

Abstract
Akt is a Ser/Thr protein kinase that regulates cell growth and metabolism and is considered a therapeutic target for cancer. Regulation of Akt by membrane recruitment and post-translational modifications (PTMs) has been extensively studied. The most well-established mechanism for cellular Akt activation involves phosphorylation on its activation loop on Thr308 by PDK1 and on its C-terminal tail on Ser473 by mTORC2. In addition, dual phosphorylation on Ser477 and Thr479 has been shown to activate Akt. Other C-terminal tail PTMs have been identified, but their functional impacts have not been well-characterized. Here, we investigate the regulatory effects of phosphorylation of Tyr474 and O-GlcNAcylation of Ser473 on Akt. We use expressed protein ligation as a tool to produce semisynthetic Akt proteins containing phosphoTyr474 and O-GlcNAcSer473 to dissect the enzymatic functions of these PTMs. We find that O-GlcNAcylation at Ser473 and phosphorylation at Tyr474 can also partially increase Akt’s kinase activity toward both peptide and protein substrates. Additionally, we performed kinase assays employing human protein microarrays to investigate global substrate specificity of Akt, comparing phosphorylated versus O-GlcNAcylated Ser473 forms. We observed a high similarity in the protein substrates phosphorylated by phosphoSer473 Akt and O-GlcNAcSer473 Akt. Two Akt substrates identified using microarrays, PPM1H, a protein phosphatase, and NEDD4L, an E3 ubiquitin ligase, were validated in solution-phase assays and cell transfection experiments.
Graphical Abstract
Akt (Akt1) is a Ser/Thr kinase and a key component of the PI3K signaling pathway, which propagates extracellular signals to promote cellular functions including growth, proliferation, and survival.1,2 Many of Akt’s downstream substrates have been identified including PRAS40 and GSK3.2,3 These substrates, along with all canonical Akt protein substrates, have Arg residues at the minus 3 and 5 positions relative to the phosphorylated Ser or Thr (pSer/pThr).2,4 Akt hyperactivation drives phosphorylation of its downstream substrates and has been implicated in contributing to cancer growth. There are several ATP-competitive and allosteric Akt inhibitors in clinical development as oncology therapeutics.5–7
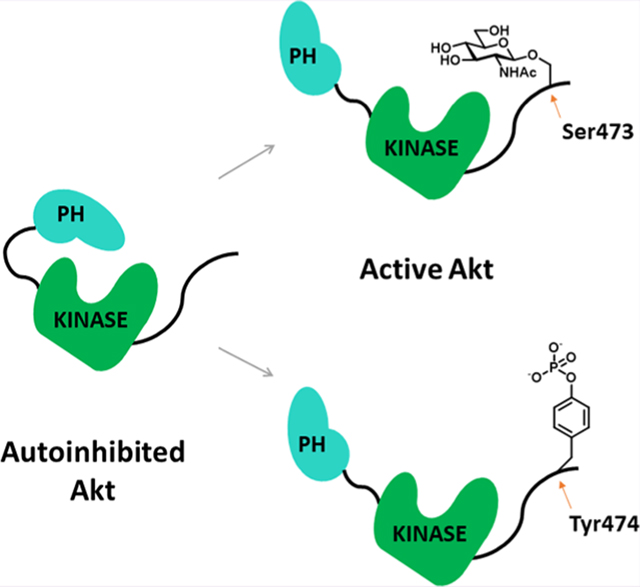
Akt is a 480 amino acid kinase comprising an N-terminal PH (pleckstrin homology) domain followed by a catalytic kinase domain and a disordered C-terminal tail that is subject to post-translational modifications (PTMs).2,8,9 In response to extracellular stimuli, Akt activation involves its recruitment to the plasma membrane by increased levels of phosphatidyl inositol 3,4,5-triphosphate (PIP3).10 At the plasma membrane, Akt is phosphorylated on its activation loop at Thr308 by PDK1 and its C-terminus at Ser473 by mTORC2 in the classical signaling pathway.11–13 These phosphorylation events induce an Akt structural change that relieves intramolecular autoinhibition mediated by PH domain-kinase domain interactions, leading to an active kinase domain conformation and substrate phosphorylation.9,14,15
Beyond the standard mTORC2-catalyzed phosphorylation of Ser473 on Akt, other C-terminal PTMs have been reported including O-GlcNAcylation (glycosylation with N-acetylglucosamine) at Ser47316 and phosphorylation at Tyr474,17 Ser477, and Thr479.18 Dual phosphorylation of Ser477/Thr479 has been demonstrated to partially stimulate Akt kinase activity,19,20 but the effects of Tyr474 phosphorylation and Ser473 O-GlcNAcylation have been only indirectly characterized. In previous work, mutation of Tyr474 to Phe in Akt showed diminished catalytic activity, which suggested that Tyr474 phosphorylation may be activated.17 In another study, cells treated with N-acetyl-glucosamine showed reduced Akt activity leading to the hypothesis that O-GlcNAcylation of Ser473 blocks phosphorylation of this residue and inhibits Akt.16 Nevertheless, site-directed mutagenesis of Tyr to Phe and changes in the growth conditions of cells may have non-specific effects that prevent elucidating the individual regulatory roles of phosphorylation at Tyr474 and O-GlcNAcylation of Ser473. Moreover, studies have identified additional O-GlcNAcylation sites in Akt at Thr430 and Thr479, which was reported to activate Akt by increasing Ser473 phosphorylation,21 or at Thr308, which were suggested to inactivate Akt by preventing phosphorylation.22 Many studies have reported that the interplay between phosphorylation and O-GlcNAcylation involves an “on” or “off” effect23 related to mutual exclusion of these modifications on the same site.
To assess the impact of Tyr474 phosphorylation and Ser473 O-GlcNAcylation directly, we utilize here expressed protein ligation (EPL) to install these PTMs in a specific and stoichiometric fashion. Previously employed for Ser473 and Ser477/Thr479 phosphorylation incorporation, this method involves a chemoselective reaction between a recombinant protein thioester and a peptide with an N-terminus Cys residue (N-Cys) to allow for precise C-tail PTM installation.19 Beyond Akt, EPL has been used broadly to investigate the role of PTMs in regulating protein structure and function.24–26
With the desired semisynthetic Akt forms in hand, we investigated their catalytic properties with peptide and protein substrates, comparing their steady-state kinetic parameters to those of the non-C-tail phosphorylated and pSer473 semisynthetic Akts.19,27 In addition, we have used protein microarrays to characterize the proteome-wide substrate specificity for Akt with pSer473 and Akt with O-GlcNAcSer473 and disclose the findings below.
METHODS
Generation of Semisynthetic Akt Proteins.
