

Abstract
Egg laying in the nematode worm Caenorhabditis elegans is a two-state behavior modulated by internal and external sensory input. We have previously shown that homeostatic feedback of embryo accumulation in the uterus regulates bursting activity of the serotonergic HSN command neurons that sustains the egg-laying active state. How sensory feedback of egg release signals to terminate the egg-laying active state is less understood. We find that Gαo, a conserved Pertussis Toxin-sensitive G protein, signals within HSN to inhibit egg-laying circuit activity and prevent entry into the active state. Gαo signaling hyperpolarizes HSN, reducing HSN Ca2+ activity and input onto the postsynaptic vulval muscles. Loss of inhibitory Gαo signaling uncouples presynaptic HSN activity from a postsynaptic, stretch-dependent homeostat, causing precocious entry into the egg-laying active state when only a few eggs are present in the uterus. Feedback of vulval opening and egg release activates the uv1 neuroendocrine cells which release NLP-7 neuropeptides which signal to inhibit egg laying through Gαo-independent mechanisms in the HSNs and Gαo-dependent mechanisms in cells other than the HSNs. Thus, neuropeptide and inhibitory Gαo signaling maintain a bi-stable state of electrical excitability that dynamically controls circuit activity in response to both external and internal sensory input to drive a two-state behavior output.
Keywords: GPCR, G protein, calcium, circuit, C. elegans, serotonin, neuropeptide, neuron, muscle, behavior
Introduction
A major goal of neuroscience is to understand how sensory signals control neural circuit activity and changes in animal behavior. Such sensory feedback informs when a behavior should begin, how long it should continue, and when it should end. Extensive evidence has shown that neuromodulators like serotonin and neuropeptides signal through G protein coupled receptors to remodel neural circuit activity and drive behavior state transitions (Jiang et al. 2001; Goulding et al. 2008; Taghert and Nitabach 2012; Oikonomou et al. 2019). Yet, there is no neural circuit in any organism for which we know precisely how each signaling event contributes to sensory modulation of a behavior. Small neural circuits typically found in invertebrates combine anatomical simplicity with genetic and experimental accessibility, allowing for a complete understanding of the molecular and physiological basis for a behavioral output (Marder 2012).
The Caenorhabditis elegans female egg-laying behavior circuit is ideally suited to study how sensory signals modulate circuit functions that underlie decision-making. As shown in Figure 1A, the circuit is comprised of two hermaphrodite specific neurons (HSNs) that synapse onto the egg-laying vulval muscles (White et al. 1986; Shen et al. 2004; Li et al. 2013). During ∼2 minute active states, rhythmic HSN Ca2+ activity releases serotonin and neuropeptides that signal to promote the excitability of the muscles, driving ejection of ∼4–6 eggs in sequence from the uterus into the environment (Waggoner et al. 1998; Shyn et al. 2003; Zhang et al. 2008; Collins et al. 2016; Brewer et al. 2019). External and internal sensory inputs regulate the onset of egg laying (Horvitz et al. 1982; Trent 1982; Sawin 1996; Aprison and Ruvinsky 2014; Ravi et al. 2018a), and genetic studies have identified neuropeptides, receptors, and two antagonistic heterotrimeric G proteins that signal to regulate HSN activity, neurotransmitter release, and egg laying (Schafer 2006; Ringstad and Horvitz 2008; Koelle 2018; Banerjee et al. 2017; Fernandez et al. 2020). Gαq signals through the conserved PLCβ and Trio RhoGEF effector pathways to promote neurotransmitter release and egg laying (Brundage et al. 1996; Lackner et al. 1999; Miller et al. 1999; Bastiani et al. 2003; McMullan et al. 2006; Williams et al. 2007; McMullan and Nurrish 2011). Because phorbol ester DAG-mimetics such as PMA rescue synaptic transmission defects of Gαq signaling mutants (Lackner et al. 1999; Williams et al. 2007), DAG production and recruitment of UNC-13 and/or protein kinase C effectors are thought to mediate the Gαq dependent modulation of synaptic transmission (Yawo 1999; Wierda et al. 2007; Lou et al. 2008). Gαq signaling is opposed by Gαo which signals to inhibit neurotransmitter release (Koelle and Horvitz 1996; Miller et al. 1999; Nurrish et al. 1999), vulval muscle activity (Shyn et al. 2003), and egg laying (Mendel et al. 1995; Segalat et al. 1995). How Gαo signaling antagonizes Gαq, Rho, and DAG signaling is not clear. Even though Gαo mediates signaling by numerous neurotransmitters in diverse animals including mammals (Jiang et al. 2001), direct effectors for Gαo have not yet been identified. In C. elegans, Gαo mutants resemble animals with too much Gαq signaling showing increased UNC-13 localization to synapses and hyperactive egg-laying behavior defects, resembling treatment with phorbol esters (Miller et al. 1999; Nurrish et al. 1999; Jose and Koelle 2005). Because synaptic transmission could be modulated by upstream changes in cell and circuit electrical excitability and/or by downstream effects on DAG that potentiate synaptic vesicle fusion, what is needed are direct measurements of how discrete changes in G protein and effector signaling affect cell and circuit activity and their consequences on animal behavior.
Figure 1.

Gαo signaling maintains the inactive egg-laying behavior state. (A) Cartoon of how identified and unidentified neuropeptides released from AWC (Fenk and de Bono 2015), BAG (Ringstad and Horvitz 2008), uv1 (Banerjee et al. 2017), and other sensory cells bind to G-protein coupled receptors expressed on HSN command neurons (green) which signal via Gαo (red) or Gαq (black) effector pathways to modulate serotonin and NLP-3 neuropeptide release (Tanis et al. 2008; Brewer et al. 2019). The egg-laying vulval muscles (orange) express receptors for serotonin (Carnell et al. 2005; Dempsey et al. 2005; Hobson et al. 2006; Fernandez et al. 2020) and possibly NLP-3 which signal to promote vulval muscle excitability and egg laying (B) Scatter plots of the first egg-laying event in wildtype (grey), null goa-1(sa734) mutants (orange), hypomorphic loss-of-function goa-1(n1134) mutants (red), egl-10(md176) null mutants (purple), and transgenic animals expressing Pertussis Toxin (blue) or GOA-1Q205L in the HSNs (pink). Error bars show 95% confidence intervals for the mean from ≥10 animals. Asterisks indicate P≤0.0001 (one-way ANOVA with Bonferroni correction for multiple comparisons). (C) Representative raster plots showing temporal pattern of egg laying during three hours in wild-type (black), hypomorphic loss-of-function goa-1(n1134) mutant (red), and egl-10(md176) null mutant animals (purple), along with transgenic animals expressing Pertussis Toxin (blue) or GOA-1Q205L in the HSNs (pink). Vertical lines indicate single egg-laying events. (D) Scatter plots of egg accumulation in the uterus in wild-type animals (grey), transgenic animals expressing Pertussis Toxin in the HSNs (blue), transgenic animals expressing of GOA-1Q205L in HSNs from the tph-1 promoter (HSN:: GOA-1Q205L, +; red), or in animals that had subsequently lost the GOA-1Q205L-expressing transgenes (HSN:: GOA-1Q205L, –; red open circles). Error bars indicate 95% confidence intervals for the mean. Asterisks indicate P < 0.0001; n.s. indicates P = 0.2567 (one-way ANOVA with Bonferroni’s correction; n ≥ 30 animals per genotype with five independent transgenic and nontransgenic lines analyzed).
Here, we explore how Gαo inhibits C. elegans egg-laying circuit activity and behavior. Our data reveal that Gαo signaling reduces the electrical excitability of the HSN command neurons to promote the inactive state while eggs are produced. Feedback of egg accumulation in the uterus then switches the circuit into active states where rhythmic circuit activity drives sequential egg-laying events. Thus, modulation by inhibitory Gαo signaling allows for proper induction of motor behavior circuit upon the alignment of optimal external and internal sensory conditions.
Materials and methods
Nematode culture and developmental staging
Caenorhabditis elegans hermaphrodites were maintained at 20°C on nematode growth medium (NGM) agar plates with Escherichia coli OP50 as a source of food as described (Brenner 1974). For assays involving young adults, animals were age-matched based on the timing of completion of the L4 larval molt. All assays involving adult animals were performed using age-matched adult hermaphrodites 20–40 hours past the late L4 stage. Table 1 lists all strains used in this study and their genotypes.
Table 1.
Strain names and genotypes for all animals used in this study (behavior assays and calcium imaging)
| Strain | Feature | Genotype | Reference |
|---|---|---|---|
| N2 | Bristol wild-type strain | Wild type | Brenner (1974) |
| DA521 | EGL-4 (protein kinase G) gain-of-function mutant. Hyperactive egg laying, sluggish locomotion | egl-4(ad805) IV | Raizen et al. (2006) |
| DG1856 | GOA-1 (Gαo) null mutant, hyperactive egg laying | goa-1(sa734) I | Robatzek and Thomas (2000) |
| IZ1236 | NLP-7 overexpression (nlp-7 promoter) | ufIs118 | Banerjee et al. (2017) |
| KG421 | GSA-1 (Gαs) gain-of-function mutant. Hyperactive locomotion | gsa-1(ce81) I | Schade et al. (2005) |
| KG518 | ACY-1 (Adenylate Cyclase) gain-of-function mutant. Hyperactive locomotion | acy-1(ce2) III | Schade et al. (2005) |
| KG532 | KIN-2 (protein kinase A inhibitory regulatory subunit) loss-of-function mutant. Hypersensitive to stimuli | kin-2(ce179) X | Schade et al. (2005) |
| KG744 | PDE-4 (phosphodiesterase) loss-of-function mutant, Hyperactive locomotion | pde-4(ce268) II | Charlie et al. (2006a) |
| LX75 | egl-10(md176) mutant strain for transgene production, multi-vulva at 20 °C in the absence of lin-15(+) rescue transgene | egl-10(md176) V; lin-15(n765ts) X | Patikoglou and Koelle (2002) |
| LX755 | HSN and NSM expressing GFP + EGL-10 (tph-1 promoter) in egl-10 mutant background. Less egg-laying defective than LX764 | egl-10(md176) V; lin-15(n765ts) X vsEx437 | This study |
| LX764 | HSN and NSM expressing GFP only (tph-1 promoter) in egl-10 mutant background. Egg-laying defective | egl-10(md176) V; lin-15(n765ts) X vsEx439 | This study |
| LX849 | HSN and NSM activated GOA-1(Q205L), tph-1 promoter, egg laying defective | vsIs49; lin-15(n765ts) X | Tanis et al. (2008) |
| LX850 | HSN and NSM Pertussis Toxin, tph-1 promoter, hyperactive egg laying | vsIs50; lin-15(n765ts) X | Tanis et al. (2008) |
| LX1832 | Strain for transgene production, blue-light insensitive, multi-vulva at 20°C in the absence of lin-15(+) rescue transgene. | lite-1(ce314) lin-15(n765ts) X | Gurel et al. (2012) |
| LX1918 | Vulval muscle GCaMP5, mCherry (unc-103e promoter, unc-54 3’ UTR) | vsIs164 lite-1(ce314) lin-15(n765ts) X | Collins et al. (2016) |
| LX1919 | Vulval muscle GCaMP5, mCherry (unc-103e promoter, unc-54 3’ UTR) | vsIs165; lite-1(ce314) lin-15(n765ts) X | Collins et al. (2016) |
| LX2004 | HSN GCaMP5, mCherry (nlp-3 promoter, nlp-3 3’UTR) | vsIs183 lite-1(ce314) lin-15(n765ts) X | Collins et al. (2016) |
| LX2007 | HSN GCaMP5, mCherry (nlp-3 promoter, nlp-3 3’UTR) | vsIs186 lite-1(ce314) lin-15(n765ts) X | Collins et al. (2016) |
| MIA26 | No HSNs | egl-1(n986dm) V | Garcia and Collins (2019) |
| MIA36 | EGL-4 (protein kinase G) gain-of-function mutant. Hyperactive egg laying, sluggish locomotion | egl-4(mg410) IV | Hao et al. (2011) |
| MIA123 | No HSNs in lite-1, lin-15 background for crossing with transgenes | egl-1(n986dm) V; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA144 | VC Tetanus Toxin (minimal lin-11 promoter enhancer, unc-54 3’UTR) | keyIs33; lite-1(ce314) lin-15(n765ts) X | Kopchock et al. (2021) |
| MIA210 | GOA-1 (Gαo) reduced-function mutant; HSN GCaMP, mCherry | goa-1(n1134) I; vsIs183 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA214 | GOA-1 (Gαo) reduced-function mutant; vulval muscle GCaMP5, mCherry | goa-1(n1134) I; vsIs164 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA216 | Increased Gαo signaling; HSN GCaMP5, mCherry | egl-10(md176) V; vsIs183 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA218 | HSN and NSM Pertussis Toxin in blue-light insensitive, lin-15 multi-vulva background | vsIs50 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA227 | HSN and NSM Pertussis Toxin (tph-1 promoter); HSN GCaMP5, mCherry (nlp-3 promoter) | vsIs186; vsIs50 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA245 | HSN and NSM Pertussis Toxin (tph-1 promoter); vulval muscle GCaMP5, mCherry (unc-103e promoter) | vsIs50 X vsIs165; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA256 | Vulval muscle mCherry (ceh-24 promoter, unc-54 3’UTR) | keyEx45; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA257 | Vulval muscle mCherry + Pertussis Toxin (ceh-24 promoter, unc-54 3’UTR) | keyEx46; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA258 | Vulval muscle mCherry + Activated GOA-1(Q205L) (ceh-24 promoter, unc-54 3’UTR) | keyEx47; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA259 | uv1 cells mCherry (tdc-1 promoter, ocr-2 3’ UTR) | keyEx48; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA260 | uv1 cells mCherry + Pertussis Toxin (tdc-1 promoter, ocr-2 3’ UTR) | keyEx49; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA261 | uv1 cells mCherry + Activated GOA-1(Q205L) (tdc-1 promoter, ocr-2 3’ UTR) | keyEx50; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA262 | Vulval muscle mCherry + Tetanus toxin (ceh-24 promoter, unc-54 3’UTR) | keyEx51; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA263 | GOA-1 (Gαo) null mutant; HSN GCaMP5, mCherry | goa-1(sa734) I; vsIs183 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA277 | Increased Gαo signaling in HSN; HSN GCaMP5, mCherry | vsIs49/+, +/vsIs183 lite-1(ce314) lin-15(n765ts) X (trans-heterozygote) | This study |
