

Abstract
The junctophilin family of proteins tether together plasma membrane (PM) and endoplasmic reticulum (ER) membranes, and couple PM- and ER-localized calcium channels. Understanding in vivo functions of junctophilins is of great interest for dissecting the physiological roles of ER-PM contact sites. Here, we show that the sole Caenorhabditis elegans junctophilin JPH-1 localizes to discrete membrane contact sites in neurons and muscles and has important tissue-specific functions. jph-1 null mutants display slow growth and development due to weaker contraction of pharyngeal muscles, leading to reduced feeding. In the body wall muscle, JPH-1 colocalizes with the PM-localized EGL-19 voltage-gated calcium channel and ER-localized UNC-68 RyR calcium channel, and is required for animal movement. In neurons, JPH-1 colocalizes with the membrane contact site protein Extended-SYnaptoTagmin 2 (ESYT-2) in the soma, and is present near presynaptic release sites. Interestingly, jph-1 and esyt-2 null mutants display mutual suppression in their response to aldicarb, suggesting that JPH-1 and ESYT-2 have antagonistic roles in neuromuscular synaptic transmission. Additionally, we find an unexpected cell nonautonomous effect of jph-1 in axon regrowth after injury. Genetic double mutant analysis suggests that jph-1 functions in overlapping pathways with two PM-localized voltage-gated calcium channels, egl-19 and unc-2, and with unc-68 for animal health and development. Finally, we show that jph-1 regulates the colocalization of EGL-19 and UNC-68 and that unc-68 is required for JPH-1 localization to ER-PM puncta. Our data demonstrate important roles for junctophilin in cellular physiology, and also provide insights into how junctophilin functions together with other calcium channels in vivo.
Keywords: membrane contact site, esyt-2, ryanodine receptor/unc-68, VGCC/egl-19
Introduction
Membrane contact sites (MCSs) are regions of close contact, generally within 10–30 nm, between organelles or between an organelle and the plasma membrane (PM). MCSs were first described between the endoplasmic reticulum (ER) and PM in muscle cells by electron microscopy over 60 years ago (Porter and Palade 1957). MCSs have now been found for most organelles in many organisms (Lang et al. 2015; Valm et al. 2017). MCSs are maintained by protein tethers that bind to opposing membranes simultaneously and hold them in close proximity. Different types of MCSs are organized by distinct protein tethers, many of which are conserved from yeast to mammals (Phillips and Voeltz 2016). Recent studies have begun to uncover their functions. For example, oxysterol-binding proteins (OSBPs) facilitate exchange of PM-localized phosphatidylinositol 4-phosphate (PI4P) for ER-localized cholesterol (Mesmin et al. 2013), and binding of ER-localized calcium sensor Stim1 to PM-localized calcium channel Orai1 triggers the entry of extracellular calcium to the ER to replenish calcium stores (Hirve et al. 2018). Genetic analysis suggests many MCS tethering proteins act redundantly. For example, studies of ER-PM contact sites in yeast showed that full separation of the ER from the PM is only achieved when six genes encoding MCS proteins are deleted (Manford et al. 2012). Similarly, in Caenorhabditis elegans, enlarged lysosomes and endosomes were observed only when knocking out all four obr genes encoding OSBP homologs (Kobuna et al. 2010). It is thus necessary to identify new experimental models or paradigms to tease apart the functions of individual MCS proteins and to dissect their interaction network in vivo.
The junctophilin (JPH) family of proteins was first identified based on their localization to muscle ER-PM contact sites in a screen using monoclonal antibodies raised against ER vesicles enriched for ER-PM junctions (Takeshima et al. 2000). Junctophilins are characterized by an N-terminal domain consisting of eight membrane occupation and recognition nexus (MORN) motifs, which bind to the PM, and a C-terminal transmembrane domain, which anchors the protein to the ER. Mammals have four junctophilins (JPH1 through 4) that are differentially expressed in excitable cells. JPH1 and JPH2 are expressed in skeletal and cardiac muscle (Nishi et al. 2000; Takeshima et al. 2000) and the smooth muscle surrounding arteries (Pritchard et al. 2019; Saeki et al. 2019). JPH3 and JPH4 are broadly expressed in neurons of the brain and many parts of the nervous system (Nishi et al. 2000, 2003; Takeshima et al. 2000). Studies of genetic knockout mice have provided some evidence for their functions. Cardiomyocytes from JPH2 knockout mice have fewer ER-PM contacts, and skeletal muscle from JPH1 knockout mice have abnormal ER morphology and fewer ER-PM contacts (Takeshima et al. 2000; Ito et al. 2001). In addition to tethering together ER and PM membranes, junctophilins bind to ER- and PM-localized calcium channels and facilitate their colocalization at ER-PM contact sites in mouse cardiomyocytes, skeletal muscle, and cultured hippocampal neurons (Van Oort et al. 2011; Nakada et al. 2018; Sahu et al. 2019). Junctophilin-mediated ER-PM tetheringis reported to promote efficient excitation-contraction coupling in heart and skeletal muscle (Takeshima et al. 2000; Ito et al. 2001; Van Oort et al. 2011; Nakada et al. 2018) and regulate action potential frequency in neurons (Moriguchi et al. 2006; Kakizawa et al. 2007; Sahu et al. 2019). Unlike mammals, invertebrates have a single junctophilin (Garbino et al. 2009). In D. melanogaster, the sole junctophilin was shown to have roles in muscle contraction and neural development (Calpena et al. 2018).
Caenorhabditis elegans has a single junctophilin gene named jph-1 (Yoshida et al. 2001; Garbino et al. 2009). Here, we show that JPH-1 protein localizes to punctate structures in muscles and neurons. In muscles, JPH-1 colocalizes with the ER-localized UNC-68/RyR calcium channel and PM-localized EGL-19/Cav1 calcium channel, and is also necessary for the colocalization of the two channels. Through characterization of jph-1 null mutants and tissue-specific rescue experiments, we defined roles of jph-1 in pharyngeal and body muscles. We also observed a cell nonautonomous effect of jph-1 in axon regeneration after injury. In neurons, JPH-1 colocalizes with the ER-PM contact site protein extended-synaptotagmin 2 (ESYT-2) in the soma, and is present near presynaptic release sites. jph-1 modulates synaptic transmission and genetically antagonizes the effects of esyt-2. Genetic double-mutant analyses reveal differential interactions between jph-1 and unc-68/RyR and two PM-localized voltage-gated calcium channels (VGCCs) for animal development and health. Lastly, we show that precise localization of JPH-1 in both neurons and muscles depends on unc-68. These data support critical roles of junctophilin in cellular function and animal development.
Materials and methods
Caenorhabditis elegans genetics
Wild-type C. elegans is the N2 Bristol variant (Brenner 1974). Strains were maintained under standard conditions on Nematode Growth Media (NGM) plates seeded with Escherichia coli OP50 bacteria. New strains were constructed using standard procedures, based on a combination of visual identification of phenotypes, such as uncoordinated (Unc) movement, and genotyping for specific alleles. Strains are listed in Supplementary Table S1; primers for genotyping are listed in Supplementary Table S4.
Molecular biology and transgenesis
We cloned jph-1 cDNAs from wild-type N2 mRNAs, first using primers YJ12558 5′-GACGTAGGTGTGTCAGCAG-3′ and YJ12559 5′-CCTGAGGAGAAGTGTGTCTG-3′ in the 5ʹUTR and 3ʹUTR of jph-1, followed by a second round of amplification using primers YJ12560 5′-ATGAATGGAGGCAGATTTGAC-3′ and YJ12561 5′-CTACGAAGAAGACTTCTTCTTCTTC-3′ targeting the start and stop codons. We obtained two amplified products, which were cloned into pCR8 vectors. Sanger sequencing analysis of these clones revealed a 2.2-kb cDNA encoding JPH-1 isoform A, and a 2.4-kb cDNA encoding JPH-1 isoform B. The coding region of JPH-1B was then amplified using primers YJ12560 5′-ATGAATGGAGGCAGATTTGAC-3′ and YJ12562 5′-CTAATATGTGAGGGTGTGTACCG-3′ and cloned into a pCR8 vector. The 4.5-kb jph-1 promoter was amplified from wild-type genomic DNA using the primers YJ12563 5′-TGTTCTGCCATTACCAGCCCG-3′ and YJ12564 5′-TTCCCATTTGCCGTACTGCTG -3′. All expression constructs were generated either by Gateway recombination (Invitrogen/Thermo Fisher Scientific), Gibson assembly (New England Biolabs), or restriction enzyme digest and ligation. All expression clones were sequenced to ensure sequence fidelity.
We generated transgenic lines by microinjection, as described (Mello et al. 1991). Plasmids, fosmids, coinjection markers, and injection concentrations are listed in Supplementary Table S2.
Single-copy insertion transgenes with ju designation were generated on Chromosome IV at cxTi10882, following a previously published protocol (Andrusiak et al. 2019). Briefly, we injected N2 hermaphrodites with four plasmids, one containing GFP-cDNA flanked by homology arms and expressing a hygromycin resistance gene (HygR), pCZGY2750 expressing Cas9 and an sgRNA targeting cxTi10882, and pCFJ90 Pmyo-2-mCherry (Addgene plasmid 19327) and pCFJ104 Pmyo-3-mCherry (Addgene plasmid 19328)(Frøkjær-Jensen et al. 2008) as coinjection markers. F1 animals from injected P0 parents were treated with hygromycin (Hyg). Among the survivors, we looked for the absence of coinjections markers to identify animals with genomic insertion, which was further verified by PCR genotyping using primers YJ10503, YJ10504, and YJ10686 (wild type 562 bp, insertion 744 bp). Single-copy insertion transgenes nuTi144 and nuTi456 were generated by using a modified Mos1 transposon, following a previously published protocol (Frøkjær-Jensen et al. 2014).
