

Abstract
Misfolding and aggregation of alpha-synuclein (α-synuclein) with concomitant cytotoxicity is a hallmark of Lewy body related disorders such as Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Although it plays a pivotal role in pathogenesis and disease progression, the function of α-synuclein and the molecular mechanisms underlying α-synuclein-induced neurotoxicity in these diseases are still elusive. Many in vitro and in vivo experimental models mimicking α-synuclein pathology such as oligomerization, toxicity and more recently neuronal propagation have been generated over the years. In particular, cellular models have been crucial for our comprehension of the pathogenic process of the disease and are beneficial for screening of molecules capable of modulating α-synuclein toxicity. Here, we review α-synuclein based cell culture models that reproduce some features of the neuronal populations affected in patients, from basic unicellular organisms to mammalian cell lines and primary neurons, to the cutting edge models of patient-specific cell lines. These reprogrammed cells known as induced pluripotent stem cells (iPSCs) have garnered attention because they closely reproduce the characteristics of neurons found in patients and provide a valuable tool for mechanistic studies. We also discuss how different cell models may constitute powerful tools for high-throughput screening of molecules capable of modulating α-synuclein toxicity and prevention of its propagation.
Keywords: cellular model, dopaminergic neurons, oligomers, Parkinson’s disease, α-Synuclein
It is well-established that mutations in, and multiplications of, the SNCA gene encoding alphαasynuclein (α-synuclein) cause Parkinson’s disease (PD) (Stefanis 2012), a neurodegenerative disorder that presents clinically with a collection of motor impairments referred to as parkinsonism as well as non-motor symptoms such as sleep disorder, depression, gastrointestinal disturbances, and often dementia (Chaudhuri et al. 2006; Langston 2006). Related neurological disorders with symptoms of parkinsonism include Parkinson’s disease dementia (PDD), dementia with Lewy bodies, and multiple system atrophy (Jellinger 2003). These disorders, also known as alpha-synuclein opathies, are neuropathologically characterized by the accumulation of α-synuclein in cytoplasmic inclusions known as Lewy bodies (LBs) in vulnerable neuronal and glial populations. The first identification of a genetic cause for PD was made more than 20 years ago, when Polymeropoulos and colleagues identified a mutation in the SNCA gene in an Italian family with an autosomal dominant form of PD (Polymeropoulos et al. 1997; Nussbaum 2017). Soon after, histological studies in post mortem brains of idiopathic PD and dementia with Lewy bodies patients revealed that α-synuclein is a major constituent of LBs, strengthening the case for α-synuclein’s pivotal role in the pathogenesis of PD and related disorders (Spillantini et al. 1997; Goedert et al. 2017). Since then, various mutations in and multiplications of SNCA have been discovered and have been directly linked to disease progression and severity (Devine et al. 2011). Subsequently, experimental in vitro and in vivo models involving overexpression of wild-type or mutant α-synuclein have been developed in an effort to model the disease pathology of α-synuclein aggregation and toxicity (Kirik et al. 2002; Lo Bianco et al. 2002; Giasson et al. 2002).
Cumulative evidence implicates a causative role of α-synuclein aggregation in neurodegeneration (Irizarry et al. 1998; Chartier-Harlin et al. 2004; Winner et al. 2011). However, the molecular mechanisms underlying cytotoxicity in PD are still elusive, hampering the development of disease modifying therapies. α-synuclein is a soluble presynaptic protein that may exist as a natively unfolded monomer (Fauvet et al. 2012; Binolfi et al. 2012; Burre et al. 2013; Theillet et al. 2016) or a functional tetramer (Bartels et al. 2011; Gurry et al. 2013; Selkoe et al. 2014; Dettmer et al. 2015). Under pathological circumstances, α-synuclein forms aggregates via the assembly of soluble oligomeric intermediates that mature into the insoluble amyloid fibrils found in LBs. Whether such cytoplasmic inclusions contribute to neuronal death or protect cells from the toxic effects of misfolded proteins remains controversial. The current hypothesis in the field suggests that α-synuclein pre-fibrillar forms represent the toxic species, making them the subject of intense investigations (Conway et al. 2000; Outeiro et al. 2008; Villar-Pique et al. 2016). Furthermore, it is now widely recognized that α-synuclein aggregates can spread throughout the central nervous system via cell-to-cell propagation, possibly in a prion-like manner (Kordower et al. 2008; Masuda-Suzukake et al. 2013; Recasens and Dehay 2014) driving disease progression. Taken together, it appears that lowering α-synuclein levels and/or eliminating toxic α-synuclein species in cells, will be an attractive target for therapeutics to halt disease progression (Nasstrom et al. 2011; Brundin et al. 2017). Above all, the availability of reliable experimental models is essential to garner a deeper understanding of the mechanisms associated with α-synuclein-mediated toxicity and, most importantly, to aid development and validation of future pharmacological interventions.
Mechanistic aspects of a disease often emerge from studies at the cellular and subcellular level. Mammalian cell lines and unicellular organisms such as yeast have already provided valuable insights into the pathophysiology of synucleinopathies and are key translational approaches prior to validation in preclinical animal models and in human specimens (Alberio et al. 2012). Cellular models have tremendous advantages over in vivo approaches as they are fast and reproducible and importantly, are a cost-effective tool. They are also readily amenable to genetic modifications and pharmacological manipulations, making the direct targeting of specific cellular processes involved in disease feasible. With a central role for α-synuclein in disease, several cellular models that mimic important aspects of α-synuclein biology, such as aggregation and toxicity, have been developed over the years and have contributed to many advances in our comprehension of the pathogenesis of PD and other synucleinopathies (Lazaro et al. 2017). Although cell models are simplified systems which do not fully reproduce neuronal networks or recapitulate the complexity of the diseases, they are powerful tools to unravel pathophysiological mechanisms at play in neurodegeneration as well as being useful for high-throughput screening of new therapeutic compounds (Schule et al. 2009).
Herein, we review α-synuclein cell-based models that are currently available and discuss their contribution to the understanding of molecular neurodegeneration and how these models may shed light on new drug discovery in synucleinopathies.
