

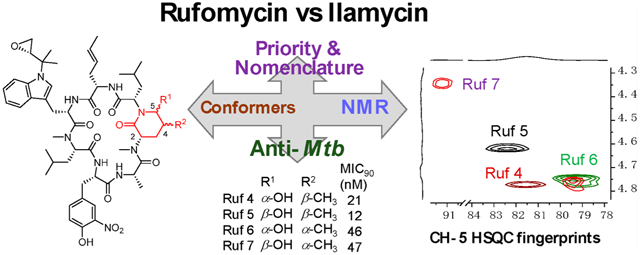
Abstract
Rufomycin and ilamycin are synonymous for the same class of cyclopeptides, currently encompassing 33 structurally characterized isolates and 9 semisynthetic derivatives. Elucidation of new structures prioritized the consolidation of the names and established the structures of four diastereoisomeric rufomycins with a 2-piperidinone, named rufomycins 4–7, including full 1H/13C NMR assignments. The characteristic HSQC cross-peak for the CH-5, the hemiaminal carbon in amino acid #5, allows assignment of the stereocenters C-4 and C-5 within this ring. Semisynthetic derivatives (rufomycinSS 1, 2, and 3) were prepared from a rufomycins 4 and 6 mixture to validate the structural assignments. Based on the X-ray crystal structures of rufomycins 2 and 4, considering the NMR differences of rufomycins 7 vs 4–6 compared to rufomycinSS 1 vs 2 and 3, and taking into account that two major conformers, A and B, occur in both rufomycinSS 2 and 3, structural modeling was pursued. Collectively, this paper discusses the NMR spectroscopic differences of the stereoisomers and their possible 3D conformers and correlates these with the anti-Mycobacterium tuberculosis activity. In addition, a look at the history prioritizes names and numbering schemes for this group of antibiotics and leads to consolidated nomenclature for all currently known members, natural and semisynthetic derivatives, and serves to accommodate future discoveries.
Graphical Abstract
In natural product research, it is common for two, or more, different groups to discover the same metabolite(s) and give them different names; the synonym of sophoretin for the very common flavonoid quercetin may serve as just one example. This can lead to muddied literature, confusion, and wrongly attributed credit. It is particularly prevalent among microbial products, where, for example, the single active pharmaceutical ingredient of fidaxomicin is tiacumicin B,1 lipiarmycin A3,2 and clostomicin B1,3 the same molecule but each produced by a different organism. This has now risen as a problem with respect to rufomycins/ilamycins. Takeda Chemical Industry was the first to publish them as rufomycins with a Japanese PCT filing in February 1960,4 supported by the production claims from a deposited new streptomycete (Streptomyces atratus nov. sp.).5,6 Almost two years after the patent filing, the Institute for Microbial Chemistry (IMC) published essentially the same complex, but called them ilamycins,7 which came from a poorly characterized and currently unavailable micro-organism, which they called Streptomyces islandicus. Three other groups working on these complexes have referred to them as rufomycins: Eli Lilly and Company,8–10 the University of Tokyo,11 and our Institute for Tuberculosis Research.12 In 2017, researchers obtained, from the South China sea, an organism that they characterized as S. atratus and which also produced these compounds; despite this, the authors referred to them, incorrectly, as ilamycins.13
These cyclic heptapeptides, almost invariably, have an isoprenyl group attached to the ring nitrogen of tryptophan, and this C5 residue is commonly oxidized, predominantly by epoxidation of the olefin. The most potent (anti-Mycobacterium tuberculosis [anti-Mtb]) members arise by post-NRPS oxidation of each of the diastereotopic terminal methyls of the N-methylleucine in amino acid position 5 (AA5). At the aldehyde level, these oxidized species interact with the nitrogen of AA6 to form hemiaminals, of which only three have been structurally characterized and none with respect to conformation. This might reflect challenges that arise from the fact that the configuration at the hemiaminal carbon is interchangeable in hydroxylic solvents, whereas that at the adjacent carbon, where it depends on from which terminal methyl the aldehyde originates, is stable. In the more anti-Mtb active series, the present study separated all four of the resulting diastereomers to allow for full NMR characterization. Moreover, three semisynthetic (rufomycinSS series) n-butyl aminal derivatives were prepared, thereby locking the stereochemistry at that carbon. While these derivatives tended to exist in multiple conformations in CD3OD, this occurred at sufficiently different concentrations to allow for complete NMR analyses of all major conformers.
Finally, to give appropriate credit to prior work and create a systemic nomenclature, this study looked at a detailed history of the discovery of rufomycins, which now appear to be available only from several strains of a single species, Streptomyces atratus. The authors have been unable to obtain S. islandicus or S. macrosporeus DSM-12818 from ATCC and IMC for the former and DSMZ for the latter. Collectively, this study establishes a consistent structural, stereochemical, and nomenclatural framework for this class of promising anti-Mtb agents.
RESULTS AND DISCUSSION
Naming Clarifications for Rufomycins vs Ilamycins.
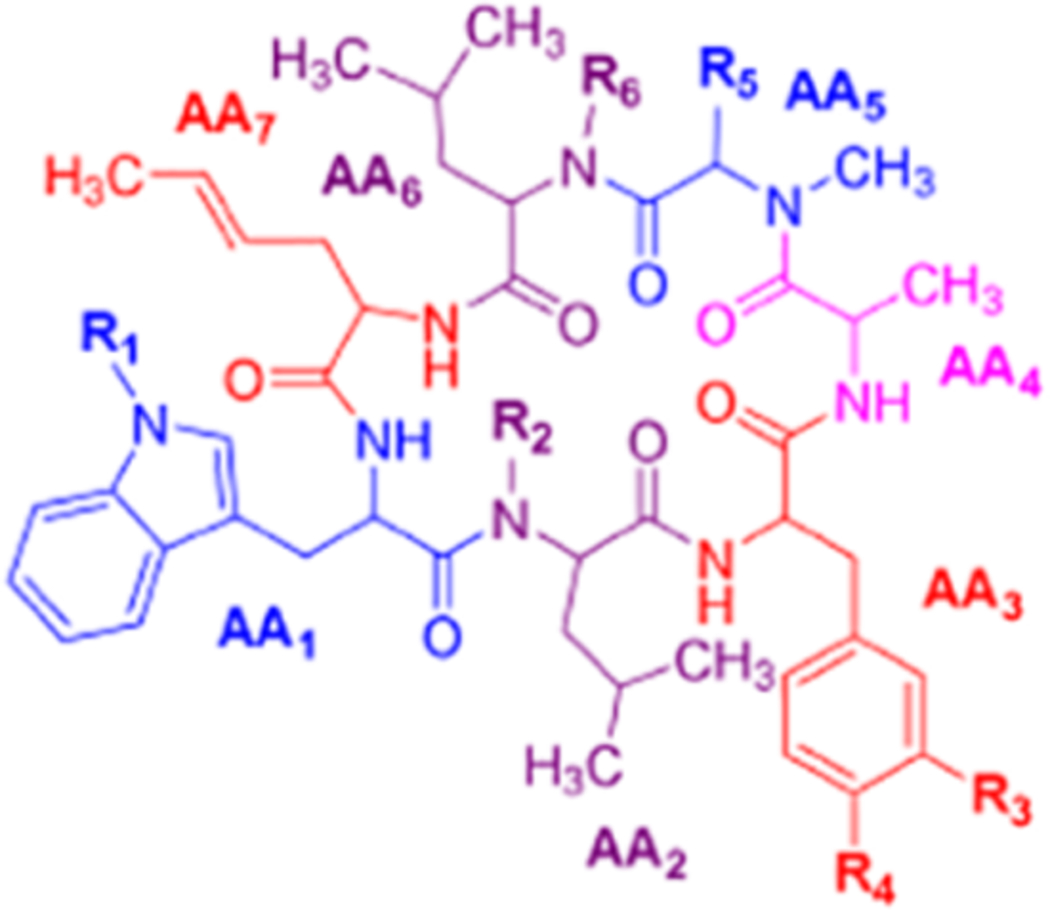
Table 1 is assembled to correlate the different names given to the rufomycin/ilamycin natural products and to give credit to the various scientists who discovered these compounds and elucidated their structures. There is clear realization that at the time of their initial discovery the science and use of NMR in structure determination was not yet well developed. Hence, many of the early structures proposed on the basis of elemental analysis, chemical degradation and derivatization, and IR and UV spectroscopy have been revised subsequently. The table is assembled in chronological order of groups making discoveries (in columns) and lists the compounds discovered (in rows). In the last column, we suggest a preferred consolidated name for what is currently believed to be the correct structure. These names will be used throughout this article, and they should help obviate confusion in the future. Furthermore, to minimize the ambiguity around atom numbering, especially while listing the NMR spectral assignments, and to rationalize this key task, we thought it would be appropriate to number the amino acids in the order they are assembled by the NRPS as AA1, AA2, etc., and the atoms in each amino acid as per IUPAC.
Table 1.
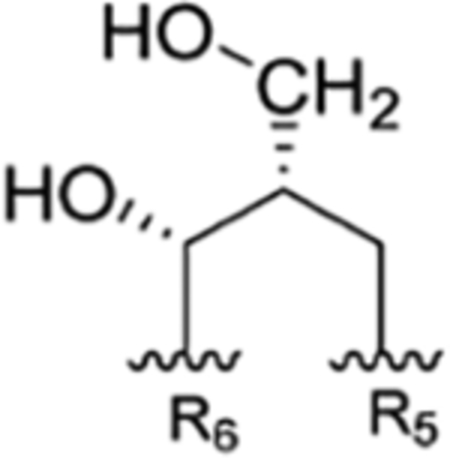
Structures, Synopsis of Published Names with References, and Preferred Consolidated Names of the Rufomycin Family of Cyclopeptidesa
| Index |
|
Discovering Research Groups and References | Preferred Consolidated Namea | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
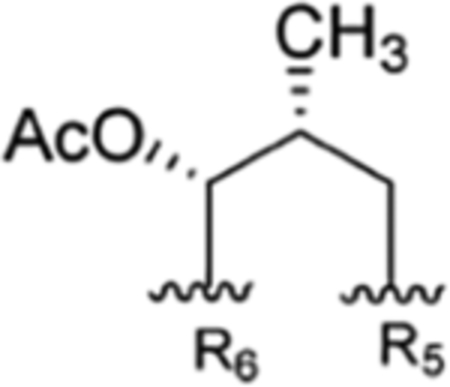
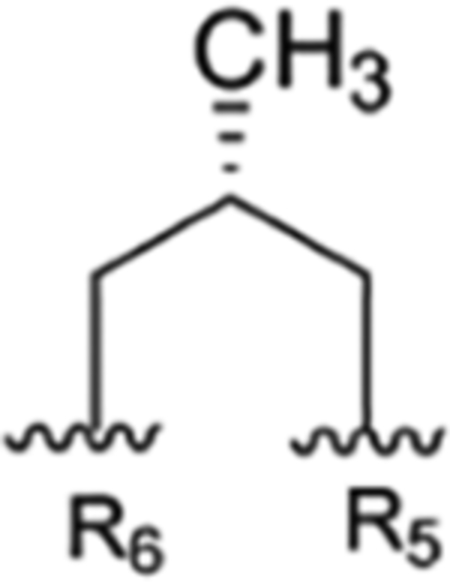
| R1 | R2 | R3 | R4 | R5 | R6 | Takeda | IMC | Eli Lilly | SCSIO, CAS | ITR | ||
| 1 |
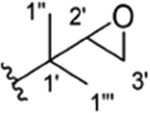
|
CH3 | NO2 | OH | IsoBu | H | rufomycin A4,5 | ilamycin B224 | Compound 28 | RUFV20 | no recommendation due to unclear C-2′configuration in R1 | |
| 2 |
|
CH3 | NO2 | OH | IsoBu | H | rufomycin B4,5 | ilamycin B16,24,25,27,28 | Compound 18 | ilamycin B113 | 1123 | rufomycin 1 |
| 3 |
|
CH3 | NO2 | OH |
|
H | rufomycin A4,5 | ilamycin14,22,24 | Compound 3# | no recommendation due to unclear C-2′configuration in R1 | ||
| 4 |
|
CH3 | NO2 | OH |
|
ilamycin22 | Compound 48 | RUF I20 rufomycin I21 1223 |
no recommendation due to unclear C-2′configuration in R1 | |||
| 5 |
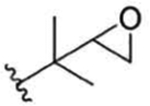
|
CH3 | NO2 | OH |
|
Compound 58 | no recommendation due to unclear C-2′confiquration in R1 | |||||
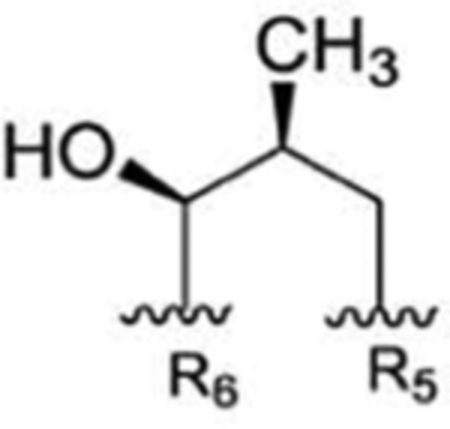
| 6 |
|
CH3 | NO2 | OH |
|
Compound 68 | RUF II20 | no recommendation due to unclear C-2′configuration in R1 | ||||
| 7 |
|
CH3 | NO2 | OH |
|
H | Compound 78 | rufomycin VII21 RUF VII20’21 |
no recommendation due to unclear C-2′configuration in R1, R5 | |||
| 8 |
|
H | NO2 | OH | IsoBu | H | RUF IV20 | no recommendation due to unclear C-2′configuration in R1 | ||||
| 9 |
|
CH3 | NO2 | OH | IsoBu | H | ilamycin B213 | 1023 | rufomycin 2 | |||
| 10 |
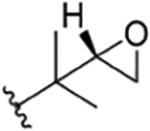
|
CH3 | NO2 | OH |
|
H | rufomycin 3# | |||||
| 11 |
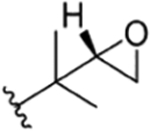
|
CH3 | NO2 | OH |
|
Compound 48 | ilamycin C113 | RUF I20 rufomycin I21 1223 |
rufomycin 4 | |||
| 12 |
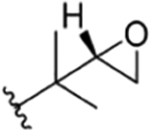
|
CH3 | NO2 | OH |
|
Compound 58 | ilamycin C213 | rufomycin 5 | ||||
| 13 |
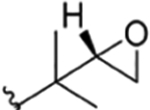
|
CH3 | NO2 | OH |
|
Compound 68 | The present study | rufomycin 6 | ||||
| 14 |
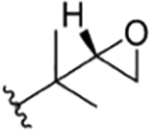
|
CH3 | NO2 | OH |
|
The present study | rufomycin 7 | |||||
| 15 |
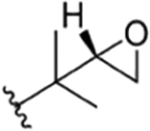
|
CH3 | NO2 | OH |
|
H | rufomycin 8# | |||||
| 16 |
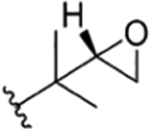
|
CH3 | NO2 | OH |
|
H | ilamycin D13 | rufomycin 9 | ||||
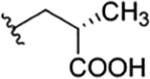
| 17 |
|
CH3 | NO2 | OH |
|
ilamycin E113,32 ilamycin E34 |
Structure revised in the present study | rufomycin 21 | ||||
| 18 |
|
CH3 | NO2 | OH |
|
ilamycin E213 | rufomycin 22 | |||||
| 19 |
|
CH3 | NO2 | OH |
|
H | ilamycin F13,32 | rufomycin 23 | ||||
| 20 |
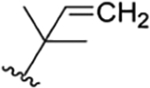
|
CH3 | H | OH | IsoBu | H | RUF VI20 rufomycin NBZ423 |
rufomycin 51 | ||||
| 21 |
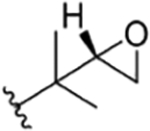
|
H | NO2 | OH | IsoBu | H | rufomycin NBZ623 | rufomycin 52 | ||||
| 22 |
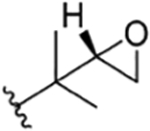
|
