

Abstract
The objective of this study is to validate a placebo pill response predictive model - a biosignature - that classifies chronic pain patients into placebo-responders (predicted-PTxResp) and non-responders (predicted-PTxNonR), and test whether it can dissociate placebo and active treatment responses. The model, based on psychological and brain functional connectivity, was derived in our previous study and blindly applied to current trial participants. 94 chronic low back pain (CLBP) patients were classified into predicted-PTxResp or predicted-PTxNonR and randomized into no-treatment, placebo treatment, or naproxen treatment. To monitor analgesia, back pain intensity was collected twice a day: 3 weeks baseline, 6 weeks of treatment, 3 weeks of washout. 89 CLBP patients were included in the intent-to-treat analyses and 77 CLBP in the per-protocol analyses. Both analyses showed similar results. At the group level, the predictive model performed remarkably well, dissociating the separate effect sizes of pure placebo response and pure active treatment response, and demonstrating that these effects interacted additively. Pain relief was about 15% stronger in the predicted-PTxResp compared to the predicted-PTxNonR receiving either placebo or naproxen, and the predicted-PTxNonR successfully isolated the active drug effect. At a single subject level, the biosignature better predicted placebo non-responders, with poor accuracy. One component of the biosignature (dorsolateral prefrontal cortex-precentral gyrus functional connectivity) could be generalized across three placebo studies and in two different cohorts - CLBP and osteoarthritis pain patients. This study shows that a biosignature can predict placebo response at a group level in the setting of a randomized controlled trial.
1. Introduction
The placebo effect refers to an improvement in symptoms after receiving an inert or sham treatment [8]. Potential utility of a placebo response lies squarely within the clinical setting, now universally embedded in the blinded randomized control trial (RCT). In everyday clinical practice, approximately 50% of internists and rheumatologists prescribe placebo treatments on a regular basis [59], and their response size is continuously increasing [60]. Thus, this study is driven by the idea that the predictability of a placebo response could become an invaluable clinical tool, providing better-tailored individualized treatments, as well as enabling novel RCT designs to help facilitate treatment discoveries [7,17,24,38].
A large number of studies have revealed the underlying mechanisms of the placebo effect [12,20,26,42,43,68]. Yet, most of these studies were performed under strict laboratory settings and their findings may not translate to clinical populations with chronic symptoms. In RCTs, patients receiving placebo treatments show consistent improvements, especially in the realm of mental health and pain [28,37]. Although the exact nature of these improvements may not be specific to the consumption of the inert treatment (see [33], [31]), mechanistic and efficacy studies of placebo have now been conducted in various clinical pain conditions, including irritable bowel syndrome [37,64,65], back pain [14,29], migraine [36], orofacial pain [16], and osteoarthritis (OA) [57,58]. Still, the extent of response predictability and dissociation of placebo effects from those of active treatment within the RCT setting remain to be demonstrated.
Multiple reasons suggest that the placebo response in RCTs may deviate from those observed in controlled experiments. For instance, the placebo response in RCTs where active treatments are expected to provide sustained pain relief for chronic pain over weeks or even months may be different from what is observed when acute pain is evoked for a few minutes in the laboratory. Moreover, long-lasting placebo response in chronic pain patients is likely to depend on the patients’ history of treatment, related psychological factors, and the complex interplay between brain regions modified by chronic pain [52,62,63]. Only a few studies have examined the neurobiology of placebo response within the settings of RCTs in fibromyalgia [51], OA [30,58], and chronic low back pain (CLBP) [29,61]. Here, we specifically tested if placebo response can be predicted by designing a RCT where CLBP patients were either classified as placebo responders or non-responders based on a previously-identified biosignature predicting placebo response [61] (phase 1), which included psychological and reting state brain functional connectivity. In the current study (phase 2), we blindly applied the biosignature to the new group of participants without any parameter manipulations prior to randomization. The predicted-placebo-responders and non-responders were then allocated to placebo treatment, naproxen treatment, or no-treatment arms. By examining responses of stratified participants with the three possible treatment arms, we could test the biosignature model validity at both group and individual level, and dissociate between placebo and active treatment responses. In a post-hoc analysis we tested generalizability of model components to other placebo trials and to chronic osteoarthritis patients.
2. Methods
The study is based on two independent data sets. The first data set was collected to identify predictors of clinical placebo, Phase 1 [61], and build a biosignature (a discovery study). In phase 2, current study, we test the validity of the biosignature using an unbiased approach. The data collection did not overlapped between the Phase 1 and Phase 2 studies. Even though the psychological and functional connectivity components composing the biosignature were reported previously [61], the final minimal model was not included. Here, we report (see supplement methods) the details of the final minimal biosignature model derived fully from Phase 1 data analysis, both for transparency and to enable others to use the model in future studies. The Phase 1 based biosignature model was built and its parameters fixed prior to the any data collection or analysis in Phase 2. The fixed-parameter biosignature was used on every subject who entered to Phase 2 study to stratify them as placebo responders or nonresponders, prior to randomization into different treatment arms. Thus, there is no leakage between Phase 1 and Phase 2 results, ensuring that this study is an unbiased validity test for the placebo biosignature. Below we expound on data collection and analysis used in Phase 2.
2.1. Participants (Study Population) of Phase 2 study
The randomization of patients occurred between September 2016 and April 2018. Note: the trial was officially registered in December 2016, about 12 weeks after the randomization of the first patient. At that time, 19/94 patients were randomized and one patient completed the study. All patients were however classified (predicted PTxResp or predicted PTxNonR) and randomized to placebo treatment, medication treatment, or no treatment, PTx, MTx, or NoTx respectively, prior to the unblinding of the study in 2018. The trial was conducted in Chicago (USA) and the data were analysed by E.V.P. (located in Montreal, Canada) who was not involved in the data aquisition and/or the supervision of the RCT. The data analyses were performed by EVP only after the study was unblinded. 181 participants with chronic low back pain (CBP) were initially recruited from the general population and clinical referrals via hospital databases and advertising in the community, as in [61]. Fig. 1 show the CONSORT flow diagram.
fig. 1. CONSORT flow diagram.
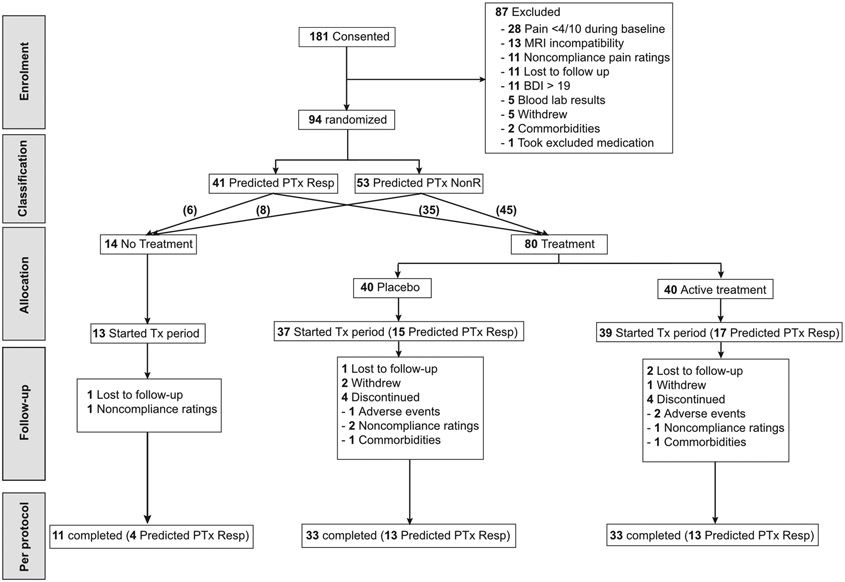
Disposition of all study participants from study entry to study completion. Of the 181 patients that consented and entered into the study, 87 screen-failed between visit 1 and visit 2 due to reasons listed in the figure. The remaining 94 patients were classified, based on the biosignature model outcome, as responders, predicted-PTxResp (n=41), or non-responders, predicted-PTxNonR (n=53), and randomized into a no-treatment (n=14) or treatment (n=80) group according to a block design, ensuring approximately equal representation of predicted-PTxResp in each arm. Of the 80 treated individuals, 40 were allocated to placebo treatment and 40 were allocated to active treatment (Naproxen + Esomeprazole) in a double-blind fashion. From these, 13 individuals in the no-treatment arm, 37 individuals in the placebo arm, and 39 participants in the active treatment arm started the treatment period and were included in the intent-to-treat analyses. There were 11 participants in the no-treatment arm, 33 individuals in the placebo arm, and 33 participants in the active treatment arm that successfully completed all 6 visits and were subsequently analyzed in the per-protocol analyses. Patients were either lost to follow-up or discontinued for reasons specified in the diagram.
