

Abstract
Although many studies have examined quantitative trait variation across many species, only a small number of genes and thereby molecular mechanisms have been discovered. Without these data, we can only speculate about evolutionary processes that underlie trait variation. Here, we review how quantitative and molecular genetics in the nematode Caenorhabditis elegans led to the discovery and validation of 37 quantitative trait genes over the past 15 years. Using these data, we can start to make inferences about evolution from these quantitative trait genes, including the roles that coding versus noncoding variation, gene family expansion, common versus rare variants, pleiotropy, and epistasis play in trait variation across this species.
Discovering the mechanisms of trait variation one gene at a time
Over the past two decades, the pace of discoveries of the genes and mechanisms underlying trait variation has increased because of advances in whole-genome sequencing and mixed-effects model approaches in quantitative genetics. Studies have identified the number and effects of loci that impact diverse traits measured in livestock, crops, model species, and humans, but only a small number of genes and molecular mechanisms have been validated in any species. This limitation exists because it is difficult (or impossible) to experimentally validate the roles of genes in quantitative traits in many species, despite compelling evidence for numerous candidate genes. These data can help elucidate models for how traits change over time and the evolutionary principles underlying these changes. Therefore, researchers interested in evolution need to identify the genes and mechanisms that cause phenotypic differences across populations. However, most species have high levels of genetic diversity that make the mapping of many small effect loci and validation of specific genes difficult, if not impossible [1]. Additionally, the literature is filled with numerous examples of quantitative trait loci (QTL) (see Glossary) that have been identified but specific genes and alleles have not been validated using precise genomic manipulations, making inferences about the molecular mechanisms of trait variation guesses, at best. Several species can mitigate these limitations and enable discoveries of the genes and mechanisms, contributing significant progress towards understanding the causes of trait variation across populations.
A little more than a decade ago, the roundworm nematode Caenorhabditis elegans emerged as a powerhouse for the discovery of genes and variants that underlie quantitative trait variation [2]. As of the writing of this review, 37 quantitative trait genes (QTGs) have been discovered and validated using precise genomic edits in defined genetic backgrounds. From that significant list, researchers have gone even further to define 24 quantitative trait variants (QTVs), elucidating the molecular mechanisms of quantitative trait variation (Table 1, Key table, Figure 1, Key figure, and Table S1 in the supplemental information online). Genetic experiments testing the role of a gene in a quantitative trait must be performed to make this connection from phenotypic variation to a QTG. The C. elegans hermaphroditic mating system and selfing lifestyle facilitate these types of experiments because genome-wide variation is relatively low and homozygous strains are easy to construct. Additionally, C. elegans are easily grown in the laboratory and have a compact and defined genome in contrast to most other metazoan species. Importantly, recent advances in CRISPR-Cas9 genome editing enabled the creation of edits to specific genomic sites [3]. These edited strains are often paired with sensitive high-throughput assays to measure subtle effects on phenotype [4–6], making genetic causality definable in a metazoan model. Beyond genome editing, other methods are made easier by selfing and further enable rapid gene identification and testing, including fine mapping the phenotypic variation using additional genetic markers and narrowing mapped intervals using near-isogenic lines (NILs). Recent discoveries of the species origins, the structure of the genome, and inferences of its natural niche [7–10] have set the context to help understand how evolution has shaped this species. The confluence of these advantages have brought C. elegans to the forefront of quantitative genetics.
Key Table: Table 1.
List of QTGs and QTVs discovered in C. elegansa
| Phenotype | Mapping type | QTG | QTV | Refs |
|---|---|---|---|---|
| Natural wild isolate alleles | ||||
| Genetic perturbation (vulval induction, gene expression) | Linkage mapping | amx-2 | NA | [50,51] |
| PolyQ aggregation | Linkage mapping | atg-5 | NA | [150] |
| Drug response (albendazole) | Association mapping | ben-1 | NA | [70] |
| Drug response (arsenic trioxide) | Association mapping; linkage mapping | dbt-1 | C78S | [76] |
| Orsay virus sensitivity | Association mapping | drh-1 | Deletion | [101] |
| Dauer formation | Linkage mapping | eak-3 | Deletion | [41] |
| Aggregation; bordering | Linkage mapping | exp-1 | NA | [90] |
| Drug response (abamectin); pathogen response (Streptomyces avermitilis) | Association mapping; linkage mapping | glc-1 | Deletion | [8,65] |
| Drug response (proprionate) | Association mapping | glct-3 | G19stop; F19fs | [74] |
| Matricidal hatching | Linkage mapping | kcnl-1 | V530L | [100] |
| Male tail morphology | Linkage mapping | mab-23 | C38F | [47,48] |
| Embryonic lethality | Linkage mapping |
peel-1
zeel-1 |
Deletion | [113] |
| Nictation | Linkage mapping | piRNA | NA | [93] |
| Male-male plugging behavior | Linkage mapping | plep-1 | V278D | [92] |
| Copulatory plugging; embryonic lethality | Linkage mapping | plg-1 | TE insertion | [96] |
| Telomere length | Association mapping | pot-2 | NA | [108] |
| RNAi sensitivity | BSA; linkage mapping | ppw-1 | NA | [103,104] |
| Competitive fitness | BSA | rcan-1 | CNV | [95] |
| Drug response (amsacrine, bleomycin, bortezomib, carmustine, cisplatin, etoposide, puromycin, silver); gene expression | Linkage mapping | scb-1 | NA | [67,68] |
| Temperature-induced sterility | BSA; linkage mapping | set-24 | Deletion | [20] |
| Drug response (zinc) | Association mapping; linkage mapping | sqst-5 | Deletion | [6] |
| Pheromone response (dauer formation) | Association mapping | srg-37 | Deletion | [39] |
| Pheromone sensitivity | Linkage mapping | srx-43 | NA | [86] |
| Drug response (abamectin); stress resistance (H2O2); fitness; gene expression | BSA | sti-1 | NA | [73] |
| Embryonic lethality | BSA |
sup-35
pha-1 |
NA | [98] |
| Drug response (etoposide, amsacrine) | Association mapping; linkage mapping | top-2 | Q797M | [75] |
| Temperature response (body size) | Linkage mapping | tra-3 | F96L | [38] |
| Food foraging response | Linkage mapping | tyra-3 | NA | [81] |
| Laboratory-adapted alleles | ||||
| Genetic perturbation (movement); body size | Linkage mapping | col-182 | Deletion | [49] |
| Oxygen sensing; oxygen response | Linkage mapping | glb-5 | Insertion | [84,85] |
| Vulval induction | Linkage mapping | nath-10 | I746M | [37] |
| Clumping; pathogen avoidance (Pseudomonas aeruginosa, Staphylococcus aureus); pathogen response (Bacillus thuringiensis); fecundity; body size; oxygen response | Linkage mapping | npr-1 | V215F | [17,63,64,84,85,94] |
| Reproductive timing; lifespan; dauer formation; growth rate; fecundity | Linkage mapping | nurf-1 | Deletion | [18,19] |
| Competitive fitness | BSA | rcan-1 | CNV | [95] |
| Dauer formation | Linkage mapping | scd-2 | G985R; G1174E | [43] |
| Dauer formation | Linkage mapping |
srg-36
srg-37 |
Deletion | [44] |
Abbreviations: BSA, bulk-segregant analysis; CNV, copy number variation; NA, not available; QTG, quantitative trait genes; QTV, quantitative trait variants; TE, transposable elements.
Figure 1.
