

ABSTRACT
Saliva is a widely used biological sample, especially in pediatric research, containing a heterogenous mixture of immune and epithelial cells. Associations of exposure or disease with saliva DNA methylation can be influenced by cell-type proportions. Here, we developed a saliva cell-type DNA methylation reference panel to estimate interindividual cell-type heterogeneity in whole saliva studies. Saliva was collected from 22 children (7–16 years) and sorted into immune and epithelial cells, using size exclusion filtration and magnetic bead sorting. DNA methylation was measured using the Illumina MethylationEPIC BeadChip. We assessed cell-type differences in DNA methylation profiles and tested for enriched biological pathways. Immune and epithelial cells differed at 181,577 (22.8%) DNA methylation sites (t-test p < 6.28 × 10−8). Immune cell hypomethylated sites are mapped to genes enriched for immune pathways (p < 3.2 × 10−5). Epithelial cell hypomethylated sites were enriched for cornification (p = 5.2 × 10−4), a key process for hard palette formation. Saliva immune and epithelial cells have distinct DNA methylation profiles which can drive whole-saliva DNA methylation measures. A primary saliva DNA methylation reference panel, easily implemented with an R package, will allow estimates of cell proportions from whole saliva samples and improve epigenetic epidemiology studies by accounting for measurement heterogeneity by cell-type proportions.
KEYWORDS: DNA methylation, saliva, epigenetics, cell type reference, cellular heterogeneity
Introduction
DNA methylation is an important mechanism regulating gene expression in both normal development and disease progression[1]. During cellular differentiation, developmental genes are silenced and other cell-type-specific genes are activated via altered DNA methylation[2], which results in a unique DNA methylation profile for each cell type[3]. Disease processes or environmental exposures can also differentially alter DNA methylation patterns in cells and tissues [4–6]. Epigenetic associations with exposures or disease outcomes are typically assessed in bulk tissues such as blood and saliva. Changes in tissue level DNA methylation profiles, however, could be caused by varying cellular responses. For example, a disease or exposure could shift average DNA methylation across all cells, shift the proportions of cell types, or shift DNA methylation in a particularly susceptible cell type [7–9]. Bulk tissues are comprised of complex cell-type mixtures and have DNA methylation profiles that are vulnerable to alterations from diseases and exposures.
Accounting for cell-type proportions is essential in bulk tissue DNA methylation studies. Cell-type proportions can mediate or confound differences in DNA methylation associated with exposures or diseases. For example, initial widespread age-related DNA methylation associations in blood were later largely attributed to age-related differences in immune cell-type proportions[10]. To estimate cell-type composition in bulk tissue using DNA methylation measures, cell-type-specific DNA methylation reference profiles are often used. DNA methylation reference panels are available for immune cells in primary adult blood and cord blood, as well as epithelial and fibroblast cell types; however, a primary saliva DNA methylation reference panel is not available for children [11–13]. Saliva is a commonly used tissue as a non-invasive source of biological material in epigenetic epidemiology studies, particularly in children. A primary DNA methylation reference panel method for estimating cell-type proportions in saliva is needed.
The use of saliva as a surrogate tissue in epigenetic epidemiology studies is increasing. A search of PubMed using the terms ‘saliva AND (epigenetics or DNA methylation) NOT chemistry’ showed that between 1994 and 2019 there have been 213 papers published that used saliva as the source material for DNA methylation studies. Over this time period, the number of papers has trended upwards. Saliva offers an easier collection strategy, particularly for vulnerable populations, and is noted as a source of DNA methylation with similar quality to blood or other difficult to access tissues [14–16]. One study found that they were able to obtain more DNA from saliva compared to blood and the data quality was high from both tissue types[16]. The use of saliva for epigenetic studies is expected to continue to increase, which highlights the need for a saliva-specific cell-type investigation.
Saliva includes a heterogenous mixture of immune and epithelial human cells. Resident immune macrophage cells are present in oral tissues, and immune cells can leave the bloodstream and enter the oral cavity [17,18]. The keratinized epithelium is found in areas of the oral cavity that experience mechanical forces[19]. Keratinocytes, a large (30–100 µm diameter) epithelial cell covering the hard palette, undergo cornification as a mechanism of programmed cell death[20]. Both immune and epithelial cells contribute to the overall DNA methylation profile of saliva and must be considered for the cell-type proportion estimation[21]. Inter-individual differences in saliva cell composition may lead to wide variations in DNA methylation profiles, independent of exposure or disease status[22]. Cell-type deconvolution methods estimate proportions of cell types from bulk tissue using differentially methylated sites in the genome[23]. A saliva-based DNA methylation reference panel adapted for deconvolution would improve the biological interpretability of existing and future epigenetic studies in saliva by improving the cell-type proportion estimates.
To assist epigenetic epidemiology studies, the objective of this study was to provide a DNA methylation reference panel for cell-type proportion estimation in children’s saliva. Our goals were: 1) Develop methods to enrich for cell types from children’s saliva; 2) Characterize and quantify differences in DNA methylation profiles of children’s saliva cell types; and 3) Apply our new cell-type reference panel to estimate cell proportions in whole saliva and compare our new cell-type reference panel to an existing method for estimating cell proportions.
Materials and methods
Study sample and saliva collection
Children between the ages of 7 and 17 years were eligible for the current study. We recruited a convenience sample of 22 children from schools in Ann Arbor, Michigan. Parents were contacted via email. We obtained written and informed consent from a parent or guardian and verbal assent from the child. This study was approved by the University of Michigan Institutional Review Board (HUM00154853). We collected the demographic data (child’s sex, age, race, and whether the child was sick in the last 3 days) via an anonymous, written survey. The sick children had minor symptoms and did not miss school.
Prior to saliva collection, participants did not eat or drink for 30 minutes. Unstimulated saliva was collected into an empty 15 mL tube (Falcon, CAT# 14–959-53A). Between 1.75 and 6.5 mL of saliva were obtained per participant. Samples were stored at room temperature before processing, and storage time ranged from 1 to 18 hours. Six of these participants additionally provided saliva samples directly into the Oragene kits (DNA Genotek, CAT# OG-250), a common method used for collecting samples for genomic research in the field.
Saliva processing & cell enrichment
The Oragene kit samples were mixed using a 1 mL pipette and a 500 µL aliquot was removed and stored on ice in a microcentrifuge tube (Corning, CAT# 3621) until DNA extraction. Saliva samples collected in Falcon tubes were processed into three components: composite ‘Whole’ saliva, enriched ‘Epithelial’ cells, and enriched ‘Immune’ cells. The overall workflow depicting the isolation of the cell fractions is shown in (Figure 1(a)). Saliva cells were washed by diluting the samples up to 14 mL with DPBS (Gibco, CAT# 14190144) and centrifuging at 500 g for 5 minutes at 4°C. The supernatant was removed, and the pellet was resuspended in 10 mL DPBS. The samples were centrifuged at 350 g for 5 minutes at 4°C, and the supernatant was removed.