2.2. Inclusions/exclusions criteria
For safety precautions, clinical measurements taken from blood draw at Visit 1 must have been within the pre-specified healthy range (as determined by the standards utilized by Northwestern University Feinberg School of Medicine Laboratory Services Department), and all participants must have passed the MRI safety screening requirements at each scanning visit. The following is the complete list of inclusion and exclusion criteria.
2.2.1. Inclusion Criteria:
History of low back pain for a minimum of 6 months with or without signs and symptoms of radiculopathy
Male or female, between the ages of 18 and 75 years, with no racial or ethnic restrictions
Must have a Visual Analog Scale (VAS) pain score of 5 mm (of 10 mm maximum) at the screening visit (for which 0 mm = no pain, and 10 mm = worst pain imaginable);
Must be able to read and speak English and be willing to read and understand instructions as well as questionnaires;
Must be in generally stable health;
Must sign an informed consent document after a complete explanation of the study, documenting that they understand the purpose of the study, procedures to be undertaken, possible benefits, potential risks, and are willing to participate
Must have, on average, 4/10 units (on phone app 0-10 Likert score) of pain over the course of a two-week period prior to visit 1; rounding up from 3.5/10 is permissible (as in Phase 1). The initial criterion was a score of 5/10 (4.5 rounded up), but this was changed following the completion of Phase 1 (and prior to the start of Phase 2) to facilitate recruitment.
Must be willing to complete daily smart phone/computer app ratings.
2.2.2. Exclusion Criteria:
Low back pain associated with any systemic signs or symptoms, e.g., fever, chills;
Evidence of rheumatoid arthritis, ankylosing spondylitis, acute vertebral fractures, fibromyalgia, history of tumor in the back;
Other comorbid chronic pain or neurological conditions;
Involvement in litigation regarding their back pain, having a disability claim, receiving workman's compensation, or seeking any of the above as a result of their low back pain;
Diagnosis of current depression or psychiatric disorder requiring treatment, or such a diagnosis in the previous 6 months;
Beck Depression Inventory (BDI) Ia score greater or equal to 19 for two consecutive completions; if the first score meets this criteria, the participant must be re-tested before his/her next visit, but if the second score does not meet this criteria, the participant will be included and followed closely throughout the study
Use of therapeutic doses of antidepressant medications (i.e., tricyclic depressants, SSRIs, SNRIs; low doses used for sleep may be allowed);
Significant other medical disease such as unstable diabetes mellitus, congestive heart failure, coronary or peripheral vascular disease, chronic obstructive lung disease, or malignancy;
History of gastrointestinal ulcer during the past year;
History of myocardial infarction in the past year;
Uncontrolled hypertension;
Renal insufficiency;
Allergic to, or non-tolerant of, NSAIDs;
History of aspirin-sensitive asthma;
Current use of recreational drugs or history of alcohol or drug abuse;
Any change in medication for back pain in the last 30 days, only applicable for visit 1
High dose opioid prophylaxis, as defined as > 50mg morphine equivalent/day;
Any medical condition that in the investigator's judgment may prevent the individual from completing the study or put the individual at undue risk;
In the judgment of the investigator, unable or unwilling to follow protocol and instructions;
Evidence of poor treatment compliance, in the judgment of the investigator;
Intra-axial implants (e.g. spinal cord stimulators or pain pumps);
All exclusion criteria for MR safety: any metallic implants, brain or skull abnormalities, tattoos on large body parts, and claustrophobia;
Pregnancy, or inability to use an effective form of contraception in women of child-bearing age;
Diabetes (Type I or Type II);
Lactose intolerance or sensitivity to lactose, a precaution for using lactose filled placebo pills.
2.3. Study Design and Procedures
This study was conducted in the setting of a clinical randomized control trial (RCT) consisting of 6 visits spread over approximately 12 weeks. The design was set up to stratify the CBP patients as either predicted placebo responder or predicted placebo non-responder prior to randomizing them into either no treatment, placebo treatment, or active medication treatment. The prediction of placebo response was then compared to the actual response to either placebo treatment or active treatment.
2.3.1. Visit 1 (week -3): Consent, screening, and baseline pain/mood monitoring (45 min)
The participants were evaluated for initial inclusion/exclusion criteria after completing the informed consent process. A medical/pain history and physical examination, including vitals, was completed. Blood was drawn for a baseline screen to obtain a complete blood count, chemistry panel, liver function tests, drug tests, and pregnancy tests if female. Participants were then asked to discontinue their current pain medications ~14 days (2 weeks) prior to their baseline scanning visit and take only acetaminophen rescue medication provided at the visit during this time. Patients were instructed to rate their pain and mood with the phone app two times a day for the duration of the study. Participants completed the questionnaires and were scheduled for visit 2.
2.3.2. Visit 2 (week -1): Baseline MRI (90 min).
Approximately 14 days after Visit 1, participants were invited for their second visit. If patients reported an average VAS score of > 4/10 (rounded) over the course of the baseline rating period and no changes in clinical status were reported or found, they were invited to complete the questionnaires and undergo fMRI procedures (anatomical and functional scans). Use of rescue medication was documented and patients were queried about changes to health experienced since their last visit. They were asked to continue to provide phone app ratings and return in approximately one week for visit 3. Rescue medication was dispensed in sufficient quantities until the next visit.
2.3.3. Interim between Visit 2 and Visit 3: Stratification of patients
The patients’ scans were analyzed between Visit 2 and 3 by extracting measurements of interest from the brain (determined a priori by results from Phase 1) and fitting them to a four-parameter prediction model using an automated pipeline (as shown in fig. S1). Each patient was stratified into either predicted placebo responders or predicted placebo non-responders. This information was given to a member of the Northwestern Research Pharmacy ensuring that the study medication was randomized according to block and group assignment. A one-week interim period was chosen to allow enough time for selected study staff to run the prediction analysis and send this information to the pharmacy, as well as provide another week of additional baseline pain ratings. The duration of this period may have been slightly longer or shorter depending upon how quickly the model was run.
2.3.4. Visit 3 (week 0): Start of Treatment Period (30-45 min)
After prediction and stratification, participants were invited to return for Visit 3. A set of questionnaires was administered, the use of rescue medication was documented, patients were queried about changes to health experienced since the last visit, and vitals were also collected. Participants were randomized to active treatment, placebo, or no treatment groups in a blind fashion (double for treatment and partial for no treatment). An independent assessor escorted the participant to the pharmacy at the end of the visit where they picked up their study medications, if assigned to a treatment arm. This was to ensure that the remaining study staff remained blind to treatment versus no-treatment group assignment.
The placebo group received two placebo capsules bid, and the active treatment group received one naproxen capsule (500mg) and one esomeprazole capsule (20 mg) bid for the treatment period. Study medication was dispensed in sufficient quantities until the next visit. All participants in all groups (including the no treatment) received rescue medication in sufficient quantities to last until the next visit. Patients were asked to continue their daily pain/mood ratings and were invited to come back 3 weeks later for a follow-up visit.
2.3.5. Visit 4 (week 3): Continuing Assessment (30-45 min).
This visit was completed entirely by the independent assessor as there were no clinical data collected at this visit. Adherence was assessed using pill counting for those in a treatment group and patients were queried about any side effects experienced. Study medication and rescue medication was dispensed in sufficient quantities until the next visit, as described in Visit 3. They were asked to continue using the phone/computer app and to come back in 3 weeks for their end-of treatment visit.
2.3.6. Visit 5 (week 6): End of treatment/Start of washout (60-90 min):
Participants underwent the same brain scanning session that they had on Visit 2, and all questionnaires, adverse events assessment, and treatment/rescue medication adherence procedures were performed as in previous visits. Participants were asked to stop the study medication (if applicable) and to start the washout period. They were only provided rescue medication at this visit. They were asked to continue rating their pain and mood on the app and invited to return for their final visit.