CRISPR-Cas9 gene editing
We generated the jph-1(ju1683) and jph-1(ju1684) deletion alleles using two CRISPR RNAs (crRNAs): 5′-CCGTCCGGTAACACCTATCA-3′ and 5′-ACGACGTTGACCAGCAAGAC-3′ (Integrated DNA Technologies) targeting jph-1 exon 1 and exon 9, respectively (Supplementary Table S4). The crRNAs were injected into wild-type hermaphrodites with purified Cas9 (MacroLabs, University of California, Berkeley), trans-activating crRNA (tracrRNA) and dpy-10 crRNA, as described (Paix et al. 2015). We selected small and slow-growing Unc animals resembling jph-1(ok2823) mutants, as we were unable to isolate animals based on Dpy or Rol phenotypes, possibly because the jph-1 crRNA was more efficient than the dpy-10 crRNA. We identified ju1683 and ju1684 as deletions in jph-1 by PCR genotyping with flanking primers YJ12565 5′-GACGACGGCGGAACCTATG-3′ and YJ12566 5′-TCAGGTACGTTCTAGTCGGT-3′ (Supplementary Table S4).
GFP11 knock-in alleles unc-68(nu664) and egl-19(nu674) were generated by injecting wild-type hermaphrodites with 75 ng/μl pDD162 expressing Cas9, 36 ng/μl pRB1017-derived guide RNA, and 75 ng/μl of a PCR product of seven copies of GFP11 flanked by 1 kb of wild-type sequence 5ʹ and 3ʹ of the cut site. mCherry11 (Feng et al. 2019) knock-in allele unc-68(nu628) was generated by injecting wild-type hermaphrodites with 75 ng/μl pDD162 expressing Cas9, 36 ng/μl pRB1017-derived guide RNA, and 75 ng/μl of a PCR product of four copies of mCherry11 flanked by 1 kb of wild-type sequence 5ʹ and 3ʹ of the cut site. Guide RNAs were selected using the CRISPR guide RNA selection tool (http://genome.sfu.ca/crispr/). The plasmid pGW28 (36ng/μl) containing sgRNA targeting unc-58) and a repair oligo (AF-JA-76) were also co-injected as a co-conversation marker (El Mouridi et al. 2017).
Animal growth assessment
Adult hermaphrodite animals were placed on seeded NGM plates and allowed to lay eggs for 2 h, after which they were removed. The plates were kept at 20°C and observed daily to determine the time it took the offspring to reach the fourth larval (L4) stage.
Brood size assay
L4 hermaphrodite animals were individually placed on seeded NGM plates and moved to new plates daily. Two days after a parent animal was placed on a plate, the number of hatched offspring was counted. This was continued until parent animals laid no more eggs or died. The number of hatched offspring produced per parent animal was totaled to calculate brood size.
Fluorescence microscopy
Animals were generally immobilized in a drop of M9 solution with or without 30 mM muscimol or 10 mM levamisole on a 4% agar pad or 10% agarose pad. Most confocal fluorescence images were collected with a 63× objective lens using either a Zeiss LSM800 or LSM710 confocal microscopes, with Z-stacks at 0.5- or 1 µm intervals. Animals coexpressing UNC-68::mCherry and EGL-19::GFP (Figure 7A) were immobilized with latex beads (Kim et al. 2013), and confocal fluorescence images were collected with a 60× objective lens using a Nikon A1R confocal microscope, with Z-stacks at 0.15 µm intervals. Maximum intensity projections were prepared using Fiji (ImageJ). Images of GFP-labeled touch neurons [Pmec-7-GFP(muIs32)] in wild-type and jph-1(0) animals were taken on a Zeiss Axio Imager A2 compound scope at 10× magnification under identical settings.
Figure 7.

JPH-1 and calcium channels require each other for localization. (A) jph-1 is required for UNC-68 and EGL-19 colocalization in body wall muscle. Maximum intensity projection of animal with split-mCherry knock-in unc-68 (nu628) and split-GFP knock-in egl-19 (nu674) expressing muscle mCherry1-10 and GFP1-10 [Ppat-10-mCherry1-10::SL2::GFP1-10(nuTi456)] in wild-type (left) and jph-1(0) (right) animals. (B and C) Single-plane confocal images of GFP::JPH-1A expressed under the jph-1 promoter [Pjph-1-GFP::JPH-1A(juEx7999)] in wild type (WT), unc-2(e55), and unc-68(e540) backgrounds. WT and unc-2(e55) images were taken at 2% laser power and unc-68(e55) was taken at 4.5% laser power to compensate for the slight variation in expression level. (B) In the body wall muscle, JPH-1A localizes to rows of puncta in WT and unc-2(e55) animals, while in unc-68(e540) animals JPH-1A puncta are less distinct. (C) In neurons of the head ganglia, JPH-1A localizes to reticulate structures surrounding the nucleus and forms puncta in the cell periphery of WT and unc-2(e55) animals, but not unc-68(e540) mutants. Arrows mark some of the GFP::JPH-1A puncta. Scale bar, 5 µm.
Brightfield microscopy
Images depicting gross body morphology (Figure 1C) were taken by immobilizing animals in a drop of M9 solution on a 10% agarose pad and imaging on a Leica DMi8 microscope under brightfield settings at 10× magnification with an Andor iXon Ultra camera. Images depicting animal movement crawling on NGM petri plates (Figure 4A) were taken on a Zeiss M2 stereodissecting microscope with a Nikon DS-Qi1Mc camera.
Figure 1.

jph-1 expresses two isoforms that differ at their C-termini and is required for animal development. (A) Illustration of jph-1 spliced isoforms and deletion alleles. Exons are dark gray boxes, introns are black lines, and UTRs are light gray boxes. ok2823 is a 637-bp deletion, ju1683 is a 3891-bp deletion, and ju1684 is a 3858-bp deletion with a 13-bp insertion. (B) Illustration of C. elegans JPH-1 proteins predicted from isolated cDNA sequences in comparison to human JPH proteins. Dark gray boxes indicate MORN repeats and white boxes indicate transmembrane domains. The striped box at the C-terminus of JPH-1B indicates the 35 amino residues predicted from the cDNA. These 35 amino acids are not predicted to form a transmembrane region or low-complexity domain using Pfam (El-Gebali et al. 2019), the TMHMM Server v 2.0 (http://www.cbs.dtu.dk/services/TMHMM/), or SMART (Letunic and Bork 2018). A BLASTp search of these 35 amino acids against all published Caenorhabditis genomes (Caenorhabditis.org) also revealed no significant hits with a low e-value threshold of 1.0. Gene accession numbers are: JPH-1A (NP_492193.2), Human JPH1 (NP_001304759.1), JPH2 (NP_065166.2), JPH3 (NP_065706.2), and JPH4 (NP_001139500.1). Pairwise sequence alignments were performed between C. elegans JPH-1A and human JPH1, JPH2, JPH3, and JPH4 using MUSCLE (Madeira et al. 2019) and the Percent Identity Matrix was viewed to find percent identity. To determine conservation between MORN repeats, we concatenated all eight 14 amino acid MORN repeats into one sequence for each protein and then performed pairwise sequence alignments using MUSCLE. Sequence identity ranges from 69% to 77% when comparing only MORN sequences in C. elegans JPH-1A and human JPH1 through 4. (C) Bright field images of L4 stage animals of genotypes indicated. Compared to wild-type animals, jph-1(ju1683) animals are small, thin, and pale, all of which was rescued by transgenic expression of a fosmid containing genomic jph-1 (juEx3390), JPH-1A expressed under the jph-1 promoter [Pjph-1-GFP::JPH-1A(juSi387)], or JPH-1A expressed in the pharyngeal muscle [Pmyo-2-GFP::JPH-1A(juEx8041)]. Scale bar 100 µm.
Figure 4.
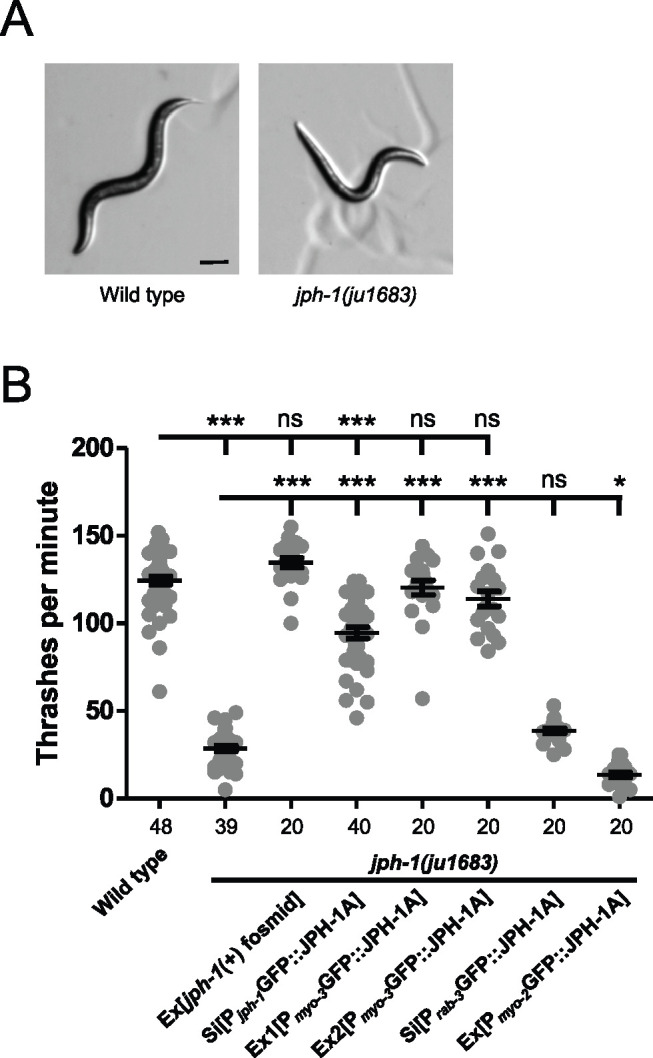
jph-1 is required in the body wall muscle for locomotion. (A) L4 stage wild-type animals exhibit smooth sinusoidal movement and posture while jph-1(ju1683) animals assume unusually straight body positions (shown here) and unusually tight sinusoidal or curled positions. Scale bar, 100 µm. (B) jph-1(ju1683) null mutants thrash less frequently than wild-type N2 animals. Thrashing rate was rescued by expression of a fosmid containing jph-1 (juEx3390) and partially rescued by expression of JPH-1A by the jph-1 promoter [Pjph-1-GFP::JPH-1A(juSi387)]. Expression of JPH-1A in body wall muscle [Pmyo-3-GFP::JPH-1A(Ex1: juEx8023 Ex2: juEx8023)] rescued thrashing rate, but expression in neurons [Prab-3-GFP::JPH-1A(juSi389)] did not. Expression of JPH-1A in the pharyngeal muscle [Pmyo-2-GFP::JPH-1A(juEx8041)] slightly decreased thrashing rate. Number of animals per genotype indicated below X-axis tick marks. Data are shown as individual data points and mean ± SEM. Statistics: One-way ANOVA with Tukey’s post-test. ns, not significant, ***P < 0.001.