| MIA278 | HSN/NSM gpb-1 and gpc-2 overexpression + GFP (tph-1 promoter) | keyEx52; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA279 | HSN/NSM empty + GFP (tph-1 promoter) | keyEx53; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA282 | PDE-2 (phosphodiesterase) loss-of-function mutant | pde-2(qj6) | Fujiwara et al. (2015) |
| MIA291 | Increased Gαo signaling in HSN (tph-1 promoter); vulval muscle GCaMP5, mCherry (unc-103e promoter) | vsIs49; vsIs165 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA293 | NLP-7 overexpression (nlp-7 promoter); HSN GCaMP5, mCherry (nlp-3 promoter) | ufIs118; vsIs183 lite-1(ce314) lin-15(n765ts) X | Banerjee et al. (2017) and This study |
| MIA294 | NLP-7 overexpression (nlp-7 promoter); goa-1(Gαo) null mutant; HSN GCaMP5, mCherry (nlp-3 promoter) | ufIs118; goa-1(sa734) I; vsIs183 lite-1(ce314) lin-15(n765ts) X | Banerjee et al. (2017) and This stud |
| MIA295 | GOA-1 (Gαo) mutant, vulval muscle GCaMP5, mCherry (unc-103e promoter) | goa-1(sa734) I; vsIs164 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA331 | GOA-1 (Gαo) null mutant; EGL-4 (protein kinase G) null mutant | goa-1(sa734) I; egl-4(n479) IV | This study |
| MIA345 | GOA-1 (Gαo) null mutant; no HSNs | goa-1(sa734) I; egl-1(n986dm) V | This study |
| MIA359 | EGL-47 null mutant; HSN GCaMP5, mCherry (nlp-3 promoter) | egl-47(ok677) V; vsIs183 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA360 | NLP-7 overexpression (nlp-7 promoter); HSN and NSM Pertussis Toxin (tph-1 promoter); HSN GCaMP5, mCherry (nlp-3 promoter) | ufIs118; vsIs50; vsIs186 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA366 | NLP-7 overexpression [nlp-7 promoter; EGL-47 null mutant; HSN GCaMP5, mCherry (nlp-3 promoter)] | ufIs118; egl-47(ok677) V; vsIs183 lite-1(ce314) lin-15(n765ts) X | This study |
| MIA369 | No HSNs; HSN and NSM Pertussis Toxin (tph-1 promoter); HSN GCaMP5, mCherry (nlp-3 promoter) | egl-1(n986dm) V; vsIs50; vsIs186 lite-1(ce314) lin-15 (n765ts) X | This study |
| MIA370 | VC Tetanus Toxin (lin-11 enhancer/promoter); HSN and NSM Pertussis Toxin (tph-1 promoter); HSN GCaMP5, mCherry (nlp-3 promoter) | keyIs33; vsIs186; vsIs50 keyIs33; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA371 | GOA-1 (Gαo) null mutant; VC Tetanus Toxin (lin-11 enhancer/promoter) | goa-1(sa734) I; keyIs33; lite-1(ce314) lin-15(n765ts) X | This study |
| MIA425 | HSN Pertussis Toxin rescued with HSN GOA-1Q205L (tph-1 promoter) | keyEx82; vsIs50; lin-15(n765ts) | This study |
| MT1073 | EGL-4 (protein kinase G) loss-of-function mutant. Egg-laying defective, roaming locomotion | egl-4(n478) IV | L'Etoile et al. (2002) |
| MT1074 | EGL-4 (protein kinase G) null mutant. Egg-laying defective, roaming locomotion | egl-4(n479) IV | L'Etoile et al. (2002) |
| MT2426 | GOA-1 (Gαo) reduced-function mutant, hyperactive egg laying | goa-1(n1134) I | Segalat et al. (1995) |
| MT8504 | Increased Gαo signaling due to mutation in RGS protein, EGL-10 | egl-10(md176) V | Koelle and Horvitz (1996) |
| RB850 | EGL-47 null mutant | egl-47(ok677) V | Moresco and Koelle (2004) and C. elegans Deletion Mutant Consortium (2012) |
Plasmid and strain construction
Calcium reporter transgenes
HSN Ca2+
HSN Ca2+ activity was visualized using LX2004 vsIs183 [nlp-3::GCaMP5::nlp-3 3'UTR + nlp-3::mCherry::nlp-3 3'UTR + lin-15(+)] lite-1(ce314) lin-15(n765ts) X strain expressing GCaMP5G and mCherry from the nlp-3 promoter as previously described (Collins et al. 2016). To visualize HSN Ca2+ activity in Gαo signaling mutants, we crossed LX2004 vsIs183 lite-1(ce314) lin-15(n765ts) X males with MT2426 goa-1(n1134) I, DG1856 goa-1(sa734) I, or MT8504 egl-10(md176) V hermaphrodites, and the fluorescent cross-progeny were allowed to self, generating MIA210 goa-1(n1143) I; vsIs183 X lite-1(ce314) lin-15 (n765ts) X, MIA263 goa-1(sa734) I; vsIs183 X lite-1(ce314) lin-15 (n765ts) X, and MIA216 egl-10(md176) V; vsIs183 lite-1(ce314) lin-15(n765ts) X strains, respectively. To visualize how NLP-7 signals to inhibit HSN Ca2+ activity and egg laying, we crossed IZ1236 ufIs118 hermaphrodites (Banerjee et al. 2017) with LX2004 males. The fluorescent cross-progeny were selfed, generating MIA293 ufIs118; vsIs183 lite-1(ce314) lin-15(n765ts) X. To test the requirement for Gαo signaling in NLP-7 inhibition, we crossed N2 males into MIA263 hermaphrodites, and the goa-1(sa734)/+ I; vsIs183 lite-1(ce314) lin-15 (n765ts) X heterozygous males obtained were then crossed with MIA293 hermaphrodites to generate MIA294 ufIs118; goa-1(sa734) I; vsIs183 lite-1(ce314) lin-15(n765ts) X. In order to test whether EGL-47 was a potential receptor for NLP-7, we crossed LX2004 males into RB850 egl-47(ok677) V hermaphrodites. The fluorescent cross-progeny were allowed to self and homozygous egl-47(ok677) mutant animals were identified by duplex PCR genotyping with the following oligos: CGTCACTTTTCCGTTGCTCTCTCATG, GTAGCGGGAAAGATGGCAAGAAGTCG, and TCGGTGAAACTCATTGTGCTCATTGTGC, creating MIA359 egl-47(ok677) V; vsIs183 lite-1(ce314) lin-15(n765ts) X. Wild-type N2 males were then crossed to MIA359 hermaphrodites, and the male egl-47(ok677)/+ V; vsIs183 lite-1(ce314) lin15(n765ts) X cross progeny were then crossed with MIA293 hermaphrodites. The cross-progeny were selfed and genotyped for the egl-47(ok677) V deletion mutant, generating MIA366 ufIs118; egl-47(ok677) V; vsIs183 lite-1(ce314) lin-15(n765ts) X.
During the course of crossing the vsIs50 transgene expressing the catalytic subunit of Pertussis Toxin in the HSNs from the tph-1 promoter (Tanis et al. 2008), we noticed linkage to the X chromosome. As such, LX850 vsIs50 lin-15(n765ts) X males were crossed with LX1832 lite-1(ce314) lin-15(n765ts) X hermaphrodites, the nonMuv progeny were selfed, and homozygous lite-1(ce314) nonMuv animals were kept, generating the strain MIA218 vsIs50 lite-1(ce314) lin-15(n765ts) X. MIA218 males were then crossed with LX2007 vsIs186; lite-1(ce314) lin-15(n765ts) X; the cross-progeny were selfed, generating MIA227 vsIs186; vsIs50 lite-1(ce314) lin-15(n765ts) X. To test whether Gαo acts in HSN to mediate NLP-7 inhibition, we crossed wild-type N2 males into LX2007 vsIs186; lite-1(ce314) lin-15 (n765ts) X hermaphrodites, and the heterozygous vsIs186/+; lite-1(ce314) lin-15 (n765ts) X cross-progeny were mated with IZ1236 ufIs118 hermaphrodites. The fluorescent cross-progeny was selfed, creating MIA358 ufIs118; vsIs186 lite-1(ce314) lin-15(n765ts) X. Wild-type N2 males were then crossed into MIA227 hermaphrodites to generate heterozygous vsIs186/+; vsIs50 lite-1(ce314) lin-15(n765ts) X males which were then crossed into MIA358 hermaphrodites. The fluorescent cross-progeny were selfed, generating MIA360 ufIs118; vsIs50; vsIs186; lite-1(ce314) lin-15(n765ts) X. Heterozygous vsIs186/+; vsIs50 lite-1(ce314) lin-15(n765ts) X males were also crossed into MIA123 egl-1(n986dm) V; lite-1(ce314) lin-15(n765ts) X hermaphrodites, the fluorescent cross-progeny were selfed, and Egl nonMuv animals were selected, generating MIA369 egl-1(n986dm) V; vsIs50; vsIs186 lite-1(ce314) lin-15 (n765ts) X. The presence of the egl-1(n986dm) allele was confirmed with snip-SNP PCR genotyping using oligonucleotides CTCTGTTCCAGCTCAAATTTCC and GTAGTTGAGGATCTCGCTTCGGC followed by NciI digestion.
In order to visualize HSN Ca2+ activity in transgenic animals expressing a constitutively active mutant GOA-1Q205L protein that increases Gαo signaling in the HSN neurons, LX2004 vsIs183 lite-1(ce314) lin-15(n765ts) X males were crossed with LX849 vsIs49; lin-15(n765ts) X hermaphrodites. We noted linkage between the vsIs49 transgene and the X chromosome. As such, we selected a strain MIA277 with trans-heterozygous vsIs49 and vsIs183 transgenes [lite-1(ce314) vsIs49 X/lite-1(ce314) vsIs183 X] for Ca2+ imaging. The MIA277 strain was maintained by picking phenotypically egg-laying defective adult animals which show GCaMP5G/mCherry expression.
Vulval Muscle Ca2+
Vulval muscle Ca2+ activity was recorded in adult animals using LX1918 vsIs164 [unc-103e::GCaMP5::unc-54 3'UTR + unc-103e::mCherry::unc-54 3'UTR + lin-15(+)] lite-1(ce314) lin-15(n765ts) X strain as described (Collins et al. 2016). To visualize vulval muscle activity in Gαo signaling mutants, LX1918 males were crossed with MT2426 goa-1(n1134) I and DG1856 goa-1(sa734) I hermaphrodites, and the fluorescent cross-progeny were selfed, generating MIA214 goa-1(n1134) I; vsIs164 lite-1(ce314) lin-15(n765ts) X and MIA295 goa-1(sa734) I; vsIs164 lite-1(ce314) lin-15(n765ts) X strains, respectively. To visualize vulval muscle activity in transgenic animals expressing the catalytic subunit of Pertussis Toxin in the HSN neurons (Tanis et al. 2008), MIA218 vsIs50 lite-1(ce314) lin-15(n765ts) X males were crossed with LX1919 vsIs165 [unc-103e::GCaMP5::unc-54 3'UTR + unc-103e::mCherry::unc-54 3'UTR + lin-15(+)]; lite-1(ce314) lin-15(n765ts) X hermaphrodites, and the cross progeny were selfed, generating MIA245 vsIs50; vsIs165; lite-1(ce314) lin-15(n765ts) X. To visualize vulval muscle activity in transgenic animals expressing a constitutively active mutant GOA-1Q205L protein which increases Gαo signaling in the HSN neurons (Tanis et al. 2008), LX849 vsIs49; lin-15(n765ts) X males were crossed with LX1919 vsIs165; lite-1(ce314) lin-15(n765ts) X hermaphrodites and the fluorescent cross-progeny were selfed, generating MIA291 vsIs165; vsIs50 lite-1(ce314) lin-15(n765ts) X.
Transgenes used to manipulate Gαo signaling and synaptic transmission in specific cells of the egg-laying circuit
HSN neurons
To test for rescue of Pertussis Toxin behavior effects through expression of GOA-1, the HSN::PTX strain (LX850) was injected with pJM70C [tph-1::goa-1(Q205L); 80 ng/µl (Tanis et al. 2008)] along with pCFJ90 [myo-2::mCherry; 5 ng/µl (Frokjaer-Jensen et al. 2008)] and pKMC42 [tph-1::mCherry; 80 ng/µl]. Five independent transgenic lines expressing mCherry were used for behavioral assays. Siblings that had lost the transgenic array were used as controls. One transgenic line, MIA425 [keyEx82; vsIs50; lin-15(n765ts)], was kept. To produce a HSN (and NSM)-specific GPB-1 expressing construct, the gpb-1 cDNA fragment was amplified from pDEST-gpb-1 (Yamada et al. 2009) using the following oligonucleotides: 5′-GAGGCTAGCGTAGAAAAAATGAGCGAACTTGACCAACTTCGA-3′ and 5′-GCGGGTACCTCATTAATTCCAGATCTTGAGGAACGAG-3′. The ∼1 kb DNA fragment was digested with NheI/KpnI and ligated into pJT40A (Tanis et al. 2008) to generate pBR30. To produce an HSN (and NSM)-specific GPC-2 expressing construct, the gpc-2 cDNA fragment was amplified from worm genomic DNA using the following forward and reverse oligonucleotides: 5′-GAGGCTAGCGTAGAAAAAATGGATAAATCTGACATGCAACGA-3′ and 5′-GCGGGTACCTTAGAGCATGCTGCACTTGCT-3′. The ∼250 bp DNA fragment was digested with NheI/KpnI and ligated into pJT40A to generate pBR31. To co-overexpress the βγ G protein subunits in the HSN neurons, we injected pBR30 (50 ng/µl), pBR31 (50 ng/µl), and pJM60 [ptph-1::GFP] (80 ng/µl) (Moresco and Koelle 2004) into the LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl), generating five independent extrachromosomal transgenic lines which were used for behavioral assays. One representative transgenic strain, MIA278 [keyEx52; lite-1(ce314) lin-15(n765ts)], was kept. To generate a control strain for comparison in the egg-laying assays, we injected pJM66 [ptph-1::empty] (100 ng/µl) (Tanis et al. 2008) and pJM60 (80 ng/µl) into the LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl) generating five independent extrachromosomal control transgenes which were used for behavioral assays. One representative transgenic strain, MIA279 [keyEx53; lite-1(ce314) lin-15(n765ts)], was kept. Strains expressing either GFP along with EGL-10 or GFP alone in the HSNs from the tph-1 promoter were a generous gift of Jessica Tanis and Michael Koelle. Briefly, plasmids pJT35A (100 ng/µl) expressing egl-10 coding sequences from a cDNA and pJM60A (70 ng/µl) expressing GFP coding sequences, both from the long (3.1 kb) tph-1 promoter, along with pL15EK (50 ng/µl) were injected into the LX75 egl-10(md176) V; lin-15(n765ts) X mutant strain to generate LX755 egl-10(md176) V; lin-15(n765ts) X vsEx437 [tph-1p::GFP, tph-1p::egl-10]. Control animals were generated by injecting LX75 with pJM66 (100 ng/µl) expressing nothing and pJM60A (70 ng/µl) expressing coding sequences for GFP, both from the long (3.1 kb) tph-1 promoter, along with pL15EK (50 ng/µl), generating LX764 egl-10(md176) V; lin-15(n765ts) X vsEx439 [tph-1p::empty, tph-1p::GFP]. To test how loss of HSNs affected the hyperactive egg-laying behavior of goa-1(sa734) null mutants, we mated wild-type N2 males into MIA26 egl-1(n986dm) V hermaphrodites, and the heterozygous egl-1(n986dm)/+ male cross-progeny were then crossed into DG1856 goa-1(sa734) I hermaphrodites. Their cross-progeny were selfed, and the presence of the egl-1(n986dm) and goa-1(sa734) were confirmed by genotyping, creating MIA345 goa-1(sa734) I; egl-1(n986dm) V.