2.3.7. Visit 6 (week 9): Final visit/End of washout (30-45 min):
Adherence to rescue medication was assessed, potential changes to health were documented, and vitals were collected. A short battery of questionnaires was administered, and participants were compensated for all app ratings completed during the study.
2.4. Interim periods (smart phone pain rating):
During weeks in between visits, participant data were tracked with the assistance of a smart phone/computer application designed for the study (available through a secure website). Depending upon preference, an app was installed onto the patients’ phones or provided to them via a link to access on their personal computers; in the case that a participant did not have a smart phone or easy access to a computer/internet, we provided him/her with a smart phone with the app already installed. Participants were given a login and were asked to use this application twice a day, morning and evening, for study duration; if applicable, they were asked to rate immediately after taking their medication for treatment to better ensure compliance with both tasks. The app asked patients to rate their pain severity and their mood on Likert scales, and probed them as to whether or not they took their medication. This procedure was exactly the same as in Phase 1.
To encourage compliance, patients received 25 cents for each pain entry. Responses and payments were logged automatically into a secure database that only study personnel had access to; if participants missed more than 3 consecutive ratings in a row, research personnel called or texted them to remind them to rate or verify that their app is still working (research personnel were automatically alerted via email by a program designed by the lab). The independent assessor designated for this study checked participants ratings bi-weekly to verify that ratings were being completed. The phone data were preprocessed the same way as in Phase 1 ([61]): the last pain entry was kept if multiple pain entries occurred within 30 minutes and missing entries were not replaced.
2.5. Randomization
All randomization (and blinding, see next section) was completed by the Northwestern University Clinical and Translational Sciences Institute (NUCATS). We used a randomized block designed where patients were first divided into homogenous blocks of predicted-PTxResp and predicted-PTxNonR in which the randomization ratio was 3 PTx: 3 MTx: 1 NoTx. This randomization ensured that predicted-PTxResp and predicted-PTxNonR were balanced across the 3 arms. The overall sample size that was randomized was n = 94: n = 40 PTx (43% predicted-PTxResp), n = 40 MTx (46% predicted-PTxResp), and n=14 NoTx (39% predicted-PTxResp). The trial was stopped after all the expected participants were enrolled and completed the study.
2.6. Blinding Procedures
Although this is listed as a double-blinded, randomized control trial, it can more accurately be described as a 3-arm randomized controlled trial in which two arms were double-blind and the third arm consisted of a no-treatment group that was unblinded but had a blinded assessor. All of the personnel conducting the study, collecting study endpoints, and analyzing data, including the PI, were blinded to the allocation of participants to study arm and to treatment. All pills were identically encapsulated by pharmacists (who are not involved in any other aspect of the study) to ensure a double blind for study participants who received treatment and all study personnel other than the designated assessor. All pills were identical in size but were encapsulated using two colors– blue and bi-colored. This was to help the participant remember to take two different tablets at each time and thus reduce the possibility of error. The only person knowing which capsules contained active vs placebo was the unblinded pharmacist responsible for ordering the medication, re-encapsulating the agent, and filling the numbered medication vials with the appropriate kind and number of capsules. This person had no other involvement with study staff.
2.7. Brain Imaging
Brain imaging scans were used only to extract resting state functional connectivity as specified in the biosignature, which were entered into the model and used to stratify subjects as placebo responders or not responders.
Brain imaging data were acquired with a Siemens Magnetom Prisma 3 Tesla. High-resolution T1-weighted brain images were collected using integrated parallel imaging techniques (PAT; GRAPPA) representing receiver coil-based data acceleration methods. The acquisition parameters were: isometric voxel size = 1 × 1 × 1 mm, TR = 2300 ms, TE = 2.40 ms, flip angle = 9°, acceleration factor of 2, base resolution 256, slices = 176, and field of view (FoV) = 256 mm. The encoding directions were from anterior to posterior, and the time of acquisition was 5 min 21 s.
Blood oxygen level-dependent (BOLD) contrast-sensitive T2*-weighted multiband accelerated echo-planar-images were acquired for resting-state fMRI scans. The acquisition parameters were: TR = 555 ms, TE = 22.00 ms, flip angle = 47°, base resolution = 104, 64 slices with a multiband acceleration factor of 8 (8 × 8 simultaneously acquired slices) with interleaved ordering. High-spatial resolution was obtained using isomorphic voxels of 2 × 2 × 2 mm, and signal-to-noise ratio was optimized by setting the field of view (FoV) to 208 mm. Phase encoding direction was from posterior to anterior. The time of acquisition lasted 10 min 24 s, during which 1110 volumes were collected. Patients were instructed to keep their eyes open and to remain as still as possible during acquisition. The procedure was repeated two times, but all functional connectivity was extracted only from the first run. The parameters were the same as in Phase 1 and additional details are presented in [61].
2.7.1. Preprocessing of functional images
The pre-processing was exactly the same as in Phase 1 and the details are presented in [61]. Briefly, we used FMRIB Software Library (FSL) and in-house software. The effect of intermediate to large motion was initially removed using fsl_motion_outliers. Time series of BOLD signal were filtered with a Butterworth band-pass filter (0.008 Hz < f < 0.1 Hz) and a non-linear spatial filter (using SUSAN tool from FSL; FWHM = 5 mm). Following this, we regressed the six parameters obtained by rigid body correction of head motion, global signal averaged overall voxels of the brain, white matter signal averaged overall voxels of eroded white matter region, and ventricular signal averaged overall voxels of eroded ventricle region. These nine vectors were filtered with the Butterworth band-pass filter before being regressed from the time series. Finally, noise reduction was completed with Multivariate Exploratory Linear Optimized Decomposition into Independent Components (MELODIC tool in FSL) that identified components in the time series that were most likely not representing neuronal activity. Components representing motion artefact were identified if a ratio between activated edge (one voxel) and all activated regions on a spatial component was >0.45, or if ratio between activated white matter and ventricle and whole-brain white matter and ventricles was >0.35. Moreover, noisy components were identified if the ratio between high frequency (0.05–0.1) and low frequency (0.008–0.05) was >1. This ICA regression process was kept very conservative so that only components obviously related to motion or noise were removed.
The functional image registration was optimized according to a two-step procedure. All volumes were averaged within each patient to generate a contrast image that was linearly registered to the MNI template and averaged across patients to generate a common template specific to our CBP patients. All pre-processed functional images were then non-linearly registered to this common template using FNIRT tool from FSL. The registered brains were visually inspected to ensure optimal registration.
2.8. Questionnaires
Multiple questionnaires were administered at Visit 1 to assess pain, measure mood, and make the predictions about placebo response. Pain measurements included: McGill Pain Questionnaire; Pain Detect; Pain Catastrophizing; Oswestry Disability Index. Psychological factors for the predictive biosignature model included; Multidimensional Assessment of Interoceptive Awareness (MAIA); Emotion Regulation Questionnaire (ERQ) (see supplementary methods for coding error in the ERQ subscale). The Loss Aversion Questionnaire was also administered but not used. The Beck Depression Index 1a (BDI) was administered because of inclusion criteria.
2.9. Generalizing the results to osteoarthritis patients
Post-hoc exploratory analyses, testing for generalizability of components of the biosignature, included a re-analysis of a RCT conducted in osteoarthritis patients comparing duloxetine treatment to placebo [58]. Here, we extracted the functional connectivity between the DLPFC-PreCG and DLPFC-PAG using the preprocessed resting state functional connectivity as in [58]. Details about the RCT, the sequence of acquisitions, the preprocessing of the data are presented in openly available [58]. The raw data are also available on http://www.openpain.org/.
2.10. Data quantification and statistical analysis
Statistical analyses were performed using Matlab R2019b and SPSS version 24. Primary outcome measure was twice daily ratings of current back pain intensity, averaged over a week, and compared between and within stratification and treatment groups. Biosignature model output was a probability of placebo response, P(resp/biosignature), which returned a 0.0-1.0 continuous value, where P(resp)>0.5 was classified as identifying a placebo responder and P(resp)≤ 0.5 as nonresponder (see supplementary methods). One-way analysis of variance (ANOVA) was use to contrast demographic characterisitcs between stratification and treatment groups, followed with post-hoc test without correction for repeated measures. Biosignature factor values between stratification groupings was tested for differences using 2-way t-tests. 2-way t-tests were also used to compare functional connectivity values between responders and non-responders in different placebo studies. Permutation test was used to define individual subject responses to treatment, see [61]. 2-way repeat-measures ANOVAs were used with time (repeat measure) and grouping as independent factors, and percent change in pain intensity relative to baseline as the dependent parameter. The post-hoc tests were one-tail (our model specifically predicted analgesia in the predicted-responders) and Bonferroni corrected for 2 comparisons (predicted responders Vs predicted non-responders and no treatment; significance was set at p < 0.05). 2-way repeat-measures analysis of covariance was used to examine raw pain response properties, with baseline pain as covariate, time (repeat measure) and grouping as independent factors and raw pain intensity as the dependent parameter. Individual subject response predictability was based on permutation based classification, followed by receiver operator curve (ROC) calculation and area under the curve (AUC) measurement to determine overall accuracy of of matching prediction with observed outcomes.
