

Abstract
Intracardiac paragangliomas most commonly arise from the left atrium and are often infiltrative and densely adherent to surrounding structures. Given their rarity, only scattered reports exist in the literature and standardized perioperative and surgical management is not well established. We describe a case of a 60-year-old woman with a mildly functioning intracardiac paraganglioma in which division of the superior vena cava improved exposure and enabled a complex limited resection. Further, we provide an overview of the diagnostic workup, perioperative medical management, surgical approach, and surveillance strategy in patients with these challenging tumors.
Keywords: Intracardiac paraganglioma, Cardiac tumor, Mediastinal mass, SDHC mutation
Introduction
Catecholamine-secreting tumors, when found outside of the adrenal gland, are termed paragangliomas (PGLs). Of these, primary cardiac PGLs are extremely rare and more prevalent in women [1]. Functioning tumors are often discovered during the workup of persistent hypertension, palpitations, presyncope or syncope. Non-functioning tumors are typically discovered incidentally or present with compressive symptoms.
Optimal treatment is complete surgical resection. 10-year survival in patients who underwent complete surgical resection is reported to be as high as 84% versus 50% in patients who underwent only a partial resection [4]. There is no role for radiotherapy in the neoadjuvant or adjuvant settings.
Cardiac PGLs most commonly arise from the left atrium, and unlike pheochromocytomas in the adrenal gland, are often infiltrative and densely adherent to surrounding structures. The majority of published cases were resections via median sternotomy with cardiopulmonary bypass, although posterolateral thoracotomy incisions have also been used [5]. Complete resection requires the en bloc excision of involved structures, and the use of auto-transplantation or orthotopic heart transplantation has been described. We describe a case in which transection of the superior vena cava and left atriotomy provided excellent exposure enabling a complex limited reconstruction.
Case report
A 60-year-old woman with syncope was found to have a 4.7 × 3.4 cm posterior mediastinal mass on CT scan (Fig. 1a). Prior to presenting to our clinic, she underwent a right thoracoscopy converted to thoracotomy. The mass was found to be intrapericardial and was biopsied without complication, revealing atypical epithelioid cells with neuroendocrine differentiation suggestive of a paraganglioma. Imaging