Pharyngeal pumping assays
To count pumping rate, day-1 adult animals on seeded plates were observed through dissection stereomicroscopes. We counted the number of grinder movements in 20 s twice per animal and took the average. Counting was done while animals were on the OP50 bacterial lawn to prevent variations in pumping rate caused by food availability.
To measure pumping strength, we adapted a published protocol that used serotonin to stimulate pumping in immobilized animals (Trojanowski and Fang-Yen 2015). We prepared 8% agarose pads with 8 mM serotonin (H7752, Sigma Aldrich), placed animals in an M9 drop on the pad, and immediately placed a cover slip on top. We began imaging when animals started pumping (about 0–10 min after animals were placed in the M9 drop). Imaging was performed on a Leica DMi8 microscope at 40× magnification. Twenty-second videos were taken at 100 ms/frame for a total of 200 frames per animal. Videos were then analyzed in Fiji (ImageJ). The distance from the grinder to an arbitrary point on the pharyngeal lumen was measured in the frame immediately preceding pump initiation (Figure 3B, left image). The distance from the grinder to the same point was measured in the frame when the grinder had moved to its fullest extent (Figure 3B, right image). The difference between these two measurements is the distance moved by the grinder in one pump. We took the average of the first five pumps in each video, although in three instances wild-type animals only pumped three or four times during the video. The distance moved by the grinder was divided by the length of the pharynx (Figure 3B, left image) to normalize to animal size.
Figure 3.

jph-1 is required in the pharyngeal muscle for normal rate and strength of pumping. (A) jph-1 is required for normal pharyngeal pumping rate. jph-1(ju1683) mutants had reduced pumping rate, which was rescued by expression of JPH-1A by the jph-1 promoter [Pjph-1-GFP::JPH-1A(juSi387)] but not by expression in the pharyngeal muscle [Pmyo-2-GFP::JPH-1A(juEx8041)]. eat-4(ky5) loss-of-function mutants had reduced pumping rate, as previously reported (Lee et al. 1999). Number of animals per genotype indicated above X-axis tick marks. Data are shown as individual data points and mean ± SEM. Statistics: Nonparametric Kruskal–Wallis test with Dunn’s multiple comparison test. ns, not significant, *P < 0.05, **P < 0.01. (B) Pumping strength was determined by the distance moved by the grinder. The image on the left shows the head of the animal just before the pump is initiated, with the grinder position indicated by the arrow. The image on the right shows the animal mid-pump when the grinder has moved to its fullest extent. The distance moved by the grinder between the two images was normalized to the total length of the pharynx to quantify pumping strength. Scale bar, 25 µm. (C) Quantification of pharyngeal pumping strength. jph-1(ju1683) mutants had substantially reduced grinder movement, which was rescued by expression of JPH-1A by the jph-1 promoter [Pjph-1-GFP::JPH-1A(juSi387)] or in the pharyngeal muscle [Pmyo-2-GFP::JPH-1A(juEx8041)]. Number of animals per genotype indicated below X-axis tick marks. Data are shown as individual data points and mean ± SEM. Statistics: One-way ANOVA with Tukey’s post-test. ns, not significant, ***P < 0.001.
Thrashing assay
Individual L4 animals were placed in 1 µl drops of M9 on a glass dissection plate. We counted the number of thrashes performed by the animal in 1 min. We considered a single thrash to be one sufficiently large movement of the animal’s head or tail back and forth, with the head or tail not necessarily crossing the center of mass.
Aldicarb and levamisole assays
To test aldicarb sensitivity, 15 day-1 adult animals were transferred to fresh plates containing 0.5 mM or 1 mM aldicarb. Animals were scored for paralysis every 30 min by gently touching the animal with a platinum wire. For levamisole sensitivity, 15 day-1 adult animals were transferred to fresh plates containing 1 mM levamisole. Animals were scored for paralysis every 15 min by gently touching the animal with a platinum wire. Final sample size for each assay was 13–15 animals due to some animals crawling off the plate. Drug sensitivity was quantified from three independent experiments.
Laser axotomy of PLM axons
We cut PLM axons and quantified the length of regrown axons as previously described (Wu et al. 2007). Briefly, GFP-labeled PLM axons [Pmec-7-GFP(muIs32)] of L4 animals were cut 40 µm anterior to the soma by a femtosecond laser on a spinning-disk confocal microscope and immediately imaged. Animals were recovered onto seeded NGM plates and the regrown axon was imaged 24 h later on a Zeiss LSM510 or LSM800 confocal microscope. Axon regrowth length was quantified by measuring the distance between the soma and cut site in the image taken immediately after axotomy, measuring the distance between the soma and the end of the regrown axon in the image taken 24 h later, and taking the difference. All strains were normalized to wild type by dividing the length grown by each axon by the average regrowth length of the same-day wild type control.
Statistical analysis
We used Prism (GraphPad Software) for all statistical analyses except for Fisher’s exact test, for which we used the online tool QuickCalcs (Graphpad Software). To compare regrowth between experiments with different control means, we normalized each experimental data point by dividing it by its control means. Statistical tests and sample sizes are indicated in Figures or Figure legends.
Data availability
Reagents are listed in Supplementary Tables S1–S4, which are available in figshare: https://doi.org/10.25386/genetics.14417696.
Strains and plasmids are available upon request.
Results
jph-1 expresses two isoforms
A previous study described a jph-1 cDNA that encodes a protein of 747 amino acids (Yoshida et al. 2001). In the process of obtaining jph-1 cDNA for our own study, we obtained two cDNAs of 2.2 and 2.4 kb in size, amplified using primers flanking the start and stop codons (Figure 1A). The 2.2-kb cDNA matches the previously reported jph-1 cDNA, which we designated isoform A (Yoshida et al. 2001). The 2.4-kb cDNA retains intron 7 and would encode a protein with the 138 C-terminal amino acids of isoform A, which contains the transmembrane domain, replaced by 35 unrelated amino acids (Figure 1B, designated isoform B). The 35 C-terminal amino acids do not contain a predicted transmembrane domain, nor conserved domains or low complexity regions. A BLASTp search of all published Caenorhabditis genomes also found no significant hits for the 35 amino acid sequence. Furthermore, a BLASTn search showed that although intron 7 can be aligned in 10 out of 26 published Caenorhabditis genomes (Supplementary Figure S1A), the predicted translated peptide has low amino acid conservation and variable sequence length due to intronic stop codons (Supplementary Figure S1B). The absence of conserved motifs and the lack of conservation between species suggest that these 35 amino acids in isoform B may not be important for the function of jph-1. JPH-1 isoform A shares between 39% and 42% overall sequence identity with human JPH1 through 4, with higher sequence homology in the MORN repeats, which target junctophilin to the PM (Takeshima et al. 2000).
jph-1 is required for normal development
To define the function of jph-1, we generated two null (0) alleles, ju1683 and ju1684, using CRISPR-Cas9 editing. Both alleles delete the entire coding sequence of both jph-1 isoforms (Figure 1A). These two alleles show indistinguishable phenotypes in all analyses; therefore, we generally present the quantification data for ju1683 (Table 1). By gross body morphology, jph-1(0) mutant animals are smaller and thinner than stage-matched wild-type animals (Figure 1C). jph-1(0) mutants develop more slowly compared to wild-type animals (Table 1). jph-1(0) mutants have reduced fertility, with a brood size (52 ± 25, n = 12) about 20% of that in wild-type animals (279 ± 19, n = 10). Transgenic expression of a fosmid containing the entire jph-1 locus rescued the developmental defects (Table 1). These observations indicate that jph-1 is necessary for proper animal development.
Table 1.
Summary of growth phenotypes of jph-1 mutants and relevant transgenic animals
| Genotype | Transgene | Days to L4 (20°C) | Body size at L4 |
|---|---|---|---|
| Wild type | None | 2 | Normal |
| jph-1(ju1683) | None | 3 to 4 | Small |
| jph-1(ju1683) | Fosmid with genomic jph-1a | 2 | Normal |
| jph-1(ju1683) | JPH-1Ab | 2 | Normal |
| jph-1(ju1683) | JPH-1Bc | 3 to 5 | Small |
| eat-4(ky5) | None | 2 | Normal |
| jph-1(ju1683) | JPH-1A in pharyngeal muscleb | 2 | Normal |
| jph-1(ju1683) | JPH-1A in body wall muscleb | 3 to 4 | Small |
| jph-1(ju1683) | JPH-1A in neuronsb | 3 to 4 | Small |
| jph-1(ok2823) | None | 3 to 4 | Small |
| jph-1(ok2823) | Fosmid with genomic jph-1a | 2 | Normal |
A fosmid containing the entire genomic locus of jph-1 [WRM0622aB02(juEx3390) or WRM0623aF07(juEx3392)].
Transgenic expression of JPH-1A under its own promoter [Pjph-1-GFP::JPH-1A(juSi387 or juEx7999)], in pharyngeal muscle [Pmyo-2-GFP::JPH-1A(juEx8041)], in body wall muscle [Pmyo-3-GFP::JPH-1A(juEx8022 or juEx8023)], in neurons [Prab-3-GFP::JPH-1A(juSi389)].
JPH-1B [Pjph-1-GFP::JPH-1B(juEx8037 or juEx8038)].
jph-1 is expressed in muscles and neurons, and its function requires the transmembrane domain
A previously reported jph-1 transcriptional reporter showed expression in most muscles and some neurons in the head (Yoshida et al. 2001). We made a similar transcriptional reporter using a 4.5-kb jph-1 promoter to express GFP (Supplementary Figure S2A). We confirmed GFP expression in all hermaphrodite muscle types, including body wall, pharyngeal, vulval, uterine, stomatointestinal, anal sphincter, and anal depressor muscles, with the exception of the contractile gonadal sheath (Supplementary Figure S2A). We also observed expression in many neurons from head to tail.