2.10.1. Intent-to-treat analyses
The intent to treat analyses included all participants that were randomized. However, four participants could not be used because of the absence of pain entries after visit 2 (dropped out after randomization but prior to receiving treatment) and one additional participant was unable to use the phone to enter pain ratings. All other participants (n = 89) were included in the intent-to-treat analyses. For the group analyses, the missing values were replaced by the last pain entry at the time of dropout. For the individual analyses (permutation test to determine Resp or NonR; fig. S4), data collected from participants were included up to the point of dropout. The results of the intent-to-treat analyses were compared to the per-protocol analyses.
2.10.2. Per-protocol analyses
The per-protocol analyses included participants that completed the study. There were 80 patients that completed all 6 visits but three of them did not enter any pain entries during the washout period. These were excluded and the per-protocol analyses included a total of 77 patients.
Data availability:
All our previous data (including Phase 1 data) are currently available at http://openpain.org/. Phase 2 numerical data and the raw data will also be made available at the time of publication, and codes used for permutation testing in Phase 1 and Phase 2 are available at https://github.com/EtienneVP.
3. Results
3.1. Participants, stratification, and treatment groups
This neuroimaging-based study is the second phase (Phase 2) of a research program aiming at testing a biosignature predicting placebo response in CBP patients in the setting of a randomized control trial (RCT). Here, 181 CLBP patients were assessed for eligibility, from which 94 were randomized (CONSORT flowchart presented in fig. 1). From these, 89 were used in intent-to-treat analysis, 77 completed the study and were used in per-protocol analysis. The Institutional Review Board of Northwestern University approved the study and all participants signed a consent form at Visit 1.
In Phase 2, we applied the biosignature identified in the first phase of the trial (Phase 1) to classify the CLBP patients as placebo responders (predicted-PTxResp; n = 41) or non-responders (predicted-PTxNonR; n = 53). Homogenous blocks of predicted-PTxResp and predicted-PTxNonR patients were then randomized into: a placebo treatment arm (PTx, n=40); an active medication treatment arm (MTx, n=40), a nonsteroidal anti-inflammatory drug (naproxen); or a no treatment arm (NoTx, n=14) controlling for spontaneous improvement of symptoms contingent on exposure to the healthcare setting. From these, five participants withdrew before starting the treatment period and were excluded from analyses. The patients then started a six-week treatment period at visit 3 (visit 3 - visit 5), followed by a three-week washout period (visit 5 - visit 6). Throughout the trial, pain intensity was measured twice a day using a smartphone app, as in Phase 1.
In the first phase of this research program (Phase 1), we derived psychological and neurophysiological features determining placebo response in CLBP patients by contrasting placebo responders (PTxResp) with non-responders (PTxNonR) relative to a no-treatment group (NoTx) based on brain functional connectivity and personality (fig. 2a; details about the blinding of the analyses and the development of the predictive model are presented in supplementary material and ClinicalTrails.gov NCT02013427). A logistic regression was used to identify four features determining placebo response in patients from Phase 1 prior to conducting Phase 2 study (fig. 2b-c). In the current Phase 2 (fig. 2d), we applied the same model blindly to a new group of patients using an automated pipeline to extract relevant functional connectivity features of resting state (fig. 2e) that were combined with the questionnaire scores (fig. 2f). The patients were classified as predicted placebo responders (predicted-PTxResp, if P(resp/biosignature)>0.5) or non-responders (predicted-PTxNonR, if P(resp/biosignature)<0.5) prior to randomization, thus preventing model flexibility [44] and minimizing bias. Therefore, we used two independent RCTs: Phase 1 was used to identify the features and Phase 2 was used to test the validity of this fixed and pre-defined biosignature in a new group of participants. The phase 2 patients were studied for a longer time-period, and randomization to treatment types provided the opportunity to study the interaction between placebo pills and an active analgesic treatment across predicted-PTxResp and predicted-PTxNonR.
fig. 2. Biosignature for placebo response prediction and its propeties in Phase 2.

a-c. The biosignature for placebo response was developed in Phase 1 [61]. a. Design: A RCT comprising 6 visits to the lab and lasting about 8 weeks was used to develop a biosignature of placebo pill response. Patients were exposed to either no treatment (NoTx), placebo treatment (PTx), or an active medication treatment (MTx; not analyzed). All Phase 1 analyses were performed blindly using 3 randomized codes; only one of them correctly labeling the responders, non-responders, and no treatment (details presented in supplementary material). Contrasting between placebo responders (PTxResp) and non-responders (PTxNonR) identified predictive features (number of subjects in each group indicated in the boxes). b-c. Derived biosignature was based on four independent features: functional connectivity between the dorsolateral/ventrolateral prefrontal cortex (DLPFC/VLPFC) with the precentral gyrus (Pre-CG; c) and the DLPFC with periaqueductal grey (PAG; c); and capacities of awareness (MAIA-emotion) and regulation of emotions (ERQ-subscore; details about the model are presented in supplementary material). d. Current phase 2 study design: consisted of six visits to the lab and lasted about 12 weeks during which patients were exposed to NoTx, PTx, or MTx lasting 6 weeks. The biosignature from phase 1 was used to stratify patients as a predicted-PTxResp (if P(resp)>0.5) or predicted-PTxNonR (if P(resp<0.5) before randomization. e-f. Each of the four components of the biosignature distinguished between predicted-PTxResp and predicted-PTxNonR in Phase 2 participants. * p<0.05, ***p<0.001.
3.2. Predictability of responses at the group level
Two examples of patients’ daily ratings are displayed in fig. 3a. The distribution of probability of placebo response was bimodal: the median probability of placebo response within predicted-PTxResp was close to 1 and close to 0 in predicted-PTxNonR (fig. 3b). Our model predicted that 37/89 (42%) of the participants included in the intent to treat analyses would respond to placebo. The relationship between extent of pain relief (%change from baseline to last week of treatment) and predicted probability of placebo response indicated that the model performance is better in identifying non-responders than responders (fig. 3b). Figure 3b shows that the distribution of pain relief was also bimodal where a group of patients showed close to 0% change in pain intensity. Thus, pain relief for predicted-PTxNonR was more clustered around 0% change in pain at the end of the treatment period, but the distribution of pain relief for predicted-PTxResp did not show a clear relationship with predicted probability of placebo response.
fig. 3. Interaction between placebo predictability, treatment types, and pain relief.

a. Two examples of pain entries in one non-responder (NonR, top panel) and one responder (Resp). b. The pain relief (%change from baseline to last week of treatment) is displayed with respect to the probability of placebo response provided by the biosignature for PTx and MTx. c. The %change from baseline are displayed between the three treatment arms. Each bin represents pain entries averaged over approximately one week of pain entries. d. The %change from baseline are displayed between the Predicted PTxResp, Predicted PTxNonR, and no treatment (NoTx). e. The %change from baseline are displayed between the Predicted PTxResp, Predicted PTxNonR receiving either placebo (PTx) or NoTx. f. The %change from baseline are displayed between the Predicted PTxResp, Predicted PTxNonR receiving either active drug (MTx) or NoTx. ITT: Intent to treat; * p < 0.05; ** p<0.01, post hoc comparison are one-tail and Bonferroni corrected for 2 comparisons (significance established at p < 0.05). Error bars are standard errors.
