

ABSTRACT
The adaptive T cell immune response requires cellular plasticity to generate distinct subsets with diverse functional and migratory capacities. Studies of CAR T cells have primarily focused on a limited number of phenotypic markers in blood, representing an incomplete view of CAR T cell complexity. Here, we adapted mass cytometry to simultaneously analyze trafficking and functional proteins expression in CD19 CAR T cells across patients’ tissues, including leukapheresis T cells, CAR product, CAR T cells in peripheral blood, bone marrow, and cerebrospinal fluid post infusion and correlate them with phenotypes. This approach revealed spatiotemporal plasticity of CAR T cells. Patients’ CAR product revealed upregulation in many trafficking and activation molecules compared to leukapheresis T cells as baseline. Including statistically significant upregulation in CD4 and CD8 integrin-β7, CD4 granzyme B, and CD11a as well as CD8 CD25 and CD95. Moreover, patients’ tissues showed spatiotemporal alteration in trafficking, activation, maturation, and exhaustion features, with a distinct signature in the central nervous system niche. Compared to peripheral blood samples, cerebrospinal fluid samples were statistically significant enriched in CD4 and CD8 trafficking and memory phenotype proteins integrin β7, CCR7, CXCR4, and CD8 CD69. Our data provide a potential framework to remodel CAR T cells and enhance immunotherapy efficacy.
KEYWORDS: CAR T cells, mass cytometry, spatiotemporal plasticity, trafficking, activation, maturation
Introduction
T cell adaptive immune response requires cellular plasticity to generate distinct subsets with diverse functional and migratory capacities. Following their ontogeny in the thymus, naïve T cells continually circulate in secondary lymphoid tissues, aiming to recognize antigens presented by major histocompatibility complex molecules expressed on the surface of antigen-presenting cells by their T cell receptors. Once activated, naïve T cells expand and differentiate into short-lived, effector T cells, which express unique trafficking receptors that enable them to migrate to target sites. With antigen clearance, a small subset of these primed T cells gives raise to diverse long-lived memory sub-populations with distinct migratory ability.1,2 By expressing unique patterns of trafficking molecules, T cells interact with tissue-specific vascular endothelia for preferential recruitment to distinct tissues. These interactions between trafficking molecules and their ligands allow T cells to migrate from the circulation in a multistep cascade of tethering, rolling, activation, and arrest. Initially, T cells tether and roll on the endothelium by binding with endothelial cells via selectins and some integrins. Later, integrins on T cells are activated by chemoattractants on endothelial surfaces that bind to their specific G protein-coupled receptors (GPCR). Finally, activated integrins mediate T cell arrest followed by transmigration into homing sites.3 Moreover, the profile of trafficking molecules expressed by T cells is determined by their state of activation and differentiation, as well as their initial tissue site of antigen priming.4
Chimeric antigen receptors (CARs) are synthetically engineered receptors that redirect the specificity, effector function, and metabolism of transduced T cells.5–8 In contrast to endogenous T cell, CAR T cell therapy differs in several aspects. Firstly, in order to generate CAR T cells, polyclonal T cells in leukapheresis products are activated and stimulated using anti-CD3/CD28 beads, followed by transduction with a lentiviral vector that encodes CAR, and continues expansion by cytokines via the common gamma chain.9 Secondly, patients are preconditioned with lymphodepletion chemotherapy which creates a favorable immune environment for adoptively transferred CAR T cells, improve their in vivo expansion, subsequent persistence, and clinical activity.10 Finally, once infused, CAR T cells migrate, proliferate, and exert antitumor activity, in medullary and extra medullary sites, independently of the major histocompatibility complex.6,7 Given these unique properties, better understanding of CAR T cells integrative biology could further enhance immunotherapy efficacy.
To address this need, we used cytometry by time of flight (CyTOF) which enables high dimensional, in-depth, proteomic analysis of immune cells at the single-cell level.11,12 We designed an integrative panel with antibodies specific for CAR, cell lineage, activation, maturation, trafficking, and exhaustion markers to investigate the spatiotemporal landscape of CD19 CAR T cells, across time, location, and surface as well as intracellular proteins. This analysis revealed spatiotemporal plasticity of CAR T cells. CAR T cell product showed increased expression of trafficking and activation molecules, and patients’ CAR T cells from peripheral blood, bone marrow (BM), and cerebrospinal fluid (CSF) showed spatiotemporal alteration in trafficking, activation, maturation, and exhaustion proteins, with distinct signature in the CSF niche. To our knowledge, our study is the first to provide a spatiotemporal landscape of CAR T cell therapy in patients.
Results
Mass cytometry permits in-depth single-cell analysis of migration, activation, maturation, and exhaustion in human T cell subsets
T cells function requires coordinated interplay between activation, maturation, and migration. Therefore, we sought to analyze key molecules in these processes (Figure S1A and Table S1). Since cryopreservation may affect expression of cellular adhesion molecules13 such as CD62L due to protein shedding,14 we obtained fresh human whole blood from three healthy donors, depleted non-T cells, and identified six canonical T cell subsets: naïve, stem-cell memory, central memory, effector, effector memory, and effector memory CD45RA+ (Table S2). Next, we interrogated the proteomic landscape inter- and intra-subsets and plotted their expression patterns (Figures 1(a, b)). Our analysis recapitulated known biology but also provided new insights into the relative expression and distribution of these molecules. For example, bimodal distribution was observed in CXCR3, CD49d central memory CD8 cells and in CD27 effector and effector memory T cells (Figure 1(a)), and sub-analysis revealed positive relationship between CXCR3 and CD49d expression and inverse relationship between CD27 and Granzyme B expression, respectively (Figure S2). Indicating that central memory and effector memory T cells, based on the well-accepted phenotypical definition, are heterogeneous and represent different functional subgroups. Furthermore, an inverse relative expression and distribution of CD127 were captured in CD4 (high) and CD8 (low) effector memory cells (Figure 1(a)).
Figure 1.

Multiplexed single cell profile of activation, maturation, trafficking, and immune checkpoint of human T cells.
(A) Normalized (99.9th percentile) violin plots of all assessed proteins across T cells subsets. Black dots indicate population means. Bimodality indicated by arrows. *p < .05, **p < .01, ***p < .001 and ****p < .0005 indicate significant difference. (B) Normalized (99.9th percentile) median expression of all assessed proteins across T cell subsets. (C) Overlay of manually gated T cell subsets on optSNE embedding. Each color designates a subset ID. (D) Overlay of unsupervised clustering by FlowSOM of T cells on optSNE embedding. Each color designates a cluster ID. (E) Normalized (99.9th percentile) median expression of all assessed proteins across T cells for manually undefined clusters. TN = naïve T cells; TSCM = stem cell memory T cells; TCM = central memory T cells; TEFF = effector T cells; TEM = effector memory T cells; TEMRA = effector memory CD45RA+ T cells. Data are representative of three independent experiments.