A baculovirusinsect cell expression system was used to express the Akt-Mxeintein-CBD fusion construct (Akt aa2–459) as described previously.19 Bacmid was transfected (Cellfectin II reagent, Thermo Fisher Scientific) into Sf21 insect cells, and baculovirus was produced as previously described.28 The resulting baculovirus was used to infect Sf9 insect cells with a multiplicity of infection (M.O.I.) of 5.0. To obtain in vivo Thr308 phosphorylation, Akt was co-expressed with GST-PDK1 in Sf9 insect cells with a M.O.I. of 1.0. Cells were cultured in media (Sf-900 II SMF, Gibco) for 36 h at 27 °C. Subsequently, 25 nM okadaic acid (Cell Signaling Technology–CST) was added, and the cells were allowed to grow for an additional 16 h, harvested (700 g, 10 min, 4 °C), flash-frozen in liquid nitrogen, and stored at −80 °C.
The cells from 200 mL culture were lysed in a 20 mL Dounce homogenizer in 10 mL of lysis buffer (50 mMHEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1% Triton X-100, and 1 mM PMSF) containing one dissolved protease inhibitor tablet and one dissolved phosphatase inhibitor tablet (Thermo Fisher Scientific). The lysate was centrifuged (17,500g, 40 min, 4 °C), and the supernatant was incubated with rotation in a 5 mL bed of cellulose (Sigma) for 1 h at 4 °C. The lysate was filtered from the cellulose and then passed through a column containing 5 mL of chitin resin (NEB). The resin was then washed with 150 mL of washing buffer (50 mM HEPES pH 7.5, 500 mM NaCl, and 0.1% Triton X-100), incubated overnight in cleavage buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 300 mM MESNA (sodium mercaptoethylsulfonate), 1 mM phenylmethylsulfonylfluoride (PMSF), and 15% glycerol) at room temperature. The cleavage buffer containing Akt thioester protein was eluted from the resin and concentrated by ultrafiltration using an Amicon 10 kDa-molecular weight cutoff (MWCO) filter. The N-terminal Akt protein fragments containing a C-terminal thioester were shown to be >90% pure by SDSPAGE and 0.4 mg was reacted with the synthetic N-Cys containing C-terminal Akt peptides (CVDSERRPHFPQFSYSASGTA), containing the desired phosphorylation or O-linked N-acetylglucosamine (O-GlcNAc) modifications, in ligation buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 1 mM TCEP, 0.5 mM PMSF, and 10% glycerol) at 2 mM final concentration for 5 h at RT and then maintained overnight at 4 °C. The ligation conversions were assessed by Coomassie-stained SDS-PAGE and typically shown to be between 70 and 90%. The semisynthetic Akt proteins were purified by size exclusion chromatography on a Superdex 200 column (Cytiva) with Akt storage buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM beta-mercaptoethanol, 0.2 mM PMSF, 1 mM EDTA, and 10% glycerol) where they were shown to be monomeric. The purified fractions were combined, concentrated, aliquoted, and then stored at −80 °C. Typical yields of the purified semisynthetic Akts were about 2 mg/L insect cell culture.
Cell-Based Assays.
Akt1/2 KO HCT116 cells29 were acquired from the Johns Hopkins Genetic Resources Core Facility (GRCF) and maintained in McCoy’s 5A medium (Corning) supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin/streptomycin (GIBCO) at 37 °C with 5% CO2. Cells were seeded in 6-well plates and transfected at 70% confluency. For the NEDD4L experiments, cells were transfected with 0.5 μg of pcDNA3.1-Flag-HA-Akt WT or pcDNA3.1-Flag-HA-Akt D274A and 1 μg of pcDNA3.1-His-NEDD4L C942S. Plasmids were complexed with 6 μL of Lipofectamine 3000 and 3 μL of P3000 reagent in Opti-MEM medium and incubated with the cells for 48 h. For the PPM1H experiments, cells were transfected with 0.25 μg of pcDNA3.1-Flag-Akt WT or 0.5 μg of pcDNA3.1-Flag-Akt D274A and 1 μg of pcMV5D-HA-PPM1H H153D. Plasmids were complexed with 6 μL of Lipofectamine 3000 and 3 μL of P3000 reagent in Opti-MEM medium and incubated with the cells for 30 h. For pT450 validation experiments, 1.5 μg of pcDNA3.1-Flag-HA- Akt WT, T450A, T450D, and T450E was transfected. Plasmids were complexed with 3 μL of Lipofectamine 3000 and 3 μL of P3000 reagent in Opti-MEM medium and incubated with the cells for 24 h. Cells were washed with PBS, lysed by adding 200 μL of RIPA buffer (CST) containing 1× complete protease inhibitor tablet and 1 mM PMSF, and placed on a shaker for 10 min at 4 °C. Cell lysate was removed from the plate with a cell scraper and spun for 10 min at 4 °C. Total protein concentration was measured with a BCA assay (Thermo Fisher Scientific). For PPM1H experiments, we incubated 100 μL of cell lysate with Pierce HA magnetic beads (Thermo Fisher Scientific) for 30 min at room temperature for immunoprecipitation (IP) of HA-PPM1H. We then washed the magnetic beads 2× with 300 μL of TBS-T and one last time with water before eluting with 50 μL of gel loading buffer. For NEDD4L experiments, 6 μg of total protein was loaded per well on SDS-PAGE. For PPM1H experiments, 5 μg of cell lysate was loaded to blot for Akt and GAPDH and 3× the volume from the elution buffer from the IP to blot for HA-tag and Akt phosphorylated substrate. Membrane transfer and western blotting were carried out as described above with 1:2500 dilution for primary antibodies: Akt, NEDD4L; 1:2000 dilution for phospho S342 and S448 NEDD4L; 1:1000 for HA-tag (Cell Signaling Technology #3724) and Akt phosphorylated substrate antibody; and 1:5000 dilution for anti-GAPDH.
Intact Mass Spectrometry Analysis of Full-Length Akt Proteins.
Akt proteins in storage buffer were diluted to 100 ng/μL in 50 mM ammonium bicarbonate, pH = 8, and centrifuged at 16,300g for 1 min. Supernatants were transferred to low-binding liquid chromatography vials, and approximately 100 ng of protein was injected onto a Vanquish Flex LC interfaced to a Q Exactive mass spectrometer (Thermo Fisher Scientific). Proteins were separated by reversed-liquid-phase chromatography at a flow rate of 0.3 mL/min on an MAbPac RP 3 μm 2.1 × 100 mm column (Thermo Fisher #088647) with 0.5 min isocratic hold at 5% mobile phase B, followed by 2 min isocratic hold at 20% B, and finally a linear gradient from 20 to 70% B (0.1% (v/v) formic acid in acetonitrile) over 5 min, where mobile phase A was 0.1% (v/v) formic acid in water. Electrospray ionization using a HESI-II probe was carried out in positive mode with a sheath gas flow rate of 40 arbitrary units, auxiliary gas flow rate of 5 arbitrary units, auxiliary gas temperature of 150 °C, sweep gas flow rate of 1 arbitrary unit, capillary temperature of 250 °C, and spray voltage of 3.5 kV. The S-lens RF level was set to 50, and solvent clusters were dissociated by in-source collision-induced dissociation at 10 eV. Analysis with the Q Exactive (Tune version 2.1) was carried out in the full MS mode. Four microscans per duty cycle were acquired with a resolution of 17,500 (FWHM at m/z 200) and an m/z scan range of 700–1700. The AGC target was set to 1,000,000 with a maximum ion injection time of 50 milliseconds. For each protein, MS scans were averaged across the chromatographic peak, and the resulting spectra were deconvoluted using UniDec software.30
RESULTS AND DISCUSSION
Semisynthetic Preparation of Akt with Phosphorylation and O-GlcNAcylation at the C-Terminal Tail.