CH3 | NO2 | OH |
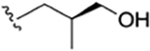
|
H | rufomycin NBZ123 | rufomycin 11 | ||||
| 23 |
|
CH3 | NO2 | OH |
|
H | rufomycin NBZ223 | rufomycin 10 | ||||
| 24 |
|
CH3 | NO2 | OH |
|
H | rufomycin NBZ523 | rufomycin 12 | ||||
| 25 |
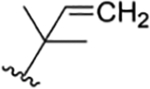
|
CH3 | NO2 | OH |
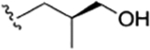
|
H | rufomycin NBZ323 | rufomycin 24 | ||||
| 26 |
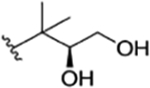
|
CH3 | NO2 | OH |
|
rufomycin NBZ723 | rufomycin 41 | |||||
| 27 |
|
CH3 | NO2 | OH |
|
rufomycin NBZ823 | rufomycin 13 | |||||
| 28 |
|
CH3 | NO2 | OH | IsoBu | H | 1323 | rufomycin 42 | ||||
| 29 |
|
CH3 | NO2 | OH |
|
H | ilamycin G33 | rufomycin 43 | ||||
| 30 |
|
CH3 | NO2 | OH |
|
H | ilamycin H33 | rufomycin 25 | ||||
| 31 |
|
CH3 | NO2 | OH |
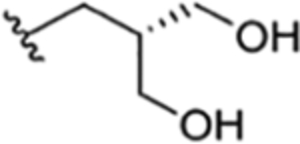
|
H | ilamycin I33 | rufomycin 26 | ||||
| 32 |
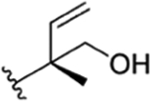
|
CH3 | NO2 | OH |
|
ilamycin J33 | rufomycin 44 | |||||
| 33 |
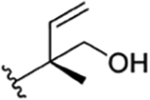
|
CH3 | NO2 | OH |
|
ilamycin K33 | rufomycin 45 | |||||
| 34 |
|
CH3 | NO2 | OH |
|
H | ilamycin L33 | rufomycin 46 | ||||
| 35 |
|
CH3 | NO2 | OH |
|
H | ilamycin M33 | rufomycin 27 | ||||
| 36 |
|
CH3 | NH-CHO | OH |
|
H | ilamycin N33 | rufomycin 53 | ||||
| 37 |
|
CH3 | H | OH |
|
H | ilamycin O33 | rufomycin 54 | ||||
| 38 |
|
CH3 | NO2 | OH |
|
ilamycin P33 | rufomycin 28 | |||||
| 39 |
|
CH3 | NO2 | OH |
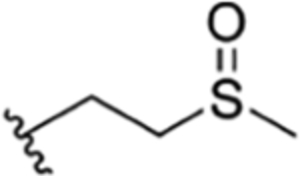
|
H | ilamycin Q33 | rufomycin 29 | ||||
| 40 |
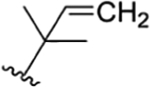
|
CH3 | NO2 | H |
|
ilamycin R33 | rufomycin 55 | |||||
| Semisynthetic derivatives (rufomycinSS) | ||||||||||||
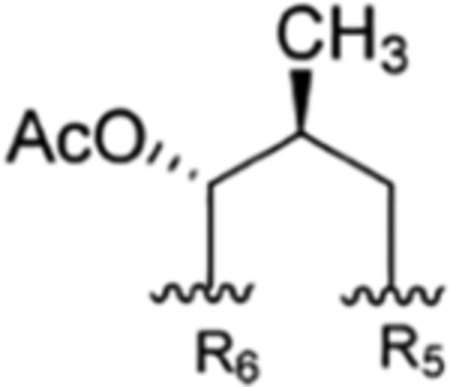
| 1 |
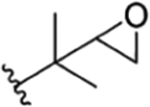
|
CH3 | NO2 | OAC |
|
Compound 88 | rufomycinSS 8 | |||||
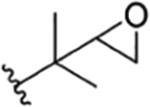
| 2 |
|
CH3 | NO2 | OAC |
|
Compound 98 | rufomycinSS 9 | |||||
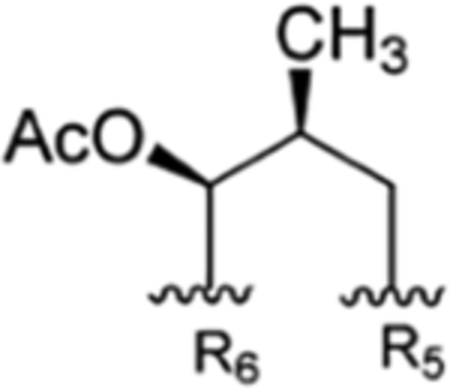
| 3 |
|
CH3 | NO2 | OH |
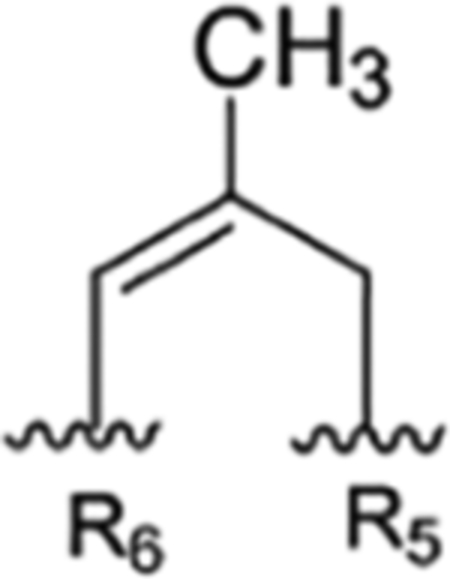
|
Compound 108 | rufomycinSS 10 | |||||
| 4 |
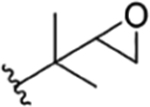
|
CH3 | NO2 | OH |
|
Compound 118 | rufomycinSS 11 | |||||
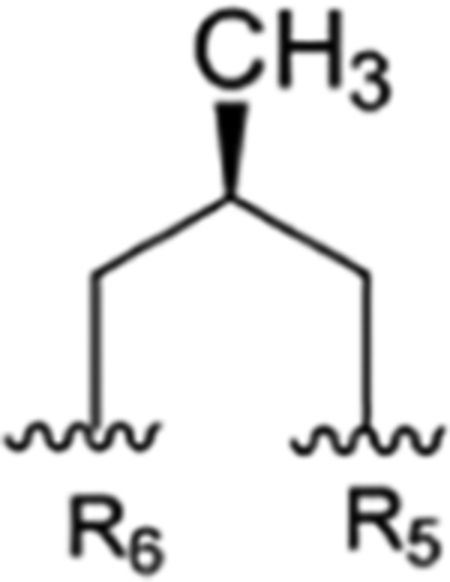
| 5 |
|
CH3 | NO2 | OAC |
|
Compound 128 | rufomycinSS 12 | |||||
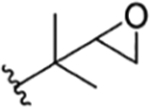
| 6 |
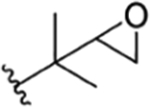
|
CH3 | NO2 | OH |
|
Compound 138 | rufomycinSS 13 | |||||
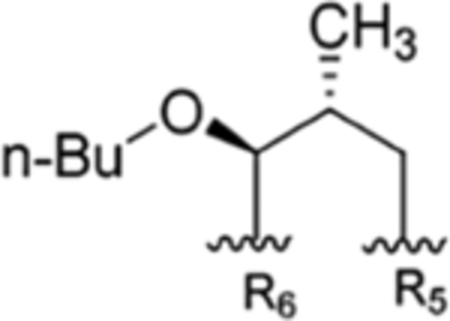
| 7 |
|
CH3 | NO2 | OH |
|
The present study | rufomycinSS 1 | |||||
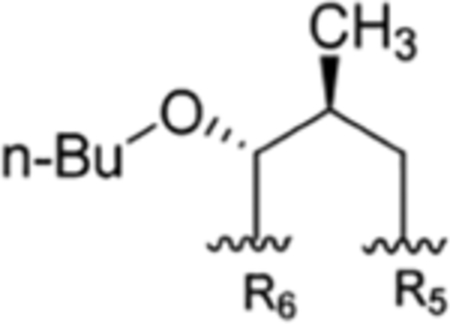
| 8 |
|
CH3 | NO2 | OH |
|
The present study | rufomycinSS 2 | |||||
| 9 |
|
CH3 | NO2 | OH |
|
The present study | rufomycinSS 3 | |||||
The numbering used has been derived by that of the Eli Lilly scientists who were the first to discover and structurally elucidate significant numbers of this family of heptapeptides. With the exception of rufomycin 1, an olefin, which was the first of the family to be structurally characterized, the numbers up to 20 have been reserved for epoxides on the isoprenyl group of AA1, numbers 21–40 for the corresponding olefins, 41–50 for other oxidations of this group, and above 50 for variants involving changes that precede the assembly via the NRPS. The semisynthetics (rufomycinSS) have been numbered by us and according to the Eli Lilly group the only groups that have been published on semisynthetics.
While these compounds have not been isolated, nor are any spectroscopic data for them available, their transitory existence is not in doubt. Rufomycin 12 is probably not a natural product but rather an artifact of the isolation as suggested.29 The same may be the case for rufomycin 30.
Biological Significance and Chemistry.
Rufomycins comprise a family of cyclic heptapeptides, which basically contain leucine (Leu), two N-methylleucines (NMeLeu), alanine (Ala), and three nonproteinogenic amino acids, N-dimethylallyl(epoxy)tryptophan, trans-2-crotylglycine (TrcGly), and m-nitrotyrosine (mNO2Tyr).4–6,14–19
The first report of the anti-Mtb property of rufomycins can be traced back to 1960 (US 3,655,879 citing Japanese 35/4,033, filed Feb 8, 1960).4 Our previous study also revealed that rufomycin 4 showed potent anti-Mtb activity with an MIC value of ~0.020 μM.20 Rufomycin 4 represents a special type of rufomycin structure with an in-chain hydroxy-methylpiperidinone moiety. It is one of four diastereomers that arise as tautomers of the two aldehydes obtained by oxidation of the diastereotopic terminal methyls of leucine AA5 and are formed by nucleophilic addition of the nitrogen of leucine AA6 to these carbonyls. This type of rufomycin structure is difficult to define because (a) several conformers were observed due to the flexible nature of the macrocycle; (b) the absolute configuration of C-4 bearing a methyl group in AA5 can be either R or S depending on which of the diastereotopic methyl groups of AA5 (leucine) is oxidized; (c) the hemiaminal carbon can be either R or S; and (d) this last feature is interchangeable due the nature of hemiaminals. Theoretically, in analogy to the open-chain aldehyde of hexoses, ring-opened products can also be observed, which predominantly exist as hemiacetals. As far as we know, three related diastereomers have been reported, including rufomycin 4 (AA5, 4S,5R), rufomycin 5 (AA5, 4S,5S), and rufomycin 6 (AA5, 4R,5R).8–10,13 Crystal structures of the rufomycin 4/ClpC1-NTD complex were obtained during our previous study,21 which not only confirmed the absolute configuration of rufomycin 4 (syn. ilamycin C1 and rufomycin A) but also showed a tight fit in a pocket of ClpC1-NTD with the hydroxyl-methyl-methylamidopiperidinone functionality. Thus, this function is considered as an important part of the anti-Mtb pharmacophore of rufomycins.
In the present study, rufomycin 4 and its related three diastereomers (rufomycins 5–7) were isolated and extensively studied. The unreported diastereomer, rufomycin 7, with AA5 4R,5S configuration, was identified as the most unstable diastereomer, showing quite different NMR patterns from those of rufomycins 4–6. To further understand the reason that such different NMR data were observed among rufomycins 4–7, a one-step reaction on a mixture of rufomycins 4 and 6 was carried out to protect the OH group with an n-butyl group and hence lock the stereochemistry at the now aminal carbon. Three corresponding semisynthetic compounds, rufomycinSS 1–3, were isolated, and their structures assigned from 1D and 2D NMR data. Both rufomycinSS 2 and 3 showed three conformers, and the major two conformers varied considerably in their 1H and 13C NMR data. The consistent trends of ΔδC and ΔδH between the two conformers in rufomycinSS 2 and 3 are similar to those seen in the spectra of rufomycins 4 and 7. The NMR differences of these compounds are believed to be caused by conformational changes. Here, NMR differences, possible 3D conformations, and correlated anti-Mtb activities of rufomycins and these semisynthetic derivatives are discussed.
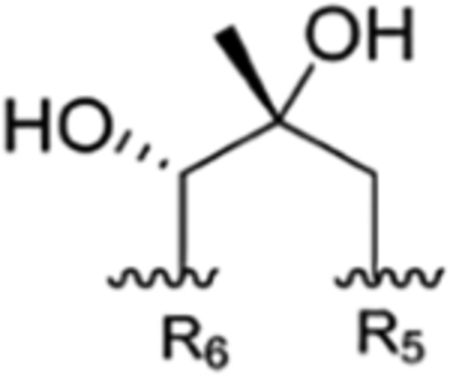
Rufomycin Piperidinone Diastereomerism.
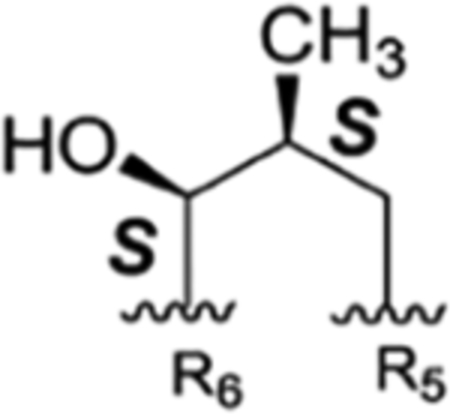
The rufomycins 4–7 were identified as four diastereomers, arising from different orientations of the hydroxyl and methyl groups in the hydroxy-methylpiperidinone moiety. The absolute configurations of C-4 and C-5 in AA5 of rufomycins 4–7 could be deduced from ROESY data, as the absolute configuration of AA5-C-2 was assigned as S, corresponding to the l-NMeLeu residue based on the X-ray crystal data and biosynthetic considerations.10,21 Rufomycin 4 was assigned the (AA5, 4S,5R) absolute configuration based on the ROESY correlations of H3-1′/H-2 and H-5 (Figure 2) and further confirmed by the cocrystal structures of rufomycin 4 and ClpC1-NTD.21 Thus, rufomycin 4 is the congener previously identified as ilamycin, compound 4, and ilamycin C1.8,13,22 HPLC analysis (Figure 3) showed rufomycins 4 and 5 are interconvertible, indicating rufomycin 5 to be an epimer of rufomycin 4 at the hemiaminal carbon, based on the expected property of hemiaminals. The assigned 4S,5S (AA5) absolute configuration for rufomycin 5 was validated by the ROESY correlations (Figure 2) of H-2/H-3β; of H-4/H-3α and H-5; and of H-5/H3-1′. Rufomycin 6, with the absolute configuration 4R,5R (AA5), was established by the ROESY cross-peaks (Figure 2) of H-2/H-3β and H-4 and of H-5/H3-1′ and H-4. Finally, the fourth diastereoisomer, rufomycin 7, which has not been reported previously, was isolated and identified in this study. Rufomycin 6 was found to convert slowly to a small amount of rufomycin 7 (~5%) under aqueous conditions. Rufomycin 7, the most unstable diastereomer, quickly epimerized to rufomycin 6 (~50%) during concentration. After being purified by semipreparative HPLC, rufomycin 7 was concentrated under a stream of forced air and freeze-drying to reduce interconversion. The 1H NMR spectra (Figure S22–1, Supporting Information) showed the coexistence of both rufomycins 6 and 7 (~2:5). The ROESY correlations (Figure 2) between H-2 and H-4 and between H-5 and H3-1′ and H-3α confirmed that rufomycin 7 had a 4R,5S (AA5) absolute configuration.
Figure 2.
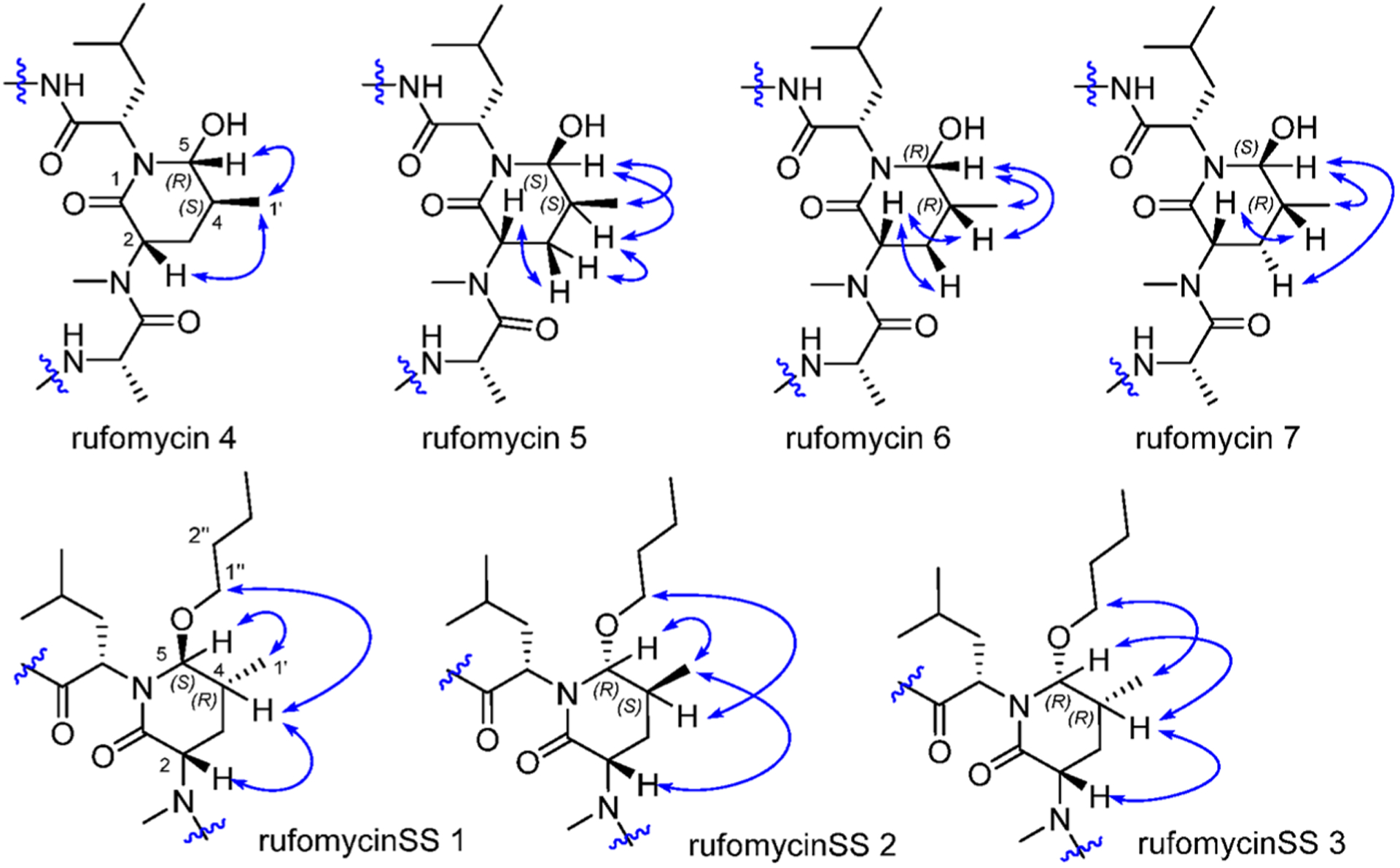
ROESY correlations within the 2-piperidinone of rufomycins 4–7 and rufomycinSS 1–3.