Vulval muscles
pJT40A Ptph-1::Pertussis Toxin (Tanis et al. 2008) was digested with NheI/KpnI and ligated into pBR3 (Pceh-24::mCherry) to generate pBR20. pBR20 [Pceh-24::Pertussis Toxin] (10 ng/µl) and pBR3 [Pceh-24::mCherry] (10 ng/µl) were injected into the LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavioral assays. One representative transgenic strain, MIA257 [keyEx46; lite-1(ce314) lin-15(n765ts)], was kept. To produce vulval muscle-specific GOA-1(Q205L), the coding sequence of GOA-1(Q205L) was recovered from pJM70C (Tanis et al. 2008) after digestion with NheI/SacI and ligated into pKMC188 [Punc-103e::GFP; (Collins and Koelle 2013)] generating pKMC268 [Punc-103e::goa-1(Q205L)]. However, because the unc-103e promoter also directs expression in neurons that might indirectly regulate egg laying, GOA-1(Q205L) coding sequences were removed from pKMC268 by digesting with NheI/NcoI and ligated into pBR3 to generate pBR21. pBR21 [Pceh-24::GOA-1Q205L] (10 ng/µl) and pBR3 [Pceh-24::mCherry] (10 ng/µl) were injected into the LX1832 lite-1(ce314) lin-15(n765ts) X animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavior assays. One representative transgenic strain, MIA258 [keyEx47; lite-1(ce314) lin-15(n765ts)], was kept. To generate control strains for comparison in egg-laying assays, pBR3 [Pceh-24::mCherry] (20 ng/µl) was injected into the LX1832 lite-1(ce314) lin-15(n765ts) X animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavioral assays. One representative control transgenic strain, MIA256 [keyEx45; lite-1(ce314) lin-15(n765ts)], was kept. To produce a vulval muscle-specific Tetanus Toxin transgene, Tetanus Toxin coding sequences were amplified from pAJ49 (Pocr-2::Tetanus toxin) (Jose et al. 2007) using the following oligonucleotides: 5′-GAGGCTAGCGTAGAAAAAATGCCGATCACCATCAACAACTTC-3′ and 5′-GCGCAGGCGGCCGCTCAAGCGGTACGGTTGTACAGGTT-3′. The DNA fragment was digested with NheI/NotI and ligated into pBR6 to generate pBR27. To block any possible neurotransmitter release from the vulval muscles, pBR27 (10 ng/µl) and pBR3 (10 ng/µl) was injected into the LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavior assays. One representative transgenic strain, MIA262 [keyEx51; lite-1(ce314) lin-15(n765ts)], was kept.
uv1 neuroendocrine cells
To generate a uv1 cell-specific Pertussis Toxin transgene, pBR20 (Pceh-24::Pertussis Toxin) was digested with NheI/NcoI and the coding sequences of Pertussis Toxin were then ligated into pAB5 (Ptdc-1::mCherry::ocr-2 3’UTR) to generate pBR25. pBR25 [Ptdc-1::Pertussis Toxin] (10 ng/µl) and pAB5 [Ptdc-1::mCherry] (5 ng/µl) were injected into LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavioral assays. One representative transgenic strain, MIA260 [keyEx49; lite-1(ce314) lin-15(n765ts)], was kept. To generate a uv1 cell-specific GOA-1(Q205L) transgene, pKMC268 [Punc-103e::GOA-1(Q205L)] was digested with NheI/NcoI and the coding sequences of GOA-1(Q205L) were then ligated into pBR25 to generate pBR26. To increase Gαo signaling in uv1 cells, we injected pBR26 [Ptdc-1::GOA-1Q205L] (10 ng/µl) and pAB5 [Ptdc-1::mCherry] (5 ng/µl) into the LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavioral assays. One transgenic strain MIA261 [keyEx50; lite-1(ce314) lin-15(n765ts)] was kept. To generate a control strain for comparison in our egg-laying assays, pAB5 [Ptdc-1::mCherry] (15 ng/µl) was injected into the LX1832 lite-1(ce314) lin-15(n765ts) animals along with pLI5EK (50 ng/µl) to generate five independent extrachromosomal transgenes which were used for behavioral assays. One representative transgenic strain, MIA259 [keyEx48; lite-1(ce314) lin-15(n765ts)], was kept.
VC neurons
Strains MIA144 keyIs33 and MIA146 keyIs46 expressing Tetanus Toxin in the VC neurons from the lin-11 promoter/enhancer have been described (Kopchock et al. 2021). To eliminate HSNs in these VC-silenced animals, wild-type N2 males were crossed into MIA26 egl-1(n986dm) V hermaphrodites, and the heterozygous egl-1(n986dm)/+ males were then crossed to MIA146 keyIs46; lite-1(ce314) lin-15(n765ts) X hermaphrodites, and the cross-progeny were then selfed, generating the homozygous strain MIA173 keyIs46; egl-1(n986dm) V; lite-1(ce314) lin-15(n765ts) X. To test whether synaptic transmission from the VCs was required for the hyperactive egg-laying behavior of Gαo signaling mutants, wild-type N2 males were crossed to MIA227 vsIs186; vsIs50 lite-1(ce314) lin-15(n765ts) X hermaphrodites and the resulting heterozygous cross-progeny were then crossed MIA144 hermaphrodites, allowed to self, generating MIA370 vsIs186; keyIs33; vsIs50 lite-1(ce314) lin-15(n765ts) X. To test how complete loss of GOA-1 affected egg laying in VC-silenced animals, we crossed wild-type N2 males into DG1856 goa-1(sa734) I hermaphrodites, and the heterozygous goa-1(sa734)/+ cross-progeny were then mated with MIA144 hermaphrodites. The cross-progeny were allowed to self, generating MIA371 goa-1(sa734) I; keyIs33; lite-1(ce314) lin-15(n765ts) X. The presence of goa-1(sa734) was confirmed by Sanger sequencing.
Fluorescence imaging
Ratiometric Ca2+ imaging
Ratiometric Ca2+ recordings were performed on freely behaving animals mounted between a glass coverslip and chunk of NGM agar, as previously described (Collins and Koelle 2013; Li et al. 2013; Collins et al. 2016; Ravi et al. 2018b). Briefly, recordings were collected on an inverted Leica TCS SP5 confocal microscope using the 8 kHz resonant scanner at ∼20 fps at 256 × 256 pixel resolution, 12-bit depth and ≥2X digital zoom using a 20x Apochromat objective (0.7 NA) with the pinhole opened to ∼20 µm. GCaMP5G and mCherry fluorescence was excited using a 488 nm and 561 nm laser lines, respectively. Adult recordings were performed 24 hours after the late L4 stage. After staging, animals were allowed to adapt for ∼30 minutes before imaging. During imaging, the stage and focus were adjusted manually to keep the relevant cell/pre-synapse in view and in focus.
Ratiometric analysis (GCaMP5: mCherry) for all Ca2+ recordings was performed after background subtraction using Volocity 6.3.1 as described (Ravi et al. 2018a). The egg-laying active state was operationally defined as the period one minute prior to the first egg-laying event and ending one minute after the last (in the case of a typical active phase where 3–4 eggs are laid in quick succession). However, in cases where two egg-laying events were apart by >60 s, peaks were considered to be in separate active phases and any transients observed between were considered to be from an inactive state. In animals where we observed no Ca2+ peaks during the entire recording, the total duration of the recording was considered as one, long, inter-transient interval. In animals where we observed a single Ca2+ transient, the duration from the start of the recording to the time of the Ca2+ transient and the time from the Ca2+ transient to the end of the recording were defined as inter-transient intervals. Animals overexpressing NLP-7 become highly egg-laying defective, and the embryonic expression of mCherry from the nlp-3 promoter in accumulated eggs interfered with the standard image segmentation protocol based on the mCherry channel. For these animals, unambiguous Ca2+ transient-dependent changes in GCaMP5 fluorescence localized at the HSN cell body were scored manually, and the frequency of such events were calculated as the number of observed events per unit recording time, typically 10–20 minutes.
Behavior assays and microscopy
Animal sterilization
Animals were sterilized using Floxuridine (FUDR) as described (Mitchell et al. 1979; Collins et al. 2016; Ravi et al. 2018a). Briefly, 100 µl of 10 mg/ml FUDR was applied to OP50 seeded NGM plates. Late L4 animals were then staged onto the FUDR plates, and the treated adults were imaged ∼24 hours later.
Egg laying assays
Unlaid eggs were quantitated as described (Chase et al. 2004). Staged adults were obtained by picking late L4 animals and culturing them for 30–40 hours at 20°C. The percentage of early-stage eggs laid were quantified as described (Koelle and Horvitz 1996). Thirty staged adults were placed on a thin lawn of OP50 bacteria on a NGM agar plate (Brenner 1974) and allowed to lay eggs for 30 minutes. This was repeated with new sets of staged animals until a total of at least 100 laid eggs were analyzed. Each egg was examined under a Leica M165FC stereomicroscope and categorized into the following categories: eggs which have 1 cell, 2 cell, 3–4 cell, 5–8 cell, and embryos with >8 cells. Eggs with eight cells or fewer were classified as “early stage.”
Long-term recording of egg-laying behavior
Egg-laying behavior was recorded at 4–5 frames per second from 24-hour adults after transfer to NGM plates seeded with a thin lawn of OP50 bacterial food using a Leica M165FC stereomicroscope and camera (Grasshopper 3, 4.1 Megapixel, USB3 CMOS camera, Point Grey Research). N2 wild-type and hyperactive egg-laying mutant strains (MT2426 and LX850) were recorded for 3 hours, and the egg-laying defective strains MT8504 and LX849 were recorded for 8–10 hours. Average duration of active and inactive states from these recordings were calculated as described (Waggoner et al. 1998, 2000).
Electrophysiology
Electrophysiological recordings were carried out on an upright microscope (Olympus BX51WI) coupled with an EPC-10 amplifier and Patchmaster software (HEKA), as previously described (Yue et al. 2018; Zou et al. 2018). Briefly, day 2 adult worms were glued on the surface of Sylgard-coated coverslips using the cyanoacrylate-based glue (Gluture Topical Tissue Adhesive, Abbott Laboratories). A dorsolateral incision was made using a sharp glass pipette to expose the cell bodies of HSN neurons for recording. The bath solution contained (in mM) 145 NaCl, 2.5 KCl, 5 CaCl2, 1 MgCl2, and 20 glucose (325–335 mOsm, pH adjusted to 7.3). The pipette solution contained (in mM) 145 KCl, 5 MgCl2, 5 EGTA, 0.25 CaCl2, 10 HEPES, 10 glucose, 5 Na2ATP and 0.5 NaGTP (315–325 mOsm, pH adjusted to 7.2). The resting membrane potentials were tested with 0 pA holding under the Current Clamp model of the whole-cell patch. The high Cl− content of the intracellular solution used may contribute to the relatively depolarized membrane potentials we observed (Liu et al. 2018).
Experimental design and statistical analysis
Sample sizes for behavioral assays followed previous studies (Chase et al. 2004; Collins and Koelle 2013; Collins et al. 2016). No explicit power analysis was performed before the study. Statistical analysis was performed using Prism v.6-8 (GraphPad). Behavior experiments were typically performed at least twice on separate days. Ca2+ transient peak amplitudes and inter-transient intervals were pooled from data collected from multiple animals over several days (typically ∼10 animals recorded per genotype/condition per experiment with ∼2–3 recordings obtained per genotype per recording session). No animals or data were excluded. Individual P-values are indicated in each Figure legend, and all tests were corrected for multiple comparisons (Bonferroni for ANOVA and Fisher exact test; Dunn for Kruskal-Wallis). For clarity, data not expected (or shown) to be normally distributed are presented in cumulative distribution plots rather than standard scatterplots.
Data availability
The authors affirm that all data necessary for confirming the conclusions of this article are represented fully within the article and its tables and figures. Requests for strains, plasmids, and ratiometric fluorescence recordings used to generate Ca2+ traces can be directed to KMC. Supplementary material is available at figshare: https://doi.org/10.25386/genetics.14627469.
Results
Reduced inhibitory Gαo signaling leads to premature egg laying and decreases the duration of egg-laying inactive states
To better understand how inhibitory Gαo signaling contributes to egg-laying behavior state transitions, we used mutants and transgenes to manipulate Gαo signaling in specific cells of the egg-laying circuit and analyzed the consequences on cell Ca2+ activity and behavior. We found that Gαo signaling inhibits the onset of egg laying. We performed a “time to first egg lay” assay in wild-type animals and in mutants with excessive or reduced Gαo signaling. As previously shown, wild-type animals release their first embryo ∼6–7 hours after becoming adults (McMullen et al. 2012; Ravi et al. 2018a). Animals bearing Gαo loss-of-function or null mutations laid their eggs much earlier, 3–4 hours after becoming adults (Figure 1B). goa-1(n1134), a hypomorphic mutant predicted to lack the conserved N-terminal myristoylation and palmitoylation sequence, and goa-1(sa734), an early stop mutant predicted to be a molecular null (Segalat et al. 1995; Robatzek and Thomas 2000), showed similarly precocious onset of egg laying (Figure 1B). This phenotype was shared in transgenic animals where Gαo function was inhibited just in HSNs through the cell-specific expression of Pertussis Toxin (Tanis et al. 2008). Because the timing of this first egg-laying event requires serotonin along with HSN function and activity (Ravi et al. 2018a), these results suggest that Gαo normally signals in HSN to inhibit neurotransmitter release and thereby delay the first egg-laying active state (Figure 1B). To test the effects of increased Gαo signaling, we analyzed the behavior of egl-10(md176) mutants which lack the major RGS protein that terminates Gαo signaling by promoting Gαo GTP hydrolysis (Koelle and Horvitz 1996). egl-10(md176) mutants showed a strong and significant delay in the onset of egg laying, laying their first egg ∼15 hours after reaching adulthood (Figure 1B). This delay of egg-laying phenotype was shared in transgenic animals expressing the constitutively active Gαo (Q205L) mutant specifically in the HSNs (Tanis et al. 2008) and resembled animals developmentally lacking HSNs (Ravi et al. 2018a), consistent with Gαo signaling in HSN to inhibit neurotransmitter release that normally drives the onset of egg laying.