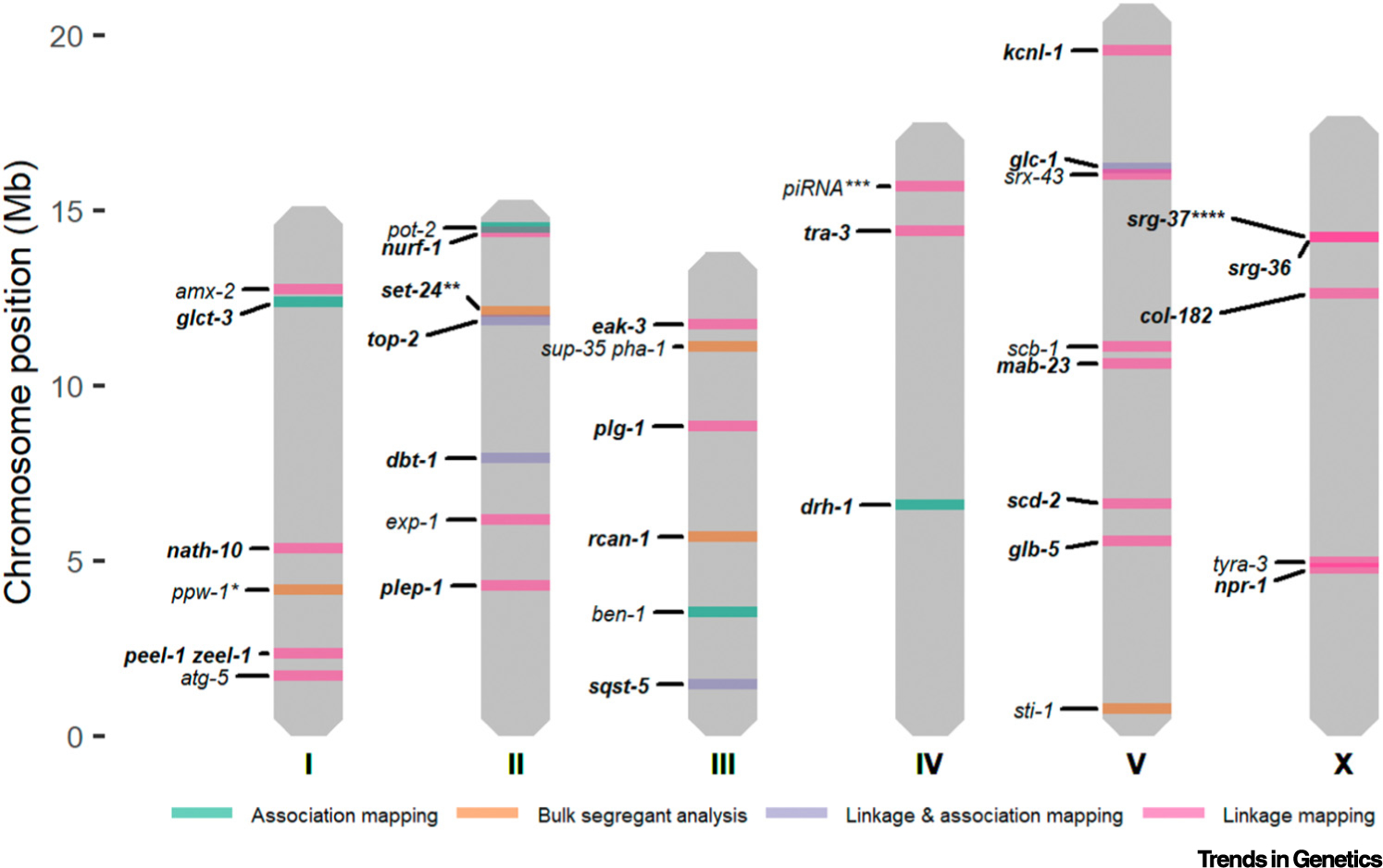
Key figure: Overview of quantitative trait gene (QTG) chromosome positions
The colors represent the mapping technique(s) that were used for quantitative trait loci (QTL) mapping: bulk-segregant analysis (BSA) (orange); linkage mapping (pink); genome-wide association (GWA) mapping (green); linkage and GWA mapping (purple). The genes in italics represent the QTGs and genes in bold italics represent the QTVs. *ppw-1 was also mapped using linkage mapping [104]. **set-24 was detected by combining linkage mapping and BSA [20]. ***The role of piRNAs was tested using a prg-1 deletion [93]. ****srg-37 was also mapped using GWA mapping [39]. Figure was created using ggplot2 in R.
The C. elegans community has identified numerous QTL (Table 1, and Table S1 in the supplemental information online) that underlie life history traits such as reproduction [5,11–21], lifespan and aging [18,22–36], body size and development [5,12–15,17,18,21,27,32,37–49], and abundances of gene transcripts [24,50–59], proteins [60], and metabolites [61]. Behavioral traits studied include pathogen responses [17,62–65], stress responses [4–6,14,34,36,55,56,65–80], responses to environmental perturbations, such as food [26,81–83], oxygen [84,85], pheromones [39,44,86], and temperature [12,13,31,38,46,52,56,87–89], and other nematode behaviors [23,90–107]. In addition to these traits, genomic features such as telomere length [108] and transposable elements [109] as well as geographical [8,110] and climate variables [110] have been used as quantitative traits for QTL mapping. In this review, we will focus on how the strains and methods of C. elegans quantitative genetics have defined 37 genes that underlie quantitative trait variation and how these data can answer fundamental questions about evolution at the molecular level.
Innovations in linkage mapping drive the discovery of genes and variants
Quantitative genetics mappings use three complementary approaches: linkage mapping, bulk-segregant analysis (BSA), and genome-wide association (GWA) mapping. Although BSA has been shown to be a fast, powerful, and effective tool to identify QTL [20,49,73,95,98], linkage mapping is the most popular method for the detection of QTL in C. elegans. In this approach, investigators leverage statistical power to detect QTL using a large number of recombinant lines generated from a cross between two or more phenotypically and genotypically diverse strains.
In the past 10 years, 59 linkage mapping studies discovered 22 genes underlying differences in one or more quantitative traits (Table 1 and Figure 1). Many of the underlying datasets are available using WormQTL2i [111]. The rapid accumulation of QTGs over the past 10 years highlights the growth in the C. elegans quantitative genetics field and the application of genome-editing technologies. Many QTGs were discovered using three recombinant panels derived from the laboratory-adapted Bristol strain, N2, and the genetically diverse Hawaiian strain, CB4856 [5,52,91]. The first panel of 80 recombinant inbred lines (RILs) was generated in 2006 [52], which led to the discovery of the first C. elegans QTG [38]. A few years later, a second panel of 239 recombinant inbred advanced intercross lines (RIAILs) was created; this intercrossing scheme created more recombination events and thereby enhanced mapping resolution [91,112]. However, after the generation of this RIAIL panel, researchers discovered that many of these lines contain the N2 allele at the peel-1 zeel-1 incompatibility locus on chromosome I [113,114]. Additionally, multiple studies found that the laboratory-derived N2 alleles of the genes npr-1, glb-5, nath-10, and col-182 have strong pleiotropic effects (Box 1) [49,115]. To reduce the effects of the genetic incompatibility between the N2 and CB4856 strains and the large pleiotropic effect of the N2 npr-1 allele, Andersen and colleagues generated a second RIAIL panel in which all 359 lines harbor the natural npr-1 allele (from CB4856) and a transposon insertion into the peel-1 gene [5]. Besides these RIL and RIAIL panels, a number of NIL panels were constructed using the N2 and CB4856 strains as parental lines [23,88,102] and used to map QTL [22,23,45,46,56,63,88,90]. QTL can be validated and fine mapped using NILs, and genetic causality can be tested using CRISPR-Cas9 genome editing of candidate genes (Box 2). Together, all of these N2xCB4856 panels led to the discovery of 16 QTGs [6,17,38,49,63–65,67,68,75,76,81,84,85,90,93,96,104,113] that underlie traits such as toxin responses [6,65,67,68,75,76], nictation [93], and RNAi sensitivity [103,104] (Table 1 and Figure 1).
Box 1. Laboratory-derived allele mapping.
Many biologists use model organisms in laboratory experiments. Typically, once individuals are isolated from the wild, reference strains are defined and grown in the laboratory for many generations. Although laboratory environments are created to optimize growth, this novel environment nevertheless is a strong selective force that can confound interpretations of experiments relevant to evolutionary biologists, typically interested in natural traits. Therefore, it is useful to identify the beneficial QTVs that are responsible for adaptation to the laboratory so that their influence can be controlled.
Additionally, these QTVs can be used to study the molecular mechanisms of adaptive evolution. In C. elegans, many of these genetic changes can be identified because of a lucky historical accident [115]. The reference strain, N2, which is used by the majority of C. elegans researchers, was grown in the lab for hundreds of generations over the decade before methods of long-term cryopreservation were developed and N2 was cryopreserved. Before that time, a culture of the N2 ancestor strain was grown independently for over five decades and eventually cryopreserved as the strain called LSJ2. Because of the self-fertilizing reproduction mode, each of the laboratory mutations that occurred in the N2 or LSJ2 lineages were readily fixed and can be identified by sequencing these strains. Approximately 300 variants distinguish these two strains.
These two strains were used to demonstrate that a QTV in the npr-1 gene, originally identified as a natural genetic variant that regulates feeding behavior, arose after isolation from the wild and increased the fitness of the N2 strain in laboratory conditions [84]. Mapping of phenotypic differences between the N2 and LSJ2 strains using a RIL panel generated between these two strains led to the identification of a number of additional beneficial QTVs in the glb-5, nurf-1, rcan-1, srg-36, and srg-37 genes [18,44,84,95,141,148]. These QTVs affect a number of behavioral, developmental, and reproductive traits, from feeding behaviors on bacterial lawns, to behavioral and developmental responses to pheromones, to reproductive output and lifespan. This work demonstrates the immense effects laboratory growth can have on animals and is important to consider when using laboratory strains to map natural trait differences.
Box 2. From QTL to validated QTG or QTV.
