

Abstract
Background:
Unintended weight loss and decreased body mass indexes (BMIs) are common symptoms of individuals with manifest HD. It is unknown at what point during disease progression weight loss starts to accelerate relative to a healthy individual’s weight and when recommended interventions should be initiated to have the strongest impact on patient care.
Objective:
The objective of this study was to identify a point in time relative to age at motor onset when the decline in weight in HD starts to accelerate relative to a non-HD population. The relationship between initiation of weight loss interventions and changes in weight loss was also explored.
Methods:
Participants from the fifth version of the Enroll-HD study were identified for this research. Linear mixed-effects piecewise regression models were used to estimate the point in time relative to the reported age of motor onset in which BMI started to decline in participants with HD compared to healthy non-HD controls. A post-hoc descriptive analysis was performed to look at when nutritional supplements and swallow therapy were initiated in participants with HD relative to motor onset.
Results:
BMI decline in the HD group began to accelerate compared to controls approximately 5.7 years after the reported age of motor onset (95% CI: 4.7–6.9). The average initiation times of swallow therapy and nutritional supplements were 7.7 years (SD=5.5 years) and 6.7 years (SD=6.5 years) after motor onset, respectively.
Conclusions:
Our findings suggest a potential point for intervention of nutrition programs or therapies used to prevent future weight loss.
Keywords: Huntington Disease, Disease Progression, Body-Weight Trajectory, Weight Loss
INTRODUCTION
Huntington’s disease (HD) is an inherited disease characterized by striatal atrophy, cortical grey and white matter loss, and cortico-striatal loop dysfunction leading to motor, cognitive, and functional impairments.[1–6] However, additional symptoms occur in HD and are not readily explained by the observed neurodegeneration.[2, 3] Specifically, unintended weight loss and decreased body mass indexes (BMIs) are common symptoms of individuals with manifest HD.[7, 8] It has been suggested that weight loss in HD may be influenced by metabolic and mitochondrial disturbances[9–11] and a recent study has also suggested that mutant huntingtin in peripheral tissues may be sufficient to cause metabolic abnormalities independent of neurodegeneration.[12] Additionally, chorea associated with HD may contribute to an increased calorie deficit leading to increased weight loss. A higher BMI has also been associated with a slower rate of clinical progression.[13, 14] It is unknown at what point during disease progression weight loss starts to become more clinically relevant relative to a healthy individual’s weight. Current recommendations for treating weight loss in HD include assessment by a dietician or nutritionist, addressing abilities that may affect weight such as swallowing, behavior, or mood, and using high-calorie and high-protein food supplements or feeding tubes when more severe weight loss is identified.[15] However, it is unclear when in the disease course of HD these recommended interventions should be initiated to have the strongest impact on patient care. Anecdotally, practitioners caring for patients with HD seem to initiate weight-maintaining interventions only after clinically relevant weight loss has already begun. Maintaining weight prophylactically or at the earliest signs of unintended weight loss may help slow disease progression in HD and maintain a higher quality of life.
Using the largest cohort of participants with HD, we identified a point relative to disease onset where BMI starts to decline relative to a healthy control population. Three sensitivity analyses were also performed based on CAG length, sex, and initial major symptom of HD. We also compared the start dates of treatments affecting weight loss relative to disease onset to the change point identified. Such a study is critically important to better understand how weight changes in HD and to identify when the most beneficial time for implementation of treatments or interventions may be.
MATERIALS AND METHODS
Participants
Participants from the fifth version of the Enroll-HD study were identified for this research study. Enroll-HD is a global, multi-center, observational study.[16] All sites are required to obtain and maintain local ethical approval. There are more than 21,000 participants enrolled, consisting of both genetic mutation carriers and controls. Controls consist of family controls (family members or individuals not related to carriers by blood, such as spouses) and genotype negative controls (first or second degree relative who is known not to carry the HD expansion mutation). Genetic mutation carriers can be split into premanifest HD (preHD) and manifest HD. Within the Enroll-HD database, participants with manifest HD have an estimated age of motor onset. At the time of enrollment into Enroll-HD, some participants were preHD and converted to manifest HD, some participants already had manifest HD at the time of enrollment, and some participants were preHD at their baseline visit and remain in the preHD group at the time of this analysis. The current analysis included participants who had presumably pheno-converted and had a reported age of motor onset, as well as non-HD controls (Figure 1).
Figure 1: Participant Inclusion Flow Chart.
