

Abstract
A nickel-catalyzed asymmetric diarylation reaction of vinylarenes enables the preparation of chiral α,α,β-triarylated ethane scaffolds, which exist in a number of biologically active molecules. The use of reducing conditions with aryl bromides as coupling partners obviates the need for stoichiometric organometallic reagents and tolerates a broad range of functional groups. The application of an N-oxyl radical as a ligand to a nickel catalyst represents a novel approach to facilitate nickel-catalyzed cross-coupling reactions.
Keywords: alkenes, aryl bromides, asymmetric catalysis, diarylation, nickel
Alkenes are versatile functional groups that enjoy some of the most successful asymmetric catalytic transformations, such as hydrogenation[1] and dihydroxylation.[2] Hydrocarbo-functionalization[3] and 1,2-dicarbofunctionalization reactions of alkenes[4,5] have emerged as compelling approaches to rapidly increase molecular complexity by forming vicinal disubstitution patterns and tertiary carbon centers. Asymmetric alkene 1,2-dicarbofunctionalization would enable the efficient construction of new C–C bonds while simultaneously introducing stereocenters,[6] but this strategy has been restricted to a handful of precedents, with the scope limited to intramolecular variants[7] and diene substrates.[8] The groups of Liu and others have reported intermolecular asymmetric trifluoromethylation and cyanation reactions of vinylarenes,[9] but an intermolecular asymmetric diarylation has not been reported previously.
The α,α,β-triarylated ethane motif is present in a number of important pharmaceutical compounds and biologically active molecules.[10] Examples include carboxylic acid 2, a molecule tested as a bone-selective estrogen receptor ligand,[11] indole derivative 3, an inhibitor of human α-thrombin,[12] and 4, which is an inhibitor of phosphodiesterase 4 (PDE 4A; Scheme 1).