Given the robustness of CyTOF to perform high-dimensional protein analysis on the single-cell level, we then sought to identify T cells not captured by canonical manual gating. By dimensionality reduction and visualization of the healthy donor samples using optSNE,15 several populations of cells that were not well defined by the canonical definitions were discovered (Figure 1(c), black). Automated clustering based on the phenotyping markers (Table S2) was performed with FlowSOM,16 and revealed these undefined populations as six distinct clusters (clusters 4, 6, 7, and 11–13) (Figure 1(d)). Then, clusters were overlaid with the manually gated populations to confirm that these six clusters account for the undefined populations (Figure 1(e)). Clustering revealed four subpopulations of CD3 positive double negative (CD4 and CD8) T cells (clusters 6, and 11–13), with variable phenotypic expression, one subpopulation of double positive T cells (cluster 7), and one subpopulation of CD4 + T cells (cluster 4) which had unique phenotype (CD45RA-, CD45ROhigh, CD62Lhigh, CCR7-, CD27-, CXCR3low), supporting a subgroup of central memory T cells, which has never been defined before (Figure 1(e)). Overall, these demonstrate the suitability and robustness of our methodology to study the relationship between trafficking, activation, and maturation proteins, and our data provide a more detailed landscape for interrogating T cell subsets.
CAR T cells exhibit temporal plasticity in vitro
We extended our approach and investigated the temporal relationships between CAR T cell trafficking, activation, maturation, effector function, and exhaustion in vitro. We generated CD19 CAR T cells with PMBCs collected from three healthy donors, following CD3/CD28 activation and lentiviral transduction. To assess CD19 CAR T cells functionality we analyzed for degranulation ability upon co-culture with CD19+ or CD19- targets. We found significantly elevated levels of CD107a+ cells in CD19 CAR T cells upon CD19+ tumor stimulation (Figure S3A). Maximum degranulation remained the same as indicated by the degranulation ability after stimulation with LCL OKT3, which engages the entire T cell receptor (TCR), suggesting that the tumor-specific and intrinsic capacity of effector function was not affected by activation and stimulation using anti-CD3/CD28 beads and continues expansion by cytokines via the common gamma chain (Figure S3B). Then, the resultant CD19 CAR T cells were activated again with anti-CD19 microbeads up to 7 days, and longitudinally analyzed (Figures 2(a), S4A and S4B). This approach has been previously used to study antigen mediated CAR T cell signaling, expansion, metabolism, and maturation.5,17 CAR T cells express truncated human epidermal growth factor protein as part of the transduced chimeric antigen receptor construct (Figure S1A).18 The inclusion of huEGFRt in the CAR construct allows for clear identification and analysis of CAR T cells from non-CAR T cells using anti-CD3 and anti-huEGFRt metal-tagged antibodies. Through manual gating, we identified CAR T cells as CD3+/EGFR+ cells and non-CAR T cells as CD3+/EGFR- cells (Figure S1B). The longitudinal expression pattern revealed statistically significant upregulation in many proteins in CAR T cell products compared to PMBCs. Including, CD4 and CD8 activation and maturation markers CD95, granzyme B, and CD4 CD25, as well as and in CD4 and CD8 trafficking molecules integrin-β7, CD49d, and CXCR3, along with CD4 CD69, and CD8 CD11a. Yet, downregulation in homeostatic cytokine receptor CD127(IL-7Rα) in both CD4 and CD8 lineages. The statistically significant elevation persisted on days 1 and 7 post CD19 beads stimulation in CD4 and CD8 CD95, CD11a, CXCR3, in addition to CD4 CD49d and CD8 integrin-β7 (Figures 2(b, c)). Notably, CD69 expression, a marker of early activation and tissue-resident memory T cells,19 developed bimodal distribution on day 7 post CD19 beads stimulation (Figure 2(b)).
Figure 2.
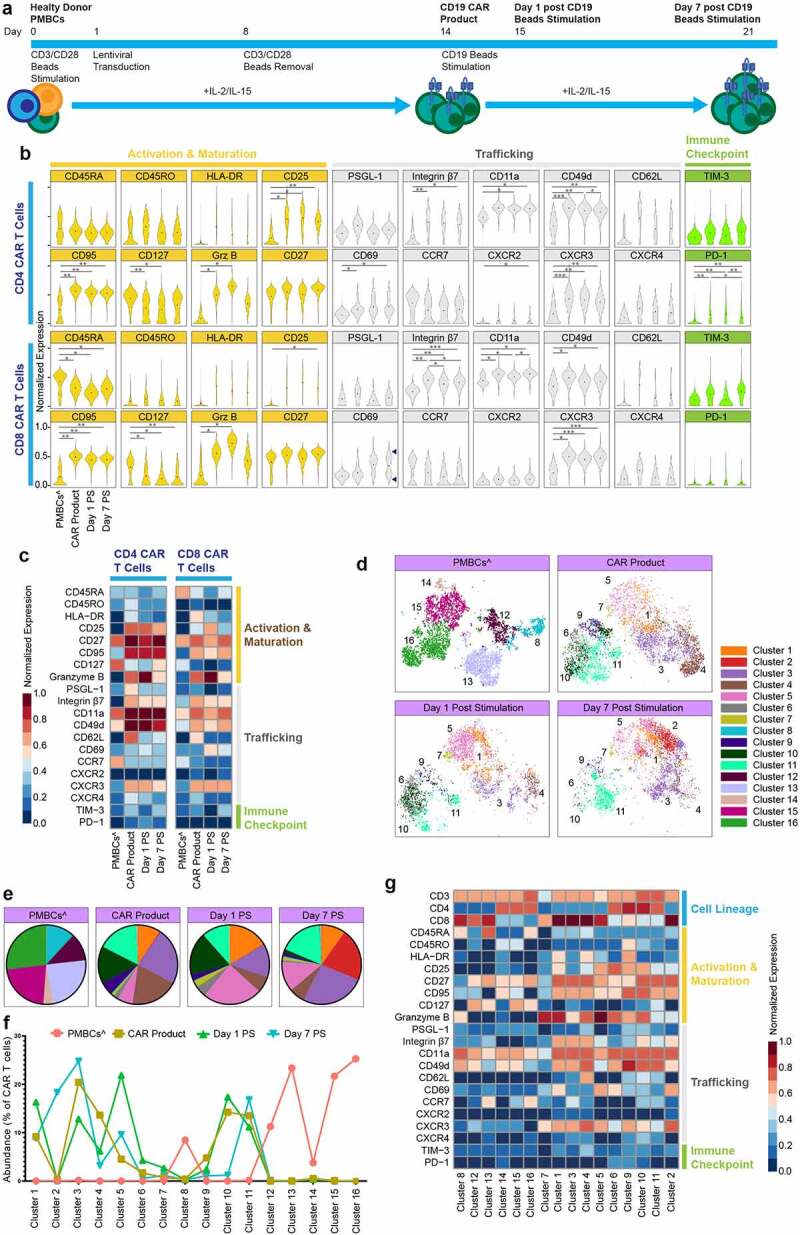
CAR T cells exhibit longitudinal plasticity in vitro.