Figure 3.
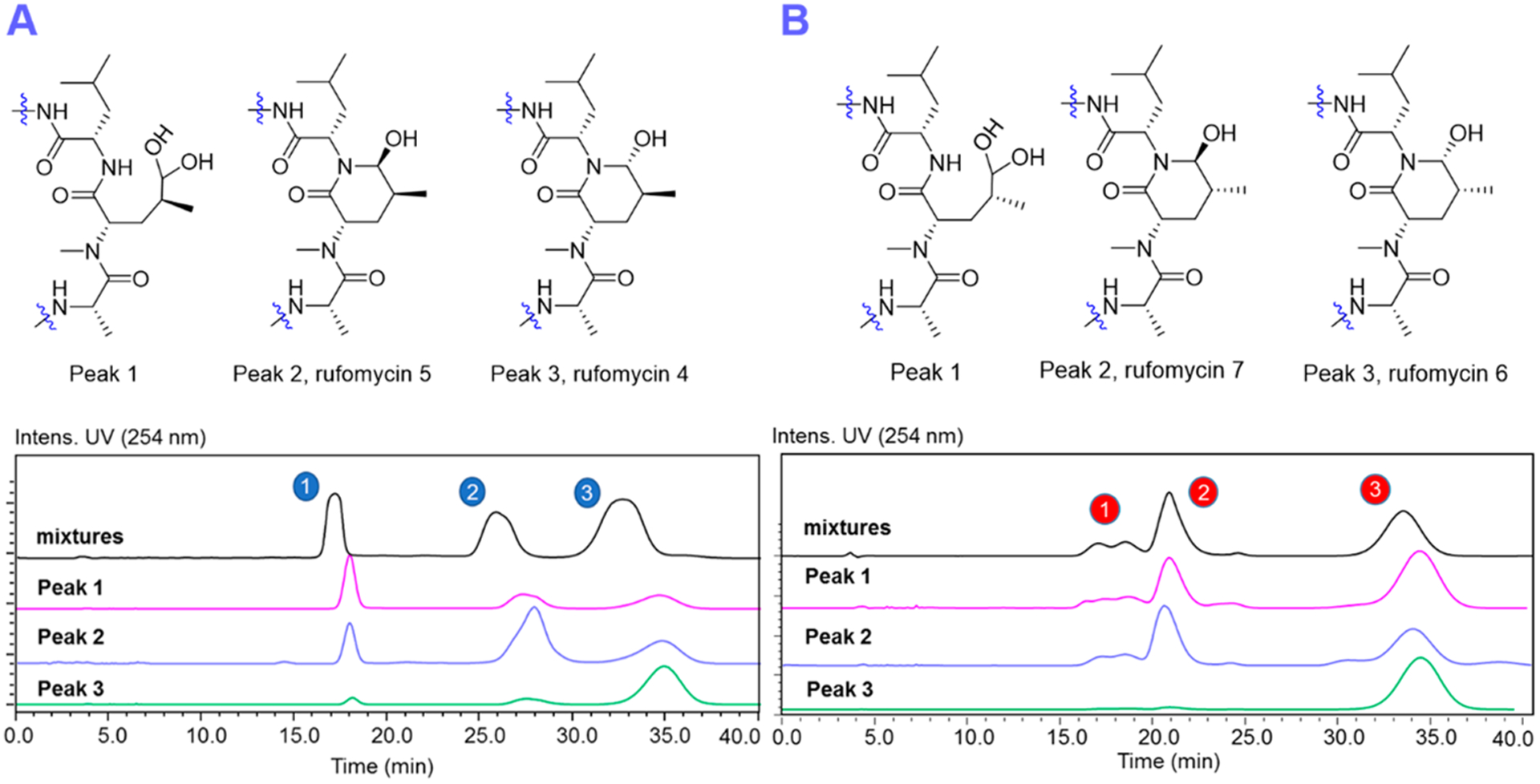
HPLC chromatograms (RP-18; 55% ACN/H2O, 2.8 mL/min for the anomers and their corresponding ring-opened forms (aldehyde hydrate) of rufomycins 4/5 (A) and rufomycins 6/7 (B).
Chromatographic Characteristics.
The HPLC chromatograms (Figure 3A) of rufomycins 4 and 5 showed three major peaks, of which peaks 1–3 were assigned, in their elution order, as follows: First is the hemiaminal ring-opened intermediate (rufomycin 3 hydrate), followed by rufomycin 5 and rufomycin 4. Reinjection of each of the materials collected as those three single peaks showed the same three peaks again (Figure 3A), indicating that the underlying three molecular species form a thermodynamic equilibrium. This equilibration slightly favors rufomycin 4 (third peak). Similar to the mutarotatory property of glucose, it involves the two major pyranoid forms, two minor furanoid forms, and traces (0.02%) of the open-chain form (see masterorganicchemistry.com/2017/08/17/mutarotation). The hemiaminal of the rufomycins displays analogous equilibration behavior and forms with a higher likelihood of its open-chain forms (free aldehyde and/or hydrate). Figure 4 summarizes how these rufomycins interconvert between open- and ring-forms by forming a hemiaminal, an aldehyde, and an aldehyde hydrate (under aqueous conditions) or hemiacetals in alcohols. The HSQC spectra of the materials from LC peaks 1–3 (Figure 5), acquired in CD3OD, clearly showed the signals of two molecular species (δH 4.26, d, J = 5.3 Hz; δC 103.3; and δH 4.35, d, J = 4.2 Hz; δC 102.8). This was indicative of the presence of two ring-opened species and can be explained by the presence of two diastereomeric trideuteromethyl hemiacetals formed by reaction of the aldehyde function with both protons and deuterons present in the NMR solvent, CD3OD, with residual HDO.
Figure 4.
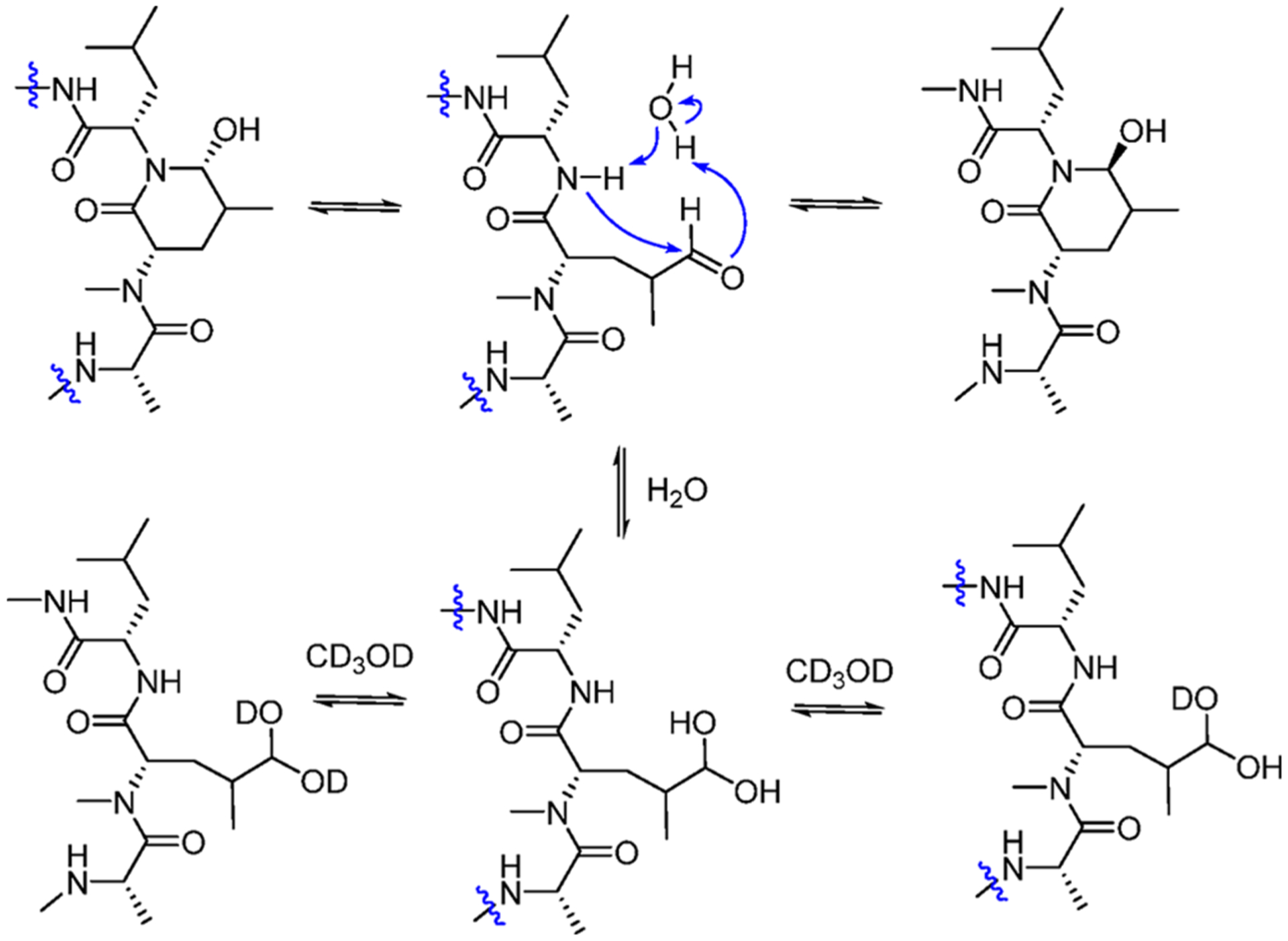
Putative mechanisms for the interconversion between the hemiaminal, aldehyde-amide, and (deuterated) hydrate forms of the rufomycins 4–7.
Figure 5.
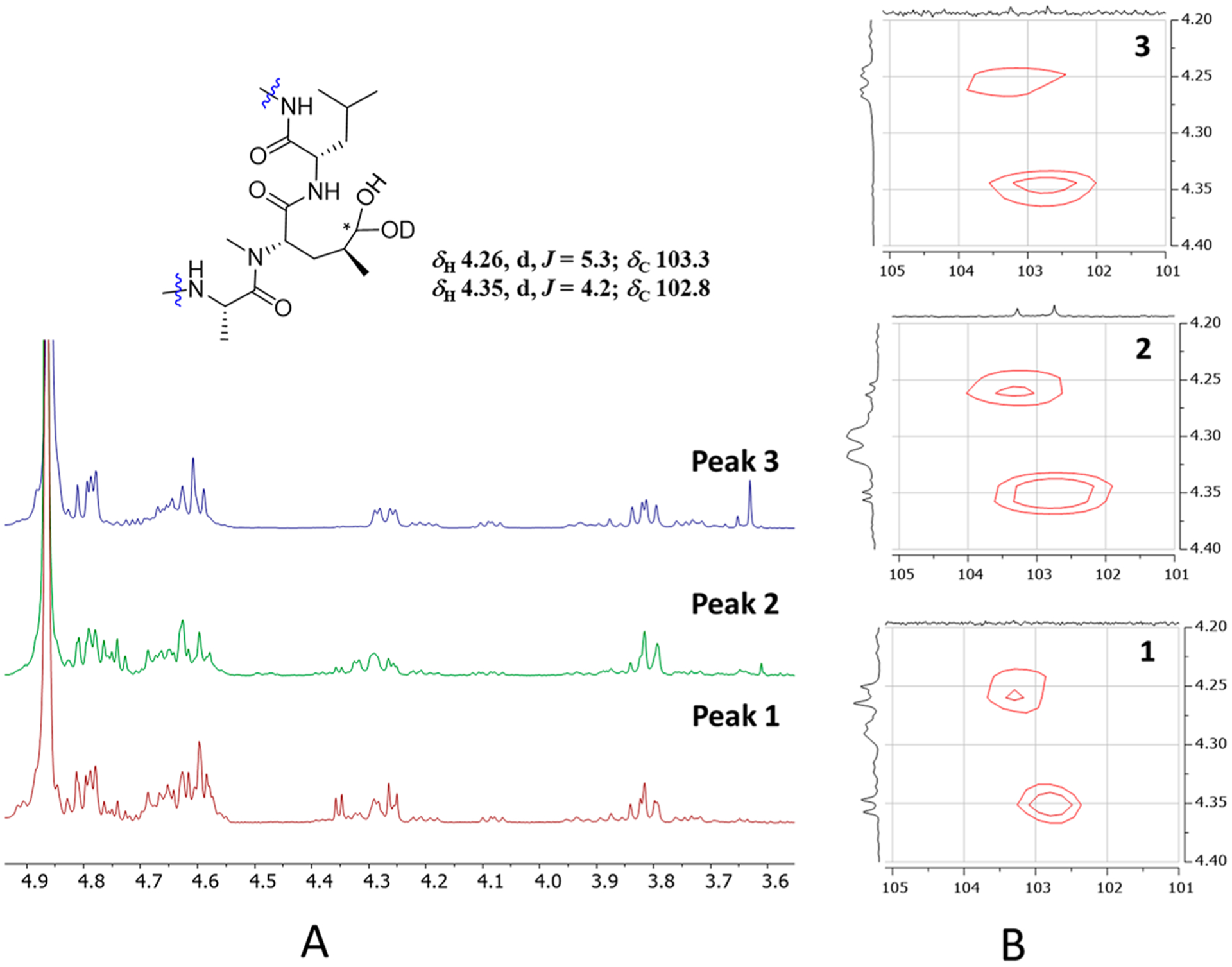
1H NMR (A) and HSQC (B) spectra of the three related LC peaks (Figure 3) of the mutarotatory rufomycins 4 and 5.
In HPLC, rufomycins 6 and 7 behaved similarly to rufomycins 4 and 5. Three peaks representing the hemiaminals and ring-opened products, rufomycin 7 and rufomycin 6, were observed (Figure 3B). The HSQC spectra (Figure S50, Supporting Information) of rufomycins 6 and 7 also showed two signals (δH 4.28, d, J = 5.8 Hz; δC 103.4; and δH 4.37, d, J = 4.7 Hz; δC 102.8) for the ring-opened products. Compared to rufomycin 7, rufomycin 6 was identified as the more stable epimer that predominates in equilibrium.
NMR Spectroscopic Characteristics of Rufomycins 4–7.
Comparison of the 1H and 13C NMR data of rufomycins 4–7 revealed that major differences were found around the hemiaminal (hydroxy-methylpiperidinone) moiety (Figures 6 and 7). The 1H and 13C NMR data of rufomycins 4–6 varied within a reasonable range; however, those of rufomycin 7 were distinctly different. The three most different carbon signals C-2 (AA6), C-5 (AA5), and NCH3 (AA5) showed 10.2, 9.5, and 7.7 ppm differences, respectively, from corresponding carbons in the spectrum of rufomycin 4 (Figure 6). Two hydrogens of rufomycin 7 showed dramatic differences from corresponding hydrogens in the spectrum of rufomycin 4 with a ΔδH value of 1.5 and 1.8 ppm for H-2 (AA6) and H-2 (AA5) (Figure 7). Comparison of the 13C NMR data revealed that the four diastereoisomers rufomycins 4–7 varied at the characteristic carbon C-5 (AA5). The HSQC cross-peaks for CH-5 (AA5) occurring in distinctly separate regions may be useful to assign the configurations of two stereocenters at C-4 and C-5 of the AA5 residue.
Figure 6.
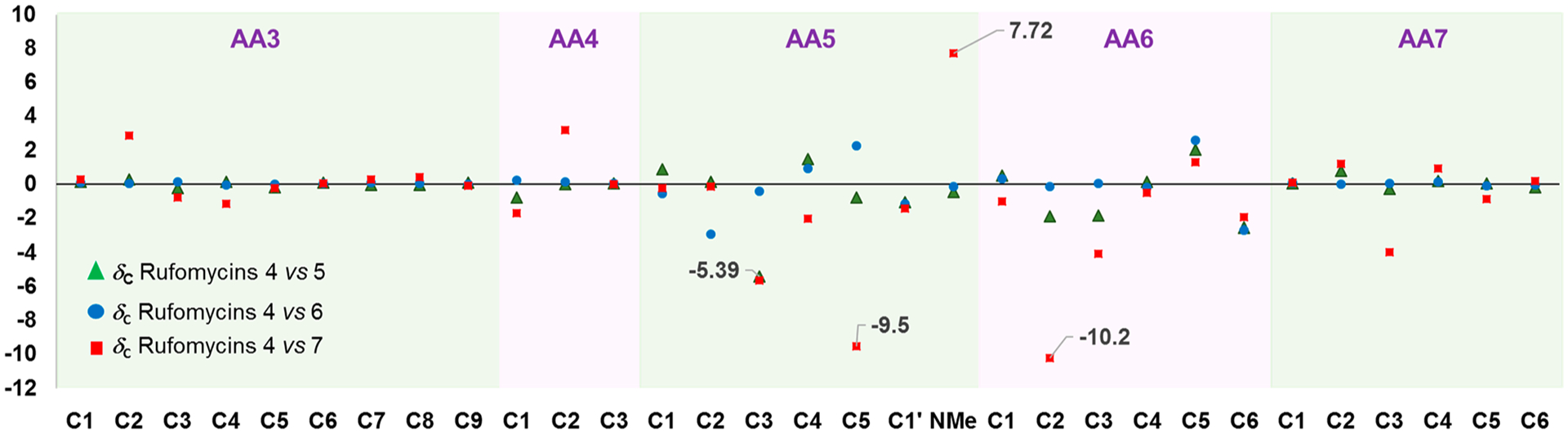
13C NMR chemical shift differences (ΔδC) between the anomeric compound pairs of rufomycins 4/5, 4/6, and 4/7 in selected regions. The X- and Y-axes represent the carbon number and the ΔδC values (in ppm), respectively.