[13] While many biological studies have been performed with racemic samples, access to enantioenriched compounds would facilitate the assessment of their efficacy. These molecules could be synthesized by asymmetric hydrogenation[14] or stereospecific[15]/stereoconvergent[16] cross-coupling, but these methods would require preparation of the substrates in multiple steps. We herein report an asymmetric diarylation of vinylarenes as an alternative approach to access chiral α,α,β-triarylated ethane structures from readily available vinylarenes. This method could be used to rapidly build a library of triarylated molecules for drug discovery. The use of an aryl bromide as the coupling partner under reducing conditions obviates the need for pregenerating air-sensitive organometallic coupling reagents,[17] and thus the process features a broad scope with good functional group compatibility.
Scheme 1.
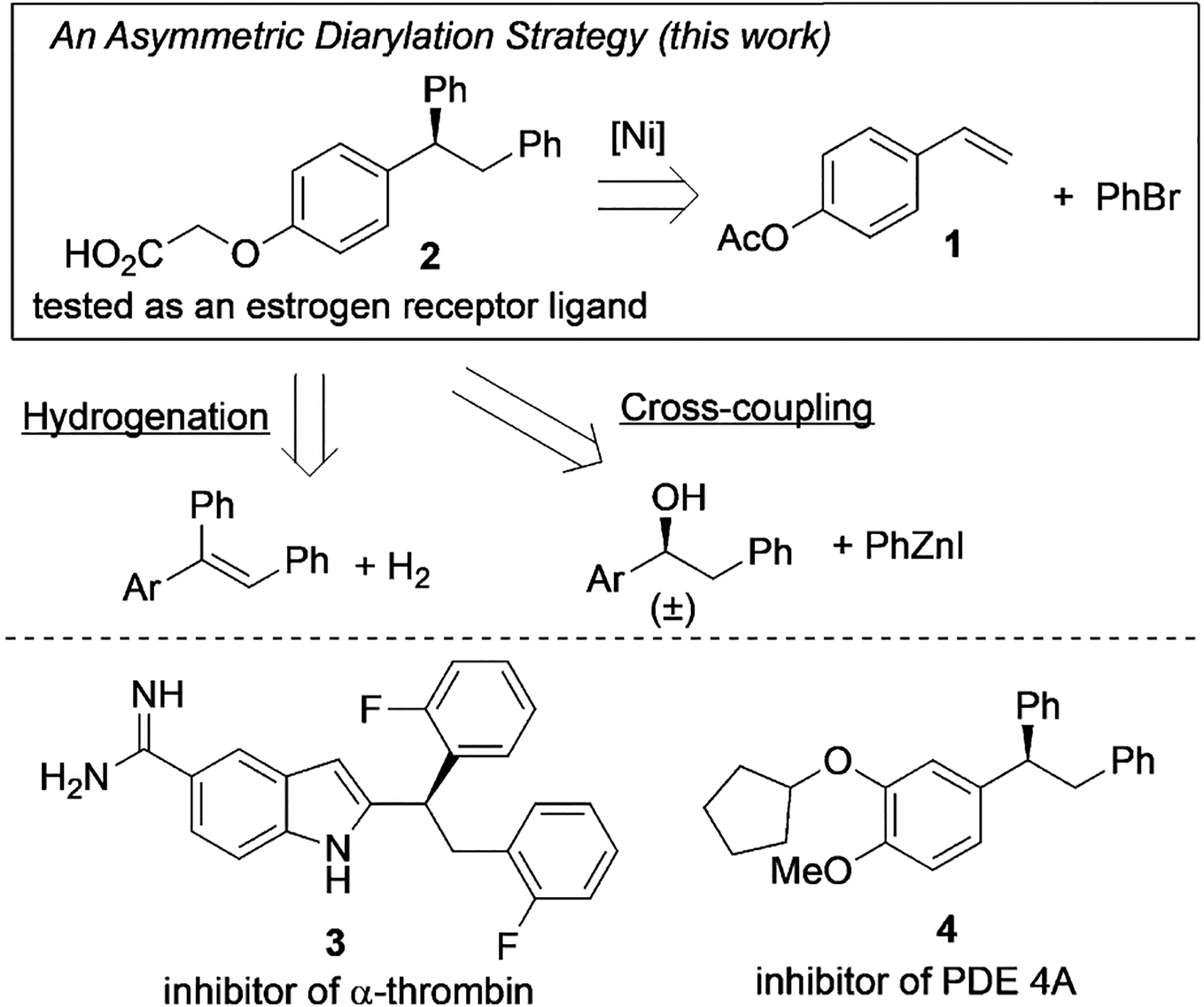
Strategies for preparing chiral α,α,β-triarylated ethane scaffolds and relevant target molecules.
The development of this asymmetric diarylation capitalized on the hypothesis that Ni catalysts can enable new modes of stereocontrol for alkene functionalization by radical addition (Scheme 2).[18] The first irreversible enantiodifferentiating step would determine the enantioselectivity.[19] In traditional asymmetric alkene functionalization reactions, such as the Pd-catalyzed asymmetric Heck reaction, the enantiodetermining step is either alkene coordination (step i) or migratory insertion (step ii).[20] Ni catalysts can initiate radical formation from alkyl[21] or aryl halides,[22] and the resulting organic radicals could add to alkenes.[23] Through this pathway, carbofunctionalization would be expected to have different enantiodetermining steps, such as radical capture by Ni (step v) or reductive elimination (step vi).[24]
Scheme 2.
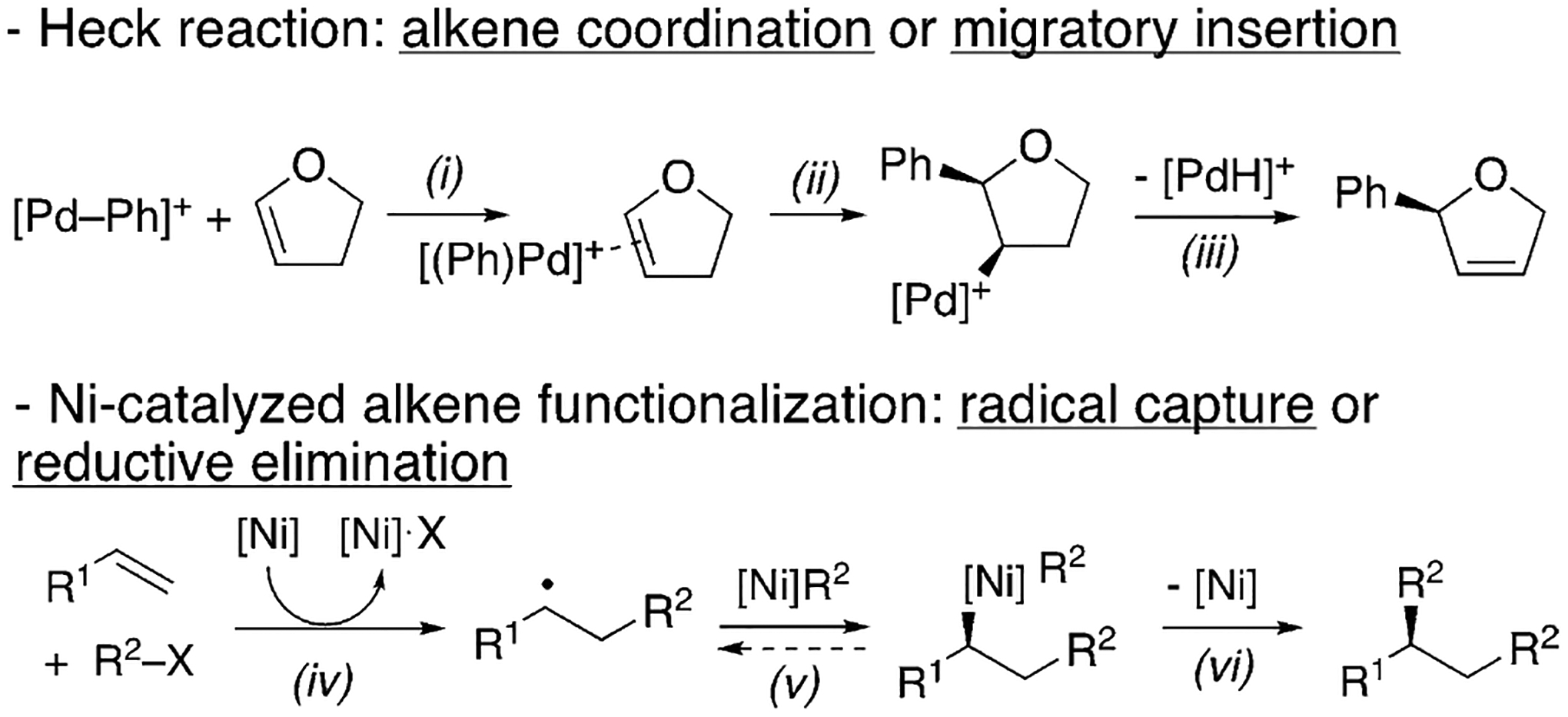
Enantiodetermining steps for alkene carbofunctionalization.