(A) Experimental workflow (n = 3 healthy donors). (B) Normalized (99.9th percentile) violin plots of all assessed proteins across CAR T cells longitudinally. Black dots indicate population means. Bimodality indicated by arrows. *p < .05, **p < .01, ***p < .001 and ****p < .0005 indicate significant difference. (C) Normalized (99.9th percentile) median expression of all assessed proteins across CAR T cells longitudinally. (D) Overlay of unsupervised clustering by FlowSOM of CAR T cells on optSNE embedding, separated longitudinally. Each color designates a cluster ID. (E) Pie charts of mean cluster abundance longitudinally. Colors correspond to the clusters shown in the optSNE embedding in panel D. (F) Abundance of each of the 16 clusters determined by FlowSOM clustering. (G) Normalized (99.9th percentile) median expression of all assessed proteins across CAR T cells clusters. ^non-CAR T cells. PMBCs = peripheral blood mononuclear cells; PS = post stimulation
To further explore the temporal changes and identify distinct cell clusters and their combinatorial expression patterns, we used FlowSOM, which identified 16 distinct clusters (Figure 2(d)). Cluster abundance was dynamic over time (Figures 2(e, f)). For example, PBMC T cells were dominated by 6 clusters (clusters 8, and 12–16) and CAR product samples were comprised of 8 distinct clusters (clusters 1, 3–6, and 9–11); CAR T cells on day 7 post stimulation have a unique cluster (cluster 2) compared to earlier timepoints. Next, we applied a standard scaled expression matrix to demonstrate the molecular identity of these clusters (Figure 2(g)). For example, the PMBC samples’ dominant clusters 12, 13, 15, and 16 were marked by low expression of activation and trafficking molecules, CD25, CD69, integrin β7, CXCR3, CD62L and elevated expression of CD127. While the CAR T product dominant clusters 3,4, 10, and 11 were marked by high expression in majority of trafficking and activation molecules (Figure 2(g)). Together, these data demonstrated CAR T cells’ temporal plasticity, and provided insights regarding CAR T cell plasticity following CD19 antigen encounter.
Patients’ CAR T cells show remarkable spatiotemporal plasticity
In addition to temporal-related changes, T cells are also influenced by the tissue microenvironment in which they reside. To further investigate the spatiotemporal dynamics of CAR T cells in vivo we analyzed clinical samples from three patients treated with CD19 CAR T cells for B cell hematological malignancies (Table 1). Clinical samples were analyzed ex vivo utilizing our CAR T cell detection method which enables the direct detection of CAR T cells amongst the background of the broader immune system of patients, with no need for ex vivo manipulation. For each patient, we sampled the leukapheresis T cells, CAR product, PMBCs on days 7 and 28, BM on day 28 and cerebrospinal fluid on day 7 (one patient) or 28 (two patients) post CAR T cell infusion (Figure S1C). Analysis of patients’ peripheral blood following CAR T infusion showed that CAR T cells reached peak counts on day 7 and declined thereafter (Figure S5A), while B cell aplasia was achieved by day 14 and persisted at day 28 (Figures S5B and S5C). Consistent with our in vitro model, patients’ CAR product revealed upregulation in many trafficking and activation molecules compared to leukapheresis T cells as baseline. Including statistically significant upregulation in CD4 and CD8 integrin-β7, CD4 granzyme B and CD11a as well as CD8 CD25 and CD95. Remarkably, the human tissue samples showed diverse spatiotemporal landscapes. For example, compared to peripheral blood samples, CSF samples were statistically significant enriched in CD4 and CD8 trafficking and memory proteins integrin β7, CCR7, CXCR4 and CD8 CD69 (Figures 3(a, b)). Notably, day 28 BM samples showed bimodal distribution of CXCR4, integrin β7, CD62L, CCR7 and PD-1, suggesting the existence of two distinct CAR T cell subpopulations in the BM niche with (possibly) distinct phenotypic features (Figure 3(c)). Next, we investigated whether we could establish a relationship between patients’ different tissue samples. We applied Spearman correlation analysis to the mean protein expression level of each marker in our panel on the CD4 and CD8 CAR T cells in each timepoint (Figure 3(d)). This demonstrated significant positive correlation between CAR T product and the post-infusion timepoints, especially with CSF and day 28 BM samples. Furthermore, CSF and BM CAR T cells exhibited strong correlation, suggesting that tissue microenvironment can shape CAR T cells; consistent with our preclinical studies which showed that CSF shifts CAR T cells to memory function.20
Table 1.
Patients’ demographic and clinical characteristics
| Patient | Age upon CAR T cell infusion | Gender | Diagnosis | Disease evaluation prior to CAR T cell infusion |
Disease evaluation on day 28 post infusion |
||
|---|---|---|---|---|---|---|---|
| BM | CNS | BM | CNS | ||||
| 1 | 54 years | Male | B-ALL | Positive | Negative | CRi, MRD negative | Negative |
| 2 | 36 years | Female | B-ALL | Positive | Negative | CRi, MRD negative | Negative |
| 3 | 48 years | Female | NHL, DLBCL | Negative | Positive | CRi, MRD negative | Stable disease |
ALL = acute lymphocytic leukemia; NHL = non-Hodgkin lymphoma; DLBCL = diffuse large B-cell lymphoma; CSF = cerebrospinal fluid; CRi = complete remission with incomplete hematologic recovery; MRD = minimal residual disease
Figure 3.

Patients CAR T cells reveal spatiotemporal plasticity.
(A) Normalized (99.9th percentile) violin plots of all assessed proteins across CAR T cells spatiotemporally. Black dots indicate population means. Bimodality indicated by arrows. *p < .05, **p < .01 and ***p < .001 indicate significant difference. (B) Normalized (99.9th percentile) median expression of all assessed proteins across CAR T cells spatiotemporally. (C) Normalized (99.9th percentile) median expression of all assessed proteins across CXCR4low/high CAR T cells on bone marrow day 28 samples. ***p < .001 indicate significant difference. (D) Correlograms showing upper triangle of Spearman correlation matrix of CAR T cells spatiotemporally. Positive correlations are shown in red. The size of the circles is proportional to the correlation value. ^non-CAR T cells. LPh = leukapheresed T cells; PB = peripheral blood; BM = bone marrow; CSF = cerebrospinal fluid
We next examined the differences between CAR T cells and non-CAR T cells sharing identical microenvironment longitudinally, by comparing the two subgroups in the product, and peripheral blood on days 7 and 28 post-infusion (Figure S6). In the product, similar findings were found for most proteins, besides CD4 and CD8 CD25, a marker of activation, as well as CD8 integrin-β7, CD45RO, and CD4 granzyme B which were significantly elevated in the CAR+ group. Day 7 peripheral blood CD4 and CD8 CAR+ cells, showed significantly elevated expression of trafficking proteins integrin-β7, CXCR3 and immune checkpoint proteins TIM-3 and PD-1. Interestingly, day 28 peripheral blood CAR- cells showed significantly elevated expression of trafficking proteins CD4 integrin-β7, CCR7, CD62L, and CXCR4, while CD4 and CD8 CAR+ cells maintained significantly elevated expression of PD-1. These data indicate that in products, successfully transduced CAR T cells have a higher activation state and that following infusion, during the expansion phase, CAR+ cells have higher trafficking molecules, compared to CAR-, which might facilitate better trafficking to medullary and extra medullary disease sites. Altogether, our data shed light on the intrinsic properties of CAR T cells along production and post infusion.