Figure 7.
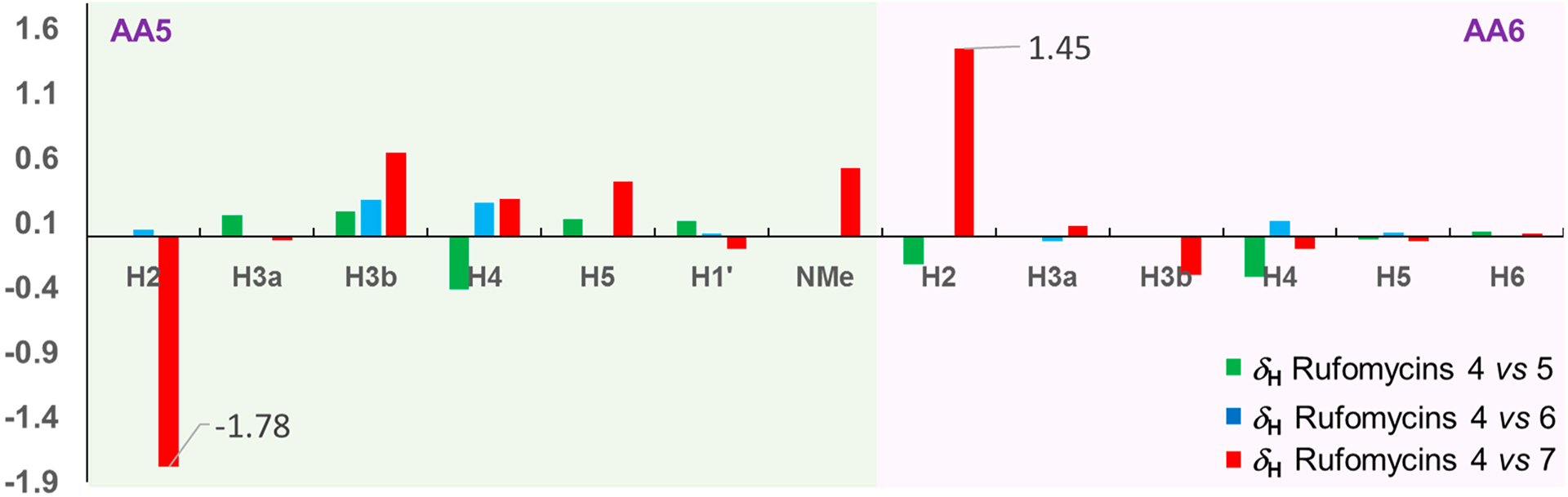
1H NMR chemical shift differences (ΔδH) between the compound pairs of rufomycins 4/5, 4/6, and 4/7 in selected regions. The X- and Y-axes represent the proton number and ΔδH values (in ppm), respectively.
The Case of Ilamycins E1 (Rufomycin 21) and E2 (Rufomycin 22): Need for Revisions.
Comparing the NMR signals of AA5-CH-5 of the purported analogues rufomycins 4–7 with those of ilamycins E1 and E2 revealed that the data reported for ilamycin E1 did not match those of rufomycin 4. The configurations at AA5 C-4 and C-5 of ilamycin E1 (C-32 and C-33 per ref 13) were deduced by comparing their chemical shifts with those of ilamycin C1 (rufomycin 4).5–7 However, this led to the observation that the chemical shifts were inconsistent and that the NMR spectra were also acquired in different solvents. Subsequent close examination of the 1H NMR and HSQC spectra of ilamycin E1 (Figures 55 and 58 in the Supporting Information of ref 13) revealed that the analyte was a mixture of at least two different molecules. While the major compound, whose NMR spectral data were tabulated in the publication (AA5 CH-5: δC 79.4 and δH 4.80 in Supplementary Table 2 of ref 13), is most comparable with rufomycin 6 (AA5 CH-5: δC 79.4 and δH 4.76), the minor compound, with an HSQC cross-peak at δC ~82/δH 4.7 ppm, tallies well with rufomycin 4, which exhibits an HSQC cross-peak at δC 81.7 and δH 4.76 (1H NMR: br d, J = 3.7 Hz) assigned to CH-5 in AA5. This led to the conclusion that ilamycin E1 as reported recently is in fact a mixture of two cyclopeptides that exhibit the same configurations at C-4 and C-5 in AA5 as rufomycin 6 (therefore assigned as the major component in “ilamycin E1”) and rufomycin 4 (the minor component).
In the present study, rufomycin 21 was isolated and showed identical 1H and 13C NMR data (Figures S51 and S52, Supporting Information) with those reported for ilamycin E1 in ref 13. Our HSQC spectra of rufomycin 21 showed a distinct cross-peak (δC 79.4 and δH 4.76, br d, J = 2.6 Hz) for CH-5 in the AA5 residue, which is nearly identical to that observed in the spectrum of rufomycin 6 (δC 79.4 and δH 4.76, br d, J = 2.1 Hz; Chart S2, Supporting Information). Thus, the structure assigned to ilamycin E1 in ref 13 should be revised to contain a 4R,5R absolute configuration in AA5. This revision can be further corroborated by the obvious NOESY correlation of H-2/H-4 in AA5 (Figure S57, Supporting Information) in rufomycin 21. Originally, the presumed mutarotation of the hydroxy-methylpiperidinone moiety was assigned in ref 13 by comparison of HPLC profiles of the interconverting species (compounds 6 and 7 in Supplementary Figures S34 and S21 in ref 13), designated as ilamycins E1 and E2.
The authors of ref 13 did describe the phenomenon of interconversion between ilamycins E1 and E2, which suggests ilamycin E1 should have a 4R,5S absolute configuration. However, based on our experience, rufomycins 4 and 6 are difficult to separate. Thus, the “peaks” for ilamycin E1 (compound 6 in Supplementary Figures S34 and S21 in ref 13) may not only contain the interconverted component from ilamycin E2 (4R,5S) but also the main component with (4R,5R), a compound analogous to rufomycin 6. Hence, its occurrence, as evidenced by NMR spectra, is not surprising.
Despite this need for revision, the configurations at C-4 and C-5 (AA5) of ilamycin E2 (rufomycin 22) remain unchanged (4S, 5S, equivalent to β-OH, β-CH3, as per the drawing). The HSQC data of ilamycin E2 (δC 82.5 and δH 4.65, br d, J = 1.6 Hz, CH-5 in AA5; see Table 2 and Figure S56 of the Supporting Information of ref 13) concur with rufomycin 5 (δC 82.5 and δH 4.63, br d, J = 2.6 Hz) and are indicative of identical configurations at C-4 and C-5 of AA5 in both ilamycin E2 and rufomycin 5.
Table 2.
13C NMR (150 MHz) and 1H NMR (600 MHz) Data of Rufomycins 4–7 in CD3OD
| rufomycin 4 | rufomycin 5 | rufomycin 6 | rufomycin 7 | |||||
|---|---|---|---|---|---|---|---|---|
| no. | δ C | δH, mult. (J in Hz) | δ C | δH, mult. (J in Hz) | δ C | δH, mult. (J in Hz) | δ C | δH, mult. (J in Hz) |
| AA1-1 | 174.2 | 174.3 | 174.2 | 174.0 | ||||
| 2 | 51.7 | 4.87, dd (9.3, 6.5) | 51.9 | 4.90, m | 51.6 | 4.87, dd (9.7, 5.9) | 51.5 | 4.96, m |
| 3 | 29.2 | 3.22, m | 29.1 | 3.22, m | 29.2 | 3.22, m | 29.2 | 3.23, m |
| 4 | 125.9 | 7.18, s | 125.8 | 7.15, s | 125.8 | 7.17, s | 125.7 | 7.17, s |
| 5 | 109.1 | 109.0 | 109.0 | 109.0 | ||||
| 6 | 130.6 | 130.5 | 130.6 | 130.6 | ||||
| 7 | 119.8 | 7.52, br d (7.9) | 119.8 | 7.51, m | 119.8 | 7.51, br d (7.9) | 119.5 | 7.53, m |
| 8 | 120.5 | 7.06, m | 120.5 | 7.06, m | 120.5 | 7.04, m | 120.5 | 7.04, m |
| 9 | 122.6 | 7.12, m | 122.6 | 7.13, m | 122.6 | 7.11, m | 122.6 | 7.13, m |
| 10 | 114.6 | 7.76, br d (8.5) | 114.6 | 7.76, m | 114.5 | 7.75, br d (8.5) | 114.7 | 7.77, m |
| 11 | 137.1 | 137.0 | 137.0 | 137.1 | ||||
| 1′ | 59.2 | 59.2 | 59.1 | 59.2 | ||||
| 2′ | 58.9 | 3.24, dd (4.1, 2.7) | 58.9 | 3.23, dd (4.1, 2.6) | 58.9 | 3.23, dd (4.1, 2.6) | 58.9 | 3.24, dd (4.1, 2.6) |
| 3′a | 46.0 | 2.81, dd (−4.6, 2.7) | 46.0 | 2.80, dd (−4.6, 2.6) | 46.0 | 2.81, dd (−4.6, 2.7) | 46.0 | 2.81, dd (−4.6, 2.6) |
| 3′b | 2.85, dd (−4.6, 4.1) | 2.85, dd (−4.6, 4.1) | 2.85, dd (−4.6, 4.1) | 2.86, dd (−4.6, 4.1) | ||||
| 1″ | 23.2 | 1.51, s | 23.2 | 1.49, s | 23.2 | 1.49, s | 23.2 | 1.51, s |
| 1″′ | 25.0 | 1.66, s | 25.0 | 1.65, s | 25.0 | 1.64, s | 25.0 | 1.66, s |
| AA2-1 | 169.9 | 169.8 | 169.8 | 170.0 | ||||
| 2 | 59.6 | 4.27, dd (10.7, 3.8) | 59.5 | 4.31, dd (11.2, 3.2) | 59.6 | 4.27, dd (10.8, 3.6) | 60.0 | 4.46, dd (11.0, 3.2) |
| 3a | 37.7 | 1.51, m | 37.6 | 1.50, m | 37.7 | 1.50, m | 37.7 | 1.47 |
| 3b | −0.51, m | −0.65, m | −0.49, m | −0.65, m | ||||
| 4 | 25.6 | 0.94, m | 25.5 | 0.94, m | 25.5 | 0.93, m | 25.5 | 0.99, m |
| 5 | 21.5 | 0.10, d (6.6) | 21.3 | 0.07, d (6.6) | 21.4 | 0.09, d (6.6) | 21.2 | 0.15, d (6.6) |
| 6 | 23.3 | 0.42, d (6.6) | 23.4 | 0.38, d (6.6) | 23.3 | 0.42, d (6.6) | 23.3 | 0.38, d (6.6) |
| NMe | 29.4 | 2.34, s | 29.3 | 2.30, s | 29.4 | 2.34, s | 29.2 | 2.13, s |
| AA3-1 | 171.7 | 171.6 | 171.6 | 171.4 | ||||
| 2 | 57.1 | 4.66, m | 56.8 | 4.68, m | 57.0 | 4.66, m | 54.2 | 4.66, m |
| 3a | 38.3 | 3.07, dd (−14.2, 5.7) | 38.5 | 3.06, dd (−14.1, 5.8) | 38.2 | 3.08, dd (−14.1, 5.9) | 39.1 | 3.13, m |
| 3b | 2.87, m | 2.86, m | 2.87, m | 2.87, m | ||||
| 4 | 130.1 | 129.9 | 130.1 | 131.2 | ||||
| 5 | 126.4 | 7.82, d (2.2) | 126.6 | 7.77, d (2.2) | 126.4 | 7.83, d (2.2) | 126.7 | 7.92, d (2.2) |
| 6 | 135.5 | 135.4 | 135.5 | 135.5 | ||||
| 7 | 154.4 | 154.4 | 154.3 | 154.1 | ||||
| 8 | 121.2 | 7.06, d (8.6) | 121.3 | 7.06, d (8.6) | 121.2 | 7.06, d (8.6) | 120.8 | 7.03, d (8.6) |
| 9 | 138.9 | 7.38, dd (8.6, 2.2) | 138.8 | 7.36, dd (8.6, 2.2) | 138.9 | 7.38, dd (8.6, 2.2) | 139.0 | 7.36, dd (8.6, 2.2) |
| AA4-1 | 172.7 | 173.5 | 172.5 | 174.4 | ||||
| 2 | 47.9 | 4.80, q (6.6) | 47.8 | 4.80, q (6.6) | 47.7 | 4.79, q (6.6) | 44.7 | 5.04, q (6.6) |
| 3 | 17.9 | 1.28, d (6.6) | 17.8 | 1.29, d (6.6) | 17.8 | 1.27, d (6.6) | 17.9 | 1.35, d (6.6) |
| AA5-1 | 171.3 | 170.4 | 171.9 | 171.6 | ||||
| 2 | 60.2 | 3.82, dd (9.9, 7.1) | 60.1 | 3.81, d (9.0) | 63.1 | 3.77, dd (11.2, 7.1) | 60.4 | 5.60, m |
| 3α | 26.4 | 2.55, ddd (−13.4, 10.1, 3.9) | 31.8 | 1.71, m | 26.8 | 2.27, m | 32.0 | 1.90, m |
| 3β | 1.87, m | 2.36, m | 1.86, m | 1.90, m | ||||
| 4 | 35.1 | 2.22, m | 33.7 | 2.63, m | 34.2 | 1.96, m | 37.2 | 1.93, m |
| 5 | 81.7 | 4.76, br d (3.70) | 82.5 | 4.63, br d (2.6) | 79.4 | 4.76, br d (2.6) | 91.2 | 4.34, br d (7.9) |
| 1′ | 16.3 | 1.10, d (7.2) | 17.4 | 0.98, d (6.7) | 17.5 | 1.08, d (6.8) | 17.8 | 1.20, d (5.8) |
| NMe | 38.3 | 3.24, s | 38.8 | 3.23, s | 38.4 | 3.24, s | 30.6 | 2.71, s |
| AA6-1 | 173.4 | 172.9 | 173.1 | 174.4 | ||||
| 2 | 55.0 | 5.24, dd (11.9, 4.3) | 56.9 | 5.46, dd (11.0, 4.1) | 55.1 | 5.24, dd (11.1, 5.4) | 65.2 | 3.97, dd (9.19, 6.1) |
| 3a | 35.8 | 1.89, ddd (−14.9, 11.9, 3.6) | 37.6 | 1.89, m | 35.8 | 1.93, m | 39.9 | 1.81, m |
| 3b | 1.96, ddd (−14.9, 10.8, 4.3) | 1.95, m | 1.96, m | 2.26, m | ||||
| 4 | 25.6 | 1.51, m | 25.5 | 1.82, m | 25.8 | 1.39, m | 26.2 | 1.61, m |
| 5 | 21.3 | 0.93, d (6.5) | 21.8 | 0.95, d (6.5) | 21.3 | 0.90, d (6.5) | 22.6 | 0.97, d (6.5) |
| 6 | 23.9 | 1.01, d (6.5) | 23.8 | 0.97, d (6.5) | 23.9 | 1.01, d (6.5) | 23.2 | 0.99, d (6.5) |
| AA7-1 | 173.4 | 173.3 | 173.3 | 173.3 | ||||
| 2 | 54.3 | 4.61, m | 53.4 | 4.78, m | 54.3 | 4.63, m | 53.1 | 4.70, m |
| 3a | 35.3 | 2.78, m | 35.6 | 2.65, m | 35.2 | 2.79, m | 39.3 | 2.64, m |
| 3b | 2.62, m | 2.60, m | 2.62, m | 2.34, m | ||||
| 4 | 127.7 | 5.59, m | 127.5 | 5.62, m | 127.5 | 5.57, m | 126.7 | 5.48, m |
| 5 | 129.4 | 5.61, m | 129.3 | 5.62, m | 129.4 | 5.63, m | 130.3 | 5.52, m |
| 6 | 18.3 | 1.65, m | 18.5 | 1.69, m | 18.4 | 1.65, m | 18.2 | 1.61, m |
Structural Identification of the Semisynthetic Derivatives RufomycinSS 1–3.