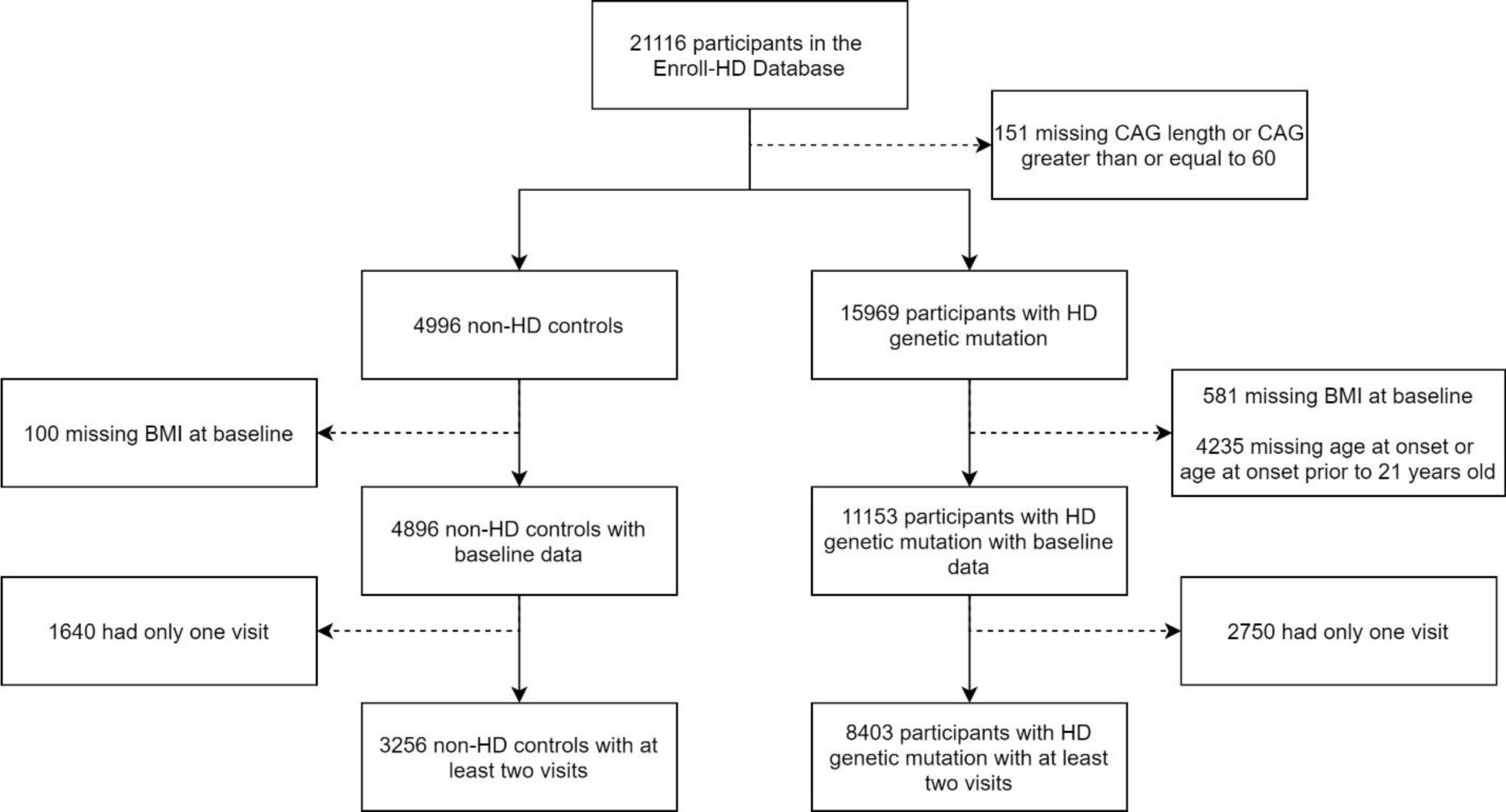
Figure 1 presents the implementation of inclusion and exclusion criteria for all participants in this study.
The CAG repeat length for HD participants was restricted to those between 36 and 59 and the age at motor onset had to be greater than 21 years old. This removes individuals with presumed juvenile-onset HD who may develop different symptoms [17] and may have different nutritional needs compared to those with adult-onset HD.[18,19] All participants were required to have a valid BMI measure and age at their baseline visit and a minimum of two study visits to be included in this study. Therefore, at baseline, some participants were already classified as having manifest HD while others were preHD at their baseline visit, but later converted to manifest HD while they were enrolled in the study.
Statistical Analysis
For the primary objective of this study, we used linear mixed-effects piecewise regression models to estimate the point in time relative to the reported age of motor onset in which BMI started to decline in participants with HD compared to healthy non-HD controls. A similar method has been used in Alzheimer’s disease to identify the onset of cognitive decline.[20] A profile likelihood approach, which identifies the change point using maximum likelihoods for the data observed, was used in this study. One knot was included in the model which allows for the change in BMI to differ at some time in the specified interval relative to disease onset. The model used for analysis is shown below for participant i at observation j:
In this model, the Group variable is 1 for participants with a CAG repeat length of 36 or greater and 0 for healthy controls. The variable ‘AgeDx’ is the reported age at motor diagnosis for participants with HD. The variable τ represents the change point, or the number of years relative to motor onset when BMI begins to decline. For the spline term, the value was 0 for non-HD controls or for a participant with HD at any observation where the age was less than the sum of the age at motor onset and the τ term. The spline term allows the trends of BMI to change in participants with HD at some point relative to motor onset. Models were fit over a range of τ values of 0.1-year increments starting at five years prior to motor onset and going to 10 years after motor onset. A quadratic spline term was tested in the model but removed due to non-significant effect. We centered age around 52 years old, the mean age of all participants included in the analysis, to allow for interpretation of the intercept. Random intercepts and slopes were also included to account for correlation between measurements of each individual and the intra-individual correlation in the rate of BMI change over time. This model allows for the groups to differ in both average BMI at baseline and the rate of change in BMI over time. Additionally, a diagonal covariance matrix was used. The optimal change point was chosen based on the model with the largest log likelihood of all models evaluated. After identifying the optimal change point, 1,000 bootstrap dataset samples with replacement were drawn and the change point identification process was repeated to calculate a confidence interval for the identified optimal change point.