Most quantitative genetics mappings detect QTL, but progress often stops when QTL cannot be narrowed nor validated to discover specific QTG. Although this process to determine genetic causality is not easy, several genetic tools have enabled many C. elegans QTL to be validated at the level of QTG, and even QTV, for a variety of quantitative traits (see Table 1 and Figure 1 in main text). Most C. elegans studies validate and narrow QTL using NILs [4,17–20,34,36,37,40–42,44,50,61,63,65,67,69,71,74–76,81,84,86,87,90,92,93,95,100,101,104,149]. Any differences in phenotype between the NIL and the parental strain with the same genetic background can be attributed to the introgression of the QTL or the interaction of the introgression with the genetic background from the opposite genotype. If a NIL validates the QTL effect, several approaches exist to narrow the interval to a list of candidate genes to test for genetic causality. First, knowledge about (predicted) gene functions is commonly used to identify candidate genes [6,39,41,65,75,86,101,108]. Second, researchers often prioritize genes with variants in the coding sequence that are predicted to have an impact on gene function [6,18,20,37,41,65,67,70,76,92,93,98,101,150]. Finally, genes with expression variation, even if there are no variants in the coding sequence, can be prioritized as candidate genes [6,67,68,73].
Because causal relationships between genetic variation and phenotypic variation require empirical tests of necessity and sufficiency, specific genes or variants must be tested. Although it is tempting to use gene deletions or RNAi in the laboratory strain background to test for phenocopy of a quantitative trait, these techniques are biased towards the N2 strain background and assume that loss-of-function variation has caused the trait difference. With the establishment of CRISPR-Cas9 genome editing, gene-specific deletions can be created for quantitative complementation or reciprocal hemizygosity tests to establish a causal QTG [6,18,67]. Alternatively, allele replacement experiments can be used to edit a single nucleotide in any genetic background and identify a causal QTV [18,67,70,74–76,100] (see Table 1 and Figure 1 in main text).
Other strains have also been used to generate RIL panels to investigate natural variation that can be independent of the N2 and CB4856 variation [14,20,36,37,42,44,86,92] (Table 2). These panels were often made from strains that are divergent in a particular trait, and have led to the discovery of the role of nath-10 in vulval induction [37], plep-1 in plugging behavior [92], srx-43 and srx-44 in pheromone sensitivity [86], and set-24 in temperature-induced sterility [20]. Additionally, custom-made recombinant panels can harbor a particular mutation in a genetic background, allowing for the identification of modifier loci (Box 3) [37,50,92]. Regardless of the strain composition, linkage mapping continues to be an extremely powerful method for identifying QTGs in C. elegans.
Table 2.
Overview of QTL mapping populationsa
| Genetic background | Parental strains | Refs |
|---|---|---|
| Recombinant inbred lines (RILs) | ||
| N2xBO | N2xBO | [28] |
| RC301xBO | RC301xBO | [35] |
| CB4857xBO | DR1345xRW7000 | [33] |
| N2xCB4856 | N2xCB4856 | [52,72] |
| N2xCB4853 | N2xDR1350 | [14] |
| N2xCB4856 | AX613xCB4856 | [85] |
| N2xCB4856 | CB5362xCB4856 tra-2(ar221); xol-1(y9) | [105] |
| N2xLSJ2 | CX12311xLSJ2 | [44] |
| N2xAB1 | JU605xJU606 | [37] |
| AB2xCB4856 | QG5xQX1199 | [92] |
| N2xCB4856 | MT2124xCB4856 | [50] |
| MY14xCX12311 | MY14xCX12311 | [86] |
| N2xLSJ2 | CX12311xLSJ2 | [18] |
| MY10xJU1395 | MY10xJU1395 | [20] |
| N2xMY16 | N2xMY16 | [42] |
| JU1200xJU751 | JU1200xJU751 | [41] |
| Recombinant inbred advanced intercross lines (RIAILs) | ||
| N2xCB4856 | N2xCB4856 | [91,113] |
| N2xCB4856 | QX1430xCB4856 | [5] |
| Multi-parent recombinant inbred lines (mpRILs) | ||
| CeMEE | CeMEE RILs | [15] |
| JU1511xJU1926xJU1931xJU1941 | JU1511xJU1926xJU1931xJU1941 | [27] |
| Introgression line populations | ||
| NILs - CB4856 > N2 | N2xCB4856 | [23] |
| NILs - CB4856 > N2 | QG613xQG590; QG614xQG591 | [102] |
| CSS - CB4856 > N2 | N2xCB4856 | [88] |
| NILs - BO > CB4857 | DR1345xRW7000 | [34] |
| Wild isolates | ||
| NA | NA | [10,116] |
Abbreviations: CSS, chromosome substitution strain; NA, not available; QTL, quantitative trait loci; NIL, near-isogenic line.
Box 3. Fixed mutations in mapping panels.
One of the benefits of C. elegans is the availability of a plethora of defined mutant strains (covering most of known N2 reference genome genes) from the Caenorhabditis Genetics Center [151]. These characterized mutations can be used to create a RIL population to discover modifier loci present in genetic backgrounds different from the laboratory reference strain (as reviewed by [152]). This strategy has been successfully applied to create populations used to identify glb-5 as a modifier of npr-1-dependent trait differences [85], nath-10 as a modifier of vulval induction and germ line development differences [37], amx-2 as a modifier of Ras pathway signaling differences [50], and plep-1 as a modifier of male–male copulatory plugging differences [92].
The use of fixed mutations in different genetically diverse strains has shown that the effects of a mutation are dependent on genetic background. This result suggests that genetic modifiers are common across different natural strains. For example, it was shown that the AB1 strain was less sensitive to Ras pathway perturbations than the commonly used laboratory strain N2 [153]. This trait difference was mapped to nath-10, where further validation in the N2 strain showed that a sensitizing mutation alone did not affect vulval induction, but the effect could be revealed only in the presence of a receptor tyrosine kinase let-23(sy1) mutation [37].
An expanding wild isolate panel facilitates investigations of population-wide trait variation
Although linkage mapping and BSA have proven invaluable tools for C. elegans quantitative geneticists, the major innovation of the past decade was the introduction of GWA mapping [91].
GWA mapping takes advantage of the breadth of natural genetic diversity that exists among genetically distinct individuals. Like other mapping techniques, GWA mapping aims to identify functional variants that contribute to phenotypic diversity. The strength of this approach is in its ability to leverage the breadth of phenotypic variation present across the species to identify common QTVs. The C. elegans Natural Diversity Resource (CeNDR) [10,116] catalogues and distributes all wild strains and genome-wide variation data. CeNDRii remains a vital resource for the C. elegans community to facilitate GWA mappings and population genomic analyses.
Performing GWA mapping studies in C. elegans requires an understanding of the genetic composition of the species-wide population. Early studies to characterize the genetic variation in C. elegans at a global scale discovered large blocks of shared haplotypes across four of the six chromosomes, likely explained by one or more recent strong selective sweeps [8]. Extensive linkage disequilibrium, particularly in the center of chromosomes, limits QTL resolution using GWA mapping. Additionally, many strains are genetically similar and can be grouped into distinct isotypes. GWA mapping analysis with several strains from the same isotype inappropriately increases the effects of these nearly genetically identical strains. Large-scale collection efforts over the past decade have led to a species-wide collection of 1378 strains comprising 540 distinct isotypes. Along with these additional strains, the catalogued genetic diversity has increased, particularly in strains collected from the Hawaiian islands and the neighboring Pacific region [7,10]. However this increased genetic diversity decreases linkage disequilibrium, making the localization of QTL more difficult, particularly in punctuated regions of the genome with extreme genetic diversity [117].
In total, association mapping led to the discovery of nine QTGs, including seven with QTVs, that underlie quantitative trait variation (Table 1 and Figure 1). In one such example, a natural deletion in the pheromone receptor gene, srg-37, was found to cause variation in the dauer pheromone response [93]. In a study of Orsay virus sensitivity, a locus in the center of chromosome IV was linked to variation in viral load. This locus was later fine-mapped to a natural deletion in the gene drh-1, a homolog of the mammalian RIG-I gene family [101]. These examples, and others, provide important insights into the pathways and molecular mechanisms that cause natural variation across wild populations.
Although linkage mapping, BSA, and GWA mapping have each had considerable success mapping QTL and QTGs, each mapping approach has its drawbacks when used in isolation. In some studies, a combination of both linkage and GWA mapping has been used to narrow genomic intervals by analyzing QTL that overlap between methods [6,65,67,75,76]. Alternatively, multi-parent recombinant inbred line (mpRIL) panels (Figure 2A) have become important quantitative genetic tools in other model organisms such as mice [118,119], Drosophila melanogaster [120], and Arabidopsis thaliana [121]. These populations capture genetic diversity within the species without sacrificing the power of recombinants to detect and localize QTL. In C. elegans, two mpRIL panels have been developed: the CeMEE and the mpRIL panel [15,27,122]. The CeMEE panel is a 16-parent experimental evolution panel that, after crossing, was exposed to more than 100 generations of experimental evolution and subsequent inbreeding [15,122]. Alternatively, the mpRIL panel was generated from four parental strains with genotypic and phenotypic variation [123] isolated in close geographic proximity [27]. In addition to simply mapping more QTL across a variety of traits, wider adoption and generation of new mpRIL could help to address several out-standing questions in quantitative trait variation and the evolution of diverse phenotypes.