Figure 1.
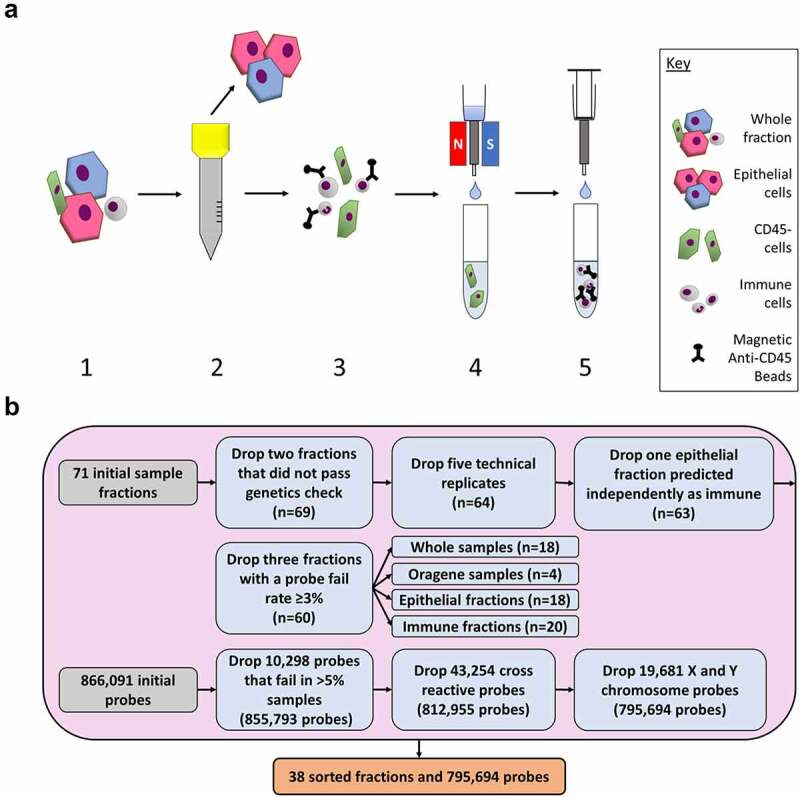
Diagram of experimental workflow. (a) Saliva samples were sorted using size exclusion filtration and antibody-based magnetic bead methods. 1) Whole saliva samples were collected from participants. Samples were diluted and centrifuged. 2) The sample was passed through a 30 µm filter. Cells captured on the filter were then rinsed into a separate collection tube. 3) The small cells that passed through the filter were mixed with CD45+ magnetic antibody beads to label the immune cells. 4) The sample was passed through a magnetized column which captured the immune cells in the column. Unlabelled cells flowed through into the collection tube. 5) The magnet was removed, and the immune cells and magnetic beads were eluted into a new collection tube. (b) Quality control checks were used on the DNA methylation data from the whole samples, Oragene samples, epithelial fractions, and immune fractions. Samples were dropped before the probes. A total of 38 fractions and 795,694 probes were used in this analysis.
Fresh Bovine Serum Albumin (BSA) Rinsing Buffer was prepared each processing day with final concentrations of 0.5% BSA Fraction V (Gibco, CAT# 15260037), 2 mM EDTA (Lonza, CAT# 51201), and DPBS. The BSA Rinsing Buffer sat on ice for a minimum of 10 minutes to degas. Cell pellets were resuspended in 3 mL BSA Rinsing Buffer. The count and percent viability of cells were recorded using the LUNA-FL™ Dual Fluorescence Cell Counter (Logos Biosystems; South Korea) with Acridine Orange/Propidium Iodide stain (Nexcelom Bioscience, CAT# CS201065 ML). Brightfield images of the samples were also taken using an EVOS-XL microscope (Advanced Microscopy Group; Bothell, Washington). A 500 µL aliquot of washed saliva was removed and stored on ice as the ‘Whole’ saliva fraction.
The remaining washed sample (~2.5 mL) was passed through a 30 µm filter to capture large epithelial cells. Following filtration, the original collection tube was washed using 1 mL BSA Rinsing Buffer, which was also passed through the filter. The filter was washed with an additional 1 mL BSA Rinsing Buffer. The cells captured in the 30 µm filter were rinsed off into a 50 mL tube (Corning, CAT# 14–432-22), which we term the epithelial fraction, using 2 mL BSA Rinsing Buffer. An additional 5 mL BSA Rinsing Buffer was added to the epithelial fraction and the tube temporarily stored on ice.
The small cell filtrate was filtered through a second 30 µm filter into a new tube to remove any remaining epithelial cells or cellular aggregates. The first filtrate tube and filter were washed again using 1 mL BSA Rinsing Buffer each. The filtered small cells were prepared for magnetic bead antibody selection according to the Miltenyi instructions. We used magnetic separation with CD45 MicroBeads (Miltenyi, CAT# 130–045-801) to obtain one fraction of CD45+ immune cells and one of CD45- discard. The bead-sorted immune fractions were centrifuged at 350 g for 5 minutes at 4°C. The whole and epithelial fractions were centrifuged at 500 g for 5 minutes at 4°C. At the end of the sample processing, there were three fractions per sample: Whole, epithelial, and immune. In addition, there were six Oragene kit samples of whole saliva.
Cell lysis, DNA extraction & quantitation, DNA methylation measurement
Large keratinocyte epithelial cells present in saliva were difficult to lyse and particular care was taken to prepare those cells. Whole, epithelial, immune, and Oragene fractions were resuspended in 500 µL Buffer ATL (Qiagen, CAT# 69,504). They were added to tissue disruptor bead tubes (MP Biomedicals, CAT# 116913100). The FastPrep-24 Tissue and Cell Homogenizer (MP Biomedicals; Irvine, CA) were used twice sequentially at a speed of 6.0 m/s for 35 seconds with the QuickPrep Adapter. The samples rested on ice for 5 minutes between shakes. To inactivate proteins, 20 µL proteinase K (Qiagen, CAT# 69504) was added to all samples. Whole, epithelial, and immune fractions were incubated on a heat block for at least 1 hour at 56°C. The Oragene fractions were heated for 2 hours according to the DNA Genotek prepIT protocol[24].
Genomic DNA was extracted from lysed cells using the DNeasy Blood & Tissue Kit (Qiagen, CAT# 69504) following the manufacturer’s protocol. DNA was eluted using two rounds of 50 µL Buffer AE each. We processed 72 total fractions (22 whole, 22 epithelial, 22 immune, and 6 Oragene kits) for DNA extraction.