To investigate the impact of Ser473 O-GlcNAcylation and Tyr474 phosphorylation on Akt regulation, we employed EPL to generate the semisynthetic Akt forms. We adapted the strategy used previously for making C-terminal Ser/Thr phospho-forms of Akt (Figure 1A). In this approach, a recombinant thioester Akt fragment (aa1–459) is prepared in a baculovirus/insect cell expression system using an intein followed by MESNA treatment. In this case, Thr308 loop phosphorylation was installed by co-expression with PDK1 and the addition of the phosphatase inhibitor okadaic acid.19 Separately, N-Cys peptides (aa460–480) containing O-GlcNAcSer473 or pTyr474 +/- pSer473 or pSer477/pThr479 (Figures 1B and S1) were generated using solid-phase peptide synthesis. We also synthesized C-tail peptides lacking PTMs and containing pSer473/Y474F (Figures 1B and S1) for comparative activity analysis. Ligation of the corresponding peptides and recombinant thioester fragments was carried out, affording 70–90% conversion to the desired products (Figure S2). All ligation reactions were carried out using recombinant Akt core fragments produced from the same batch of baculovirus and insect cells to ensure reliable comparisons since there can be variability in the insect cell-produced protein among different preparations.27 For Akt with O-GlcNAcylation on Ser473, the ligation conversion was slightly lower than with the phosphopeptides (Figure S2), possibly related to the reduced solubility of the O-GlcNAc peptide. The semisynthetic proteins were further purified by size exclusion chromatography, which removed the remaining unligated peptides and revealed that each Akt form produced behaved as a monomer. The purity and quantity of semisynthetic proteins were assessed by Coomassie-stained SDS-PAGE (Figure S2). A subset of the semisynthetic proteins, selected because they were the focus of our enzymatic studies, were subjected to western blot with pSer473, pTyr, and O-GlcNAc antibodies, which provided evidence that the desired PTMs were successfully added (Figure 1C). The relatively modest western blot band intensity observed with the O-GlcNAc antibody, although specific to the O-GlcNAcSer473 Akt form (Figure 1C), is consistent with previous signal-to-noise levels using this antibody with other semisynthetic proteins.31 Residual unligated Akt is unlikely to show significant kinase activity, as described previously.19
Figure 1.

Semisynthetic Akt with specific C-tail PTMs patterns. (A) Scheme of the EPL reaction showing truncated Akt (aa1–459) with a C-terminal thioester and a peptide with an N-terminal cysteine. Upon ligation these form an amide bond and yield a full-length Akt protein with its native sequence. (B) Sequence of C-tail peptides with the corresponding PTMs in bold. (C) Western blot of selected semisynthetic Akt proteins with various PTMs at the C-tail. Antibodies: total Akt (1:5000), Akt pSer473 (1:5000), pTyr (4G10) (1:500), and O-GlcNAc (RL2) (1:500). The broader/thicker band for O-GlcNAcS-er473 Akt (total Akt) may reflect a mix with unligated Akt.
We also performed intact mass spectrometry analysis of representative semisynthetic Akts, and this confirmed the presence of the expected Akt forms within a distribution of masses that showed a range of additional phosphorylations (Figure S3). This mass heterogeneity is consistent with prior work on recombinant Akt produced in insect cells which show multiple phosphorylations that localize to the PH-kinase linker and Thr450 and perhaps elsewhere.27,32 Notwithstanding the observed heterogeneity, the mass spectra of the O-GlcNAc samples showed the expected increased mass shifts in the set of Akt peaks relative to the non-O-GlcNAc containing forms, providing additional evidence for their structural identities.
It has recently been reported that Akt produced using EPL showed nearly undetectable Thr450 phosphorylation, as assessed by label-free LC-MS/MS quantification of proteolytic Akt digests, leading the authors to question the reliability of the semisynthetically produced Akts since pThr450 is proposed to be important for Akt stability.33,34 We therefore analyzed the pThr450 levels in representative semisynthetic Akts produced in this study and previously and two commercial recombinant full-length Akt proteins by western blot with anti-pThr450 Ab (Figure S4A). Validation of the pThr450 Ab specificity was achieved using a C-terminally truncated Akt (aa2–443) and a cell transfection experiment with full-length T450A Akt expressed in HCT116 colon cancer Akt1/2 knockout cells (Figure S4B). These experiments revealed that pThr450 levels were quite similar among the semisynthetic and recombinant full-length Akt forms, supporting the reliability of the semisynthetic approach used to produce Akts employed in this study. It is possible that the differences between our findings and those recently reported34 about Thr450 phosphorylation relate to the use of a truncated and heavily mutated PH domain-kinase linker used in the latter study.
Kinetic Characterization of the Differentially Modified Semisynthetic Akt Proteins.