Figure 2. Powerful approaches to quickly identify the genes and molecular mechanisms underlying quantitative trait variation.
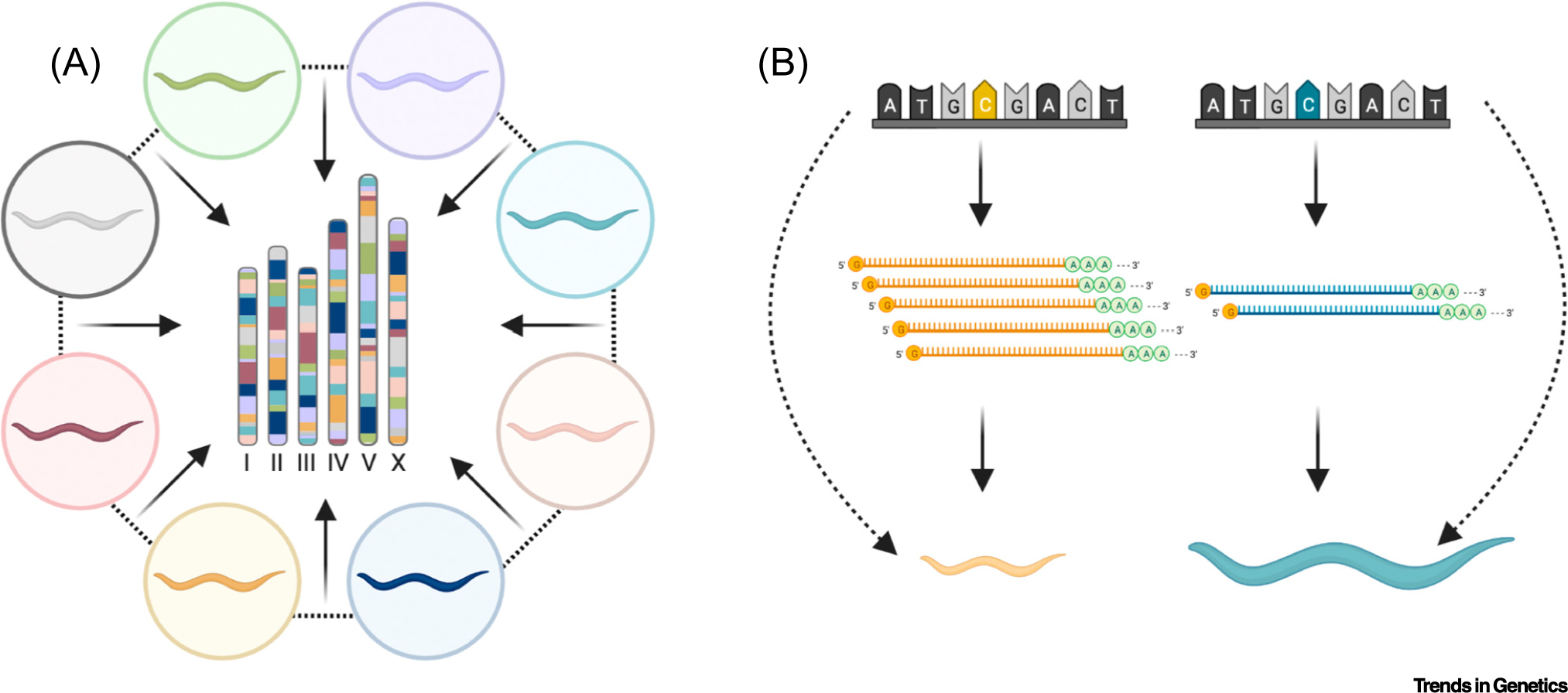
(A) A schematic of a hypothetical multi-parent recombinant cross is shown. The eight colored nematodes along the outside represent the parental strains in the cross. The genome of one hypothetical line is shown in the center of the cross with bars to represent chromosomes colored by the genetic background retained from each parental strain. (B) A mediation model where phenotypic variation (animal size) between strains (color) can be explained by variation in gene expression caused by a genetic variant. This figure was created using BioRender.com.
Validated QTGs and QTVs provide insights into evolution
Each of the 37 C. elegans QTGs discovered in the past 15 years (Table 1 and Figure 1) individually reveal molecular mechanisms for how phenotypic diversity is shaped, offering clues into how this species has evolved. Together, this set of experimentally validated QTGs give researchers numerous examples to connect quantitative trait variation to understanding evolutionary principles. The high confidence in these QTGs ensures that any conclusions drawn from these data are not influenced by false positive QTL or wishful thinking. By investigating these genes, we can begin to make suppositions about the variants most commonly underlying trait variation important for evolutionary change.
Validated QTVs confer fitness advantages in specific environments
Most validated QTVs fall into two groups: common variants with small effects or rare variants with large effects [124]. Of the 24 QTVs identified in C. elegans, 11 are common or present in more than 5% of isotypes (CeNDR, Table S1 in the supplemental information online). Of these 11 QTVs, three were identified using GWA mapping alone, four using linkage mapping alone, and four using both mapping methods. For example, multiple common alleles have been correlated with toxin response differences [6,65,74–76,101]. This result suggests that these alleles have been maintained over many generations and the predicted fitness costs of harboring such alleles are likely to be small. The remaining 13 QTVs are rare alleles across the C. elegans population and were identified exclusively using linkage mapping, which fits expectations about the power to detect these loci when parent strains harbor rare variants. These rare QTVs fall into two groups: nine laboratory-derived alleles (Box 1) and four alleles detected in wild populations. The wild rare alleles are associated with severe detrimental effects on life-history traits. For example, males with the mab-23(e2518) allele are unable to reproduce [47,48], and the set-24(mfP23) allele causes sterility after prolonged exposure to 25°C [20]. It is possible that their fitness effects are only present in specific environments (e.g., the rare variant eak-3 confers fitness advantages under stressful conditions by increasing dauer formation), suggesting that these alleles might have been selected in specific environments [41], as illustrated by the rare laboratory-derived QTVs that confer fitness advantages in that environment (Box 4). Overall, we still need more research into the natural ecology of C. elegans to understand how any discovered alleles or genes are influenced by selection [125].
Box 4. Experimental evolution and QTVs.
Experimental evolution uses controlled laboratory manipulations to investigate evolutionary processes. It can test the role of selection and genetic drift on changes in allele frequencies under specific environmental conditions. C. elegans is particularly useful for experimental evolution because its short generation time (approximately 3 days) and high brood size (greater than 200 offspring per individual) enables multiple-generation experiments with large population sizes.
After identification of a QTV, one common use of experimental evolution in C. elegans is to test the role of selection in the spread or loss of QTVs in a population [18,37,70,86,95,141,148,154–156]. Typically, two strains with homozygous genotypes for alternate alleles compete against each other for multiple generations in specific environments. By measuring the change in allele frequencies over the course of an experiment, the relative fitness of the two strains can be estimated. Such experiments have been used to demonstrate that mutations that are fixed during laboratory growth increase the fitness of reference strains in laboratory environments (Box 1).
These measurements can also be used to study the role that environment plays on fitness effects of QTVs. By comparing the relative fitness of two strains in different environments, empirical evidence can support the role of selection in the spread of alleles. For example, these experiments have been used to show that the increased use of anthelmintic drugs are responsible for the spread of resistance alleles [70,154]. Similar experiments have provided evidence that balancing selection could maintain different alleles that explain alternative foraging strategies induced by pheromones released by conspecifics [86]. By modifying the distribution of food, a QTV could either be beneficial or detrimental, leading the authors to propose that environmental heterogeneity in C. elegans natural environments creates balancing selection at this locus. Interestingly, many regions of the C. elegans genome show signatures of balancing selection, suggesting many loci could follow similar patterns [10].
Additionally, genetic manipulation can be used in these experiments to test specific evolutionary hypotheses. One elegant example took advantage of genotypes with different outcrossing rates [157], exposing C. elegans strains to pathogens that killed their hosts in a matter of days. Although the QTVs responsible for resistance to these pathogens were not identified, these and subsequent experiments [158,159] provided support that recombination between QTVs is important for adaptation to novel conditions.
The combination of high-throughput sequencing with competition experiments, also known as evolve and resequence, has been widely used in other species to identify regions of the genome with adaptive alleles. The use of evolve and resequence has lagged in C. elegans, likely because of its partially selfing mating system, but recent work has spurred development of this approach in Caenorhabditis nematodes (reviewed in [160,161]). Recently, a technique called ceX-QTL was developed to map QTVs that affect fitness in specific conditions [73]. The ceX-QTL technique uses bulk selection on millions of recombinant animals that compete against each other for multiple generations. QTVs that segregate between two strains of C. elegans and influence fitness in laboratory conditions were identified. This technique, and other types of evolve and resequence approaches, will likely become more popular with C. elegans researchers in the future.