To understand how Gαo signaling controls the normal two-state pattern of egg laying, we performed long-term recordings of adults as they transitioned into and out of the active states in which clusters of several eggs are typically laid. We defined intra-cluster intervals as the time elapsed between consecutive egg-laying events within a single active state (e.g., intervals <4 minutes) and inter-cluster intervals as the time elapsed between different active states (e.g., intervals >4 minutes), as described (Waggoner et al. 1998; Collins and Koelle 2013). As expected, wild-type animals displayed a two-state pattern of egg laying with multiple egg-laying events clustered within brief, ∼2-minute active states about every 20–30 minutes (Figure 1C and Table 2). Animals bearing the goa-1(n1134) hypomorphic mutation entered active states two- to threefold more frequently, often laying single eggs during active states separated by only ∼12–13 minutes (Figure 1C and Table 2), as previously shown (Waggoner et al. 2000). We found the duration of inactive states in animals expressing Pertussis Toxin in the HSN neurons was indistinguishable from the goa-1(n1134) mutant, indicating that Gαo signals in HSN to reduce the probability of entering the active state (Figure 1C and Table 2). Gαo is likely the only major target for Pertussis Toxin in the HSNs regulating egg laying, as transgenic re-expression of GOA-1Q205L from an HSN-specific promoter rescues the hyperactive egg-laying defect of Pertussis Toxin expressing animals (Figure 1D). This rescue was qualitatively similar to that seen when GOA-1Q205L was expressed in HSNs of goa-1(n1134) mutant animals (Tanis et al. 2008). Loss of inhibitory Gαo signaling led to active states in which 1-2 embryos were laid almost immediately after they were positioned in the uterus next to the vulval opening. As a result, successive egg-laying events were often rate-limited by egg production, explaining why the average intra-cluster intervals were typically double that seen in wild-type animals (Figure 1C and Table 2). Indeed, goa-1 null mutants including goa-1(sa734) have a strongly reduced brood size (36 progeny) compared to goa-1(n1134) hypomorphic mutants (150 progeny) (Segalat et al. 1995), making quantitative measures of intra- and inter-cluster intervals in such mutants challenging. However, analyzing the timing of egg-laying events in animals with too much Gαo signaling supports our interpretation that Gαo does not play a significant role in regulating the timing of egg laying within the active state. Both egl-10(md176) mutants with a global increase of Gαo signaling or transgenic animals expressing the Gαo(Q205L) mutant specifically in HSNs show the same intra-cluster time constant as wild-type animals (Table 2). In contrast, both egl-10(md176) mutant animals or Gαo(Q205L) gain-of-function mutation in the HSNs showed prolonged inactive periods of 258 and 67 minutes, respectively (Figure 1C and Table 2), resembling animals lacking HSNs altogether (Waggoner et al. 2000). Interestingly, animals without HSNs (Waggoner et al. 1998) or with too much inhibitory Gαo signaling (Figure 1C and Table 2) still lay multiple eggs within discrete active states, consistent with our results showing that a stretch-dependent homeostat can maintain the active state even when neurotransmitter release from the HSN is inhibited (Collins et al. 2016; Ravi et al. 2018a). These results support a model where Gαo signaling in HSN does not modulate patterns of egg laying within active states. Instead, Gαo acts to depress entry into the egg-laying active state.
Table 2.
Egg-laying behavior measurements in animals with altered Gαo signaling
| Genotype | Intra-cluster interval (1/λ1) | Inter-cluster interval (1/λ2) | Eggs laid per active state | Unlaid eggs per animal |
|---|---|---|---|---|
| Average (min) (95%CI range) | Average (min) (95%CI range) | Average ± 95%CI | Average ± 95%CI | |
| Wild type | 0.42 | 22.11 | 2.5 ± 0.5 | 15.9 ± 1.5 |
| (0.33–0.51) | (21.19–23.09) | |||
| goa-1(n1134) | 1.31 | 12.88* | 1.2 ± 0.1‡ | 2.2 ± 0.3 |
| (0.94–1.67) | (12.31–12.75) | |||
| HSN::Pertussis Toxin | 1.69 | 12.42* | 1.1 ± 0.07‡ | 5.0 ± 0.4 |
| (1.17–2.20) | (12.09–12.75) | |||
| egl-10(md176) | 0.64 | 258.6* | 2.5 ± 0.2 | 45.9 ± 3.3 |
| (0.49–0.79) | (231.26–293.42) | |||
| HSN::GOA-1Q205L | 0.35 | 66.97* | 2.2 ± 0.2 | 36.8 ± 3.8 |
| (0.24–0.47) | (64.14–70.02) |
Long-term behavior recordings were used to extract features of egg-laying active and inactive behavior states for the indicated genotypes, as described (Waggoner et al. 1998). “*” indicates significant differences compared to wild-type animals (P < 0.0001, Kruskal-Wallis test with Dunn’s correction for multiple comparisons), “‡” indicates significant differences compared to wildtype (P < 0.0001, one-way ANOVA with Bonferroni correction).
Gαo signaling inhibits HSN Ca2+ activity to promote the inactive behavior state
Previous work has shown that Gαo signals to depress neurotransmitter release (Miller et al. 1999; Nurrish et al. 1999). Whether Gαo signals to inhibit HSN electrical excitability that might be evident in changes in cell Ca2+ activity, or instead signals downstream of Ca2+ to regulate steps in UNC-13-dependent docking, priming, and vesicle fusion, has not been tested directly. To address how Gαo signals to inhibit HSN neurotransmitter release, we performed ratiometric Ca2+ imaging in our panel of Gαo signaling mutants as they entered spontaneous egg-laying active states. Freely behaving animals bearing the goa-1(n1134) hypomorphic or goa-1(sa734) null mutations showed a clear change in HSN Ca2+ activity from two-state burst firing to more tonic firing (Figure 2A, Supplementary Videos S1–S3). Complete loss of inhibitory Gαo signaling caused a significant increase in the frequency of HSN Ca2+ transients (Figure 2, B and C). We were surprised that the goa-1(n1134) mutants, which show strongly hyperactive egg-laying behavior indistinguishable from that of goa-1(sa734) null mutants, showed only a modest and statistically insignificant increase in HSN Ca2+ activity compared to wild-type (Figure 2C). However, our results with goa-1(n1134) match previous findings (Shyn et al. 2003). The goa-1(n1134) hypomorphic mutant is expected to have residual Gαo signaling activity in that its major defect is the absence of a proper membrane anchor sequence (Mumby et al. 1990). These results show that Gαo signaling depresses HSN presynaptic activity, but that the hyperactive egg-laying phenotypes observed in goa-1(n1134) mutants are not a result of dramatic changes in presynaptic HSN Ca2+ activity.
Figure 2.

Gαo signaling inhibits HSN neuron Ca2+ activity and burst firing. (A) Representative GCaMP5: mCherry (ΔR/R) ratio traces showing HSN Ca2+ activity in freely behaving wild-type (black), goa-1(n1134) loss-of-function mutant (green), goa-1(sa734) null mutant (blue), and egl-10(md176) null (purple) mutant animals, along with transgenic animals expressing the activated GOA-1(Q205L) in the HSN neurons (pink) during an egg-laying active state. Arrowheads indicate egg-laying events. (B) Cumulative distribution plots of instantaneous Ca2+ transient peak frequencies (and inter-transient intervals) in wild-type (black open circles), goa-1(n1134) (green filled triangles), goa-1(sa734) (blue squares), egl-10(md176) mutants (purple open squares) along with transgenic animals expressing the activated GOA-1(Q205L) in the HSN neurons (pink open circles). Asterisks indicate P < 0.0001 (Kruskal-Wallis test with Dunn’s correction for multiple comparisons). (C) Scatter plots show average Ca2+ transient frequency (per min). Error bars indicate 95% confidence intervals for the mean. Asterisk indicates P < 0.0001; pound (#) indicates P ≤ 0.0079; n.s. indicates P > 0.05 (one-way ANOVA with Bonferroni correction for multiple comparisons).
We next tested how increased inhibitory Gαo signaling affects HSN activity. Both egl-10(md176) mutants and transgenic animals expressing the activated GOA-1(Q205L) in HSNs showed a significant and dramatic reduction in the frequency of HSN Ca2+ transients, with single HSN Ca2+ transients occurring several minutes apart (Figure 2, A and B). The rare egg-laying events seen in animals with increased Gαo signaling mostly followed single HSN Ca2+ transients, not multi-transient bursts typically seen in wild-type animals (Figure 2, A and C). In one egl-10(md176) animal, we observed an egg-laying event that was not accompanied by an HSN Ca2+ transient. This suggests that elevated Gαo signaling is sufficient to silence the HSNs, and that, in this case, egg laying becomes HSN-independent. In support of this model, previous work has shown that egl-10(md176) mutants respond to serotonin but are resistant to the serotonin reuptake inhibitor imipramine (Trent et al. 1983). Alternatively, (or additionally) Gαo signaling may function to depress coordinated activity between the gap-junctioned, contralateral HSNs, whose Ca2+ activity we were unable to observe simultaneously because our confocal imaging conditions only captures one HSN at a time.
To determine how disruption of inhibitory Gαo signaling in HSN affects its activity, we recorded HSN Ca2+ transients in transgenic animals expressing Pertussis Toxin in the HSNs. Gαo-silenced HSNs showed a dramatic increase in the frequency of HSN Ca2+ transients, leading to a nearly constitutive, tonic Ca2+ activity like that observed in goa-1(sa734) null mutants (Figure 3, A–C; Supplementary Video S4). While control animals showed an average HSN Ca2+ transient frequency of about ∼0.4 transients per minute, animals expressing Pertussis Toxin in HSN showed an average 1.9 transients per minute, a significant increase (Figure 3C). These results suggest that even under normal growth conditions, unidentified neuromodulators signal through Gαo-coupled receptors on HSN to inhibit Ca2+ activity.
Figure 3.

Inhibitory Gαo signaling in HSN is required for two-state Ca2+ activity and facilitates modulation by feedback of egg accumulation. (A) Representative GCaMP5: mCherry (ΔR/R) ratio traces showing HSN Ca2+ activity in untreated (fertile) wild-type animals (black open circles), FUDR-sterilized wild-type animals (black filled circles), untreated (fertile) animals expressing Pertussis Toxin (PTX) in the HSN neurons (blue open squares), and in FUDR-sterilized transgenic animals expressing PTX in the HSNs (blue filled squares). Arrowheads indicate egg-laying events. (B) Cumulative distribution plots of instantaneous Ca2+ transient peak frequencies (and inter-transient intervals) in untreated and FUDR-treated animals. Asterisks indicate P < 0.0001 (Kruskal-Wallis test with Dunn’s test for multiple comparisons). (C) Scatter plots show average Ca2+ transient frequency (per min). Error bars indicate 95% confidence intervals for the mean. Asterisks indicate P < 0.0001 (one-way ANOVA with Bonferroni’s test for multiple comparisons). Data from 10 animals were used for each strain for analysis.
We have previously shown that burst Ca2+ activity in the command HSN neurons is initiated and sustained by a stretch-dependent homeostat. In chemically or genetically sterilized animals, burst Ca2+ activity in HSN is largely eliminated (Ravi et al. 2018a). As such, we were surprised to observe high frequency Ca2+ transients in animals with reduced Gαo signaling because these animals typically retain few (1 to 3) eggs in the uterus at steady state, conditions that normally eliminate HSN burst firing. We hypothesized that the stretch-dependent homeostat was not required to promote HSN Ca2+ activity in Gαo signaling mutants. To test this, we chemically sterilized transgenic animals expressing Pertussis Toxin in the HSNs with Floxuridine (FUDR), a blocker of embryogenesis (Mitchell et al. 1979), and recorded HSN Ca2+ activity. Wild-type animals treated with FUDR showed a dramatic decrease in the frequency of HSN Ca2+ activity and an elimination of burst firing (Figure 3, A–C), as we have previously shown (Ravi et al. 2018a). Sterilized transgenic animals expressing Pertussis Toxin in the HSNs showed only a slight reduction in HSN Ca2+ frequency (Figure 3, A and B). Both fertile and sterile Pertussis Toxin-expressing animals had dramatically and significantly increased HSN Ca2+ transient frequency (∼1.9/minute), indicating their HSNs no longer responded to the retrograde signals of egg accumulation arising from the stretch-homeostat. One explanation for this result could be that in fertile wild-type animals, feedback of egg accumulation elevates HSN excitability above a firing threshold that overcomes endogenous inhibitory Gαo signaling. Together, these results support a model where Gαo signals in HSN to depress cell electrical excitability which allows for the proper two-state pattern of HSN Ca2+ activity that responds to homeostatic feedback of egg accumulation.
Presynaptic Gαo signaling inhibits postsynaptic vulval muscle activity
To test how changes in inhibitory Gαo signaling affect the postsynaptic vulval muscles, we recorded Ca2+ activity in the vulval muscles of goa-1(n1134) mutant and Pertussis Toxin expressing transgenic animals. Both wild-type and goa-1(n1134) mutants showed increased vulval muscle Ca2+ transients as the animals entered the egg-laying active state (Figure 4, A and B). goa-1(n1134) mutants also showed elevated muscle activity compared to wild-type control animals during inactive states when no eggs were laid (Figure 4, A and B), as previously shown (Shyn et al. 2003). Surprisingly, Ca2+ activity during egg-laying active states was not significantly different in goa-1(n1134) mutant animals (Figure 4, A and B; compare Supplementary Videos S5 and S6), a result consistent with Gαo signaling primarily to prolong the inactive state. These results suggest Gαo signals to inhibit circuit activity during the egg-laying inactive state and/or that loss of Gαo elevates circuit activity that prolongs the active state beyond the brief ∼2–3 minutes window during which eggs are typically laid (Waggoner et al. 1998). Recordings of vulval muscle Ca2+ activity in transgenic animals expressing Pertussis Toxin in the presynaptic HSNs show a dramatic and significant increase in vulval muscle Ca2+ transient frequency during both active and inactive states (Figure 4, C and D; Supplementary Video S7), confirming Gαo is required in HSN to inhibit neurotransmitter release.