To characterize the kinetic parameters of the semisynthetic Akt proteins, we performed direct, radioactive kinase assays using γ-32P-ATP and a well-characterized synthetic GSK3 peptide substrate (RSGRARTSS-FAEPGGK) modified with an N-ε-biotin-lysine for avidin trapping.19 Initially, we screened semisynthetic Akts with fixed amounts of ATP and peptide substrate at two different Akt concentrations to demonstrate that the reaction velocities were approximately linear with respect to Akt concentrations (Figure S5A,B). We then measured apparent kcat and Km with varying concentrations of ATP (Figures 2A and S6). Since saturation with ATP was not achieved for many of the modified Akt forms, kcat and Km values are considered apparent. Nonetheless, kcat /Km could be accurately determined and was used as a measure of catalytic activity (Figure 2B). As reported recently for these enzyme batches,27 the kcat /Km for pSer473 Akt (A1, 0.24 μM−1 min−1) was ~10-fold higher than that of pSer477/pThr479 Akt (A2, kcat /Km 0.027 min−1 μM−1) and much greater (~100-fold) than that of non-C-terminally modified Akt (A3, 0.0026 μM−1 min−1). Unexpectedly, catalytic effciencies of pTyr474 Akt (A4, kcat /Km 0.044 μM−1 min−1) and O-GlcNAcSer473 Akt (A8, kcat/Km 0.053 μM−1 min−1) were ~20-fold greater than those of non-C-terminally modified Akt (A3, kcat /Km 0.0026 min−1 μM−1) and ~5-fold lower than those of pSer473-Akt (A1, kcat /Km 0.24 μM−1 min−1). pTyr474 (A4) Akt and O-GlcNAcSer473 (A8) forms display higher ATP Km values relative to pSer473 Akt (A1) (Figure 2B). To fully characterize these enzymes, we measured the Km of peptide substrate and found that the values are in the range of ~2 μM for pTyr474 (A4) and O-GlcNAcSer473 (A8) Akt (Figure S7). These are in agreement with previous work,19 in which it was reported that the absence or presence of C-terminal modifications did not have a large impact on peptide Km. Additionally, we measured the kinase activity of Y474F in the context of pSer473 as this mutation in Akt was shown to be inhibitory in cellular studies.17 Notably, incorporating Y474F in the context of pSer473 (A6, kcat /Km 0.047 μM−1 min−1) led to a ~5-fold reduction in activity compared to pSer473 alone (A1, kcat /Km 0.24 μM−1 min−1). This suggests a possible role for the sidechain phenol group in Tyr474 as a promoter of full kinase activation in the pSer473 Akt form (A1). Our results are consistent with a prior proposal that Tyr474 phosphorylation is partially activating based on cellular experiments using Tyr474 transfection.17 However, that study used Y474F to deduce the impact of Tyr474 phosphorylation, which we show can impact the degree of activation by pSer473, limiting its value for interpreting PTM effects. Our observation that O-GlcNAcylation at Ser473 can partially activate Akt counters previous reports about the effects of this PTM.16,22 It is understandable why the prior studies deduced that Ser473 O-GlcNAcylation would be inhibitory since their measurements were relative to the more robust activity of pSer473 and the observations were based on complex cellular assays. However, the level of Akt activity associated with O-GlcNAcSer473 may still be physiologically meaningful depending on the cellular circumstances. For example, if growth factors are low and glucose levels are high, driving up UDP-N-acetyl-glucosamine, the co-substrate for O-GlcNAc transferase, would lead to a concomitant increase in O-GlcNAcylation of proteins.35 Under such circumstances, O-GlcNAcSer473 Akt might contribute to phosphorylating particular substrates in cells.
Figure 2.

Kinetic characterization of semisynthetic Akt proteins. (A) Representative steady-state kinetic plots of v/[E] vs [ATP] with 20 μM GSK3 peptide substrate. Enzyme concentrations employed are 5 nM FL-Akt-pThr308/pSer473 (A1), 10 nM FL-Akt-pThr308/O-GlcNAcSer473 (A8), 10 nM FL-Akt- pThr308/pTyr474 (A4), and 100 nM non-phosphorylated FL-Akt-pThr308 (A3). The reactions were carried out at 30 °C for 10 min. (B) Enzymatic parameters for the semisynthetic full-length Akt proteins are shown ±SE; n = 2. (*represents apparent measurements) (**data published previously,27 shown together for ease of comparison).
Given the observed Akt kinase activation associated with pTyr474 and O-GlcNAcSer473 modifications, we investigated the catalytic effects of various combinations of C-tail PTMs (Figure 2B) that could theoretically occur on Akt. Akt with both pSer473 and pTyr474 (A5) showed reduced activity (kcat/Km 0.019 μMT−1 min−1) relative to the singly modified forms (Al, kcat/Km 0.24 μM−1 min−1 and A4, kcat/Km 0.044 μM−1 min−1), indicating antagonistic actions of the PTMs. Likewise, pTyr474/pSer477/pThr479 Akt (A7) had lower activity (kcat/Km 0.01 μM−1 min−1) than either pTyr474 Akt (A4, kcat/Km 0.044 μM−1 min−1) or pSer477/pThr479 (A2, kcat/Km 0.027 μM−1 min−1) forms. O-GlcNAcSer473 with pSer477/pThr479 (A9) showed a moderately higher activity (kcat/Km 0.11 μM−1 min−1) relative to the O-GlcNAcSer473 (A8, kcat/Km 0.053 μM−1 min−1) and pSer477/pThr479 (A2, kcat/Km 0.027 μM−1 min−1) states, indicating a degree of cooperativity among these PTMs.
Previous studies have demonstrated that the Gln218 side chain in Akt serves as a hydrogen bond donor to the phosphate of pSer473, and this interaction plays a role in driving the active conformation of this modified Akt form.19,36 We therefore prepared the Q218A mutants of the O-GlcNAcSer473 and pTyr474 semisynthetic Akts. In both cases, Gln mutation abolished the activation associated with these C-terminal PTMs in a comparable manner to Q218A’s impact on pSer473, affording activities similar to non-C-terminally modified Akt (Figure S8). These results suggest that Gln218 plays a key role in mediating the activation of Akt by both C-terminal Ser473 O-GlcNAcylation and Tyr474 phosphorylation.
To gain further insights into the functional properties of O-GlcNAc S473 Akt (A8), we performed a fluorescence anisotropy binding assay to measure its PH domain affinity to PIP3 using a soluble analogue (di-C6-PIP3) (Figure S9A). O-GlcNAcSer473 Akt (A8) showed a KD of 0.13 ± 0.02 μM for PIP3, within ~2-fold to the KD previously determined for pSer473 Akt (A1) (KD of 0.20 ± 0.07 μM) and non-C-terminally phosphorylated Akt (A3)19 (KD of 0.23 ± 0.05 μM). This suggests that the O-GlcNAc functionality at Ser473 does not drive a major conformational change in the PIP3-PH domain interaction relative to that of the standard Akt forms. It has previously been reported that O-linked N-acetylglucosamine transferase (OGT), the enzyme that transfers O-GlcNAc to Ser/Thr residues, may be recruited to membranes via PIP3.37 Consequently, we attempted to use fluorescence anisotropy to measure the affinity of OGT (Figure S9B) for soluble PIP3. Although there was a concentration-dependent increase in anisotropy in the range measured (Figure S9C), there was no evidence of saturation at the highest OGT concentration tested (15 μM), indicating that if OGT binds PIP3, it does so very weakly.
PRAS40 is a Substrate for Akt Proteins with Different C-Tail PTMs.