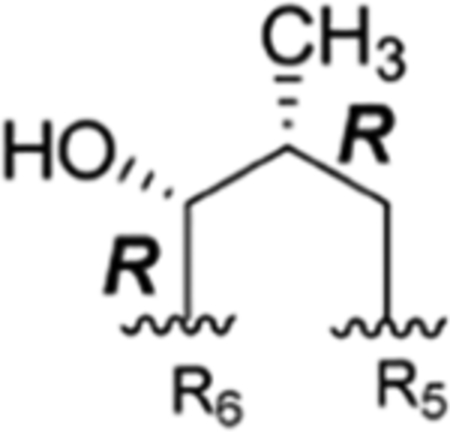
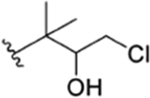
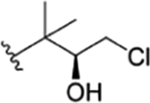
The interconvertible property of rufomycins 4–7 makes the assignment of NMR data challenging; thus, OH-protected compounds rufomycinSS 1–3 were semi-synthesized from a rufomycins 4 and 6 mixture. Treatment of this mixture with catalytic HCl in n-butanol afforded rufomycinSS 1–3. The molecular formula C58H84ClN9O12 for rufomycinSS 1 was assigned by the (+)-HRMS (ESI) ion at m/z 1134.6032 [M + H]+ (calcd 1134.6001). O-Alkylation and epoxide opening reactions took place on the HO-5 group in the AA5 residue and the epoxide moiety of AA1. The opened epoxide moiety with chloride attached at AA1-C-3′ was verified by the HMBC correlations (Figure S33, Supporting Information) from both AA1-H3-1″ (δH 1.62, s) and AA1-H3-1″′ (δH 1.75, s) to AA1-C-2′ (δC 76.5) and from AA1-H2-3′ (δH 2.86, dd, J = −11.3, 2.0 Hz; δH 3.30, dd, J = −11.3, 9.8 Hz) to AA1-C-1′ (δC 63.2) and AA1-C-2′ (δC 76.5). The HO group at AA5 C-5 was substituted by an n-butyl group, and this was verified by the HMBC from H-5 (δH 4.44, d, J = 8.1 Hz) to the assigned α-carbon (C-1″) of the butyl group (δC 65.5). The similar coupling constant of 8.1 Hz between AA5-H-4 and -H-5 of rufomycinSS 1 to the corresponding one in rufomycin 7 (J = 7.9 Hz) suggested a 4R,5S absolute configuration for rufomycinSS 1. The assignment was further validated by the ROESY correlations of H-2 and H2-1″/H-4 and of H-5/H3-1′ (Figure 2). RufomycinSS 2, sharing the molecular formula C58H84ClN9O12 with that of rufomycinSS 1, was verified by the (+)-HRMS (ESI) and 13C NMR data (Table 4). RufomycinSS 2 was assigned the same planar structure as that of rufomycinSS 1 by TOCSY and HMBC cross-peaks (Figures S38 and S40, Supporting Information).
Table 4.
13C NMR (150 MHz) Data of RufomycinSS 1–3 in CD3OD
| rufomycinSS 1 | rufomycinSS 2 conformer A | rufomycinSS 2 conformer B | rufomycinSS 3 conformer A | rufomycinSS 3 conformer B | |
|---|---|---|---|---|---|
| no. | δ C | δ C | δ C | δ C | δ C |
| AA1-1 | 174.00 | 174.19 | 174.35 | 174.10 | 174.10 |
| 2 | 51.44 | 51.59 | 50.98 | 51.60 | 51.22 |
| 3 | 28.92 | 29.03 | 28.72 | 29.00 | 28.74 |
| 4 | 126.19 | 126.23 | 125.92 | 126.19 | 126.17 |
| 5 | 109.15 | 109.23 | 109.55 | 109.18 | 109.31 |
| 6 | 130.86 | 130.96 | 131.09 | 130.88 | 130.94 |
| 7 | 119.69 | 120.05 | 119.99 | 120.00 | 119.88 |
| 8 | 120.63 | 120.58 | 120.49 | 120.56 | 120.55 |
| 9 | 122.73 | 122.67 | 122.62 | 122.66 | 122.66 |
| 10 | 115.03 | 114.83 | 114.76 | 114.80 | 114.82 |
| 11 | 136.64 | 136.64 | 136.65 | 136.59 | 136.57 |
| 1′ | 63.20 | 63.21 | 63.16 | 63.15 | 63.17 |
| 2′ | 76.50 | 76.58 | 76.56 | 76.52 | 76.48 |
| 3′ | 46.87 | 46.93 | 47.00 | 46.93 | 47.00 |
| 1″ | 23.00 | 23.20 | 23.20 | 23.22 | 23.02 |
| 1″′ | 26.61 | 26.45 | 26.45 | 26.45 | 26.28 |
| AA2-1 | 169.95 | 169.99 | 169.90 | 169.83 | 170.52 |
| 2 | 59.87 | 59.81 | 59.85 | 59.72 | 59.97 |
| 3 | 37.71 | 37.64 | 37.84 | 37.60 | 37.74 |
| 4 | 25.54 | 25.63 | 25.74 | 25.55 | 25.70 |
| 5 | 21.21 | 21.64 | 21.93 | 21.57 | 21.62 |
| 6 | 23.37 | 23.20 | 23.14 | 23.22 | 23.35 |
| N-Me | 29.21 | 29.23 | 29.15 | 29.23 | 29.25 |
| AA3-1 | 171.29 | 171.80 | 171.60 | 171.62 | 171.47 |
| 2 | 54.22 | 57.30 | 54.70 | 57.21 | 54.50 |
| 3 | 39.21 | 38.89 | 38.60 | 39.02 | 38.44 |
| 4 | 131.18 | 129.90 | 131.68 | 129.77 | 131.57 |
| 5 | 126.70 | 126.42 | 126.71 | 126.38 | 126.66 |
| 6 | 135.52 | 135.61 | 135.57 | 135.56 | 135.50 |
| 7 | 154.13 | 154.35 | 154.11 | 154.29 | 154.07 |
| 8 | 120.86 | 121.21 | 120.82 | 121.17 | 120.79 |
| 9 | 138.99 | 138.91 | 139.08 | 138.92 | 139.03 |
| AA4-1 | 174.80 | 172.47 | 174.25 | 172.35 | 174.32 |
| 2 | 44.68 | 48.14 | 44.74 | 48.05 | 44.78 |
| 3 | 17.86 | 17.96 | 17.93 | 17.93 | 17.91 |
| AA5-1 | 173.00 | 171.18 | 170.70 | 171.47 | 170.92 |
| 2 | 60.21 | 60.00 | 57.15 | 63.46 | 59.70 |
| 3 | 32.04 | 24.73 | 26.56 | 26.94 | 28.74 |
| 4 | 32.42 | 30.01 | 29.70 | 34.83 | 34.71 |
| 5 | 97.34 | 88.94 | 95.89 | 88.12 | 95.00 |
| 1′ | 17.91 | 15.58 | 14.99 | 17.50 | 17.31 |
| NMe | 30.75 | 38.28 | 30.44 | 38.21 | 30.32 |
| 1″ | 65.53 | 69.26 | 69.47 | 73.59 | 74.47 |
| 2″ | 32.73 | 33.27 | 33.14 | 33.50 | 33.50 |
| 3″ | 20.32 | 20.51 | 20.54 | 20.21 | 20.46 |
| 4″ | 14.63 | 14.27 | 14.20 | 14.42 | 14.33 |
| AA6-1 | 174.00 | 172.80 | 173.39 | 172.43 | 172.94 |
| 2 | 66.44 | 54.80 | 69.60 | 54.52 | 69.58 |
| 3 | 40.02 | 35.80 | 41.05 | 35.66 | 40.81 |
| 4 | 26.16 | 25.47 | 26.14 | 25.70 | 26.25 |
| 5 | 22.79 | 21.18 | 22.60 | 21.42 | 22.23 |
| 6 | 23.06 | 23.94 | 23.20 | 23.93 | 23.83 |
| AA7-1 | 173.00 | 173.48 | 173.10 | 173.48 | 173.15 |
| 2 | 53.00 | 53.90 | 53.06 | 53.61 | 53.06 |
| 3 | 39.26 | 34.80 | 38.90 | 35.05 | 38.68 |
| 4 | 126.77 | 127.30 | 126.83 | 127.14 | 126.93 |
| 5 | 130.28 | 129.35 | 130.22 | 129.47 | 130.15 |
| 6 | 18.18 | 18.38 | 18.12 | 18.45 | 18.16 |
Analysis of the 1H and 13C NMR data (Figures S36 and S58, Supporting Information) showed that rufomycinSS 2 possesses three conformers in the ratio ~11:7:2. The two major conformers had major differences in the 1H and 13C NMR data, which were fully assigned from 2D NMR data. The minor conformer was not assigned due to the low amount. The two major conformers varied mostly at AA6-CH-2 (δC 54.8 and δH 5.28, dd, J = 12.0, 4.1 Hz for conformer A; δC 69.6 and δH 3.74, dd, J = 7.5, 7.5 Hz for conformer B), AA5-CH-5 (δC 88.9 and δH 4.41, br s for conformer A; δC 95.9 and δH 4.33, br s for conformer B), and AA5-NCH3 (δC 38.3 and δH 3.22, s for conformer A; δC 30.4 and δH 2.73, s for conformer B). Both of the major conformers showed clear ROESY correlation cross-peaks (Figure 2) of AA5-H3-1′ (δH 1.12 for conformer A; δH 1.14 for conformer B)/AA5-H-2 (δH 3.85 for conformer A; δH 5.45 for conformer B) and AA5-H-5 and of AA5-H2-1″ (δH 3.46 and 3.53 for conformer A; δH 3.52 and 3.72 for conformer B)/AA5-H-4 (δH 2.44 for conformer A; δH 2.37 for conformer B), verifying a 4S,5R absolute configuration for rufomycinSS 2. RufomycinSS 3, another diastereoisomer of rufomycinSS 1 and 2, showed similar NMR patterns with three conformers (~5:2:1, Figures S43 and S58) to those of rufomycinSS 2. The two major conformers varied mainly at AA6-C-2, AA5-C-5, and −NCH3 with ΔδC of 15.1, 6.9, and 7.9 ppm, respectively, similar to those of rufomycinSS 2. The ROESY cross-peaks (Figure 2) of H-2 and H-5/H-4 and H3-1′/H2-1″(AA5) in both conformers of rufomycinSS 3 were assigned a 4R,5R absolute configuration. The ΔδC and ΔδH between two major conformers in rufomycinSS 2 and 3 are similar, both showing the highest ΔδC values at AA6-C-2 (ΔδC ~15 ppm), AA5-C-5 (ΔδC ~7 ppm), and –NCH3 (ΔδC ~8 ppm) and the most variant ΔδH values at AA6-H-2 (ΔδH ~1.6 ppm) and AA5-H-2 (ΔδH ~1.6 ppm), exhibiting similar property trends to those between rufomycins 4 and 7 (Figures 6–8).
Figure 8.
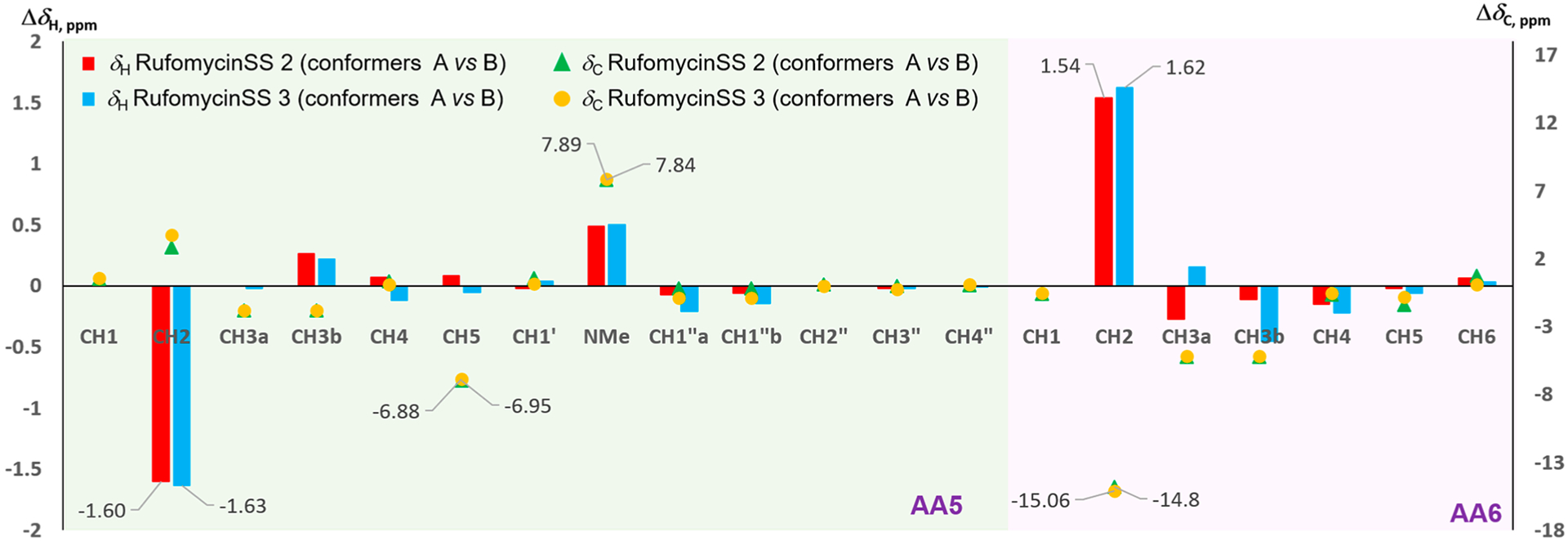
ΔδH and ΔδC values between conformers A and B of rufomycinSS 2 and 3 in selected regions. The X- and Y-axes represent the nuclei number and Δδ values (left Y-axis for ΔδH and right Y-axis for ΔδC in ppm), respectively.
Possible Conformers of Rufomycins.
Based on this information, when C-4 and C-5 of AA5 ring are S,R (rufomycin 4 and rufomycinSS 2, respectively), R,R (rufomycin 6 and rufomycinSS 3, respectively), or S,S (rufomycin 5), the corresponding structures prefer conformation A over B, which exhibit dramatic differences in their NMR spectra. However, rufomycin 7 and rufomycinSS 1 belonging to the R,S series of C-4/5 diastereomers prefer conformation B and show special NMR resonance patterns when compared to the other three diastereoisomers. The crystal structures of rufomycins 421 and 513 provided some indication of the possible main conformation: both revealed that two of the seven carbonyl groups, those of AA4 and AA7, are pointed inward, toward the center of the macrocycle. Transannular hydrogen bonds between AA7-CO and the −NH groups of the Ala and m-NO2Tyr residues and between AA4-CO with the NH group of AA7 residue were predicted based on their close spatial distance, suggesting a high potential for the low-energy conformer A. The in-chain piperidinone moiety lies orthogonally below the macrocycle, sandwiched between two bulky groups (the side chains of AA6 and AA7). Comparison of the NMR data of conformers A and B showed obvious differences of the resonance from AA5-NCH3. This shielded methyl in conformer B (ΔδH 0.5 and ΔδC 8 ppm) as compared to those in conformer A turned out to have similar chemical shifts (δH ~2.7 and δC ~30 ppm) to those of rufomycins without a piperidinone.13,23
Thus, despite the flexibility differences between the solid state (crystals) and solution, the X-ray diffraction studies of these rufomycins provided valuable information. The crystal structures of ilamycins B2, D, and F13 (rufomycins 1, 9, and 23, respectively) and rufomycin 11,23 which do not contain a piperidinone, all showed a similar conformation of the macrocycle. For these compounds the carbonyl of AA3 rather than that of AA4 lies inside the ring. Thus, conformer B for rufomycinSS 2 and 3, similar to the major conformer for rufomycin 7 and rufomycinSS 1, was proposed as drawn (Figure 9). Compared to conformer A, several distinctly different geometries of conformer B were observed, explaining dramatic NMR differences between conformers A and B. In conformer B, AA4 carbonyl and AA5-NCH3 were cis-oriented. This N-methyl group was thus placed in the shielding region of the aromatic ring of the m-NO2Tyr residue, resulting in its high frequency of both 1H and 13C NMR signals in conformer B compared to corresponding signals in conformer A. The NOESY correlations (Figure 9, Figure S34, Supporting Information) of AA5-H-2/AA5-NCH3 and AA4-H-2, of AA4-H-2/AA5-H-3, and of AA5-NCH3/AA6-H3-5 and -H3-6 in conformer B differ from the NOESY correlations between AA5-NCH3/-H-2, AA4-H-2, and -H3-3 in conformer A, validating the geometric relationships of conformers A and B. Another obvious difference observed was the different spatial relationship between the piperidinone ring and the macrocycle. In conformer A, the piperidinone is lying orthogonally below the macrocycle and is close to two bulky groups (AA6 and AA7). The 21-membered macrocyclic ring is not flat but shaped like two butterfly wings that bend symmetrically around an axis crossing AA4-NH–AA7-C-2. The inside space of the macrocycle is highly crowded with strong steric effects. In conformer B, the orientation of the piperidinone was flipped, now lying on the inside of the macrocycle “butterfly”. The spatial environments of AA6-H-2, AA5-H-2, and AA4-H-2 were thus varied between conformers A and B. In conformer A, AA6-H-2 was placed in the valley of the macrocycle, getting strong steric effects from the crowded environment. Conversely, this hydrogen in conformer B was placed outside that valley, and AA5-H-2 and AA4-H-2 in conformer B are turned to the crowded environment, resulting in upfield frequency shifted AA5-C-2, AA4-C-2, and AA6-H-2 and low-field frequency shifted AA6-C-2, AA5-H-2, and AA4-H-2 as fitting the observed differences between conformers A and B in rufomycinSS 2 and 3. Flipping the piperidinone also releases its steric hindrance between the bulky groups of AA6 and AA7, and it may explain the lower frequencies of AA7-C-3, AA6-C-3, and AA5-C-5 in conformer B as compared to those in conformer A.
Figure 9.