Lessons learned from the baker’s yeast Saccharomyces cerevisiae
The baker’s yeast Saccharomyces cerevisiae has been used for thousands of years for industrial applications, including baking bread. Therefore, our understanding of the biology and genetics of this organism is vast, and many of its characteristics can be exploited for the study of basic biological processes that are conserved among all eukaryotes. In particular, the detailed understanding of molecular machines involved in protein folding and degradation, and the identification of yeast proteins with prion behavior led to the use of S. cerevisiae (herewith referred to as yeast) as a living test tube with which to investigate the molecular underpinnings of neurodegenerative diseases associated with protein misfolding and aggregation. At around the same time that multiplications of the SNCA gene were linked with familial forms of PD, heterologous expression of human α-synuclein in yeast resulted in two key aspects of synucleinopathies, dose-dependent cytotoxicity and inclusion formation (Outeiro and Lindquist 2003). Strikingly, the toxic effects of α-synuclein resulted from an impairment in intracellular trafficking, altered lipid metabolism, and increased levels of oxidative stress (Outeiro and Lindquist 2003). Several other groups have exploited the yeast toolbox to further dissect the molecular mechanisms underlying α-synuclein toxicity and aggregation (Zabrocki et al. 2005). Importantly, using powerful genetic screens, modifiers of toxicity and aggregation have been identified (Willingham et al. 2003; Cooper et al. 2006; Liang et al. 2008; Su et al. 2010), and further validated, in primary neuronal cultures and in vivo, in multicellular organisms such as Caenorhabditis elegans, drosophila, and mice (Tardiff et al. 2014). From the genetic screens, endoplasmic reticulum (ER) to Golgi trafficking and membrane and lipid metabolism alterations have emerged as highly conserved pathways affected by α-synuclein in the cell (Zabrocki et al. 2008). Despite initial skepticism, these basic studies in yeast have provided tremendous insight into the biology and pathobiology of α-synuclein and other PD-associated genes (Buttner et al. 2008; Sampaio-Marques et al. 2012; Tardiff et al. 2014; Dhungel et al. 2015), and have enabled the identification of several small molecules that are capable of alleviating α-synuclein -induced cytotoxicity (Fleming et al. 2008; Su et al. 2010).
More recently, yeast has proven a useful tool to study the effect of PD-associated mutations in α-synuclein (Lazaro et al. 2014; Lazaro et al. 2016) and of post-translational modifications, such as phosphorylation (Tenreiro et al. 2014; Mbefo et al. 2015; Kleinknecht et al. 2016; Tenreiro et al. 2017; Bras et al. 2018), sumoylation (Shahpasandzadeh et al. 2014), or glycation (Vicente Miranda et al. 2017). The development of effective therapies for synucleinopathies has been a tremendous challenge, and continues to be an awesome task. Yeast alone will not be the solution, but together with more complex models, it has already proven to be an invaluable tool/model organism in which to investigate the basic biology and pathobiology of α-synuclein (Tenreiro and Outeiro 2010; Menezes et al. 2015).
Non-neuronal cell models of α-synuclein overexpression
Since mutations in and multiplications of SNCA are considered causative for some cases of PD, numerous cellular models based on overexpression have been generated in the last two decades aimed at understanding the contribution of α-synuclein to the disruption of cellular processes (Table 1). Indeed, several biochemical pathways known to be affected in synucleinopathies including mitochondrial dysfunction, increased apoptosis, and oxidative stress, as well as defects in protein degradation machinery, were all discovered in cellular overexpression models (Outeiro et al. 2008; Klucken et al. 2012; Lazaro et al. 2014). In patients with SNCA multiplications, increased expression of wild-type α-synuclein is sufficient to cause parkinsonism, and rare point mutations seem to increase α-synuclein aggregation leading to neurodegeneration (Devine et al. 2011). Consequently, generating cellular models where α-synuclein accumulates and forms oligomers has been considered a useful strategy. Two of the most commonly used immortalized human non-neuronal cell lines in this context are human embryonic kidney 293 (HEK293) and human H4 neuroglioma (H4) lines. These cells are easily transfected with transient and constitutive (stable) overexpression of human wild-type or mutated α-synuclein widely reported in both cases (Tabrizi et al. 2000; McLean et al. 2001; Outeiro et al. 2008; Lazaro et al. 2016). Increased expression of wild-type α-synuclein can be detected in a short period of time and most importantly, α-synuclein positive inclusions are often formed, depending on the specific paradigm. Interestingly, time-lapse imaging performed in an HEK293 model illustrated how cells formed and accumulated aggregated forms of α-synuclein (Opazo et al. 2008). These cell lines are also suitable to study the effect of α-synuclein mutations. Among them, the A53T or A30P point mutations are known to cause familial early onset PD with A53T being the most highly penetrant and widely studied of the mutations. Interestingly, these two mutations show greater toxicity in cellular models than wild-type α-synuclein. Thus it seems plausible that these mutations render α-synuclein more susceptible to aggregation (Lazaro et al. 2014).
Table 1.
Cell lines available to study α-synuclein toxicity and aggregation with their associated advantages and disadvantages
| Cellular model | Uses | Advantages | Disadvantages | |
|---|---|---|---|---|
| Yeast | Synuclein overexpression; small molecule screens; synuclein post translational modifications | Easy genetic manipulations; easy to culture; ideal for high-throughput screening | Basic model; Limited translational relevance; | |
| Non-neuronal | HEK293 | Overexpression of mutant and wildtype synuclein; effects of toxins on synuclein toxicity; co-expression studies; tracking of aggregate formation | Easy to culture; facile transfection; labelling techniques can be used to monitor aggregate formation; suitable for high-throughput screens | No dopaminergic phenotype; limited endogenous asyn expression and functionality |
| H4 | ||||
| Primary neurons | Brain-region specific preparations; seeding with pre-formed fibrils; propagation of α - syn between neurons; overexpression of mutant and wildtype synuclein; mutant forms from transgenic animals | Dissection to culture neurons from specific brain regions of interest; can be used to study catecholamine neurotransmission; opportunity to study α-syn in physiological setting | Difficult to prepare and maintain; variability in cell- type composition between preps | |
| Differentiated immortalized cells | PC12 | Generation of dopaminergic neurons; overexpression of mutant and wildtype synuclein; α -syn transmission studies; high- throughput screening | Can be differentiated to dopaminergic phenotype; | Non-human; cancer derived |
| SH-SY5Y | Human cell-line; can be differentiated to dopaminergic phenotype | Cancer-derived; inconsistent differentiation | ||
| LUHMES | Difficult to culture and transfect | |||
| Patient derived | Fibroblasts/PBMCs | Patient specific cell lines; genetic mutants and gene edited isogenic controls; overexpression of mutant and wildtype synuclein, brain- region specific differentiation | Can be generated from individual patients non- invasively; isogeneic controls can be developed with gene editing; can be differentiated to different cell types; no ethical concerns | Difficult to transfect; loss of cell aging; difficult to maintain; lentivirus needed for generation |
| iPSCs | ||||
| iNeurons | ||||
Illustrations adapted from Servier Medical Art (https://smart.servier.com) under Creative Commons License.