For our catalyst development, we employed para-acetoxystyrene (1) as the model substrate and PhBr as the electrophilic coupling partner (Table 1). Initial optimization showed that the use of 10 mol % Ni(DME)Br2 as the catalyst and Zn powder as the reductant resulted in the conversion of 1 into the desired diphenylation product, which gave 5 upon hydrolysis, an appealing precursor to 2. Replacing Zn with Mg and TDAE (tetrakis(dimethylamino)ethylene) both gave product 5 with modest ee (see the Supporting Information, Table S1). When PhZnCl was used in such reactions instead of PhBr, the reactions failed to afford any alkene arylation product (Scheme S1). These results rule out organozinc reagents as intermediates, suggesting that zinc merely serves as a reductant.
Table 1:
Catalyst optimization: Effects of ligands and solvents.[a]
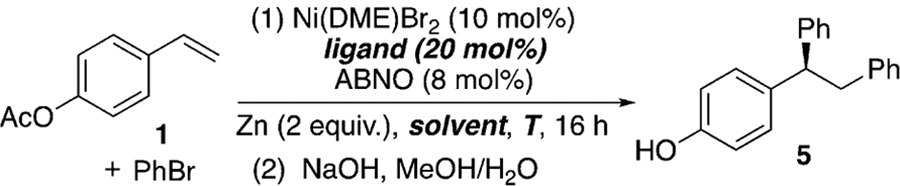
| |||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T [°C] | Yield [%] | ee [%] |
| 1 | iPr-biOx | DMPU | 25 | 52 | 83 |
| 2 | iBu-biOx | DMPU | 25 | 69 | 89 |
| 3 | Cy-biOx | DMPU | 25 | 41 | 87 |
| 4 | tBu-biOx | DMPU | 25 | 28 | 0 |
| 5 | Ph-biOx | DMPU | 25 | 25 | 0 |
| 6 | 4-hept-biOx | DMPU | 25 | 64 | 73 |
| 7 | indane-biOx | DMPU | 25 | 52 | 64 |
| 8 | Ph-box | DMPU | 25 | 0 | – |
| 9 | iPr-pyrox | DMPU | 25 | 30 | 28 |
| 10 | iPr-pybox | DMPU | 25 | 5 | – |
| 11 | iBu-biOx | DMPU | 10 | 38 | 90 |
| 12 | iBu-biOx | DMPU/THF (3:1) | 10 | 74 | 90 |
| 13 | iBu-biOx | DMPU/THF (1:1) | 10 | 90 | 91 |
| 14 | iBu-biOx | DMPU/THF (1:3) | 10 | 28 | 91 |
Reaction conditions: 1 (0.2 mmol, 1 m), PhBr (4 m). Yields determined by 1H NMR spectroscopy using mesitylene as an internal standard; ee values determined by HPLC analysis on a chiral stationary phase.
![[a]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/29a0/8864584/b9921331e0f9/nihms-1779912-f0004.jpg)
Bi-oxazoline (biOx) ligands have recently been extensively utilized in nickel-catalyzed asymmetric coupling reactions,[16,25] and were shown to be effective with ABNO as an additive, whose role will be discussed below in Table 2 (Table 1, entries 1–7). The substituents on the biOx ligand surprisingly had only a minor effect on the ee, except for tBu-biOx and Ph-biOx, which resulted in significantly lower yields and no enantioselectivity (entries 4 and 5). This observation could be attributed to the bulky substituents preventing coordination, as is evident from the different reaction color. Other common N-ligands frequently used in asymmetric Ni catalysis, such as bisoxazoline (box), pyridine-bisoxazoline (pybox), and pyridine-oxazoline (pyrox) ligands, resulted in low conversion (entries 8–10). Lowering the temperature led to slightly higher ee, but also a lower yield (entry 11). The low yield could result from inefficient mixing, owing to the high viscosity of N,N’-dimethylpropyleneurea (DMPU) at low temperatures. A mixture of DMPU and THF in a 1:1 ratio ameliorated this viscosity issue, improving the yield to 90% with an ee of 91% (entry 13).