Figure 4.

Gαo signals in HSN to reduce excitatory modulation of the vulval muscles. (A) Representative GCaMP5: mCherry (ΔR/R) ratio traces showing vulval muscle Ca2+ activity in wild-type (black) and goa-1(n1134) loss-of-function mutant animals (red). Egg-laying events are indicated by arrowheads, and egg-laying active states are indicated by dashed green lines. (B) Cumulative distribution plots of instantaneous vulval muscle Ca2+ transient peak frequencies (and inter-transient intervals) in wild-type (black circles) and goa-1(n1134) mutant animals (red squares) in the inactive and active egg-laying states (filled and open, respectively). (C) Representative GCaMP5: mCherry (ΔR/R) ratio traces showing vulval muscle Ca2+ activity in untreated (circles) and FUDR-treated (cross) wild-type animals (black) along with untreated (filled) or FUDR-treated (open) transgenic animals expressing Pertussis Toxin in the HSNs (blue squares). Egg-laying events are indicated by arrowheads, and egg-laying active states are indicated by green dashed lines. (D) Cumulative distribution plots of instantaneous vulval muscle Ca2+ transient peak frequencies (and inter-transient intervals) in wildtype and in transgenic animals expressing Pertussis Toxin in the HSN neurons, with or without FUDR treatment (black cross or blue square), during the inactive and active egg-laying states (filled and open, respectively). Asterisks indicate P < 0.0001 (Kruskal-Wallis test with Dunn’s test for multiple comparisons).
We have previously shown that egg accumulation promotes vulval muscle excitability during the active state while sterilization reduces it to that seen during the inactive state (Collins et al. 2016; Ravi et al. 2018a). Because FUDR-sterilization failed to reduce HSN Ca2+ activity in Pertussis Toxin expressing animals (Figure 3C), we predicted that vulval muscle Ca2+ activity would remain similarly elevated in these animals after sterilization. To our surprise, FUDR treatment significantly reduced, but did not completely eliminate, the elevated vulval muscle Ca2+ activity in animals expressing Pertussis Toxin in HSN (Figure 4, C and D). This result suggests egg accumulation and/or germline activity is still required for full vulval muscle activity even when HSN Ca2+ activity is dramatically increased. However, because these animals lay eggs almost as soon as they are made, the degree of uterine stretch necessary to induce the active state must be markedly reduced. This supports a model where both the stretch-dependent homeostat and Gαo signaling dynamically interact to regulate egg-laying behavior states.
Gαo signaling modulates the HSN resting membrane potential
Reduction of inhibitory Gαo signaling strongly increased HSN Ca2+ activity and burst firing, prompting us to investigate whether Gαo signals to modulate HSN electrical excitability. We recorded the resting membrane potential of the HSN neurons in animals with altered Gαo signaling using the whole-cell patch clamp technique (Figure 5A), as described (Yue et al. 2018). As shown in Figure 5B, transgenic animals expressing Pertussis Toxin specifically in the HSNs had significantly depolarized HSNs (−14.75 mV) compared to the wild-type parental strain (−21.8 mV). Similarly, hypomorphic goa-1(n1134) loss-of-function mutants had more depolarized resting potentials (−17.9 mV) compared to HSNs from recorded wild-type animals (−21.1 mV). In contrast, the resting membrane potential of HSNs in egl-10(md176) Gαo RGS protein mutant animals with a global increase in Gαo signaling (Koelle and Horvitz 1996) was significantly hyperpolarized (−40.8 mV) compared to wild-type control animals (Figure 5B). To confirm this hyperpolarization arose from changes in Gαo signaling in HSN, we transgenically expressed EGL-10 from cDNA in the HSNs of egl-10(md176) mutant animals. Re-expression of EGL-10 raised the membrane potential to −18.7 mV from −30.4 mV. The hyperpolarization of HSNs in egl-10(md176) mutants explains the reduced frequency of HSN Ca2+ transients and their strong defects in egg-laying behavior. These results show that Gαo signals in the HSNs to promote membrane polarization, reducing cell electrical excitability, Ca2+ activity, and neurotransmitter release.
Figure 5.

Gαo signaling depresses HSN resting membrane potential and may inhibit egg-laying behavior in parallel to βγ, cGMP, and cAMP signaling pathways. (A) Cartoon of the glued worm preparation used for patch clamp electrophysiology of HSN. (B) Scatter plots show resting membrane potentials in (left) transgenic animals expressing either nothing (blue open squares) or Pertussis Toxin in HSN (blue filled squares). Asterisk indicates P < 0.0001 (Student’s t-test). (center) Resting membrane potentials of wild-type control animals (grey hexagons), goa-1(n1134) loss-of-function mutants (red triangles), and egl-10(md176) null mutants (purple filled circles). Asterisks indicate P ≤ 0.0347 (one-way ANOVA with Bonferroni correction for multiple comparisons). (right) Resting membrane potentials in transgenic egl-10(md176) animals expressing in HSN either GFP alone (green open circles) or GFP + egl-10 cDNA driven by the HSN-specific tph-1 promoter (green filled circles). Asterisks indicate P < 0.0001 (Student’s t-test). Error bars indicate 95% confidence intervals for the mean. N ≥ 7 animals recorded per genotype. (C) Scatter plots show average number of eggs retained by wild-type animals (gray filled circles), egl-10(md176) null mutants (orange open circles), and in transgenic animals expressing either Gβ (GPB-2) and Gγ (GPC-1) subunits (green filled circles) or nothing (blue filled circles) in HSN from the tph-1 gene promoter along with GFP. Error bars indicate means with 95% confidence intervals. Asterisk indicates P < 0.0001; n.s. indicates P > 0.05 (one-way ANOVA with Bonferroni correction for multiple corrections). (D) Top, cartoon of how Gαo signaling might interact with cAMP and cGMP signaling pathways. Gene names for C. elegans orthologs tested here are indicated in parentheses. Middle, scatterplots show average number of eggs retained by wildtype (grey), egl-10(md176) (red open circles), goa-1(n1134) and goa-1(sa734) Gαo loss of function (red filled circles) mutants, and in animals with altered cAMP effector signaling (green): gsa-1(ce81) Gαs gain-of-function, acy-1(ce2) Adenylate Cyclase gain-of-function, kin-2(ce179) protein kinase A (PKA) inhibitory regulatory subunit loss-of-function, pde-4(ce268) Phosphodiesterase (PDE) loss-of-function, pde-2(qj6) Phosphodiesterase (PDE) null; altered cGMP effector signaling (pink): egl-4(ad805) and egl-4(mg410) protein kinase G (PKG) gain-of-function (pink filled circles), egl-4(n478) and egl-4(n479) loss-of-function mutants (pink open circles). Mean totals below ∼10 eggs indicate hyperactive egg laying while totals above ∼20 eggs indicate egg-laying behavior defects. Bottom, bar graphs indicate percent of embryos laid at early stages of development. Animals laying ≥50% embryos at early stages are considered hyperactive. Error bars indicate ± 95% confidence intervals for the mean proportion. N.D. indicates the stages of eggs laid was not determined because those mutants are egg-laying defective (Egl). Asterisk indicates highlighted significant differences (P ≤ 0.0001; Fisher Exact Test with Bonferroni correction for multiple comparisons).
Inhibition of egg laying by Gαo is not replicated by elevated βγ expression
Receptor activation of Gαi/o heterotrimers releases βγ subunits which have previously been shown to bind to activate specific K+ channels and inhibit Ca2+ channels (Reuveny et al. 1994; Herlitze et al. 1996). RNAi-mediated knockdown of the GPB-1 β subunit in HSN reduces egg-laying behavior (Esposito et al. 2007). An increase in free βγ subunits in Gαo mutants might explain their hyperactive egg-laying behavior phenotypes. To test if βγ over-expression in HSN would increase egg laying, we transgenically overexpressed the sole nonRGS Gβ subunit, GPB-1 (Jansen et al. 1999), and the broadly expressed Gγ subunit, GPC-2 (Yamada et al. 2009), under the tph-1 promoter along with GFP. βγ over-expression in HSN did not cause any significant differences in steady-state egg accumulation (Figure 5C). The number of eggs stored in-utero in these animals (13.0 ± 1.1 eggs) was comparable to wild-type animals (15.7 ± 1.2 eggs) and less than egl-10(md176) mutant animals (44.5 ± 2.3 eggs). These results suggest that Gαo signals to inhibit HSN activity in a distinct manner from simple titration or release of βγ subunits.
Egg-laying behavior is dysregulated in cAMP and cGMP signaling mutants
Gαo is in the Gαi/o class of G proteins that bind to and inhibit nucleotide cyclases, which function to reduce cAMP and cGMP levels and their subsequent activation of protein kinases (Kobayashi et al. 1990; Zhang and Pratt 1996; Matsubara 2002; Ghil et al. 2006). The hyperactive egg-laying behavior phenotypes of animals with reduced Gαo function could be explained by a failure to properly terminate cAMP and/or cGMP signaling (Figure 5D, top). To test this, we analyzed egg-laying behavior in strains bearing mutations that increase Gαs and cAMP effector signaling (Reynolds et al. 2005; Schade et al. 2005; Charlie et al. 2006a, 2006b). Animals carrying gsa-1(ce81) gain-of-function mutations in Gαs are predicted to increase signaling accumulate fewer eggs compared to wild-type animals (Figure 5D, middle). Because a reduction in steady-state egg accumulation could be caused by indirect effects of these mutations on egg production or brood size, we also examined the developmental age of embryos laid. As previously reported (Bany et al. 2003), loss of inhibitory Gαo signaling causes embryos to be laid precociously, before they reach the 8-cell stage (Figure 5D, bottom). Gαs gain-of-function mutant animals do not show a corresponding increase in early-stage embryos that are laid, suggesting the reduction in egg accumulation observed in Gαs gain-of-function mutant animals is indirect. In contrast, gain-of-function acy-1 Adenylate Cyclase mutations or loss-of-function pde-4 phosphodiesterase mutations, both predicted to result in increased cAMP signaling, cause animals to accumulate fewer eggs and lay them at earlier stages (Figure 5D). Similarly, kin-2 mutations that disrupt an inhibitory regulatory subunit of protein kinase A showed a modest but significant hyperactive egg-laying phenotype. Together, these results indicate that Gαs, cAMP, and protein kinase A signaling promote egg-laying behavior. Because these mutant phenotypes were not nearly as strong as animals carrying Gαo mutants, Gαo likely signals to inhibit egg laying via other effectors.
Extensive work has shown that the cGMP-dependent protein kinase G, EGL-4, regulates egg laying in C. elegans (Fujiwara et al. 2002; L'Etoile et al. 2002; Raizen et al. 2006; Hao et al. 2011). Mutations which increase EGL-4 activity cause hyperactive egg laying and release of early stage embryos, while elimination of EGL-4 signaling causes egg-laying defects and significant egg accumulation (Figure 5D, middle). To determine whether Gαo and protein kinase G regulate egg laying in a shared pathway, we performed a genetic epistasis experiment. goa-1(sa734); egl-4(n479) double null mutants accumulate very few eggs (Figure 5D, middle), resembling the goa-1(sa734) null mutant. However, the low brood size of the goa-1(sa734) mutant could prevent accurate measurement of these animal’s egg-laying defects. To address this, we measured the stage of eggs laid as indicated above. Loss of the EGL-4 protein kinase G strongly and significantly suppressed the hyperactive egg-laying behavior of Gαo null mutants (Figure 5D, bottom). goa-1(sa734); egl-4(n479) mutants laid 33% of their embryos at early stages compared to 88% for the goa-1(sa734) single mutant, an intermediate egg-laying phenotype. These results are consistent with cGMP and/or protein kinase G signaling acting downstream of or parallel to Gαo in the proper regulation of egg-laying behavior, but their precise relationship remains unclear at this point.
Gαo signals in the vulval muscles and uv1 neuroendocrine cells to inhibit egg laying
In addition to HSN, GOA-1 is expressed in all neurons of the reproductive circuit, the egg-laying vulval muscles, and the uv1 neuroendocrine cells (Mendel et al. 1995; Segalat et al. 1995; Jose et al. 2007), raising questions as to how Gαo signaling functions in those other cells to regulate egg-laying behavior. Previous work has shown that GOA-1(Q205L) was sufficient to rescue the hyperactive egg-laying behavior of goa-1(n1134) mutants when expressed in the HSNs but had little effect when expressed in the VCs or vulval muscles (Tanis et al. 2008). Similarly, transgenic expression of Pertussis Toxin in the VCs or vulval muscles failed to modify egg-laying behavior strongly (Tanis et al. 2008). Previous work failing to identify a function for Gαo in the vulval muscles used a modified Nde-box element from the ceh-24 promoter that drives expression more efficiently in the vm1 muscle cells compared to the vm2 muscles innervated by the HSN and VC neurons (data not shown). We find that expression of Pertussis Toxin in both vm1 and vm2 vulval muscles from a larger region of the ceh-24 gene promoter (Harfe and Fire 1998; Ravi et al. 2018a) also failed to cause any significant changes in the steady-state egg accumulation (Figure 6A). Conversely, expression of the activated GOA-1(Q205L) mutant in the vulval muscles from the same promoter did cause a modest but significant egg-laying defect, with animals accumulating 24.1 ± 2.0 eggs compared to mCherry-expressing control transgenic animals (13.2 ± 0.7 eggs). This egg-laying defect was significantly weaker than egl-10(md176) mutants (46.0 ± 3.3 eggs) or transgenic animals expressing GOA-1(Q205L) just in the HSNs (36.8 ± 3.8 eggs). We do not believe this modest egg-laying defect was caused by transgene expression outside of the vulval muscles, as blocking synaptic transmission via transgenic expression of Tetanus Toxin from the same ceh-24 promoter showed no such egg-laying defect (Figure 6B). Collectively, these results show that Gαo does not play a significant role in suppressing vulval muscle excitability under steady-state conditions but that activated Gαo can signal in these cells to induce a mild but significant inhibition of cell activity and egg-laying behavior.
Figure 6.