Nucleic acids were quantified using the NanoDrop 2000 c (Thermo Scientific; Waltham, MA). Samples with a minimum of 250 ng nucleic acid (22 whole, 22 epithelial, 22 immune, and 5 Oragene samples) were submitted to the Epigenomics Core at the University of Michigan for analysis using the Infinium MethylationEPIC BeadArray (Illumina, CAT# WG-317-1003). The Epigenomics Core used a Qubit 2.0 Fluorometer (Life Technologies; Carlsbad, CA) to measure the DNA concentration. Samples with a minimum 200 ng DNA were used for DNA methylation measurements. To examine the reliability of the DNA methylation measurements of the epithelial cells, duplicates of six epithelial fractions were run as technical replicates. The 71 samples which passed the minimum 200 ng DNA threshold were subjected to sodium bisulphite conversion and cleaning (EZ-96 DNA Methylation™ Kit, Zymo Research) according to the manufacturer’s instructions. Samples were then randomized and loaded onto a single MethylationEPIC BeadArray plate that contains probes to measure the DNA methylation at specific CpG sites. Fluorescence was measured using the iScan System (Illumina; San Diego, CA) at the Advanced Genomics Core at the University of Michigan.
DNA methylation data preprocessing
EPIC BeadArray IDAT image files were processed and control metrics were assessed using the ewastools package[25]. Background correction was performed using noob [26] in the minfi package[27]. Sex, predicted from the DNA methylation data, was compared to the survey demographic data. To ensure fractions derived from the same participant had the same genotype, SNPs were compared between fractions from each participant. Detection p-values were calculated to identify failed probes. Samples with >3% probes exceeding the detection p-value = 0.01 were dropped. For a preliminary estimate of the relative amounts of the epithelial and immune cell fractions in each sample, cell-type proportions were estimated for each sample using a reference panel generated from ENCODE and adult white blood cell data, implemented in ewastools. Based on this analysis, we excluded one epithelial cell fraction that was estimated to be >70% immune cells. In total, seven samples were excluded (control metrics n = 1, sex comparison n = 0, genotype comparison n = 2, probe detection n = 3, cell distribution n = 1) (Figure 1(b)). To quantify the variation in measurements, the mean centred Pearson correlations of beta values (ratio between 0 and 1 of methylated and unmethylated alleles) were calculated for the five remaining epithelial technical replicate pairs (r = 0.83, 0.88, 0.92, 0.94, 0.96). The paired sample from the technical replicates with the higher probe fail rate was excluded.
DNA methylation was measured at 866,091 sites. Following sample exclusion, probes with >5% of samples with detection p-values >0.01 were dropped (n = 10,298). Cross-reactive probes (n = 43,254) and sex chromosome probes (n = 19,681) were also dropped [28,29]. The quality controlled DNA methylation data contained 795,694 probes from 18 epithelial, 20 immune, 18 whole saliva, and 4 Oragene samples (Figure 1(b)).
Statistical and bioinformatic analysis
We calculated sample descriptive statistics on demographic and laboratory measures. For continuous variables (age, cell count, cell viability, sample volume), minimum, maximum median, and mean were calculated. For categorical variables (sex, race, illness status), count and frequency were provided.
To visualize DNA methylation distributions by cell type, density plots were constructed. To summarize variations in the DNA methylation data, principal component analysis was conducted. The principal components explaining cumulatively at least 90% of variance in the sample were examined. Principal components were tested for association with demographic and laboratory covariates using ANOVA tests for categorical variables and Pearson correlation tests for continuous variables.
To test for differences in DNA methylation between all 18 epithelial and 20 immune cell samples, unpaired t-tests were used at each DNA methylation site. For a sensitivity analysis, we conducted a paired t-test between the 17 pairs of epithelial and immune cell fractions at each DNA methylation site. To account for multiple comparisons and identify probes that were significantly different by cell type, we used the Bonferroni significance level (p < 6.28 × 10−8) threshold calculated as the alpha level of 0.05 divided by 795,694 probes. Among the significant probes, the average methylation at each probe was calculated and plotted in a histogram. The 500 most statistically different probes by p-values were plotted in a heatmap using unbiased hierarchical clustering to group samples by similarity in probe profiles. Global DNA methylation was calculated for each sample by averaging DNA methylation across all 795,694 probes. Linear regression was used to test the association between the categorical variable, cell type (exposure), and global DNA methylation (outcome). We tested the 10,000 most statistically different probes for enrichment in gene ontology biological processes using the missMethyl package[30]. Pathways with less than five annotated genes were excluded. Gene ontologies were constructed for both hypomethylated and hypermethylated probes.
To estimate saliva cell-type proportions in each sample using our saliva reference dataset, we integrated our new immune and epithelial sorted cell data into the ewastools package[25]. ewastools [31,32] is a convenient R package for Illumina DNA methylation array preprocessing including cell-type estimation, which has been available through GitHub since 2018 (https://github.com/hhhh5/ewastools). Reference datasets are normalized and processed prior to package integration, which greatly reduces computation time for the end user, as well as provides more consistent cell proportion estimates across different datasets. Cell types were then estimated using the Houseman algorithm [11] as applied by the estimateLC function with the constrained parameter.
To compare our new sorted saliva reference panel to datasets of similar cell types, we examined ENCODE epithelial cell data and adult white blood cell data [33–35]. The ENCODE dataset contained DNA methylation data derived from eleven epithelial tissues around the body (Supplemental Table 1) [33,35]. These eleven epithelial tissues matched the ones in the HEpiDISH package that used ENCODE data to estimate epithelial and fibroblast cell proportions, and used sorted adult white blood cells to estimate immune proportions[13]. In an exploratory analysis, we examined the fibroblast cell data from ENCODE and observed a median of 0% fibroblasts in our whole saliva samples (data not shown), and thus restricted our analysis to the ENCODE epithelial cells. The adult white blood cell DNA methylation data were derived from seven magnetic bead sorted cell types (neutrophils, CD4+ T cells, CD8+ T cells, B cells, eosinophils, monocytes, and natural killer cells) donated by six men[36]. We pre-processed the ENCODE and adult white blood cell data using the methods described above (Supplemental Figure 1). To understand the sample clustering, we conducted principal component analysis across all datasets.
To benchmark our new saliva DNA methylation cell-type reference dataset, we compared whole-saliva DNA methylation-based cell-type proportions estimated using our new saliva reference dataset to those estimated using ENCODE epithelial data and adult white blood cell data [33–35] (Supplemental Figure 2). From whole-saliva sample DNA methylation measures, we estimated cell-type proportions using both reference panels with the ewastools function estimateLC, constrained at zero and one. Next, we calculated the variance explained by the estimated cell-type proportions using linear regression at each probe and calculated the R2 values. Across all probes, we calculated the R2 median as the median variance of DNA methylation values explained by estimated cell-type proportions.
For sensitivity analysis, we compared the matched estimated cell proportions from Oragene and whole saliva samples from the same three people when both paired samples were available. In an exploratory analysis, to assess whether sick children (n = 3) had a different proportion of immune cells compared to healthy children, we conducted a two-sided t-test between the proportion of immune cells estimated in whole saliva samples from the sick and healthy children. To make a qualitative comparison of the reference panels, we estimated the cell proportions in the whole saliva samples using the ENCODE and adult white blood cell reference panel as well as our new saliva reference panel, both implemented in ewastools. To compare the immune proportion estimates from each reference panel, we calculated a Pearson correlation and a root mean square error (RMSE). We visualized the matched estimates with the sample brightfield image.
