpesviruses as well. Using a mutant library, Hwang et al. identified ORF36 as an anti-IFN MHV68 protein. The authors demonstrated that ORF36 interacted with IRF3 and that this binding interrupted IFN-β promoter activity [158]. Like KSHV, ORF36 is the MHV68 viral protein kinase. The authors went on to test the MHV68 ORF36 homologues of KSHV, EBV, human cytomegalovirus (HCMV), and HSV, and found that these proteins also interfered with IFN-β promoter activity, suggesting a common innate immunity defense across herpesviruses. The MHV68 large tegument protein, ORF64, contains DUB activity, a feature which is also conserved across herpesviruses. This DUB activity was shown to be important for suppressing type I IFN signaling upon MHV68 infection, as well as for the establishment of latency [159]. This inhibition of IFN production by MHV68 ORF64 is reminiscent of the observed inhibition of RIG-I-mediated IFN production by KSHV ORF64, as described above [67]. Finally, the MHV68 protein ORF54 is a viral dUTPase, an enzyme that is conserved across many different viruses. MHV68 ORF54, as well as KSHV ORF54, were shown to inhibit type I IFN activity. Interestingly, this type I IFN inhibition was shown to occur independently of dUTPase enzymatic activity, suggesting two distinct and unrelated roles of MHV68 ORF54 in the viral lifecycle [160]. Mechanistically, ORF54 achieved this IFN modulation by decreasing expression of IFNAR, which then led to decreased levels of phosphorylated STAT1. Notably, MHV68 ORF54 was shown in the same study to be necessary for the successful establishment of the latent phase of the viral lifecycle. Thus, evasion of type I IFN signaling is critical not only for innate immune escape but also for latency establishment, two hallmarks of herpesvirus infection.
MHV68, similar to KSHV and EBV, employs additional mechanisms by which to escape antiviral NF-κB signaling and the actions of type I IFN. MHV68 subverts antiviral cytokine production by targeting p65/RelA. MHV68 infection increases the activity of the kinase IKKβ, which then phosphorylates p65/RelA at Ser468, resulting in proteasomal degradation of p65/RelA and decreased cytokine production [161] (see Figure 1). Interestingly, this observed phenotype was dependent on MAVS, as cells and animals lacking MAVS exhibited increased cytokine production following MHV68 infection. However, the precise roles that MHV68 proteins hold in mediating this observed disruption of NF-κB signaling is unclear. A follow-up study sought to answer this question using an NF-κB reporter screen consisting of over twenty MHV68 proteins, and identified RTA (ORF50) as a sole hit. Further experimentation revealed that RTA directly interacted with p65/RelA and mediated its ubiquitination via an E3 ligase domain, which subsequently targeted p65/RelA for degradation by the proteasome [162]. Finally, MHV68 ORF11 was shown to interact with TBK1, leading to disrupted TBK1/IRF3 interactions and a functional decrease in downstream IRF3 signaling, including decreased IFN-β production [163].
Not all innate immune evasion presents as a battle between virus and host. Mandal et al. made the surprising discovery that MHV68 senses type I IFN concentrations in the host cell and, when elevated, undergoes transcriptional repression to enforce latency establishment. The MHV68 latent gene M2 was found to contain an ISRE capable of being bound by IRF2. Increased IFN-α/β signaling promotes activation of IRF2, which can then bind the ISRE present in the M2 promoter, resulting in decreased M2 transcription and decreased viral reactivation [164]. The authors speculate that this IFN sensing provides an advantage to MHV68, allowing for reversion to active viral replication when the levels of IFN-α/β are low in the cell and thus conducive to the production of progeny virions. Given the presence of ISREs in EBV latency promoters [165] and the ability of KSHV to sense IFN-α levels within the host cell [130], this observed cooperation between MHV68 and type I IFN signaling could very well be conserved across gammaherpesviruses.
Lastly, NLRs can also be targeted by MHV68. Upon observing that deletion of caspase-1 and caspase-11 from mice did not affect MHV68 pathogenesis, Cieniewicz et al. examined whether MHV68 infection downregulated the inflammasome response. Results showed that MHV68 inhibited transcription of IL-1β, and that this inhibition was mediated by a protein other than RTA [166].
In addition to the above discussed mechanisms of gammaherpesvirus innate immune evasion, these viruses, as well as all herpesviruses, inherently evade the host immune response by nature of their lifecycle. Although the detailed mechanisms of latency are beyond the scope of this review, the latent phase of the herpesvirus lifecycle effectively prevents sustained detection of the virus by the immune system. These viruses maintain long-term persistence in hosts by replicating their DNA alongside cellular DNA using host cell machinery, and only produce a limited subset of genes needed to maintain viral persistence during this time. By establishing these latency programs, herpesviruses can intrinsically avoid immune surveillance while successfully preserving their genome.
Current challenges.
Like gammaherpesvirus recognition by the innate immune system, our understanding of how gammaherpesviruses evade host immune detection has considerably increased over the last 15 years. We now understand at the mechanistic level the roles that many gammaherpesvirus proteins play in subverting the initial host cell response to infection. However, most of these studies were performed in cells, either in the context of transfection or viral infection, and our knowledge of how these viral proteins function in the context of an infected human and the subsequent implications of this for disease progression remains severely lacking. The use of MHV68 and accompanying mouse models as a proxy for human gammaherpesvirus infection and pathogenesis have yielded important insights into this understudied area, but much more work is needed. Recently advanced humanized mouse models for KSHV and EBV infection are one tool that can be utilized to help address these questions [167–169]. As will be discussed in more detail in the next section, the inability of the immune system to fully clear KSHV and EBV infection, in large part due to the plethora of immune evasion mechanisms these viruses have evolved, is a major driver of KSHV− and EBV-associated malignancies, including cancer. Although we have a solid baseline understanding of how the innate immune system detects invasion of these viruses and the viral clearance mechanisms involved, a more detailed understanding of the interplay between innate immunity and gammaherpesvirus infection, establishment of latency, and development of lymphoproliferative disorders can help guide future therapeutics for these diseases.
Clinical relevance of innate immunity to gammaherpesvirus infection
Infection with gammaherpesviruses, like all herpesviruses, is lifelong, in part due to long-term maintenance of the viral genome in host cells and an inability of the immune system to detect and clear the virus while it is maintained in the latent phase. There are several steps of the viral life cycle that can be targeted in an effort to deter herpesvirus persistence in host cells, including initial infection and latency establishment. As these viruses employ an arsenal of virally-encoded proteins to avoid innate immune detection during these phases, immunomodulatory viral proteins therefore represent therapeutic targets for prevention and treatment of gammaherpesvirus-associated malignancies. In this section, we discuss the implications of innate immunity for both pathogenesis as well as drug development in the context of human gammaherpesvirus infection.