Three sensitivity analyses were performed. The first assessed the optimal change point for groups based on their CAG repeat length. Participants with HD were grouped into those with CAG lengths between 36 and 39, those with CAG lengths between 40 and 43, those with CAG lengths between 44 and 46, those with CAG lengths between 47 and 50, and those with CAG lengths between 51 and 59. Each CAG group was compared with the non-HD controls using the modeling described above to identify the optimal change point for each group. The second sensitivity analysis was a sex stratified analysis. An optimal change point was identified for both males and females using the modeling methods described above. The final sensitivity analysis assessed the difference in change point based on the participant’s initial major symptom of HD. Motor, cognitive, and psychiatric initial major symptoms were included, and an optimal change point was identified compared with non-HD controls.
A post-hoc descriptive analysis was performed to look at when nutritional supplements that may be used to prevent weight loss and swallow therapy were initiated in participants with HD relative to motor onset. Swallow therapy was determined using the non-pharmacologic data collected through the Enroll-HD study. Participants are given a list of nine potential therapies and report their use at each visit. Nutritional supplements with an ingredient name containing “carbohydrates”, “protein”, or “fat” were of interest. Medications and supplements with those ingredients from the nutritional supplement and pharmacologic therapy data files were identified. Both supplements and swallow therapy had a listed day of initiation relative to the baseline study visit. This value was used to calculate the age at which an individual initiated the intervention-of-interest. The difference between age at which an intervention was initiated and age at motor onset was then calculated. The age at which participants with HD initiated an intervention aimed at improving weight loss was investigated relative to the presumed age that weight loss begins to decline in patients with HD compared to controls from the primary analysis. R version 4.0.3 was used for all analyses and a p-value of 0.05 was used to indicate statistical significance.
RESULTS