suggested the mass received blood supply via bronchial arteries. She did not have hypertension or other symptoms of catecholamine excess. However, further workup revealed plasma normetanephrines to be slightly elevated at 0.97 nmol/L (normal < 0.90) and the alpha-blocker terazosin (1 mg qhs) was initiated. Genetic testing revealed a pathogenic SDHC mutation (c.397C>T, p.Arg133*), and further imaging with Ga68-DOTATATE scan demonstrated a single avid lesion (SUV 13) with no evidence of distant disease (Fig. 1b) and cardiac magnetic resonance (MRI) demonstrated the mass arising from the left atrium with arterial hyperenhancement (Fig. 1c).
Fig. 1.
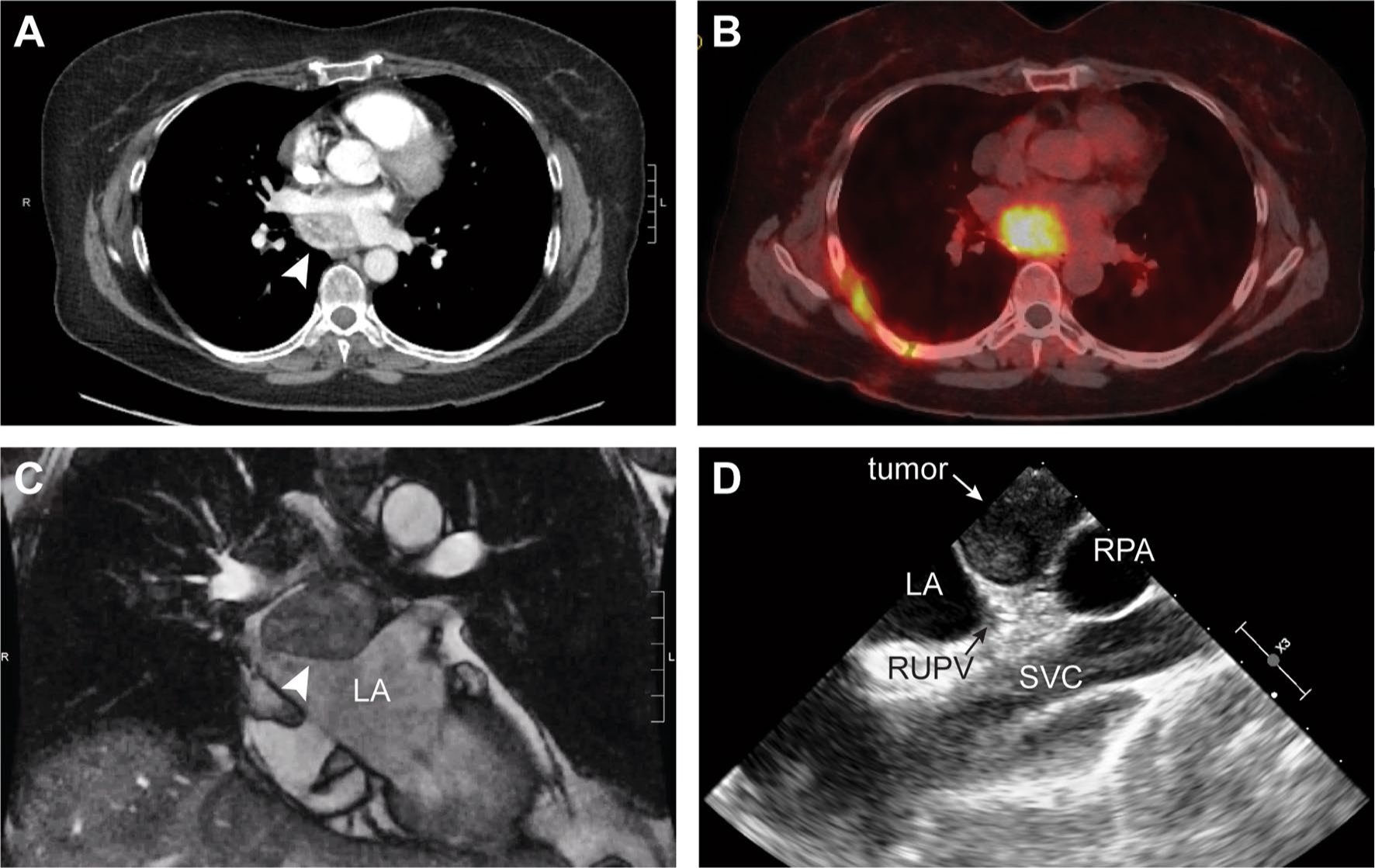
Pre-operative imaging diagnostics: a Computed tomography (CT) with IV contrast demonstrating a 4.7 × 3.4 cm enhancing mediastinal mass (arrow). b Positive Ga68-DOTATATE scan (SUV 13) demonstrating a single avid lesion with no distant disease, c Cardiac magnetic resonance imaging demonstrating mass (arrow) arising from the LA with arterial hyperenhancement and T2 hyperintensity, and d Transesophageal echo (TEE) demonstrating tumor with LA compression. LA, left atrium; RPA, right pulmonary artery; RUPV, right upper pulmonary vein; SVC, superior vena cava
Intraoperative transesophageal echocardiogram showed the tumor compressing the left atrium (Fig. 1d). She underwent median sternotomy, aortic and bicaval cannulation, and initiation of cardiopulmonary bypass (Supplementary Video 1). The superior vena cava (SVC) was transected (Fig. 2) allowing the heart to be rotated towards the left. A left atriotomy was performed and the tumor was resected en bloc with the posterior left atrial wall, close to the ostia of the right pulmonary veins. Complex reconstruction of the left atrium was performed with de-cellularized bovine pericardium. The SVC was repaired and the patient was weaned from bypass. Pathologic examination showed a completely excised 4.5 cm tumor (Fig. 3a) composed of epithelioid cells with abundant cytoplasm exhibiting nested growth in a richly vascular stroma (Fig. 3b). By immunohistochemistry, the tumor expressed the neuroendocrine markers chromogranin A (Fig. 3c), synaptophysin and INSM-1 (Fig. 3d), with peripheral sustentacular cells positive for S100 (Fig. 3e) and SOX-10. The patient had an unremarkable post-operative course, and adjuvant therapy is not indicated. Follow-up will include annual visits with blood pressure monitoring, MRI chest, plasma metanephrines and Chromogranin A, and her family members will be screened for the mutation.