All developmental defects of jph-1(0) were rescued by expression of N-terminally GFP-tagged JPH-1A under the control of the 4.5-kb jph-1 promoter (Supplementary Figure S2B) as a multicopy extrachromosomal array or a single-copy insertion line (Table 1, Figure 1C). This result indicates that GFP-tagged JPH-1A can perform the developmental functions of jph-1 and that the 4.5-kb promoter provides sufficient tissue specificity for jph-1 function. In contrast, transgenic expression of GFP::JPH-1B, the isoform lacking the C-terminal transmembrane domain, under the same promoter, did not show rescuing activity (Supplementary Figure S2C, Table 1). This analysis supports a conclusion that the transmembrane domain is necessary for the function of JPH-1.
JPH-1A localizes to subcellular puncta and colocalizes with the ER-PM contact site protein ESYT-2 in neurons
We observed that the functional GFP-tagged JPH-1A showed a punctate subcellular pattern in muscles and neurons. In body wall muscle, GFP::JPH-1A localizes to rows of puncta that follow the obliquely striated pattern of the muscle (Figure 2, A and B). In the pharyngeal muscle, JPH-1A localizes to puncta radiating from the pharyngeal lumen and lining the pharynx periphery (Figure 2A). We observed broad expression in neurons including the head and tail ganglia, the bundled neuronal processes of the nerve ring, the ventral nerve cord neurons, and touch receptor neurons (Figure 2, A–E). In neuronal somas of the head ganglia (Figure 2C), tail ganglia (Figure 2D), and ventral nerve cord (Figure 2E), GFP::JPH-1A shows a reticulate localization pattern and forms bright puncta near the periphery of the soma. This localization is observed in newly hatched L1 animals and adults, suggesting that the localization is established prior to hatching and maintained into adulthood (Supplementary Figure S2B).
Figure 2.

JPH-1A colocalizes with calcium channels UNC-68 and EGL-19 in muscles and MCS protein ESYT-2 in neurons. (A–D) Confocal images of GFP::JPH-1A expressed under the jph-1 promoter as a single-copy insertion [Pjph-1-GFP::JPH-1A(juSi387)] in L4 stage animals. (A) Maximum intensity projection of the head showing GFP::JPH-1A expression in body wall muscle, pharyngeal muscle, and neurons. Arrow indicates nerve ring. (B) Single plane image of body wall muscle. JPH-1A localizes to rows of dots that run parallel to muscle striations. (C) Single plane image of head ganglia neurons. JPH-1A in neuronal somas is excluded from the nucleus and is concentrated in puncta. Arrowheads indicate some of the neurons expressing GFP::JPH-1A. (D) Single plane image of tail ganglia. Arrowheads indicate neurons expressing GFP::JPH-1A. Arrow indicates PLM soma. Asterisks mark body wall muscle. (E) Maximum intensity projection of GFP::JPH-1A [Pjph-1-GFP::JPH-1A(juEx7999)] in the ventral nerve cord in an L4 stage jph-1(ju1683) animal. Arrowheads indicate neuronal somas. Fluorescent blobs outside the cells are autofluorescent particles in the gut. (F–H) JPH-1A colocalizes with UNC-68 in body wall muscle. Single plane confocal images of an L4 stage animal with split-GFP knock-in unc-68 (nu664) expressing muscle GFP1-10 [Pmyo-3-GFP1-10(nuTi144)] and mKate2::JPH-1A expressed under the jph-1 promoter [Pjph-1-mKate2::JPH-1A(juEx8103)]. (I–K) JPH-1A colocalizes with EGL-19. Single plane confocal images of an L4 stage animal with split-GFP knock-in egl-19 (nu674) expressing muscle GFP1-10 [Pmyo-3-GFP1-10(nuTi144)] and mKate2::JPH-1A expressed under the jph-1 promoter [Pjph-1-mKate2::JPH-1A(juEx8103)]. (L–N) JPH-1A localizes to ER-PM contact sites labeled by ESYT-2 in the neuronal soma. Single plane confocal images of an L4 animal expressing mKate2-tagged ESYT-2 under the mec-4 touch neuron-specific promoter [Pmec-4-mKate2::ESYT-2(juIs540)] and GFP::JPH-1A under the jph-1 promoter [Pjph-1-GFP::JPH-1A(juSi387)]. PLM soma outlined by dashed line. (O) Close-up of panel N showing partial colocalization of JPH-1A and ESYT-2 in the PLM soma. (P) Close up of the box in Panel N shows that both ESYT-2 and JPH-1A are in the PLM axon. In all images, anterior is to the left, dorsal is up. Scale bars, 5 µm.
Junctophilins generally function to couple calcium channels between the ER and PM, including ER-localized ryanodine receptors (RyRs) and PM-localized L-type calcium channels (Landstrom et al. 2014). In C. elegans, unc-68 encodes the RyR and egl-19 encodes the Cav1 VGCC α1-subunit (Maryon et al. 1996; Lee et al. 1997). We generated split-GFP knock-in alleles for both unc-68 and egl-19 and visualized the subcellular localization of UNC-68 and EGL-19 by expressing muscle GFP1-10. We also coexpressed mKate2::JPH-1A under the jph-1 promoter. In the body wall muscle, both GFP::UNC-68 and GFP::EGL-19 localize to rows of puncta, which nearly completely overlap with mKate2::JPH-1A puncta (Figure 2, F–K). The observed colocalization is consistent with JPH-1 and the two calcium channels being targeted to ER-PM contact sites in muscle cells.
To determine if the neuronal puncta of JPH-1A represent MCSs, we analyzed animals coexpressing GFP::JPH-1A with a reporter expressing mKate2::ESYT-2 in touch receptor neurons. E-Syt (extended-synaptotagmin) proteins are conserved tethering proteins at ER-PM contact sites (Giordano et al. 2013). We showed previously that C. elegans ESYT-2 is expressed broadly in neurons and colocalizes with an ER marker at the cell periphery (Kim et al. 2018). In the PLM soma, GFP::JPH-1A puncta colocalized with mKate2::ESYT-2 (Figure 2, L–P), suggesting that JPH-1A clusters at ER-PM contact sites in neuronal somas. We also examined GFP-tagged JPH-1B and observed a mostly diffuse localization in the muscles and neurons (Supplementary Figure S2C), consistent with the transmembrane domain being critical for JPH-1 subcellular localization. The lack of jph-1(0) rescuing activity by JPH-1B suggests that the transmembrane domain is important for its localization and function.
jph-1 regulates pharyngeal muscle contraction
The gross phenotypes of jph-1(0) mutants broadly resemble those of mutants with feeding defects in that they are small, thin, pale, and take longer to reach adulthood than wild-type animals (Avery and Horvitz 1989; Avery 1993). Our observation that jph-1 is expressed in the pharynx suggests that the jph-1(0) phenotype may be due to defects in feeding-related function. Caenorhabditis elegans eat by drawing bacteria into their mouth using pharynx pumping and crushing the bacteria with their grinder (Avery and You 2012). We measured pumping rate by counting grinder movements and found that jph-1(0) mutants had a lower pumping rate than wild-type animals (Figure 3A). Pharyngeal muscle contraction is regulated by glutamatergic transmission. Loss of function in eat-4, encoding the sole glutamate transporter, causes reduced pumping rate (Lee et al. 1999). We found that jph-1(0) mutants had a similar pumping rate to eat-4(ky5) mutants (Figure 3A). However, since eat-4(ky5) animals are not as small as jph-1(0) mutants (Table 1), reduced pumping rate alone cannot account for the starved appearance of jph-1(0) mutants. We next quantified pumping strength by measuring the distance moved by the grinder in one pump (Figure 3B, Materials and Methods). jph-1(0) mutants had significantly weaker pumping strength than either wild type or eat-4 mutants (Figure 3C). To test if reduced pharyngeal muscle activity was causing the starved appearance of jph-1(0) mutants, we expressed JPH-1A specifically in the pharyngeal muscle using the myo-2 promoter. Pharyngeal muscle expression of JPH-1A restored pumping strength to wild-type levels (Figure 3C). Importantly, it also rescued the small body size and delayed development of jph-1(0) mutants (Table 1, Figure 1C). These observations indicate that JPH-1A is required for proper pharyngeal muscle function, which ultimately impacts gross organismal development.
jph-1 is required in the body wall muscle for locomotion
On solid surfaces, wild-type C. elegans crawl by sinusoidal body undulations (Figure 4A). In contrast, jph-1(0) mutants adopt unusual extended or curled postures during locomotion, move slowly, and are frequently immobile, consistent with previous observations of jph-1 RNAi-treated animals (Yoshida et al. 2001). When placed in liquid, C. elegans swim by moving their entire bodies side-to-side to produce alternating C-shaped conformations (Gjorgjieva et al. 2014). By counting thrashing frequency, we observed that jph-1(0) mutants exhibit far fewer thrashes per minute than wild-type animals (Figure 4B). Furthermore, jph-1(0) mutants would often thrash only the head without moving the tail, suggesting a failure of muscle contraction to propagate to the tail. A fosmid containing genomic jph-1 fully rescued locomotion on both solid surfaces and in liquid (Figure 4B, Table 1). JPH-1A driven by the jph-1 promoter rescued locomotion defects and thrashing frequency, although not as well as the fosmid transgene, possibly because the fosmid transgene contains more regulatory elements for efficient expression than the promoter-cDNA expression construct (Figure 4B). JPH-1B did not discernably improve movement on solid surfaces, supporting the importance of the transmembrane domain for JPH-1 function. Expression of JPH-1A in body wall muscle, but not pharyngeal muscle or neurons, restored full-body thrashing in liquid and sinusoidal movement on solid surfaces (Figure 4B). These data indicate that jph-1 is required in the body wall muscle for animal movement and suggest that it may be involved in both muscle contraction and propagation of a signal for contraction between muscle cells.