Enzymatic analysis of the Akt semisynthetic proteins described here and in previous studies have relied on GSK3 peptide as a model substrate.19,27 As the first step toward characterizing Akt forms with a full-length protein substrate, we selected the well-established Akt substrate, PRAS40.2 In previous work, PRAS40 has been shown to be efficiently phosphorylated by Akt on Thr246, which regulates PRAS40’s interaction with the mTORC1 complex.2 Using western blots, we compared the levels of PRAS40 phosphorylation by pSer473 (A1), pS477/p479 (A2), O-GlcNAcSer473 (A8), pTyr474 (A4), and non-C-terminally modified (A3) Akt forms (Figure S10A). It should be noted that these reactions were carried out with 2 mM ATP, a concentration believed to be typical of the physiological range.38 These measurements revealed that pSer473 Akt (A1) was the most active PRAS40 kinase, mildly greater than the O-GlcNAcSer473 (A8) and pTyr474 (A4) Akt forms and considerably faster than pS477/p479 Akt (A2) (Figure S10B). As expected, non-C-terminally modified Akt (A3) showed near-background levels of PRAS40 phosphorylation under these conditions (Figure S10B). Overall, these kinetic results roughly correlate with the more quantitatively rigorous radioactive kinase assays with the GSK3 peptide substrate.
Comparison of Kinase Activity Between pSer473 Akt and O-GlcNAcSer473 Akt Using Human Protein Microarrays.
Given the unexpected relatively high catalytic activity of O-GlcNAcSer473 Akt (8), we wanted to compare the kinase activity of the modified Akt isoforms pSer473 (A1) and O-GlcNAcSer473 (A8) using a diverse pool of substrates. To do this, we employed human proteome microarrays (HuProt),39 which comprise over 20,000 purified human proteins in full length and spatially arrayed in duplicates on a glass slide. HuProt microarrays have been used previously to determine kinase substrate preferences for casein kinase 2 (CK2) and other protein kinases.25,39 Akt kinase assays on HuProt were performed using 5 mM ATP and 0.5 μM Akt at room temperature for 2 h. Note that 5 mM ATP, higher than that used in the PRAS40 experiments, is still in the physiological range38 and was selected to maximize the number of potential substrates identified. To detect protein phosphorylation on HuProt, we first attempted to use radioisotopes for product labeling,25 but we found that the signal was too weak to be reliable. To overcome this, we employed a phospho-Akt substrate antibody that recognizes the sequence around the Ser or Thr phosphorylated by Akt (R/KXR/KXXS*/T*) and a secondary fluorescence antibody for detection, which provided better signal to noise ratios in our HuProt analysis. We scanned each HuProt to obtain the fluorescence intensity of each spot (protein) based on GenePix software quantitation. We defined positive hits as the proteins with a fluorescence intensity greater than three standard deviations (SD) above the median from each HuProt experiment. Then, we subtracted “false positive” hits that appeared on the control HuProt not exposed to Akt and used visual inspection to further assess if the intensity measured using software correlated with the intensity observed in the images. Based on these criteria, we identified 67 total hits with 63 shared between the pSer473 (A1) and O-GlcNAcSer473 (A8) Akt forms and four selective for pSer473 Akt (A1) (Figure S11A).
The full details of these hits and their signal intensities are displayed in Figure S12. Out of the four protein pSer473 Akt (A1) selective substrates, PPM1H showed the largest difference between the two kinase forms (Figure S12B). We decided to further investigate PPM1H as an Akt substrate due to its reported ability to counteract a kinase, LRKK2, that when overactivated causes Parkinson’s disease.40 In addition, we decided to validate NEDD4L, which was a hit for both pSer473 Akt (A1) and O-GlcNAcSer473 Akt (A8) and functions to regulate cellular homeostasis with implications in renal disease, blood-brain barrier permeability, and hypertension.41–43
Validation of the Protein Substrates PPM1H and NEDD4L with pSer473 and O-GlcNAcSer473 Akt Forms.
To validate PPM1H as a substrate for Akt, we expressed and purified wild-type (WT) PPM1H along with its catalytically dead mutant (H153D)40,44 and tested these as substrates in solution-phase assays with varying concentrations of pSer473 Akt (A1) and O-GlcNAcSer473 Akt (A8). PPM1H phosphorylation was analyzed using western blots (Figures 3A and S13A). We observed that both pSer473 Akt (A1) and O-GlcNAcSer473 Akt (A8) can phosphorylate PPM1H in a concentration-dependent manner. Furthermore, pSer473 Akt (A1) is a more potent PPM1H kinase than O-GlcNAcSer473 Akt (A8), consistent with the protein microarray data. Interestingly, H153D PPM1H demonstrated enhanced phosphorylation by both Akt forms.
Figure 3.

Akt phosphorylates WT and dead PPM1H in a concentration-dependent manner. (A) 1 μM WT or dead (H153D) PPM1H as a substrate for pSer473 Akt (A1) and O-GlcNAcSer473 Akt (A8) kinases at 50, 100, and 200 nM. (n = 2). (B) WT, dead (H153D), S124A, and S124A/H153D PPM1H as substrates for pSer473 Akt (A1) at 100 nM. Both assays were carried out using 2 mM ATP at 30 °C for 20 min and analyzed by western blot. 250 ng of PPM1H was loaded per well. Membranes were blotted with the phospho Akt substrate antibody (R/KXR/KXXS*/T*) (1:1000) total Akt antibody (1:5000) and signal was quantified to show that Akt phosphorylates PPM1H in a concentration-dependent manner (Figure S13). (n = 3). In the PPM1H (coomassie) image, BSA used in this assay is shown as the band right above PPM1H.
As catalytically dead PPM1H appeared to be a better substrate for both Akt forms tested, we hypothesized that PPM1H phosphatase activity might be capable of targeting Akt for dephosphorylation. To investigate this further, we performed kinase assays with pre-incubation of reaction components at various times and temperatures prior to initiating with ATP. We observed that pre-incubation of WT PPM1H (Figure S14A) but not dead PPM1H (Figure S14B) with pSer473 Akt (A1) led to reduced phosphorylation on Ser473 of A1 over time, and concomitant with this, A1’s ability to phosphorylate PPM1H decreased. We observed that Akt with O-GlcNAcylation at Ser473 (A8) was also less efficient at phosphorylating PPM1H over the time course (Figures S14A and S13B), and this was not observed for the catalytically dead PPM1H (Figures S14B and S13C). These results suggest that besides depleting Ser473 phosphorylation, PPM1H may remove other phosphorylations that are important for maintaining Akt’s active conformation (e.g., pThr308 or pThr450).2 These observations lead us to speculate that there could be reciprocal regulation of Akt activity by PPM1H phosphatase action. Indeed, PPM1H is related to the well-established Akt pSer473 phosphatase PHLPP1 and so it was notable that we found PPM1H to possess a similar activity.45 In previous work, PHLPP1 was shown to weakly dephosphorylate Rab10, a PPM1H substrate, in cells which supports the idea that these phosphatases may have comparable substrate preferences.40 Moreover, PPM1H may be capable of auto-dephosphorylation, which could be an additional explanation for the increased overall phosphorylation observed for dead PPM1H compared to the WT. To prevent PPM1H dephosphorylation, we used the catalytically dead PPM1H protein as a substrate to determine the Km values and observed a ~2-fold lower Km when comparing pSer473 Akt (A1) to O-GlcNAcSer473 Akt (A8)(Figure S15).