Fig. 2.
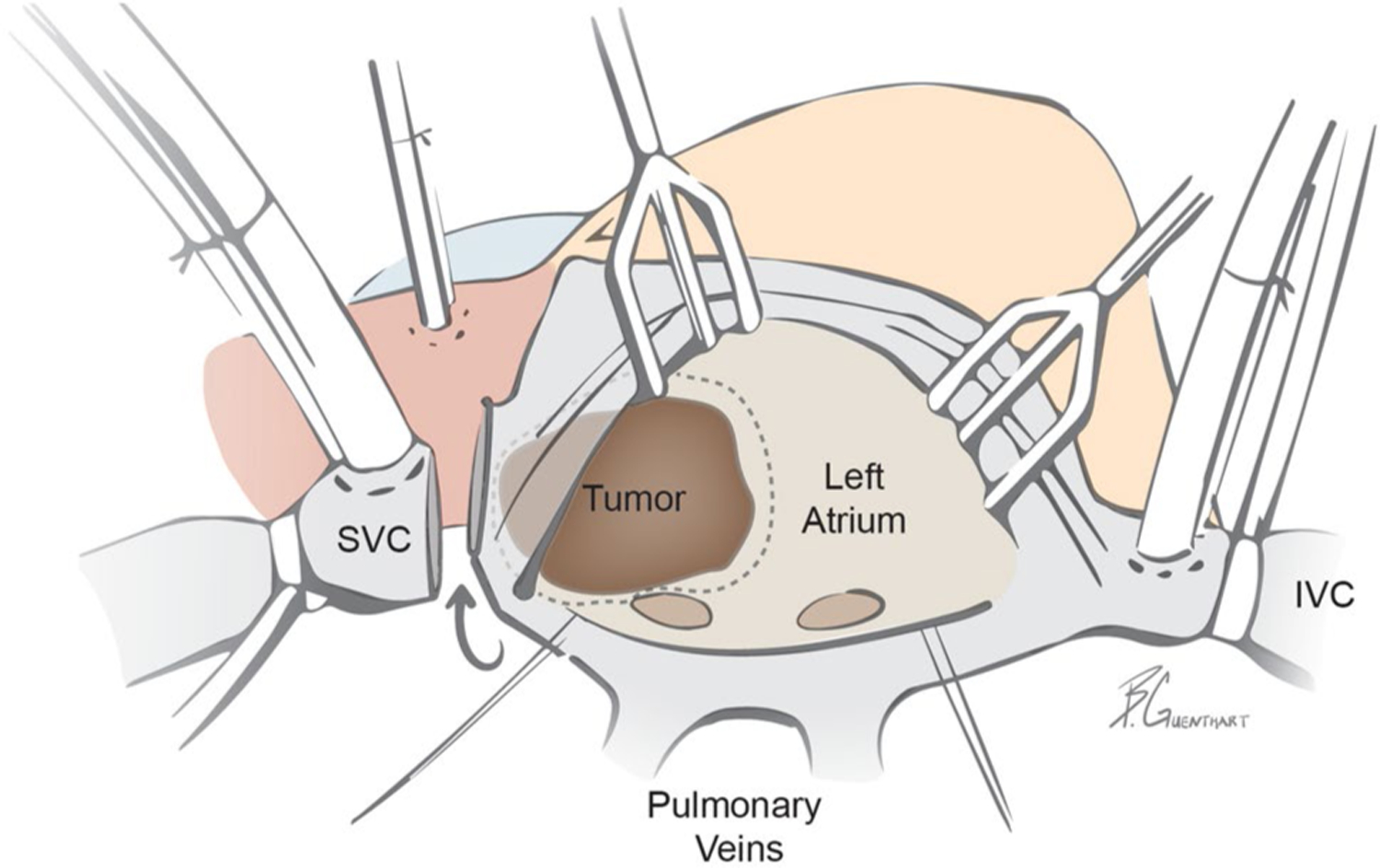
Schematic of surgical approach via median sternotomy utilizing aortic and bicaval cannulation for cardiopulmonary bypass: SVC transection allowing for heart rotation and exposure and en bloc excision of tumor and posterior left atrium (dotted line represents resection plane). Subsequently, the posterior left atrium was reconstructed with de-cellularized bovine pericardium sewn into the pulmonary venous ostia. Additional surgical detail highlighted in Supplementary Movie 1. IVC, inferior vena cava, SVC, superior vena cava
Fig. 3.