Even though overexpression of wild-type or mutated α-synuclein in cells seems to recapitulate aspects of synucleinopathies, one major drawback in these models is the low level of toxicity associated with α-synuclein overexpression. A second limitation is the absence of abundant α-synuclein aggregates to mimic the major pathologic features observed in diseased post mortem brains. Many of these cell models exhibit small inclusions that are Thioflavin S positive or resistant to protease digestion, but only a few will actually develop larger aggregates which share properties of LBs found in patients. Thus, additional insults have commonly been used to challenge the cells and create a toxic environment. For example, the frequency of intracytoplasmic inclusions increases when toxins such as 1- methyl-4- phenylpyridinium (MPP+), rotenone, or proteasome inhibitor are applied to α-synuclein overexpressing cells (McLean et al. 2001; Lee et al. 2002). Importantly, co-expression with specific proteins facilitates the formation of more mature aggregates. Co-expression of synphilin-1, an α-synuclein interacting protein, can induce the formation of inclusions in H4 cells (McLean et al. 2001) as well as HEK293 cells (Engelender et al. 1999; O’Farrell et al. 2001; Tanaka et al. 2004). Lastly, the brain specific protein, p25a, has also been identified as a stimulator of α-synuclein aggregation in vitro (Lindersson et al. 2005; Ejlerskov et al. 2013).
Tracking α-synuclein oligomers in vitro
A large body of evidence indicates that the assembly of toxic oligomeric species of α-synuclein may be one of the key processes underlying the induction of pathology and spread of synucleinopathies. Understanding how these aggregates form and assessing their impact on neuronal function will contribute to the development of therapeutic targets to prevent disease progression. It is well known that in vitro induced α-synuclein oligomers ectopically applied to cell cultures are formed because of overexpression of α-synuclein, induce cell death and toxicity (Chen et al. 2007; Danzer et al. 2007; Tetzlaff et al. 2008). However, the lack of sensitive in situ detection methods has hindered the study of oligomeric α-synuclein species. Therefore tremendous efforts have been made to generate cell lines that allow tracking and visualization of aggregation in living cells (Fig. 1). Innovative technologies using biosensors from fluorescence labeling to protein complementation assays (PCA) have been developed to monitor α-synuclein/α-synuclein interaction. Intracellular α-synuclein oligomerization was visualized for the first time in H4 cells using a highly sensitive molecular fluorescence lifetime imaging microscopy by fusing a small epitope tag to α-synuclein (Klucken et al. 2006). A similar approach was used in HEK293 cells when α-synuclein was tagged with a six amino acid PDZ binding motif and co-expressed with the corresponding PDZ domain fused with enhanced green fluorescent protein (Opazo et al. 2008) and in contrast to traditional approaches with fusion proteins, provided higher sensitivity. Lastly, fluorescent-based biosensor cell lines have allowed the detection and the quantification of α-synuclein seeding activity. α-synuclein ‘seeds’ isolated from post mortem brain tissue with pathologically confirmed synucleinopathy, were found to trigger aggregation in HEK293T cells stably expressing α-synuclein-yellow fluorescent protein (YFP) resulting in a fluorescent readout that can be visualized and quantified using regular confocal microscopy (Prusiner et al. 2015). Likewise, Holmes and colleagues developed a HEK-based biosensor cell line that takes advantage of a Förster resonance energy transfer readout for seeding that can be detected and quantified using flow cytometry (Holmes and Diamond 2017).
Fig. 1.
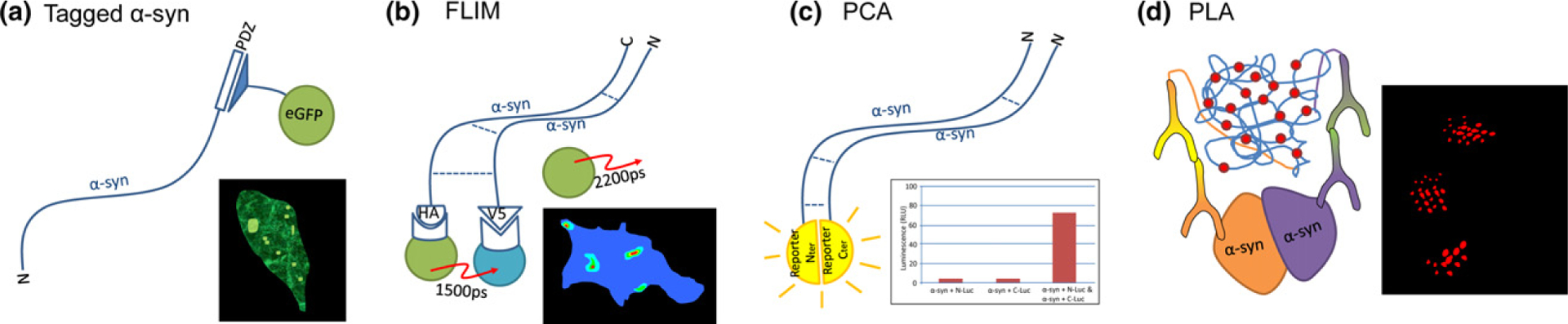
A number of methods have been developed to visualize aggregating α-synuclein in cellular models. These methods include the indirect (a) where synuclein containing a PDZ binding motif epitope is co-expressed with the corresponding PDZ domain fused to eGFP, sensitively labeling α-synuclein, with aggregates presenting as bright puncta (Opazo et al, 2008). Multimeric conformations of α-synuclein are more directly visualized using fluorescence lifetime imaging microscopy (FLIM) (b) by expressing α-synuclein containing small epitope tags. These can be targeted with FRET donor and acceptor secondary antibodies where the proximity of the interaction between synuclein molecules determined as the fluorescence lifetime of the donor fluorophore (Klucken et al. 2006). More recently (c) protein complementation assay (PCA) approaches have been used where synuclein tagged with either N- or C- terminal portions of a split reporter (fluorescent or bioluminescent) is expressed and the reporter signal used as a proxy for dimeric or higher order oligomeric species (Moussaud et al, 2015). In order to label endogenous α-synuclein aggregates, PLA can be employed (d) where proximity-dependent rolling circle amplification of oligonucleotide-labeled antibodies generates a signal to mark α-synuclein aggregates (Roberts et al, 2015)
Another method to study α-synuclein/α-synuclein interactions that has been developed is based on the principle of bimolecular protein-fragment complementation (Remy and Michnick 1999; Kerppola 2008). PCA have been adapted to enable rapid and non-destructive reporting of α-synuclein oligomerization in living cells by the fusion of α-synuclein molecules with the inactive C-terminal or N-terminal fragments of a fluorescent (ie, YFP) (fPCA) or luminescent (ie. humanized Gaussia luciferase) reporter bioluminescent PCA [bioluminescent PCA (bPCA)] (Outeiro et al. 2008). The functional fluorophore or bioluminescent protein is reconstituted upon α-synuclein/α-synuclein interactions and acts as a surrogate reporter for the formation of oligomers that can be detected with readouts such as fluorescence microscopy, flow cytometry, or photometric measurement (Fig. 1c). α-synuclein -fPCA and bPCA have been widely used to confirm a major role for multimeric species in cytotoxicity and disease propagation (Outeiro et al. 2008; Danzer et al. 2012; Lazaro et al. 2014; Jiang et al. 2017). Moreover, this technology allows the investigation of early stages of α-synuclein aggregation, at a time before larger oligomers formation occured. Recently, stable cell lines co-expressing α-synuclein PCA using bioluminescent and fluorescence reporters, were developed and used in high-throughput screening (HTS) to identify inhibitors of α-synuclein oligomerization (Moussaud et al. 2015). Lastly, because the spread of α-synuclein from neuron to neuron is now considered an important step in PD pathogenesis, PCA can be applied to develop model systems where cell-to-cell transmission can be quantitatively analyzed. Using co-cultures, cells expressing α-synuclein tagged with either the N-or C-terminal fragment of a reporter allow the identification of cells where α-synuclein has been transferred from cell to cell by monitoring the signal of the reconstituted complete reporter (Bae et al. 2014). Using α-synuclein-bPCA, Danzer and colleagues found that α-synuclein oligomers are present in secreted extracellular vesicles that are taken up by recipient cells inducing toxicity (Danzer et al. 2012).