Most validated QTGs are members of gene families
It has been hypothesized that paralogous genes or genes that are part of a functionally redundant gene family might offer a source of variation across populations because the genes can diverge without strongly affecting function [126]. Because of the ever-growing collection of C. elegans strains [10,116], the rapidly increasing availability of high-quality nematode genomes [127], and recent developments in evolutionary biology and comparative genomics [128], we can begin to determine how often quantitative trait variation is caused by differences in gene families. Of the 37 QTGs identified in C. elegans, 27 genes had one or more paralogs (Table S1 in the supplemental information online) [129,130], providing strong empirical data that, as genes increase in copy number, they can functionally diverge and cause trait variation. By contrast, it is estimated that about 6000 genes (or 32% of the genome) have at least one paralog [131], indicating a highly significant enrichment of QTGs belonging to a gene family (Fisher’s exact test; P < 0.00001). This result supports the duplication-divergence model, where new genes come from copies of pre-existing genes [132]. In one example, researchers mapped variation in propionate sensitivity to a putative glucuronosyltransferase that is part of an expanded gene family specific to C. elegans [74]. Importantly, new results show that hyper-divergent regions of the C. elegans genome contain environmental-response genes that are genes not found in the N2 reference genome and members of C. elegans specific expanded gene families [10]. The validated QTGs that are members of gene families suggest that quantitative trait variation is likely focused in hyper-divergent regions and must be characterized using long-read genome sequencing to define strain- or species-specific genes. As studies into the natural ecology of C. elegans continue, it will be important to investigate how these expanded and variable gene families contribute to fitness in the niche.
Noncoding variation is responsible for organism-level trait differences
Most known QTVs are large-effect protein-coding variants that cause phenotypic differences (Table 1). However, noncoding variation might be more evolutionarily important [133–135].
Numerous studies across several species suggest that genetic variation impacts gene expression [136,137]. However, it is often unclear how these gene expression differences translate to trait variation. Again, C. elegans offers six examples (eak-3, exp-1, prg-1, scb-1, srx-43, and tyra-3) in which noncoding variation is stated to be correlated with trait differences [41,67,68,81,86,90,93]. Furthermore, several gene expression QTL (eQTL) studies have discovered thousands of differentially expressed genes that are largely controlled by genetic factors [24,51–56,58,59]. Colocalization of eQTL and organism-level QTL could suggest that a single genetic variant underlies both [6,17,51,68]. Techniques such as mediation analysis can make statistical connections between genetic variation, variation in an intermediate trait such as gene expression, and variation in complex organism-level phenotypes (Figure 2B). This technique was successfully used to suggest that scb-1 affects responses to several chemotherapeutics [68] and that sqst-5 affects differential responses to exogenous zinc [6]. The effects of both loci were subsequently validated using genome editing. In addition to providing another resource for candidate gene prioritization within a QTL interval (separate from evaluating protein-coding variation), mediation analysis can help to identify the mechanism by which genetic variation causes trait variation. This technique is especially powerful to establish candidate genes whose expression is controlled by loci far from the regulated gene, as most fine-mapping techniques only consider genes within the QTL confidence interval. In the case of tyra-3 and exp-1 [81,90], phenotypic differences could be explained by gene expression, but eQTL for neither gene are detected, suggesting that whole-organism gene expression data might not always be sufficient to identify expression differences at single-cell resolution [59]. To date, most eQTL datasets in C. elegans have been generated from two-parent recombinant lines (specifically, N2xCB4856 recombinants). Therefore, a genome-wide analysis of gene expression in wild isolates or other mpRILs could provide an unprecedented resource for studying the role of regulatory variation in quantitative traits [58].
C. elegans mapping studies are just beginning to define the complexity of many quantitative traits
Although many early quantitative genetics studies in C. elegans identified mostly single, large-effect loci [2,138], technological advancements coupled with the collection of more genetically distinct wild isolates led to increases in the power to detect more QTL with ever smaller effects. Many quantitative traits map to at least two independent loci and some traits have five or more QTL. One large-scale QTL mapping study of nematode responses to 16 diverse toxins identified 82 QTL from 47 traits; a third of these traits mapped to two or more loci [69]. Strikingly, most of these QTL had small effect sizes, explaining less than 10% of the phenotypic variation in the mapping panel. Several studies used NILs to validate small-effect loci, demonstrating that small effects can be studied in C. elegans with the right tools and a sensitive assay [66,69,82].
Current mapping populations and studies detect some of the loci underlying quantitative trait variation, but we can use estimates of heritability to understand the levels of complexity for most traits. The total fraction of trait variation explained by genetic variation in a population can be estimated by calculating broad-sense heritability. When compared with an estimate of narrow-sense heritability, which accounts for all additive effects, the so-called missing heritability can be estimated as the difference between the total genetic effect and the additive effects [1]. One explanation for this discrepancy can be explained by nonadditive effects, including epistasis. For C. elegans, only five studies have estimated both broad- and narrow-sense heritability [15,39,69,74,76] and most trait variation is additive, as observed in yeast [139]. To more broadly impact our understanding of quantitative trait variation, these estimates should be calculated for every quantitative trait mapping and the data organized in a central repository. In this way, we can make more significant inferences about the loci underlying quantitative trait variation. Central data repositories like WormQTL2i and CeNDRii can facilitate these analyses [111,116].
Genetic architectures of quantitative traits can be affected when a single gene underlies multiple trait differences (pleiotropy [140]) and the varying contributions of genetic interactions among QTL (epistasis). In C. elegans, we have evidence of pleiotropic QTGs in amx-2, mab-23, nath-10, nict-1, npr-1, nurf-1, scb-1, and top-2 [17,18,37,41,48,50,51,68,75,93,141]. Many of these pleiotropic genes affect life history traits or toxin responses. The gene scb-1, for example, underlies variation in responses to amsacrine, bleomycin, carmustine, and cisplatin, demonstrating that a single gene can affect sensitivities to multiple chemotherapeutics [67,68]. Effects of epistatic loci on phenotypic variation are more difficult to define, as most of the QTL detected so far appear to be largely additive [54,56,66,69,80]. Although this result is consistent with what is observed in many other species [139,142–146], most mapping panels are underpowered to detect epistatic loci. Despite this obstacle, several cases of epistasis have been reported in C. elegans [19,56,61,63,66,67,82,84,87,104]. Although the current tools available to the quantitative genetics community are still best suited to identify single, large-effect QTVs [138], these examples of more complex architectures suggest that we are beginning to fill in the gap in our understanding of quantitative trait variation. Additionally, as the number of QTGs and QTVs grow, we can apply these results to investigations of the C. elegans natural ecology and niche to understand better the roles and trade-offs that pleiotropy and epistasis have on evolution of this species.
Concluding remarks
Better tools and newer technologies have led to an increase in the number of QTL detected and QTVs validated in C. elegans over the past decade. These discoveries have led to a better understanding of the molecular mechanisms underlying quantitative trait variation. Importantly, by synthesizing a large, experimentally validated dataset of QTGs and QTVs, we can begin to learn more about how traits can evolve in natural populations (see Outstanding questions). Although we can gain significant insights from studying C. elegans, it remains to be investigated how and if these conclusions can be applied more broadly to non-selfing species that lack the strong influence of genetic drift and linkage disequilibrium caused by selfing. Furthermore, we need to learn more about the ecological context of this species, so we can also learn to emulate the natural conditions in the laboratory and test the effects of natural alleles empirically [125,147]. The applications of QTGs and QTVs to knowledge about its niche and direct sources of selection will be critical to understand the tempo and mode of evolution at a mechanistic level. Regardless, in the continuing quest to connect QTL to specific QTVs, the implementation of newer, more powerful mapping methods like BSA and mpRILs will likely add to our current knowledge of the molecular mechanisms underlying quantitative trait variation.
Outstanding questions.
Can larger and more diverse cross panels improve our understanding of trait variation across natural populations?
What is the contribution of regulatory variation to quantitative trait variation?
Will mapping intermediate phenotypic traits (e.g., metabolites and gene expression) in combination with techniques such as mediation analysis define more QTG and lead to specific regulatory variant discovery?
How large is the role of epistasis in quantitative trait variation and can these results impact estimates of missing heritability?
Are pleiotropic loci common and how strongly do they influence evolutionary processes?
What is the role of specific environments (niches) and functional redundancy in expanded gene families in the selection of QTGs?
How can QTGs and QTVs be combined with the ecological context and niche to understand evolutionary processes?
Do the evolutionary conclusions inferred from C. elegans hold true for other non-selfing species?
Supplementary Material
Highlights.
Innovations in quantitative trait loci mapping and genome editing have led to the discovery and validation of 37 genes and variants underlying phenotypic variation in C. elegans.
Numerous recombinant panels and a large collection of wild strains make C. elegans a formidable model to understand quantitative trait variation.
Most of the identified quantitative trait genes have paralogs, providing evidence that gene duplication events are important for shaping quantitative traits.
Pleiotropy is relatively common among C. elegans quantitative trait genes.