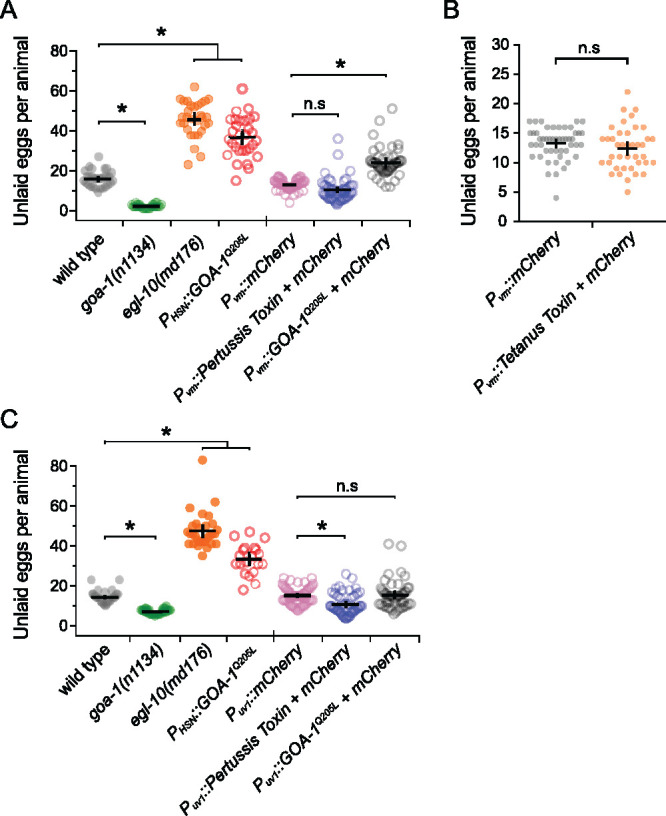
Altered Gαo signaling in the vulval muscles and uv1 neuroendocrine cells causes only modest effects on egg laying. (A) Scatter plots show average number of eggs retained by wild-type (grey), goa-1(n1134) mutants (green), egl-10(md176) null mutant animals (orange) along with transgenic animals expressing GOA-1Q205L in the HSNs (red open circles) compared to transgenic animals expressing mCherry only (pink open circles), Pertussis Toxin (blue open circles), or GOA-1Q205L (black open circles) in the vulval muscles from the ceh-24 gene promoter. (B) Scatter plots show average number of eggs retained in transgenic animals expressing mCherry only (gray) or Tetanus Toxin along with mCherry (orange) in the vulval muscles (vm) using the ceh-24 gene promoter. Error bars indicate means with 95% confidence intervals. (C) Scatter plots show average number of eggs retained by wild-type (grey), goa-1(n1134) mutant (green), egl-10(md176) null mutant (orange) animals, and transgenic animals expressing GOA-1Q205L in the HSNs (red) compared to transgenic animals expressing mCherry only (pink), Pertussis Toxin (blue), or GOA-1Q205L (black open circles) in the uv1 neuroendocrine cells from the tdc-1 gene promoter. Four or five independent extrachromosomal arrays were generated for each transgene in (A–C) and ∼10 animals bearing each extrachromosomal array were analyzed. Error bars indicate 95% confidence intervals for the mean. Asterisks indicate P < 0.0001; n.s. indicates P > 0.05 (one-way ANOVA with Bonferroni’s correction for multiple comparisons or Student’s t test).
The uv1 cells express the neurotransmitter tyramine along with NLP-7 and FLP-11 neuropeptides which inhibit egg laying (Alkema et al. 2005; Collins et al. 2016; Banerjee et al. 2017). Based on the function of Gαo signaling in inhibiting neurotransmitter release in neurons, we would expect that loss of Gαo function in uv1 would enhance their excitability, promoting release of inhibitory tyramine and neuropeptides, causing a reduction of egg laying. Surprisingly, previous work has shown that transgenic expression of Pertussis Toxin in uv1 cells increased the frequency of early-stage eggs that are laid, similar to the blocking of neurotransmitter release by Tetanus Toxin (Jose et al. 2007). A caveat of these experiments is that the ocr-2 gene promoter used for transgene expression in uv1 also expresses in the utse (uterine-seam) associated cells and head sensory neurons (Jose et al. 2007). To test whether Gαo functions specifically in uv1 to regulate egg laying, we used the tdc-1 gene promoter (Alkema et al. 2005) along with the ocr-2 3’ untranslated region (Jose et al. 2007) to drive expression more specifically in uv1. Transgenic expression of Pertussis Toxin in uv1 caused a mild but significant decrease in steady-state egg accumulation (10.9 ± 1.5 eggs) compared to mCherry-expressing control animals (15.3 ± 1.2 eggs), indicating Gαo signaling facilitates uv1-dependent inhibition of egg-laying behavior (Figure 6C). We also tested how increased Gαo signaling in uv1 affects egg laying. Transgenic expression of the activated GOA-1(Q205L) mutant in uv1 cells caused no quantitative differences in egg accumulation (15.5 ± 2.1 eggs) (Figure 6C). Together, these results show that Gαo has a limited role in regulating egg-laying behavior in the vulval muscles or uv1 neuroendocrine cells, unlike the strong phenotypes observed when we genetically manipulate Gαo function in HSN.
NLP-7 inhibition of HSN activity and egg-laying requires the EGL-47 receptor and Gαo
Multiple neuropeptides and receptors have been identified to inhibit egg laying by signaling through Gαo-coupled receptors expressed on HSN (Figure 1A). Recent work has identified NLP-7 neuropeptides, synthesized in the VC neurons and uv1 neuroendocrine cells, as potential ligands for EGL-47 receptor signaling through Gαo (Moresco and Koelle 2004; Banerjee et al. 2017). Animals overexpressing the NLP-7 neuropeptide are egg-laying defective, accumulating 39.6 ± 3.4 eggs in the uterus (Figure 7A). To test how NLP-7 inhibits egg laying, we crossed NLP-7 over-expressing transgenes into goa-1 mutant animals and evaluated their egg-laying behavior phenotypes. As shown in Figure 7A, the hypomorphic goa-1(n1134) loss-of-function mutant significantly suppressed these egg-laying defects, as previously shown (Banerjee et al. 2017). However, the suppression by goa-1(n1134) was incomplete; NLP-7 over-expressing, goa-1(n1134) double mutant animals retained more eggs than the goa-1(n1134) single mutant (Table 2) or even wild-type control animals (Figure 7A). To confirm whether Gαo was required for NLP-7 inhibition, we tested the goa-1(sa734) null mutant and found it fully suppressed the egg-laying defects caused by NLP-7 over-expression. To confirm this epistatic relationship, we measured the stage of embryos laid by these animals. goa-1(sa734) null mutant animals over-expressing NLP-7 laid ∼100% of their embryos at early stages, a level not significantly different goa-1(sa734) single mutant animals (Figure 7B). Together, these results show that NLP-7 neuropeptides cannot inhibit egg-laying behavior in the absence of Gαo function.
Figure 7.

NLP-7 neuropeptides signal through EGL-47 and Gαo to inhibit egg-laying outside of the HSNs. (A) NLP-7 signals through Gαo to inhibit egg-laying behavior. Scatter plots show average number of eggs retained by wild-type (gray circles), goa-1(sa734) mutants (blue open circles), NLP-7 over-expressing (OE) transgenics in the wild-type (pink triangles or orange squares), and in NLP-7 over-expressing transgenics in the goa-1(n1134) (pink open triangles) and goa-1(sa734) null mutant background (orange open squares). Data in orange squares are from animals that also carry the vsIs183 transgene used for HSN Ca2+ imaging. Mean totals below ∼10 eggs indicate hyperactive egg laying while totals above ∼20 eggs indicate egg-laying defective behavior defects. Error bars indicate 95% confidence intervals for the mean. Asterisks indicate P < 0.0001 (one-way ANOVA with Bonferroni correction for multiple comparisons). N ≥ 30 animals for each strain. (B) Measure of early stage eggs laid by wild-type (black), goa-1(sa734) null mutants (blue), and goa-1(sa734) null mutants over-expressing NLP-7 neuropeptides (orange). Both goa-1(sa734) mutant strains also carry the vsIs183 transgene used for HSN Ca2+ imaging. (C) NLP-7 over-expression silences HSN Ca2+ activity. Representative GCaMP5/mCherry ratio traces showing HSN Ca2+ activity in wild-type (black), goa-1(sa734) null mutants, and NLP-7 overexpressing transgenic animals in the wild-type (pink) and goa-1(sa734) null mutant backgrounds (orange). Arrowheads indicate egg-laying events. (D) Scatter plots show HSN Ca2+ peaks per minute measurements in wild-type (black), goa-1(sa734) null mutants, or egl-47(ok677) null mutant animals in the absence or presence of NLP-7 over-expression (OE). Error bars indicate 95% confidence intervals for the mean for N ≥ 10 animals; n.s. indicates P > 0.05 while asterisk indicates P ≤ 0.0001 (one-way ANOVA with Bonferroni correction for multiple comparisons). (E) NLP-7 inhibition of egg laying is not exclusive to HSN silencing. Scatter plots show the average number of eggs retained by wild-type animals, transgenic animals expressing Pertussis Toxin in HSN, and egl-47(ok677) mutant animals in the absence or presence of NLP-7 over-expression (OE) or the absence of HSNs. Error bars indicate 95% confidence intervals for the mean. Asterisks indicate P < 0.0001 (one-way ANOVA with Bonferroni correction for multiple comparisons); N ≥ 30 animals for each strain. (F) Measure of early stage eggs laid by wild-type or transgenic animals expressing Tetanus Toxin the VC neurons (black outlined boxes) in animals expressing Pertussis Toxin in the HSNs (green) or in goa-1(sa734) animals missing Gαo (blue). Error bars indicate ± 95% confidence intervals for the mean proportion. Asterisk indicates highlighted significant differences (P ≤ 0.0001) while n.s. indicates P > 0.05 (Fisher Exact Test with Bonferroni correction for multiple comparisons).
Because the HSNs appear to be the principal sites of inhibitory Gαo signaling, we tested how NLP-7 over-expression affects HSN Ca2+ activity. As expected, over-expression of NLP-7 strongly inhibited HSN Ca2+ activity (Figure 7, C and D), consistent with the strong egg-laying defects of these animals. To our surprise, loss of Gαo in these animals did now show high frequency HSN Ca2+ transient activity, despite showing the strong hyperactive egg-laying behavior of goa-1(sa734) single mutants (Figure 7, C and D). This result shows that the hyperactive egg-laying behavior of Gαo null mutants is unlinked to the increase in HSN Ca2+ activity we observe. Previous work showed NLP-7 inhibition of HSN activity and egg laying requires the EGL-47 receptor (Banerjee et al. 2017). To show directly whether NLP-7 inhibition of HSN activity requires the EGL-47 receptor, we recorded HSN Ca2+ activity in egl-47(ok677) null mutants. Unexpectedly, animals lacking EGL-47 had significantly fewer HSN Ca2+ transients than wild-type animals whether or not NLP-7 was over-expressed (Figure 7D). These results show that although NLP-7 signals to silence HSN Ca2+ activity, it does not require EGL-47 or Gαo function to do so.
To confirm whether NLP-7 inhibition of egg laying requires EGL-47 and Gαo function outside of HSN, we compared egg-laying behavior in NLP-7 transgenic animals where Gαo and synaptic transmission were blocked in defined cells of the egg-laying circuit. As expected from our Ca2+ imaging experiments, NLP-7 over-expressing animals accumulated a similar number of eggs as egl-1(n986dm) animals lacking HSNs (35.7 ± 1.4 eggs vs 33.4 ± 0.9 eggs Figure 7E). Transgenic expression of Pertussis Toxin in HSNs or loss of the EGL-47 receptor weakly suppressed egg accumulation in NLP-7 over-expressing animals (Figure 7E). The suppression of NLP-7 egg-laying defects by Pertussis Toxin expression was specific, as egl-1(n986dm) animals developmentally lacking HSNs showed no comparable decrease in egg retention and showed a mild but significant increase (Figure 7E), possibly through co-expression of Pertussis Toxin from the transgene in the NSMs (Tanis et al. 2008). Thus, NLP-7 inhibits egg laying, in part, through an inhibitory G protein like Gαo in HSN and the EGL-47 receptor. Because whole-animal, but not HSN-specific, elimination of Gαo function completely suppressed NLP-7-induced egg-laying defects (Figure 7A), these data show that NLP-7 signals to inhibit egg laying through Gαo-coupled receptors on cells other than HSN. The cholinergic VC neurons also make extensive synapses onto the vulval muscles, suggesting they may act alongside the HSNs to promote vulval muscle contractility and egg laying (White et al. 1986; Waggoner et al. 1998). To test whether enhanced VC neurotransmitter release was driving the hyperactive egg-laying behavior of Gαo signaling mutants, we transgenically expressed Tetanus Toxin in the VC neurons using a cell-specific promoter (Bany et al. 2003). Loss of VC synaptic transmission had no effect on egg-laying behavior by goa-1(sa734) null mutants or animals expressing Pertussis Toxin in HSN (Figure 7F). As previously reported (Mendel et al. 1995; Segalat et al. 1995), we find that loss of Gαo bypasses the HSNs, as goa-1(sa734); egl-1(n986dm) double mutants developmentally missing the HSNs still lay their eggs at early stages of development (Figure 7F). Together, these results show that NLP-7 neuropeptides and Gαo signal to inhibit egg-laying behavior through cellular targets other than the VCs and HSNs.
Discussion
Using a combination of genetic, imaging, physiological, and behavioral approaches, we found that the conserved G protein, Gαo, signals to depress activity in the C. elegans egg-laying circuit, preventing entry into the active behavior state. Neuropeptides, released in response to aversive external sensory input and feedback of successful egg release, activate inhibitory receptors and Gαo which signal to reduce cell electrical excitability and neurotransmitter release. Without inhibitory Gαo signaling, the presynaptic HSN command neurons remain electrically excited, rendering them resistant to inhibitory sensory input and feedback from the stretch-dependent homeostat. As a result, animals lacking Gαo enter the egg-laying active state twice as frequently as wild-type animals and in environments that would normally be unfavorable for egg laying. Thus, inhibitory Gαo signaling allows for a two-state pattern of circuit activity that can be properly gated by external and internal sensory input.