jph-1 promotes axon regeneration cell nonautonomously
We previously characterized a different jph-1 mutation, jph-1(ok2823), for its role in axon regeneration (Kim et al. 2018). jph-1(ok2823) is a small deletion removing part of the fourth intron to the sixth exon (Figure 1A). By analyzing cDNA isolated from jph-1(ok2823) animals, we found that jph-1(ok2823) could generate a protein truncated after the seventh MORN repeat. The gross morphology and movement of jph-1(ok2823) animals is similar to jph-1(0), except jph-1(ok2823) animals grow slightly more quickly and are slightly larger than jph-1(0) animals. Gross defects of jph-1(ok2823) are fully rescued by the jph-1 fosmid transgene (Table 1). We had observed that PLM axons of jph-1(ok2823) animals display reduced axon regeneration after laser-induced axon injury and a higher rate of fusion between the regrowing axon stump and the axon fragment, which we call axon fusion (Kim et al. 2018). We tested if PLM axon regeneration is similarly affected in jph-1(0) mutants. Like jph-1(ok2823) animals, touch receptor neurons of jph-1(0) mutants have normal morphology (Supplementary Figure S3A), indicating that jph-1 is not required for axon outgrowth during development. After laser injury of PLM axons, jph-1(0) mutants exhibited strongly reduced axon regeneration, significantly different from both wild type and jph-1(ok2823) (Figure 5, A and B). Expression of JPH-1A under the jph-1 promoter fully rescued the regeneration defect, indicating that jph-1 is required for axon regrowth after injury. However, while jph-1 is expressed in PLM neurons (Figure 2), expression of JPH-1A specifically in touch neurons did not rescue axon regrowth (Figure 5B). Furthermore, knocking down GFP::JPH-1A specifically in touch neurons of jph-1(0) animals through Degron-mediated degradation of GFP::JPH-1A (Wang et al. 2017) did not reduce axon regeneration (Supplementary Figure S3B). Instead, expression of JPH-1A in pharyngeal muscle, which rescued the growth and size of the animal (Table 1), rescued axon regrowth (Figure 5B). Together, these data indicate that jph-1 regulates axon regeneration cell nonautonomously and suggest that nutrient intake may influence axon regeneration.
Figure 5.
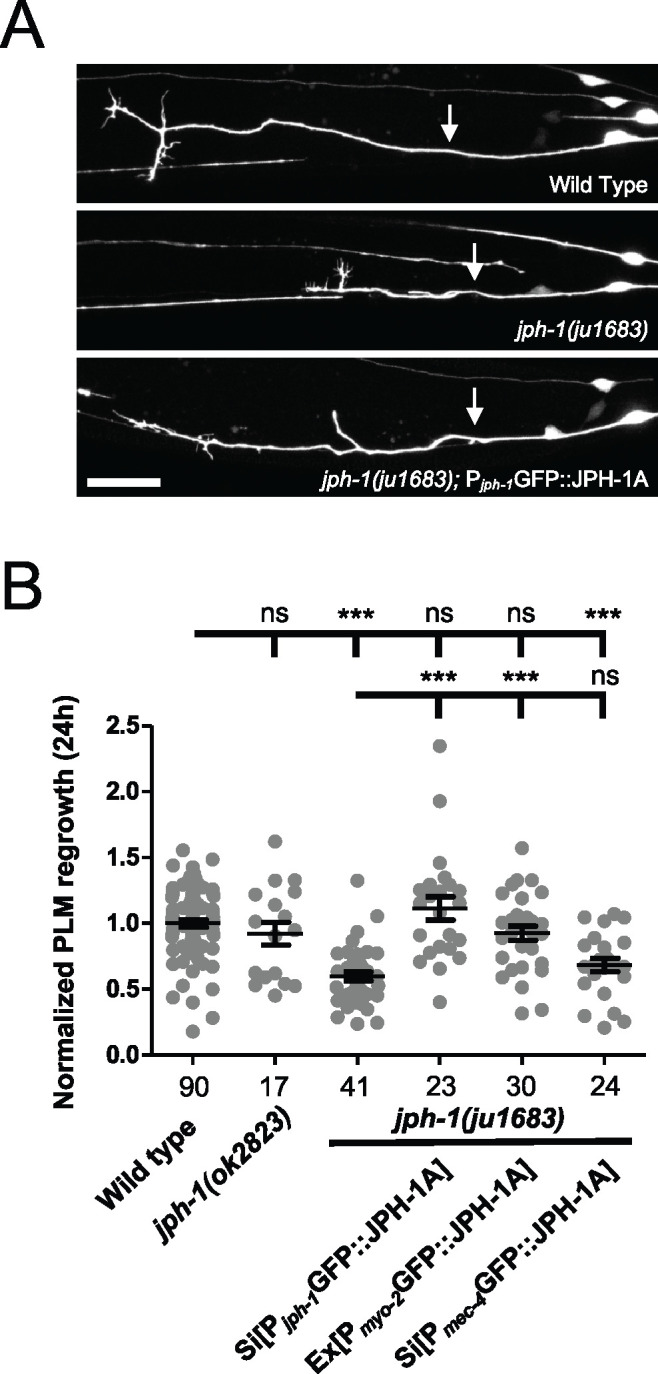
jph-1 promotes touch neuron PLM axon regeneration cell nonautonomously. (A) Representative confocal images of PLM axon regrowth 24-h post-axotomy in animals expressing the touch neuron marker Pmec-7-GFP(muIs32). Genotype in the bottom image is jph-1(ju1683); Pjph-1-GFP::JPH-1A(juSi387). Anterior is to the left, dorsal is up. Arrows indicate the site of axon injury. Scale bar, 20 µm. (B) jph-1 is required in the pharyngeal muscle for touch neuron axon regeneration. Distance regrown by PLM axon 24-h post-injury, normalized to wild-type regrowth. jph-1(ok2823) axon regrowth was not significantly different from wild type [Pmec-7-GFP(muIs32)]. jph-1(ju1683) animals had significantly reduced regrowth. Expression of JPH-1A by the jph-1 promoter [Pjph-1-GFP::JPH-1A(juSi387)] or in the pharyngeal muscle [Pmyo-2-GFP::JPH-1A(juEx8041)] rescued the reduced regrowth of jph-1(ju1683) mutants. Expression of JPH-1A in the touch receptor neurons [Pmec-4-GFP::JPH-1A(juSi388)] did not rescue axon regeneration. Number of animals per genotype indicated below X-axis tick marks. Data are shown as individual data points and mean ± SEM. Statistics: Nonparametric Kruskal–Wallis test with Dunn’s multiple comparison test. ns, not significant, ***P < 0.001.
While we were able to replicate the increased axon fusion of jph-1(ok2823) mutants, we did not observe an increase in axon fusion in injured PLM axons in jph-1(0) mutants (Supplementary Figure S3C). We considered if the enhanced axon fusion observed in jph-1(ok2823) animals might be caused by the production of an abnormal protein. To test this, we made a construct fusing GFP to jph-1 cDNA isolated from jph-1(ok2823) animals, named GFP::JPH-1(ok2823). In contrast to the subcellular punctate pattern of full-length JPH-1A, GFP::JPH-1(ok2823) was found in the nucleus of many neurons and body wall muscles (Supplementary Figure S3D). As described earlier, jph-1(ok2823) animals are slightly healthier than jph-1(0) animals. Therefore, two explanations can be made for the increased axon fusion of jph-1(ok2823) mutants: either that jph-1(ok2823) is a partial loss of function and fusion is more likely when axon regrowth is only mildly impaired, or that jph-1(ok2823) produces a protein with altered activity that enhances axon fusion.
jph-1 contributes to neuromuscular synaptic transmission
By colocalization with reporters for motor neurons in the ventral cord, we found that jph-1 is expressed in cholinergic motor neurons (Supplementary Figure S4A). To assess synaptic localization of JPH-1 in these neurons, we analyzed animals coexpressing endogenously tagged vesicular acetylcholine transporter UNC-17::mKate2 (ot907) (Pereira et al. 2019) and Prab-3-GFP::JPH-1A. In the dorsal nerve cord and sublateral cord, JPH-1A mostly borders UNC-17::mKate2-enriched presynaptic sites (Supplementary Figure S4, B and C). To examine if jph-1 plays a role in synaptic transmission, we focused our study on the neuromuscular junction, where pharmacological assays can assess neuromuscular transmission. Release of acetylcholine from ventral cord motor neurons stimulates body wall muscle contraction in C. elegans (Rand 2007). The acetylcholinesterase inhibitor aldicarb is widely used to assess neuromuscular transmission. Aldicarb causes the accumulation of acetylcholine at the neuromuscular junction, which leads to muscle hypercontraction and paralysis (Miller et al. 1996). After 2 h of exposure to 1 mM aldicarb, nearly all wild-type animals were paralyzed (Figure 6A). In contrast, 70–80% of jph-1(0) mutants were still moving, suggesting that these animals have impaired synaptic transmission at the neuromuscular junction. Aldicarb resistance was confirmed using a second jph-1(0) allele (Supplementary Figure S5A) and expression of a fosmid containing jph-1 genomic DNA rescued the aldicarb resistance of jph-1(0) mutants (Figure 6A). We attempted to discern whether jph-1 acts pre- or postsynaptically. However, we observed that while overexpression of JPH-1A in body wall muscles fully rescued locomotion of jph-1(0), the transgenes caused aldicarb resistance when expressed in wild-type animals. Similar overexpression of JPH-1A in neurons in wild-type animals also caused aldicarb resistance, despite no detectable behavioral abnormality. As a control, overexpression of JPH-1A in pharyngeal muscles fully rescued the pumping and growth deficits of jph-1(0) and did not cause aldicarb resistance in wild-type animals. Thus, while the precise cellular action by which JPH-1 affects synaptic transmission at the neuromuscular junction remains to be addressed, these observations imply a dosage-sensitive function of JPH-1 in body wall muscles and neurons.
Figure 6.