To further validate PPM1H as a substrate for Akt, we used HCT116 Akt1/2 knockout (KO) cells transfected with either WT or D274A (catalytically dead) Akt46 and PPM1H H153D (Figure 4A). The results of these experiments suggest that WT Akt, but not catalytically dead Akt, phosphorylates PPM1H in this cell line (Figures 4A,B and S16).
Figure 4.

Akt phosphorylates PPM1H in HCT116 Akt1/2 KO cells. (A) Western blot analysis of HCT116 Akt1/2 KO cells transfected with either WT (left) or D274A (catalytically dead, right) Akt and PPM1H (H153D) showing three different replicates. Anti-Akt (1:2000) and Anti-GAPDH (1:5000) were blotted from cell lysates. Anti-HA-tag (1:1000) and Anti-phospho Akt substrates (1:1000) were blotted from lysates that were immunoprecipitated with HA-tag magnetic beads (see the Methods). (B) Bar plot represents quantification of band intensity of WB for phosphorylated Akt substrate forms normalized to total HA-tag bands intensity for each band. Data represent five biological replicates with n = 2 or 3 for each. The P-value is 0.0128.
NEDD4L was also identified as a substrate for Akt in our protein microarray screen, and it has been suggested previously to be targeted by Akt.47 We performed kinase assays to compare phosphorylation levels of NEDD4L catalyzed by pSer473 Akt (A1) versus O-GlcNAcSer473 Akt (A8) and observed that A1 is ~1.6× more efficient at phosphorylating NEDD4L than A8 (Figure S17), consistent with the mild selectivity observed with the protein microarray results. To identify the sites of NEDD4L phosphorylation, we employed mutagenesis and site-specific phospho-NEDD4L antibodies using pSer473 Akt (A1). We observed that pSer473 Akt (A1) phosphorylates NEDD4L at both Ser342 and Ser448 (Figure 5A). To analyze the potential of Akt to phosphorylate NEDD4L in cells, we used HCT116 Akt1 / 2 knockout (KO) cells transfected with either WT or D274A (catalytically dead) Akt46 and NEDD4L C942S (Figure 5B). The results of these experiments suggest that WT Akt could phosphorylate NEDD4L at S342 and S448 in this cell line (Figure 5B,C) and agrees with previous reports of NEDD4L phosphorylation by Akt at S342.47 Although other reports suggest that S448 in NEDD4L is phosphorylated exclusively by Sgk,48 Akt and Sgk are close relatives within the AGC family of kinases. Our cell-based assays suggest that WT Akt can phosphorylate NEDD4L at S342 and S448. Substrate selectivity of Sgk and Akt may depend on factors such as subcellular localization and other levels of regulation because NEDD4L is suggested to promote ubiquitination and degradation of Sgk but not Akt.49
Figure 5.

Akt phosphorylates NEDD4L (A) Western blot analysis of kinase assays using 100 nM pSer473 Akt (A1) and 1 μM NEDD4L. Assays were carried out using 2 mM ATP at 30 °C for 20 min 200 ng of NEDD4L was loaded per well. Membranes were blotted with the phospho Akt substrate antibody (R/KXR/KXXS*/T*) (1:1000), total Akt antibody (1:5000), NEDD4L, and phospho S342 and phospho S448 (1:2000). (B) Western blot analysis of HCT116 Akt1/2 KO cells transfected with either WT (left) or D274A (catalytically dead, right) Akt and NEDD4L (C942S) showing three different replicates. Anti-Akt(1:2000), pS342 NEDD4L (1:2000), NEDD4L (1:2000), pS448 NEDD4L (1:3000), and anti-GAPDH (1:5000). (C) Bar plot represents quantification of band intensity of WB for phospho NEDD4L forms normalized to total NEDD4L for each band. Data represent four biological replicates with n = 3 for each. P-values are 0.0037 and 0.0063, respectively.
SUMMARY
Our studies extend the EPL technique to introduce a wider range of PTMs into Akt. This method overcomes the intrinsic challenges of accessing such complex materials for biochemical analysis. We hope that this study will encourage future applications of EPL to study the precise impact of PTMs in proteins and complement more classical genetic approaches. With the various semisynthetic Akt forms produced here, we made the unexpected observations that O-GlcNAcylation at Ser473 and phosphorylation at Tyr474 can partially activate Akt kinase activity toward peptide and protein substrates. It is possible that these two PTMs can influence each other in regulating Akt, and this should be investigated in future studies. We have also used protein microarrays to study the pattern of protein phosphorylation by O-GlcNAcSer473 Akt (A8) versus pSer473 Akt (A1) and observed a high degree of overlap in phosphorylated substrates. It will be interesting to apply protein microarrays to future studies and see if substrate selectivity holds true with a wider range of Akt forms. Two proteins identified using the microarrays, NEDD4L and PPM1H, were further validated as likely Akt substrates using solution-phase assays and cell transfection experiments. To the best of our knowledge, PPM1H has not been previously identified as an Akt substrate. Moreover, although NEDD4L has been suggested to be phosphorylated by Akt in prior work, here, we obtain evidence that Ser448 of NEDD4L may be a novel Akt phosphorylation site. These findings can pave the way to delineating novel cellular functions of Akt in signaling.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Alessi for PPM1H plasmids, D. Kim for PRAS40 plasmid, and Thermo Fisher Scientific’s Aspire Program for RL2 antibody. P.A.C. and A.S. are grateful for funding from NIH grant CA74305 and the corresponding diversity supplement. N.C. thanks NCI for funding support from K22CA241105. B.A.P. is supported by an NIH postdoctoral fellowship (F32 CA259214). This work was partially funded by R01 GM136148 (S.B.G.) and R01 GM114537 (M.R.P.). The Q-Exactive mass spectrometer used here was obtained with NIH grant GM62437S1.
Footnotes
The authors declare the following competing financial interest(s): Heng Zhu is a cofounder and equity holder in the company CDI, which fabricated the protein microarrays used here.Philip Cole has been a consultant for Scorpion Therapeutics.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.1c00632
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00632.
MALDI or ESI spectra for peptides, Coomassie-stained gels and intact mass analysis for all semisynthetic proteins, pT450 antibody validation, western blot images of various kinases assays with PPM1H and PRAS40 and protein microarray list of common and selective hits, Plasmids, additional experimental methods describing peptide synthesis, expression, and purification for all the proteins utilized, and reaction conditions for kinase assays (PDF)
Contributor Information
Antonieta L. Salguero, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, United States; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States
Maggie Chen, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, United States; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
Aaron T. Balana, Department of Chemistry, University of Southern California, Los Angeles, California 90089, United States
Nam Chu, Department of Cancer Biology and Genetics, and the Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio 43210, United States.