Cancers associated with gammaherpesvirus infection.
As mentioned above, KSHV-associated cancers include Kaposi’s sarcoma (KS), multicentric Castleman’s disease (MCD), and primary effusion lymphoma (PEL), and EBV-associated cancers include Burkitt’s lymphoma (BL) and nasopharyngeal carcinoma (NPC). In addition to cancer, infection with KSHV can lead to an inflammatory cytokine syndrome termed KICS, classified by increased viral burden and elevated levels of cytokines including IL-6 and IL-10 [170]. Although some treatment options exist for these malignancies, including highly active antiretroviral therapy (HAART) for KS and PEL patients co-infected with HIV; various chemotherapeutic and immunotherapeutic regimens for PEL, MCD, and BL patients; and surgery followed by radiation therapy for NPC patients, we are still in dire need of novel therapeutic options for many of these viral diseases. Resistance to chemotherapy arises frequently, and many of these cancers are highly aggressive. Current frontline treatments target the cancer after it has formed, but preventing the expansion of the viral reservoir and the formation of cancer represents a more ideal, albeit challenging, strategy. This can potentially be achieved in part by crippling the virus’s ability to evade immune detection during the establishment or maintenance of infection, as will be discussed shortly.
Innate immunity and pathogenesis.
Activation of innate immune signaling pathways can induce gammaherpesvirus reactivation as well as excessive production of cytokines, both of which have been associated with increased pathogenesis in humans. Reactivation of herpesviruses from latency allows for the production of infectious virus particles which can then establish new infections within the same host or in a different host. Additionally, lytic gene expression and viral replication have been associated with KSHV-induced cancers, although the biological significance of these findings related to oncogenesis remains to be elucidated [171, 172]. A study using a panel of TLR agonists revealed that signaling through TLR7/8 results in KSHV lytic reactivation following latency in PEL. Interestingly, only TLR7/8 agonists were shown to have this effect, as ligands for other TLRs such as LPS (TLR4), CpG DNA (TLR9), Poly IC (TLR3), and flagellin (TLR5) did not induce viral reactivation [10]. These findings are of particular significance since TLR7/8 senses ssRNA, a component of many human viral pathogens. Indeed, although not a canonical human virus, infection of PEL cells with VSV caused KSHV reversion from latency, suggesting that infection with other viruses that stimulate TLR7/8 signaling can be a risk factor for gammaherpesvirus reactivation [10]. A similar study using MHV68 revealed that signaling through TLRs 3, 4, 5, and 9, but not TLR7/8, resulted in viral reactivation in B cells. Furthermore, TLR4 and TLR9 activation also induced MHV68 virion production in vivo, reflecting the observations made in cell culture [173]. Taken together, these two studies show that signaling through TLRs can cause gammaherpesvirus reactivation from latency; however, the efficiency of this process for each individual TLR might be virus-specific.
Overabundance of cytokines in a host, whether proinflammatory or anti-inflammatory, can result in increased disease burden. In the context of KSHV infection, two examples of this are MCD and KICS. Although distinct and separate disorders, both MCD and KICS are characterized by excessive IL-6 and IL-10 production with inflammatory signatures [174], and this cytokine overexpression most likely drives the development and progression of these diseases. As such, treatment of MCD and KICS patients with anti-IL6 or anti-IL6R antibodies is currently under investigation [175, 176]. It would also be of interest to test anti-IL10 antibodies in KICS patients as well, given its unique upregulation and reflection of disease severity [175]. Additionally, NF-κB signaling may be vital to PEL survival, as this pathway was shown to be active in both PEL cell culture and primary tumor samples, and blocking the NF-κB pathway with small molecule drugs resulted in decreased IL-6 production and increased apoptosis of PEL [177]. Therefore, in sum, aberrant immune signaling can contribute to pathogenesis in KSHV-associated malignancies.
Innate immunity and therapeutics.
As gammaherpesviruses rely on immune evasion genes for propagation and survival, these genes can be targeted for therapeutic purposes. This strategy can be approached from two directions, either by targeting viral immunomodulatory genes directly as a treatment strategy, or by deleting these genes in the design of viral vaccines. One attractive target across all herpesviruses is the viral protein kinase. Indeed, maribavir, a drug against the HCMV protein kinase UL97, has been tested in clinical trials for treatment of resistant HCMV infection in the context of transplantation [178]. Data suggest that the KSHV protein kinase (vPK) plays a role in tumorigenesis, as mice expressing vPK exhibited increased B cell proliferation and lymphoma development compared to control mice [179]. Thus, inhibition of vPK in the context of KSHV-associated malignancies may be beneficial. Additionally, inhibitors against EBV immunomodulatory proteins are currently being identified and developed. Computational approaches can be used to determine structural areas of a viral protein that would be most beneficial to target, and this has been undertaken for EBV EBNA1, with results yielding insights into how to improve future anti-EBV drug design [180]. Indeed, this approach recently led to the generation and characterization of several novel EBNA1 inhibitors with efficacy both in vitro and in vivo. These inhibitors decreased growth of EBV-positive, but not EBV-negative, cell lines, and significantly reduced tumor burden and increased survival in multiple xenograft mouse models, including those derived from NPC patient tissue [181]. Similarly, a screen for novel inhibitors against EBV BPLF1, a conserved herpesviral DUB, was recently performed. Results of the screen yielded ten hits that blocked the DUB activity of BPLF1 and, intriguingly, the most promising drug was not toxic to cells at any concentration tested and exhibited a dose-dependent decrease in EBV infectivity [182]. Although not a comprehensive compilation, these select studies demonstrate the potential of designing small molecule drugs to inhibit the functions of key gammaherpesvirus proteins that modulate the immune environment and drive oncogenesis.
Currently, no approved vaccine against gammaherpesvirus infection or associated cancer exists. However, deletion of immune evasion genes represents a promising viral vaccine development strategy. An ideal gammaherpesvirus vaccine would prevent the establishment of latency in a host; if the initial viral infection could be cleared by the immune system, then viral persistence could not occur, and cancer development could likely be avoided. This could theoretically be achieved by altering and/or deleting the viral genes responsible for innate immune evasion and latency establishment in a way that still allows for the vaccine strain to elicit robust immune responses in the host (reviewed in [183]). Currently, this is being tested in MHV68 models as a proxy for human gammaherpesvirus infection, and recent results have been promising. Removal of ORF10, ORF36, ORF54, and K3 (anti-IFN and anti-MHC genes) as well as the latency genomic region (including mLANA) from MHV68 resulted in a vaccine strain that could not effectively replicate nor go latent. The modified virus did, however, activate innate immune responses in mice as well as establish protective cellular and humoral immunity against WT virus challenge in vivo [184]. These data are encouraging, and support the feasibility of designing live attenuated vaccines against human gammaherpesvirus infection.