jph-1 and esyt-2 null mutants are aldicarb resistant and exhibit mutual suppression. (A) jph-1(0) animals are resistant to aldicarb compared to wild-type animals. Aldicarb resistance was rescued by expression of a fosmid containing jph-1 genomic DNA (juEx3390). Statistical significance shown between jph-1(0) and jph-1; Ex[jph-1(+) fosmid]. (B) esyt-2(0) animals are resistant to aldicarb compared to wild-type animals. Aldicarb resistance was rescued by pan-neuronal expression of GFP::ESYT-2 [Prab-3-GFP::ESYT-2(juEx8112)]. Statistical significance shown between esyt-2(0) and esyt-2; Ex[Prab-3-GFP::ESYT-2]. (C) jph-1(0); esyt-2(0) double mutants exhibit a wild-type response to aldicarb. Statistical significance shown between jph-1(0) and jph-1; esyt-2. (D) Expression of esyt-2 genomic DNA (juEx7581) restores aldicarb resistance to jph-1(ju1683); esyt-2(ju1409) double mutants. Statistical significance shown between jph-1; esyt-2 and jph-1; esyt-2; Ex[esyt-2 gDNA]. 1 mM aldicarb. Thirteen to 15 animals tested per genotype per trial, n = 3 trials. Data are shown as individual data points and mean ± SEM. Statistics: One-way ANOVA with Tukey’s post-test. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
jph-1 antagonizes esyt-2 to modulate neuromuscular synaptic transmission
Like JPH-1, the ER-PM contact site protein ESYT-2 is broadly expressed in neurons (Kim et al. 2018). We assessed synaptic localization of ESYT-2 in the cholinergic motor neurons. We found that GFP::ESYT-2 puncta were clustered around UNC-17::mKate2-labeled presynaptic sites in the dorsal cord, and were more discrete than those of GFP::JPH-1A (Supplementary Figure S4D). Like jph-1(0) animals, esyt-2(0) mutants were also aldicarb resistant, which was rescued by a transgene containing the full-length esyt-2 genomic DNA (Supplementary Figure S5B). We then transgenically overexpressed esyt-2 cDNA in neurons and found that such expression fully rescued the aldicarb resistance of esyt-2(0) and did not cause any effects in a wild-type background (Figure 6B). These data support a conclusion that esyt-2 acts in neurons to modulate neuromuscular synaptic transmission.
To address how jph-1 might interact with esyt-2, we constructed double mutants of jph-1(0) and esyt-2(0). While esyt-2(0) animals are superficially wild-type, jph-1(0); esyt-2(0) mutants do not show improved growth or locomotion over the jph-1(0) mutant, suggesting that the esyt-2 mutation does not compensate for the loss of jph-1 in muscles. However, by aldicarb assay, we found that jph-1(0); esyt-2(0) double mutants paralyzed at a wild-type rate (Figure 6C), suggesting that jph-1(0) and esyt-2(0) mutations had mutually suppressed each other. We observed this mutual suppression using a second allele of jph-1(0) and esyt-2(0) (Supplementary Figure S5C). Moreover, expression of wild-type esyt-2 in the jph-1(0); esyt-2(0) double mutants restored aldicarb resistance (Figure 6D). Taken together, these results suggest that jph-1 and esyt-2 have antagonistic roles in neuromuscular synaptic transmission.
jph-1 promotes animal health and development in parallel with unc-68/RyR and VGCCs
Our observation that JPH-1A colocalizes with UNC-68/RyR and the VGCC α1-subunit EGL-19 raised the possibility of direct interaction between them. We thus next investigated genetic interactions between jph-1 and calcium channels in C. elegans. unc-68 is expressed in neurons and muscles. unc-68(e540) null mutants resemble jph-1(0) mutants as both are small, slow growing, and show incomplete flaccid paralysis (Maryon et al. 1996). Previous studies suggested that the slow growth of unc-68(0) is caused by defects in pharyngeal function, similar to our finding for jph-1 (Maryon et al. 1998). However, unc-68(0) mutants have darker pigmentation and grew more quickly than jph-1(0) mutants (Table 2), suggesting that unc-68(0) mutants have milder defects in pharyngeal function (Avery 1993). We found that jph-1(0); unc-68(0) double mutants grew more slowly than either jph-1(0) or unc-68(0) single mutants (Table 2). Expressing JPH-1A under the jph-1 promoter in jph-1(0); unc-68(0) double mutants partially restored animal growth, confirming that the exacerbated slow growth was due to the double mutant. jph-1(0); unc-68(0) double mutants also exhibited more severe locomotion defects than either single mutant (Table 2). The enhanced slow growth and movement defects in the jph-1(0); unc-68(0) mutant indicate that the jph-1(0) and unc-68(0) mutations are at least partially additive. Therefore, while jph-1 and unc-68 may have overlapping roles, jph-1 has functions independent of unc-68. egl-19 is expressed in both muscles and neurons, and egl-19 null mutants are embryonic lethal (Lee et al. 1997). We therefore used a partial loss-of-function mutation, egl-19(ad1006lf), to test genetic interactions with jph-1. Animals homozygous for the egl-19(ad1006lf) mutation are long, thin, and flaccid, move slowly, and display weak pumping (Lee et al. 1997). We were unable to obtain viable jph-1(0); egl-19(lf) double mutants, suggesting that jph-1 becomes crucial when egl-19 function is impaired. We also constructed double mutants of jph-1(0) with the gain-of-function mutation egl-19(ad695gf). Animals with egl-19(ad695gf) are short due to body wall muscle hypercontraction (Lainé et al. 2014) but otherwise appear normal in overall growth rate and movement. We found that jph-1(0); egl-19(gf) animals lived to adulthood, but grew more slowly than jph-1(0) single mutants (Table 2). These observations suggest that when egl-19 activity is impaired or altered, jph-1 becomes critical, possibly because it couples the mutant EGL-19 to ER-localized calcium channels that can amplify the voltage-gated calcium signal.
Table 2.
Summary of growth rates and movement of double mutants of jph-1(0) with calcium channel and esyt-2 mutants
| Genotype | Days to L4 (20°C) | Movement |
|---|---|---|
| jph-1(ju1683) | 3 to 4 | Partial flaccid paralysis |
| unc-68(e540) | 2 to 3 | Partial flaccid paralysis |
| egl-19(ad1006lf) | 2 | Partial flaccid paralysis |
| egl-19(ad695gf) | 2 | Normal |
| unc-2(e55) | 2 | Paralyzed |
| unc-2(zf35gf) | 2 | Hyperactive |
| esyt-2(ju1409) | 2 | Normal |
| jph-1(ju1683); unc-68(e540) | 4 to 6 | Severe flaccid paralysis |
| jph-1(ju1683); unc-68(e540); juEx7999 [Pjph-1-GFP:: JPH-1A] | 3 to 4 | Partial flaccid paralysis |
| jph-1(ju1683); egl-19 (ad1006lf) | Lethal | |
| jph-1(ju1683); egl-19 (ad695gf) | 4 to 5 | Partial flaccid paralysis |
| jph-1(ju1683); unc-2(e55) | 4 to 7 | Paralyzed |
| jph-1(ju1683); unc- 2(zf35gf) | 5 to 7 | Partial flaccid paralysis |
| jph-1(ju1683); esyt-2 (ju1409) | 3 to 5 | Partial flaccid paralysis |
All alleles are null unless otherwise annotated as gain of function (gf) or partial loss of function (lf).
The non-L-type VGCC α1-subunit unc-2, orthologous to CACNA1A, is predominantly expressed in neurons and localizes to presynaptic terminals (Mathews et al. 2003; Saheki and Bargmann 2009). unc-2(e55) null mutants exhibit sluggish movement but normal development and growth (Schafer et al. 1996; Mathews et al. 2003). We found that jph-1(0); unc-2(0) double mutants grew substantially more slowly than jph-1(0) single mutants, with some animals taking up to 7 days to reach the L4 stage (Table 2). Furthermore, jph-1(0); unc-2(0) mutants were sterile as adults. We also tested the unc-2(zf35gf) gain-of-function mutation, which causes the channel to open at a lower membrane potential and results in hyperactive locomotion but otherwise normal growth and development (Huang et al. 2019). jph-1(0); unc-2(gf) double mutants displayed similarly severe growth defects as jph-1(0); unc-2(0) double mutants. Our data show that while the unc-2 mutations alone have no effect on growth rate, they strongly exacerbate the jph-1(0) growth defect, and this synergistic interaction suggests that jph-1 and unc-2 function cooperatively.
Altogether, the observed genetic interactions support a conclusion that jph-1 and RyR and VGCC channels have overlapping, but partially independent, roles for animal development.
jph-1 is necessary for colocalization of UNC-68/RyR and EGL-19/VGCC, and JPH-1A subcellular localization also depends on unc-68/RyR
Studies of junctophilins in other animals suggest that localization of junctophilins and their interacting partners to ER-PM contact sites may depend on each other (Golini et al. 2011; Nakada et al. 2018). We thus tested if UNC-68 and EGL-19 localization depends on jph-1. We generated a split-mCherry knock-in allele for unc-68, which enabled us to observe UNC-68 and EGL-19 simultaneously using the split-GFP egl-19 knock-in allele. In combination with coexpression of mCherry1-10 and GFP1-10 in the body wall muscle, we observed that in wild-type animals UNC-68::mCherry and EGL-19::GFP colocalized to longitudinal rows of puncta (Figure 7A). In jph-1(0) the organized punctate pattern of UNC-68 and EGL-19 was abolished (Figure 7A), and UNC-68 expression was also noticeably decreased (Figure 7A). These data show that jph-1 is required for the colocalization of UNC-68 and EGL-19, and suggests that jph-1 may have an additional role in stabilizing UNC-68.
We also tested if JPH-1A localization depends on calcium channels and esyt-2. In the body wall muscle of wild-type animals, JPH-1A localizes to longitudinal rows of puncta (Figure 7B). In unc-68 mutants, JPH-1A puncta were less distinct and often connected to neighboring puncta (Figure 7B). In wild-type neurons, JPH-1A has a reticulate pattern with bright puncta at the cell periphery (Figure 7C). In unc-68 mutants, while the reticulate pattern of JPH-1 remained, the bright puncta were absent (Figure 7C). The perturbation of puncta in both muscles and neurons of unc-68(0) animals suggests that unc-68 is required for anchoring JPH-1A in puncta. JPH-1A localization was unchanged from wild type in unc-2 and esyt-2 mutants (Figure 7, B and C, Supplementary Figure S6), indicating that these genes are not required for JPH-1A localization.