To further explore the spatiotemporal dynamics, we employed FlowSOM which identified 21 distinct clusters (Figure 4(a)). Quantitation of clusters frequency and protein expression revealed significant heterogeneity, individual clusters showed different patterns of expansion and contraction by time and place (Figures 4(b–d)). For example, leukapheresis T cells were solely enriched in clusters 1–2 marked by CD45RA CD27, and CCR7 expression, while CAR product was enriched in clusters 9,11, and 13 marked by CXCR3, integrin β7 trafficking molecules and granzyme B expression. Furthermore, day 7 peripheral blood samples were solely enriched in clusters 19–20 marked by the expression of the activation markers HLA-DR, CD25, and granzyme B, in addition to the trafficking proteins CD49d, CD62L, and CXCR3. Notably, cluster 12 which had low abundance in the leukapheresis T cells and was depleted in the CAR product, dominated day 7 peripheral blood samples, and persisted to a lesser degree in all tissue samples. Moreover, clusters 3 and 4 that were enriched in the CSF expressed high CXCR3, CXCR4, CCR7, integrin β7, CD62L, and CD49b, which may attribute to their migration between blood and CSF.
Figure 4.
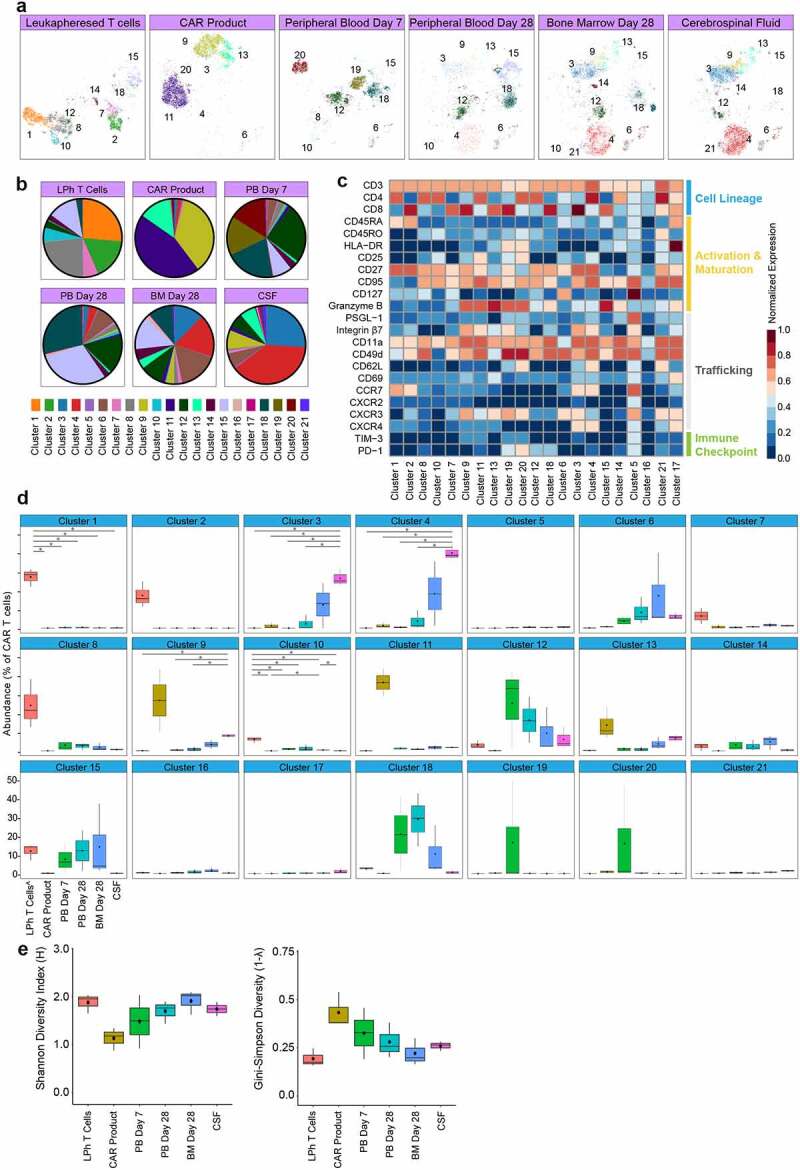
Clusters diversity and abundance in patients CAR T cells.
(A) OptSNE embeddings of FlowSOM clusters in CAR T cells separated spatiotemporally. Each color designates a cluster ID. (B) Pie charts of mean cluster abundance spatiotemporally. Colors correspond to the clusters shown in panel A. (C) Normalized (99.9th percentile) median expression of all assessed proteins of CAR T cells across clusters. (D) Abundance of each of the 21 clusters determined by FlowSOM clustering spatiotemporally. Black dots indicate population medians. Horizontal lines indicate population mean. The lower and upper hinges of the boxes correspond to the 25th and 75th percentiles and the whiskers represent the 1.5 × inter-quartile range extending from the hinges. (E) Shannon’s H and Gini-Simpson diversity indices calculated for all samples spatiotemporally. The lower and upper hinges of the boxes correspond to the 25th and 75th percentiles and the whiskers represent the 1.5 × inter-quartile range extending from the hinges. ^non-CAR T cells. LPh = leukapheresed T cells; PB = peripheral blood; BM = bone marrow; CSF = cerebrospinal fluid
To provide more quantitative measures of the spatiotemporal CAR T cell diversity and evenness, we applied metrics used in population ecology to the clinical samples CAR T cell clusters. We calculated both Shannon’s diversity index, which measures cluster diversity by factoring both the number of total clusters and cluster distribution, and the Gini-Simpson index which measures inequality of a given sample so that the higher the Gini-Simpson index, the more unequal the distribution of individual clusters in the sample. Based on the 21 clusters identified by FlowSOM, we observed that (i) CAR product had the lowest diversity and highest degree of unevenness. (ii) blood samples had longitudinal increase in diversity and evenness. (iii) CSF and day 28 tissue samples showed similar diversity and evenness as leukapheresed cells (Figure 4(e)). Overall, our findings show cluster enrichment in CAR product and increasing diversities in CAR T cell clusters in the first 28 days after adoptive transfer.
CAR T cells in the cerebrospinal fluid niche have distinct properties
As mentioned above, CSF samples showed unique characteristics, and therefore we further interrogated CAR T cells in the CSF niche. We asked whether our analysis is influenced by patients’ heterogeneity, sampling time, type of B cell malignancy and central nervous system (CNS) disease status (Figure 5(a)). Sub-analysis of CD4:CD8 ratio, protein expression and cluster distribution patterns did not differ between patients (Figures 5(b-e)). All patients’ CSF samples were enriched in memory phenotype proteins, activation markers, and displayed a distinct trafficking signature; CD25, CD27, granzyme B, integrin β7, CD11a, CD49d, CD62L, CD69, CCR7, CXCR3, CXCR4 were upregulated while PD-1 was diminished (Figures 5(d) and 5(e)), and showed similar clusters abundance across different patients, with clusters 3–4 dominating (Figure 5(c)). Furthermore, clusters 3–4 showed significant abundance in comparison to other tissue samples, highlighting phenotypic dominance in the CSF (Figure 4(d)). Next, we performed optSNE analysis that captured the dominance and co-expression of the memory, activation, and trafficking proteins mentioned above. Interestingly, it revealed a subset of CAR T cells with effector markers marked by elevated granzyme B and low memory markers CD27, CCR7, CD62L, 45RA, suggesting that the CSF niche contains both memory and effector functions (figure 5(f)). Collectively, these findings demonstrate that CAR T cells in the CSF niche have distinct properties independent of clinical or sampling heterogeneity. Of note, our findings reveal CAR T cells migration to the CSF as early as day 7 post infusion.