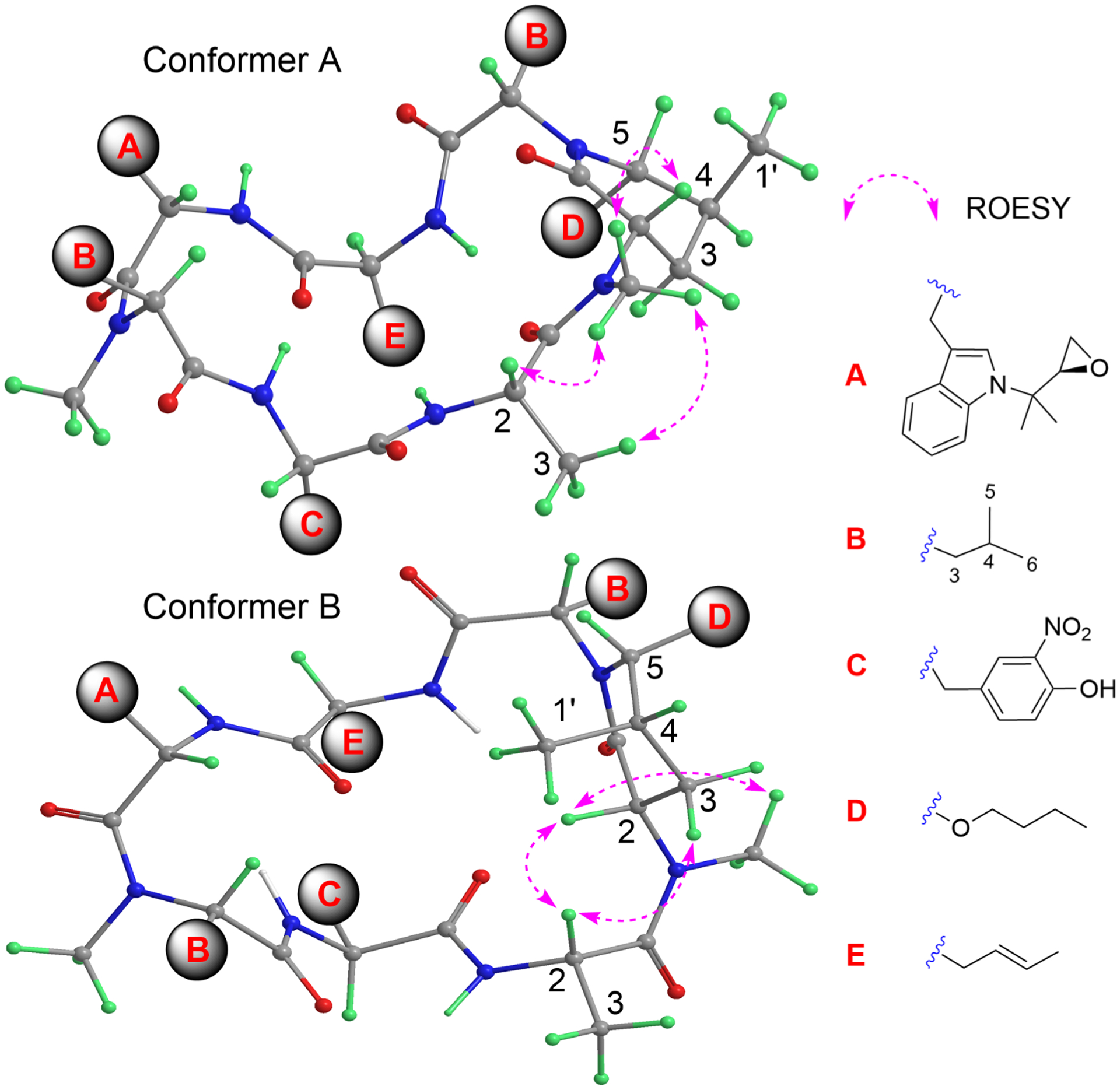
Plausible 3D structures for conformers A and B of rufomycinSS 2, along with selected ROESY correlations. The conformers were drawn based on reported X-ray crystallographic structures of the rufomycins, and the observed ROESY correlations were fitted into these structures.
Anti-Mycobacterium tuberculosis Bioactivity Profiles and Structure–Activity Relationship (SAR) Considerations.
Anti-Mtb activities and binding affinities (KD) with both caseinolytic protein C1 (ClpC1) N-terminal domain and the full-length protein by surface plasmon resonance of rufomycins 4–7 and rufomycinSS 1–3 were measured (Table 5). Based on the nature of hemiaminal, rufomycins 4 and 5 and rufomycins 6 and 7 are supposed to be similar mixtures of corresponding epimers in the test medium. Similar KD and MIC values for rufomycins 4 and 5 and for rufomycins 6 and 7 supported this presumption and suggested that rufomycins with an S configuration at C-4 in AA5 are preferred for anti-Mtb activity. Surprisingly, rufomycinSS 1–3 varied considerably in their anti-Mtb activities, and the MICs support our hypothesis that an S configuration at C-4 in the AA5 residue is preferred. RufomycinSS 2 showed strong anti-Mtb activity with an MIC value of 48 nM, similar to that of rufomycins 4–7. However, rufomycinSS 1 showed only moderate anti-Mtb activity, with an MIC value of 769 nM, and rufomycinSS 3 was less active, with an MIC value of 4.3 μM. The KD values for rufomycinSS 1 and 2, representing their weak and strong bond affinities with the ClpC1 target, are consistent with their high and low MIC values, respectively. However, the favorable KD value (211 nM for NTD) for rufomycinSS 3 was not reflected in its high MIC value (4.3 μM) in anti-Mtb susceptibility testing, thereby indicating that permeability or other molecular properties are sources of complexity in the overall SAR of rufomycins.
Table 5.
Anti-Mtb Activities (MIC90, nM) and Surface Plasmon Resonance (SPR) Data (KD, nM) of Rufomycins 4–7 and RufomycinSS 1–3a
| compound | MIC90 | KD NTD | KD FL |
|---|---|---|---|
| rufomycin 4 | 21 | 32 | 31 |
| rufomycin 5 | 12 | 29 | 27 |
| rufomycin 6 | 46 | 183 | 178 |
| rufomycin 7 | 47 | 186 | 209 |
| rufomycinSS 1 | 769 | 602 | 724 |
| rufomycinSS 2 | 48 | 27 | 29 |
| rufomycinSS 3 | 4300 | 211 | 171 |
| RMP | 28 | NT | NT |
| INH | 463 | NT | NT |
NT = not tested; RMP = rifampin; INH = isoniazid; NTD = N-terminal domain; FL = full-length.
In this study, four diastereoisomers, rufomycins 4–7, and three semisynthetic rufomycins, rufomycinSS 1–3, were characterized, their 1H and 13C NMR were fully assigned, and their anti-Mtb activity was evaluated. The 2-piperidone played an important role in their potency, and this was related to their preferred conformation. Two major conformations, A and B, were assigned based on crystal structures and NMR data. Different configurations of the hydroxy and methyl groups in the 2-piperidone varied the preference for conformations and are related to their anti-Mtb activities. As a conclusion, the S configuration at C-4 in amino acid 5 provided a better anti-Mtb activity than the R configuration.
Early History and Naming Priority.
Around 1960, two Japanese groups, Takeda Chemical Industries (Takeda) and the Institute for Microbial Chemistry (IMC), independently discovered the same antimycobacterial antibiotics. The former obtained the substances from a Streptomycete isolated from a soil sample collected at the Kinokawa riverside in Wakayama Prefecture6 and called them rufomycins. The IMC group obtained the antibiotics from a Streptomycete isolated from a sample collected on Oshima island and called them ilamycins.7
The first available public disclosure was the filing of Japanese patent application number 35/4,033 on February 8, 1960, and this was quoted as priority for the United States filing on February 7, 1961, issued as US 3,655,879 assigned to Takeda Chemical Industries Ltd., claiming rufomycins A and B (rufomycins 2 and 1, respectively) and processes for producing them from Streptomyces atratus nov. sp.4 The antibiotics were characterized in this filing by their physical appearance, solubility in several solvents, reaction with various test reagents, elemental analyses, infrared and UV spectra, and optical rotations. No structure or partial structure was assigned at that time. The biology describing the S. atratus source was that published in ref 6, and the strain was deposited in the Institute for Fermentation, Osaka, as IFO-3897, as well as in the ATCC in Washington, DC, as ATCC-14046 as the type culture for this novel species. The chemical description of rufomycins was published in a sequential paper, which also notes that the rufomycins were studied in a mouse tuberculosis model, and they were almost as efficacious as streptomycin. This experiment was the first to demonstrate in vivo activity of these compounds and, hence, their antituberculosis efficacy rather than in vitro anti-Mtb activity. Furthermore, ilamycins were subsequently compared, and it was commented that “their properties closely resembled those of the rufomycins”.6
Early Efforts in Structure Determination.
Three structural features gave early workers major problems. The exact structures of the two different isoprenyl groups attached to tryptophan were sorted out from IR evidence not until the 11th publication on structures in November 1964.24 The diastereotopic nature of the two terminal methyl groups of leucine AA5 and the tautomeric hemiaminals from the oxygenation of each of these methyls to the aldehydes did not become apparent until two decades later, although by May 1965 three isomers here were assigned as a hemiaminal (without stereochemistry) and two aldehydes only differing in a hydrogen bond.22 The first NMR study, 60 MHz, published in May 1964 was incorrectly interpreted,18 and it was not until 1971 that a high-field NMR was obtained on the unoxidized rufomycin 1 (syn. ilamycin B1, rufomycin B). This was followed in 1974 by an X-ray crystallographic analysis of a derivative of the same compound.14,16 The configuration of the epoxide of rufomycin 2 (syn. rufomycin A, ilamycin B2) was not elucidated until 2017.13 This leads to two entries in Table 1 for most compounds with the epoxide, initial ones with undefined stereochemistry for this substructure and then a later one with the currently accepted stereochemistry including absolute configurations.
Both groups, Takeda and IMC, were clearly aware of the near-coincident discoveries, and despite the fact that they each made presentations at the same meetings (Symposium on the Chemistry of Natural Products at Tokyo University, October 1963; Japan Antibiotic Research Association, 127th, 128th, 129th, 135th, and 139th meetings from January 1962 to mid 1964), the publications on ilamycins from IMC gave only vague hints of identity between the ilamycins and rufomycins, or perhaps more importantly of the producing strains. In refs 7, 19, and 25 they named the ilamycins producer as Streptomyces islandicus, but they do not appear to have deposited this culture nor to have described it thoroughly. In his 1962 article, Shibata says that the description Takita gives of S. islandicus “resembles” that of S. atratuş6 but “it is incomplete and therefore is impossible to compare the strain with S. atratus in detail”. ATCC, in response to a query, said that they did not know of, nor could they find a source for, Streptomyces islandicus.
In several smaller communications on ilamycins that appeared from 1962 through 1965,7,18,19,22,24–28 the chronologically second was a note on the fermentation yields and monitoring these by a susceptible organism designated Sarcina X.26 Most of the following articles described chemical degradative steps toward the structure determination of ilamycins. The first degradation was to isolate AA-5 with an oxidized terminal methyl, incorrectly named l-γ-formyl-N-methyl-norvaline.19 By September 1963,27 structures were proposed for ilamycin and ilamycin B, but lacked the isoprenyl group on the tryptophan, and the authors did not appreciate the hemiaminal interaction between the aldehydic carbonyl described in a previous paper19 and the adjacent amide nitrogen.
Next came an identification of the unusual amino acid l-2-amino-4-hexenoic acid.28 Then came the appreciation that ilamycin B was, in fact, two compounds and the presence of a five-carbon unit on the tryptophan moiety. Unfortunately, the proposed structure for ilamycin B2 was incorrect.18 In the following month’s issue of the Journal of Antibiotics, the group proposed new structures of ilamycin B2 (rufomycin 2) and ilamycin as containing leucine and an oxidized terminal methyl (aldehyde) of leucine in position 5, respectively. These structures shared yet another and incorrect version of the tryptophan N-alkyl group.25 This was corrected for ilamycin B1 (rufomycin 1) with a little help from the Takeda group (see later) to formulate ilamycin B1 as having an unoxidized isoprenyl at the tryptophan ring nitrogen and having the olefin of this group oxidized to an epoxide in both ilamycin (rufomycin 22) and ilamycin B2. Both ilamycins B1 and B2 have the fifth amino acid simply as leucine, whereas ilamycin is formulated with one of the terminal methyls of that leucine as oxidized to an aldehyde. There was still no appreciation of its hemiaminal interaction with the adjacent amide nitrogen.24 This oversight was corrected by the isolation of two more minor ilamycins, C1 and C2 (rufomycins 4 and 5): both of these seemed to closely resemble ilamycin, with differences associated with the aldehyde function, which was explained by formulating ilamycin as a hemiaminal.22 Several years passed before a 300 MHz NMR study confirmed the structure of ilamycin B1.14 The final coup de grâce in the structure of ilamycin B1 came in 1974 with an X-ray diffraction study of the p-bromobenzoate ester located at the m-nitrotyrosine phenol.16
The Takeda group had followed up on their initial patent application with a second filed in Japan as 36/3,150 on January 31, 1961 (priority cited in ref 29 claiming “water-soluble derivatives of rufomycin” and outlining preparation of the sodium hemisuccinate of rufomycin A, the sodium hemisulfate of rufomycin A, and the ammonium, sodium, and potassium salts of the phosphates of both rufomycins A and B (rufomycins 2 and 1). More impressively, the authors reported on an efficacy study in Mtb H37Rv i.v. infected mice, with daily s.c. administered rufomycins A and B ammonium phosphates and dihydrostreptomycin as control. The rufomycin A salt was comparably as effective as dihydrostreptomycin, and the rufomycin B salt was slightly less effective.
As part of their structure elucidation effort, the Takeda group reduced the pseudoaldehyde function with sodium borohydride, and they were able to selectively cleave the cyclic peptide at that amino acid, eventually proposing a structure for rufomycin A (rufomycin 2), which as in the IMC19 is identical to that proposed for ilamycin: both cases simply point to the isoprenyl group.17 Later that year, the Takeda group published the correct structure for rufomycin B (rufomycin 1) and commented that “rufomycin A and rufomycin B resemble ilamycin and ilamycin B1, respectively”.15 Only one other publication on rufomycins seems to have come out of the Takeda group: in 1968, a study on the effects of feeding d,l-leucine to the producing organism was made by Eiji Higashide, and although new compounds were produced, they were not fully characterized.30
Almost Three Decades Later.
The compounds next sprung into the literature with three Eli Lilly patents, all filed in the U.S. on June 21, 1999. In all cases, although they call them rufomycin factors, the text refers to individual “factors” as compounds 1, 2, 3, etc. The smaller of these patents, entitled “Process for the isolation of rufomycin factors”, describes the discovery of a new producer of rufomycins, Streptomyces macrosporeus, which had been deposited in the Deutsche Sammlung von Mikroorganismen and Zellkulturen GmbH (Mascheroder Weg 1b, D-38124 Braunschweig, Germany) with the accession number DSM-12818. This application8 described the discovery of rufomycins 1 and 2, both with unoxidized leucine in position 5 and with the olefin and the epoxide in the isoprenyl group, respectively (i.e., identical to Takeda’s rufomycins B and A, respectively). It very pointedly disclaims any aldehyde, but does describe three diastereomeric hemiaminals from the aldehydes derived from rufomycin 2. (These corresponded to rufomycins 4–6 described above.) Several derivatives of two of these hemiaminals were prepared, and the carboxylic acid corresponding to oxidation of the aldehyde was isolated from the fermentation. A Plackett–Burman analysis of the fermentation conditions led to an optimized 150 L yield of >600 mg/L total rufomycins. This application specifically claimed the pH conditions used in the isolation process.
Another Eli Lilly patent9 claims the use of rufomycins for the treatment of multidrug-resistant diseases, specifically cancer and malaria. All compounds mentioned in Lambooy’s patent application,8 both natural products and semisynthetics, are included with the same exclusion of the aldehyde. Comprehensive hydrogen and carbon NMR assignments are listed for 13 such compounds. In addition, a theoretical set of 160 further derivatives is claimed. The third Eli Lilly patent10 is almost identical to their second application but claims these compounds as antituberculosis agents.
Almost Six Decades after the Initial Discovery.
The next relevant publication of the rufomycins came from the University of Tokyo, published on August 3, 2017.11 It described the cytochrome P450 involved in the biosynthesis of m-nitrotyrosine in rufomycins. It also speculated on the biosynthesis of the 2-amino hexenoic acid after identifying the rufomycin biosynthetic locus in S. atratus ATCC-14046. A sophisticated mutation and in vitro biochemical study concluded that in this organism nitration occurs prior to NRPS synthesis of the heptapeptide.
An almost coincidentally published article of August 30, 2017, on the biosynthesis of ilamycins with anti-Mtb properties states that the compounds came from a strain of S. atratus.13 The reported biosynthetic analysis was similar to a prior study11 and was augmented with labeled substrate feeding to various orf knockouts. In these two papers, the labeling of the orfs in the biosynthetic gene cluster was alphabetical, and in each case orfs K and L are transporter genes but differed by the NRPS gene, which is labeled S in this paper and T in Tomita’s. The different depictions of the biosynthetic gene cluster do not allow defining how this difference occurs. However, source organism and compound naming in ref 13 contained several inconsistencies.
First, while the likely existence of minor congeners can justify giving otherwise identical microbial metabolites from different species different names, application of this naming convention would have required a demonstration that S. atratus was different from S. islandicus. Such evidence could conceivably only have been provided by the IMC. However, as rufomycin remained the priority name based on the patent filing on February 8, 1960, and considering that an S. atratus strain was used, the study13 should have described the compounds as rufomycins. Second, the 2017 publication13 generated further confusion by stating that the ilamycins were isolated from several Streptomyces in the early 1960s to 1970s, citing three references, all to S. islandicus, whereas only two such strains had been described, and designation of S. islandicus as a distinct species was already questionable. Third, by referring to three Eli Lilly patents and stating that the ilamycins were reisolated in 2000 as rufomycins from another S. macrosporeus (DSM-12818), the 2017 report13 implied that the Eli Lilly group was the initial discoverers of the rufomycins, which is not the case. Strikingly, the investigated strain of S. atratus13 was not from a soil but from a deep-sea sediment sample collected below 3500 m depth at a very different location.