Acknowledgments
We would like to thank Jan Kammenga, Lisa van Sluijs, Robyn Tanny, and Sam Widmayer for helpful comments on the manuscript. E.C.A. and K.S.E. received support from the NSF-Simons Center for Quantitative Biology at Northwestern University (awards Simons Foundation/SFARI 597491-RWC and the National Science Foundation 1764421). E.C.A. also received support from a National Science Foundation CAREER Award. K.S.E. also received support from the Cell and Molecular Basis of Disease Training grant (T32-GM008061). P.T.M. received support from NIH R01GM114170. M.H.W. was supported by NIH R01AA026658. M.G.S. was supported by NWO domain Applied and Engineering Sciences VENI grant (17282).
Glossary
- Broad-sense heritability
the total fraction of trait variation explained by genetic variation in a test population
- Bulk-segregant analysis (BSA)
a QTL mapping method in which pools of recombinant individuals are whole-genome sequenced after phenotypic selection to identify loci using allele frequency skews
- Epistasis
interactions between alleles that cause phenotypic effects greater than observed for the individual alleles alone
- Expression QTL (eQTL)
transcript abundances are quantitative traits that can be mapped in recombinant or natural populations. These QTL can be divided into two types by whether the physical position gene in the expression trait is nearby (local) or far away (distant) from the QTL position
- Genome-wide association (GWA) mapping
a quantitative trait mapping method where genetic markers segregating across a wild population are correlated with phenotypic variation in that same population
- Genetic causality
experimental determination of a direct role between a genetic difference and a phenotypic difference
- Haplotype
a genomic region with linked allelic variation
- Isotype
a collection of wild strains (typically from the same geographic location) that share greater than 99.97% of their genetic variants
- Linkage mapping
a quantitative trait mapping method where genetic markers segregating across a recombinant line panel are correlated with phenotypic variation in that same panel
- Mediation analysis
determination of a role of an intermediate variable (mediator) in a direct effect process (e.g., the mediating role of gene expression variation in the direct effect of genetic variation on phenotypic variation in a different trait)
- Multi-parent recombinant inbred lines (mpRIL)
a collection of homozygous strains generated after inbreeding recombinants from a cross between more than two genetically divergent parent strains. Can be used for linkage mapping
- Narrow-sense heritability
the fraction of trait variation explained by additive genetic variation in a test population
- Near-isogenic line (NIL)
a strain that harbors a region of the genomes from one genetic background in the presence of a different genetic background
- Pleiotropy
the effect of a single allele on multiple distinct traits
- Quantitative trait gene (QTG)
a gene in which genetic variation has been shown to directly impact phenotypic variation
- Quantitative trait locus (QTL)
a genomic interval in which genetic variation has been shown to be correlated with phenotypic variation
- Quantitative trait variant (QTV)
a variant (e.g., single-nucleotide or insertion-deletion variant) that has been shown to directly impact phenotypic variation
- Recombinant inbred lines (RILs)
a collection of homozygous strains generated after inbreeding recombinants from a cross between two or more genetically divergent parent strains. Can be used for linkage mapping
- Recombinant inbred advanced intercross lines (RIAILs)
a collection of homozygous strains generated after inbreeding recombinants from a cross between two or more genetically divergent parent strains. Unlike recombinant inbred lines, they have undergone additional rounds of crossing before inbreeding to increase recombination breakpoints and mapping resolution. Can be used for linkage mapping
Footnotes
Declaration of interests
No interests are declared.
Supplemental information
Supplemental information associated with this article can be found online at https://doi.org/10.1016/j.tig.2021.06.005.
Resources
References
- 1.Boyle EA et al. (2017) An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaertner BE and Phillips PC (2010) Caenorhabditis elegans as a platform for molecular quantitative genetics and the systems biology of natural variation. Genet. Res. 92, 331–348 [DOI] [PubMed] [Google Scholar]
- 3.Frøkjær-Jensen C (2013) Exciting prospects for precise engineering of Caenorhabditis elegans genomes with CRISPR/Cas9. Genetics 195, 635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elvin M et al. (2011) A fitness assay for comparing RNAi effects across multiple C. elegans genotypes. BMC Genomics 12, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen EC et al. (2015) A powerful new quantitative genetics platform, combining Caenorhabditis elegans high-throughput fitness assays with a large collection of recombinant strains. G3 5, 911–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans KS et al. (2020) Natural variation in the sequestosome-related gene, sqst-5, underlies zinc homeostasis in Caenorhabditis elegans. PLoS Genet. 16, e1008986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crombie TA et al. (2019) Deep sampling of Hawaiian Caenorhabditis elegans reveals high genetic diversity and admixture with global populations. Elife 8, e50465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersen EC et al. (2012) Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat. Genet. 44, 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulenburg H and Félix M-A (2017) The natural biotic environment of Caenorhabditis elegans. Genetics 206, 55–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee D et al. (2021) Balancing selection maintains hyper-divergent haplotypes in Caenorhabditis elegans. Nat. Ecol. Evol. 5, 794–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimond C et al. (2019) Natural variation and genetic determinants of Caenorhabditis elegans sperm size. Genetics 213, 615–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutteling EW et al. (2007) Environmental influence on the genetic correlations between life-history traits in Caenorhabditis elegans. Heredity 98, 206–213 [DOI] [PubMed] [Google Scholar]
- 13.Gutteling EW et al. (2007) Mapping phenotypic plasticity and genotype-environment interactions affecting life-history traits in Caenorhabditis elegans. Heredity 98, 28–37 [DOI] [PubMed] [Google Scholar]
- 14.Harvey SC et al. (2008) Quantitative genetic analysis of life-history traits of Caenorhabditis elegans in stressful environments. BMC Evol. Biol. 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noble LM et al. (2017) Polygenicity and epistasis underlie fitness-proximal traits in the Caenorhabditis elegans multiparental experimental evolution (CeMEE) panel. Genetics 207, 1663–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Z et al. (2019) Identification of specific nuclear genetic loci and genes that interact with the mitochondrial genome and contribute to fecundity in Caenorhabditis elegans. Front. Genet. 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen EC et al. (2014) A variant in the neuropeptide receptor npr-1 is a major determinant of Caenorhabditis elegans growth and physiology. PLoS Genet. 10, e1004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Large EE et al. (2016) Selection on a subunit of the NURF chromatin remodeler modifies life history traits in a domesticated strain of Caenorhabditis elegans. PLoS Genet. 12, e1006219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Large EE et al. (2017) Modeling of a negative feedback mechanism explains antagonistic pleiotropy in reproduction in domesticated Caenorhabditis elegans strains. PLoS Genet. 13, e1006769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frézal L et al. (2018) Natural genetic variation in a multigenerational phenotype in C. elegans. Curr. Biol. 28, 2588–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knight CG et al. (2001) Testing life-history pleiotropy in Caenorhabditis elegans. Evolution 55, 1795–1804 [DOI] [PubMed] [Google Scholar]
- 22.Stastna JJ et al. (2015) Genotype-dependent lifespan effects in peptone deprived Caenorhabditis elegans. Sci. Rep. 5, 16259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doroszuk A et al. (2009) A genome-wide library of CB4856/N2 introgression lines of Caenorhabditis elegans. Nucleic Acids Res. 37, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viñuela A et al. (2010) Genome-wide gene expression regulation as a function of genotype and age in C. elegans. Genome Res. 20, 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viñuela A et al. (2012) Aging uncouples heritability and expression-QTL in Caenorhabditis elegans. G3 2, 597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Z et al. (2015) Compatibility between mitochondrial and nuclear genomes correlates with the quantitative trait of lifespan in Caenorhabditis elegans. Sci. Rep. 5, 17303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snoek BL et al. (2019) A multi-parent recombinant inbred line population of C. elegans allows identification of novel QTLs for complex life history traits. BMC Biol. 17, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks A and Johnson TE (1991) Genetic specification of life span and self-fertility in recombinant-inbred strains of Caenorhabditis elegans. Heredity 67, 19–28 [DOI] [PubMed] [Google Scholar]
- 29.Ebert RH 2nd et al. (1993) Longevity-determining genes in Caenorhabditis elegans: chromosomal mapping of multiple noninteractive loci. Genetics 135, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebert RH 2nd et al. (1996) Defining genes that govern longevity in Caenorhabditis elegans. Dev. Genet. 18, 131–143 [DOI] [PubMed] [Google Scholar]
- 31.Shook DR et al. (1996) Mapping quantitative trait loci affecting life history traits in the nematode Caenorhabditis elegans. Genetics 142, 801–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shook DR and Johnson TE (1999) Quantitative trait loci affecting survival and fertility-related traits in Caenorhabditis elegans show genotype-environment interactions, pleiotropy and epistasis. Genetics 153, 1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayyadevara S et al. (2003) Genetic loci modulating fitness and life span in Caenorhabditis elegans: categorical trait interval mapping in CL2a x Bergerac-BO recombinant-inbred worms. Genetics 163, 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vertino A et al. (2011) A narrow quantitative trait locus in C. elegans coordinately affects longevity, thermotolerance, and resistance to paraquat. Front. Genet. 2, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayyadevara S et al. (2001) Genetic mapping of quantitative trait loci governing longevity of Caenorhabditis elegans in recombinant-inbred progeny of a Bergerac-BO x RC301 interstrain cross. Genetics 157, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ayyadevara S et al. (2014) Rec-8 dimorphism affects longevity, stress resistance and X-chromosome nondisjunction in C. elegans, and replicative lifespan in S. cerevisiae. Front. Genet. 5, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duveau F and Félix M-A (2012) Role of pleiotropy in the evolution of a cryptic developmental variation in Caenorhabditis elegans. PLoS Biol. 10, e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kammenga JE et al. (2007) A Caenorhabditis elegans wild type defies the temperature-size rule owing to a single nucleotide polymorphism in tra-3. PLoS Genet. 3, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee D et al. (2019) Selection and gene flow shape niche-associated variation in pheromone response. Nat. Ecol. Evol. 3, 1455–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webster AK et al. (2019) Population selection and sequencing of wild isolates identifies a region on chromosome III affecting starvation resistance. G3 9, 3477–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Billard B et al. (2020) A natural mutational event uncovers a life history trade-off via hormonal pleiotropy. Curr. Biol. 30, 4142–4154 [DOI] [PubMed] [Google Scholar]