Figure 5.
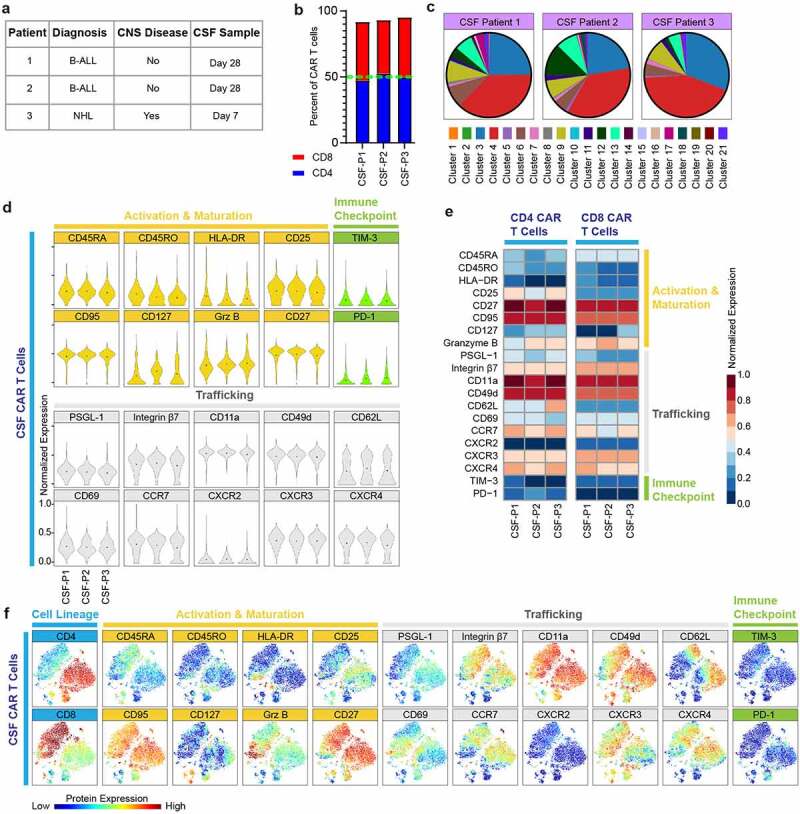
CAR T cells in the CSF niche have distinct properties regardless of patient and sample heterogeneity.
(A) Patient and sample characteristics (n = 3 patients). (B) CSF CD4 to CD8 CAR T cell ratio by sample. (C) Pie charts of mean cluster abundance across CSF samples. Each color designates a cluster ID. Clusters correspond to the protein expression shown in Figure 4c. (D) Normalized (99.9th percentile) violin plots of all assessed proteins by CAR T cells across CSF samples. Black dots indicate population means. (E) Normalized (99.9th percentile) median expression of all assessed proteins of CAR T cells across CSF samples. (F) OptSNE plots showing the expression patterns of all assessed proteins by CAR T cells across CSF samples.
Healthy donor T cell subsets correlate with patients CAR T cells
Since CAR T cells are genetically modified T cells with distinct properties, we next asked to evaluate the phenotypic similarities between patients’ tissue CAR T cells (Figures 3(a) and 4(c)) and T cell subsets in healthy donor PBMCs, as captured by the first part of our study (Figure 1(a)). Spearman correlation analysis (Figure 6) demonstrated positive correlation between leukapheresed T cells and all subsets. Whereas peripheral blood samples correlated the most with effector and effector memory phenotype, notably on day 28 post infusion. Furthermore, CSF samples were most similar to central/stem cell memory phenotype, mirroring our findings of enriched memory markers in the CSF niche, and BM CAR T cells, correlated with both effector and memory phenotypes, highlighting the role of the BM as a T cell memory niche.21 Altogether, these findings revealed remarkable diversification within patients CAR T cells and their spatiotemporal relationship to T cells specialization within the human immune system.
Figure 6.
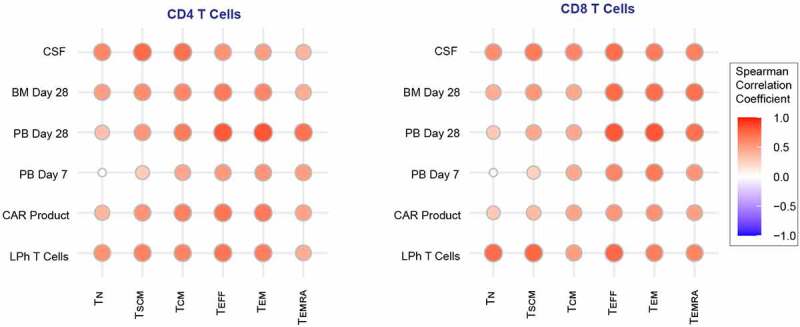
Healthy donor T Cell subsets correlate with patient CAR T cells.
Correlograms showing Spearman correlation matrix among CAR T cells in tissue samples and healthy donor T cell subsets. Positive correlations are shown in red. The size of the circles is proportional to the correlation value.LPh = leukapheresed T cells cells; PB = peripheral blood; BM = bone marrow; CSF = cerebrospinal fluid; TN = naïve T cells; TSCM = stem cell memory T cells; TCM = central memory T cells; TEFF = effector T cells; TEM = effector memory T cells; TEMRA = effector memory CD45RA+ T cells.
Discussion
In summary, mass cytometry enabled us for the first time, to our knowledge, to provide insights into the spatiotemporal plasticity of CAR T cell therapy. We identified diverse functional and tissue-specific CAR T cell expression profiles and provide a potential framework to remodel CAR T cells to enhance immunotherapy efficacy.
We show that CAR T products, regardless of their origin (healthy donors or patients), had upregulated trafficking and activation molecules. CAR T cell production includes ex vivo activation and stimulation using anti-CD3/CD28 beads and continues expansion by IL-2/IL-15 via the common gamma chain. Activated T cells upregulate many integrins and adhesion molecules, enabling their rolling and adhesion to vessel walls, although the relationship between T cell stimulation via chemokine receptors or TCR activation and migration is well studied,22–24 little is known about CAR T cells activation and trafficking. Agreeing with our data, a recent study using single cell RNA sequencing of successfully transduced CAR T cells vs un-transduced T cells of the same product, reported CAR T cells to be enriched in pathways related to T cell migration and activation; including integrin, actin cytoskeleton, and RhoA signaling that is elicited when TCR is engaged during T cell activation.25 Furthermore, RNA sequencing combined with CITE-sequencing revealed that leukapheresed cells from patients with long-term CAR T-cell persistence were enriched in lymphocyte activation and migration pathways.26 These findings provide insights into the close relationship, and potential role, of activation and trafficking molecules in CAR T cell production and clinical function.