Although a powerful tool, PCA requires the use of tagged-proteins and does not discriminate between dimers and larger multimeric species of aggregates. To overcome these issues, the use of the newly developed proximity ligation assay (PLA) represents an alternative strategy (Fig. 1d). PLA has attracted a lot of attention because it allows the investigation of protein interactions at the endogenous level without the need for tagging or overexpression of proteins (Soderberg et al. 2006; Dettmer and Bartels 2015). A pair of oligonucleotide-labeled secondary antibodies (PLA probes) generate a signal only when the two PLA probes are bound in close proximity. The signal from each detected pair of PLA probes is then amplified and visualized as an individual fluorescent spot. In an elegant study, Roberts and colleagues (Roberts et al. 2015) developed the α-synuclein -PLA and demonstrated the sensitivity of the technique to detect α-synuclein oligomers with minimal recognition of monomeric and higher molecular weight species.
Primary neurons
An alternative to immortalized cell lines is the use of primary neurons prepared from embryonic or early post-natal mouse or rat pups. These cultures closely simulate a neuronal environment and may yield more physiologically significant results. They also offer the possibility to isolate neurons from specific brain regions allowing enrichment of specific neuronal populations. In synucleinopathies, and in particular PD, neuronal cell loss is observed in dopamine (DA) producing neurons and to lesser extent in cholinergic neurons. Therefore preparation of primary DAergic neurons from the ventral mesencephalon of mice has been extensively used to closely model this pathological hallmark of PD (Dryanovski et al. 2013; Gaven et al. 2014). Overexpression of wild-type or mutated forms of α-synuclein can be easily achieved in these models using transfection techniques (Tonges et al. 2014; Hassink et al. 2018) or neurons overexpressing α-synuclein can be directly cultured by preparing primary neurons from α-synuclein transgenic animals (Li et al. 2013). Recently recombinant α-synuclein has been utilized to generate small seeds of pre-formed α-synuclein fibrils (PFFs) and induce aggregates with characteristics of those found in diseased brains (Volpicelli-Daley et al. 2014). The addition of PFFs to primary neurons leads to the recruitment of endogenous α-synuclein into LB-like aggregates that are insoluble in detergent, hyperphosphory-lated, ubiquitinated, and have a filamentous ultrastructure when examined using electron microscopy (Volpicelli-Daley et al. 2014). This model system provides researchers with an opportunity to examine aggregation of α-synuclein from early formation to spread throughout the neuron, and ultimately neuronal death. In addition, intercellular trafficking of internalized α-synuclein seeds in primary neurons has been characterized using microfluidic devices that allow fluidic separation of neuronal soma from axonal projections. Freund et al. used live cell imaging to show the transfer of fluorescent α-synuclein PFFs to a second-order neuron (Freundt et al. 2012) and fluorescently labeled PFFs have also recently been utilized to image the internalization of α-synuclein seeds (Karpowicz et al. 2017; Jiang et al. 2017). Recently, mechanistic studies using PFFs revealed that the immune receptor Lag3 is a receptor for PFF α-synuclein in neurons initiating α-synuclein fibrils endocytosis, transmission and toxicity (Mao et al., 2016). While other receptors for α-synuclein have been proposed based on proteomics studies, none have been validated functionally to date (Shrivastava et al. 2015; Bieri et al. 2018).
Differentiated dopaminergic cell-model
Many of the concepts discussed above are common to the multiple neurodegenerative diseases classified as synucleinopathies. Although cell death is a common key pathologic feature, the selective loss of DAergic neurons from the substantia nigra is specific to PD. DA deficit observed in PD patients underlies the three cardinal motor symptoms tremor, rigidity and akinesia which can be substantially improved by DA replacement therapy. Over the years, efforts to develop DAergic cell lines have expanded as an alternative strategy to obtain faithful cellular models of PD. The SH-SY5Y neuroblastoma and the PC12 pheochromocytoma cell lines bear many similarities to the neuronal populations affected in PD and are widely used to unravel neurodegenerative mechanisms. Indeed, PC12 and SH-SY5Y cells have the ability to differentiate into neurons after sequential exposure to retinoic acid or brain derived neurotrophic factor respectively. Upon differentiation, changes in morphology and function are observed with the extension of neuron-like processes and production and release of catecholamines. These DAergic-like neuronal cell lines are very similar to mesencephalon-derived primary neurons with the advantage that they can be continuously expanded and are less labor intensive (Westerink and Ewing 2008; Xicoy et al. 2017). Transient and constitutive (stable) overexpression of wildtype or mutant α-synuclein in PC12 and SH-SY5Y shows cytotoxicity and can affect cell survival (Stefanis et al. 2001; Matsuzaki et al. 2004; Vekrellis et al. 2009). Remarkably, in a stable PC12 cell line expressing non-toxic levels of wild-type human α-synuclein or A30P mutant, impaired DA release is observed (Larsen et al. 2006). Addition of extracellular α-synuclein from PD brain lysates in differentiated SH-SY5Y also triggers the formation of α-synuclein aggregates (Xin et al. 2015). Finally, Desplats and colleagues demonstrated that α-synuclein is transmitted to neighboring cells via endocytosis and forms Lewy body–like inclusions that displayed ubiquitin immunoreactivity and were thioflavin S positive in a differentiated SH-SY5Y model (Desplats et al. 2009). Collectively, these studies demonstrate the usefulness of DAergic neuron-like cell lines as a predictive tool to mimic a PD-like phenotype in vitro.