In addition to targeting viral immune evasion genes for prevention and/or treatment of gammaherpesvirus infection and their associated cancers, host proteins that these viruses rely on to modulate the surrounding immune landscape in their favor can also be pursued as novel therapeutic targets. An example of this is the cellular protein exportin 1 (XPO1 or CRM1) during KSHV infection. Meng and Gao demonstrated that inhibition of XPO1 resulted in decreased viral replication due to increased IRF3 activity and elevated ISG levels [185], suggesting that XPO1 subdues innate immune responses towards KSHV. EBV LMP1 upregulates expression of cellular cyclooxygenase-2 (COX-2) in NPC in an NF-κB dependent manner, and this increase in COX-2 expression was shown to drive increased VEGF expression, which is associated with angiogenesis [186]. Therefore, targeting COX-2 could potentially decrease NPC tumor burden. Indeed, further studies have shown that COX-2 is associated with NPC proliferation, metastasis, and drug resistance in patients [187, 188] although, to date, the use of COX-2 inhibitors in NPC treatment has not yet been assessed. Similarly, KSHV infection increases COX-2 expression, and treating KSHV-associated diseases with anti-inflammatory agents such as COX-2 inhibitors is also of interest [189, 190]. Finally, enhancing the activity of host immune genes through exogenous IFN-α administration has been used as treatment for HIV-positive KS and PEL patients [191, 192].
Current challenges.
Design of gammaherpesvirus viral vaccines and host immune gene inhibitors are still in their infancy but hold promise for the improved treatment of these viral infections and their associated cancers. One consideration when designing these inhibitors is the potential for off-target toxicity. Targeting host proteins could have detrimental effects on normal cellular functions, and dosing manipulation or controlled administration could help alleviate this potential pitfall. A great challenge in vaccine design in general is balancing an inability to cause pathogenesis while at the same time inducing a robust enough immune response to provide long-term protection in the host. Deleting immune evasion genes is an attractive approach for viral vaccine design, as it reduces virulence while allowing for recognition and response by both the innate and adaptive arms of the immune system. The most daunting challenge that remains for gammaherpesvirus (and all herpesvirus) treatment is the ability to prevent the establishment of latency upon initial infection. If this can be achieved, great strides can be made in reducing the burden of herpesvirus-associated cancers in the human population. Although latency may not be absolutely required for gammaherpesvirus-associated oncogenesis [193], successfully hindering viral persistence in the host will undoubtedly improve the prognosis for pan-herpesvirus infection and related malignancies.
Conclusion and future perspectives
Gammaherpesviruses are pervasive pathogens that establish lifelong infections in nearly all adults. Importantly, immune control of these cancer-associated viruses is critical to the prevention of oncogenesis. Innate immune signaling plays an important role in controlling viral replication not only during de novo infection but also during chronic latent infection throughout the lifetime of the host [194]. As such, gammaherpesviruses have evolved extensive mechanisms by which to avoid innate immune detection and thus persist in host cells. Chronic gammaherpesvirus infection is therefore defined by a delicate balance between viral recognition by innate immune sensors and viral subversion of these antiviral pathways. Many of these activation and evasion mechanisms are shared among gammaherpesviruses, while others are more virus-specific (Figure 4). Consequently, targeting both the viral and cellular immunomodulatory proteins these viruses rely on to successfully establish infection represents a promising therapeutic approach for better controlling and possibly preventing chronic gammaherpesvirus infection and their related malignancies.
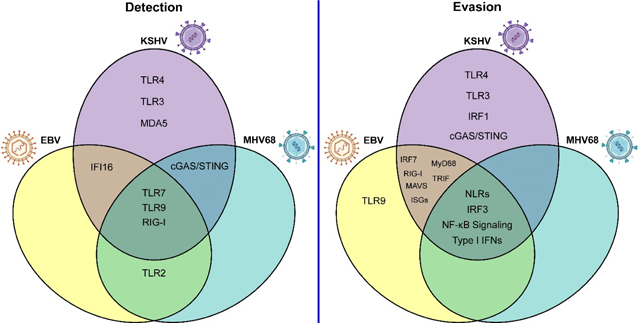
Fig. 4. Shared and differential innate immune sensing of and evasion by gammaherpesviruses.

(A) Sensing of gammaherpesvirus infection occurs through shared and differential innate immune pathways. (B) Similarly, gammaherpesviruses utilize conserved and unique mechanisms to circumvent antiviral innate immune signaling. Importantly, this figure summarizes only the detection and evasion strategies discussed in this review, and the relationships between gammaherpesviruses and innate immune signaling pathways remain incompletely characterized. TLR, Toll-like receptor; IFN, interferon; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; RIG-I, retinoic acid-inducible gene I; MAVS, mitochondrial antiviral-signaling protein; ISGs, interferon stimulated genes; IFI16, interferon gamma-inducible protein 16; MyD88, myeloid differentiation primary response 88; TRIF, TIR domain-containing adaptor inducing IFN-β; NF-κB, nuclear factor kappa B; IRF, interferon regulatory factor.
Building upon the robust knowledge base of innate immune detection of gammaherpesviruses, future areas of study can apply these findings towards advancing our ability to diagnose, treat, and prevent disease associated with these viruses. Several gammaherpesvirus-driven diseases have clear genetic associations related to the innate immune response [195, 196], and the geographic distribution of other gammaherpesvirus-related pathologies suggests additional genetic components that have yet to be characterized. Therefore, the development of biomarker-based diagnositics may enhance our ability to identify individuals at risk of developing EBV− and KSHV-associated diseases.
Currently available antiviral therapies for the control of herpesvirus infection are focused on the conserved viral DNA polymerase to prevent lytic replication. However, these drugs have variable levels of efficacy across individuals and the development of resistance is of concern. Furthermore, most gammaherpesvirus-associated diseases are not driven by robust lytic replication and, as such, drugs that enhance immune detection of latently infected cells should be prioritized.
Finally, arguably the greatest clinical challenge regarding herpesvirus infection is the design of efficacious vaccines that not only induce a robust and protective immune response in the host but also prevent the establishment of latency. Initial efforts on this front should focus on vaccines that prevent the development of disease in at-risk individuals, as was done with shingles vaccines that prevent reactivation of varicella zoster virus in aging populations.
Overall, our knowledge surrounding innate immune sensing of gammaherpesviruses and the strategies by which these viruses circumvent host cell detection has greatly advanced in the last two decades. The translation of these findings may allow for better diagnostic, thearapeutic, and preventative strategies that will ultimately reduce the substantial health burden of these viruses.