As a sensitivity analysis, to compare our saliva reference panel to the ENCODE and adult white blood cell reference panel, we split our saliva epithelial and immune fractions into training and testing subsets. We randomly selected 70% of the epithelial fractions (n = 12) and 70% of the immune fractions (n = 14) as our ‘training’ subset. We created a reference panel based on these 26 fractions and used it to estimate the cell proportions in the remaining 30% of fractions (n = 6 epithelial and n = 6 immune). Then, we used the ENCODE and adult white blood cell reference panel to estimate the cell proportions of the 12 ‘testing’ fractions. Finally, we compared the cell proportions estimated from the training subset reference panel with the cell proportions estimated using the ENCODE and adult white blood cell reference panel.
To compare our saliva reference panel and the ENCODE and adult white blood cell reference panel on independent adult and child saliva datasets and test the usability of our saliva reference panel on adult saliva samples, we downloaded publicly available data from the Genome Expression Omnibus accession numbers GSE111631 (adult) and GSE138279 (child) [13,37]. We processed the datasets using the same method as our saliva data (Supplemental Figure 3). We estimated the cell proportions of each sample using the ENCODE and adult white blood cell reference panel as well as our new saliva reference panel, both implemented in ewastools.
As an exploratory analysis, we estimated specific immune cell types in the whole saliva samples, using the adult white blood cell reference panel[36], implemented in ewastools [25]. The distribution of these immune cell types in whole saliva was then compared to the expected ranges in healthy pediatric peripheral blood [38–43].
All of our data are currently available on the Genome Expression Omnibus (GSE #GSE147318), ExperimentHub (EH4539, EH4540), and Bioconductor (package name: BeadSorted.Saliva.EPIC), so other researchers can use our reference panel with the cell-type estimation method of their choice. Saliva cell-type estimation can be implemented directly through the ewastools package or with any other cell-type estimator package. All DNA methylation data preprocessing and analyses were conducted in R statistical software (version 3.6). Code to reproduce preprocessing and analyses is available (https://github.com/bakulskilab).
Results
Study sample description
Saliva samples were collected from 22 participants. One participant was excluded due to insufficient DNA for measurement at all fractions. Of the 21 participants with DNA methylation data, 15 were male and 10 were non-Hispanic white (Table 1). Three of the 21 participants were reported to be sick at the time of sample collection. The mean age was 11.8 years with a range of 7.9–16.9 years. From each participant, we collected a mean 3.1 mL of unstimulated saliva. Collected saliva cell counts ranged from 720,000 to 34,000,000 cells per whole sample. Cell viability ranged from 5.1% to 81.5% and the median viability was 69.2%. Following microscopic evaluation, large, flat, and geometric epithelial cells were easily differentiated from the small, round immune cells (Figure 2). These microscopy images highlight the interindividual heterogeneity in cell size, shape, and proportions in saliva samples.
Table 1.
Descriptive statistics for participants (n = 21). Saliva was collected and 18 whole saliva samples, 18 epithelial fractions, 20 immune fractions, and 4 Oragene kit whole samples passed DNA methylation quality control measures
| Mean (sd) | Count (%) | Range | |
|---|---|---|---|
| Sex (male) | 15 (71.4) | ||
| Race | |||
| White | 10 (47.6) | ||
| Black | 1 (4.8) | ||
| Biracial | 10 (47.6) | ||
| Sick | 3 (14.3) | ||
| Age (years) | 11.8 (2.7) | (7.9, 16.9) | |
| Cell count+ | 6.3 × 106 (9.8 × 106) | (7.2 × 105, 3.4 × 107) | |
| Viability (%)+ | 69.2 (16.7) | (5.1, 81.5) | |
| Volume (mL)+ | 3.5 (1.4) | (1.9, 6.5) |
+Median used due to non-normal distribution
Figure 2.
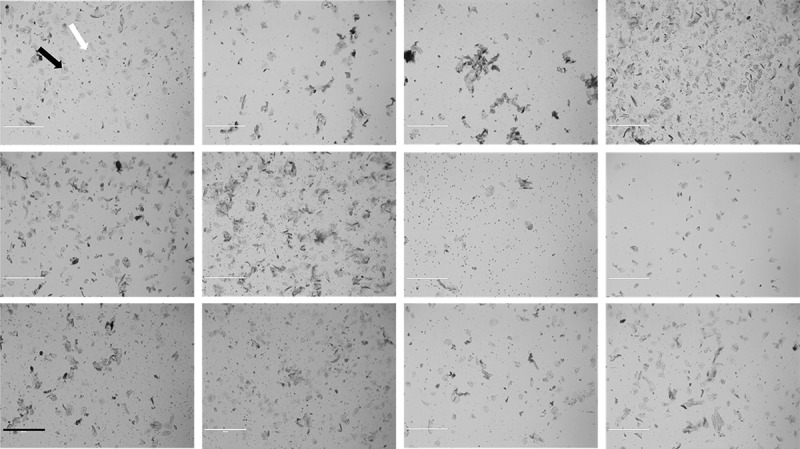
Representative sample of saliva diversity from 12 participants. Images were taken using a brightfield microscope at 4x (EVOS-xl) following the resuspension in BSA Rinsing Buffer. The black arrow points to an epithelial cell and the white arrow points to an immune cell. Scale bar = 400 μm.
Assessment of differences in DNA methylation between saliva cell types
Following quality control, DNA methylation from 18 epithelial cell fractions, 20 immune cell fractions, 18 whole samples, and four Oragene kits were included in this analysis. A total of 795,694 probes were analysed (Figure 1(b)). The mean global DNA methylation of the immune cells was 57.5% (standard error: 0.2%). The mean global DNA methylation of epithelial cells was 53.2% (standard error: 0.2%) (Supplemental Figure 4). Immune cells had 4.3% (standard error: 0.3%) higher mean global methylation compared to the epithelial cells (p = 2 × 10−16). The density plot reflects the expected bimodal DNA methylation distribution as measured by probes (Figure 3(a)). A principal component analysis of the immune and epithelial cell DNA methylation data showed that the first principal component of the DNA methylation data explained 80.8% of the variance and was associated with cell type (p = 1.7 × 10−15) (Figure 3(b)). The second principal component explained 4.6% of the variance in the DNA methylation data and was associated with participant age (p = 0.004), saliva sample cell viability (p = 0.004), and participant sex (p = 0.03).
Figure 3.