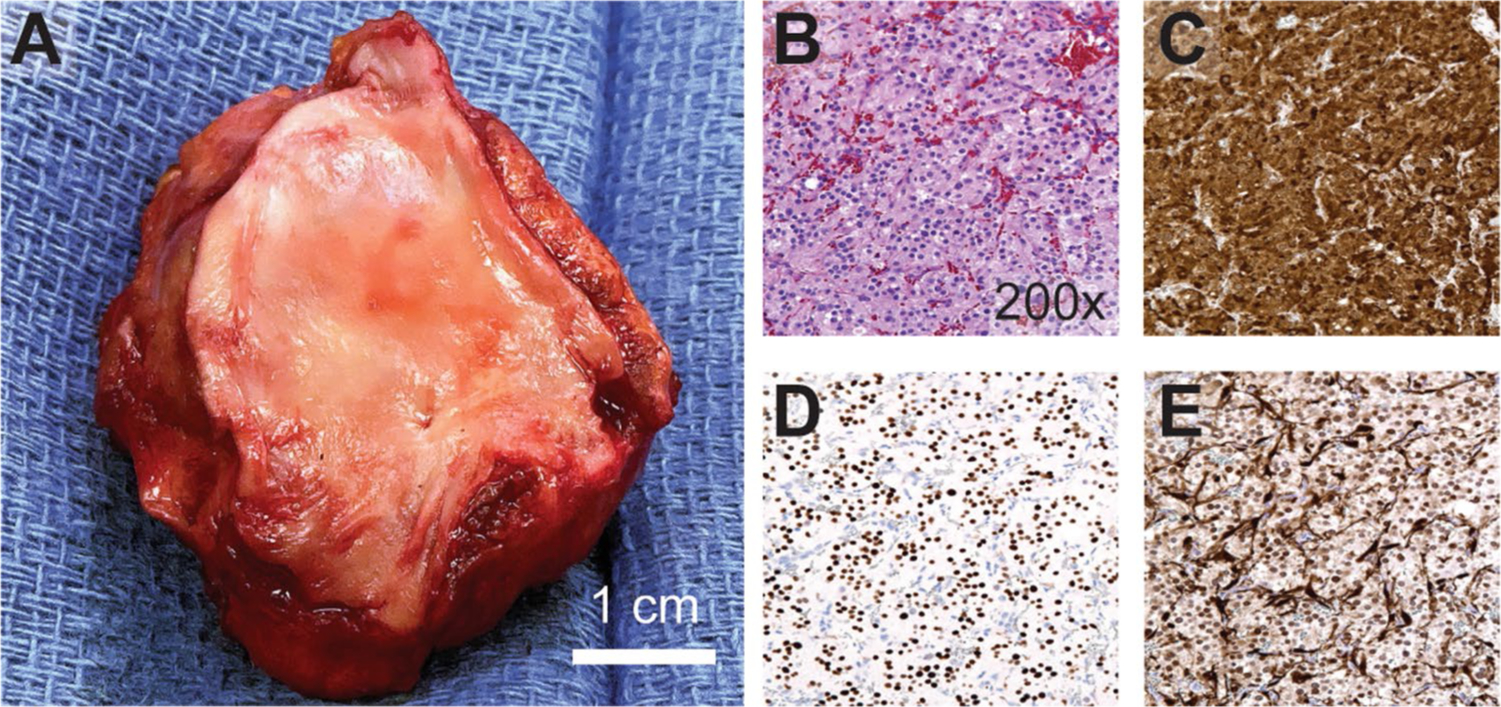
Pathologic assessment and immunohistochemistry: a Gross imaging of the specimen with en bloc portion of LA and underlying tumor mass. Histologic features including: b packed “Zellballen” growth of epithelioid tumor cells on hematoxylin and eosin stain, c strong and diffuse expression of chromogranin A, d INSM-1 stain highlighting chief cell nuclei, e S100 stain highlighting sustentacular cell nuclei at the periphery of cell nests (darkly staining)
Discussion
Paragangliomas are rare tumors with variable clinical presentation. They are usually diagnosed by the combination of biochemical analysis and various imaging modalities (Table 1). Biopsy should be avoided due to the potential for triggering a catecholamine surge or bleeding. Computed tomography (CT) is the first imaging modality of choice due to its excellent spatial resolution. Cardiac magnetic resonance imaging (MRI) is used to further delineate tumor location, and degree of invasion to assist in operative planning. MRI is superior to CT and echocardiography and should be obtained routinely to avoid the potential pitfall of mistaking these tumors for a posterior mediastinal mass. A Ga-68 DOTATATE PET/CT should also be obtained to rule out metastatic disease and/or additional tumors [2], and coronary angiography may be necessary to delineate the tumor’s vascular supply (coronary, bronchial or intercostal arteries). Genetic testing should be performed in all patients; familial mutations in the succinate dehydrogenase enzyme (SDH) gene are reported to be present in as many 25–50% of cases with the subgroup SDHB mutation associated with a higher risk of malignancy [3].