Next, in a 2019 paper, our group reported studies on rufomycin from yet another strain of S. atratus, strain MJM3502 from the Myongji University ECUM collection.12 Further studies greatly increased the recognized importance of these compounds by determining their mode of action at a recently discovered and unutilized target in killing both M. tuberculosis and M. abscessus.20 Examination of rufomycin-resistant strains showed that it binds to the N-terminal domain of ClpC1 and, thereby, shuts down critical protein degradation processes within the mycobacterial cell. Two other 2019 papers31,32 reported on the action of “ilamycins” against triplenegative breast cancer. One of the reports32 links “ilamycin C” to both S. atratus and S. islandicus as producers and, by referring to the 2017 paper,13 potentially implies the discovery of anti-TB/-Mtb activity that had been known for a long time (see ref 4 and introduction). The claim in the same report13 that the bioactive “ilamycin C” was 97.8% pure is puzzling, as there is no “ilamycin C”, but ilamycins C1 and C2 that were separated in 196522 and as rufomycins (compounds 4 and 5) by the Eli Lilly group.8–10 The analogous problem exists in the other paper on “ilamycin E”,31 where the previously described two compounds, ilamycins E1 and E2, are postulated as differing from C1 and C2, respectively, in that the former pair have an olefin in the isoprenyl group, whereas the latter have an epoxide. No purity of “ilamycin E” is claimed. As the structure of “ilamycin E1” claimed in ref 13 has to be revised based on evidence from the present study, this raises a major question as to exactly what compound or compounds were used in this study.
Most recently, in 2020, our group reported on more studies of MJM3502 as the source of eight new rufomycins, as well as five known ones in a comprehensive isolation, structural elucidation, and biological activity study.23 This study revealed that, at least in this producer, the oxidation of the diastereotopic methyls of AA5 is not stereospecific, as the two diastereomeric oxidation products were obtained and separated. Several of these rufomycins had lower MICs against M. abscessus than rufomycins 4/6. Moreover they also showed potential for anti-TB activity with a good selectivity of >100 (anti-Mtb/cytotocity (Vero cells)).23
A recent reference to ilamycins33 concerns the isolation of 12 compounds from a 200 L fermentation of an S. atratus strain, where the gene immediately prior to the NRPS gene was knocked out. While not providing references, this report perpetuates the aforementioned misconceptions about (a) the S. atratus ATCC 14046 vs S. islandicus origins in the 1960s, despite referencing IMC publications, and (b) seven rufomycins including the ilamycins B1, B2, A, C1, C2, and D as being isolated from S. macrosporeus DSM-12818. In addition, the allegedly new anti-TB activity of the cyclopeptides and new mechanistic claims described in the study33 had previously been published in detail,20 including evidence for a then novel anti-TB mode of action of the rufomycins acting at ClpC1.
Conclusions at the Interface of Nomenclature, Structural Rigor, and Drug Discovery.
The above systematic analysis of the literature plus the collective analytical evidence for known and four newly presented cyclopeptides led to three key conclusions about the nomenclature of these compounds, discussed in the following:
The naming priority is clearly established based on both the timeline and the identity of the producing organism including taxonomic evidence. When isolated from Streptomyces atratus or S. macrosporeus, these heptapeptides are rufomycins.
The confusion generated by subsequent authors not doing a careful check of the history of these antibiotics leads to an unnecessary fragmentation and undue shortage of credit to the authors of the priority report.
Proper reference to all applicable literature and respect of their priority is of the essence in both scientific conduct and intellectual property protection.
The fact that S. atratus is currently the only available species known to produce these compounds and that the type strain of this Streptomycete was deposited by Takeda reinforce their claim as the priority discoverers of the compounds. Thus, rufomycin is the valid priority name of the discussed class of cyclopeptide antibiotics. This claim is based on the priority of the first public disclosure and most specifically for the public availability of the producing strain. There is no valid availability of S. islandicus and hence no valid source today of ilamycins. These heptapeptides have been isolated only from S. atratus and S. macrosporeus and at IMC from a poorly described and unavailable Streptomycete.
The rufomycin/ilamycin paradox is a prototypical example of several aspects that have to come together to advance natural product based drug discovery: (i) specific naming, including the proper use of the priority producing organism; (ii) full attention to complex stereochemistry, including isomeric equilibria such as [hemi]aminals and conformation; (iii) rigor of analytical characterization including purity and structure. All three are concurrently necessary for the development of valid SAR, rigorous structure elucidation, and correct intellectual property claims. In fact, unless all nomenclature, analytical, purity, and documentation elements work together productively, an otherwise promising class of hit compounds might well fail to develop into a valid drug lead and further translation into efficacious therapy. Rigorously characterized and defined rufomycins are integral to the ongoing collaborative NIAID-funded Center of Excellence in Translational Research and continue to hold promise as candidates for translation into early stage clinical trials.
EXPERIMENTAL SECTION
General Experimental Procedures.
UV spectra were either extracted from a Shimadzu SPD-M20A PDA detector on UHPLC or acquired on a Cary 5000 UV–vis–NIR spectrophotometer. IR spectra were acquired on a Thermo Scientific Nicolet 6700 with an ATR probe. ESIMS/MS spectra were carried out by using a Bruker Impact II quadrupole time-of-flight (q-TOF) equipped with a Shimadzu UHPLC (Kyoto, Japan). The ion source was operated in the positive electrospray ionization mode using a capillary voltage of 4.0 kV; nebulizer and drying gas (N2) at 0.4 bar and 4.0 L/min, respectively; drying temperature of 225 °C; and mass scan range set from m/z 50 to 2000. The separation was performed on a CORTECS C18 (100 × 3.0 mm, 2.7 μm) UPLC column. Data were collected and processed by the Data Analysis 4.4 software (Bruker Daltonik GmbH, Germany). All 1D/2D NMR spectra were acquired on a JEOL (JEOL Resonance Inc., Peabody, MA, USA) ECZ 400 MHz or Bruker Ultrashield 600 Plus with an AVANCE III console 600 MHz spectrometer (Bruker, Billerica, MA, USA). The acquired spectra were processed using the Mnova NMR software package (v.12.0.4, MestReLab Research S.L., A Coruña, Spain). Sephadex LH-20 (Pharmacia, Uppsala, Sweden) and silica gel (ICN EcoChrom 32–63, 60 Å) were used for column chromatography (CC). Semipreparative HPLC was performed on a Shimadzu HPLC (Kyoto, Japan) connected to a PDA detector (Shimadzu, model SPD-20A) and equipped with a Kinetex EVOC18 (250 × 10 mm, S-5, 100 Å) column. TLC was analyzed by a UV detector and vanillin–sulfuric acid spray (3 g vanillin, 95 mL ethanol, and 1.5 mL sulfuric acid). All solvents used were obtained from Fisher Scientific (Fair Lawn, NJ, USA) or Sigma-Aldrich (St. Louis, MO, USA).
Strain Material.
The strain MJM3502 was obtained from the Extract Collection of Useful Microorganisms (ECUM) at Myongji University, Republic of Korea. The Streptomyces strain MJM3502 was shown to be 99% identical to Streptomyces atratus (NRRL B-16927; identical to the ATCC strain) through classification using the 16S rDNA sequence and phylogenetic analysis.12,23 Strain MJM3502 showed similar morphology to S. atratus NRRL: B-16927 with gray to pale yellow aerial mycelium on ISP2–ISP4 medium, and the growth was robust. However, in ISP5 medium, the growth was poor compared to S. atratus NRRL-B-16927.
Extraction and Isolation.
MJM3502 whole broth (300 L) was treated the same way as previously reported,23 affording the 3502 ethyl acetate (3502 EA) fraction. A rufomycin-enriched fraction (28 g) was obtained from the 3502 EA extract by silica gel CC with n-hexane/EA (5:5) and ethyl acetate as eluent. The rufomycin-enriched fraction was further chromatographed on silica gel using a gradient elution of n-hexane/acetone, 6:1, 5:1–4:1, 3:1–2:1, 1:1, 1:2, and 1:5) to give six fractions (A–F). About 6 g of rufomycin 4 and 6 mixtures was enriched from fraction C (10 g) by a series of columns packed with silica gel (CHCl3/MeOH) and Sephadex LH-20 (MeOH or EtOH), and rufomycins 4–7 and 21 (10 mg) were obtained by semipreparative HPLC (60% ACN in H2O, 2.5 mL/min) from the remaining material of fraction C.
Rufomycin 4:
pale yellow, amorphous solid; UV (MeOH) λmax 220, 282, 358 nm; IR (ATR) νmax 3251, 2950, 2361, 2092, 1627, 1539, 1456, 1314, 1180, 966, 741 cm−1; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) see Table 2; (+)-HRESIMS m/z 1042.5632 [M + H]+ (calcd for C54H76N9O12+, 1042.5608).
Rufomycin 5:
pale yellow, amorphous solid; UV (MeOH) λmax 223, 282, 359 nm; IR (ATR) νmax 3273, 2956, 2359, 2085, 1628, 1539, 1456, 1314, 1250, 966, 740 cm−1; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) see Table 2; (+)-HRESIMS m/z 1042.5606 [M + H]+ (calcd for C54H76N9O12+, 1042.5608).
Rufomycin 6:
pale yellow, amorphous solid; UV (MeOH) λmax 225, 282, 360 nm; IR (ATR) νmax 3292, 2957, 2359, 1628, 1538, 1456, 1314, 1256, 1208, 1082, 835, 740 cm−1; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) see Table 2; (+)-HRESIMS m/z 1042.5601 [M + H]+ (calcd for C54H76N9O12+, 1042.5608).
Rufomycin 7:
pale yellow, amorphous solid; UV (MeOH) λmax 220, 282, 358 nm; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) see Table 2; (+)-HRESIMS m/z 1042.5599 [M + H]+ (calcd for C54H76N9O12+, 1042.5608).
Semisynthesis of RufomycinSS 1–3.
A mixture of rufomycins 4 and 6 (50 mg, 0.048 mmol) was dissolved in 4 mL of n-butanol. To this solution was added 20 μL of 12 M hydrogen chloride, and the reaction mixture was stirred at room temperature for 12 h. The reaction solvent was then removed under reduced pressure, and the residue was purified by semipreparative HPLC (68% ACN in H2O, 2.5 min/mL) to give rufomycinSS 1 (6.1 mg, 11.0%, 19 min), rufomycinSS 2 (4.9 mg, 9.0%, 27 min), and rufomycinSS 3 (15.0 mg, 27.5%, 28 min).
RufomycinSS 1:
pale yellow, amorphous solid; UV (MeOH) λmax (log ε) 211 (4.1), 269 (3.8), 357 (1.5) nm; IR (ATR) νmax 2956, 2330, 2087, 1626, 1538, 1456, 1254, 1173, 1080, 966, 737 cm−1; 1H NMR (CD3OD, 600 MHz), see Table 3; 13C NMR (CD3OD, 150 MHz), see Table 4; (+)-HRESIMS [M + H]+ m/z 1134.6032 (calcd for C58H85ClN9O12+, 1134.6001).
Table 3.
1H NMR (600 MHz) Data of RufomycinSS 1–3 in CD3OD
| rufomycinSS 1 | rufomycinSS 2 conformer A | rufomycinSS 2 conformer B | rufomycinSS 3 conformer A | rufomycinSS 3 conformer B | |
|---|---|---|---|---|---|
| no. | δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) |
| AA1-2 | 4.90, dd (10.6, 5.1) | 4.88, m | 5.22, t (7.7) | 4.85, m | 5.06, dd (8.6, 6.9) |
| 3 | 3.18−3.22, m | 3.21, m | 3.13, m; 3.20, m | 3.21, m | 3.18, m |
| 4 | 7.06, s | 7.12, s | 7.19, s | 7.11, s | 7.19, s |
| 7 | 7.51, br d (7.6) | 7.55, br d (7.9) | 7.57, br d (7.9) | 7.55, br d (7.9) | 7.57, br d (7.9) |
| 8 | 7.08, m | 7.06, m | 7.08, m | 7.06, m | 7.08, m |
| 9 | 7.15, m | 7.15, m | 7.15, m | 7.13, m | 7.15, m |
| 10 | 7.66, br d (8.5) | 7.66, br d (8.5) | 7.66, br d (8.5) | 7.65, br d (8.5) | 7.66, br d (8.5) |
| 2′ | 4.47, dd (9.8, 2.0) | 4.48, dd (9.8, 1.9) | 4.49, dd (9.7, 2.0) | 4.49, dd (9.8, 1.8) | 4.47, dd (9.8, 2.0) |
| 3′a | 2.86, dd (−11.3, 2.0) | 2.93, dd (−11.3, 1.8) | 2.87, dd (−11.3, 2.0) | 2.93, dd (−11.3, 1.8) | 2.86, dd (−11.3, 2.0) |
| 3′b | 3.30, dd (−11.3, 9.8) | 3.29, dd (−11.3, 9.8) | 3.30, dd (−11.3, 9.8) | 3.30, dd (−11.3, 9.8) | 3.30, dd (−11.3, 9.8) |
| 1″ | 1.62, s | 1.66, s | 1.66, s | 1.65, s | 1.62, s |
| 1′″ | 1.75, s | 1.77, s | 1.77, s | 1.76, s | 1.77, s |
| AA2-2 | 4.49, m | 4.37, dd (10.6, 4.1) | 4.70, m | 4.39, dd (10.6, 3.8) | 4.57, dd (10.2, 4.1) |
| 3a | 1.46, m | 1.56, m | 1.56, m | 1.55, m | 1.58, m |
| 3b | −0.74, m | −0.34, m | −0.00, m | −0.39, m | −0.21, m |
| 4 | 0.94, m | 0.99, m | 1.13, m | 0.99, m | 1.13, m |
| 5 | 0.11, d (6.7) | 0.17, d (6.7) | 0.34, d (6.7) | 0.16, d (6.7) | 0.29, d (6.7) |
| 6 | 0.35, d (6.7) | 0.45, d (6.7) | 0.50, d (6.7) | 0.44, d (6.7) | 0.48, d (6.7) |
| N-Me | 2.12, s | 2.34, s | 2.16, s | 2.31, s | 2.16, s |
| AA3-2 | 4.65, dd (11.6, 3.9) | 4.70, m | 4.66, m | 4.73, m | 4.67, m |
| 3a | 3.12, dd (−14.0, 3.9) | 3.02, m | 3.15, m | 3.03, m | 3.15, m |
| 3b | 2.82, dd (−14.0, 11.6) | 2.82, m | 3.06, m | 2.81, m | 3.03, m |
| 5 | 7.90, d (2.2) | 7.85, d (2.2) | 7.96, d (2.2) | 7.85, d (2.2) | 7.94, d (2.2) |
| 8 | 7.04, d (8.6) | 7.08, d (8.6) | 7.06, d (8.6) | 7.07, d (8.6) | 7.07, d (8.6) |
| 9 | 7.35, dd (8.6, 2.2) | 7.39, dd (8.6, 2.2) | 7.42, dd (8.6, 2.2) | 7.38, dd (8.6, 2.2) | 7.39, dd (8.6, 2.2) |
| AA4-2 | 5.04, q (6.6) | 4.76, q (6.6) | 4.97, q (6.6) | 4.77, q (6.6) | 4.99, q (6.6) |
| 3 | 1.35, d (6.6) | 1.27, d (6.6) | 1.35, d (6.6) | 1.28, d (6.6) | 1.34, d (6.6) |
| AA5-2 | 5.68, dd (11.5, 5.9) | 3.85, dd (12.1, 6.6) | 5.45, m | 3.75, m | 5.38, m |
| 3α | 1.92, m | 2.59, m | 2.33, m | 2.31, m | 2.09, m |
| 3β | 1.88, m | 1.85, m | 1.85, m | 1.77, m | 1.79, m |
| 4 | 2.29, m | 2.44, m | 2.37, m | 1.99, m | 2.11, m |
| 5 | 4.44, d (8.1) | 4.41, br s | 4.33, br s | 4.50, br s | 4.55, br s |
| 1′ | 1.18, d (6.6) | 1.12, d (6.8) | 1.14, d (6.2) | 1.15, d (6.8) | 1.11, d (6.2) |
| NMe | 2.72, s | 3.22, s | 2.73, s | 3.22, s | 2.72, s |
| 1″a | 3.38, m | 3.46, m | 3.53, m | 3.52, m | 3.73, m |
| 1″b | 3.58, m | 3.53, m | 3.59, m | 3.72, m | 3.86, m |
| 2″(H2) | 1.52, m | 1.60−1.64, m | 1.60−1.64, m | 1.55, m; 1.64, m | 1.55, m; 1.64, m |
| 3″(H2) | 1.41, m | 1.39, m | 1.41, m | 1.39, m | 1.41, m |
| 4″ | 1.02, m | 0.95, m | 0.95, m | 0.94, m | 0.95, m |
| AA6-2 | 3.85, dd (8.5, 6.7) | 5.28, dd (12.0, 4.1) | 3.74, t (7.6) | 5.39, dd (11.7, 4.5) | 3.77, m |
| 3a | 1.88, m | 1.76, m | 2.03, m | 1.87, m | 1.72, m |
| 3b | 2.18, ddd (−14.4, 8.9, 5.8) | 1.97, m | 2.08, m | 1.98, m | 2.43, m |
| 4 | 1.61, m | 1.48, m | 1.63, m | 1.40, m | 1.62, m |
| 5 | 0.99, d (6.5) | 0.94, d (6.5) | 0.96, d (6.5) | 0.91, d (6.5) | 0.97, d (6.5) |
| 6 | 0.97, d (6.5) | 1.03, d (6.7) | 0.97, d (6.6) | 1.02, d (6.7) | 0.99, d (6.6) |
| AA7-2 | 4.70, m | 4.64, m | 4.69, m | 4.71, m | 4.72, m |
| 3a | 2.31, m | 2.43, m | 2.36, m | 2.43, m | 2.37, m |
| 3b | 2.65, m | 3.02, m | 2.65, m | 3.07, m | 2.67, m |
| 4 | 5.49, m | 5.48, m | 5.48, m | 5.50, m | 5.50, m |
| 5 | 5.55, m | 5.60, m | 5.52, m | 5.63, m | 5.55, m |
| 6 | 1.61, m | 1.64, m | 1.58, m | 1.65, m | 1.65, m |
RufomycinSS 2:
pale yellow, amorphous solid; UV (MeOH) λmax (log ε) 215 (4.0), 288 (4.1), 356 (1.7) nm; IR (ATR) νmax 2956, 2353, 2104, 1627, 1538, 1424, 1314, 1256, 1077, 966, 738 cm−1; 1H NMR (CD3OD, 600 MHz), see Table 3; 13C NMR (CD3OD, 150 MHz), see Table 4; (+)-HRESIMS [M + H]+ m/z 1134.6004 (calcd for C58H85ClN9O12+, 1134.6001).