There were 8,403 participants with HD and 3,256 non-HD participants included in the primary analysis. Table 1 shows the age, sex, and BMI values at baseline for participants with HD and non-HD controls. There were significant differences in both age and BMI at baseline between the two groups. The table also describes the CAG length and age at motor onset for the participants with HD. Using the model described above, we found that BMI in the HD group began to decline approximately 5.7 years after the reported age of motor onset. Figure 2 is a graphical depiction of the log likelihood values plotted against values of the change point (τ) with the peak at 5.7 years. The 95 percent confidence interval calculated using the bootstrapped samples is 4.7 to 6.9 years after motor onset. The results of the final model are found in Table 2. To visualize the model, a plot was created using the effect estimates from the model with male, non-HD controls as the control group and male participants with HD who had an age at motor onset of 46 years as the comparison group (Figure 3). Raw data was plotted with the results from the model for individuals who meet the criteria of each group listed above.
Table 1.
| HD N = 8403 | Non-HD N = 3256 | P-Value | |
|---|---|---|---|
|
| |||
| Age, mean (SD) | 50.9 (12.4) | 47.6 (14.6) | < 0.001 |
| Sex – Male, N (%) | 4026 (47.9) | 1236 (38.0) | < 0.001 |
| CAG, mean (SD) | 43.6 (3.1) | 20.2 (3.7) | < 0.001 |
| BMI, mean (SD) | 25.1 (4.9) | 27.9 (6.2) | < 0.001 |
| Age at Motor Onset, mean (SD) | 46.6 (11.4) | NA | NA |
Figure 2: Identification of Optimal Change Point Using Log Likelihood.
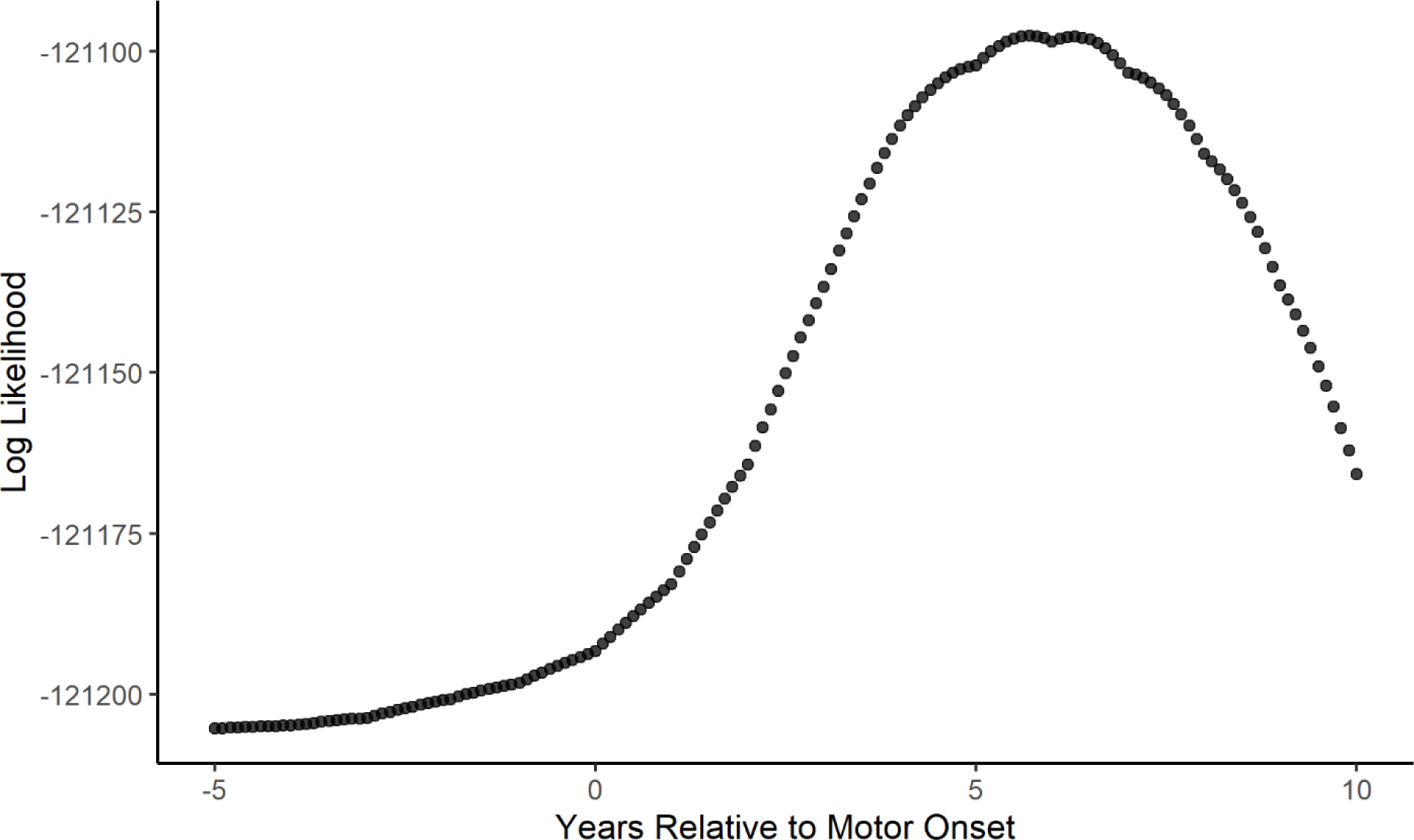
Figure 2 displays a graph of the log likelihood value for each potential change point in years relative to motor onset. The optimal change point is identified as the point with the greatest log likelihood value.
Table 2.
| Variable | Estimate | 95% Confidence Interval | p-value |
|---|---|---|---|
|
| |||
| Intercept | 28.77 | (28.63, 28.91) | < 0.001 |
| Age | 0.048 | (0.039, 0.057) | < 0.001 |
| Age2 | −0.0037 | (−0.0045, −0.0029) | < 0.001 |
| Sex | 0.69 | (0.61, 0.77) | < 0.001 |
| Group | −3.50 | (−3.66, −3.34) | < 0.001 |
| Age*Group | −0.020 | (−0.0079, −0.032) | 0.024 |
| Age2*Group | 0.0030 | (0.0021, 0.0039) | < 0.001 |
| Spline | −0.16 | (−0.18, −0.14) | < 0.001 |
Figure 3: Trajectories of BMI Over Time using the Optimal Change Point Model.
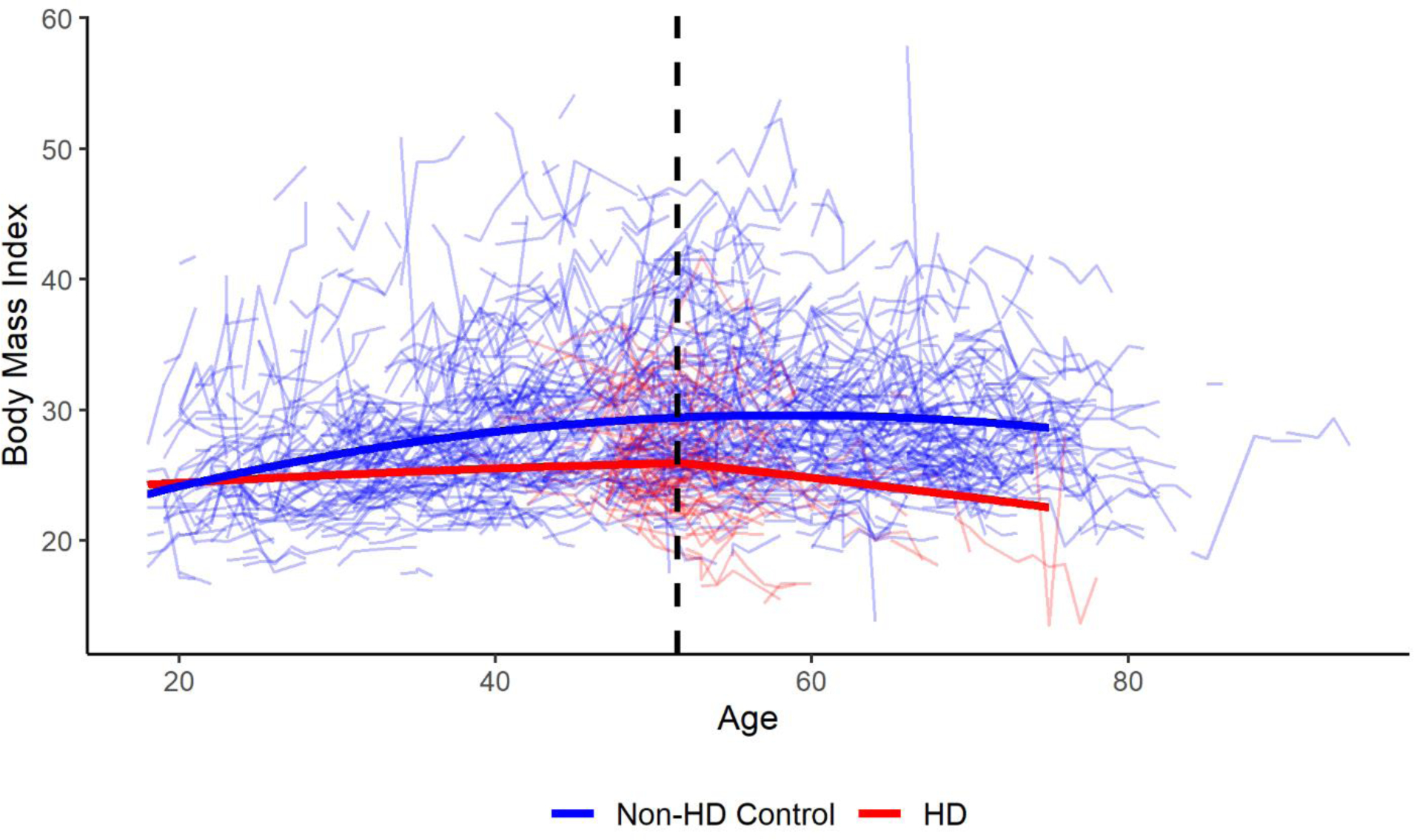
Figure 3 presents a graph of the optimal change point model for male participants with HD and non-HD controls. Male HD participants had an age at motor onset of 46 years. Raw data are also plotted in the background and the change point is indicated with a dashed black line.