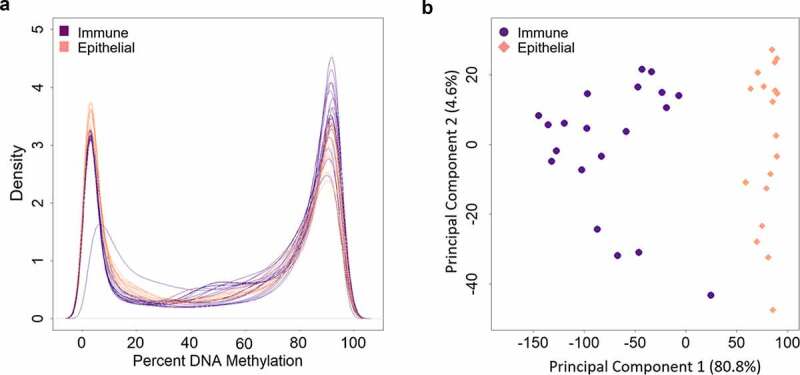
Density plot and principal component analysis of DNA methylation measures painted cell types. A) Density plots of DNA methylation of all CpG sites, displaying DNA methylation distributions by cell type (immune cells in purple and epithelial cells in pink). Each line represents one cell fraction. Beta values were converted to percentages. B) DNA methylation principal components one and two, colored by immune cells in purple and epithelial cells in pink. Principal component 1 explained 80.8% of the variance in the DNA methylation data. Principal component 2 explained 4.6% of the variance in the DNA methylation data.
DNA methylation levels at each site were compared between epithelial and immune cells using unpaired t-tests. We identified 181,577 (22.8% of all probes) differentially methylated sites between epithelial and immune cells (p < 6.28 × 10−8). In a paired t-test analysis of 17 epithelial cells and 17 immune cells, we identified 111,922 (14.1% of all probes) differentially methylated sites (p < 6.28 × 10−8). Among the genome-wide significantly differentially methylated sites, 72.2% had higher DNA methylation in immune cells relative to epithelial cells (Figure 4(a)). Among the differentially methylated sites, the average magnitude of DNA methylation difference was 32.4%. A 27.8% of sites were hypomethylated in immune cells relative to epithelial cells. The highest magnitude differences were observed at cg07110356 in the MPO gene (Myeloperoxidase) with 70.2% higher methylation in epithelial cells, compared to immune cells (p = 8.2 × 10−18) (Supplemental Table 2) and at cg17804342 in RGS10 gene (Regulator of G Protein Signalling 10) with 68.6% higher DNA methylation in immune cells compared to epithelial cells (p = 7.6 × 10−20). The 500 most statistically differentially methylated sites between immune and epithelial cells were analysed by unbiased hierarchical clustering and visualized by heatmap (Figure 4(b)). Saliva sample fractions clustered by cell type. Among these 500 sites, 93.6% had higher DNA methylation in immune cells relative to the epithelial cells (Supplemental Table 2).
Figure 4.
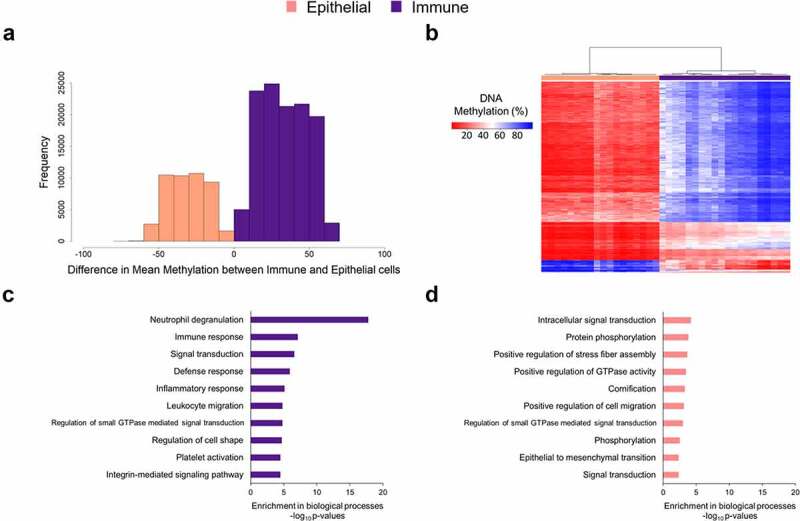
DNA methylation differences between immune and epithelial cell types. (a) Among DNA methylation sites differentially methylated between immune and epithelial cells (p < 10−8, 164,793 sites), histogram of the magnitude of DNA methylation difference. The x-axis is percent methylation, and the reference group was epithelial cells. The values were calculated as percent methylation of immune minus that of epithelial. (b) The top 500 most differentially methylated sites between immune and epithelial cells by p-value (t-test) are plotted in heatmap rows (red indicates lower DNA methylation, blue indicates higher DNA methylation). In heatmap columns, unbiased hierarchical clustering of samples was performed (immune fractions in purple and epithelial fractions in pink). (c) Bar chart of the gene ontology biological processes enriched (minimum p < 3.2 × 10−5) among genes hypomethylated in immune cells, relative to epithelial cells. (d) Bar chart of the gene ontology biological processes enriched (minimum p < 4.7 × 10−3) among genes hypomethylated in epithelial cells, relative to immune cells.
We tested for enriched gene ontology biological pathways in the differentially methylated sites between immune and epithelial cells. Sites with lower DNA methylation in immune cells were mapped to genes enriched for immune pathways such as neutrophil degranulation (p = 1.6 × 10−18), immune response (p = 7.6 × 10−8), and leukocyte migration (p = 1.5 × 10−5) (Figure 4(c)). Twelve of the pathways were enriched for hypomethylated sites (FDR < 0.05) (Supplemental Table 3). Although no epithelial cell pathways were significant (FDR < 0.05) (Supplemental Table 4), sites with lower DNA methylation in epithelial cells mapped to general cell activity pathways such as intracellular signal transduction (p = 6.3 × 10−5), protein phosphorylation (p = 1.6 × 10−4), and positive regulation of stress fibre assembly (p = 2.2 × 10−4) (Figure 4(d)). Cornification, a key process for hard palate formation, was also enriched for differentially methylated genes in epithelial cells (p = 5.2 × 10−4).