We grouped pain relief time courses into four different arrangements to test the influence of predicted placebo response on placebo pill use and on active drug treatment. First the data is segregated only by treatment type (NoTx, PTx, MTx), where each treatment arm was composed of equal numbers of randomized CLBP participants of both types (predicted-PTxNonR and predicted-PTxResp). The interaction group*time was not significant due to small sample size (repeated measure ANOVA(8.78,377.71), F=1.42; p=0.18), but we observed the expected orderly response: close to zero pain relief in NoTx (3.61 ± 13.66% (mean ± SD)), pain relief of 12.75 ± 28.55% in PTx, and pain relief of 22.49 ± 31.54% in MTx (fig. 3c). When the data are grouped by predicted-PTxNonR and predicted-PTxResp irrespective of treatment type received and compared to NoTx (fig. 3d), we observe a significant group*time interaction (repeated measure ANOVA(9.33,401.19), F=3.44; p<0.001). The predicted-PTxNonR exhibited minimal pain relief (11.21 ± 25.10%, not different from NoTx group), while the predicted-PTxResp experienced 26. 74 ± 34.73% pain relief, a significant decrease compared to NoTx (p= 0.02) and predicted-PTxNonR (p= 0.01). Thus, the biosignature categorization more than doubles backpain relief effect size in predicted-PTxResp irrespective of treatment type, confirming validity of predicted-placebo-response at this group level.
We next separated the data by type of treatment and tested the group*time interaction. For PTx treatment (fig. 3e) (repeated measure ANOVA(10.04,235.99), F=3.15; p<0.001), CLBP participants categorized as predicted-PTxNonR exhibited minimal pain relief with PTx and matched the NoTx response, while those categorized as predicted-PTxResp experienced 21.25 ± 35.78% pain relief, lower than predicted-PTxNonR (p= 0.01) and NoTx groups (p= 0.03). These results specifically confirm the validity of our model for placebo pill ingestion by appropriately segregating placebo-responders from non-responders. Given that the predicted-PTxNonR showed no pain relief, the pain relief observed in predicted-PTxResp may be considered the magnitude of the pure placebo response, for this specific CLBP population, and within the context of this trial design (neutral instructions and unbiased classification of predicting placebo responders).
For MTx treatment, time by group interaction was significant (repeated measure time*group ANOVA(7.65,187.39), F=2.07; p=0.04) (fig. 3f). The CLBP participants categorized as predicted-PTxNonR exhibited 15.46 ± 28.19% pain relief with MTx, which approximately doubled in predicted-PTxResp, 31.58 ± 34.10 (significantly lower than NoTX, p = 0.03). The findings show that our biosignature could consistently predict what is believed to represent the placebo response across both placebo and active treatment arms. Given that predicted-PTxNonR in PTx showed no change in pain (fig. 3e), it is reasonable to suggest that the MTx responses in predicted-PTxNonR identifies the pure pharmacological effect of the MTx, which seems slightly less than the pure placebo effect size. Overall, these results isolate the pure effect sizes for placebo and for an analgesic drug treatment, and demonstrate approximate additivity of pain relief in predicted-PTxResp for placebo and active treatment responses.
After treatment termination, participants continued to rate their back pain for 3 more weeks (washout period). We expected to observe distinct time constants for extinction of pain relief for PTx and MTx treatments. Consistent with this notion, we observe extinction for PTx treatment, but not for MTx, when data was segregated only by treatment type (fig. 3c). Surprisingly, however, the data segregated by biosignature over the last week of the treatment and the 3-week observation period after cessation of pill ingestion, as revealed by a time by group interaction (repeated ANOVA F(10.17, 213,61) = 2.65; p = 0.004). Post hoc compairons showed that the predicted-PTxResp receiving PTx, and both predicted-PTxNonR and predicted-PTxResp groups receiving MTx resisted extinction (return to baseline or to NoTX pain levels; fig. 3e-f). Only the predicted-PTxNonR for PTx treatment showed rapid small extinction effect after cessation of pill ingestion (even though this group showed no pain relief during PTx treatment, p = 0.005; Bonferroni corrected for 5 post-hoc comparisons), exhibiting a trend for a nocebo effect (worse pain than NoTx). The persistence of post-treatment pain relief was also observed in Phase 1 study [61]. Return to baseline during washout would have strengthen the argument that observed effects are causal to the treatment. One reason for a lack of washout may be non-specificty of observed results. The latter worry is however counterbalanced by the NoTx arm showing no change in pain during this whole observation period, which in turn favors the alternative interpretation that pain relief, when maintained for multiple weeks, creates an extinction resistant state independent of the type of treatment used.
Additional analyses were performed to ensure the robustness of our findings at the group level. Examining the raw scores indicated that the averaged baseline pain intensity was higher in the NoTx group but did not differ between predicted-PTxResp and predicted-PTxNonR or the PTx and MTx arms (Table 1). Fig. S2 shows the raw pain ratings, including baseline. Testing a group*time interaction when entering pain intensity at baseline as covariate showed that group differences were still detected with raw scores (repeated measure ANCOVA(15.46,320.68), F = 4.71; p = 0.002). We secondly performed per-protocol analyses and showed that the results were almost identical with those from the intent-to-treat analyses (repeated measure ANOVA(19.72,355.00), F = 2.29; p = 0.002; fig. S3).
Table 1:
Mean and standard deviation from the mean (std) are presented for the baseline pain intensity measured using the phone app, pain duration (weeks), age, and sex. Group comparisons show no differences between the groups for pain duration, age or sex. A one-way ANOVA however showed that baseline levels of pain intensity were different between the groups (F(84,4) = 3.23; p = 0.02). Uncorrected post-hoc comparisons showed that baseline was higher or marginally higher than all other groups (all p’s < 0.07). There were no significant differences between Predicted PTxResp and PTxNonR in either PTx or MTx.
| No treatment |
Predicted PTxNonR
receiving PTx |
Predicted PTxResp
receiving PTx |
Predicted PTxNonR
receiving MTx |
Predicted PTxResp
receiving Mtx |
|
|---|---|---|---|---|---|
| Mean pain intensity (std)* | 7.00 (1.52) | 5.98 (1.22) | 5.25 (1.15) | 6.20 (1.25) | 6.00 (1.43) |
| Mean pain duration (std) | 458.67 (489.71) | 475.55 (127.32) | 263.44 (291.50) | 396.60 (380.33) | 323.24 (406.01) |
| Age | 57.00 (11.57) | 60.78 (8.77) | 54.98 (16.37) | 55.77 (10.89) | 51.26 (11.65) |
| Sex(women) | 4/13 | 10/22 | 14/22 | 5/15 | 8/17 |
3.3. Predictability of responses at the individual level
We next tested the diagnostic value of our biosignature at the individual level. A permutation test between pain entries during baseline and during treatment (cutoff of p<0.05) was used to identify the actual treatment responders (Resp) and non-responders (NonR), as in Phase 1, see [61]. We computed the receiver operating characteristics (ROC) curves to determine if our biosignature showed good discrimination value. The area under the ROC curve (AUC) was 0.64 (95% confidence interval [C.I], 0.51 to 0.77; p=0.04) when combining all patients receiving treatment, AUC was 0.61 (95% C.I, 0.42 to 0.80; p=0.26) in the PTx, and AUC was 0.67 (95% C.I, 0.50 to 0.84; p=0.07) in the MTx (fig. S4). Although some of these results were statistically trending (MTx), or significant (pooling all participants), AUCs between 0.6 and 0.7 are normally considered as having a poor, but above chance, diagnostic value.
3.4. Generalizability of placebo predicting biosignature
We finally used univariate statistics to explore which features best segregated Resp from NonR in PTx and MTx. These exploratory analyses were performed a posteriori and only compared Resp (n=36) from nonR (n=40) that received and completed treatment per-protocol. There were no group differences between Resp and NonR in psychological factors (all p’s>0.22) or DLPFC-PAG connectivity (all p’s>0.08). The functional coupling of the DLPFC-PreCG was however stronger in Resp in both PTx (t(31)=2.10, p = 0.048) and MTx (t(31)=2.62, p=0.01; fig. 4a). This result was further generalized in a re-analysis of placebo response, compared to duloxetine response in OA patients [58], where Resp also showed stronger DLPFC-PreCG connectivity compared to NonR (t(34)=2.23, p=0.03; see supplementary material). Fig. 4b shows that this unique brain feature segregated Resp from NonR with AUCs of 0.75 for PTx (95% C.I, 0.56 to 0.93; p=0.02), 0.75 for MTx (95% C.I, 0.57 to 0.92; p=0.03), and 0.70 for the re-analysis of placebo response in OA data (95% C.I, 0.53 to 0.88; p=0.04). The result seen in OA data is an unbiased out-of-sample validation of DLPFC-PreCG functional connectivity predicting placebo responders in a data where participants are distinct and the placebo trial design is also distinct. Therefore we can conclude that the DLPFC-PreCG coupling represents a potential candidate biosignature for predicting placebo response across multiple kinds of patient cohorts and for two distinct active treatment types.
fig. 4. Placebo responders show stronger DLPFC-PreCG connectivity prior to trial exposure across three clinical trials.