- 42.Torres Cleuren YN et al. (2019) Extensive intraspecies cryptic variation in an ancient embryonic gene regulatory network. eLife 8, e48220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reiner DJ et al. (2008) C. elegans anaplastic lymphoma kinase ortholog SCD-2 controls dauer formation by modulating TGF-beta signaling. Curr. Biol. 18, 1101–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGrath PT et al. (2011) Parallel evolution of domesticated Caenorhabditis species targets pheromone receptor genes. Nature 477, 321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green JWM et al. (2013) Genetic mapping of variation in dauer larvae development in growing populations of Caenorhabditis elegans. Heredity 111, 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Donnell MP et al. (2018) Rictor/TORC2 mediates gut-to-brain signaling in the regulation of phenotypic plasticity in C. elegans. PLoS Genet. 14, e1007213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hodgkin J and Doniach T (1997) Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics 146, 149–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lints R and Emmons SW (2002) Regulation of sex-specific differentiation and mating behavior in C. elegans by a new member of the DM domain transcription factor family. Genes Dev. 16, 2390–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noble LM et al. (2020) The ancestral Caenorhabditis elegans cuticle suppresses rol-1. G3 10, 2385–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmid T et al. (2015) Systemic regulation of RAS/MAPK signaling by the serotonin metabolite 5-HIAA. PLoS Genet. 11, e1005236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sterken MG et al. (2017) Ras/MAPK modifier loci revealed by eQTL in Caenorhabditis elegans. G3 7, 3185–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y et al. (2006) Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2, e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y et al. (2010) Global genetic robustness of the alternative splicing machinery in Caenorhabditis elegans. Genetics 186, 405–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rockman MV et al. (2010) Selection at linked sites shapes heritable phenotypic variation in C. elegans. Science 330, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snoek BL et al. (2017) Contribution of trans regulatory eQTL to cryptic genetic variation in C. elegans. BMC Genomics 18, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sterken MG et al. (2020) Dissecting the eQTL micro-architecture in Caenorhabditis elegans. Front. Genet. 11, 501376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Francesconi M and Lehner B (2014) The effects of genetic variation on gene expression dynamics during development. Nature 505, 208–211 [DOI] [PubMed] [Google Scholar]
- 58.Snoek BL et al. (2021) The genetics of gene expression in a C. elegans multi parental recombinant inbred line population. bioRxiv Published online March 4, 2021. 10.1101/2021.03.04.433879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ben-David E et al. (2021) Whole-organism eQTL mapping at cellular resolution with single-cell sequencing. Elife 10, e65857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh KD et al. (2016) Natural genetic variation influences protein abundances in C. elegans developmental signalling pathways. PLoS One 11, e0149418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao AW et al. (2018) Natural genetic variation in identified genomic loci controlling metabolite levels. Genome Res. 28, 1296–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balla KM et al. (2015) A wild C. elegans strain has enhanced epithelial immunity to a natural microsporidian parasite. PLoS Pathog. 11, e1004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakad R et al. (2016) Contrasting invertebrate immune defense behaviors caused by a single gene, the Caenorhabditis elegans neuropeptide receptor gene npr-1. BMC Genomics 17, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddy KC et al. (2009) A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans. Science 323, 382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghosh R et al. (2012) Natural variation in a chloride channel subunit confers avermectin resistance in C. elegans. Science 335, 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernstein MR et al. (2019) Tightly linked antagonistic-effect loci underlie polygenic phenotypic variation in C. elegans. Evol. Lett. 3, 462–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brady SC et al. (2019) A novel gene underlies bleomycin-response variation in Caenorhabditis elegans. Genetics 212, 1453–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans KS and Andersen EC (2020) The gene scb-1 underlies variation in Caenorhabditis elegans chemotherapeutic responses. G3 10, 2353–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evans KS et al. (2018) Shared genomic regions underlie natural variation in diverse toxin responses. Genetics 210, 1509–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hahnel SR et al. (2018) Extreme allelic heterogeneity at a Caenorhabditis elegans beta-tubulin locus explains natural resistance to benzimidazoles. PLoS Pathog. 14, e1007226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zamanian M et al. (2018) Discovery of genomic intervals that underlie nematode responses to benzimidazoles. PLoS Negl. Trop. Dis. 12, e0006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez M et al. (2012) Genetic variation for stress-response hormesis in C. elegans lifespan. Exp. Gerontol. 47, 581–587 [DOI] [PubMed] [Google Scholar]
- 73.Burga A et al. (2019) Fast genetic mapping of complex traits in C. elegans using millions of individuals in bulk. Nat. Commun. 10, 2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Na H et al. (2020) Natural variation in a glucuronosyltransferase modulates propionate sensitivity in a C. elegans propionic acidemia model. PLoS Genet. 16, e1008984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zdraljevic S et al. (2017) Natural variation in a single amino acid substitution underlies physiological responses to topoisomerase II poisons. PLoS Genet. 13, e1006891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zdraljevic S et al. (2019) Natural variation in C. elegans arsenic toxicity is explained by differences in branched chain amino acid metabolism. eLife 8, e40260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Swinderen B et al. (1998) Common genetic determinants of halothane and isoflurane potencies in Caenorhabditis elegans. Anesthesiology 89, 1509–1517 [DOI] [PubMed] [Google Scholar]
- 78.van Swinderen B et al. (1997) Quantitative trait loci controlling halothane sensitivity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 94, 8232–8237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Evans KS and Andersen EC (2020) The cadmium-responsive gene, cdr-6, does not influence Caenorhabditis elegans responses to exogenous zinc. microPublication Biol. 2020, 33005884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Evans KS et al. (2021) Two novel loci underlie natural differences in Caenorhabditis elegans abamectin responses. PLoS Pathog. 17, e1009297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bendesky A et al. (2011) Catecholamine receptor polymorphisms affect decision-making in C. elegans. Nature 472, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Glater EE et al. (2014) Multigenic natural variation underlies Caenorhabditis elegans olfactory preference for the bacterial pathogen Serratia marcescens. G3 4, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harvey SC (2009) Non-dauer larval dispersal in Caenorhabditis elegans. J. Exp. Zool. B Mol. Dev. Evol. 312B, 224–230 [DOI] [PubMed] [Google Scholar]
- 84.McGrath PT et al. (2009) Quantitative mapping of a digenic behavioral trait implicates globin variation in C. elegans sensory behaviors. Neuron 61, 692–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Persson A et al. (2009) Natural variation in a neural globin tunes oxygen sensing in wild Caenorhabditis elegans. Nature 458, 1030–1033 [DOI] [PubMed] [Google Scholar]
- 86.Greene JS et al. (2016) Balancing selection shapes density-dependent foraging behaviour. Nature 539, 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gaertner BE et al. (2012) More than the sum of its parts: a complex epistatic network underlies natural variation in thermal preference behavior in Caenorhabditis elegans. Genetics 192, 1533–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Glauser DA et al. (2011) Heat avoidance is regulated by transient receptor potential (TRP) channels and a neuropeptide signaling pathway in Caenorhabditis elegans. Genetics 188, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghosh R et al. (2015) Genetics of intraspecies variation in avoidance behavior induced by a thermal stimulus in Caenorhabditis elegans. Genetics 200, 1327–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bendesky A et al. (2012) Long-range regulatory polymorphisms affecting a GABA receptor constitute a quantitative trait locus (QTL) for social behavior in Caenorhabditis elegans. PLoS Genet. 8, e1003157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rockman MV and Kruglyak L (2009) Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet. 5, e1000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Noble LM et al. (2015) Natural variation in plep-1 causes male-male copulatory behavior in C. elegans. Curr. Biol. 25, 2730–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee D et al. (2017) The genetic basis of natural variation in a phoretic behavior. Nat. Commun. 8, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Bono M and Bargmann CI (1998) Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94, 679–689 [DOI] [PubMed] [Google Scholar]