Table 2:
Catalyst optimization: Effect of an N-oxyl additive.[a]
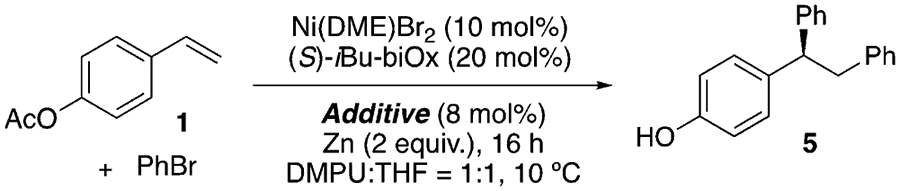
| |||||
|---|---|---|---|---|---|
| Additive | Yield [%] | ee [%] | Additive | Yield [%] | ee [%] |
| – | 78 | 26 | tBu2NO | 63 | 60 |
| TEMPO | 60 | 21 | 2-Me-AZADO | 71 | 57 |
| 4-oxo-TEMPO | 50 | 29 | AZADO | 76 | 89 |
| 4-OAc-TEMPO | 64 | 33 | ABNO | 90 | 91 |
| 4-OH-TEMPO | 82 | 46 | NO | 39 | 54 |
Reaction conditions: 1 (0.2 mmol, 1 m), PhBr (4 m). Crude reaction mixtures were treated with aqueous NaOH to saponify the acetate. Yields determined by 1H NMR analysis using mesitylene as an internal standard; ee values determined by HPLC analysis on a chiral stationary phase.
During catalyst optimization, we noticed that the N-oxyl radical additive exerted a significant effect on the enantioselectivity (Table 2). Examining a number of N-oxyl radical derivatives, we used multivariate analysis[26] to deconvolute the parameters that affect the ee. Several parameters were computed to describe the electronic and steric effects of various N-oxyl radicals, including the SOMO energy (ESOMO), the redox potential, the IR stretching frequency of the corresponding R2N+=O (υN+=O), the polarizability, and the buried volume (% Vbur; Table S2). In particular, % Vbur was obtained by submitting the structures of (bipyridine)Ni(N-oxyl) after geometry optimization to SambVca 2.0.[27] When we applied linear regression modeling to relate the enantioselectivity (expressed as ΔΔG‡) and the computed parameters, % Vbur exhibited a dominant effect, providing a linear correlation with an R2 value of 0.8 when plotted directly against ΔΔG‡ (Figure S3). The most sterically unhindered N-oxyl additives, ABNO and AZADO, gave the greatest ee. A stronger correlation (R2 = 0.96) was obtained when ESOMO and υN+ = O were included along with % Vbur as descriptors (Figure S4). Examining the stoichiometry of Ni/ABNO revealed that it is critical to apply a Ni/ABNO ratio of 1:1 to 1:0.8 to obtain high yield and ee, as a slight excess of ABNO led to complete inhibition of the diarylation reaction. While characterizing the precise nature of the observed N-oxyl effect requires further investigation, the required 1:1 ratio of Ni/ABNO is consistent with it serving as a ligand.[28]
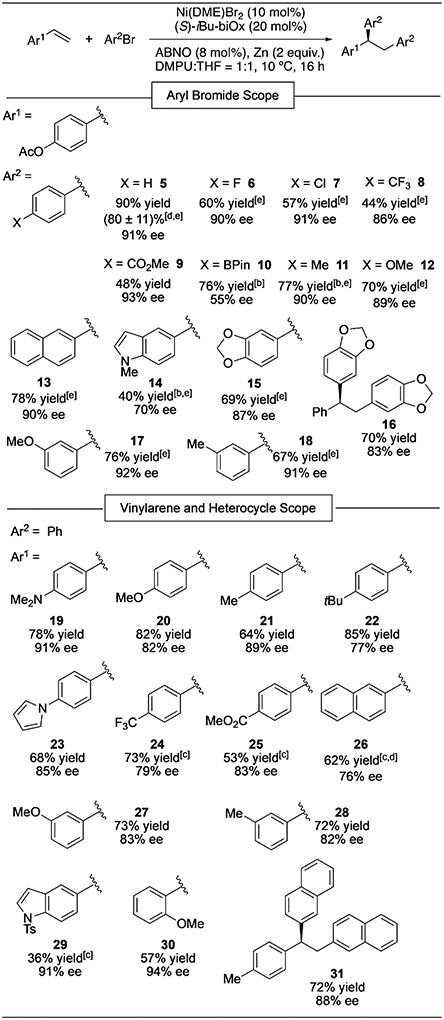
With optimized reaction conditions established, we explored the scope of the asymmetric diarylation (Table 3). The enantioselectivities generally appeared to be insensitive to the electronic nature of the electrophile. The low ee of 10 may be attributed to an interaction of Ni with the boronic ester. The reaction conditions are compatible with a number of functional groups, including fluoride, chloride, ester, amine, and indole moieties. When bromobenzene was used as the electrophile and the vinylarene component was varied, a wide variety of electron-rich and electron-deficient vinylarenes underwent diphenylation to give α,α,β-triarylethane products (19–30). Changing the electronic nature of the vinylarene component by using various para-substituted styrenes seemed to have a modest effect on enantioselectivity. However, the observed enantiomeric ratios of the products do not show a linear free energy relationship with the Hammett parameters for the corresponding para substituents (R2 = 0.17, Figure S6). Simple disubstituted olefins, such as α- and β-methylstyrene, gave no diarylation product under the optimized conditions. Performing the diphenylation of 1 on a 1.0 mmol scale furnished 5 in 71% yield upon isolation and 92% ee. When the reaction was conducted on the bench using standard Schlenk techniques, 5 was formed in 66% yield and 90% ee. The catalyst, once activated, decomposes in air, and thus the reaction requires an inert atmosphere.