a. The red rectangle illustrates the initial finding in Phase 1. Patients from Phase 2 responding to either PTx or MTx showed stronger connectivity between the DLPFC-PreCG. Results were generalized in osteoarthritis patients (OA), where placebo responders also showed stronger DLPFC-PreCG connectivity. b. The AUC showed the sensitivity and specificity of the DLPFC-PreCG to classify placebo responders (Resp) and placebo non-responders (NonR). * p<0.05; ** p<0.01 two-tailed.
4. Discussion
This study had two main objectives: to validate a biosignature for predicting placebo response in RCT and to test whether placebo and active treatment effects could be dissociated from each other. We provide evidence for such a biosignature, as the predicted-PTxResp receiving PTx showed stronger pain relief compared to both the predicted-PTxNonR receiving PTx and the NoTx group. Moreover, the biosignature isolated pure active treatment effects and showed additivity of pain relief; demonstrating both model validity and utility at the group level. We also demonstrate that pure placebo responses are sustained over multiple weeks after withdrawing treatment, highlighting the role of learning and related brain adaptations to the pain observed in CLBP. At the single subject level, our model performed poorly but above chance. However, post-hoc analyses revealed that the DLPFC-PreCG functional network was sufficient in predicting placebo responders from non-responders with moderate efficacy across three different clinical trials.
Multiple components of present observations require further discussion: 1) unbiased application of the biosignature; 2) dissociation of pure placebo from pure active treatment effects, and demonstration of their additive interactions; 3) the lack of deviation from baseline pain in the NoTx group for both studies; 4) biosignature-based classification leads to a robust dichotomous outcome at a group level, independent of treatment type, including minimal pain relief in the predicted-PTxNonR and a clinically meaningful analgesia in the predicted-PTxResp group; and finally 5) observed results suggest the predicted-PTxNonR and the predicted-PTxResp posses distinct characteristics.
1) An important contribution of our study to the placebo literature, and to the more general field of developing appropriate biomarkers, is that the previously-identified biosignature was applied blindly to classify participants prior to randomization. Although the risk of failure is higher in such experimental designs, the approach has the advantage of reducing model flexibility, decreasing likelihood of overfitting, and minimizing analysis bias. Thus, we demonstrate that placebo pill response is a priori predictable in RCT, which enhances its usefulness as a viable treatment option, even in open-labeled placebo trials [14]. Moreover, identifying responders apriori empowers clinical trial design: better-balanced allocation of participants, decreasing placebo effect size by using placebo run-in designs [27], or eliminating the placebo confound by excluding predicted-responders during enrollment, in turn targeting pure active treatments.
2) The use of three different arms permitted leveraging our biosignature across multiple contexts. We reasoned that the addition of a no-treatment arm could control for the natural history of patients enrolling in the RCT [32], and an active treatment arm could isolate pain relief specific to each treatment type. Thus, identifying placebo non-responders may reveal pure pharmacological effect of tested drug, as these subjects are expected to demonstrate zero placebo response when exposed to an active drug treatment. On the other hand, proper a-priori identification of placebo responders exposed to placebo pills also allows for the isolation of the pure placebo response. Our results show that the pure active treatment effect corresponded to about 10% decrease and the pure placebo-specific effect to 15-20% decrease in backpain. Thus, in line with Benedetti’s concept [9], we could partition a commonly dispensed analgesic into its pure pharmacological and psychological (placebo) components.
The only other study where pure placebo and pharmacological effects were separated compared open-label administration of various analgesics to hidden procedures, in healthy subjects [3]. Overall their results indicate ~10% decrease in drug efficacy when administration was hidden, thus identifying the effect size for psychological influences. Even though this effect seems similar to ours, the two studies diverge in methods, pain conditions and duration of drugs used. The pure placebo effect we see under neutral instructions should be regarded as the minimum possible which may be enhanced by various conditioning paradigms [54].
It is informative to compare observed effect sizes for pure placebo, obtained under a doubleblind with neutral instructions and unbiased predictors (PUP), with corresponding clinical literature. Two open label placebo (OLP) RCTs in CLBP [14,41] show back pain decrease by about 15-30%. This OLP effect size should be even larger, assuming around 50% of the participants may have been nonresponders. The PUP and the OLP closely match in design and treatment duration, except OLP participants were explicitly instructed about the power of placebo. These positive instructions seems to have increased the effect size in OLP by 4 times that of PUP, consistent with evidence that instructions induce large hypoalgesic effects [6,11,18,23].
NSAIDs including naproxen are the most common treatment option for CLBP, their efficacy is extensively studied and exhibit superiority over placebo [15,25,50]. A recent Cochrane review (6 trials, 1354 participants) reports NSAIDs more effective than placebo only by 3.3%, [25]. This value is even less than our estimate of the pure pharmacological effect for naproxen (~10%) but lies within its uncertainty range, and is nicely consistent with our observation that segregating participants by biosignature is more informative than by treatment type.
Additivity of placebo and drug effects were expected since placebo is based on central mechanisms, whereas naproxen is mainly active in peripheral tissue [13]. Our earlier study of placebo predictability examined placebo in comparison to duloxetine, a centrally-acting SSRI [47], and showed a multiplicative interaction. Thus, together the two studies highlight the concept that placebo-drug interactions are also drug-type (site of action) dependent.
3) Under the current study design, in both phase 1 and phase 2 studies, we observed no statistically meaningful change in pain in the NoTx arm over many weeks. This is in contrast with meta-analytic results [32], probably mainly due to repeated assessment of current pain which should diminish recall biases [55], [10,48], as well as inclusion of at least 1-week of baseline pain assessment. If one assumes that NoTx pain does not differ in intensity from initial enrollment, then the placebo effect size can be directly estimated from extant literature. For example, in a state-of-the-art RCT (n=111 treated with duloxetine, n=120 with placebo, for 13 weeks), report a 30% decrease in backpain with duloxetine in contrast to a 20% decrease with placebo [53]. Presuming a null no-treatment effect (and for simplicity falsely assuming additivity, see above), one can conclude that the efficacy of pure placebo is twice as large as pure duloxetine. Moreover, if ~50% of these participants were placebo non-responders, then placebo hypoalgesia would be four times larger than that of duloxetine. With such knowledge, would physicians opt to prescribe duloxetine rather than placebo for treating CLBP, especially if they could readily identify placebo responders apriori and consequently minimize exposure to active drug side effects?
4-5) In principle the derived biosignature (a Bayesian model, similar to [12]) is agnostic regarding underlying mechanisms. However, differential properties of identified subgroups suggest novel mechanistic insights. At the group level, we observe no pain relief with PTx treatment in predicted-PTxNonR, rapid extinction post-treatment showing a small nocebo effect, which was consistent at the individual level. In contrast, the predicted-PTxResp showed robust pain relief with PTx exposure at the group level, no extinction post-treatment cessation after either PTx or MTx, and a wide distribution of pain relief at the individual level. Our biosignature should be capturing the accumulated priors (learning and expectation influences of past history), and everyone received the same neutral instructions; given this, the analgesia distribution differences between predicted-PTxNonR and predicted-PTxResp can thus only be due to differential responses to environmental or experiential influences. Placebo hypoalgesia studies emphasize the contribution of classical conditioning [2,18,49,66,67], expectation conditioning [21,22,39,40], and social observational/instructional [5,19,35,43,56,67] learning mechanisms, which have been probed extensively regarding underlying circuitry [12,20,26,42,43,68] (for differential contributions see [4]). More recent data demonstrate that operant learning (an influence consequent to exposure) also contributes to placebo hypoalgesia, and operant placebo effects show resistance to extinction [1]. The latter may account for the differences between predicted-PTxNonR and predicted-PTxResp, but will require future studies.
Regarding our brain imaging results, the involvement of the DLPFC in placebo response has been previously demonstrated [45], [34,46,69]. Here, we extend this knowledge by showing that DLPFC functional connectivity in chronic pain patients predisposed them to respond to placebo pills; both in CLBP patients with napoxen, and OA patients with duloxetine, highlighting its potential for generalizability.