- 95.Zhao Y et al. (2020) A spontaneous complex structural variant in rcan-1 increases exploratory behavior and laboratory fitness of Caenorhabditis elegans. PLoS Genet. 16, e1008606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palopoli MF et al. (2008) Molecular basis of the copulatory plug polymorphism in Caenorhabditis elegans. Nature 454, 1019–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Snoek LB et al. (2014) Widespread genomic incompatibilities in Caenorhabditis elegans. G3 4, 1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ben-David E et al. (2017) A maternal-effect selfish genetic element in Caenorhabditis elegans. Science 356, 1051–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alcorn MR et al. (2016) Heterotaxy in Caenorhabditis: widespread natural variation in left-right arrangement of the major organs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 20150404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vigne P et al. (2021) A single-nucleotide change underlies the genetic assimilation of a plastic trait. Sci. Adv. 7, eabd9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ashe A et al. (2013) A deletion polymorphism in the Caenorhabditis elegans RIG-I homolog disables viral RNA dicing and antiviral immunity. eLife 2, e00994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bernstein MR and Rockman MV (2016) Fine-scale crossover rate variation on the Caenorhabditis elegans X chromosome. G3 6, 1767–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tijsterman M et al. (2002) PPW-1, a PAZ/PIWI protein required for efficient germline RNAi, is defective in a natural isolate of C. elegans. Curr. Biol. 12, 1535–1540 [DOI] [PubMed] [Google Scholar]
- 104.Pollard DA and Rockman MV (2013) Resistance to germline RNA interference in a Caenorhabditis elegans wild isolate exhibits complexity and nonadditivity. G3 3, 941–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chandler CH (2010) Cryptic intraspecific variation in sex determination in Caenorhabditis elegans revealed by mutations. Heredity 105, 473–482 [DOI] [PubMed] [Google Scholar]
- 106.Farhadifar R et al. (2020) Stoichiometric interactions explain spindle dynamics and scaling across 100 million years of nematode evolution. eLife 9, e55877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sterken MG et al. (2021) Punctuated loci on chromosome IV determine natural variation in Orsay virus susceptibility of Caenorhabditis elegans strains Bristol N2 and Hawaiian CB4856. J. Virol. 95, e02430–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cook DE et al. (2016) The genetic basis of natural variation in Caenorhabditis elegans telomere length. Genetics 204, 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Laricchia KM et al. (2017) Natural variation in the distribution and abundance of transposable elements across the Caenorhabditis elegans species. Mol. Biol. Evol. 34, 2187–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Evans KS et al. (2017) Correlations of genotype with climate parameters suggest Caenorhabditis elegans niche adaptations. G3 7, 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Snoek BL et al. (2020) WormQTL2: an interactive platform for systems genetics in Caenorhabditis elegans. Database 2020, baz149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rockman MV and Kruglyak L (2008) Breeding designs for recombinant inbred advanced intercross lines. Genetics 179, 1069–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Seidel HS et al. (2008) Widespread genetic incompatibility in C. elegans maintained by balancing selection. Science 319, 589–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seidel HS et al. (2011) A novel sperm-delivered toxin causes late-stage embryo lethality and transmission ratio distortion in C. elegans. PLoS Biol. 9, e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sterken MG et al. (2015) The laboratory domestication of Caenorhabditis elegans. Trends Genet. 31, 224–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cook DE et al. (2016) CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 45, D650–D657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee D et al. (2020) Balancing selection maintains ancient genetic diversity in C. elegans. bioRxiv Published online July 25, 2020. 10.1101/2020.07.23.218420 [DOI] [Google Scholar]
- 118.Churchill GA et al. (2004) The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 36, 1133–1137 [DOI] [PubMed] [Google Scholar]
- 119.Churchill GA et al. (2012) The Diversity Outbred mouse population. Mamm. Genome 23, 713–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.King EG et al. (2012) Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 22, 1558–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kover PX et al. (2009) A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 5, e1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Noble LM et al. (2021) Gene-level quantitative trait mapping in Caenorhabditis elegans. G3 Genes|Genomes|Genetics 11, jkaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Volkers RJM et al. (2013) Gene-environment and proteindegradation signatures characterize genomic and phenotypic diversity in wild Caenorhabditis elegans populations. BMC Biol. 11, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gibson G (2012) Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Petersen C et al. (2015) Why we need more ecology for genetic models such as C. elegans. Trends Genet. 31, 120–127 [DOI] [PubMed] [Google Scholar]
- 126.Harris RM and Hofmann HA (2015) Seeing is believing: dynamic evolution of gene families. Proc. Natl. Acad. Sci. U. S. A. 112, 1252–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.International Helminth Genomes Consortium (2019) Comparative genomics of the major parasitic worms. Nat. Genet. 51, 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kapli P et al. (2020) Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 21, 428–444 [DOI] [PubMed] [Google Scholar]
- 129.Howe KL et al. (2017) WormBase ParaSite - a comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 215, 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Howe KL et al. (2016) WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 44, D774–D780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Woollard A (2005) Gene duplications and genetic redundancy in C. elegans. In WormBook (The C. elegans Research Community, ed.), WormBook; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang J (2003) Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298 [Google Scholar]
- 133.Stern DL and Frankel N (2013) The structure and evolution of cis-regulatory regions: the shavenbaby story. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hoekstra HE and Coyne JA (2007) The locus of evolution: evo devo and the genetics of adaptation. Evolution 61, 995–1016 [DOI] [PubMed] [Google Scholar]
- 135.Chan YF et al. (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jansen RC and Nap JP (2001) Genetical genomics: the added value from segregation. Trends Genet. 17, 388–391 [DOI] [PubMed] [Google Scholar]
- 137.Brem RB and Kruglyak L (2005) The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc. Natl. Acad. Sci. U. S. A. 102, 1572–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rockman MV (2012) The QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution 66, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bloom JS et al. (2013) Finding the sources of missing heritability in a yeast cross. Nature 494, 234–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paaby AB and Rockman MV (2013) The many faces of pleiotropy. Trends Genet. 29, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xu W et al. (2019) Evolution of yin and yang isoforms of a chromatin remodeling subunit precedes the creation of two genes. eLife 8, e48119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hill WG et al. (2008) Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 4, e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mäki-Tanila A and Hill WG (2014) Influence of gene interaction on complex trait variation with multilocus models. Genetics 198, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ehrenreich IM (2017) Epistasis: searching for interacting genetic variants using crosses. G3 7, 1619–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang J et al. (2010) Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bloom JS et al. (2015) Genetic interactions contribute less than additive effects to quantitative trait variation in yeast. Nat. Commun. 6, 8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barrett RDH and Hoekstra HE (2011) Molecular spandrels: tests of adaptation at the genetic level. Nat. Rev. Genet. 12, 767–780 [DOI] [PubMed] [Google Scholar]
- 148.Zhao Y et al. (2018) Changes to social feeding behaviors are not sufficient for the fitness gains of the C. elegans N2 reference strain. Elife 7, e38675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Félix M-A and Duveau F (2012) Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC Biol. 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Alexander-Floyd J et al. (2020) Unexpected cell type-dependent effects of autophagy on polyglutamine aggregation revealed by natural genetic variation in C. elegans. BMC Biol. 18, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Daul AL et al. (2019) The Caenorhabditis genetics center (CGC) and the Caenorhabditis elegans natural diversity resource. In The Biological Resources of Model Organisms (Jarret RL and McCluskey K, eds), pp. 69–94, CRC Press [Google Scholar]
- 152.Kammenga JE (2017) The background puzzle: how identical mutations in the same gene lead to different disease symptoms. FEBS J. 284, 3362–3373 [DOI] [PubMed] [Google Scholar]
- 153.Milloz J et al. (2008) Intraspecific evolution of the intercellular signaling network underlying a robust developmental system. Genes Dev. 22, 3064–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dilks CM et al. (2020) Quantitative benzimidazole resistance and fitness effects of parasitic nematode beta-tubulin alleles. Int. J. Parasitol. Drugs Drug Resist. 14, 28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morran LT et al. (2009) Mutation load and rapid adaptation favour outcrossing over self-fertilization. Nature 462, 350–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yeh S-D et al. (2018) The mutational decay of male-male and hermaphrodite-hermaphrodite competitive fitness in the androdioecious nematode C. elegans. Heredity 120, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Morran LT et al. (2011) Running with the Red Queen: host-parasite coevolution selects for biparental sex. Science 333, 216–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wernick RI et al. (2019) Sex and mitonuclear adaptation in experimental Caenorhabditis elegans populations. Genetics 211, 1045–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Slowinski SP et al. (2016) Coevolutionary interactions with parasites constrain the spread of self-fertilization into outcrossing host populations. Evolution 70, 2632–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Teotónio H et al. (2017) Experimental evolution with Caenorhabditis nematodes. Genetics 206, 691–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gray JC and Cutter AD (2014) Mainstreaming Caenorhabditis elegans in experimental evolution. Proc. Biol. Sci. 281, 20133055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.