Table 3:
Substrate scope of the asymmetric diarylation of vinylarenes.[a]
|
Reaction conditions: Vinylarene (0.2 mmol, 1 m), ArBr (4 m). Yields determined by 1H NMR analysis using mesitylene as an internal standard; ee values determined by HPLC analysis on a chiral stationary phase. The absolute stereochemistry was assigned for 29 based on X-ray crystallography, while those of all other products were assigned by analogy.
Vinylarene (0.5 m).
With 20 mol % Ni(DME)Br2, 40 mol % iBu-biOx, and 16 mol % ABNO.
Yield of isolated product. The error bar is based on four duplicate experiments.
The product was isolated as the corresponding phenol after deacylation with aqueous NaOH.
Subsequently, we carried out preliminary studies to probe factors that control the enantioselectivity. We first sought to examine the ligand/Ni ratio. The ee of the product exhibits a linear correlation with the ee of the ligand (Figure S7).[29] This observation suggests that the ligand/Ni ratio is 1:1 in the enantiodetermining step, although excess ligand is required to stabilize the Ni catalyst in solution.
It is generally accepted that Ni-catalyzed cross-coupling reactions proceed through radical intermediates.[30,31] Several observations provide evidence for the formation of radical intermediates at the benzylic position: 1) inhibition of the reaction by excess N-oxyl, which could be attributed to its interaction with organic radicals; 2) formation of benzylic dimers, such as 32, with several substrates [Eq. (1)];[32] 3) selective formation of trans-diphenylation product 33 in 70% ee with indene [Eq. (2)]. A difunctionalization proceeding through olefin migratory insertion has been reported to form cis products.[33] The trans diastereoselectivity observed here could arise from a cis-migratory insertion, followed by reversible radical ejection that scrambles the benzylic stereocenter as the coordination favors the less hindered face (step vi, Scheme 3). This proposal serves as experimental support for Kozlowski and Molander’s computational study.[24] The observation of 70% ee for 33 could rule out free phenyl radical addition to indene. In Scheme 3, we present a plausible mechanism. Detailed mechanistic studies are underway to fully elucidate the role of the N-oxyl radical, the sequence of reagent activation, and the identity of the enantiodetermining step.
Scheme 3.
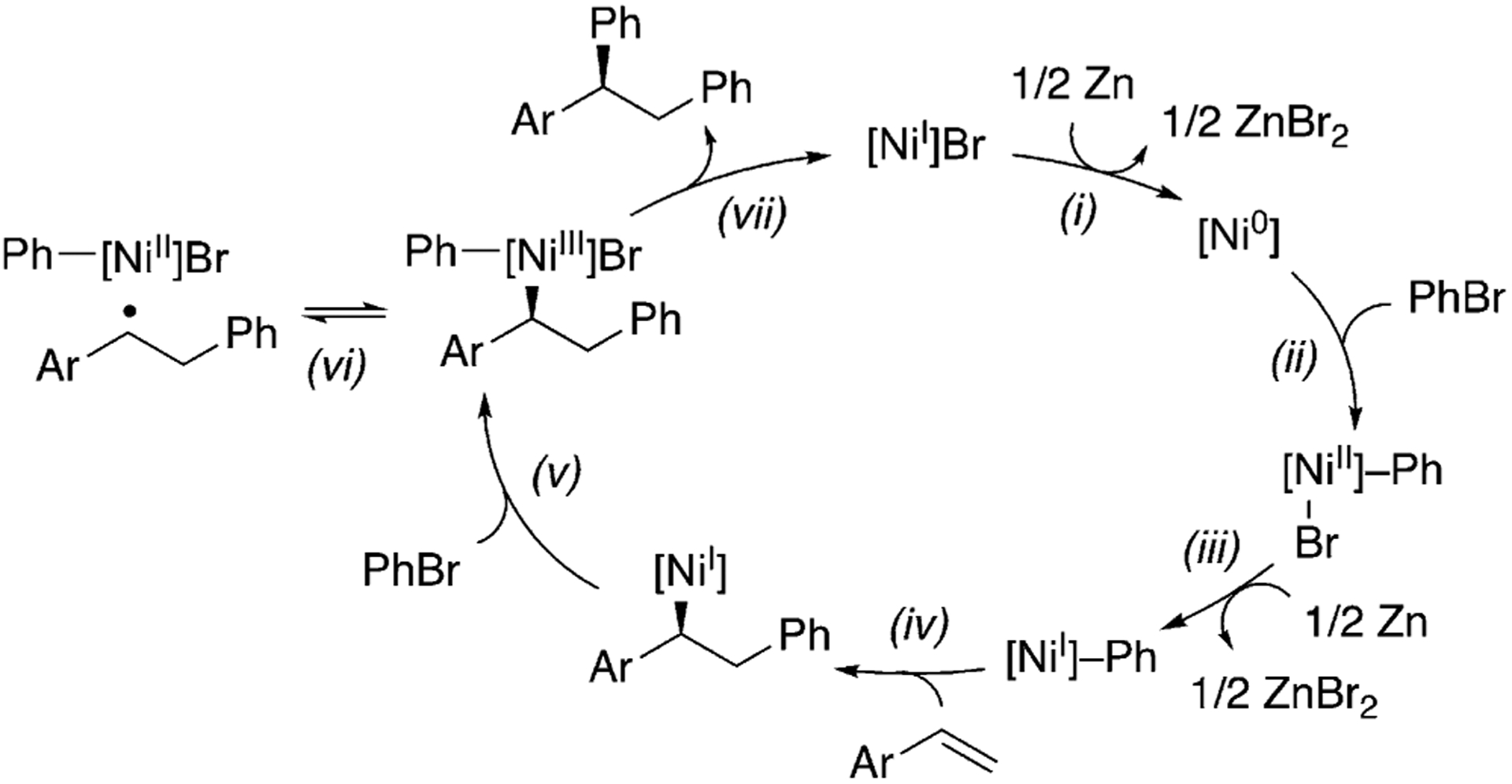
Proposed mechanism.
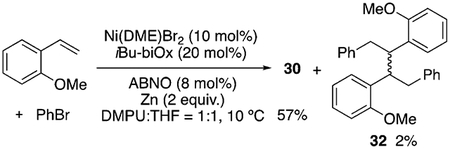 |
(1) |
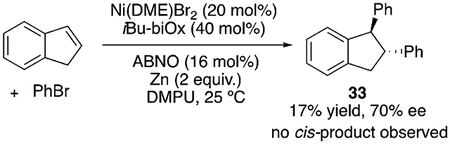 |
(2) |
In summary, we have developed a nickel-catalyzed asymmetric diarylation reaction of vinylarenes. The reaction represents a compelling method for preparing chiral α,α,β-triarylated ethane scaffolds. The use of reducing conditions with aryl bromides as the coupling partner obviates the need for stoichiometric organometallic reagents and tolerates a broad range of functional groups. During catalyst development, we discovered that the use of an auxiliary N-oxyl ligand, in conjunction with a chiral Ni catalyst, enabled the formation of dicarbofunctionalized products with high enantioselectivity. We anticipate that this insight could inform catalyst development for other nickel-catalyzed cross-coupling reactions.