RufomycinSS 3:
pale yellow, amorphous solid; UV (MeOH) λmax (log ε) 214 (3.9), 265 (3.5), 357 (0.2) nm; IR (ATR) νmax 2956, 2372, 2088, 1624, 1537, 1423, 1319, 1255, 1059, 964, 737 cm−1; 1H NMR (CD3OD, 600 MHz), see Table 3; 13C NMR (CD3OD, 150 MHz), see Table 4; (+)-HRESIMS [M + H]+ m/z 1134.6036 (calcd for C58H85ClN9O12+, 1134.6001).
MICs against M. tuberculosis.
The MIC was defined as the minimum concentration of the compound required to achieve a reduction in fluorescence by 90% relative to the untreated bacterial controls. The anti-TB activity was determined by the microplate Alamar Blue assay as previously described.20
Analysis of Binding Affinity to Mycobacterial ClpC1-NTD and FL by Surface Plasmon Resonance (SPR).
These binding assays were performed using a previously reported method.20,21 Kinetic rate constants (ka and kd) were determined by fitting the double-reference data globally to the 1:1 Langmuir model embedded in the Biacore T200 evaluation software (v3.0). KD values were then calculated from the two rate constants (KD = kd/ka). Smaller KD values represent tighter binding affinities.
Supplementary Material
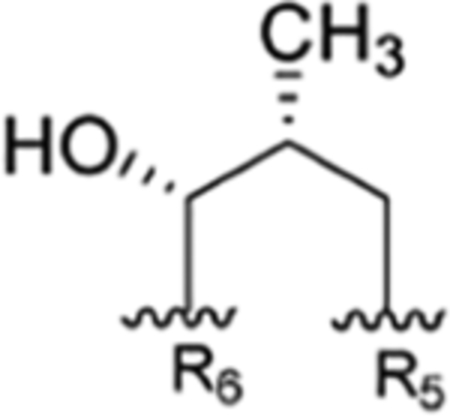
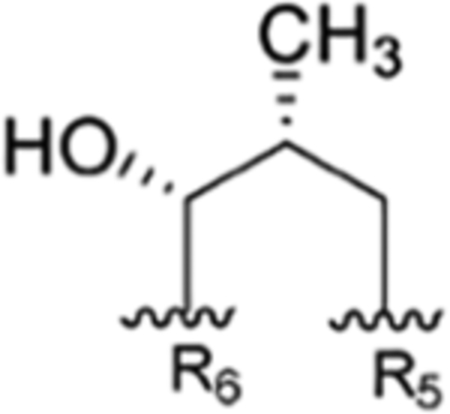
Figure 1.
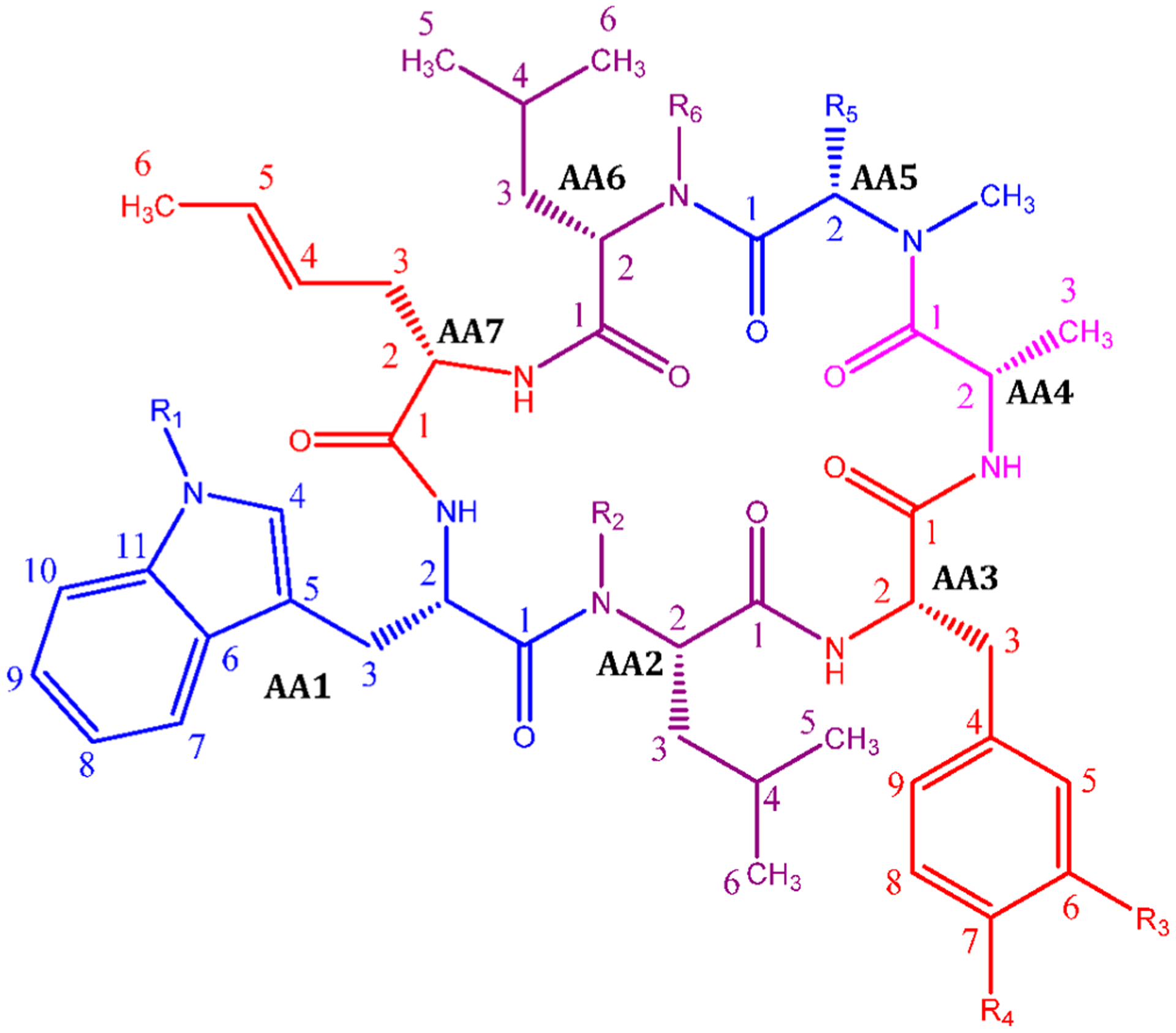
General structure of the rufomycins with consolidated numbering scheme. Individual amino acids are labeled by their NRPS loading sequence. Within each amino acid, atom labels follow IUPAC schemes.
ACKNOWLEDGMENTS
This study was funded in part by grant U19AI142735 from NIAID/NIH and Project No. PJ01564001 (Cooperative Research Program for Agriculture Science and Technology Development) from the Rural Development Administration, Republic of Korea.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.1c00198.
1H and 13C NMR and IR spectra of rufomycins (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jnatprod.1c00198
The authors declare no competing financial interest.
Raw NMR data The original NMR data (FIDs) are made available at DOI: 10.7910/DVN/O87I9V
This paper represents part 41 of the series on Residual Complexity and Bioactivity (see http://go.uic.edu/residualcomplexity).
NOTE ADDED IN PROOF
Following acceptance of this manuscript, the authors became aware of an article (Kazmaier, U.; Junk, L. Marine Drugs, 2021, 19, 446) reviewing the synthesis of cyclopeptide antibiotics. This led us to a second article (Cheng,Y.Y.; Tang, S. B.; Guo, Y.; Ye, T. Org. Lett., 2018, 10, 6166–6169) that describes the synthesis of Ilamycin E1 and Ilamycin F. These two papers further exemplify one of the core messages of the present article, viz the need for consolidated naming for an actively pursued chemical class. The Kazmaier and Junk article always refers to them as “Ilamycina/Rufomycins”. Ilamycin E1 when first described was clearly a mixture of two related congeners. Inspection of the SI of both ref 13 and Cheng et al’s papers indicates that their synthesis produced the major component of the natural product isolated, but as this article points out not the claimed compound.
Contributor Information
Bin Zhou, Institute for Tuberculosis Research and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Prabhakar S. Achanta, Department of Pharmaceutical Sciences, College of Pharmacy and Pharmacognosy Institute, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States
Gauri Shetye, Institute for Tuberculosis Research, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Shao-Nong Chen, Institute for Tuberculosis Research, Department of Pharmaceutical Sciences, College of Pharmacy, and Pharmacognosy Institute, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States;.
Hyun Lee, Institute for Tuberculosis Research and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States; Biophysics Core at Research Resources Center, University of Illinois at Chicago, Chicago, Illinois 60607, United States;.
Ying-Yu Jin, Man Bang Bio Co., Ltd., Subsidiary of Myongji University Technology Holdings Ltd., Cheoin-gu, Gyeonggi-do 17058, Republic of Korea.
Jinhua Cheng, Myongji Bioefficacy Research Center, Myongji University, Cheoin-gu, Gyeonggi-do 17058, Republic of Korea.
Mi-Jin Lee, Myongji Bioefficacy Research Center, Myongji University, Cheoin-gu, Gyeonggi-do 17058, Republic of Korea.
Joo-Won Suh, Myongji Bioefficacy Research Center, Myongji University, Cheoin-gu, Gyeonggi-do 17058, Republic of Korea.
Sanghyun Cho, Institute for Tuberculosis Research and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States;.
Scott G. Franzblau, Institute for Tuberculosis Research and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States;.
Guido F. Pauli, Institute for Tuberculosis Research, Department of Pharmaceutical Sciences, College of Pharmacy, and Pharmacognosy Institute, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States;.
James B. McAlpine, Institute for Tuberculosis Research, Department of Pharmaceutical Sciences, College of Pharmacy, and Pharmacognosy Institute, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States;.
REFERENCES
- (1).Hochlowski JE; Swanson SJ; Ranfranz LM; Whittern DN; Buko AM; McAlpine JB J. Antibiot 1987, 40, 575–588. [DOI] [PubMed] [Google Scholar]
- (2).Arnone A; Nasini G; Cavalleri B J. Chem. Soc., Perkin Trans 1 1987, 1353–1359. [Google Scholar]
- (3).Ōmura S; Imamura N; Oiwa R; Kuga H; Iwata R; Masuma R; Iwai Y J. Antibiot 1986, 39, 1407–1412. [DOI] [PubMed] [Google Scholar]
- (4).Nakazawa K; Shibata M; Higashide E; Kanzaki T; Yamamoto H; Miyake A; Ueyanagi J; Isasaki H Rufomyicn. US 3,655,879, 1961.
- (5).Shibata M; Higashide E; Yamamoto H; Nakazawa K Agric. Biol. Chem 1962, 26, 228–233. [Google Scholar]
- (6).Higashide E; Shibata M; Yamamoto H; Nakazawa K; Iwasaki H; Ueyanagi J; Miyake A Agric. Biol. Chem 1962, 26, 234–237. [Google Scholar]
- (7).Takita T; Ohi K; Okami Y; Maeda K; Umezawa HJ Antibiot. Ser. A 1962, 15, 46–48. [PubMed] [Google Scholar]
- (8).Lambooy PK Process for the Isolation of Rufomycin Factors. PCT/US2000/015044, 06/16/2000, 2000.
- (9).Kulanthaivel P; Vasudevan V Rufomycins and Derivatives thereof Useful as Inhibitors of Multi-drug Resistance Associated Protein-1 (MRP-1). PCT/US2000/015020, 06/08/2000, 2000.
- (10).Kulanthaivel P; Vasudevan V Rufomycin Derivatives Useful as Antibiotics. PCT/US2000/015038, 06/15/2000, 2000.
- (11).Tomita H; Katsuyama Y; Minami H; Ohnishi YJ Biol. Chem 2017, 292, 15859–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Choules MP; Klein LL; Lankin DC; McAlpine JB; Cho S-H; Cheng J; Lee H; Suh J-W; Jaki BU; Franzblau SG; Pauli GF J. Org. Chem 2018, 83, 6664–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ma J; Huang H; Xie Y; Liu Z; Zhao J; Zhang C; Jia Y; Zhang Y; Zhang H; Zhang T; Ju J Nat. Commun 2017, 8, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cary LW; Takita T; Ohnishi M FEBS Lett. 1971, 17, 145–148. [DOI] [PubMed] [Google Scholar]
- (15).Fujino M; Kamiya T; Iwasaki H; Ueyanagi J; Miyake A Chem. Pharm. Bull 1964, 12, 1390–1392. [DOI] [PubMed] [Google Scholar]
- (16).Iitaka Y; Nakamura H; Takada K; Takita T Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem 1974, 30, 2817–2825. [Google Scholar]
- (17).Iwasaki H; Witkop B J. Am. Chem. Soc 1964, 86, 4698–4708. [Google Scholar]
- (18).Takita T; Naganawa H; Maeda K; Umezawa H J. Antibiot. Ser. A 1964, 17, 90–91. [PubMed] [Google Scholar]
- (19).Takita T J. Antibiot. Ser. A 1963, 16, 175–177. [PubMed] [Google Scholar]
- (20).Choules MP; Wolf NM; Lee H; Anderson JR; Grzelak EM; Wang Y; Ma R; Gao W; McAlpine JB; Jin Y-Y; Cheng J; Lee H; Suh J-W; Duc NM; Paik S; Choe JH; Jo E-K; Chang CL; Lee JS; Jaki BU; Pauli GF; Franzblau SG; Cho S Antimicrob. Agents Chemother 2019, 63, No. e02204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wolf NM; Lee H; Choules MP; Pauli GF; Phansalkar R; Anderson JR; Gao W; Ren J; Santarsiero BD; Lee H; Cheng J; Jin YY; Ho NA; Duc NM; Suh JW; Abad-Zapatero C; Cho S-H ACS Infect. Dis 2019, 5, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Takita T; Naganawa H; Maeda K; Umezawa H J. Antibiot. Ser. A 1965, 18, 135–136. [PubMed] [Google Scholar]
- (23).Zhou B; Shetye G; Yu Y; Santarsiero BD; Klein LL; Abad-Zapatero C; Wolf NM; Cheng J; Jin Y; Lee H; Suh J-W; Lee H; Bisson J; McAlpine JB; Chen S-N; Cho S-H; Franzblau SG; Pauli GF J. Nat. Prod 2020, 83, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Takita T; Naganawa H; Maeda K; Umezawa H J. Antibiot. Ser. A 1964, 17, 264–265. [PubMed] [Google Scholar]
- (25).Takita T; Naganawa H; Maeda K; Umezawa H J. Antibiot. Ser. A 1964, 17, 129–131. [PubMed] [Google Scholar]
- (26).Nakayama Y; Ozawa H; Tahara K; Umezawa H J. Antibiot. Ser. A 1962, 15, 49–50. [Google Scholar]
- (27).Takita T J. Antibiot. Ser. A 1963, 16, 211–212. [PubMed] [Google Scholar]
- (28).Takita T; Naganawa H J. Antibiot. Ser. A 1963, 16, 246–246. [PubMed] [Google Scholar]
- (29).Nawa H; Nakazawa K; Miyake A; Kamiya T Water-soluble derivatives of rufomycin. 3,330,725, 07/11/1967, 1967.
- (30).Higashide E Nippon Nogei Kagaku Kaishi 1968, 42, 394–400. [Google Scholar]
- (31).Zhou W; Fang H; Wu Q; Wang X; Liu R; Li F; Xiao J; Yuan L; Zhou Z; Ma J; Wang L; Zhao W; You H; Ju J; Feng J; Chen C Int. J. Biol. Sci 2019, 15, 1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xie Q; Yang Z; Huang X; Zhang Z; Li J; Ju J; Zhang H; Ma J J. Hematol. Oncol 2019, 12, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sun C; Liu Z; Zhu X; Fan Z; Huang X; Wu Q; Zheng X; Qin X; Zhang T; Zhang H; Ju J; Ma J J. Nat. Prod 2020, 83, 1646–1657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.