Discussion
Junctophilins couple PM- and ER-localized calcium channels to efficiently trigger calcium release from the ER following membrane depolarization (Van Oort et al. 2011; Chen et al. 2013; Reynolds et al. 2013; Nakada et al. 2018). Junctophilins play key roles in excitation–contraction coupling in heart and skeletal muscles (Takeshima et al. 2000; Ito et al. 2001; Van Oort et al. 2011; Nakada et al. 2018), and are thought to regulate action potential frequency in neurons (Moriguchi et al. 2006; Kakizawa et al. 2007; Sahu et al. 2019). Here, we report that the C. elegans junctophilin JPH-1 is expressed in pharyngeal muscle, body wall muscle, and neurons, and performs important functions in each tissue. In the pharyngeal muscle, jph-1 is required for robust pumping and timely growth and development. The stunted development of jph-1(0) mutants is likely due to reduced food intake caused by weak pumping, as slow growth, small body size, and pale pigmentation is also seen in other mutants with defects in feeding-related function (Avery and Horvitz 1989; Avery 1993). In the body wall muscle, jph-1 is required for body movement and locomotion. jph-1(0) mutants move slowly and display flaccid paralysis, suggesting that the body wall muscle lacks contraction strength. Our tissue-specific rescue experiments indicate that muscle contraction in both pharyngeal and body wall muscle requires jph-1. Our findings are consistent with perturbation of junctophilin expression in other organisms. In flies, knockdown or overexpression of the sole junctophilin was shown to cause muscular deficits and cardiac dysfunction (Calpena et al. 2018). Skeletal muscle from neonatal JPH-1 knockout mice have weaker electrically stimulated contractile force (Ito et al. 2001). Thus, the role for junctophilin in muscle contraction is conserved from C. elegans to vertebrates.
The role of calcium regulation in axon regeneration in C. elegans has been widely demonstrated (Ghosh-Roy et al. 2010). unc-68/RyR promotes axon regeneration and is required for localized calcium release from the ER following axon injury (Sun et al. 2014). We previously reported that jph-1(ok2823) mutants have decreased axon regeneration (Kim et al. 2018). Here, we extended our analysis to the genetic null alleles of jph-1 and uncovered a surprising role of jph-1 in promoting axon regeneration in a cell nonautonomous manner. Our observation that the regeneration defects could be rescued by expressing jph-1 in the pharyngeal muscle implies that PLM axon regeneration may be influenced by nutrient uptake or through substances released by the pharynx. This finding raises an intriguing possibility that gut nutrients may impact neuronal injury response, a theme that shares similarities to emerging findings on the gut-brain axis in other axon regeneration studies (Kigerl et al. 2020; Lin-Moore et al. 2020). Additionally, despite jph-1(ok2823) animals largely resembling jph-1(0) in all gross phenotypes, our data suggest that the increased fusion in jph-1(ok2823) is due to either partial loss of function or an altered activity associated with the truncated protein JPH-1(ok2823) that localizes to the nucleus. Interestingly, a study in mouse found that heart stress induces cleavage of JPH-2, with the N-terminal JPH-2 fragment translocating to the nucleus where it alters transcription (Guo et al. 2018). Therefore, it is conceivable that the mutant protein produced in jph-1(ok2823) alters neuronal transcription to enhance axon fusion after injury.
Studies of mouse JPH3 and JPH4 show that double knockout of these two neuronal junctophilins causes impaired motor coordination and defects in learning and memory (Moriguchi et al. 2006; Kakizawa et al. 2007). In Purkinje cells and hippocampal neurons, JPH3 and JPH4 couple the PM-localized VGCCs, ER-localized RyRs, and PM-localization Ca2+-activated K+ channels that generate the slow afterhyperpolarization current that modulates action potential frequency (Moriguchi et al. 2006; Kakizawa et al. 2007; Sahu et al. 2019). We find that jph-1 is necessary for the colocalization of UNC-68/RyR and EGL-19, consistent with the conserved functions of junctophilins. Moreover, jph-1(0) mutants are resistant to the acetylcholinesterase inhibitor aldicarb, suggesting a role of jph-1 in modulating neurotransmission. We further uncover an intriguing genetic interaction between jph-1 and esyt-2 in synaptic transmission. Our observation that esyt-2 acts presynaptically to modulate neurotransmission reinforces the findings on Drosophila Esyt (Kikuma et al. 2017). Strikingly, we observed mutual suppression between jph-1(0) and esyt-2(0) aldicarb phenotypes, suggesting that jph-1 and esyt-2 have antagonistic roles in synaptic transmission. We were not able to discern the cell-type requirement of jph-1 due to unresolved dose-dependent effects of transgenic tissue-specific expression of jph-1. As body muscles in jph-1 mutants show disorganization of UNC-68 and EGL-19, which likely leads to muscle contraction defects, we speculate that reduced presynaptic neurotransmitter release in esyt-2(0) may be compensated by weakened muscle contraction in jph-1(0), leading to the double mutants showing a wild-type response to aldicarb. However, our speculation does not exclude other possibilities such as jph-1 and esyt-2 acting in presynaptic terminals to balance neurotransmitter release, or jph-1 modulating presynaptic release in a retrograde pathway. Future work will be necessary to unravel the mechanism of this novel interaction between esyt-2 and jph-1.
Besides a canonical role of junctophilins to couple ER-localized RyRs to PM-localized calcium channels, multiple lines of evidence suggest that jph-1 has roles independent of unc-68. First, jph-1(0) mutants grow more slowly than unc-68(0). Second, jph-1(0); unc-68(0) double mutants grow more slowly than unc-68(0) single mutants. Third, while jph-1 and unc-68 expression patterns largely overlap, jph-1 is expressed in the anterior pharynx while unc-68 is not (Maryon et al. 1998). Our observation that in jph-1(0) mutants EGL-19 is largely diffuse and the remaining puncta are sparse suggests that jph-1 is required for the localization of EGL-19 to evenly distributed puncta, and that this localization is important for EGL-19 function. In C2C12 myotubes, dual knockdown of JPH1+JPH2 caused VGCC localization to change from punctate to diffuse (Golini et al. 2011). In GLT myotubes, Cav1.1 with a point mutation that prevented binding to junctophilin had a diffuse localization (Nakada et al. 2018). Another possible unc-68-independent role for jph-1 is regulation of store-operated calcium entry, which has been described in other organisms (Hirata et al. 2006; Li et al. 2010).
Our analysis of UNC-68 and EGL-19 localization in the jph-1(0) mutant may shed light on why jph-1 becomes critical when egl-19 function is impaired, as the jph-1(0); egl-19(lf) double mutant appears to be lethal. In the absence of jph-1, UNC-68 and EGL-19 are no longer coupled, and UNC-68 is no longer able to amplify the remaining signaling dependent of egl-19. Alternatively, the localization of EGL-19 into orderly rows of puncta by jph-1 is required for its residual function. In addition, we find that JPH-1 localization depends on unc-68. In rat cardiomyocytes, RyR localizes to muscle triads before JPH-2 arrives (Ziman et al. 2010), suggesting that the targeting of junctophilins by RyR may be conserved. Mammalian JPH1 and JPH2 have been shown to directly bind RyR (Phimister et al. 2007; Woo et al. 2008; Golini et al. 2011; Van Oort et al. 2011; Beavers et al. 2013; Nakada et al. 2018). Thus, it is possible that junctophilin targeting may involve directly binding to RyR already localized at MCSs. In conclusion, our study shows that C. elegans jph-1, similar to vertebrate homologs, has broad functions in excitable cells. The conservation in function between mammalian and C. elegans junctophilins presents the opportunity for C. elegans to be used for further investigations of junctophilins.
Acknowledgments
We thank our lab members for discussion and comments.
Funding
This work was supported by grants from NIH (R01 NS093588 to A.D.C. and Y.J., R01 GM054657 to A.D.C., R01 NS32196 to J.M.K., and R37 NS035546 to Y.J.).
Conflicts of interest
None declared.
Literature cited
- Andrusiak MG, Sharifnia P, Lyu X, Wang Z, Dickey AM, et al. 2019. Inhibition of axon regeneration by liquid-like TIAR-2 granules. Neuron. 104:290–304. e8. doi:10.1016/j.neuron.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BXH, Artiles KL, Hartman PS, et al. 2014. Efficient marker-free recovery of custom genetic modifications with CRISP R/Cas9 in Caenorhabditis elegans. Genetics. 198:837–846. doi:10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L. 1993. The genetics of feeding in Caenorhabditis elegans. Genetics. 133:897–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L, Horvitz HR.. 1989. Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron. 3:473–485. doi:10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- Avery L, You Y-J.. 2012. C. elegans feeding (May 21, 2012), WormBook, ed. The C. elegans Research Community, Wormbook, doi:10.1895/wormbook.1.150.1, http://wormbook.org.