Alternatively, the Lund human mesencephalic cells are a well-established model of human DAergic neurons (Lotharius et al. 2002; Lotharius et al. 2005; Scholz et al. 2011). The cells can be differentiated within a few days into a DAergic phenotype when cultured in dibutyryl cyclic adenosinemonophosphate and glial cell-derived neurotrophic factor. They are post-mitotic neurons with electrical properties similar to those of DAergic neurons (Scholz et al. 2011) and produce DA and wild-type human α-synuclein (Lotharius et al. 2002). With these characteristics they represent a good model to mimic a PD-like phenotype and have been used to study neurodegeneration caused by α-synuclein-induced toxicity (Lotharius et al. 2005; Paiva et al. 2017). Recently they were used as a model for high-throughput screening where 1600 FDA approved drugs were screened in differentiated Lund human mesencephalic cells overexpressing α-synuclein to identify neuroprotective compounds (Höllerhage et al.2017). Also worthy of a mention in the present review is the immortalized human neural stem cell (NSC) line, ReNcell VM. Overexpression of the myc family of transcription factors in human primary cells from developing mesencephalon was used to produce a stable multipotential NSC line that can be continuously expanded in monolayer culture and presents with neuronal activity (Hoffrogge et al. 2006; Donato et al. 2007). This line can also be differentiated to exhibit a DAergic phenotype as identified using immunocy-tochemical markers, however further characterization is still necessary. Regardless, immortalized mesencephalic NSCs may well provide a promising model to studying disease mechanisms related to DAergic neurons.
Patient-derived cell lines modeling disease in a dish
As described throughout this review, numerous α-synuclein based-cellular models have been established and are available to the scientific community for studies of pathophysiological mechanisms, toxicity or for high-throughput screening of future therapeutics (Table 1). Nonetheless they represent simplified models that may not accurately recapitulate the complexity of synucleinopathies and discoveries from these models may not precisely reflect the pathogenesis seen in patients. Human skin fibroblasts are an alternative to study disease mechanisms and can be used to develop patient-specific cell lines. However this approach is limited by the fact that fibroblasts in long-term culture become senescent and undergo clonal selection. Interestingly, transduction of specific pluripotency regulators has proven to be sufficient to convert skin fibroblasts into induced pluripotency stem cells (iPSCs). The emergence of patient-derived iPSCs has opened up new possibilities to create physiologically relevant disease models in a culture dish (Piper et al. 2018). Induced pluripotent stem cells offer unprecedented and exciting opportunities to understand disease mechanisms and have potential for high-throughput drug screening to test personalized therapies. Importantly, these new models facilitate the study of patient-specific risk factors or disease-specific mutations (e.g., A53T) in relevant cell types (i.e., DAergic neurons) while comparing the function and phenotype to iPSC lines derived from healthy control individuals (Piper et al. 2018). Lastly in combination with cutting edge genome-editing technologies such as CRISPR-Cas9, disease-causing mutations can be corrected in patient-derived iPSCs or mutations can be introduced into control iPSCs to further elucidate the role of disease-causing mutations (Calatayud et al., 2017; Cobb et al. 2018).
To date several human iPSC models with mutations and genetic variations in the SNCA gene have been engineered to model specific molecular and cellular phenotypes. The generation of iPSC-derived midbrain DAergic neurons from patients with a SNCA triplication produces twice the amount of α-synuclein protein compared to healthy controls (Byers et al. 2011; Devine et al. 2011) and, importantly, the cell lines exhibit disease-related phenotypes such as increased expression of oxidative stress and protein aggregation-related genes. Several iPSCs lines also show lysosomal dysfunction induced by α-synuclein accumulation (Mazzulli et al. 2016). In SNCA triplication iPSC-derived DA neurons, α-synuclein aggregates were shown to physically interact with ATP synthase and lead to premature mitochondrial permeability transition pore (PTP) opening, making the neurons more vulnerable to cell death (Ludtmann et al. 2018). Interestingly, increases in reactive oxygen species following exogenous addition of α-synuclein oligomers were observed in SNCA triplication iPSCs (Deas et al. 2016), while another study demonstrated that the presence of SNCA triplication in iPSC-derived neural precursor cells reduces their capacity to differentiate into DA neurons, decreases neurite outgrowth, and lowers neuronal activity compared to control neurons (Oliveira et al. 2015) Additionally, microglia differentiated from SNCA triplication iPSCs had impaired phagocytosis compared to isogenic controls, suggesting that impaired microglial clearance of extracellular α-synuclein, contributes to α-synuclein accumulation and aggregation phenotype (Haenseler et al. 2017).
Patient-derived iPSCs differentiated into midbrain DA neurons carrying the A53T mutation, have been found to contain higher concentrations of α-synuclein monomers relative to tetramers when compared to the corresponding isogenic controls supporting a possible neuroprotective role of the α-synuclein conformation (Dettmer et al. 2015). Also in A53T iPSC-neurons, α-synuclein nitrosylation was found to be increased compared to isogenic control lines, while correction of the A53T mutation reversed nitrosative stress and ER stress suggesting that the mutation contributes to the aberrant phenotype (Chung et al. 2013). Increased sensitivity to environmental mitochondrial toxins with consequent nitrosative stress-induced neuronal loss has also been observed in A53T SNCA iPSC-DA neurons (Stykel et al. 2018).