T cells migration into the CNS is limited by protective barriers and requires specific molecular keys.27 In the absence of neuroinflammation, the majority of both CD4+ and CD8 + T-cells in the CSF are of a memory phenotype compared to peripheral blood, and predominantly express CD27, CCR7, CD62L, PSGL-1, CD11a, CXCR3.28,29 Furthermore, T cell activation facilitates migration of effector and memory T cells across the blood-brain barrier.30–32 In line with these findings, our study shows that CAR T cells in the CSF are enriched in activation markers, memory phenotype proteins and have a distinct trafficking/homing signature, including statistically significant upregulation of integrin β7, CCR7, CD69, and CXCR3, regardless of patient and sample heterogeneity. Moreover, spearman correlation analysis revealed strong positive correlation between the expression profile of CAR T cells in the CSF and healthy donors central/stem cell memory T cells. Consistently, we previously showed in mice, that CAR T cells conditioned by exposure to CSF in the intracerebroventricular environment had superior anti-tumor activity and memory phenotype compared with intravenous delivered CAR T cells, through metabolic reprogramming.20 The superior trait of CAR T cells in CSF prompted us to further explore the mechanism behind it and would provide the insights how to make “CSF CAR T cells” for enhanced efficacy. The chemokine receptors and memory markers CCR7 and CXCR3 are suggested to be involved in T cells adhesion to blood-brain barrier vascular endothelium and CNS migration during immune surveillance and neuroinflammation.29,33,34 Therefore, it remains to be elucidated, whether memory phenotype holds the molecular key for CAR T cell CNS migration, or migrated cells are being reprogrammed to a memory phenotype in the CNS niche.
Efficient CAR T cell trafficking is essential for effective adoptive cell therapy.35 One strategy to improve CAR-T cell trafficking involves overexpression of adhesion molecules on CAR-T cells that respond to tumor-derived chemokines or the tumor microenvironment. For example, several pre-clinical studies demonstrated that CAR T cells overexpressing CXCR1,36 CXCR2,36–38 CXCR4,39 CCR2b,40,41 and CCR4,42 enhanced trafficking and improved antitumor efficacy. Furthermore, two phase one clinical trials with modified CAR T cells are currently ongoing. Anti-epidermal growth factor receptor CAR T cells modified by CXCR5 in patients with advanced adult non-small cell lung cancer (ClinicalTrials.gov #NCT04153799), and CXCR4 modified B-cell maturation antigen CAR T cells in patients with refractory/relapsed multiple myeloma (ClinicalTrials.gov #NCT04727008). Another approach to improve CAR T cells trafficking includes infecting tumor cells with oncolytic viruses that overexpress chemokines for which CAR T cells express chemokine receptors and are responsive to. For example, the combined use of CCL5-expressing oncolytic adenovirus with GD2 CAR T cells promoted CAR T cells trafficking and tumor clearance in a preclinical neuroblastoma model.43 While the field is already acknowledging the importance of CAR T cell trafficking and activation, our work, sheds light on the endogenous spatiotemporal trafficking and function of CAR T cells and may provide additional strategies to enhance trafficking and improve antitumor efficacy of CAR T cell therapy, especially for CNS disease.
This work was not without limitations. Patients’ clinical samples, especially CSF and BM samples which require invasive procedures, are rare and precious, limiting our study to three patients. Therefore, the results cannot exclude the possibility of patient-specific features contributing to the proteomic profiles. Nevertheless, our single-cell data consisted with over 60,000 CAR T cells across six timepoints and tissues delineated the spatiotemporal landscape and identified CAR T cell plasticity in patients.
Methods
Generation of CD19 CAR T lentivirus
The lentivirus CAR construct was modified from the previously described CD19-specific scFvFc:ζ chimeric immunoreceptor44 to create a second-generation vector. The CD19CAR containing a CD28z costimulatory domain carries mutation at two sites (L235E; N297Q) within the CH2 region on the IgG4-Fc spacers to ensure enhanced potency and persistence after adoptive transfer.45 The lentiviral vector also expresses a truncated human epidermal growth factor receptor (huEGFRt), which includes a cetuximab (Erbitux) binding domain but excludes the EGF-ligand binding and cytoplasmic signaling domains. A T2A ribosome skip sequence links the codon-optimized CD19R:CD28:ζ sequence to the huEGFRt sequence, resulting in coordinate expression of both CD19R:CD28:ζ and EGFRt from a single transcript.18 The CD19CARCD28EGFRt DNA sequence (optimized by GeneArt) was then cloned into a self-inactivating (SIN) lentiviral vector, pHIV7 (gift from Jiing-Kuan Yee, Beckman Research Institute of City of Hope, Duarte, CA).
Patients samples and CAR T cell therapy
De-identified clinical samples were collected from a subset of adult patients with relapsed and refractory B-cell ALL or NHL, who received CD19-specific CAR-T cells in a phase 1 clinical trial (clinicaltrials.gov #NCT01815749). The study was conducted in accordance with the principles of the Declaration of Helsinki and with the approval of the City of Hope Internal Review Board (IRBs 13447 and 13351). Briefly, autologous CD19R(EQ)28ζ/EGFRt+ TN/MEM – enriched T Cells product was administered by intravenous infusion in a single dose on Day 0 following a 1-5-day lymphodepleting regimen. T cells were isolated from patient leukapheresis product after Ficoll separation. Peripheral blood mononuclear cell (PBMC) preparations then underwent immunomagnetic selection to derive TN/MEM enriched T cell preparations. The resulting cell preparation, highly enriched for CD62L+ TN/MEM, was activated with anti-CD3/CD28 beads. Activated TN/MEM underwent a lentiviral transduction to express the CD19R(EQ)28ζ/EGFRt+ CAR (description of the lentiviral vector is provided below), expanded in vitro to achieve cell numbers sufficient for the research participant’s planned clinical cell dose and all related product release testing, and then harvested, washed, and formulated for cryopreservation.
Healthy donor samples
De-identified peripheral blood samples from healthy human blood donors were obtained, and experimental procedures were carried out in accordance with the guidelines of City of Hope Institutional Review Board. Fresh whole-human blood in leukoreduction system chamber contents (Terumo BCT) were obtained from the City of Hope Blood Center. PBMCs were isolated via Ficoll (GE Healthcare) density gradient centrifugation. For T cell subsets studies, T cells were enriched through negative selection using magnetic beads (EasySep Human T Cell Isolation Kit, STEMCELL Technologies).