Abbreviations:
BMI: Body Mass Index
HD: Huntington’s disease
For the sensitivity analysis comparing different CAG groupings compared to controls, the change point was further from onset in those with lower CAG lengths, except for the group of those with a CAG length of 36 to 39. Figure 4 shows the trajectories for each CAG grouping using the average age at onset for that group from the data. The average age at motor onset was 59 years old for the CAG 36 to 39 group, 52 years old for the CAG 40 to 43 group, 41 years old for the CAG 44 to 46 group, 34 years old for the CAG 47 to 40 group, and 27 years old for the CAG 51 to 59 group. The change points were 9.8 years after onset for those with a CAG length between 40 and 43, 6.5 years after onset for those with a CAG length between 44 and 46, 5.3 years after onset for those with a CAG length between 47 and 50, and 5.4 for those with a CAG length between 51 and 59. For those with a CAG length of 36 to 39, the change point was 4.0 years prior to onset. One potential explanation for the change point being prior to onset in those with a CAG length of 36 to 39 is the relatively small number of participants in this group (n=274). Another possible explanation is that the mean BMI seems to decrease in the non-HD control group near the age of 65, as seen in Figure 3. For those in the CAG length of 36 to 39 group, it is more likely that participants may have motor onset after the age of 65. Consequently, it is unclear if the observed decrease in BMI that begins near the age of 65 in the CAG length of 36 to 39 group (Figure 4) is age-dependent or a consequence of the expanded CAG repeat. This may explain why the identified change point in the CAG length of 36 to 39 group occurred prior to the average age of onset. For the sensitivity analysis comparing males and females, males had a change point of 5.5 years after onset compared to 7.5 years after onset for females. For the sensitivity analysis comparing motor, cognitive, and psychiatric initial major symptoms, those with a motor initial symptom had a change point of 5.7 years after onset compared to 4.1 years after onset for those with a cognitive initial symptom and 6.5 years after onset for those with a psychiatric initial symptom.
Figure 4: Trajectories of BMI Over Timing by CAG Length using the Optimal Change Point Model.
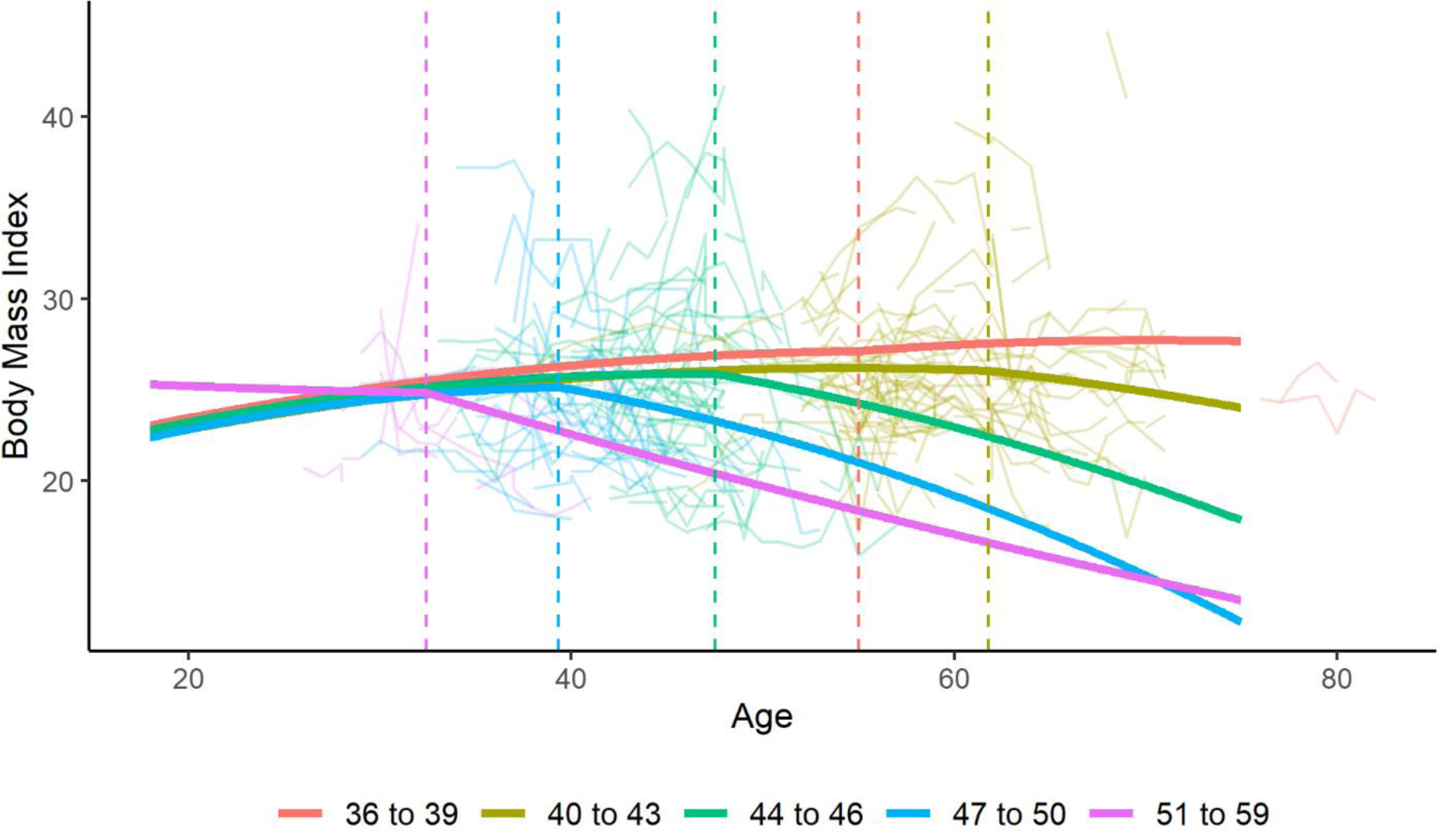
Figure 4 presents a graph of the optimal change point model for male participants stratified by CAG groupings. Age at onset varied for each group based on the average age at onset for the group from the data. Raw data are also plotted in the background. The change points are indicated with a dashed line.
Abbreviations:
BMI: Body Mass Index
CAG: Cytosine-Adenine-Guanine
There were 502 participants with HD who reported initiating swallow therapy. The average initiation of that therapy was 7.7 years after motor onset (SD=5.5 years). When removing individuals who had swallow therapy prior to their motor diagnosis, the average initiation of the therapy was 8.0 years after motor onset (SD = 5.3 years). There were only 11 participants with HD who started swallow therapy prior to their reported motor onset. There were no significant differences between those individuals and those who started therapy after onset with respect to sex, CAG length, or age at onset. However, the age at onset was approximately 6 years later in those who started therapy prior to onset than in those who started after (52.7 years compared to 46.3 years, p = 0.062). When compared to the change point identified in the previous analysis, swallow therapy was initiated approximately two years after the initial acceleration in decline of the BMI.