In summary iPSCs represent a means to derive physiologically relevant cell types from patients and healthy controls, offering promise for the testing of individualized medicine approaches. Genome-editing techniques to correct genetic variants provide optimal isogenic controls and enable investigations into mechanistic links between genotype and phenotype. As well as utilizing cells from carriers of SNCA multiplications and point mutations, sporadic PD iPSC models can be generated which appear to recapitulate most cellular phenotypes of corresponding monogenic models. Since sporadic cases account for 90% of all PD, these iPSCs models may be considered more relevant. IPSC lines can be utilized in the high-throughput screening of individuals with both genetic and sporadic forms of PD to attempt to identify novel biomarkers or therapeutic approaches.
Although it can be argued that iPSCs are the cellular model with the most translatable relevance, there are some important caveats to be considered. Firstly the epigenetic profile of iPSC may not accurately resemble that of mature neurons. Potentially related to this, α-synuclein accumulation and aggregation is not so far a common feature in iPSC-derived models unless they are maintained in culture for extremely long periods of time. Therefore, more studies must be performed where aging of iPSC-DA neurons is provoked pharmacologically or another method (Vera et al. 2016; Tagliafierro et al. 2019). Alternatively, technological advances in cell differentiation may improve our ability to directly convert patient somatic cells into specific neuronal populations (iNeurons) without altering the epigenetic profile.
Concluding remarks
The central role of α-synuclein in the neurodegenerative process of synucleinopathies has led to the generation of cellular models aiming to elucidate its contribution to the dysregulation of various cellular processes and cellular toxicity (Table 1). As reviewed here, α-synuclein cellular models offer myriad opportunities for studying pathogenic mechanisms and aid development and validation of future pharmacological interventions. Each model has the potential to be a powerful tool provided the limitations and disadvantages of such simplified models are taken into account. Finally, the availability of newer cell culture systems, such as deriving iPSCs from patient somatic cells, offers hope that we will soon be able to closely mirror the disease in a petri dish which will pave the way towards personalized medicine and thus enhance the probability of success of future clinical trials.
Acknowledgments and conflict of interest disclosure
TFO was supported by the DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB).
TFO is a guest editor of the special issue Synuclein Meeting 2019.
Abbreviations used:
- DA
dopamine
- DLB
dementia with Lewy bodies
- FLIM
fluorescence lifetime imaging microscopy
- H4
human H4 neuroglioma
- HEK293
human embryonic kidney 293
- IPSCs
induced pluripotent stem cells
- LB
Lewy bodies
- LUHMES
Lund human mesencephalic cells
- MSA
multiple system atrophy
- NSC
neural stem cell
- PCA
protein complementation assay
- PD
Parkinson’s disease
- PFFs
pre-formed fibrils
- PLA
proximity ligation assay
References
- Alberio T, Lopiano L and Fasano M (2012) Cellular models to investigate biochemical pathways in Parkinson’s disease. FEBS J. 279, 1146–1155. [DOI] [PubMed] [Google Scholar]
- Bae EJ, Yang NY, Song M, et al. (2014) Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun 5, 4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG and Selkoe DJ (2011) alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieri G, Gitler AD and Brahic M (2018) Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis 109, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binolfi A, Theillet FX and Selenko P (2012) Bacterial in-cell NMR of human alpha-synuclein : a disordered monomer by nature? Biochem. Soc. Trans 40, 950–954. [DOI] [PubMed] [Google Scholar]
- Bras IC, Tenreiro S, Silva AM and Outeiro TF (2018) Identification of novel protein phosphatases as modifiers of alpha-synuclein aggregation in yeast. FEMS Yeast Res. 18, 108. 10.1093/femsyr/foy108 [DOI] [PubMed] [Google Scholar]
- Brundin P, Dave KD and Kordower JH (2017) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Vivona S, Diao J, Sharma M, Brunger AT and Sudhof TC (2013) Properties of native brain alpha-synuclein. Nature 498, E4–6; discussion E6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner S, Bitto A, Ring J, et al. (2008) Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J. Biol. Chem 283, 7554–7560. [DOI] [PubMed] [Google Scholar]
- Byers B, Cord B, Nguyen HN, et al. (2011) SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS ONE 6, e26159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calatayud C, Carola G, Consiglio A and Raya A (2017) Modeling the genetic complexity of Parkinson’s disease by targeted genome edition in iPS cells. Curr. Opin. Gen. Dev 46, 123–131. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, et al. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Chaudhuri KR, Healy DG, Schapira AH, Institute National and for Clinical E (2006) Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 5, 235–245. [DOI] [PubMed] [Google Scholar]
- Chen L, Jin J, Davis J, Zhou Y, Wang Y, Liu J, Lockhart PJ and Zhang J (2007) Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem. Biophys Res. Commun 356, 548–553. [DOI] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Auluck PK, et al. (2013) Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MM, Ravisankar A, Skibinski G and Finkbeiner S (2018) iPS cells in the study of PD molecular pathogenesis. Cell Tissue Res. 373, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Harper JD and Lansbury PT Jr (2000) Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39, 2552–2563. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, et al. (2006) Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B and Kostka M (2007) Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci 27, 9220–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR and McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, Cremades N, Angelova PR, et al. (2016) Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid. Redox Signal 24, 376–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E and Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl Acad. Sci. USA 106, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U and Bartels T (2015) ExPLAining early synucleinopathies. Brain 138, 1449–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Soldner F, et al. (2015) Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun 6, 7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Gwinn K, Singleton A and Hardy J (2011) Parkinson’s disease and alpha-synuclein expression. Mov. Disord 26, 2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhungel N, Eleuteri S, Li LB, et al. (2015) Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on alpha-synuclein. Neuron 85, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA and Sinden JD (2007) Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-DaleyL A, Lee VM, Miller RJ, Schumacker PT and Surmeier DJ (2013) Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci 33, 10154–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH and Vilhardt F (2013) Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J. Biol. Chem 288, 17313–17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelender S, Kaminsky Z, Guo X, et al. (1999) Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet 22, 110–114. [DOI] [PubMed] [Google Scholar]
- Fauvet B, Mbefo MK, Fares MB, et al. (2012) alpha-synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem 287, 15345–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J, Outeiro TF, Slack M, Lindquist SL and Bulawa CE (2008) Detection of compounds that rescue Rab1-synuclein toxicity. Methods Enzymol. 439, 339–351. [DOI] [PubMed] [Google Scholar]