Supplementary Material
Acknowledgements
This work was supported by the National Science Foundation under Award Number DMR-1420073. The NMR cryoprobe is supported by NIH (S10 OD016343). T.D. is grateful to the Sloan Foundation (FG-2018-10354). D.A. thanks Prof. Jim Canary for sharing chiral HPLC columns, Dr. Giulio Volpin and Peter Rühmann (Trauner group) for help with purifying 14 and 26 by preparatory HPLC, and Lotus Separations for determining the ee of 24.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201900228.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].(a) Knowles WS, Angew. Chem. Int. Ed 2002, 41, 1998 – 2007; [Google Scholar]; Angew. Chem 2002, 114, 2096 – 2107; [Google Scholar]; (b) Noyori R, Angew. Chem. Int. Ed 2002, 41, 2008 – 2022; [PubMed] [Google Scholar]; Angew. Chem 2002, 114, 2108 – 2123. [Google Scholar]
- [2].Kolb HC, VanNieuwenhze MS, Sharpless KB, Chem. Rev 1994, 94, 2483 – 2547. [Google Scholar]
- [3].(a) Crossley SWM, Obradors C, Martinez RM, Shenvi RA, Chem. Rev 2016, 116, 8912 – 9000; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuang Y, Anthony D, Katigbak J, Marrucci F, Humagain S, Diao T, Chem 2017, 3, 268 – 280. [Google Scholar]
- [4].(a) Dhungana RK, KC S, Basnet P, Giri R, Chem. Rec 2018, 18, 1314 – 1340; [DOI] [PubMed] [Google Scholar]; (b) Giri R, KC S, J. Org. Chem 2018, 83, 3013 – 3022. [DOI] [PubMed] [Google Scholar]
- [5].(a) Urkalan KB, Sigman MS, Angew. Chem. Int. Ed 2009, 48, 3146 – 3149; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 3192 – 3195; [Google Scholar]; (b) You W, Brown MK, J. Am. Chem. Soc 2014, 136, 14730 – 14733; [DOI] [PubMed] [Google Scholar]; (c) Liu Z, Zeng T, Yang KS, Engle KM, J. Am. Chem. Soc 2016, 138, 15122 – 15125; [DOI] [PubMed] [Google Scholar]; (d) Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS, Science 2016, 352, 801 – 805; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Thapa S, Basnet P, Giri R, J. Am. Chem. Soc 2017, 139, 5700 – 5703; [DOI] [PubMed] [Google Scholar]; (f) García-Dominguez A, Li Z, Nevado C, J. Am. Chem. Soc 2017, 139, 6835 – 6838; [DOI] [PubMed] [Google Scholar]; (g) Shrestha B, Basnet P, Dhungana RK, Kc S, Thapa S, Sears JM, Giri R, J. Am. Chem. Soc 2017, 139, 10653 – 10656; [DOI] [PubMed] [Google Scholar]; (h) Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM, J. Am. Chem. Soc 2017, 139, 10657 – 10660; [DOI] [PubMed] [Google Scholar]; (i) Gao P, Chen LA, Brown MK, J. Am. Chem. Soc 2018, 140, 10653 – 10657; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Basnet P, Dhungana RK, Thapa S, Shrestha B, Kc S, Sears JM, Giri R, J. Am. Chem. Soc 2018, 140, 7782 – 7786; [DOI] [PubMed] [Google Scholar]; (k) Kc S, Dhungana RK, Shrestha B, Thapa S, Khanal N, Basnet P, Lebrun RW, Giri R, J. Am. Chem. Soc 2018, 140, 9801 – 9805; [DOI] [PubMed] [Google Scholar]; (l) Basnet P, Kc S, Dhungana RK, Shrestha B, Boyle TJ, Giri R, J. Am. Chem. Soc 2018, 140, 15586 – 15590; [DOI] [PubMed] [Google Scholar]; (m) Kuang Y, Wang X, Anthony D, Diao T, Chem. Commun 2018, 54, 2558 – 2561. [DOI] [PubMed] [Google Scholar]