Cell proportion estimation
DNA methylation data from epithelial cell types in ENCODE and primary adult white blood cell samples were used to estimate saliva cell proportions. DNA methylation data from these samples together with our saliva samples analysed by principal component analysis revealed samples primarily clustered by cell type and study (Supplemental Figure 5). Principal component 1 explained 47.9% of the variation in the data and was associated with cell type (p = 1.8 × 10−50) and study (p = 4.3 × 10−27) variable. Principal component 2 explained 16.1% of the variation in the data and was also associated with cell type (p = 2.3 × 10−51) and study (p = 6.1 × 10−48). We first estimated cell proportions in our saliva DNA methylation data using a reference panel constructed from ENCODE epithelial cells and adult white blood cells, implemented in ewastools [33–35]. Our saliva epithelial cell fractions were estimated to be 92.0–100.0% epithelial cells (median: 100%, IQR: 84.3%) and the immune cell fractions were 22.3–92.3% immune cells (median: 59.7%, IQR: 29.7%) (Figure 5(a)). Our whole saliva samples were estimated to be 8.3–96.2% epithelial cells (median: 81.7%, IQR: 28.3%) and 3.8–91.7% immune cells (median: 18.3%, IQR: 28.3%) (Supplemental Figure 6a). Our Oragene saliva samples were estimated to be 23.2–70.4% epithelial cells (median: 35.2%, IQR: 22.9%) and 29.6–76.7% immune cells (median: 64.8%, IQR: 22.9%). Estimated cell percentages derived from the ENCODE and adult white blood cell reference panel explained a median of 28.2% of the variation in the whole saliva DNA methylation data (Supplemental Figure 7).
Figure 5.
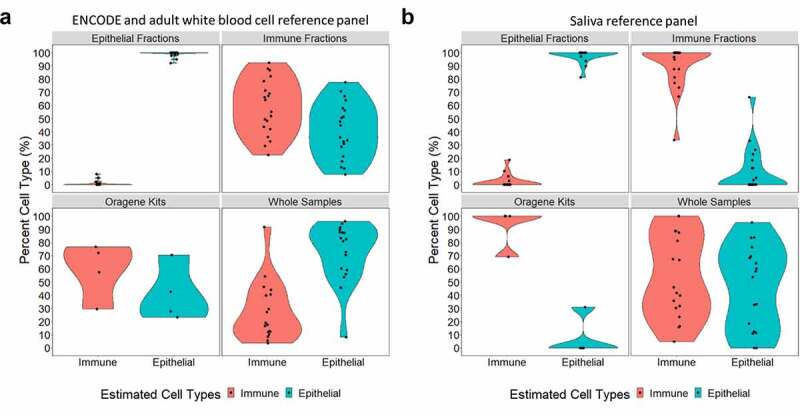
Cell-type percentages estimated in saliva samples from DNA methylation data, using two estimation methods. Violin plots are used to visualize the percent immune cells estimated in red and the percent epithelial cells estimated in blue. In both panels A and B, the upper left quadrant shows the percent cell types estimated in sorted epithelial samples (n = 18); the upper right quadrant shows the percent cell types estimated in sorted immune cell samples (n = 20), the lower left quadrant shows the percent cell types estimated in whole saliva collected in Oragene kits (n = 4); and the lower right quadrant shows the percent cell types estimated in whole saliva samples collected directly (n = 18). (a) Percent cell types were estimated using a reference panel generated from ENCODE epithelial cell DNA methylation data [29,31] and an adult white blood cell data[30], implemented through ewastools. (b) Percent cell types estimated using our new primary saliva reference panel implemented through ewastools[23].
We next estimated cell proportions in our saliva DNA methylation data using our new saliva reference data integrated into the ewastools package. The new primary saliva reference panel-derived cell proportions explained a median of 26.6% of the variation in the whole saliva sample DNA methylation data. Our saliva epithelial cell fractions were estimated to be 81.2–100.0% epithelial cells (median: 100.0%, IQR: 0.0%) (Figure 5(b)). Saliva immune cell fractions were 33.8–100.0% immune cells (median: 100.0%, IQR: 14.1%). Our whole saliva samples were estimated to be 0.0–95.1% epithelial cells (median: 56.1%, IQR: 55.3%) and 4.9–100.0% immune cells (median: 43.9%, IQR: 55.3%) (Supplemental Figure 6b). Our Oragene saliva samples were estimated to be 0.0–31.0% epithelial cells (median: 0.0%, IQR: 7.7%) and 69.0–100.0% immune cells (median: 100.0%, IQR: 7.7%). Whole saliva samples that had a high estimated proportion of immune cells correspondingly had a low estimated proportion of epithelial cells. For example, whole-saliva sample 19 had the highest estimated immune proportion and the lowest epithelial estimated proportion.
In an exploratory analysis, using our new saliva reference panel, we compared the estimated immune cell proportions in three Oragene saliva samples to three matched whole saliva samples from the same participants. The Oragene saliva samples had an average of 25.7% higher estimated immune cell proportions compared to the whole saliva samples (Supplemental Figure 6 c). Three children were reported to be sick at the time of saliva sample collection. We did not observe a significant difference in the estimated proportion of immune cells in the whole saliva samples between the sick and the healthy children (p = 0.6) (Supplemental Figure 6d).
We compared whole-saliva DNA methylation cell proportions estimated using the ENCODE epithelial and adult white blood cell reference panel to cell proportions estimated using our new primary saliva reference panel. Immune cell proportions estimated by the two methods were highly correlated (r = 0.90), but also had a high RMSE of 30.1 (Figure 6). The new saliva reference panel estimated a higher mean immune proportion (53.7%) compared to the ENCODE epithelial and adult white blood cell reference panel (27.4%). We visually compared the whole saliva cell proportions estimated from both reference panels to the brightfield images of the whole saliva samples (Supplemental Figure 8).
Figure 6.
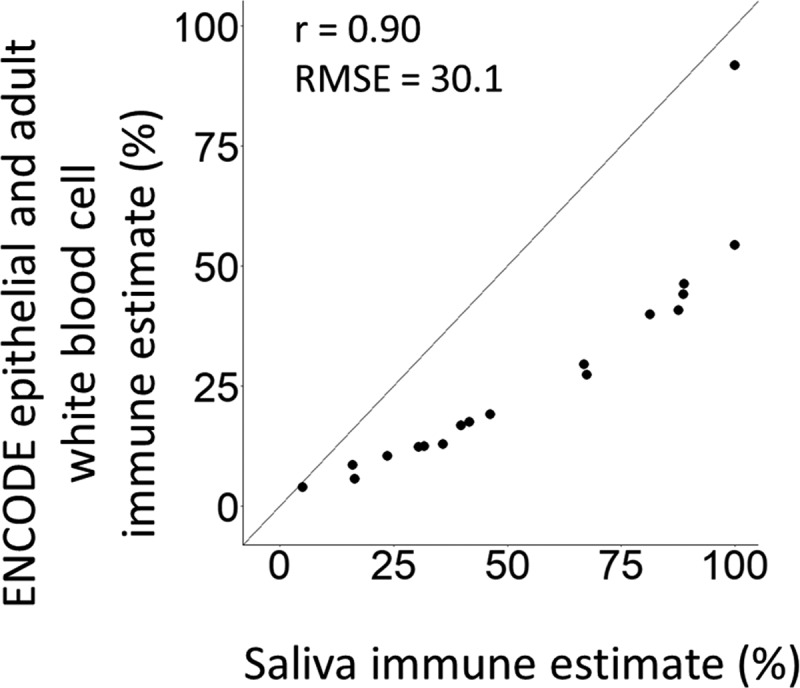
ENCODE and adult white blood cell reference panel vs saliva reference panel percent estimates from whole saliva samples. The x-axis represents the percent immune cells that was estimated using our saliva reference panel. The y-axis represents the percent immune cells that was estimated using the ENCODE and adult white blood cell reference panel. Both constrained estimates were conducted using the reference panels integrated into ewastools.[23].