Research highlights.
Gammaherpesviruses are pervasive pathogens that are associated with numerous cancers
Innate immune sensing is a critical component of the immune response to gammaherpesvirus infection throughout all stages of the viral life cycle
Cellular pattern recognition receptors such as TLRs, RLRs, and DNA sensors efficiently recognize viral constituents and induce antiviral responses
Gammaherpesviruses employ numerous mechanisms to subvert innate immune recognition and signaling
Improving our understanding of the dynamic between gammaherpesviruses and innate immunity may facilitate the development of novel therapeutic and prophylactic strategies and reduce the cancer burden associated with these pathogens
Acknowledgements
This work was supported by National Institutes of Health grants CA019014, DE028211, CA096500, CA239583, CA254564, and CA163217. PTL is supported by a fellowship from the Lymphoma Research Foundation. MCW is supported by NIH grant 5T32CA009156-44. BD is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in Infectious Disease. We apologize to those authors whose work we did not discuss.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Farrell PJ. Epstein-Barr Virus and Cancer. Annual review of pathology. 2019;14:29–53. [DOI] [PubMed] [Google Scholar]
- [2].Goncalves PH, Ziegelbauer J, Uldrick TS, Yarchoan R. Kaposi sarcoma herpesvirus-associated cancers and related diseases. Current opinion in HIV and AIDS. 2017;12:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen Q, Chen J, Li Y, Liu D, Zeng Y, Tian Z, et al. Kaposi’s sarcoma herpesvirus is associated with osteosarcoma in Xinjiang populations. Proceedings of the National Academy of Sciences of the United States of America. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shannon-Lowe C, Rickinson A. The Global Landscape of EBV-Associated Tumors. Frontiers in oncology. 2019;9:713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. The Journal of clinical investigation. 2010;120:939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chandriani S, Ganem D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 2010;84:5565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Takagi S, Takada K, Sairenji T. Formation of intranuclear replication compartments of Epstein-Barr virus with redistribution of BZLF1 and BMRF1 gene products. Virology. 1991;185:309–15. [DOI] [PubMed] [Google Scholar]
- [8].Mellinghoff I, Daibata M, Humphreys RE, Mulder C, Takada K, Sairenji T. Early events in Epstein-Barr virus genome expression after activation: regulation by second messengers of B cell activation. Virology. 1991;185:922–8. [DOI] [PubMed] [Google Scholar]
- [9].Flemington EK, Goldfeld AE, Speck SH. Efficient transcription of the Epstein-Barr virus immediate-early BZLF1 and BRLF1 genes requires protein synthesis. Journal of virology. 1991;65:7073–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hirsiger JR, Fuchs PS, Häusermann P, Müller-Durovic B, Daikeler T, Recher M, et al. Syphilis Reactivates Latent Epstein-Barr Virus Reservoir via Toll-Like Receptor 2 and B-Cell Receptor Activation. Open forum infectious diseases. 2019;6:ofz317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, et al. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood. 2001;97:3244–50. [DOI] [PubMed] [Google Scholar]
- [13].Jiang JH, Wang N, Li A, Liao WT, Pan ZG, Mai SJ, et al. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2006;37:98–103. [DOI] [PubMed] [Google Scholar]
- [14].Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. Journal of virology. 2005;79:1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS letters. 2007;581:3485–8. [DOI] [PubMed] [Google Scholar]
- [16].Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, et al. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. Journal of virology. 2007;81:13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sun CC, Thorley-Lawson DA. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. Journal of virology. 2007;81:13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Glaser R, Kutz LA, MacCallum RC, Malarkey WB. Hormonal modulation of Epstein-Barr virus replication. Neuroendocrinology. 1995;62:356–61. [DOI] [PubMed] [Google Scholar]
- [19].Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy. 2017;2:17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. Journal of immunology (Baltimore, Md : 1950). 2005;174:2942–50. [DOI] [PubMed] [Google Scholar]
- [21].Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. [DOI] [PubMed] [Google Scholar]
- [22].Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. [DOI] [PubMed] [Google Scholar]
- [23].Bauer S, Kirschning CJ, Häcker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–103. [DOI] [PubMed] [Google Scholar]
- [25].Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (New York, NY). 2004;303:1526–9. [DOI] [PubMed] [Google Scholar]
- [26].Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). The Journal of experimental medicine. 1999;189:615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. Journal of immunology (Baltimore, Md : 1950). 1999;162:3749–52. [PubMed] [Google Scholar]
- [28].Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. [DOI] [PubMed] [Google Scholar]
- [29].Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology. 2010;11:373–84. [DOI] [PubMed] [Google Scholar]
- [30].Uematsu S, Akira S. Toll-Like Receptors (TLRs) and Their Ligands. In: Bauer S, Hartmann G, editors. Toll-Like Receptors (TLRs) and Innate Immunity. Berlin, Heidelberg: Springer Berlin Heidelberg; 2008. p. 1–20. [Google Scholar]
- [31].Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science (New York, NY). 1997;278:1612–5. [DOI] [PubMed] [Google Scholar]
- [32].Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–51. [DOI] [PubMed] [Google Scholar]
- [33].Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (New York, NY). 2003;301:640–3. [DOI] [PubMed] [Google Scholar]
- [34].Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. Journal of immunology (Baltimore, Md : 1950). 2002;169:6668–72. [DOI] [PubMed] [Google Scholar]
- [35].Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003;4:161–7. [DOI] [PubMed] [Google Scholar]
- [36].Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11416–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15416–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–40. [DOI] [PubMed] [Google Scholar]
- [39].Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–8. [DOI] [PubMed] [Google Scholar]
- [40].Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. The Journal of experimental medicine. 2005;201:915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gaudreault E, Fiola SP, Olivier M, Gosselin J. Epstein-Barr Virus Induces MCP-1 Secretion by Human Monocytes via TLR2. Journal of virology. 2007;81:8016–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Michaud F, Coulombe F, Gaudreault É, Kriz J, Gosselin J. Involvement of TLR2 in Recognition of Acute Gammaherpesvirus-68 Infection. PLoS ONE. 2010;5:e13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, et al. Human Cytomegalovirus Activates Inflammatory Cytokine Responses via CD14 and Toll-Like Receptor 2. Journal of virology. 2003;77:4588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, Finberg RW. Varicella-Zoster Virus Activates Inflammatory Cytokines in Human Monocytes and Macrophages via Toll-Like Receptor 2. Journal of virology. 2005;79:12658–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proceedings of the National Academy of Sciences. 2004;101:1315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (New York, NY). 2004;303:1529–31. [DOI] [PubMed] [Google Scholar]