Generation and in vitro stimulation of CD19 CAR T cells
PBMC from three healthy donors were activated with CD3/CD28 Dynabeads (Invitrogen) at 1:3 (cells:beads ratio). CD19 CAR T cells were generated by transducing activated PBMCs the next day with a lentivirus encoding CD19R (EQ):CD28:ζ/EGFRt at an MOI of 3 and expanded in the presence of 50 U/mL rhIL-2 (CellGenix) and 0.5 ng/ml rhIL-15 (CellGenix) as previously described (PMID: 23090078, PMID: 28331616). Cultures were then supplemented with 50 U/mL rhIL-2 and 0.5 ng/mL IL-15 every 48 h up to day 14. CD19 antigen stimulation of CD19CAR T cells was carried out by Dynabeads M-270 Epoxy beads (ThermoFisher Scientific) coated with recombinant human CD19 protein (Abcam). 100 μg CD19 protein was used to coat 2.6x10^8 beads. Beads were washed following the coupling protocol from ThermoFisher Scientific (14311D). CD19 protein was added to the beads and incubated overnight (16–24 hours) at 37°C with constant mixing. Coupled beads were further washed and separated using DynaMag following wash protocol. 10x10^6 coupled beads were washed once with 3 ml 1% human serum albumin (HAS in PBS) and 3 ml RPMI media supplemented with 10% FBS. 10x10^6 CD19-coupled beads were introduced to CD19CAR T cells on day 14 at a 1:1 ratio and was supplemented with 50 U/mL rhIL-2 and 0.5 ng/mL IL-15 every 48 h for 7 more days (up to day 21).
Mass cytometry antibody panel, staining procedures, and data acquisition
Single-cell mass cytometry analysis was performed using 1x10^6 cells per sample, in clinical samples with lower number of cells (e.g. CSF) healthy donor T cells (EGFR-) were spiked in to maintain the same cell-antibody ratio in all samples (1x10^6 cells/100 µl final antibody volume). All mass cytometry antibodies used for analysis can be found in Table S1. Cells were stained according to the Cell-ID Cisplatin and Maxpar Cytoplasmic/Secreted Antigen Staining with Fresh Fix protocols (Fluidigm). Briefly, cells were thawed and resuspended in 50 μl pre-warmed serum-free medium into 15 ml polystyrene tubes for each sample and stained for viability according to the Cell-ID Cisplatin protocol (Fluidigm). Cells were then resuspended in 50 μl of Maxpar Cell Staining Buffer in 5 mL polypropylene tubes for each sample to be stained. 50 µl of the surface marker antibody cocktail was added to each tube (final volume 100 µl). Samples were mixed and incubated for 30 min at room temperature. After incubation, the samples were washed twice with Maxpar Cell Staining Buffer and centrifuged for 5 min at 800 × g. Cells were then fixed by adding 1 ml of 1X Maxpar Fix I BuffeR to each tube and incubated for 30 min. After incubation, cells were washed twice with Maxpar Perm-S Buffer and centrifuged for 5 min at 800 × g. Cells were then suspended in 50 μl of Maxpar Perm-S Buffer and incubated as described above with 50 µl of the cytoplasmic/secreted antibody cocktail (final volume 100 µl). After incubation, the samples were washed twice with Maxpar Cell Staining Buffer and centrifuged for 5 min at 800 × g. Cells were then fixed by adding 1 ml of 1.6% formaldehyde solution to each tube and incubated for 10 min. After the incubation, cells were washed twice with Maxpar Cell Staining Buffer, centrifuged for 5 min at 800 × g and incubated with Cell-ID Intercalator-Ir solution at the final concentration of 125 nM into Maxpar Fix and Perm Buffer at 4 C overnight. Before acquisition, samples were washed once with Maxpar Cell Staining Buffer and twice in Maxpar Cell Acquisition Solution and filtered through a cell strainer (Falcon). Cells were then resuspended at 1 × 10^6 cells per ml in a 1:10 dilution of EQ Four Element Calibration Beads (Fluidigm) in Maxpar Cell Acquisition Solution and acquired on a Helios mass cytometer (Fluidigm).
Data cleanup & pre-processing
After acquisition, the CyTOF data were bead normalized per manufacturer’s recommendations via CyTOF (Fluidigm) software version 7.0.8493 using Normalization Passport EQ-P13H2302_ver2. Normalized files were uploaded to OMIQ data analysis software (Omiq, Inc., Santa Clara, CA) for manual data cleanup using the Gaussian parameter cleanup method.46
Manual analysis
All manual gating was performed in OMIQ data analysis software. Manual gating was performed to classify six canonically described CD4+ and CD8 + T cell subsets in the healthy donor data (Table S2) and to classify CD4+ CAR+ and CD8+ CAR+ T cells in the in vitro and in vivo data.
Dimension reduction
Prior to dimensionality reduction, down sampling was performed in OMIQ to equalize cell counts on a per file basis for each dataset (500,000 T cells for the healthy donor samples, 2,500 CAR T cells for the in vitro samples, and 5,000 CAR T cells for the in vivo samples. Note, 5 samples of the in vivo data had less than 5,000 CAR T cells (Table 2). Following down sampling, the data was virtually concatenated in OMIQ and opt-SNE15 was performed for each down-sampled dataset (Perplexity = 30, Max Iterations = 1000, Theta = 0.5, Verbosity = 25), all markers in the CyTOF panel were selected as features.
Table 2.
Number of CAR T cells analyzed per sample
| Patient | Sample | Number of CAR T cells analyzed |
|---|---|---|
| 1 | CAR T cell product | 5000 |
| Peripheral blood day 7 post infusion | 1687 | |
| Peripheral blood day 28 post infusion | 3803 | |
| Bone marrow day 28 post infusion | 322 | |
| Cerebrospinal fluid day 28 post infusion | 4179 | |
| 2 | CAR T cell product | 5000 |
| Peripheral blood day 7 post infusion | 5000 | |
| Peripheral blood day 28 post infusion | 5000 | |
| Bone marrow day 28 post infusion | 5000 | |
| Cerebrospinal fluid day 28 post infusion | 5000 | |
| 3 | CAR T cell product | 5000 |
| Peripheral blood day 7 post infusion | 5000 | |
| Peripheral blood day 28 post infusion | 563 | |
| Bone marrow day 28 post infusion | 5000 | |
| Cerebrospinal fluid day 7 post infusion | 5000 |
Unsupervised clustering
Data clusters were classified by performing FlowSOM16 on the down-sampled data as implemented in OMIQ, with 100 clusters using Manhattan distance as the distance metric, and all markers in the panel were used as features for clustering, with the exception of the healthy donor analysis.
Statistical analysis
Manually gated and clustered population abundances, population protein median metal intensities (MMI), and single-cell protein MMI were exported from OMIQ (Omiq) and imported to R. The population protein MMIs were 99th percentile normalized using the scales package in R. Spatiotemporal differences in population protein MMIs were calculated using multiple t-tests with using the standard FDR p-value adjustment for multiple comparisons with the rstatix R packages. Violin plots and boxplots were created in R using the ggplot2 package. Heatmaps were created in R using the pheatmap package. Spearman correlation matrices were created using the Hmisc and ggcorrplot R packages. Diversity indices were performed using the diversity result function from the BiodiversityR-package and plotted using the ggplot2 package in R.