Our results inform our understanding of how G protein signaling modulates the stretch-dependent homeostat that governs egg-laying behavior. Even in “optimal” environmental conditions including abundant food, egg laying in wild-type animals typically begins ∼6 hours after the L4-adult molt upon the accumulation of 5–8 eggs in the uterus (Ravi et al. 2018a). Loss of inhibitory Gαo signaling causes precocious egg laying, with eggs being released soon after being deposited into the uterus. Animals lacking Gαo function in HSN show tonic Ca2+ transient activity even after chemical sterilization, conditions that normally inhibit HSN and vulval muscle Ca2+ activity in wild-type animals. Genetic perturbations that increase inhibitory Gαo signaling delay egg laying until ∼18 hours after the molt, like animals lacking HSNs altogether. Egg laying in animals with reduced or eliminated HSN function resumes when the accumulation of eggs activates the circuit via the stretch-dependent homeostat (Collins et al. 2016; Ravi et al. 2018a). Wild-type animals acutely exposed to aversive sensory conditions show a rapid inhibition of HSN activity and egg laying (Sawin 1996; Zhang et al. 2008; Fenk and de Bono 2015), but they continue to make and accumulate eggs in the uterus. Egg laying resumes upon removal of the inhibitory sensory stimulus or a return to food (Sawin 1996; Dong et al. 2000). Feedback of egg accumulation and release has long-term consequences on female reproductive physiology. The cGMP-dependent protein kinase G ortholog, EGL-4, regulates the expression of a novel secreted protein in the uterine epithelium whose levels correlate with egg-laying rate (Hao et al. 2011). Feedback of egg release regulates tph-1 gene expression that increases serotonin biosynthesis in the HSNs, alters sensory responses to male pheromone, and germline precursor cell proliferation (Aprison and Ruvinsky 2019a,b). Because the primary function of all adult C. elegans behaviors is survival and reproduction, we hypothesize that most or all salient sensory signals ultimately converge to modulate egg-laying circuit activity and animal fecundity.
Our work shows that Gαo also signals in cells other than HSN to inhibit egg-laying circuit activity and behavior. GOA-1 is likely expressed in all electrically excitable cells including all cells in the egg-laying circuit (Mendel et al. 1995; Segalat et al. 1995; Jose et al. 2007). Over-expression of NLP-7 neuropeptides strongly inhibits HSN Ca2+ activity and egg-laying behavior. While complete loss of Gαo suppresses egg-laying defects caused by NLP-7 overexpression, HSN Ca2+ activity remains largely inhibited. This suggests NLP-7 signals through Gαo to inhibit egg laying in cells other than HSN. Consistent with this result, goa-1 null mutants remain strongly hyperactive for egg laying even when HSN or VC synaptic transmission is blocked. Where else does Gαo function to inhibit egg laying? While Gαo is expressed in the vulval muscles, transgenic expression of Pertussis Toxin or a GTP-locked form of Gαo causes only modest egg-laying defects. Extensive work has shown that Gαo signals to inhibit release of acetylcholine from motor neurons during locomotion (Miller et al. 1999; Nurrish et al. 1999). EM reconstruction that revealed the C. elegans synaptic wiring diagram shows that the VA7 and VB6 cholinergic motor neurons that drive body wall muscle contraction for locomotion also synapse onto the vm1 vulval muscles (White et al. 1986). Recent work employing a new fluorescent reporter of acetylcholine shows rhythmic activity in the vm1 muscles (Borden et al. 2020), and this vm1 activity is precisely where we observe weak vulval muscle “twitch” Ca2+ transients (Collins et al. 2016; Brewer et al. 2019). Mutations that increase Gαs and cAMP signaling increase acetylcholine release from motor neurons and promote locomotion (Reynolds et al. 2005; Schade et al. 2005; Charlie et al. 2006a,2006b), possibly explaining why these mutations also increase egg laying. We predict that cholinergic neurons like VA7 and VB6, or their command interneurons, express neuropeptides and neurotransmitter receptors that signal through Gαo to depress acetylcholine release and excitation of the vulval muscles. Identifying where such receptors are expressed and how they signal will help explain how each cell in the circuit functions to drive discrete steps in egg laying (Brewer et al. 2019; Fernandez et al. 2020).
Our work is consistent with prior work showing Gαo inhibits synaptic transmission via modulation of presynaptic ion channels. Genetic studies in C. elegans have identified the IRK inward rectifying K+ channels, NCA Na+ leak channels, and CCA-1 T-type voltage-gated Ca2+ channels as potential targets of Gαo signaling (Yeh et al. 2008; Emtage et al. 2012; Zang et al. 2017; Topalidou et al. 2017b). Inward rectifying GIRK K+ channels are activated by release of βγ subunits (Hille 1994). Previous work has shown the IRK-1 K+ channel is expressed in HSN and is required for inhibition of egg laying by the Gαo-coupled EGL-6 neuropeptide receptor (Emtage et al. 2012). The egg-laying phenotypes of irk-1 mutant animals are not as strong as goa-1 mutants, and our results show that over-expression of βγ subunits in HSN causes little or no effect on egg laying. These results suggest Gαo signals to inhibit HSN neurotransmitter release via additional mechanism(s). NALCN Na+ leak channels are also expressed in HSN, and gain-of-function mutations increase HSN Ca2+ activity and drive hyperactive egg-laying and locomotion behaviors (Yeh et al. 2008). NALCN channels are genetically downstream of both Gαq and Gαo in the regulation of dopamine signaling and locomotion, suggesting that NALCN channels could be direct targets for modulation (Lutas et al. 2016; Topalidou et al. 2017a). Physiological experiments in mammalian neurons support this model. NALCN currents are activated by neuropeptide signaling through Gαq (Lu et al. 2009) and inhibited by dopamine and GABA signaling through Gαi/o (Philippart and Khaliq 2018). However, C. elegans knockout mutants in NALCN channel components do not show the strong behavior defects seen in Gαq and Gαo mutants, suggesting other channels act in parallel to NALCN to regulate HSN and circuit excitability. One candidate is TMC channels that drive a background Na+ leak conductance that promotes HSN and vulval muscle cell electrical excitability (Yue et al. 2018). TMC channels may also regulate cell electrical excitability as mechanosensors (Pan et al. 2018; Tang et al. 2020). Cl- channels and transporter proteins also regulate HSN activity and egg-laying behavior. CLH-3 is a swelling and hyperpolarization-activated, inwardly rectifying chloride channel that inhibits HSN activity (Branicky et al. 2014). Dynamic expression of Cl- transporter proteins in the HSNs as animals mature into egg-laying adults regulates the Cl- reversal potential (Tanis et al. 2009; Bellemer et al. 2011; Han et al. 2015). Loss of KCC-2 or ABTS-1 transporters flips the reduced egg laying behavior of egl-47(dm) mutants into hyperactive egg laying. Modulation of Cl- transporter expression or activity by G protein signaling could drive the shifts in cell excitability we observe between the inactive and active egg-laying behavior states.
The modulation of antagonistic cation and anion channels likely determines the Ca2+ dependent spiking probability of cells in the egg-laying circuit. HSN expresses L-type, P/Q-type, and T-type voltage-gated Ca2+ channels (Mathews et al. 2003; Zang et al. 2017), and each contributes to a normal serotonin response and egg-laying behavior (Schafer and Kenyon 1995; Lee et al. 1997; Kwok et al. 2006). The modest but significant changes in HSN resting membrane potential we observe in Gαo signaling mutants are expected to alter the probability of eliciting Ca2+ spiking activity. Ca2+-dependent action potentials have been found in C. elegans neurons and muscles (Gao and Zhen 2011; Liu et al. 2011, 2018) and is regulated by both L-type and T-type Ca2+ channels. T-type channels like CCA-1 can contribute to a “window current” where the channel can pass current at depolarized potentials that are insufficient to trigger channel inactivation (Williams et al. 1997; Crunelli et al. 2005; Zang et al. 2017). Activation of these window currents might allow neurons like HSN to shift from spontaneous tonic firing to high-frequency Ca2+ bursting. Future work leveraging the powerful molecular tools uniquely available in C. elegans and the egg-laying circuit along with direct physiological measurements of membrane potential will allow mechanistic insight into how neuromodulators like serotonin and neuropeptides signal through effectors to shape patterns of circuit activity that underlie distinct behavior states.
Acknowledgments
The authors thank Drs. Jessica Tanis and Michael Koelle for sharing unpublished strains. They thank Yuichi Iino for sharing plasmids. They thank Dr. Addys Bode Hernandez and Michael Scheetz for technical assistance. They also thank Drs. Qiang Liu, James Baker, Julia Dallman, Laura Bianchi, Brock Grill, Peter Larsson, Stephen Roper, along with members of the Collins lab for helpful discussions and feedback on the manuscript.
Funding
This work was funded by grants from the National Institutes of Health (NS086932) and the National Science Foundation (IOS-1844657) to K.M.C. R.J.K. 3rd was supported by a University of Miami Maytag Fellowship. J.M.K. is supported by a grant from the National Institutes of Health (NS032196). Strains used in this study have been provided to the C. elegans Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Conflicts of interest
None declared.
Literature cited
- Alkema MJ, Hunter-Ensor M, Ringstad N, Horvitz HR.. 2005. Tyramine functions independently of octopamine in the Caenorhabditis elegans nervous system. Neuron. 46:247–260. [DOI] [PubMed] [Google Scholar]
- Aprison EZ, Ruvinsky I.. 2014. Balanced trade-offs between alternative strategies shape the response of C. elegans reproduction to chronic heat stress. PLoS One. 9:e105513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprison EZ, Ruvinsky I.. 2019a. Coordinated behavioral and physiological responses to a social signal are regulated by a shared neuronal circuit. Curr Biol. 29:4108–4115.e4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprison EZ, Ruvinsky I.. 2019b. Dynamic regulation of adult-specific functions of the nervous system by signaling from the reproductive system. Curr Biol. 29:4116–4123.e4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N, Bhattacharya R, Gorczyca M, Collins KM, Francis MM.. 2017. Local neuropeptide signaling modulates serotonergic transmission to shape the temporal organization of C. elegans egg-laying behavior. PLoS Genet. 13:e1006697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bany IA, Dong MQ, Koelle MR.. 2003. Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J Neurosci. 23:8060–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiani CA, Gharib S, Simon MI, Sternberg PW.. 2003. Caenorhabditis elegans Galphaq regulates egg-laying behavior via a PLCbeta-independent and serotonin-dependent signaling pathway and likely functions both in the nervous system and in muscle. Genetics. 165:1805–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellemer A, Hirata T, Romero MF, Koelle MR.. 2011. Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J. 30:1852–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden PM, Zhang P, Shivange AV, Marvin JS, Cichon J, et al. 2020. A fast genetically encoded fluorescent sensor for faithful in vivo acetylcholine detection in mice, fish, worms and flies. bioRxiv. 2020.2002.2007.939504. [Google Scholar]
- Branicky R, Miyazaki H, Strange K, Schafer WR.. 2014. The voltage-gated anion channels encoded by clh-3 regulate egg laying in C. elegans by modulating motor neuron excitability. J Neurosci. 34:764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JC, Collins KM, Koelle MR, Olsen A.. 2019. Serotonin and neuropeptides are both released by the HSN command neuron to initiate C. elegans egg laying. PLoS Genet. 15:e1007896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, et al. 1996. Mutations in a C. elegans Gqalpha gene disrupt movement, egg laying, and viability. Neuron. 16:999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnell L, Illi J, Hong SW, McIntire SL.. 2005. The G-protein-coupled serotonin receptor SER-1 regulates egg laying and male mating behaviors in Caenorhabditis elegans. J Neurosci. 25:10671–10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Deletion Mutant Consortium. 2012. large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 (Bethesda). 2:1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlie NK, Schade MA, Thomure AM, Miller KG.. 2006a. Presynaptic UNC-31 (CAPS) is required to activate the G alpha(s) pathway of the Caenorhabditis elegans synaptic signaling network. Genetics. 172:943–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlie NK, Thomure AM, Schade MA, Miller KG.. 2006b. The Dunce cAMP phosphodiesterase PDE-4 negatively regulates G alpha(s)-dependent and G alpha(s)-independent cAMP pools in the Caenorhabditis elegans synaptic signaling network. Genetics. 173:111–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Pepper JS, Koelle MR.. 2004. Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci. 7:1096–1103. [DOI] [PubMed] [Google Scholar]
- Collins KM, Bode A, Fernandez RW, Tanis JE, Brewer JC, et al. 2016. Activity of the C. elegans egg-laying behavior circuit is controlled by competing activation and feedback inhibition. eLife. 5:e21126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KM, Koelle MR.. 2013. Postsynaptic ERG potassium channels limit muscle excitability to allow distinct egg-laying behavior states in Caenorhabditis elegans. J Neurosci. 33:761–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Toth TI, Cope DW, Blethyn K, Hughes SW.. 2005. The ‘window’ T-type calcium current in brain dynamics of different behavioural states. J Physiol. 562:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey CM, Mackenzie SM, Gargus A, Blanco G, Sze JY.. 2005. Serotonin (5HT), fluoxetine, imipramine and dopamine target distinct 5HT receptor signaling to modulate Caenorhabditis elegans egg-laying behavior. Genetics. 169:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MQ, Chase D, Patikoglou GA, Koelle MR.. 2000. Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev. 14:2003–2014. [PMC free article] [PubMed] [Google Scholar]
- Emtage L, Aziz-Zaman S, Padovan-Merhar O, Horvitz HR, Fang-Yen C, et al. 2012. IRK-1 potassium channels mediate peptidergic inhibition of Caenorhabditis elegans serotonin neurons via a G(o) signaling pathway. J Neurosci. 32:16285–16295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Schiavi ED, Bergamasco C, Bazzicalupo P.. 2007. Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene. 395:170–176. [DOI] [PubMed] [Google Scholar]
- Fenk LA, de Bono M.. 2015. Environmental CO2 inhibits Caenorhabditis elegans egg-laying by modulating olfactory neurons and evokes widespread changes in neural activity. Proc Natl Acad Sci USA. 112:E3525–E3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez RW, Wei K, Wang EY, Mikalauskaite D, Olson A, et al. 2020. Cellular expression and functional roles of all 26 Neurotransmitter GPCRs in the C. elegans egg-laying circuit. J Neurosci. 40:7475–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, et al. 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 40:1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Hino T, Miyamoto R, Inada H, Mori I, et al. 2015. The importance of cGMP signaling in sensory cilia for body size regulation in Caenorhabditis elegans. Genetics. 201:1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Sengupta P, McIntire SL.. 2002. Regulation of body size and behavioral state of C. elegans by sensory perception and the EGL-4 cGMP-dependent protein kinase. Neuron. 36:1091–1102. [DOI] [PubMed] [Google Scholar]