Our study has several limitations. The main weakness of the biosignature was poor predictability of individual participants’ magnitude of pain relief, especially in those predicted to be placebo responders. The univariate analyses suggest that some features of the model could be further optimized as the psychological factors did not distinguish responders from non responders in this new group of patients. Yet it is also possible that a substantial component of the placebo effect may simply remain unpredictable. Overall, our results demonstrate that placebo responses observed in the setting of a RCT can actually be predicted a priori in chronic pain patients at the group level, and also suggests that simpler models perform better on individual patients.
Supplementary Material
Acknowledgement:
We are thankful to Dr. Luana Colloca for her feedback on the manuscript.
Funding
Funded by NIH/NCCIH R01AT007987; ClinicalTrials.gov NCT02986334
Grant Information:
Funded by National Center for Complementary and Integrative Health AT007987. EVP was funded through Canadian Institutes of Health Research (CIHR) and Fonds de Recherche Santé Québec (FRQS).
Footnotes
Authors declare no competing interests.
References
- [1].Adamczyk WM, Wiercioch-Kuzianik K, Bajcar EA, Babel P. Rewarded placebo analgesia: A new mechanism of placebo effects based on operant conditioning. Eur J Pain 2019;23:923–935. [DOI] [PubMed] [Google Scholar]
- [2].Amanzio M, Benedetti F. Neuropharmacological dissection of placebo analgesia: expectation-activated opioid systems versus conditioning-activated specific subsystems. J Neurosci 1999;19:484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Amanzio M, Pollo A, Maggi G, Benedetti F. Response variability to analgesics: a role for non-specific activation of endogenous opioids. Pain 2001;90:205–15. [DOI] [PubMed] [Google Scholar]
- [4].Babel P. Classical Conditioning as a Distinct Mechanism of Placebo Effects. Front Psychiatry 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bajcar EA, Babel P. How Does Observational Learning Produce Placebo Effects? A Model Integrating Research Findings. Front Psychol 2018;9:2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bartels DJP, van Laarhoven AIM, Stroo M, Hijne K, Peerdeman KJ, Donders ART, van de Kerkhof PCM, Evers AWM. Minimizing nocebo effects by conditioning with verbal suggestion: A randomized clinical trial in healthy humans. PLoS One 2017;12. doi: 10.1371/journal.pone.0182959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Benedetti F. Placebo and the new physiology of the doctor-patient relationship. Physiol Rev 2013;93:1207–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Benedetti F. Placebo Effects. Oxford University Press, 2020. [Google Scholar]
- [9].Benedetti F, Pollo A, Lopiano L, Lanotte M, Vighetti S, Rainero I. Conscious expectation and unconscious conditioning in analgesic, motor, and hormonal placebo/nocebo responses. J Neurosci 2003;23:4315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Berger SE, Vachon-Presseau É, Abdullah TB, Baria AT, Schnitzer TJ, Apkarian AV. Hippocampal morphology mediates biased memories of chronic pain. NeuroImage 2018;166:86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bräscher A-K, Witthöft M, Becker S. The Underestimated Significance of Conditioning in Placebo Hypoalgesia and Nocebo Hyperalgesia. Pain Research and Management 2018;2018:e6841985. doi: 10.1155/2018/6841985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Buchel C, Geuter S, Sprenger C, Eippert F. Placebo analgesia: a predictive coding perspective. Neuron 2014;81:1223–1239. [DOI] [PubMed] [Google Scholar]
- [13].Burian M, Geisslinger G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacology & Therapeutics 2005;107:139–154. [DOI] [PubMed] [Google Scholar]
- [14].Carvalho C, Caetano JM, Cunha L, Rebouta P, Kaptchuk TJ, Kirsch I. Open-label placebo treatment in chronic low back pain: a randomized controlled trial. Pain 2016;157:2766–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chung JW, Zeng Y, Wong TK. Drug therapy for the treatment of chronic nonspecific low back pain: systematic review and meta-analysis. Pain Physician 2013;16:E685–704. [PubMed] [Google Scholar]
- [16].Colloca L, Akintola T, Haycock NR, Blasini M, Thomas S, Phillips J, Corsi N, Schenk LA, Wang Y. Prior Therapeutic Experiences, Not Expectation Ratings, Predict Placebo Effects: An Experimental Study in Chronic Pain and Healthy Participants. PPS 2020;89:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Colloca L, Barsky AJ. Placebo and Nocebo Effects. New England Journal of Medicine 2020;382:554–561. [DOI] [PubMed] [Google Scholar]
- [18].Colloca L, Benedetti F. How prior experience shapes placebo analgesia. Pain 2006;124:126–33. [DOI] [PubMed] [Google Scholar]
- [19].Colloca L, Benedetti F. Placebo analgesia induced by social observational learning. Pain 2009;144:28–34. [DOI] [PubMed] [Google Scholar]
- [20].Colloca L, Klinger R, Flor H, Bingel U. Placebo analgesia: psychological and neurobiological mechanisms. Pain 2013;154:511–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Colloca L, Miller FG. How placebo responses are formed: a learning perspective. Philos Trans R Soc Lond B Biol Sci 2011;366:1859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Colloca L, Miller FG. Role of expectations in health. Curr Opin Psychiatry 2011;24:149–55. [DOI] [PubMed] [Google Scholar]
- [23].Cuyul-Vásquez I, Barría JA, Perez NF, Fuentes J. The influence of verbal suggestions in the management of musculoskeletal pain: a narrative review. Physical Therapy Reviews 2019;24:175–181. [Google Scholar]
- [24].Enck P, Bingel U, Schedlowski M, Rief W. The placebo response in medicine: minimize, maximize or personalize? Nat Rev Drug Discov 2013;12:191–204. [DOI] [PubMed] [Google Scholar]
- [25].Enthoven WT, Roelofs PD, Deyo RA, van Tulder MW, Koes BW. Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst Rev 2016;2:CD012087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Geuter S, Koban L, Wager TD. The Cognitive Neuroscience of Placebo Effects: Concepts, Predictions, and Physiology. Annu Rev Neurosci 2017;40:167–188. [DOI] [PubMed] [Google Scholar]
- [27].Gurrell R, Dua P, Feng G, Sudworth M, Whitlock M, Reynolds DS, Butt RP. A randomised, placebo-controlled clinical trial with the α2/3/5 subunit selective GABAA positive allosteric modulator PF-06372865 in patients with chronic low back pain. Pain 2018;159:1742–1751. [DOI] [PubMed] [Google Scholar]
- [28].Haake M, MüLler H-H, Schade-Brittinger C, Basler HD, SchäFer H, Maier C, Endres HG, Trampisch HJ, Molsberger A. German Acupuncture Trials (Gerac) For Chronic Low Back Pain: Randomized, Multicenter, Blinded, Parallel-Group Trial With 3 Groups. Arch Intern Med 2007;167:1892–1898. [DOI] [PubMed] [Google Scholar]
- [29].Hashmi JA, Baria AT, Baliki MN, Huang L, Schnitzer TJ, Apkarian AV. Brain networks predicting placebo analgesia in a clinical trial for chronic back pain. Pain 2012;153:2393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hashmi JA, Kong J, Spaeth R, Khan S, Kaptchuk TJ, Gollub RL. Functional network architecture predicts psychologically mediated analgesia related to treatment in chronic knee pain patients. J Neurosci 2014;34:3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hróbjartsson A, Gøtzsche PC. Is the Placebo Powerless? New England Journal of Medicine 2001;344:1594–1602. [DOI] [PubMed] [Google Scholar]
- [32].Hrobjartsson A, Gotzsche PC. Is the placebo powerless? An analysis of clinical trials comparing placebo with no treatment. N Engl J Med 2001;344:1594–602. [DOI] [PubMed] [Google Scholar]