There were 1,138 participants with HD who used a nutritional supplement product with carbohydrates, fats, or proteins. The average time of initiation of those supplements was 6.7 years after motor onset (SD = 6.5 years). Based on our primary analysis showing that BMI decline accelerates about 5.7 years after motor onset, nutritional supplements were not initiated until about 1 year after the identified change inflection point. Of the 1,130 participants who used a nutritional supplement, 108 started a nutritional supplement prior to disease onset. These participants had a lower mean CAG length (42.9 compared to 44.2, p < 0.001) and later age at motor onset (50.1 years compared to 45.6 years, p < 0.001) relative to the participants who began taking nutritional supplements after motor onset. There was no significant difference in sex distribution between the two groups. It is unclear if these participants initiated nutritional supplements due to unintended weight loss or for other reasons. When looking only at the participants who initiated nutritional supplements after their reported motor onset, though, the average time of initiation of the supplements was 8.0 years after motor onset (SD = 5.2 years), which was more than 2 years after the beginning of acceleration in the decline of BMI.
DISCUSSION
This study used the largest cohort of participants with HD to show that acceleration in weight loss begins, on average, 5.7 years after the onset of motor symptoms of HD. Previous studies have described the pattern of weight loss in HD, but very little information exists regarding when in the disease course of HD that acceleration of weight loss begins. Therefore, these results fill a significant gap in our current knowledge regarding the potential timing of treatment for weight loss of patients with HD. Specifically, weight loss has been identified as a common symptom of HD, particularly in late stages of disease, and can lead to the use of a feeding tube or other invasive methods to prevent significant loss. Preventing or postponing the use of these measures may help increase patient quality of life and decrease the burden on patient caregivers. Our findings suggest a potential point for intervention of nutrition programs or therapies used to prevent future weight loss. Treatment information for swallow therapy and carbohydrate supplements showed that participants with HD initiated these therapies approximately two years after the time that the accelerated decline in BMI based on the change point analysis. This suggests that there may be a delay between weight loss symptoms and the initiation of treatment. Earlier intervention, even prior to the onset of acceleration in weight loss, may slow the acceleration of significant weight loss in HD and improve patient quality of life. Additionally, given that higher BMIs have been shown to be associated with slower disease progression of HD[13, 14], earlier interventions aimed at slowing unintended weight loss may actually slow the progression of HD. The results of this study also support previous work that rate of decline in BMI may be dependent on CAG length.[13] These results showed that the time from onset to acceleration in weight loss was longer for those with lower CAG lengths and shorter for those with higher CAG lengths. There were also differences in the change point based on sex with women having a longer period of time between onset and acceleration of weight loss. These results support future exploration of factors that would affect rate of weight loss to identify groups at the highest risk for more extreme declines in weight.