- Beavers DL, Wang W, Ather S, Voigt N, Garbino A, et al. 2013. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 62:2010–2019. doi:10.1016/j.jacc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calpena E, Del AV, Chakraborty M, Llamusi B, Artero R, et al. 2018. The Drosophila junctophilin gene is functionally equivalent to its four mammalian counterparts and is a modifier of a Huntingtin poly-Q expansion and the Notch pathway. DMM Dis Model Mech. 11:1–13. doi:10.1242/dmm.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Guo A, Zhang C, Chen R, Zhu Y, et al. 2013. Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc Res. 100:54–62. doi:10.1093/cvr/cvt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, et al. 2019. The Pfam protein families database in 2019. Nucleic Acids Res. 47:D427–D432. doi:10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Mouridi S, Lecroisey C, Tardy P, Mercier M, Leclercq-Blondel A, et al. 2017. Reliable CRISPR/Cas9 genome engineering in Caenorhabditis elegans using a single efficient sgRNA and an easily recognizable phenotype. G3 (Bethesda). 7:1429–1437. doi:10.1534/g3.117.040824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Varshney A, Coto Villa D, Modavi C, Kohler J, et al. 2019. Bright split red fluorescent proteins for the visualization of endogenous proteins and synapses. Commun Biol. 2:1–12. doi:10.1038/s42003-019-0589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, et al. 2014. Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat Methods. 11:529–534. doi:10.1038/nmeth.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Wayne Davis M, Hopkins CE, Newman BJ, Thummel JM, et al. 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 40:1375–1383. doi:10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbino A, Van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, et al. 2009. Molecular evolution of the junctophilin gene family. Physiol Genomics. 37:175–186. doi:10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD.. 2010. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 30:3175–3183. doi:10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, et al. 2013. PI(4,5)P(2)- dependent and Ca2+-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 153:1494–1509. doi:10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorgjieva J, Biron D, Haspel G.. 2014. Neurobiology of Caenorhabditis elegans locomotion: where do we stand? Bioscience. 64:476–486. doi:10.1093/biosci/biu058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golini L, Chouabe C, Berthier C, Cusimano V, Fornaro M, et al. 2011. Junctophilin 1 and 2 proteins interact with the L-type Ca 2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J Biol Chem. 286:43717–43725. doi:10.1074/jbc.M111.292755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Wang Y, Chen B, Wang Y, Yuan J, et al. 2018. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science (80-). 362:eaan3303. doi:10.1126/science.aan3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, et al. 2006. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J. 90:4418–4427. doi:10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirve N, Rajanikanth V, Hogan PG, Gudlur A.. 2018. Coiled-coil formation conveys a STIM1 signal from ER lumen to cytoplasm. Cell Rep. 22:72–83. doi:10.1016/j.celrep.2017.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, Pirri JK, Rayes D, Gao S, Mulcahy B, et al. 2019. Gain-of-function mutations in the UNC-2/CaV2α channel lead to excitation-dominant synaptic transmission in C. elegans. Elife. 8:1–28. doi:10.7554/eLife.45905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, et al. 2001. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 154:1059–1067. doi:10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa S, Kishimoto Y, Hashimoto K, Miyazaki T, Furutani K, et al. 2007. Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 26:1924–1933. doi:10.1038/sj.emboj.7601639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Zane K, Adams K,, Sullivan MB,, Popovich PG.. 2020. The spinal cord-gut-immune axis as a master regulator of health and neurological function after spinal cord injury. Exp Neurol. 323:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuma K, Li X, Kim D, Sutter D, Dickman DK.. 2017. Extended synaptotagmin localizes to presynaptic ER and promotes neurotransmission and synaptic growth in Drosophila. Genetics. 207:993–1006. doi:10.1534/genetics.117.300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sun L, Gabel CV, Fang-Yen C.. 2013. Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS One. 8:e53419. doi:10.1371/journal.pone.0053419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Tang NH, Piggott CA, Andrusiak MG, Park S, et al. 2018. Expanded genetic screening in Caenorhabditis elegans identifies new regulators and an inhibitory role for NAD + in axon regeneration. Elife. 7:e39756. doi:10.7554/eLife.39756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobuna H, Inoue T, Shibata M, Gengyo-Ando K, Yamamoto A, et al. 2010. Multivesicular body formation requires OSBP-related proteins and cholesterol. PLoS Genet. 6:e1001055. doi:10.1371/journal.pgen.1001055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainé V, Ségor JR, Zhan H, Bessereau JL, Jospin M.. 2014. Hyperactivation of L-type voltage-gated Ca2+ channels in Caenorhabditis elegans striated muscle can result from point mutations in the IS6 or the IIIS4 segment of the α1 subunit. J Exp Biol. 217:3805–3814. doi:10.1242/jeb.106732. [DOI] [PubMed] [Google Scholar]
- Landstrom AP, Beavers DL, Wehrens XHT.. 2014. The junctophilin family of proteins: from bench to bedside. Trends Mol Med. 20:353–362. doi:10.1016/j.molmed.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AB, John Peter Atat Walter P, Kornmann B.. 2015. ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol. 210:883–890. doi:10.1083/jcb.201502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RYN, Lobel L, Hengartner M, Horvitz HR, Avery L.. 1997. Mutations in the α1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 16:6066–6076. doi:10.1093/emboj/16.20.6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RYN, Sawin ER, Chalfie M, Horvitz HR, Avery L.. 1999. EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci. 19:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P.. 2018. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46:D493–D496. doi:10.1093/nar/gkx922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ding X, Lopez JR, Takeshima H, Ma J, et al. 2010. Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J Biol Chem. 285:39171–39179. doi:10.1074/jbc.M110.149690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Moore AT, Oyeyemi MJ, Hammarlund M.. 2020. rab-27 acts in an intestinal secretory pathway to inhibit axon regeneration in C. elegans. bioRxiv. doi:10.1101/2020.09.05.283267. [DOI] [PMC free article] [PubMed]
- Madeira F, Park YM, Lee J, Buso N, Gur T, et al. 2019. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47:W636–W641. doi:10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manford AG, Stefan CJ, Yuan HL, MacGurn JA, Emr SD.. 2012. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell. 23:1129–1140. doi:10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Maryon EB, Coronado R, Anderson P.. 1996. unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contraction. J Cell Biol. 134:885–893. doi:10.1083/jcb.134.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryon EB, Saari B, Anderson P.. 1998. Muscle-specific functions of ryanodine receptor channels in Caenorhabditis elegans. J Cell Sci. 111:2885–2895. [DOI] [PubMed] [Google Scholar]
- Mathews EA, García E, Santi CM, Mullen GP, Thacker C, Moerman DG, et al. 2003. Critical residues of the Caenorhabditis elegans unc-2 voltage-gated calcium channel that affect behavioral and physiological properties. J Neurosci. 23:6537–6545. doi:10.1523/JNEUROSCI.23-16-06537.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V.. 1991. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10:3959–3970. doi:10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser Von Filseck J, Lacas-Gervais S, Drin G, et al. 2013. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi Tether OSBP. Cell. 155:830. doi:10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, et al. 1996. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A. 93:12593–12598. doi:10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, et al. 2006. Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci U S A. 103:10811–10816. doi:10.1073/pnas.0509863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada T, Kashihara T, Komatsu M, Kojima K, Takeshita T, et al. 2018. Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proc Natl Acad Sci U S A. 115:4507–4512. doi:10.1073/pnas.1716649115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Mizushima A, Nakagawara K, Ichi, Takeshima H.. 2000. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 273:920–927. doi:10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- Nishi M, Sakagami H, Komazaki S, Kondo H, Takeshima H.. 2003. Coexpression of junctophilin type 3 and type 4 in brain. Mol Brain Res. 118:102–110. doi:10.1016/S0169-328X(03)00341-3. [DOI] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G.. 2015. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9ribonucleoprotein complexes. Genetics. 201:47–54. doi:10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Aeschimann F, Wang C, Lawson H, Serrano-Saiz E, et al. 2019. Timing mechanism of sexually dimorphic nervous system differentiation. Elife. 8:1–31. doi:10.1101/416735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Voeltz GK.. 2016. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 17:69–82. doi:10.1038/nrm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phimister AJ, Lango J, Eun HL, Ernst-Russell MA, Takeshima H, et al. 2007. Conformation-dependent stability of junctophilin 1 (JP1) and Ryanodine Receptor Type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J Biol Chem. 282:8667–8677. doi:10.1074/jbc.M609936200. [DOI] [PubMed] [Google Scholar]
- Porter KR, Palade GE.. 1957. Studies on the endoplasmic reticulum III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol. 3:269–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard HAT, Griffin CS, Yamasaki E, Thakore P, Lane C, et al. 2019. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc Natl Acad Sci U S A. 116:21874–21881. doi:10.1073/pnas.1911304116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB. 2007. Acetylcholine (January 30, 2007), Wormbook, ed. The C. elegans Research Community, Wormbook, 10.1895/wormbook.1.131.1, http://www.wormbook.org. [DOI] [Google Scholar]
- Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, et al. 2013. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res. 100:44–53. doi:10.1093/cvr/cvt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki T, Suzuki Y, Yamamura H, Takeshima H, Imaizumi Y.. 2019. A junctophilin-caveolin interaction enables efficient coupling between ryanodine receptors and BKCa channels in the Ca2+ microdomain of vascular smooth muscle. J Biol Chem. 294:13093–13105. doi:10.1074/jbc.RA119.008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, Bargmann CI.. 2009. Presynaptic CaV2 calcium channel traffic requires CALF-1 and the α2 subunit UNC-36. Nat Neurosci. 12:1257–1265. doi:10.1038/nn.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu G, Wazen RM, Colarusso P, Chen SRW, Zamponi GW, Turner RW.. 2019. Junctophilin proteins tether a Cav1-RyR2-KCa3.1 tripartite complex to regulate neuronal excitability. Cell Rep. 28:2427–2442. e6. doi:10.1016/j.celrep.2019.07.075. [DOI] [PubMed] [Google Scholar]
- Schafer WR, Sanchez BM, Kenyon CJ.. 1996. Genes affecting sensitivity to serotonin in Caenorhabditis elegans. Genetics. 143:1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens L, Félix M-A, Beltran T, Braendle C, Caurcel C, et al. 2019. Comparative genomics of 10 new Caenorhabditis species. Evol Lett. 3:217–236. doi:10.1002/evl3.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Shay J, McLoed M, Roodhouse K, Chung SH, et al. 2014. Neuronal regeneration in C. elegans requires subcellular calcium release by ryanodine receptor channels and can be enhanced by optogenetic stimulation. J Neurosci. 34:15947–15956. doi:10.1523/JNEUROSCI.4238-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K.. 2000. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 6:11–22. doi:10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- Trojanowski NF, Fang-Yen C.. 2015. Simultaneous optogenetic stimulation of individual pharyngeal neurons and monitoring of feeding behavior in intact C. elegans. In: Biron D, Haspel G, editors. C. elegans: Methods and Applications. Totowa, NJ: Humana Press. p. 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, et al. 2017. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature. 546:162–167. doi:10.1038/nature22369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, et al. 2011. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 123:979–988. doi:10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tang NH, Lara-Gonzalez P, Zhao Z, Cheerambathur DK, et al. 2017. A toolkit for GFP-mediated tissue-specific protein degradation in C. elegans. Development. 144:2694–2701. doi:10.1242/dev.150094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JS, Kim DH, Allen PD, Lee EH.. 2008. TRPC3-interacting triadic proteins in skeletal muscle. Biochem J. 411:399–405. doi:10.1042/BJ20071504. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, et al. 2007. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 104:15132–15137. doi:10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Sugimoto A, Ohshima Y, Takeshima H.. 2001. Important role of junctophilin in nematode motor function. Biochem Biophys Res Commun. 289:234–239. doi:10.1006/bbrc.2001.5951. [DOI] [PubMed] [Google Scholar]
- Ziman AP, Gómez-Viquez NL, Bloch RJ, Lederer WJ.. 2010. Excitation-contraction coupling changes during postnatal cardiac development. J Mol Cell Cardiol. 48:379–386. doi:10.1016/j.yjmcc.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Reagents are listed in Supplementary Tables S1–S4, which are available in figshare: https://doi.org/10.25386/genetics.14417696.
Strains and plasmids are available upon request.