Hanjie Jiang, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, United States; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States.
Brad A. Palanski, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, United States; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States
Hwan Bae, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, United States; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Katharine M. Wright, Department of Biophysics and Biophysical Chemistry, The Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States
Sara Nathan, Department of Biophysics and Biophysical Chemistry, The Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States.
Heng Zhu, Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States; The Center for High-Throughput Biology, The Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, United States.
Sandra B. Gabelli, Department of Biophysics and Biophysical Chemistry, The Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States; Department of Medicine, The Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, United States; Department of Oncology, The Johns Hopkins University School of Medicine, Baltimore, Maryland 21287, United States.
Matthew R. Pratt, Departments of Chemistry and Biological Sciences, University of Southern California, Los Angeles, California 90089, United States.
Philip A. Cole, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, United States; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, Baltimore, Maryland 21205, United States.
REFERENCES
- (1).Liao Y; Hung M-C Physiological Regulation of Akt Activity and Stability. Am. J. Transl. Res. 2010, 2,19–42. [PMC free article] [PubMed] [Google Scholar]
- (2).Manning BD; Toker A AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cross DAE; Alessi DR; Cohen P; Andjelkovich M; Hemmings BA Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase B. Nature 1995, 378, 785–789. [DOI] [PubMed] [Google Scholar]
- (4).Alessi DR; Barry Caudwell F; Andjelkovic M; Hemmings BA; Cohen P Molecular Basis for the Substrate Specificity of Protein Kinase B; Comparison with MAPKAP Kinase-1 and P70 S6 Kinase. FEBS Lett. 1996, 399, 333–338. [DOI] [PubMed] [Google Scholar]
- (5).Ferguson FM; Gray NS Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discovery 2018, 17, 353–377. [DOI] [PubMed] [Google Scholar]
- (6).Song M; Bode AM; Dong Z; Lee M-H AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [DOI] [PubMed] [Google Scholar]
- (7).Kostaras E; Kaserer T; Lazaro G; Heuss SF; Hussain A; Casado P; Hayes A; Yandim C; Palaskas N; Yu Y; et al. A Systematic Molecular and Pharmacologic Evaluation of AKT Inhibitors Reveals New Insight into Their Biological Activity. Br. J. Cancer 2020, 123, 542–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kannan N; Haste N; Taylor SS; Neuwald AF The Hallmark of AGC Kinase Functional Divergence Is Its C-Terminal Tail, a Cis-Acting Regulatory Module. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cole PA; Chu N; Salguero AL; Bae H AKTivation Mechanisms. Curr. Opin. Struct. Biol. 2019, 59,47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).James SR; Downes CP; Gigg R; Grove SJ; Holmes AB; Alessi DR Specific Binding of the Akt-1 Protein Kinase to Phosphatidylinositol 3,4,5-Trisphosphate without Subsequent Activation. Biochem. J. 1996, 315, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Alessi DR; James SR; Downes CP; Holmes AB; Gaffney PRJ; Reese CB; Cohen P Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Bα. Curr. Biol. 1997, 7, 261–269. [DOI] [PubMed] [Google Scholar]
- (12).Alessi DR; Andjelkovic M; Caudwell B; Cron P; Morrice N; Cohen P; Hemmings BA Mechanism of Activation of Protein Kinase B by Insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- (13).Sarbassov DD; Guertin DA; Ali SM; Sabatini DM Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- (14).Calleja V; Laguerre M; Parker PJ; Larijani B Role of a Novel PH-Kinase Domain Interface in PKB/Akt Regulation: Structural Mechanism for Allosteric Inhibition. PLoS Biol. 2009, 7, No. e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Calleja V; Alcor D; Laguerre M; Park J; Vojnovic B; Hemmings BA; Downward J; Parker PJ; Larijani B Intramolecular and Intermolecular Interactions of Protein Kinase B Define Its Activation In Vivo. PLoS Biol. 2007, 5, No. e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kang E-S;Han D;Park J;Kwak TK;Oh M-A;Lee S-A; Choi S; Park ZY; Kim Y; Lee JW O-GlcNAc Modulation at Akt1 Ser473 Correlates with Apoptosis of Murine Pancreatic μ Cells. Exp. Cell Res. 2008, 314, 2238–2248. [DOI] [PubMed] [Google Scholar]
- (17).Conus NM; Hannan KM; Cristiano BE; Hemmings BA; Pearson RB Direct Identification of Tyrosine 474 as a Regulatory Phosphorylation Site for the Akt Protein Kinase. J. Biol. Chem. 2002, 277, 38021–38028. [DOI] [PubMed] [Google Scholar]
- (18).Risso G; Blaustein M; Pozzi B; Mammi P; Srebrow A Akt/PKB: One Kinase, Many Modifications. Biochem. J. 2015, 468, 203–214. [DOI] [PubMed] [Google Scholar]
- (19).Chu N; Salguero AL; Liu AZ; Chen Z; Dempsey DR; Ficarro SB; Alexander WM; Marto JA; Li Y; Amzel LM; et al. Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis. Cell 2018, 174, 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu P; Begley M; Michowski W; Inuzuka H; Ginzberg M; Gao D; Tsou P; Gan W; Papa A; Kim BM; et al. Cell-Cycle-Regulated Activation of Akt Kinase by Phosphorylation at Its Carboxyl Terminus. Nature 2014, 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Heath JM; Sun Y; Yuan K; Bradley WE; Litovsky S; Dell’Italia LJ; Chatham JC; Wu H; Chen Y Activation of AKT by O-Linked N-Acetylglucosamine Induces Vascular Calcification in Diabetes Mellitus. Circ. Res. 2014, 114, 1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shi J; Gu J.-h.; Dai C.