To further evaluate the performance of our new saliva reference panel, we used 70% of our saliva fractions to create a new training reference panel and tested cell-type estimation on the remaining 30% to estimate the cell-type proportions (Supplemental Figure 9). We compared the cell proportions estimated using the ENCODE epithelial and adult white blood cell reference panel to the cell proportions estimated using the 70% training reference panel. Using the ENCODE epithelial and adult white blood cell reference panel, the epithelial fractions were estimated to be a median of 100.0% (IQR: 0.4%) epithelial (Supplemental Figure 9a), and using the saliva reference panel, the epithelial fractions were estimated to be a median of 99.7% (IQR: 1.5%) epithelial (Supplemental Figure 9b). Using the ENCODE and adult white blood cell reference panel, the immune fractions were estimated to be a median of 45.8% (IQR: 18.5%) immune, and using our new saliva reference panel, the immune fractions were estimated to be a median of 77.6% (IQR: 24.3%) immune.
We also estimated the cell proportions in an adult saliva dataset and an independent child saliva dataset using the ENCODE epithelial and adult white blood cell reference panel as well as our new saliva reference panel (Supplemental Figure 10). Using the ENCODE epithelial and adult white blood cell reference panel, the adult saliva samples were estimated to be 3.4–51.9% epithelial cells (median: 17.2%, IQR: 11.1%) and 48.1–96.6% immune cells (median: 82.8%, IQR: 11.1%) (Supplemental Figure 10a). Using the new saliva reference panel, the adult saliva samples were estimated to be 0.0–6.9% epithelial cells (median: 0.0%, IQR: 0.0%) and 93.1–100.0% immune cells (median: 100.0%, IQR: 0.0%) (Supplemental Figure 10b). Using the ENCODE epithelial and adult white blood cell reference panel, the independent child saliva samples were estimated to be 9.1–78.6% epithelial cells (median: 36.1%, IQR: 16.2%) and 21.4–90.9% immune cells (median: 63.9%, IQR: 16.2%) (Supplemental Figure 10 c). Using the new saliva reference panel, the independent child saliva samples were estimated to be 0.0–55.0% epithelial cells (median: 0.2%, IQR: 8.2%) and 45.0–100.0% immune cells (median: 99.8%, IQR: 8.2%) (Supplemental Figure 10d).
We estimated immune cell subtypes in saliva using the adult white blood cell reference panel [36] and compared the proportions to literature ranges of immune cells in pediatric peripheral blood (no standard saliva cell proportions were available). In the saliva immune cell fractions, Oragene samples, and whole samples, the range of estimated granulocytes was wider than the normal range in blood (Supplemental Figure 11). Saliva immune fractions were estimated to be 29–99% granulocytes, while in pediatric blood, the normal range was 38–72%. The estimated range of granulocytes in the Oragene samples was 40–85%. The estimated range of granulocytes in the whole samples was 17–99%. Saliva estimated CD4+ T-cell proportions had a similar range to pediatric blood, though saliva estimates were lower. For example, saliva immune fractions were estimated to be 0–30% CD4+ T-cells, while in pediatric blood the normal range was 31–52%. In all saliva samples, no CD8+ T-cells were estimated, though the normal range of CD8+ T-cells in paediatric blood was 18–35%. In general, the estimated range of immune cell proportions in saliva was more variable than in blood.
Discussion
Saliva is a commonly used biosample for epigenetic epidemiology studies, and especially in pediatric studies. A saliva-specific cell-type reference panel was critically needed to estimate cell-type proportions from bulk saliva DNA methylation data in children. This gap was particularly salient in light of the substantial interindividual heterogeneity in salivary cell-type composition (highlighted in the microscopy images in Figure 2). We collected whole saliva samples and Oragene kits from children and sorted the whole samples into immune and epithelial fractions based on a combination of size and antibody-based sorting, and the DNA methylation profiles of each were measured using the Illumina MethylationEPIC BeadChip. We identified substantial differences in DNA methylation patterns between the sorted cell fractions with sites enriched for logical biological pathways (e.g., immune pathways in the immune fraction, cornification in the epithelial fraction). Our datasets were integrated into the publicly available ewastools [25] and produced a saliva reference panel R data package (BeadSorted.Saliva.EPIC) [44] to facilitate cell-type proportion estimation. Future saliva DNA methylation studies will be able to easily integrate this reference panel for cell-proportion estimation into their current analytic workflow.
We compared the performance of our saliva reference panel to a reference panel we generated using ENCODE epithelial cells and adult white blood cell data. The ENCODE and adult white blood cell reference panel, on average, explained slightly more of the variance in saliva DNA methylation data compared to our saliva reference panel, implemented in ewastools. Because the surface-level oral mucosa only contains a few types of epithelial cells[19], the higher variance explained by the ENCODE and adult white blood cell reference panel could be a result of the larger number and range of epithelial cell types included from ENCODE. There was a strong, positive correlation (r = 0.90) between the whole saliva cell proportion estimates using our reference panel and estimates in the same samples using the ENCODE epithelial cells and adult white blood cells reference panel. Our saliva reference panel estimates a larger proportion of immune cells in all whole saliva samples compared to the ENCODE and adult white blood cell reference panel. Unfortunately, there is no existing external validation dataset of cell counts in saliva to compare our new saliva reference panel to the ENCODE epithelial and adult white blood cell reference panel data. However, using the new saliva reference panel, we observed a more dynamic range in the cell-type proportions estimated in whole saliva samples (Supplemental Figure 6a-b), which is consistent with the highly variable biosamples (Supplemental Figure 8). We further recommend the use of the new saliva reference panel for pediatric saliva epigenetic studies, as reference panels based on primary site-specific tissues may be more biologically relevant.
There was substantial interindividual variability in the proportions of cell types in saliva. We estimated our whole samples to have a median of 56.1% epithelial cells (Figure 5(b)), with an interquartile range of 55.3%. Similarly, another study found an interquartile range of epithelial cell saliva in children to be 46.3%, and the cell-type variability in saliva was higher in children compared to adults[45]. Large interindividual differences in saliva sample cell proportions can drive the DNA methylation profile and therefore influence results[22]. Together, these findings emphasize the importance of understanding the proportions of cell types in DNA methylation analyses, especially when using saliva.