- Freundt EC, Maynard N, Clancy EK, et al. (2012) Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol 72, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaven F, Marin P and Claeysen S (2014) Primary culture of mouse dopaminergic neurons. J. Vis. Exp e51751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ and Lee VM (2002) Neuronal alpha-synuclein opathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R and Spillantini MG (2017) The synucleinopathies: twenty years on. J. Parkinsons Dis 7, S51–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T and StultzC M (2013) The dynamic structure of alpha-synuclein multimers. J. Am. Chem. Soc 135, 3865–3872. [DOI] [PubMed] [Google Scholar]
- Haenseler W, Sansom SN, Buchrieser J, et al. (2017) A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Reports 8, 1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassink GC, Raiss CC, Segers-Nolten IMJ, van Wezel RJA, Subramaniam V, le Feber J and Claessens M (2018) Exogenous alpha-synuclein hinders synaptic communication in cultured cortical primary rat neurons. PLoS ONE 13, e0193763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffrogge R, Mikkat S, Scharf C, et al. (2006) 2-DE proteome analysis of a proliferating and differentiating human neuronal stem cell line (ReNcell VM). Proteomics 6, 1833–1847. [DOI] [PubMed] [Google Scholar]
- Höllerhage M, Moebius C, Melms J, et al. (2017) Protective efficacy of phosphodiesterase-1 inhibition against alpha-synuclein toxicity revealed by compound screening in LUHMES cells. Sci. Rep 7, 11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB and Diamond MI (2017) Cellular models for the study of prions. Cold Spring Harbor Perspec. Med 7, a024026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF and Hyman BT (1998) Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J. Neuropathol. Exp. Neurol 57, 334–337. [DOI] [PubMed] [Google Scholar]
- Jellinger KA (2003) Neuropathological spectrum of synucleinopathies. Mov. Disord 18(Suppl 6), S2–12. [DOI] [PubMed] [Google Scholar]
- Jiang P, Gan M, Yen SH, McLean PJ and Dickson DW (2017) Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci. Rep 7, 7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowicz RJ Jr, Haney CM, Mihaila TS, Sandler RM, Petersson EJ and Lee VM (2017) Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem 292, 13482–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK (2008) Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys 37, 465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ and Bjorklund A (2002) Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci 22, 2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinknecht A, Popova B, Lazaro DF, Pinho R, Valerius O, Outeiro TF and Braus GH (2016) C-terminal tyrosine residue modifications modulate the protective phosphorylation of serine 129 of alpha-synuclein in a yeast model of Parkinson’s disease. PLoS Genet 12, e1006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J, Outeiro TF, Nguyen P, McLean PJ and Hyman BT (2006) Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J. 20, 2050–2057. [DOI] [PubMed] [Google Scholar]
- Klucken J, Poehler AM, Ebrahimi-Fakhari D, et al. (2012) Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 8, 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB and Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med 14, 504–506. [DOI] [PubMed] [Google Scholar]
- Langston JW (2006) The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann. Neurol 59, 591–596. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, et al. (2006) Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci 26, 11915–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro DF, Rodrigues EF, Langohr R, et al. (2014) Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 10, e1004741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro DF, Dias MC, Carija A, Navarro S, Madaleno CS, Tenreiro S, Ventura S and Outeiro TF (2016) The effects of the novel A53E alpha-synuclein mutation on its oligomerization and aggregation. Acta Neuropathol. Commun 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro DF, Pavlou MAS and Outeiro TF (2017) Cellular models as tools for the study of the role of alpha-synuclein in Parkinson’s disease. Exp. Neurol 298, 162–171. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Shin SY, Choi C, Lee YH and Lee SJ (2002) Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J. Biol. Chem 277, 5411–5417. [DOI] [PubMed] [Google Scholar]
- Li L, Nadanaciva S, Berger Z, et al. (2013) Human A53T alpha-synuclein causes reversible deficits in mitochondrial function and dynamics in primary mouse cortical neurons. PLoS ONE 8, e85815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Clark-Dixon C, Wang S, Flower TR, Williams-Hart T, Zweig R, Robinson LC, Tatchell K and Witt SN (2008) Novel suppressors of alpha-synuclein toxicity identified using yeast. Hum. Mol. Genet 17, 3784–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindersson E, Lundvig D, Petersen C, et al. (2005) p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synuclein opathies. J. Biol. Chem 280, 5703–5715. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N and Aebischer P (2002) alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc. Natl Acad. Sci. USA 99, 10813–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK and Brundin P (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem 277, 38884–38894. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Falsig J, van Beek J, Payne S, Dringen R, Brundin P and Leist M (2005) Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J. Neurosci 25, 6329–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtmann MHR, Angelova PR, Horrocks MH, et al. (2018) alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun 9, 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, et al. (2016) Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353. 10.1126/science.aah3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM and Hasegawa M (2013) Prion-like spreading of pathological alpha-synuclein in brain. Brain 136, 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Hasegawa T, Takeda A, Kikuchi A, Furukawa K, Kato Y and Itoyama Y (2004) Histochemical features of stress-induced aggregates in alpha-synuclein overexpressing cells. Brain Res. 1004, 83–90. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L and Krainc D (2016) alpha-synuclein -induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl Acad. Sci. USA 113, 1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbefo MK, Fares MB, Paleologou K, et al. (2015) Parkinson disease mutant E46K enhances alpha-synuclein phosphorylation in mammalian cell lines, in yeast, and in vivo. J. Biol. Chem 290, 9412–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H and Hyman BT (2001) Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104, 901–912. [DOI] [PubMed] [Google Scholar]
- Menezes R, Tenreiro S, Macedo D, Santos CN and Outeiro TF (2015) From the baker to the bedside: yeast models of Parkinson’s disease. Microb. Cell 2, 262–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaud S, Malany S, Mehta A, Vasile S, Smith LH and McLeanP J (2015) Targeting alpha-synuclein oligomers by protein-fragment complementation for drug discovery in synucleinopathies. Expert Opin. Ther. Targets 19, 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasstrom T, Goncalves S, Sahlin C, Nordstrom E, Screpanti Sundquist V, Lannfelt L, Bergstrom J, Outeiro TF and Ingelsson M (2011) Antibodies against alpha-synuclein reduce oligomerization in living cells. PLoS ONE 6, e27230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum RL (2017) The identification of alpha-synuclein as the first Parkinson disease gene. J. Parkinsons Dis 7, S43–S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell C, Murphy DD, Petrucelli L, Singleton AB, Hussey J, Farrer M, Hardy J, Dickson DW and Cookson MR (2001) Transfected synphilin-1 forms cytoplasmic inclusions in HEK293 cells. Brain Res. Mol. Brain Res 97, 94–102. [DOI] [PubMed] [Google Scholar]