Table 1.
Diagnosis and management of cardiac paragangliomas
| Clinical presentation | ||||
|
Functional tumors - Hypertension, episodic sweating, palpitations, presyncope or syncope, headache, anxiety, heart failure/cardiomyopathy |
Non-functional tumors - Compression or local invasion: coronary flow, valvular function, and venous return - Myocardial ischemia, congestive heart failure, lower extremity edema, SVC syndrome dyspnea, syncope |
|||
| Diagnostic workup | ||||
|
Biochemical analysis (functional vs non-functional) - Urinary fractionated metanephrines (± dopamine) and/or - Plasma free metanephrines and Chromogranin A (CgA) Differential diagnosis: Myxoma, papillary fibroelastoma, hemangioma, osteosarcoma, angiosarcoma, malignant lymphoma, leiomyosarcoma, thrombus |
Imaging (anatomic location*, degree of invasion, vascular supply**, rule out meta-static/additional disease) - CT neck/chest/abdomen - Cardiac MRI - Ga-68 DOTATATE PET/CT*** Consider (after initiation of alpha blockade) - Coronary angiography - TTE/TEE (given retrocardiac location of most tumors sensitivity is variable) *Location: anterior mediastinum, posterior mediastinum, intracardiac (LA, RA, aortic root) **Vascular supply: coronary (RCA, LCx), bronchial, intercostal *** preferred as initial study over F-18 FDG and I-123 MIBG scans |
Biopsy - Not recommended due to potential catecholamine surge / bleeding Genetic testing - SDHx (A,B*,C,D), FH, SDHAF2, vHL, NF1, RET, TMEM127, MAX (minimum) - Family members should be offered testing if a germline mutation is present *SDHB mutation increases risk of malignancy; newer evidence suggest SDHA, SDHC, and FH also haver higher malignant potential |
||
| Perioperative management | ||||
|
Pre-op - Normalize hemodynamics (SBP< 135/90, HR< 100) - Initiate alpha blockade 2–3 weeks prior - Beta blockers should only be used with alpha blockade if needed to control HR - Consider embolization prior to surgery if blood supply via bronchial or intercostal arteries (avoid if tumor is functional as it can precipitate a crisis) |
Peri-op - Volume expansion - Avoid tumor manipulation / catecholamine release - Set up fast acting vasoactive drips that can be easily titrated to manage periods of hypertension / hypotension |
|||
| Surgical approach | ||||
|
Key: Complete surgical excision with en bloc excision of involved structures is necessary for cure and is associated with significantly lower rates of recurrence. Limited metastatic disease and/or lymphatic spread should also be treated with surgery with curative intent Incision - Median sternotomy Support - Cardiopulmonary bypass, bicaval cannulation - Left atriotomy (LA tumors) |
Exposure techniques - Division of superior vena cava, inferior vena cava, or pulmonary artery - Rotation of the heart to the left Complex reconstruction - Autologous pericardium - Decellularized bovine pericardium - Coronary bypass or reconstruction with autologous vein (in cases of coronary involvement) Alternative - Cardiac auto-transplantation vs orthotopic transplant |
|||
| Post-operative management and surveillance | ||||
|
Adjuvant therapy: - No role for adjuvant chemo/radiation* Biochemical analysis - Ensure normalization of urine/plasma levels 2–4 weeks post-operatively *AZEDRA (iobenguane I 131) FDA approved for metastatic disease |
Genetic counseling - Appropriate screening (genetic / biochemical testing) of family members in cases of familial / syndromic / metastatic disease Surveillance - Minimum 10 years in absence of germline mutation; life-long in patients with germline mutation - MRI chest annually ± DOTATATE PET/CT every 5 years - Annual clinic visit with BP monitoring, plasma-free metanephrines and CgA |
|||
Prior to surgical intervention, medical management should include the initiation of alpha blockade 2–3 weeks prior with the goal to normalize hemodynamics (SBP < 135/90, HR < 100). Beta blockade should only be used after alpha blockade if needed for heart rate control. Although some reports suggest a potential role for embolization, this is often not feasible given their highly vascular nature and is contraindicated in functional tumors as embolization can precipitate a crisis. To mitigate the potential for large fluctuations in hemodynamics during the immediate perioperative period, patients should be adequately resuscitated, excessive tumor manipulation should be avoided, and vasoactive medications should be fast acting in the form of easily titratable drips.