- [47].Munz C. Dendritic cells during Epstein Barr virus infection. Frontiers in Microbiology. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kadowaki N, Ho S, Antonenko S, De Waal Malefyt R, Kastelein RA, Bazan F, et al. Subsets of Human Dendritic Cell Precursors Express Different Toll-like Receptors and Respond to Different Microbial Antigens. Journal of Experimental Medicine. 2001;194:863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H. TLR9 Contributes to Antiviral Immunity during Gammaherpesvirus Infection. The Journal of Immunology. 2008;180:438–43. [DOI] [PubMed] [Google Scholar]
- [50].Luckhardt TR, Coomes SM, Trujillo G, Stoolman JS, Vannella KM, Bhan U, et al. TLR9-induced interferon β is associated with protection from gammaherpesvirus-induced exacerbation of lung fibrosis. Fibrogenesis & Tissue Repair. 2011;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bussey KA, Murthy S, Reimer E, Chan B, Hatesuer B, Schughart K, et al. Endosomal Toll-Like Receptors 7 and 9 Cooperate in Detection of Murine Gammaherpesvirus 68 Infection. Journal of virology. 2018;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gargano LM, Moser JM, Speck SH. Role for MyD88 Signaling in Murine Gammaherpesvirus 68 Latency. Journal of virology. 2008;82:3853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Quan TE, Roman RM, Rudenga BJ, Holers VM, Craft JE. Epstein-Barr virus promotes interferon-α production by plasmacytoid dendritic cells. Arthritis & Rheumatism. 2010;62:1693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].West JA, Gregory SM, Sivaraman V, Su L, Damania B. Activation of Plasmacytoid Dendritic Cells by Kaposi’s Sarcoma-Associated Herpesvirus. Journal of virology. 2011;85:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fiola S, Gosselin D, Takada K, Gosselin J. TLR9 Contributes to the Recognition of EBV by Primary Monocytes and Plasmacytoid Dendritic Cells. The Journal of Immunology. 2010;185:3620–31. [DOI] [PubMed] [Google Scholar]
- [56].West J, Damania B. Upregulation of the TLR3 Pathway by Kaposi’s Sarcoma-Associated Herpesvirus during Primary Infection. Journal of virology. 2008;82:5440–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science (New York, NY). 2006;314:994–7. [DOI] [PubMed] [Google Scholar]
- [58].Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. [DOI] [PubMed] [Google Scholar]
- [59].Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science (New York, NY). 2006;314:997–1001. [DOI] [PubMed] [Google Scholar]
- [60].Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, et al. Structural and functional insights into 5’-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nature structural & molecular biology. 2010;17:781–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:21010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. The EMBO Journal. 2006;25:4207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chiang JJ, Sparrer KMJ, Van Gent M, Lässig C, Huang T, Osterrieder N, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nature Immunology. 2018;19:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Duan Y, Li Z, Cheng S, Chen Y, Zhang L, He J, et al. Nasopharyngeal carcinoma progression is mediated by EBER-triggered inflammation via the RIG-I pathway. Cancer Letters. 2015;361:67–74. [DOI] [PubMed] [Google Scholar]
- [65].Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nature Immunology. 2009;10:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chiu Y-H, Macmillan JB, Chen ZJ. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell. 2009;138:576–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, et al. Inhibition of RIG-I-Mediated Signaling by Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Deubiquitinase ORF64. Journal of virology. 2011;85:10899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].West JA, Wicks M, Gregory SM, Chugh P, Jacobs SR, Zhang Z, et al. An Important Role for Mitochondrial Antiviral Signaling Protein in the Kaposi’s Sarcoma-Associated Herpesvirus Life Cycle. Journal of virology. 2014;88:5778–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhao Y, Ye X, Dunker W, Song Y, Karijolich J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nature Communications. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang Y, Dittmer DP, Mieczkowski PA, Host KM, Fusco WG, Duncan JA, et al. RIG-I Detects Kaposi’s Sarcoma-Associated Herpesvirus Transcripts in a RNA Polymerase III-Independent Manner. mBio. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Karijolich J, Abernathy E, Glaunsinger BA. Infection-Induced Retrotransposon-Derived Noncoding RNAs Enhance Herpesviral Gene Expression via the NF-κB Pathway. PLOS Pathogens. 2015;11:e1005260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang H, Ni G, Damania B. ADAR1 Facilitates KSHV Lytic Reactivation by Modulating the RLR-Dependent Signaling Pathway. Cell Reports. 2020;31:107564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Andreeva L, Hiller B, Kostrewa D, Lässig C, De Oliveira Mann CC, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature. 2017;549:394–8. [DOI] [PubMed] [Google Scholar]
- [74].Luecke S, Holleufer A, Christensen MH, Jønsson KL, Boni GA, Sørensen LK, et al. cGAS is activated by DNA in a length-dependent manner. EMBO reports. 2017;18:1707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Volkman HE, Cambier S, Gray EE, Stetson DB. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature. 2020;587:668–72. [DOI] [PubMed] [Google Scholar]
- [77].Guey B, Wischnewski M, Decout A, Makasheva K, Kaynak M, Sakar MS, et al. BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science (New York, NY). 2020;369:823–8. [DOI] [PubMed] [Google Scholar]
- [78].Sun H, Huang Y, Mei S, Xu F, Liu X, Zhao F, et al. A Nuclear Export Signal Is Required for cGAS to Sense Cytosolic DNA. Cell Rep. 2021;34:108586. [DOI] [PubMed] [Google Scholar]
- [79].Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proceedings of the National Academy of Sciences. 2015;112:E4306–E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell host & microbe. 2015;18:333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proceedings of the National Academy of Sciences. 2016;113:E1034–E43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ni G, Ma Z, Wong JP, Zhang Z, Cousins E, Major MB, et al. PPP6C Negatively Regulates STING-Dependent Innate Immune Responses. mBio. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Jeon H, Lee J, Lee S, Kang S-K, Park SJ, Yoo S-M, et al. Extracellular Vesicles From KSHV-Infected Cells Stimulate Antiviral Immune Response Through Mitochondrial DNA. Frontiers in Immunology. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].McNamara RP, Chugh PE, Bailey A, Costantini LM, Ma Z, Bigi R, et al. Extracellular vesicles from Kaposi Sarcoma-associated herpesvirus lymphoma induce long-term endothelial cell reprogramming. PLOS Pathogens. 2019;15:e1007536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Schoggins JW, Macduff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yang K, Wang J, Wu M, Li M, Wang Y, Huang X. Mesenchymal stem cells detect and defend against gammaherpesvirus infection via the cGAS-STING pathway. Scientific Reports. 2015;5:7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gram AM, Sun C, Landman SL, Oosenbrug T, Koppejan HJ, Kwakkenbos MJ, et al. Human B cells fail to secrete type I interferons upon cytoplasmic DNA exposure. Molecular Immunology. 2017;91:225–37. [DOI] [PubMed] [Google Scholar]