- [33].Hróbjartsson A, Gøtzsche PC. Placebo interventions for all clinical conditions. Cochrane Database Syst Rev 2010:CD003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huneke NTM, Brown CA, Burford E, Watson A, Trujillo-Barreto NJ, El-Deredy W, Jones AKP. Experimental Placebo Analgesia Changes Resting-State Alpha Oscillations. PLOS ONE 2013;8:e78278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hunter T, Siess F, Colloca L. Socially induced placebo analgesia: a comparison of a pre-recorded versus live face-to-face observation. Eur J Pain 2014;18:914–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kam-Hansen S, Jakubowski M, Kelley JM, Kirsch I, Hoaglin DC, Kaptchuk TJ, Burstein R. Altered placebo and drug labeling changes the outcome of episodic migraine attacks. Sci Transl Med 2014;6:218ra5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kaptchuk TJ, Kelley JM, Conboy LA, Davis RB, Kerr CE, Jacobson EE, Kirsch I, Schyner RN, Nam BH, Nguyen LT, Park M, Rivers AL, McManus C, Kokkotou E, Drossman DA, Goldman P, Lembo AJ. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ 2008;336:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kaptchuk TJ, Kelley JM, Deykin A, Wayne PM, Lasagna LC, Epstein IO, Kirsch I, Wechsler ME. Do “placebo responders” exist? Contemporary clinical trials 2008;29:587–95. [DOI] [PubMed] [Google Scholar]
- [39].Kirsch I. Response Expectancy and the Placebo Effect. Int Rev Neurobiol 2018;138:81–93. [DOI] [PubMed] [Google Scholar]
- [40].Kirsch I. Response expectancy as a determinant of experience and behavior. Am Psychol 1985;40:1189–202. [Google Scholar]
- [41].Kleine-Borgmann J, Schmidt K, Hellmann A, Bingel U. Effects of open-label placebo on pain, functional disability, and spine mobility in patients with chronic back pain: a randomized controlled trial. PAIN 2019;160:2891–2897. [DOI] [PubMed] [Google Scholar]
- [42].Klinger R, Colloca L, Bingel U, Flor H. Placebo analgesia: clinical applications. Pain 2014;155:1055–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Koban L, Jepma M, Geuter S, Wager TD. What’s in a word? How instructions, suggestions, and social information change pain and emotion. Neurosci Biobehav Rev 2017;81:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kragel PA, Koban L, Barrett LF, Wager TD. Representation, Pattern Information, and Brain Signatures: From Neurons to Neuroimaging. Neuron 2018;99:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Krummenacher P, Candia V, Folkers G, Schedlowski M, Schonbachler G. Prefrontal cortex modulates placebo analgesia. Pain 2010;148:368–74. [DOI] [PubMed] [Google Scholar]
- [46].Lui F, Colloca L, Duzzi D, Anchisi D, Benedetti F, Porro CA. Neural bases of conditioned placebo analgesia. PAIN® 2010;151:816–824. [DOI] [PubMed] [Google Scholar]
- [47].Myers J, Wielage RC, Han B, Price K, Gahn J, Paget M-A, Happich M. The efficacy of duloxetine, non-steroidal anti-inflammatory drugs, and opioids in osteoarthritis: a systematic literature review and meta-analysis. BMC Musculoskeletal Disorders 2014;15:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Redelmeier DA, Kahneman D. Patients’ memories of painful medical treatments: real-time and retrospective evaluations of two minimally invasive procedures. PAIN 1996;66:3–8. [DOI] [PubMed] [Google Scholar]
- [49].Reicherts P, Gerdes AB, Pauli P, Wieser MJ. Psychological Placebo and Nocebo Effects on Pain Rely on Expectation and Previous Experience. J Pain 2016;17:203–14. [DOI] [PubMed] [Google Scholar]
- [50].Roelofs PD, Deyo RA, Koes BW, Scholten RJ, van Tulder MW. Non-steroidal anti-inflammatory drugs for low back pain. Cochrane Database Syst Rev 2008:CD000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schmidt-Wilcke T, Ichesco E, Hampson JP, Kairys A, Peltier S, Harte S, Clauw DJ, Harris RE. Resting state connectivity correlates with drug and placebo response in fibromyalgia patients. Neuroimage Clin 2014;6:252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Seminowicz DA, Wideman TH, Naso L, Hatami-Khoroushahi Z, Fallatah S, Ware MA, Jarzem P, Bushnell MC, Shir Y, Ouellet JA, Stone LS. Effective treatment of chronic low back pain in humans reverses abnormal brain anatomy and function. J Neurosci 2011;31:7540–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Skljarevski V, Desaiah D, Liu-Seifert H, Zhang Q, Chappell AS, Detke MJ, Iyengar S, Atkinson JH, Backonja M. Efficacy and safety of duloxetine in patients with chronic low back pain. Spine 2010;35:E578–85. [DOI] [PubMed] [Google Scholar]
- [54].Stewart-Williams S, Podd J. The Placebo Effect: Dissolving the Expectancy Versus Conditioning Debate. Psychological Bulletin 2004;130:324–340. [DOI] [PubMed] [Google Scholar]
- [55].Stone AA, Obbarius A, Junghaenel DU, Wen CKF, Schneider S. High-resolution, field approaches for assessing pain: Ecological Momentary Assessment. PAIN 2021;162:4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Swider K, Babel P. The Effect of the Type and Colour of Placebo Stimuli on Placebo Effects Induced by Observational Learning. PLoS One 2016;11:e0158363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tetreault P, Baliki MN, Baria AT, Bauer WR, Schnitzer TJ, Apkarian AV. Inferring distinct mechanisms in the absence of subjective differences: Placebo and centrally acting analgesic underlie unique brain adaptations. Hum Brain Mapp 2018;39:2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tetreault P, Mansour A, Vachon-Presseau E, Schnitzer TJ, Apkarian AV, Baliki MN. Brain Connectivity Predicts Placebo Response across Chronic Pain Clinical Trials. PLoS Biol 2016;14:e1002570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tilburt JC, Emanuel EJ, Kaptchuk TJ, Curlin FA, Miller FG. Prescribing “placebo treatments”: results of national survey of US internists and rheumatologists. BMJ 2008;337:a1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tuttle AH, Tohyama S, Ramsay T, Kimmelman J, Schweinhardt P, Bennett GJ, Mogil JS. Increasing placebo responses over time in U.S. clinical trials of neuropathic pain. Pain 2015;156:2616–2626. [DOI] [PubMed] [Google Scholar]
- [61].Vachon-Presseau E, Berger SE, Abdullah TB, Huang L, Cecchi GA, Griffith JW, Schnitzer TJ, Apkarian AV. Brain and psychological determinants of placebo pill response in chronic pain patients. Nature Communications 2018;9:3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vachon-Presseau E, Centeno MV, Ren W, Berger SE, Tétreault P, Ghantous M, Baria A, Farmer M, Baliki MN, Schnitzer TJ, Apkarian AV. The Emotional Brain as a Predictor and Amplifier of Chronic Pain. J Dent Res 2016;95:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vachon-Presseau E, Tétreault P, Petre B, Huang L, Berger SE, Torbey S, Baria AT, Mansour AR, Hashmi JA, Griffith JW, Comasco E, Schnitzer TJ, Baliki MN, Apkarian AV. Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain 2016;139:1958–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vase L, Robinson ME, Verne GN, Price DD. Increased placebo analgesia over time in irritable bowel syndrome (IBS) patients is associated with desire and expectation but not endogenous opioid mechanisms. Pain 2005;115:338–47. [DOI] [PubMed] [Google Scholar]
- [65].Vase L, Robinson ME, Verne GN, Price DD. The contributions of suggestion, desire, and expectation to placebo effects in irritable bowel syndrome patients. An empirical investigation. Pain 2003;105:17–25. [DOI] [PubMed] [Google Scholar]
- [66].Voudouris NJ, Peck CL, Coleman G. Conditioned placebo responses. J Pers Soc Psychol 1985;48:47–53. [DOI] [PubMed] [Google Scholar]
- [67].Voudouris NJ, Peck CL, Coleman G. The role of conditioning and verbal expectancy in the placebo response. Pain 1990;43:121–8. [DOI] [PubMed] [Google Scholar]
- [68].Wager TD, Atlas LY. The neuroscience of placebo effects: connecting context, learning and health. Nat Rev Neurosci 2015;16:403–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wager TD, Atlas LY, Leotti LA, Rilling JK. Predicting Individual Differences in Placebo Analgesia: Contributions of Brain Activity during Anticipation and Pain Experience. J Neurosci 2011;31:439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All our previous data (including Phase 1 data) are currently available at http://openpain.org/. Phase 2 numerical data and the raw data will also be made available at the time of publication, and codes used for permutation testing in Phase 1 and Phase 2 are available at https://github.com/EtienneVP.