- [6].Cherney AH, Kadunce NT, Reisman SE, Chem. Rev 2015, 115, 9587 – 9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Yasui Y, Kamisaki H, Takemoto Y, Org. Lett 2008, 10, 3303 – 3306; [DOI] [PubMed] [Google Scholar]; (b) Cong H, Fu GC, J. Am. Chem. Soc 2014, 136, 3788 – 3791; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) You W, Brown MK, J. Am. Chem. Soc 2015, 137, 14578 – 14581; [DOI] [PubMed] [Google Scholar]; (d) Wang K, Ding Z, Zhou Z, Kong W, J. Am. Chem. Soc 2018, 140, 12364 – 12368. [DOI] [PubMed] [Google Scholar]
- [8].(a) Stokes BJ, Liao L, de Andrade AM, Wang Q, Sigman MS, Org. Lett 2014, 16, 4666 – 4669; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu X, Lin H-C, Li M-L, Li L-L, Han Z-Y, Gong L-Z, J. Am. Chem. Soc 2015, 137, 13476 – 13479; [DOI] [PubMed] [Google Scholar]; (c) Xiong Y, Zhang G, J. Am. Chem. Soc 2018, 140, 2735 – 2738. [DOI] [PubMed] [Google Scholar]
- [9].(a) Wang F, Chen P, Liu G, Acc. Chem. Res 2018, 51, 2036 – 2046; [DOI] [PubMed] [Google Scholar]; (b) Sha W, Deng L, Ni S, Mei H, Han J, Pan Y, ACS Catal 2018, 8, 7489 – 7494; [Google Scholar]; (c) Lin J-S, Li T-T, Liu J-R, Jiao G-Y, Gu Q-S, Cheng J-T, Guo Y-L, Hong X, Liu X-Y, J. Am. Chem. Soc 2019, 141, 1074 – 1083. [DOI] [PubMed] [Google Scholar]
- [10].Hills CJ, Winter SA, Balfour JA, Drugs 1998, 55, 813 – 820. [DOI] [PubMed] [Google Scholar]
- [11].(a) Ruenitz PC, Bourne CS, Sullivan KJ, Moore SA, J. Med. Chem 1996, 39, 4853 – 4859; [DOI] [PubMed] [Google Scholar]; (b) Rubin VN, Ruenitz PC, Boudinot FD, Boyd JL, Bioorg. Med. Chem 2001, 9, 1579 – 1587. [DOI] [PubMed] [Google Scholar]
- [12].Iwanowicz EJ, Lau WF, Lin J, Roberts DGM, Seiler SM, Bioorg. Med. Chem. Lett 1996, 6, 1339 – 1344. [Google Scholar]
- [13].Alexander RP, Warrellow GJ, Eaton MAW, Boyd EC, Head JC, Porter JR, Brown JA, Reuberson JT, Hutchinson B, Turner P, Boyce B, Barnes D, Mason B, Cannell A, Taylor RJ, Zomaya A, Millican A, Leonard J, Morphy R, Wales M, Perry M, Allen RA, Gozzard N, Hughes B, Higgs G, Bioorg. Med. Chem. Lett 2002, 12, 1451 – 1456. [DOI] [PubMed] [Google Scholar]
- [14].(a) Mazuela J, Norrby P-O, Andersson PG, Pàmies O, Diéguez M, J. Am. Chem. Soc 2011, 133, 13634 – 13645; [DOI] [PubMed] [Google Scholar]; (b) Tolstoy P, Engman M, Paptchikhine A, Bergquist J, Church TL, Leung AWM, Andersson PG, J. Am. Chem. Soc 2009, 131, 8855 – 8860. [DOI] [PubMed] [Google Scholar]
- [15].Yonova IM, Johnson AG, Osborne CA, Moore CE, Morrissette NS, Jarvo ER, Angew. Chem. Int. Ed 2014, 53, 2422 – 2427; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 2454 – 2459. [Google Scholar]