- [88].Tang C-HA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Research. 2016;76:2137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proceedings of the National Academy of Sciences. 2015;112:E1773–E81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nature Communications. 2017;8:14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nature Communications. 2017;8:14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol Cell. 2018;71:745–60 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nature Immunology. 2010;11:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Jin T, Perry A, Jiang J, Smith P, James, Unterholzner L, et al. Structures of the HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor. Immunity. 2012;36:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kerur N, Mohanan, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell host & microbe. 2011;9:363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Singh VV, Kerur N, Bottero V, Dutta S, Chakraborty S, Ansari MA, et al. Kaposi’s Sarcoma-Associated Herpesvirus Latency in Endothelial and B Cells Activates Gamma Interferon-Inducible Protein 16-Mediated Inflammasomes. Journal of virology. 2013;87:4417–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Orzalli MH, Deluca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proceedings of the National Academy of Sciences. 2012;109:E3008–E17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ansari MA, Dutta S, Veettil MV, Dutta D, Iqbal J, Kumar B, et al. Herpesvirus Genome Recognition Induced Acetylation of Nuclear IFI16 Is Essential for Its Cytoplasmic Translocation, Inflammasome and IFN-β Responses. PLOS Pathogens. 2015;11:e1005019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Dutta D, Dutta S, Veettil MV, Roy A, Ansari MA, Iqbal J, et al. BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-β Responses. PLOS Pathogens. 2015;11:e1005030. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [100].Iqbal J, Ansari MA, Kumar B, Dutta D, Roy A, Chikoti L, et al. Histone H2B-IFI16 Recognition of Nuclear Herpesviral Genome Induces Cytoplasmic Interferon-β Responses. PLOS Pathogens. 2016;12:e1005967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, et al. Constitutive Interferon-Inducible Protein 16-Inflammasome Activation during Epstein-Barr Virus Latency I, II, and III in B and Epithelial Cells. Journal of virology. 2013;87:8606–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pisano G, Roy A, Ahmed Ansari M, Kumar B, Chikoti L, Chandran B. Interferon-γ-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency. Virology Journal. 2017;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chihara D, Ito H, Matsuda T, Shibata A, Katsumi A, Nakamura S, et al. Differences in incidence and trends of haematological malignancies in Japan and the United States. British journal of haematology. 2014;164:536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Argirion I, Zarins KR, Ruterbusch JJ, Vatanasapt P, Sriplung H, Seymour EK, et al. Increasing incidence of Epstein-Barr virus-related nasopharyngeal carcinoma in the United States. Cancer. 2020;126:121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chijioke O, Müller A, Feederle R, Mario, Krieg C, Emmel V, et al. Human Natural Killer Cells Prevent Infectious Mononucleosis Features by Targeting Lytic Epstein-Barr Virus Infection. Cell Reports. 2013;5:1489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Pappworth IY, Wang EC, Rowe M. The Switch from Latent to Productive Infection in Epstein-Barr Virus-Infected B Cells Is Associated with Sensitization to NK Cell Killing. Journal of virology. 2007;81:474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lünemann A, Vanoaica LD, Azzi T, Nadal D, Münz C. A Distinct Subpopulation of Human NK Cells Restricts B Cell Transformation by EBV. The Journal of Immunology. 2013;191:4989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood. 2014;124:2533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Strowig T, Brilot F, Arrey F, Bougras G, Thomas D, Muller WA, et al. Tonsilar NK Cells Restrict B Cell Transformation by the Epstein-Barr Virus via IFN-γ. PLoS Pathogens. 2008;4:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sathish N, Zhu FX, Golub EE, Liang Q, Yuan Y. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi’s sarcoma-associated herpesvirus protein ORF45. The Journal of biological chemistry. 2011;286:746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lefort S, Soucy-Faulkner A, Grandvaux N, Flamand L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. Journal of virology. 2007;81:10950–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Bisson SA, Page AL, Ganem D. A Kaposi’s sarcoma-associated herpesvirus protein that forms inhibitory complexes with type I interferon receptor subunits, Jak and STAT proteins, and blocks interferon-mediated signal transduction. Journal of virology. 2009;83:5056–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Cloutier N, Flamand L. Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits interferon (IFN) beta expression by competing with IFN regulatory factor-3 for binding to IFNB promoter. The Journal of biological chemistry. 2010;285:7208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lingel A, Ehlers E, Wang Q, Cao M, Wood C, Lin R, et al. Kaposi’s Sarcoma-Associated Herpesvirus Reduces Cellular Myeloid Differentiation Primary-Response Gene 88 (MyD88) Expression via Modulation of Its RNA. Journal of virology. 2016;90:180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zhao Q, Liang D, Sun R, Jia B, Xia T, Xiao H, et al. Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. Journal of virology. 2015;89:415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. Journal of virology. 2012;86:11663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lagos D, Vart RJ, Gratrix F, Westrop SJ, Emuss V, Wong PP, et al. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell host & microbe. 2008;4:470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Yu Y, Wang SE, Hayward GS. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity. 2005;22:59–70. [DOI] [PubMed] [Google Scholar]
- [120].Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, et al. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). The Journal of biological chemistry. 2011;286:7865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ Jr., et al. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001;20:800–11. [DOI] [PubMed] [Google Scholar]
- [122].Hwang KY, Choi YB. Modulation of Mitochondrial Antiviral Signaling by Human Herpesvirus 8 Interferon Regulatory Factor 1. Journal of virology. 2016;90:506–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ. Inhibition of interferon signaling by the Kaposi’s sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. Journal of virology. 2006;80:3092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Aresté C, Mutocheluh M, Blackbourn DJ. Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. The Journal of biological chemistry. 2009;284:23272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. Journal of virology. 2007;81:8282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hwang SW, Kim D, Jung JU, Lee HR. KSHV-encoded viral interferon regulatory factor 4 (vIRF4) interacts with IRF7 and inhibits interferon alpha production. Biochemical and biophysical research communications. 2017;486:700–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, et al. The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. Journal of virology. 2013;87:798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Jacobs SR, Stopford CM, West JA, Bennett CL, Giffin L, Damania B. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 1 Interacts with a Member of the Interferon-Stimulated Gene 15 Pathway. Journal of virology. 2015;89:11572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science (New York, NY). 2011;331:330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science (New York, NY). 2002;298:1432–5. [DOI] [PubMed] [Google Scholar]