- Oliveira LM, Falomir-Lockhart LJ, Botelho MG, et al. (2015) Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 6, e1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo F, Krenz A, Heermann S, Schulz JB and Falkenburger BH (2008) Accumulation and clearance of alpha-synuclein aggregates demonstrated by time-lapse imaging. J. Neurochem 106, 529–540. [DOI] [PubMed] [Google Scholar]
- Outeiro TF and Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT and McLean PJ (2008) Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE 3, e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva I, Pinho R, Pavlou MA, et al. (2017) Sodium butyrate rescues dopaminergic cells from alpha-synuclein -induced transcriptional deregulation and DNA damage. Hum. Mol. Genet 26, 2231–2246. [DOI] [PubMed] [Google Scholar]
- Piper DA, Sastre D and Schule B (2018) Advancing stem cell models of alpha-synuclein gene regulation in neurodegenerative disease. Front. Neurosci 12, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Woerman AL, Mordes DA, et al. (2015) Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl Acad. Sci. USA 112, E5308–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A and Dehay B (2014) Alpha-synuclein spreading in Parkinson’s disease. Front. Neuroanat 8, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I and Michnick SW (1999) Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc. Natl Acad. Sci. USA 96, 5394–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RF, Wade-Martins R and Alegre-Abarrategui J (2015) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 138, 1642–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio-Marques B, Felgueiras C, Silva A, et al. (2012) SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509. [DOI] [PubMed] [Google Scholar]
- Scholz D, Poltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S and Leist M (2011) Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem 119, 957–971. [DOI] [PubMed] [Google Scholar]
- Schule B, Pera RA and Langston JW (2009) Can cellular models revolutionize drug discovery in Parkinson’s disease? Biochim. Biophys. Acta 1792, 1043–1051. [DOI] [PubMed] [Google Scholar]
- Selkoe D, Dettmer U, Luth E, Kim N, Newman A and Bartels T (2014) Defining the native state of alpha-synuclein. Neurodegener Dis. 13, 114–117. [DOI] [PubMed] [Google Scholar]
- Shahpasandzadeh H, Popova B, Kleinknecht A, Fraser PE, OuteiroT F and Braus GH (2014) Interplay between sumoylation and phosphorylation for protection against alpha-synuclein inclusions. J. Biol. Chem 289, 31224–31240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava AN, Redeker V, Fritz N, et al. (2015) alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 34, 2408–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, et al. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R and Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Stefanis L (2012) alpha-synuclein in Parkinson’s disease. Cold SpringHarb Perspect Med 2, a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D and Greene LA (2001) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci 21, 9549–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stykel MG, Humphries K, Kirby MP, Czaniecki C, Wang T, Ryan T, Bamm V and Ryan SD (2018) Nitration of microtubules blocks axonal mitochondrial transport in a human pluripotent stem cell model of Parkinson’s disease. FASEB J. 32, 5350–5364. [DOI] [PubMed] [Google Scholar]
- Su LJ, Auluck PK, Outeiro TF, et al. (2010) Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis. Model. Mech 3, 194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM and Schapira AH (2000) Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet 9, 2683–2689. [DOI] [PubMed] [Google Scholar]
- Tagliafierro L, Zamora ME and Chiba-Falek O (2019) Multiplication of the SNCA locus exacerbates neuronal nuclear aging. Hum. Mol. Genet 28, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T and Mouradian MM (2004) Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem 279, 4625–4631. [DOI] [PubMed] [Google Scholar]
- Tardiff DF, Khurana V, Chung CY and Lindquist S (2014) From yeast to patient neurons and back again: powerful new discovery platform. Mov. Disord 29, 1231–1240. [DOI] [PubMed] [Google Scholar]
- Tenreiro S and Outeiro TF (2010) Simple is good: yeast models of neurodegeneration. FEMS Yeast Res. 10, 970–979. [DOI] [PubMed] [Google Scholar]
- Tenreiro S, Eckermann K and Outeiro TF (2014) Protein phosphorylation in neurodegeneration: friend or foe? Front. Mol. Neurosci 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenreiro S, Franssens V, Winderickx J and Outeiro TF (2017) Yeast models of Parkinson’s disease-associated molecular pathologies. Curr. Opin. Genet. Dev 44, 74–83. [DOI] [PubMed] [Google Scholar]
- Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT and McLean PJ (2008) CHIP targets toxic alpha-Synuclein oligomers for degradation. J. Biol. Chem 283, 17962–17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theillet FX, Binolfi A, Bekei B, et al. (2016) Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530, 45–50. [DOI] [PubMed] [Google Scholar]
- Tonges L, Szego EM, Hause P, et al. (2014) Alpha-synuclein mutations impair axonal regeneration in models of Parkinson’s disease. Front. Aging Neurosci 6, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E and Stefanis L (2009) Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J. Neurochem 109, 1348–1362. [DOI] [PubMed] [Google Scholar]
- Vera E, Bosco N and Studer L (2016) Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep. 17, 1184–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente Miranda H, Szego EM, Oliveira LMA, et al. (2017) Glycation potentiates alpha-synuclein -associated neurodegeneration in synucleinopathies. Brain 140, 1399–1419. [DOI] [PubMed] [Google Scholar]
- Villar-Pique A, Lopes da Fonseca T and Outeiro TF (2016) Structure, function and toxicity of alpha-synuclein : the Bermuda triangle in synucleinopathies. J. Neurochem 139(Suppl 1), 240–255. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC and Lee VM (2014) Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc 9, 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerink RH and Ewing AG (2008) The PC12 cell as model for neurosecretion. Acta Physiol (Oxf) 192, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL and Muchowski PJ (2003) Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 302, 1769–1772. [DOI] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, et al. (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xicoy H, Wieringa B and Martens GJ (2017) The SH-SY5Y cell line in Parkinson’s disease research: a systematic review. Mol. Neurodegener 12, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Emadi S, Williams S, Liu Q, Schulz P, He P, Alam NB, Wu J and Sierks MR (2015) Toxic oligomeric alpha-synuclein variants present in human Parkinson’s disease brains are differentially generated in mammalian cell models. Biomolecules 5, 1634–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabrocki P, Pellens K, Vanhelmont T, Vandebroek T, Griffioen G, Wera S, Van Leuven F and Winderickx J (2005) Characterization of alpha-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J. 272, 1386–1400. [DOI] [PubMed] [Google Scholar]
- Zabrocki P, Bastiaens I, Delay C, Bammens T, Ghillebert R, Pellens K, De Virgilio C, Van Leuven F and Winderickx J (2008) Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780. [DOI] [PubMed] [Google Scholar]