Supplementary Material
Acknowledgments
L.G. is a SHARE Inc research fellow in pediatric Hematology-oncology. Research reported in this publication was supported by City of Hope Lymphoma SPORE of the National Institutes of Health under award number P50 CA107399-11
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Author contributions
L.G., S.J.F., and X.W. conceived and designed the study. L.G., V.V., and R.U. performed experiments. L.G., and E.R.H performed data analysis and generated figures. L.G. wrote the original draft. L.G., E.R.H, V.V., R.U., S.J.F., and X.W. reviewed and edited the final manuscript. X.W., and S.J.F provided funding and supervised the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Kumar BV, Connors TJ, Farber DL.. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202–13. doi: 10.1016/j.immuni.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nolz JC, Starbeck-Miller GR, Harty JT. Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions. Immunotherapy. 2011;3(10):1223–1233. doi: 10.2217/imt.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Andrian Uh, Mackay CR, von Andrian UH. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343(14):1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 4.Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. 2013;37(4):273–283. doi: 10.1152/advan.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, Norris AP, Wong CW, Urak RZ, Chang W-C, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127(24):2980–2990. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 12.Bendall SC, Simonds EF, Qiu P, Amir El AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Huckelhoven A, Hong J, Jin N, Mani J, Chen BA, Schmitt M, Schmitt A. Standardization of cryopreserved peripheral blood mononuclear cells through a resting process for clinical immunomonitoring–Development of an algorithm. Cytometry A. 2016;89(3):246–258. doi: 10.1002/cyto.a.22813. [DOI] [PubMed] [Google Scholar]
- 14.Costantini A, Mancini S, Giuliodoro S, Butini L, Regnery CM, Silvestri G, Montroni M. Effects of cryopreservation on lymphocyte immunophenotype and function. J Immunol Methods. 2003;278(1–2):145–155. doi: 10.1016/S0022-1759(03)00202-3. [DOI] [PubMed] [Google Scholar]
- 15.Belkina AC, Ciccolella CO, Anno R, Halpert R, Spidlen J, Snyder-Cappione JE. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat Commun. 2019;10(1):5415. doi: 10.1038/s41467-019-13055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87(7):636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 17.Ramello MC, Benzaid I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN, Pabón-Saldaña M, Darville L, Fang B, Rix U, et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. 2019;12(568). doi: 10.1126/scisignal.aap9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo PA, Miron M, Farber DL. Location, location, location: tissue resident memory T cells in mice and humans. Sci Immunol. 2019;4(34). doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Huynh C, Urak R, Weng L, Walter M, Lim L, Vyas V, Chang W-C, Aguilar B, Brito A, et al. The cerebroventricular environment modifies CAR T cells for potent activity against both central nervous system and systemic lymphoma. Cancer Immunol Res. 2021;9(1):75–88. doi: 10.1158/2326-6066.CIR-20-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Rosa F. Two niches in the bone marrow: a hypothesis on life-long T cell memory. Trends Immunol. 2016;37(8):503–512. doi: 10.1016/j.it.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-Integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front Immunol. 2019;10:254. doi: 10.3389/fimmu.2019.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walling BL, Kim M. LFA-1 in T cell migration and differentiation. Front Immunol. 2018;9:952. doi: 10.3389/fimmu.2018.00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dupre L, Houmadi R, Tang C, Rey-Barroso J. T lymphocyte migration: an action movie starring the actin and associated actors. Front Immunol. 2015;6:586. doi: 10.3389/fimmu.2015.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai Z, Lundh S, Kim D, Woodhouse S, Barrett DM, Myers RM, Grupp SA, Maus MV, June CH, Camara PG, et al. Single-cell multiomics dissection of basal and antigen-specific activation states of CD19-targeted CAR T cells. J Immuno Cancer. 2021;9(5):e002328. doi: 10.1136/jitc-2020-002328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen GM, Chen C, Das RK, Gao P, Chen CH, Bandyopadhyay S, Ding -Y-Y, Uzun Y, Yu W, Zhu Q, et al. Integrative bulk and single-cell profiling of pre-manufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov. 2021;11(9):2186–2199. doi: 10.1158/2159-8290.CD-20-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33(12):579–589. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, Wu L, Baekkevold ES, Lassmann H, Staugaitis SM, et al. Human cerebrospinal fluid central memory CD4+ CD4 + T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A. 2003;100(14):8389–8394. doi: 10.1073/pnas.1433000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kivisakk P, Trebst C, Liu Z, Tucky BH, Sorensen TL, Rudick RA, Mack M, Ransohoff RM. T-cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: implications for CNS trafficking. Clin Exp Immunol. 2002;129(3):510–518. doi: 10.1046/j.1365-2249.2002.01947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vajkoczy P, Laschinger M, Engelhardt B. Alpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J Clin Invest. 2001;108(4):557–565. doi: 10.1172/JCI12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laschinger M, Vajkoczy P, Engelhardt B. Encephalitogenic T cells use LFA-1 for transendothelial migration but not during capture and initial adhesion strengthening in healthy spinal cord microvessels in vivo. Eur J Immunol. 2002;32(12):3598–3606. doi: 10.1002/1521-4141(200212)32:12<3598::AID-IMMU3598>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 32.Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991;1(2):97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 33.Krumbholz M, Theil D, Steinmeyer F, Cepok S, Hemmer B, Hofbauer M, Farina C, Derfuss T, Junker A, Arzberger T. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol. 2007;190(1–2):72–79. doi: 10.1016/j.jneuroim.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 34.Muller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, Dreykluft A, Lu B, Gerard C, King NJC, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. 2007;179(5):2774–2786. doi: 10.4049/jimmunol.179.5.2774. [DOI] [PubMed] [Google Scholar]
- 35.Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics. 2016;3:16006. doi: 10.1038/mto.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, Dyson KA, Grippin AJ, Deleyrolle LP, Zhang W, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. doi: 10.1038/s41467-019-11869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, Maher J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel). 2019;11(5):674. doi: 10.3390/cancers11050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, Dong J, Zhao X, Lin X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020;50(5):712–724. doi: 10.1002/eji.201948457. [DOI] [PubMed] [Google Scholar]
- 39.Itoh-Nakadai ASY, Murasawa-Tomizawa M, Kajita H, Matsumoto T, Matsuda M, Watanabe T, Shirouzu M, Ohara O, Koseki H, Leonard S, et al. CXCR4-Expressing Anti-CD25 CAR T-cells effectively eliminate human AML cells in Vivo. Blood. 2020;136:35–36. doi: 10.1182/blood-2020-142228. [DOI] [Google Scholar]
- 40.Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, Li, B., Pan, H., Jiang, Z., Shen, X. and Liang, Z. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol. 2021;12:628906. doi: 10.3389/fimmu.2021.628906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, and Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74(18):5195–5205. doi: 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJN, et al. CD28 costimulation provided through a CD19-Specific chimeric antigen receptor enhances in Vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66(22):10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 45.Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, Starr, R., Priceman, S. and Ostberg, J.R. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757–768. doi: 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagwell CB, Inokuma M, Hunsberger B, Herbert D, Bray C, Hill B, Stelzer, G., Li, S., Kollipara, A., Ornatsky, O. and Baranov, V. Automated data cleanup for mass cytometry. Cytometry A. 2020;97(2):184–198. doi: 10.1002/cyto.a.23926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.