- [131].Yu K, Tian H, Deng H. PPM1G restricts innate immune signaling mediated by STING and MAVS and is hijacked by KSHV for immune evasion. Science advances. 2020;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ma Z, Hopcraft SE, Yang F, Petrucelli A, Guo H, Ting JP, et al. NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLoS Pathog. 2017;13:e1006350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Botto S, Gustin JK, Moses AV. The Heme Metabolite Carbon Monoxide Facilitates KSHV Infection by Inhibiting TLR4 Signaling in Endothelial Cells. Front Microbiol 2017;8:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].van Gent M, Griffin BD, Berkhoff EG, van Leeuwen D, Boer IG, Buisson M, et al. EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. Journal of immunology (Baltimore, Md : 1950). 2011;186:1694–702. [DOI] [PubMed] [Google Scholar]
- [135].Fathallah I, Parroche P, Gruffat H, Zannetti C, Johansson H, Yue J, et al. EBV latent membrane protein 1 is a negative regulator of TLR9. Journal of immunology (Baltimore, Md : 1950). 2010;185:6439–47. [DOI] [PubMed] [Google Scholar]
- [136].Cohen JI, Lekstrom K. Epstein-Barr virus BARF1 protein is dispensable for B-cell transformation and inhibits alpha interferon secretion from mononuclear cells. Journal of virology. 1999;73:7627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Valentine R, Dawson CW, Hu C, Shah KM, Owen TJ, Date KL, et al. Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol Cancer. 2010;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Chen T, Wang Y, Xu Z, Zou X, Wang P, Ou X, et al. Epstein-Barr virus tegument protein BGLF2 inhibits NF-κB activity by preventing p65 Ser536 phosphorylation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2019;33:10563–76. [DOI] [PubMed] [Google Scholar]
- [139].van Gent M, Braem SG, de Jong A, Delagic N, Peeters JG, Boer IG, et al. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014;10:e1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Saito S, Murata T, Kanda T, Isomura H, Narita Y, Sugimoto A, et al. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-κB signaling during productive replication. Journal of virology. 2013;87:4060–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Michaud F, Coulombe F, Gaudreault E, Paquet-Bouchard C, Rola-Pleszczynski M, Gosselin J. Epstein-Barr virus interferes with the amplification of IFNalpha secretion by activating suppressor of cytokine signaling 3 in primary human monocytes. PLoS One. 2010;5:e11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hahn AM, Huye LE, Ning S, Webster-Cyriaque J, Pagano JS. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. Journal of virology. 2005;79:10040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bentz GL, Liu R, Hahn AM, Shackelford J, Pagano JS. Epstein-Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-beta. Virology. 2010;402:121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Wu L, Fossum E, Joo CH, Inn KS, Shin YC, Johannsen E, et al. Epstein-Barr virus LF2: an antagonist to type I interferon. Journal of virology. 2009;83:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Wang P, Deng Y, Guo Y, Xu Z, Li Y, Ou X, et al. Epstein-Barr Virus Early Protein BFRF1 Suppresses IFN-β Activity by Inhibiting the Activation of IRF3. Front Immunol. 2020;11:513383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Geiger TR, Martin JM. The Epstein-Barr virus-encoded LMP-1 oncoprotein negatively affects Tyk2 phosphorylation and interferon signaling in human B cells. Journal of virology. 2006;80:11638–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Stewart S, Dawson CW, Takada K, Curnow J, Moody CA, Sixbey JW, et al. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Shah KM, Stewart SE, Wei W, Woodman CB, O’Neil JD, Dawson CW, et al. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene. 2009;28:3903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. 2017;8:945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Gupta S, Ylä-Anttila P, Sandalova T, Sun R, Achour A, Masucci MG. 14–3-3 scaffold proteins mediate the inactivation of trim25 and inhibition of the type I interferon response by herpesvirus deconjugases. PLoS Pathog. 2019;15:e1008146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Vilmen G, Glon D, Siracusano G, Lussignol M, Shao Z, Hernandez E, et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy. 2020:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Wang JT, Doong SL, Teng SC, Lee CP, Tsai CH, Chen MR. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. Journal of virology. 2009;83:1856–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Chang LS, Wang JT, Doong SL, Lee CP, Chang CW, Tsai CH, et al. Epstein-Barr virus BGLF4 kinase downregulates NF-κB transactivation through phosphorylation of coactivator UXT. Journal of virology. 2012;86:12176–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Haneklaus M, Gerlic M, Kurowska-Stolarska M, Rainey AA, Pich D, McInnes IB, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. Journal of immunology (Baltimore, Md : 1950). 2012;189:3795–9. [DOI] [PubMed] [Google Scholar]
- [155].Hooykaas MJG, van Gent M, Soppe JA, Kruse E, Boer IGJ, van Leenen D, et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. Journal of immunology (Baltimore, Md : 1950). 2017;198:4062–73. [DOI] [PubMed] [Google Scholar]
- [156].Lu Y, Qin Z, Wang J, Zheng X, Lu J, Zhang X, et al. Epstein-Barr Virus miR-BART6–3p Inhibits the RIG-I Pathway. Journal of innate immunity. 2017;9:574–86. [DOI] [PubMed] [Google Scholar]
- [157].Ramalingam D, Ziegelbauer JM. Viral microRNAs Target a Gene Network, Inhibit STAT Activation, and Suppress Interferon Responses. Sci Rep. 2017;7:40813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, et al. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell host & microbe. 2009;5:166–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Sun C, Schattgen SA, Pisitkun P, Jorgensen JP, Hilterbrand AT, Wang LJ, et al. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. Journal of immunology (Baltimore, Md : 1950). 2015;194:1819–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Leang RS, Wu TT, Hwang S, Liang LT, Tong L, Truong JT, et al. The anti-interferon activity of conserved viral dUTPase ORF54 is essential for an effective MHV-68 infection. PLoS Pathog. 2011;7:e1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Dong X, Feng P. Murine gamma herpesvirus 68 hijacks MAVS and IKKβ to abrogate NFκB activation and antiviral cytokine production. PLoS Pathog. 2011;7:e1002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Dong X, He Z, Durakoglugil D, Arneson L, Shen Y, Feng P. Murine gammaherpesvirus 68 evades host cytokine production via replication transactivator-induced RelA degradation. Journal of virology. 2012;86:1930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Kang HR, Cheong WC, Park JE, Ryu S, Cho HJ, Youn H, et al. Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. Journal of virology. 2014;88:6832–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Mandal P, Krueger BE, Oldenburg D, Andry KA, Beard RS, White DW, et al. A gammaherpesvirus cooperates with interferon-alpha/beta-induced IRF2 to halt viral replication, control reactivation, and minimize host lethality. PLoS Pathog. 