-l.; Gu J; Jin X; Sun J; Iqbal K; Liu F; Gong C-X O-GlcNAcylation Regulates Ischemia-Induced Neuronal Apoptosis through AKT Signaling. Sci. Rep. 2015, 5, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shi J; Wu S; Dai C-L; Li Y; Grundke-Iqbal I; Iqbal K; Liu F; Gong C-X Diverse Regulation of AKT and GSK-3/β by O-GlcNAcylation in Various Types of Cells. FEBS Lett. 2012, 586,2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Muir TW; Sondhi D; Cole PA Expressed Protein Ligation: A General Method for Protein Engineering. Proc. Natl. Acad. Sci. U.SA. 1998, 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tarrant MK; Rlio H-S; Xie Z; Jiang YL; Gross C; Culhane JC; Yan G; Qian J; Ichikawa Yj Matsuoka Tj et al. Regulation of CK2 by Phosphorylation and O-GlcNAcylation Revealed by Semisynthesis. Nat. Chem. Biol. 2012, 8, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Balana AT; Levine PM; Craven TW; Mukherjee S; Pedowitz NJ; Moon SP; Takahashi TT; Becker CFW; Baker D; Pratt M R O-GlcNAc Modification of Small Heat Shock Proteins Enhances Their Anti-Amyloid Chaperone Activity. Nat. Chem. 2021, 13, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chu N; Viennet T; Bae H; Salguero A; Boeszoermenyi A; Arthanari H; Cole PA The Structural Determinants of PH DomainMediated Regulation of Akt Revealed by Segmental Labeling. eLife 2020, 9, No. e59151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bolduc D; Rahdar M; Tu-Sekine B; Sivakumaren SC; Raben D; Amzel LM; Devreotes P; Gabelli SB; Cole P Phosphorylation-Mediated PTEN Conformational Closure and Deactivation Revealed with Protein Semisynthesis. eLife 2013, 2, No. e00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ericson K; Gan C; Cheong I; Rago C; Samuels Y; Velculescu VE; Kinzler KW; Huso DL; Vogelstein B; Papadopoulos N Genetic Inactivation of AKT 1, AKT2, and PDPK1 in Human Colorectal Cancer Cells Clarifies Their Roles in Tumor Growth Regulation. Proc. Natl. Acad. Sci. U.S.A. 2010,107,2S98–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Marty MT; Baldwin AJ; Marklund EG; Hochberg GKA; Benesch JLP; Robinson CV Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles. Anal. Chem. 2015, 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Levine PM; Galesic A; Balana AT; Mahul-Mellier AL; Navarro MX; De Leon CA; Lashuel HA; Pratt M R α-Synuclein O-GlcNAcylation Alters Aggregation and Toxicity, Revealing Certain Residues as Potential Inhibitors of Parkinson’s Disease. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lučić I; Rathinaswamy MK; Truebestein L; Hamelin DJ; Burke JE; Leonard TA Conformational Sampling of Membranes by Akt Controls Its Activation and Inactivation. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E3940–E3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Facchinetti V; Ouyang W; Wei H; Soto N; Lazorchak A; Gould C; Lowry C; Newton AC; Mao Y; Miao RQ; et al. The Mammalian Target of Rapamycin Complex 2 Controls Folding and Stability of Akt and Protein Kinase C. EMBO J. 2008, 27, 1932–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Truebestein L; Hornegger H; Anrather D; Hard M; Fleming KD; Stariha JTB; Pardon E; Steyaert J; Burke JE; Leonard TA Structure of Autoinhibited Akt1 Reveals Mechanism of PIP3Mediated Activation. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, No. e2101496118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liu K; Paterson AJ; Chin E; Kudlow JE Glucose Stimulates Protein Modification by O-Linked GlcNAc in Pancreatic Beta Cells: Linkage of O-Linked GlcNAc to Beta Cell Death. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 2820–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yang J; Cron P; Good VM; Thompson V; Hemmings BA; Barford D Crystal Structure of an Activated Akt/Protein Kinase B Ternary Complex with GSK3-Peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9, 940–944. [DOI] [PubMed] [Google Scholar]
- (37).Yang X; Ongusaha PP; Miles PDj Havstad JC; Zhang F; So WV; Kudlow JE; Michell RH; Olefsky JM; Field SJ; et al. Phosphoinositide Signalling Links O -GlcNAc Transferase to Insulin Resistance. Nature 2008, 451, 964–969. [DOI] [PubMed] [Google Scholar]
- (38).Gribble FM; Loussouarn G; Tucker SJ; Zhao C; Nichols CG; Ashcroft FM A Novel Method for Measurement of Submembrane ATP Concentration. J. Biol. Chem. 2000, 275, 30046–30049. [DOI] [PubMed] [Google Scholar]
- (39).Syu G-D; Dunn J; Zhu H Developments and Applications of Functional Protein Microarrays. Mol. Cell. Proteomics 2020, 19, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Berndsen K; Lis P; Yeshaw WM; Wawro PS; Nirujogi RS; Wightman M; Macartney T; Dorward M; Knebel A; Tonelli F; et al. PPM1H Phosphatase Counteracts LRRK2 Signaling by Selectively Dephosphorylating Rab Proteins. eLife 2019, 8, No. e50416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Manning JA; Shah SS; Nikolic A; Henshall TL; KhewGoodall Y; Kumar S The Ubiquitin Ligase NEDD4–2/NEDD4L Regulates Both Sodium Homeostasis and Fibrotic Signaling to Prevent End-Stage Renal Disease. Cell Death Discovery 2021, 12, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cui Y; Wang Y; Song X; Ning H; Zhang Y; Teng Y; Wang J; Yang X Brain Endothelial PTEN/AKT/NEDD4–2/MFSD2A Axis Regulates Blood-Brain Barrier Permeability. Cell Rep. 2021,36,109327. [DOI] [PubMed] [Google Scholar]
- (43).Ronzaud C; Loffing-Cueni D; Hausel P; Debonneville A; Malsure SR; Fowler-Jaeger N; Boase NA; Perrier R; Maillard M; Yang B; et al. Renal Tubular NEDD4–2 Deficiency Causes NCCMediated Salt-Dependent Hypertension. J. Clin. Invest. 2013, 123, 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chen MJ; Dixon JE; Manning G Genomics and Evolution of Protein Phosphatases. Sci. Signaling 2017, 10, No. eaagl796. [DOI] [PubMed] [Google Scholar]
- (45).Brognard J; Sierecki E; Gao T; Newton AC PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [DOI] [PubMed] [Google Scholar]
- (46).Ebner M; Lučić I; Leonard TA; Yudushkin I PI(3,4,5)P3 Engagement Restricts Akt Activity to Cellular Membranes. Mol. Cell 2017, 65, 416–431. [DOI] [PubMed] [Google Scholar]
- (47).Lee I-H; Dinudom A; Sanchez-Perez A; Kumar S; Cook DI Akt Mediates the Effect of Insulin on Epithelial Sodium Channels by Inhibiting Nedd4–2. J. Biol. Chem. 2007, 282, 29866–29873. [DOI] [PubMed] [Google Scholar]
- (48).Debonneville C; Flores SY; Kamynina E; Plant PJ; Tauxe C; Thomas MA; Münster C; Chraïbi A; Pratt JH; Horisberger J-D; et al. Phosphorylation of Nedd4–2 by Sgkl Regulates Epithelial Na+ Channel Cell Surface Expression. EMBO J. 2001, 20, 7052–7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhou R; Snyder PM Nedd4–2 Phosphorylation Induces Serum and Glucocorticoid-Regulated Kinase (SGK) Ubiquitination and Degradation. J. Biol. Chem. 2005, 280, 4518–4523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.