

Correctly identifying candidate drugs for protein targets is crucial for drug discovery. Despite the importance of this problem for the pharmaceutical industry, chemical screening remains a challenging task, and drug–target misidentification may contribute to failures in drug development. In their recent study, Sauer and colleagues (Holbrook‐Smith et al, 2022) demonstrate proof‐of‐concept for a new way to identify drug–target interactions using high‐throughput metabolomics, potentially paving the way towards a universal method for predicting drug–target relationships.
Subject Categories: Metabolism, Methods & Resources, Pharmacology & Drug Discovery
Correctly identifying candidate drugs for protein targets is crucial for drug discovery. In their recent work, Sauer and colleagues (Holbrook‐Smith et al, 2022) demonstrate proof‐of‐concept for a new way to identify drug–target interactions using high‐throughput metabolomics.

The advent of “omics” technologies including genomics, transcriptomics, proteomics, and metabolomics has presented an opportunity to discover drug candidates using systematic, global measurements of biomolecules. Over 20 years ago, early microarray profiling demonstrated that novel drug–target relationships could be identified by comparing the transcriptomic profiles of gene knockout and drug‐treated yeast (Hughes et al, 2000). Later efforts expanded this concept to mammalian cells treated with tens of thousands of drugs and other perturbagens (Subramanian et al, 2017). Proteomic approaches to drug target identification have also been successful at defining drug–target relationships, including thermal proteome profiling which exploits the increased resistance to heat‐induced protein unfolding that occurs upon drug binding (Savitski et al, 2014).
Notably, of all the “omic” layers, metabolomics is considered the closest to phenotype (Patti et al, 2012). Based on this idea, Holbrook‐Smith et al adopted a conceptually similar approach to that of Hughes et al, 2000. Specifically, Holbrook‐Smith et al compared the metabolomic profiles of the yeast Saccharomyces cerevisiae that had been subjected to either drug treatments or inducible overexpression of various intracellular and membrane‐bound proteins (Fig 1). If the metabolomic profiles following drug treatment and protein overexpression were highly correlated, then that drug was considered a candidate for the protein target. Testing this approach on membrane protein‐coding genes, Holbrook‐Smith et al identified and validated five novel antagonists for the G‐protein‐coupled receptor GPR1. Expanding to a larger set of 86 druggable genes, the authors further demonstrated that their method recovered true‐positive drug–target interactions with high sensitivity and specificity. Taken together, these results establish proof‐of‐concept for the identification of drug–target relationships using metabolomics.
Figure 1. Drug target identification by high‐throughput metabolomics.
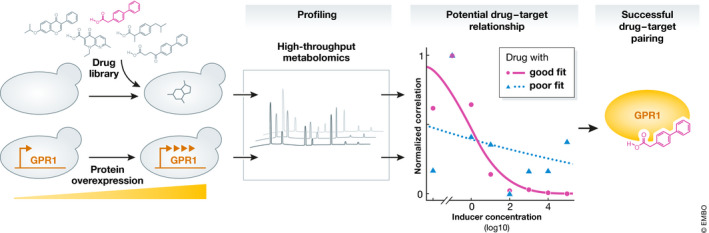
The metabolomic profiles of yeast treated with > 1,000 different drugs and yeast where a protein target had been inducibly overexpressed were collected by high‐throughput metabolomics. Candidate drugs were identified based on the similarity of the metabolomic profile of drug‐treated cells and that of cells with protein overexpression. This novel approach may represent a tool for universal prediction of drug–target relationships.
The key innovation that enables the approach of Holbrook‐Smith et al is flow injection time‐of‐flight mass spectrometry (FIA TOF MS), a high‐throughput metabolomic method which requires sample analysis time of < 1 min per sample (Fuhrer et al, 2011). This technique removes the bottleneck on the time required for MS sample analysis (~1 h per sample) in more traditional metabolomic approaches. With this method, the authors were able to profile thousands of conditions representing diverse drug treatments and protein overexpression conditions in a tractable manner, enabling the acquisition of a data set rich enough for candidate drug identification. Compared to traditional methods, this approach has several notable advantages. First, it leverages the ability of the metabolome to serve as the direct readout of cellular biochemistry. Secondly, by screening in vivo, drug candidates may be less susceptible to downstream development problems related to in vivo drug metabolism or the inability of drugs to reach intracellular targets. Lastly, this approach is amenable to membrane proteins, which are notoriously difficult to study biochemically yet represent many important drug targets.
Despite the advances of this project, significant hurdles remain before high‐throughput metabolomics becomes a tool for universal prediction of drug–target interactions. First, it will be important to show that this approach functions for drug discovery in mammalian cells. Although metabolic pathways are largely conserved between yeast and mammals, will differences in nutrient‐sensing mechanisms (González & Hall, 2017) and/or other pathways limit the applicability of this method in mammalian cells? Another question left unanswered is how media composition might impact drug target identification by metabolomics. In mammalian cells, media composition can significantly affect the response to drugs and genetic perturbations (Joly et al, 2021). As such, would the authors have found different results if their yeast had been cultured in a different media? Lastly, while FIA TOF MS enables rapid analysis of samples by omitting upstream chromatography, it is unable to resolve isomeric compounds. Are there drugs whose effects will be missed because of limitations in measuring critical metabolites? Similarly, there may be some druggable proteins whose effects on the metabolome are not captured by FIA TOF MS metabolomics. Answering these questions will be critical to realizing the potential of high‐throughput metabolomics for drug discovery.
In summary, the work of Holbrook‐Smith et al represents a significant step in expanding drug target discovery through metabolomics, the molecular “omics” layer closest to phenotype. Despite the billions of dollars poured into drug discovery, only roughly 14% of drug development programs result in FDA approval (Wong et al, 2019), although the usage of biomarkers based on stringent drug–target identification could improve the success rate of clinical trials. The success of Holbrook‐Smith et al suggests that high‐throughput metabolomics has broad potential to generate robust, whole‐genome predictions of drug–target interactions to improve drug development.
Mol Syst Biol. (2022) 18: e10914
See also: D Holbrook‐Smith et al
References
- Fuhrer T, Heer D, Begemann B, Zamboni N (2011) High‐throughput, accurate mass metabolome profiling of cellular extracts by flow injection‐time‐of‐flight mass spectrometry. Anal Chem 83: 7074–7080 [DOI] [PubMed] [Google Scholar]
- González A, Hall MN (2017) Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36: 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook‐Smith D, Durot S, Sauer U (2022) High‐throughput metabolomics predicts drug‐target relationships for eukaryotic proteins. Mol Syst Biol 18: e10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD et al (2000) Functional discovery via a compendium of expression profiles. Cell 102: 109–126 [DOI] [PubMed] [Google Scholar]
- Joly JH, Chew BTL, Graham NA (2021) The landscape of metabolic pathway dependencies in cancer cell lines. PLoS Comput Biol 17: e1008942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti GJ, Yanes O, Siuzdak G (2012) Metabolomics: the apogee of the omic triology. Nat Rev Mol Cell Biol 13: 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S et al (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346: 1255784 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK et al (2017) A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171: 1437–1452.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CH, Siah KW, Lo AW (2019) Estimation of clinical trial success rates and related parameters. Biostatistics 20: 273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]