2011;7:e1002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhang L, Pagano JS. Interferon regulatory factor 2 represses the Epstein-Barr virus BamHI Q latency promoter in type III latency. Molecular and cellular biology. 1999;19:3216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Cieniewicz B, Dong Q, Li G, Forrest JC, Mounce BC, Tarakanova VL, et al. Murine Gammaherpesvirus 68 Pathogenesis Is Independent of Caspase-1 and Caspase-11 in Mice and Impairs Interleukin-1β Production upon Extrinsic Stimulation in Culture. Journal of virology. 2015;89:6562–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Caduff N, McHugh D, Rieble L, Forconi CS, Ong’echa JM, Oluoch PO, et al. KSHV infection drives poorly cytotoxic CD56-negative natural killer cell differentiation in vivo upon KSHV/EBV dual infection. Cell Rep. 2021;35:109056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Schuhmachers P, Münz C. Modification of EBV Associated Lymphomagenesis and Its Immune Control by Co-Infections and Genetics in Humanized Mice. Front Immunol. 2021;12:640918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Romero-Masters JC, Ohashi M, Djavadian R, Eichelberg MR, Hayes M, Zumwalde NA, et al. An EBNA3A-Mutated Epstein-Barr Virus Retains the Capacity for Lymphomagenesis in a Cord Blood-Humanized Mouse Model. Journal of virology. 2020;94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Polizzotto MN, Uldrick TS, Wyvill KM, Aleman K, Marshall V, Wang V, et al. Clinical Features and Outcomes of Patients With Symptomatic Kaposi Sarcoma Herpesvirus (KSHV)-associated Inflammation: Prospective Characterization of KSHV Inflammatory Cytokine Syndrome (KICS). Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2016;62:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Rondeau NC, Finlayson MO, Miranda JL. Widespread Traces of Lytic Kaposi Sarcoma-Associated Herpesvirus in Primary Effusion Lymphoma at Single-Cell Resolution. Microbiology resource announcements. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Decker LL, Shankar P, Khan G, Freeman RB, Dezube BJ, Lieberman J, et al. The Kaposi sarcoma-associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. The Journal of experimental medicine. 1996;184:283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Gargano LM, Forrest JC, Speck SH. Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. Journal of virology. 2009;83:1474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Polizzotto MN, Uldrick TS, Hu D, Yarchoan R. Clinical Manifestations of Kaposi Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front Microbiol. 2012;3:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Caro-Vegas C, Sellers S, Host KM, Seltzer J, Landis J, Fischer WA 2nd, et al. Runaway Kaposi Sarcoma-associated herpesvirus replication correlates with systemic IL-10 levels. Virology. 2020;539:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Ramaswami R, Lurain K, Peer CJ, Serquiña A, Wang V, Widell A, et al. Tocilizumab in patients with symptomatic Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood. 2020;135:2316–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96:2537–42. [PubMed] [Google Scholar]
- [178].Papanicolaou GA, Silveira FP, Langston AA, Pereira MR, Avery RK, Uknis M, et al. Maribavir for Refractory or Resistant Cytomegalovirus Infections in Hematopoietic-cell or Solid-organ Transplant Recipients: A Randomized, Dose-ranging, Double-blind, Phase 2 Study. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2019;68:1255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Anders PM, Montgomery ND, Montgomery SA, Bhatt AP, Dittmer DP, Damania B. Human herpesvirus-encoded kinase induces B cell lymphomas in vivo. J Clin Invest. 2018;128:2519–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Gianti E, Messick TE, Lieberman PM, Zauhar RJ. Computational analysis of EBNA1 “druggability” suggests novel insights for Epstein-Barr virus inhibitor design. Journal of computer-aided molecular design. 2016;30:285–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Messick TE, Smith GR, Soldan SS, McDonnell ME, Deakyne JS, Malecka KA, et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Atkins SL, Motaib S, Wiser LC, Hopcraft SE, Hardy PB, Shackelford J, et al. Small molecule screening identifies inhibitors of the Epstein-Barr virus deubiquitinating enzyme, BPLF1. Antiviral research. 2020;173:104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Wu TT, Blackman MA, Sun R. Prospects of a novel vaccination strategy for human gamma-herpesviruses. Immunologic research. 2010;48:122–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Brar G, Farhat NA, Sukhina A, Lam AK, Kim YH, Hsu T, et al. Deletion of immune evasion genes provides an effective vaccine design for tumor-associated herpesviruses. NPJ vaccines. 2020;5:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Meng W, Gao SJ. Targeting XPO1 enhances innate immune response and inhibits KSHV lytic replication during primary infection by nuclear stabilization of the p62 autophagy adaptor protein. Cell death & disease. 2021;12:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Murono S, Inoue H, Tanabe T, Joab I, Yoshizaki T, Furukawa M, et al. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Yang G, Deng Q, Fan W, Zhang Z, Xu P, Tang S, et al. Cyclooxygenase-2 expression is positively associated with lymph node metastasis in nasopharyngeal carcinoma. PLoS One. 2017;12:e0173641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Shi C, Guan Y, Zeng L, Liu G, Zhu Y, Xu H, et al. High COX-2 expression contributes to a poor prognosis through the inhibition of chemotherapy-induced senescence in nasopharyngeal carcinoma. International journal of oncology. 2018;53:1138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Chandrasekharan JA, Huang XM, Hwang AC, Sharma-Walia N. Altering the Anti-inflammatory Lipoxin Microenvironment: a New Insight into Kaposi’s Sarcoma-Associated Herpesvirus Pathogenesis. Journal of virology. 2016;90:11020–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Chandrasekharan JA, Sharma-Walia N. Arachidonic Acid Derived Lipid Mediators Influence Kaposi’s Sarcoma-Associated Herpesvirus Infection and Pathogenesis. Front Microbiol. 2019;10:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Mitsuyasu RT. Interferon alpha in the treatment of AIDS-related Kaposi’s sarcoma. British journal of haematology. 1991;79 Suppl 1:69–73. [DOI] [PubMed] [Google Scholar]
- [192].Narkhede M, Arora S, Ujjani C. Primary effusion lymphoma: current perspectives. OncoTargets and therapy. 2018;11:3747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Ma SD, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, et al. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. Journal of virology. 2012;86:7976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Johnson KE, Aurubin CA, Jondle CN, Lange PT, Tarakanova VL. Interferon Regulatory Factor 7 Attenuates Chronic Gammaherpesvirus Infection. Journal of virology. 2020;94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Blumenthal MJ, Cornejo Castro EM, Whitby D, Katz AA, Schäfer G. Evidence for altered host genetic factors in KSHV infection and KSHV-related disease development. Reviews in medical virology. 2021;31:e2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Jouanguy E, Béziat V, Mogensen TH, Casanova JL, Tangye SG, Zhang SY. Human inborn errors of immunity to herpes viruses. Current opinion in immunology. 2020;62:106–22. [DOI] [PMC free article] [PubMed] [Google Scholar]