- [16].(a) Do H-Q, Chandrashekar ERR, Fu GC, J. Am. Chem. Soc 2013, 135, 16288 – 16291; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Poremba KE, Kadunce NT, Suzuki N, Cherney AH, Reisman SE, J. Am. Chem. Soc 2017, 139, 5684 – 5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].(a) Weix DJ, Acc. Chem. Res 2015, 48, 1767 – 1775; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gu J, Wang X, Xue W, Gong H, Org. Chem. Front 2015, 2, 1411 – 1421. [Google Scholar]
- [18].(a) Lucas EL, Jarvo ER, Nat. Rev. Chem 2017, 1, 0065; [Google Scholar]; (b) Choi J, Fu GC, Science 2017, 356, eaaf7230; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tasker SZ, Standley EA, Jamison TF, Nature 2014, 509, 299 – 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Landis CR, Halpern J, J. Am. Chem. Soc 1987, 109, 1746 – 1754. [Google Scholar]
- [20].(a) Ashimori A, Bachand B, Calter MA, Govek SP, Overman LE, Poon DJ, J. Am. Chem. Soc 1998, 120, 6488 – 6499; [Google Scholar]; (b) Henriksen ST, Norrby P-O, Kaukoranta P, Andersson PG, J. Am. Chem. Soc 2008, 130, 10414 – 10421; [DOI] [PubMed] [Google Scholar]; (c) Xu L, Hilton MJ, Zhang X, Norrby P-O, Wu Y-D, Sigman MS, Wiest O, J. Am. Chem. Soc 2014, 136, 1960 – 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Laskowski CA, Bungum DJ, Baldwin SM, Del Ciello SA, Iluc VM, Hillhouse GL, J. Am. Chem. Soc 2013, 135, 18272 – 18275. [DOI] [PubMed] [Google Scholar]
- [22].(a) Tsou TT, Kochi JK, J. Am. Chem. Soc 1979, 101, 6319 – 6332; [Google Scholar]; (b) Tsou TT, Kochi JK, J. Am. Chem. Soc 1979, 101, 7547 – 7560. [Google Scholar]
- [23].Green SA, Matos JLM, Yagi A, Shenvi RA, J. Am. Chem. Soc 2016, 138, 12779 – 12782. [DOI] [PubMed] [Google Scholar]
- [24].Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC, J. Am. Chem. Soc 2015, 137, 4896 – 4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Woods BP, Orlandi M, Huang C-Y, Sigman MS, Doyle AG, J. Am. Chem. Soc 2017, 139, 5688 – 5691. [DOI] [PubMed] [Google Scholar]
- [26].Sigman MS, Harper KC, Bess EN, Milo A, Acc. Chem. Res 2016, 49, 1292 – 1301. [DOI] [PubMed] [Google Scholar]
- [27].Falivene L, Credendino R, Poater A, Petta A, Serra L, Oliva R, Scarano V, Cavallo L, Organometallics 2016, 35, 2286 – 2293. [Google Scholar]
- [28].Isrow D, Captain B, Inorg. Chem 2011, 50, 5864 – 5866. [DOI] [PubMed] [Google Scholar]
- [29].Guillaneux D, Zhao S-H, Samuel O, Rainford D, Kagan HB, J. Am. Chem. Soc 1994, 116, 9430 – 9439. [Google Scholar]
- [30].Hu XL, Chem. Sci 2011, 2, 1867 – 1886. [Google Scholar]
- [31].Diccianni J, Katigbak J, Hu C, Diao T J. Am. Chem. Soc 2019, 141, 1788 – 1796. [DOI] [PubMed] [Google Scholar]
- [32].Gibian MJ, Corley RC, Chem. Rev 1973, 73, 441 – 464. [Google Scholar]
- [33].Logan KM, Brown MK, Angew. Chem. Int. Ed 2017, 56, 851 – 855; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 869 – 873. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.