To achieve cure, tumors must be completely excised which includes the en bloc excision of involved structures. As most PGLs arise from the left atrium, they are often best approached via median sternotomy with cardiopulmonary bypass. In our case, to avoid the need for cardiac auto-transplantation, exposure was optimized by dividing the SVC and rotating the heart to the left (Supplementary Video 1).
Following surgical resection, patients should have biochemical markers evaluated 2–4 weeks post-operatively as these would be expected to normalize in the case of complete resection. In patients presenting with familial, syndromic, or metastatic disease, genetic and biochemical screening should be offered to family members. In addition, these patients will require lifelong annual screening. In the absence of a germline mutation, patients should undergo a minimum of 10 years of screening although given the rarity of intracardiac PGLs, many clinicians will continue to screen beyond this time period. It is our preference that annual screening includes MRI of the chest (plus a DOTATATE PET/CT every 5 years) and a clinic visit for blood pressure monitoring and screening for plasma-free metanephrines and Chromogranin A.
Conclusion
Despite their slow growth, all cardiac PGLs should be treated as malignant lesions due to their tendency towards local invasion and high risk of metastatic spread and recurrence. In cases of limited disease, prompt workup and complete surgical excision are mandated.
The majority of intracardiac PGLs arise from the left atrium and are best approached via median sternotomy. En-bloc excision, followed by complex reconstruction, is feasible using cardiopulmonary bypass with bicaval cannulation, and maneuvers to increase exposure, such as transection of the superior vena cava. In rare cases, tumors may be unresectable and orthotopic transplantation should be considered.
Supplementary Material
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Conflict of interest The authors have no conflicts of interest related to the subject matter.
Compliance with ethical standards
Ethical approval The study has been approved by the Ethical Committee of our institution.
Informed Consent All the subjects involved in the present study gave their consent to participate.
Consent to publication All the subjects involved in the present study gave their consent to the publication of this document.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s11748-020-01503-2) contains supplementary material, which is available to authorized users.
References
- 1.Khan MF, Datta S, Chisti MM, Movahed MR. Cardiac paraganglioma: clinical presentation, diagnostic approach and factors affecting short and long-term outcomes. Int J Cardiol. 2013;166:315–20. [DOI] [PubMed] [Google Scholar]
- 2.Chang CA, Pattison DA, Tothill RW, et al. 68 Ga-DOTATATE and 18 F-FDG PET/CT in Paraganglioma and Pheochromocytoma: utility, patterns and heterogeneity. Cancer Imaging. 2016;16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramlawi B, David EA, Kim MP, et al. Contemporary surgical management of cardiac paragangliomas. Ann Thoracic Surg. 2012;93:1972–6. [DOI] [PubMed] [Google Scholar]
- 4.Huo JL, Choi JC, DeLuna A, et al. Cardiac paraganglioma: diagnostic and surgical challenges. J Card Surg. 2012;27:178–82. [DOI] [PubMed] [Google Scholar]
- 5.Brown ML, Zayas GE, Abel MD, Young WF Jr, Schaff HV. Mediastinal paragangliomas: the mayo clinic experience. Ann Thoracic Surg. 2008;86:946–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.