Many studies have shown that patients with HD, in any stage, have lower BMI values than healthy controls.[11, 21–23] There have also been studies that have shown patients with HD have lower BMI values even though dietary intake is similar to healthy controls.[24, 25] Data from this study also suggest that participants with HD have lower BMI values and may not have weight trajectories similar to controls prior to the acceleration in weight loss (Figure 3). This could suggest that implementing weight management strategies in the prodromal or early stages of the disease may provide additional benefit. Additional studies are required to validate these hypotheses, though. In Parkinson’s disease, weight loss has been associated with a decreased quality of life[26] and it can be hypothesized that similar effects may be seen in HD. Previous studies have identified effective diets and strategies to curb unintended weight loss in patients with neurodegenerative diseases.[27–29] Our results build on those previous studies by demonstrating that earlier validated therapies may have a positive impact on patients with HD.
This study does have limitations. First, treatment for symptoms of HD rely heavily on individual need. In this analysis, we identified the mean time at which patients with HD experience significantly accelerated weight loss compared to healthy controls. However, these results are based on a group analysis and do not necessarily reflect the needs of each individual patient with HD. Provider judgement is necessary on determining a healthy weight for each individual patient and treatments should be identified based on plans for specific patients. The results from this study emphasize that conversations about potential acceleration of weight loss may need to occur prior to signs of accelerated weight loss for potential benefit of the patient. Another limitation is that covariates that involve diet, physical activity, or body composition are not present in the Enroll-HD study which could help explain some of the results seen here. It is also unknown how the presence of psychiatric comorbidities could affect trends in BMI over time. Another limitation is all information about treatments and therapies are self-reported by study participants. It is possible that participants with HD addressed potential weight loss prior to the use of swallow therapy or nutritional supplements that are not included or recorded in the Enroll-HD study. The therapies are also not defined by set criteria, so swallow therapy may be dependent on a participant’s knowledge of what may be considered swallow therapy. Additionally, it is unknown what the indication was for both swallow therapy and supplement use. Patients with HD may have initiated swallow therapy due to dysphagia and not symptoms of weight loss. While dysphagia may affect the ability to maintain caloric intake, there are also many patients who may experience weight loss without the decrease in caloric intake or signs of dysphagia.
Despite these limitations, this study has many strengths. One of these strengths is the sample size used for analysis, as the Enroll-HD study is the largest of its kind for patients with HD. Another strength is the use of robust methods to identify unknown change points without a priori knowledge or assumptions and a bootstrapping approach to calculate unbiased standard errors and confidence intervals. Overall, the present study provides some of the first evidence about when in the disease course of HD acceleration in weight loss becomes significant. Furthermore, interventions aimed at potentially slowing or preventing further weight loss were not initiated until approximately two years after weight loss began. Future work to further assess the acceleration of weight loss in HD may focus on the identification of subgroups more likely to experience greater acceleration of weight loss, such as BMI prior to disease onset, and whether weight loss and therapy use is associated with changes in quality of life for both patients and their families. Additional work should also explore the association between weight loss and other common symptoms of HD, such as cognitive and psychiatric changes. Given that previous studies allow us to presume that maintenance of weight may slow disease progression and improve quality of life in HD, the results may provide practitioners new insights into the timing of treatment for weight loss in patients with HD.
ACKNOWLEDGEMENTS
Enroll-HD is a clinical research platform and longitudinal observational study for Huntington’s disease families intended to accelerate progress towards therapeutics; it is sponsored by CHDI Foundation, a nonprofit biomedical research organization exclusively dedicated to collaboratively developing therapeutics for HD. Enroll-HD would not be possible without the vital contribution of the research participants and their families.