- Gao S, Zhen M.. 2011. Action potentials drive body wall muscle contractions in Caenorhabditis elegans. Proc Natl Acad Sci USA. 108:2557–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J, Collins KM.. 2019. The HSN egg-laying command neurons are required for normal defecation frequency in Caenorhabditis elegans (II). MicroPubl Biol. doi: 10.17912/10.17912/micropub.biology.000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghil S, Choi JM, Kim SS, Lee YD, Liao Y, et al. 2006. Compartmentalization of protein kinase A signaling by the heterotrimeric G protein Go. Proc Natl Acad Sci USA. 103:19158–19163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding EH, Schenk AK, Juneja P, MacKay AW, Wade JM, et al. 2008. A robust automated system elucidates mouse home cage behavioral structure. Proc Natl Acad Sci USA. 105:20575–20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel G, Gustafson MA, Pepper JS, Horvitz HR, Koelle MR.. 2012. Receptors and other signaling proteins required for serotonin control of locomotion in Caenorhabditis elegans. Genetics. 192:1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Bellemer A, Koelle MR.. 2015. An evolutionarily conserved switch in response to GABA affects development and behavior of the locomotor circuit of Caenorhabditis elegans. Genetics. 199:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Xu N, Box AC, Schaefer L, Kannan K, et al. 2011. Nuclear cGMP-dependent kinase regulates gene expression via activity-dependent recruitment of a conserved histone deacetylase complex. PLoS Genet. 7:e1002065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Fire A.. 1998. Muscle and nerve-specific regulation of a novel NK-2 class homeodomain factor in Caenorhabditis elegans. Development. 125:421–429. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, et al. 1996. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 380:258–262. [DOI] [PubMed] [Google Scholar]
- Hille B. 1994. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 17:531–536. [DOI] [PubMed] [Google Scholar]
- Hobson RJ, Hapiak VM, Xiao H, Buehrer KL, Komuniecki PR, et al. 2006. SER-7, a Caenorhabditis elegans 5-HT7-like receptor, is essential for the 5-HT stimulation of pharyngeal pumping and egg laying. Genetics. 172:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD.. 1982. Serotonin and octopamine in the nematode Caenorhabditis elegans. Science. 216:1012–1014. [DOI] [PubMed] [Google Scholar]
- Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, et al. 1999. The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat Genet. 21:414–419. [DOI] [PubMed] [Google Scholar]
- Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L.. 2001. Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci USA. 98:3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose AM, Bany IA, Chase DL, Koelle MR.. 2007. A specific subset of transient receptor potential vanilloid-type channel subunits in Caenorhabditis elegans endocrine cells function as mixed heteromers to promote neurotransmitter release. Genetics. 175:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose AM, Koelle MR.. 2005. Domains, amino acid residues, and new isoforms of Caenorhabditis elegans diacylglycerol kinase 1 (DGK-1) important for terminating diacylglycerol signaling in vivo. J Biol Chem. 280:2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi I, Shibasaki H, Takahashi K, Tohyama K, Kurachi Y, et al. 1990. Purification and characterization of five different alpha subunits of guanine-nucleotide-binding proteins in bovine brain membranes. Their physiological properties concerning the activities of adenylate cyclase and atrial muscarinic K+ channels. Eur J Biochem. 191:499–506. [DOI] [PubMed] [Google Scholar]
- Koelle MR. 2018. Neurotransmitter signaling through heterotrimeric G proteins: insights from studies in C. elegans. WormBook. 1–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelle MR, Horvitz HR.. 1996. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 84:115–125. [DOI] [PubMed] [Google Scholar]
- Kopchock RJ 3rd, Ravi B, Bode A, Collins KM.. 2021. The sex-specific VC Neurons are mechanically activated motor neurons that facilitate Serotonin-induced egg laying in C. elegans. J Neurosci. 41:3635–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok TC, Ricker N, Fraser R, Chan AW, Burns A, et al. 2006. A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature. 441:91–95. [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM.. 1999. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 24:335–346. [DOI] [PubMed] [Google Scholar]
- Lee RY, Lobel L, Hengartner M, Horvitz HR, Avery L.. 1997. Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 16:6066–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Etoile ND, Coburn CM, Eastham J, Kistler A, Gallegos G, et al. 2002. The cyclic GMP-dependent protein kinase EGL-4 regulates olfactory adaptation in C. elegans. Neuron. 36:1079–1089. [DOI] [PubMed] [Google Scholar]
- Li P, Collins KM, Koelle MR, Shen K.. 2013. LIN-12/Notch signaling instructs postsynaptic muscle arm development by regulating UNC-40/DCC and MADD-2 in Caenorhabditis elegans. eLife. 2:e00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Ge Q, Chen B, Salkoff L, Kotlikoff MI, et al. 2011. Genetic dissection of ion currents underlying all-or-none action potentials in C. elegans body-wall muscle cells. J Physiol. 589:101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kidd PB, Dobosiewicz M, Bargmann CI.. 2018. C. elegans AWA olfactory neurons fire calcium-mediated all-or-none action potentials. Cell. 175:57–70.e17. [DOI] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, Schneggenburger R.. 2008. Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci. 28:8257–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Su Y, Das S, Wang H, Wang Y, et al. 2009. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature. 457:741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutas A, Lahmann C, Soumillon M, Yellen G.. 2016. The leak channel NALCN controls tonic firing and glycolytic sensitivity of substantia nigra pars reticulata neurons. eLife. 5:e15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E. 2012. Neuromodulation of neuronal circuits: back to the future. Neuron. 76:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews EA, Garcia E, Santi CM, Mullen GP, Thacker C, et al. 2003. Critical residues of the Caenorhabditis elegans unc-2 voltage-gated calcium channel that affect behavioral and physiological properties. J Neurosci. 23:6537–6545., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara H. 2002. [Angiotensin II type 2 (AT2) receptor signal and cardiovascular action]. Nihon Yakurigaku Zasshi. 119:95–102. [DOI] [PubMed] [Google Scholar]
- McMullan R, Hiley E, Morrison P, Nurrish SJ.. 2006. Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev. 20:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan R, Nurrish SJ.. 2011. The RHO-1 RhoGTPase modulates fertility and multiple behaviors in adult C. elegans. PLoS One. 6:e17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen PD, Aprison EZ, Winter PB, Amaral LA, Morimoto RI, et al. 2012. Macro-level modeling of the response of C. elegans reproduction to chronic heat stress. PLoS Comput Biol. 8:e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, et al. 1995. Participation of the protein Go in multiple aspects of behavior in C. elegans. Science. 267:1652–1655. [DOI] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, Rand JB.. 1999. Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron. 24:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DH, Stiles JW, Santelli J, Sanadi DR.. 1979. Synchronous growth and aging of Caenorhabditis elegans in the presence of fluorodeoxyuridine. J Gerontol. 34:28–36. [DOI] [PubMed] [Google Scholar]
- Moresco JJ, Koelle MR.. 2004. Activation of EGL-47, a Galpha(o)-coupled receptor, inhibits function of hermaphrodite-specific motor neurons to regulate Caenorhabditis elegans egg-laying behavior. J Neurosci. 24:8522–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby SM, Heukeroth RO, Gordon JI, Gilman AG.. 1990. G-protein alpha-subunit expression, myristoylation, and membrane association in COS cells. Proc Natl Acad Sci USA. 87:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurrish S, Segalat L, Kaplan JM.. 1999. Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron. 24:231–242. [DOI] [PubMed] [Google Scholar]
- Oikonomou G, Altermatt M, Zhang RW, Coughlin GM, Montz C, et al. 2019. The serotonergic raphe promote sleep in Zebrafish and Mice. Neuron. 103:686–701.e688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Akyuz N, Liu XP, Asai Y, Nist-Lund C, et al. 2018. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. 99:736–753.e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patikoglou GA, Koelle MR.. 2002. An N-terminal region of Caenorhabditis elegans RGS proteins EGL-10 and EAT-16 directs inhibition of G(alpha)o versus G(alpha)q signaling. J Biol Chem. 277:47004–47013. [DOI] [PubMed] [Google Scholar]
- Philippart F, Khaliq ZM.. 2018. Gi/o protein-coupled receptors in dopamine neurons inhibit the sodium leak channel NALCN. eLife. 7:e40984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raizen DM, Cullison KM, Pack AI, Sundaram MV.. 2006. A novel gain-of-function mutant of the cyclic GMP-dependent protein kinase egl-4 affects multiple physiological processes in Caenorhabditis elegans. Genetics. 173:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi B, Garcia J, Collins KM.. 2018a. Homeostatic feedback modulates the development of two-state patterned activity in a model serotonin motor circuit in Caenorhabditis elegans. J Neurosci. 38:6283–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi B, Nassar LM, Kopchock RJ 3rd, Dhakal P, Scheetz M, et al. 2018b. Ratiometric calcium imaging of individual neurons in behaving Caenorhabditis elegans. J Vis Exp. 132:56911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhi JA, et al. 1994. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 370:143–146. [DOI] [PubMed] [Google Scholar]
- Reynolds NK, Schade MA, Miller KG.. 2005. Convergent, RIC-8-dependent Galpha signaling pathways in the Caenorhabditis elegans synaptic signaling network. Genetics. 169:651–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Horvitz HR.. 2008. FMRFamide neuropeptides and acetylcholine synergistically inhibit egg-laying by C. elegans. Nat Neurosci. 11:1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robatzek M, Thomas JH.. 2000. Calcium/calmodulin-dependent protein kinase II regulates Caenorhabditis elegans locomotion in concert with a G(o)/G(q) signaling network. Genetics. 156:1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin ER. 1996. Genetic and Cellular Analysis of Modulated Behaviors in Caenorhabditis elegans. Cambridge, USA: Massachusetts Institute of Technology. [Google Scholar]
- Schade MA, Reynolds NK, Dollins CM, Miller KG.. 2005. Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the G alpha(s) pathway and define a third major branch of the synaptic signaling network. Genetics. 169:631–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer WF. 2006. Genetics of egg-laying in worms. Annu Rev Genet. 40:487–509. [DOI] [PubMed] [Google Scholar]
- Schafer WR, Kenyon CJ.. 1995. A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature. 375:73–78. [DOI] [PubMed] [Google Scholar]
- Segalat L, Elkes DA, Kaplan JM.. 1995. Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science. 267:1648–1651. [DOI] [PubMed] [Google Scholar]
- Shen K, Fetter RD, Bargmann CI.. 2004. Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell. 116:869–881. [DOI] [PubMed] [Google Scholar]
- Shyn SI, Kerr R, Schafer WR.. 2003. Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr Biol. 13:1910–1915. [DOI] [PubMed] [Google Scholar]
- Taghert PH, Nitabach MN.. 2012. Peptide neuromodulation in invertebrate model systems. Neuron. 76:82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YQ, Lee SA, Rahman M, Vanapalli SA, Lu H, et al. 2020. Ankyrin is an intracellular tether for TMC mechanotransduction channels. Neuron. 107:759–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanis JE, Bellemer A, Moresco JJ, Forbush B, Koelle MR.. 2009. The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci. 29:9943–9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanis JE, Moresco JJ, Lindquist RA, Koelle MR.. 2008. Regulation of serotonin biosynthesis by the G proteins Galphao and Galphaq controls serotonin signaling in Caenorhabditis elegans. Genetics. 178:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalidou I, Chen PA, Cooper K, Watanabe S, Jorgensen EM, et al. 2017a. The NCA-1 and NCA-2 Ion channels function downstream of Gq and Rho to regulate locomotion in Caenorhabditis elegans. Genetics. 206:265–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalidou I, Cooper K, Pereira L, Ailion M.. 2017b. Dopamine negatively modulates the NCA ion channels in C. elegans. PLoS Genet. 13:e1007032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent C. 1982. Genetic and Behavioral Studies of the Egg-Laying System of Caenorhabditis elegans. Cambridge, USA: Massachusetts Institute of Technology. [Google Scholar]
- Trent C, Tsuing N, Horvitz HR.. 1983. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 104:619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner LE, Hardaker LA, Golik S, Schafer WR.. 2000. Effect of a neuropeptide gene on behavioral states in Caenorhabditis elegans egg-laying. Genetics. 154:1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner LE, Zhou GT, Schafer RW, Schafer WR.. 1998. Control of alternative behavioral states by serotonin in Caenorhabditis elegans. Neuron. 21:203–214. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S.. 1986. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 314:1–340. [DOI] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M.. 2007. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 54:275–290. [DOI] [PubMed] [Google Scholar]
- Williams SL, Lutz S, Charlie NK, Vettel C, Ailion M, et al. 2007. Trio's Rho-specific GEF domain is the missing Galpha q effector in C. elegans. Genes Dev. 21:2731–2746., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Toth TI, Turner JP, Hughes SW, Crunelli V.. 1997. The ‘window’ component of the low threshold Ca2+ current produces input signal amplification and bistability in cat and rat thalamocortical neurones. J Physiol. 505:689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Hirotsu T, Matsuki M, Kunitomo H, Iino Y.. 2009. GPC-1, a G protein gamma-subunit, regulates olfactory adaptation in Caenorhabditis elegans. Genetics. 181:1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. 1999. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol. 515:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Ng S, Zhang M, Bouhours M, Wang Y, et al. 2008. A putative cation channel, NCA-1, and a novel protein, UNC-80, transmit neuronal activity in C. elegans. PLoS Biol. 6:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Zhao J, Li X, Fan Y, Duan D, et al. 2018. TMC proteins modulate egg laying and membrane excitability through a background leak conductance in C. elegans. Neuron. 97:571–585.e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang KE, Ho E, Ringstad N.. 2017. Inhibitory peptidergic modulation of C. elegans serotonin neurons is gated by T-type calcium channels. eLife. 6:e22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Pratt RE.. 1996. The AT2 receptor selectively associates with Gialpha2 and Gialpha3 in the rat fetus. J Biol Chem. 271:15026–15033. [PubMed] [Google Scholar]
- Zhang M, Chung SH, Fang-Yen C, Craig C, Kerr RA, et al. 2008. A self-regulating feed-forward circuit controlling C. elegans egg-laying behavior. Curr Biol. 18:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Fu J, Zhang H, Du K, Huang W, et al. 2018. Decoding the intensity of sensory input by two glutamate receptors in one C. elegans interneuron. Nat Commun. 9:4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors affirm that all data necessary for confirming the conclusions of this article are represented fully within the article and its tables and figures. Requests for strains, plasmids, and ratiometric fluorescence recordings used to generate Ca2+ traces can be directed to KMC. Supplementary material is available at figshare: https://doi.org/10.25386/genetics.14627469.