This research was supported in part through computational resources provided by The University of Iowa, Iowa City, Iowa.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
- [1].A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–83. [DOI] [PubMed] [Google Scholar]
- [2].Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. The Lancet Neurology. 2013;12(7):637–49. [DOI] [PubMed] [Google Scholar]
- [3].Paulsen JS, Long JD, Ross CA, Harrington DL, Erwin CJ, Williams JK, et al. Prediction of manifest Huntington’s disease with clinical and imaging measures: a prospective observational study. The Lancet Neurology. 2014;13(12):1193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gómez-Ansón B, Alegret M, Muñoz E, Monté GC, Alayrach E, Sánchez A, et al. Prefrontal cortex volume reduction on MRI in preclinical Huntington’s disease relates to visuomotor performance and CAG number. Parkinsonism & Related Disorders. 2009;15(3):213–9. [DOI] [PubMed] [Google Scholar]
- [5].Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. The Lancet Neurology. 2011;10(1):31–42. [DOI] [PubMed] [Google Scholar]
- [6].Unschuld PG, Joel SE, Liu X, Shanahan M, Margolis RL, Biglan KM, et al. Impaired cortico-striatal functional connectivity in prodromal Huntington’s Disease. Neuroscience Letters. 2012;514(2):204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Djoussé L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH. Weight loss in early stage of Huntington’s disease. Neurology. 2002;59(9):1325–30. [DOI] [PubMed] [Google Scholar]
- [8].Sanberg PR, Fibiger HC, Mark RF. Body weight and dietary factors in Huntington’s disease patients compared with matched controls. The Medical journal of Australia. 1981;1(8):407–9. [DOI] [PubMed] [Google Scholar]
- [9].Mochel F, Charles P, Seguin F, Barritault J, Coussieu C, Perin L, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One. 2007;2(7):e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Morea V, Bidollari E, Colotti G, Fiorillo A, Rosati J, De Filippis L, et al. Glucose transportation in the brain and its impairment in Huntington disease: one more shade of the energetic metabolism failure? Amino Acids. 2017;49(7):1147–57. [DOI] [PubMed] [Google Scholar]
- [11].Dubinsky JM. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. Journal of Huntington’s Disease. 2017;6(4):267–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lakra P, Aditi K, Agrawal N. Peripheral Expression of Mutant Huntingtin is a Critical Determinant of Weight Loss and Metabolic Disturbances in Huntington’s Disease. Scientific Reports. 2019;9(1):10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].van der Burg JMM, Gardiner SL, Ludolph AC, Landwehrmeyer GB, Roos RAC, Aziz NA. Body weight is a robust predictor of clinical progression in Huntington disease. Annals of Neurology. 2017;82(3):479–83. [DOI] [PubMed] [Google Scholar]
- [14].Myers RH, Sax DS, Koroshetz WJ, Mastromauro C, Cupples LA, Kiely DK, et al. Factors associated with slow progression in Huntington’s disease. Archives of Neurology. 1991;48(8):800–4. [DOI] [PubMed] [Google Scholar]
- [15].Bachoud-Lévi AC, Ferreira J, Massart R, Youssov K, Rosser A, Busse M, et al. International Guidelines for the Treatment of Huntington’s Disease. Frontiers in Neurology. 2019;10:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Landwehrmeyer GB, Fitzer-Attas CJ, Giuliano JD, Gonçalves N, Anderson KE, Cardoso F, et al. Data Analytics from Enroll-HD, a Global Clinical Research Platform for Huntington’s Disease. Movement Disorders Clinical Practice. 2017;4(2):212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, Barbagallo G, et al. Biological and clinical manifestations of juvenile Huntington’s disease: a retrospective analysis. Lancet Neurology. 2018;17(11):986–93. [DOI] [PubMed] [Google Scholar]
- [18].Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, EHDI Study Group, et al. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71(19):1506–13. [DOI] [PubMed] [Google Scholar]
- [19].Tereshchenko A, McHugh M, Lee JK, Gonzalez-Alegre P, Crane K, Dawson J, et al. Abnormal Weight and Body Mass Index in Children with Juvenile Huntington’s Disease. Journal of Huntington’s Disease. 2015;4(3):231–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hall CB, Lipton RB, Sliwinski M, Stewart WF. A change point model for estimating the onset of cognitive decline in preclinical Alzheimer’s disease. Statistics in Medicine. 2000;19(11–12):1555–66. [DOI] [PubMed] [Google Scholar]
- [21].Dorsey E Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study. PloS One. 2012;7(2):e29522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Robbins AO, Ho AK, Barker RA. Weight changes in Huntington’s disease. European Journal of Neurology. 2006;13(8):e7. [DOI] [PubMed] [Google Scholar]
- [23].Farrer LA, Meaney FJ. An anthropometric assessment of Huntington’s disease patients and families. American Journal of Physical Anthropology. 1985;67(3):185–94. [DOI] [PubMed] [Google Scholar]
- [24].Trejo A, Tarrats RM, Alonso ME, Boll MC, Ochoa A, Velásquez L. Assessment of the nutrition status of patients with Huntington’s disease. Nutrition (Burbank, Los Angeles County, Calif). 2004;20(2):192–6. [DOI] [PubMed] [Google Scholar]
- [25].Cubo E, Rivadeneyra J, Armesto D, Mariscal N, Martinez A, Camara RJ. Relationship between Nutritional Status and the Severity of Huntington’s Disease. A Spanish Multicenter Dietary Intake Study. Journal of Huntington’s Disease. 2015;4(1):78–85. [PubMed] [Google Scholar]
- [26].Akbar U, He Y, Dai Y, Hack N, Malaty I, McFarland NR, et al. Weight loss and impact on quality of life in Parkinson’s disease. PloS One. 2015;10(5):e0124541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Christodoulou CC, Demetriou CA, Zamba-Papanicolaou E. Dietary Intake, Mediterranean Diet Adherence and Caloric Intake in Huntington’s Disease: A Review. Nutrients. 2020;12(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aganzo M, Montojo MT, López de Las Hazas MC, Martínez-Descals A, Ricote-Vila M, Sanz R, et al. Customized Dietary Intervention Avoids Unintentional Weight Loss and Modulates Circulating miRNAs Footprint in Huntington’s Disease. Molecular Nutrition & Food Research. 2018;62(23):e1800619. [DOI] [PubMed] [Google Scholar]
- [29].Trejo A, Boll MC, Alonso ME, Ochoa A, Velásquez L. Use of oral nutritional supplements in patients with Huntington’s disease. Nutrition (Burbank, Los Angeles County, Calif). 2005;21(9):889–94. [DOI] [PubMed] [Google Scholar]