We observed that saliva from children had wider ranges of granulocytes, CD4+ T cells, and monocytes compared to the normal blood ranges for children. Granulocytes had the highest estimated proportion of immune cells in our saliva samples, similar to a study that manually counted segmented immune cells (granulocytes) in saliva from children[45]. No CD8+ T-cells were predicted in any saliva sample from the present study (Supplemental Figure 11). Using flow cytometry to sort saliva immune cells from three participants, T-cells ranged from 0.8% to 1.2%, but they did not separate out CD8+ cells[18]. Both CD4+ and CD8+ T-cells have been identified in salivary glands [46] which suggests that CD8+ T-cells are present in oral cavity tissue, though they may not migrate into saliva. Variability observed in saliva cell types from relatively healthy children could influence observed differences in DNA methylation between groups.
Many large saliva DNA methylation studies use Oragene kits to collect biosamples, including the independent child saliva dataset we analysed. Oragene kits provide long storage time, with high DNA quality and yield[47]. From our small comparison (n = 3) of matched whole samples collected in a tube without preservatives and samples collected in Oragene kits, we estimated higher immune cell proportions in the Oragene kit samples. Both our Oragene samples and the Oragene samples from the independent child saliva dataset were estimated to contain high immune cell proportions (Supplemental Figure 6c and Supplemental Figure 10d). These preliminary findings suggest that the Oragene kits may enrich for immune cell DNA. In our experience, keratinized saliva epithelial cells are considerably more resistant to lysis than immune cells. It is possible that epithelial cells are inadequately lysed, enriching for immune cell DNA, and potentially biasing DNA methylation measures. A larger and specifically designed study is needed to explore this trend.
Although our new reference panel was created using saliva samples from children, we showed that the reference panel can also be used to estimate cell-type proportions in adult saliva samples (Supplemental Figure 10). The magnitude of age-related DNA methylation changes is much smaller than the magnitude of DNA methylation differences between cell types [10,48]. Our new reference panel estimated a high proportion of immune cells in the adult saliva samples. These samples were collected in tubes that contained a preservative fluid. Future studies can investigate differences in DNA methylation measures by sample collection method and DNA extraction process.
Our study had a number of limitations. We isolated two main saliva cell types: epithelial and immune cells. In each of these types, there were likely several subtypes of cells that we grouped into one category. For example, there are several subtypes of epithelial cells that cover the oral surfaces[19]. Papanicolaou staining of buccal samples from children have identified three main epithelial cell types: intermediate squamous, non-keratinous, and keratinous superficial squamous[45]. Surface markers for flow cytometry sorting are not well characterized by normal oral epithelial cells. We initially attempted to isolate epithelial cells using flow cytometry with an antibody for epithelial cell adhesion molecule (EpCAM), a typical surface marker on epithelial cells [49–51], but oral epithelial cells do not appear to express EpCAM (data not shown). In addition, we found that the large size of saliva epithelial cells, which can be up to 100 µm in diameter, blocked the microfluidic tubes of the flow cytometry and droplet-based single-cell instruments. We also attempted nuclear isolation for single nuclei sequencing, but the recommended detergent was insufficient to lyse the epithelial cells, possibly due to their highly keratinized nature. Future studies may use other methods to isolate and profile different epithelial populations. When a gold standard saliva cell counting method is developed, a future study could compare the results of our DNA methylation-based reference panel to other methods. Although we combined saliva immune cells into one category as well, there are existing reference panels from blood that can be used to predict leukocyte proportions[36]. Future studies may generate a saliva cell-type reference panel in different study populations, including adults or participants from outside the United States. This would help inform the generalizability of these findings in US children.
Our study also has a number of strengths. While our participants were not a random sample, we included saliva samples from 21 children with 18 epithelial cell samples and 20 immune cell samples. To test for differences in cell-type proportion estimates by sampling method, we collected saliva in tubes without preservation fluid and in matched Oragene kits. During the DNA extraction procedure, we used a tissue disruptor to ensure that we lysed the epithelial cells. We identified 181,577 significantly different (p < 6.28 × 10−8) DNA methylation sites between epithelial and immune cells, highlighting that the separation procedure isolated biologically distinct cellular populations. For comparison, the commonly used adult blood reference panel based on six participants observed 37,837 sites that differentiated at least one leukocyte cell type [12,34]. We also included five technical replicates of epithelial cell fractions. The average mean centred correlation between the replicates was 0.91. The epithelial fractions, immune fractions, whole samples, and Oragene kit samples were randomized on the slides and all run on one plate to reduce batch effects. Using our new saliva reference panel, estimation of cell type proportions in saliva samples from DNA methylation arrays is improved relative to currently available cell type estimation methods (Figure 6). To improve generalizability across the widest number of available Illumina DNA methylation datasets[52], our cell-type estimation was restricted to sites overlapping between the 450k and EPIC arrays. Based on individual study needs, investigators can elect to include all sites in the EPIC array. Although we use ewastools to estimate cell counts in this paper, our reference panel can be implemented with any cell-type estimator package or software. This is the first primary saliva reference panel for cell-type proportion deconvolution in saliva. This saliva reference panel will improve epigenetic studies by providing a tool that is both appropriate and easy to use for estimating cell-type proportions for use in regression modelling approaches.
Conclusions
DNA methylation differences measured in saliva are likely influenced by the relative proportions of cells present in the sample[53]. Epithelial and immune cells from saliva have distinct DNA methylation profiles, and there is substantial interindividual heterogeneity in the cell proportions in saliva. Changes in the proportion of epithelial to immune cells will influence whole-saliva DNA methylation measurements. Our reference panel and accompanying R package (BeadSorted.Saliva.EPIC) [44] provide a new, more biologically relevant, method to better account for cell-type proportions in pediatric saliva DNA methylation research, which is an important step to account for cell-type effects in epigenetic studies.
Supplementary Material
Funding Statement
We thank the University of Michigan Epigenomics Core and Advanced Genomics Core for DNA methylation measures. Support for this research was provided by the National Institute of Environmental Health Sciences (grants P30 ES017885, R01 ES028802, U01 ES026697, R35 ES031686, R01 ES025531, R01 ES025574), the National Institute on Aging (grants R01 AG067592, R01 AG060110-01), the National Institute on Minority Health and Health Disparities (grants R01 MD011716, R01 MD013299), and the National Institutes of Health Office of the Director (grants UG3 OD023285, UH3 OD023285).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. [DOI] [PubMed] [Google Scholar]
- [2].Huang K, Fan G.. DNA methylation in cell differentiation and reprogramming: an emerging systematic view. Regen Med. 2010;5(4):531–544. [DOI] [PubMed] [Google Scholar]
- [3].Khavari DA, Sen GL, Rinn JL. DNA methylation and epigenetic control of cellular differentiation. Cell Cycle. 2010;9(19):3880–3883. [DOI] [PubMed] [Google Scholar]
- [4].Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. [DOI] [PubMed] [Google Scholar]
- [5].Liu L, Wylie RC, Andrews LG, et al. Aging, cancer and nutrition: the DNA methylation connection. Mech Ageing Dev. 2003;124(10–12):989–998. [DOI] [PubMed] [Google Scholar]
- [6].Zeilinger S, Kühnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5). DOI: 10.1371/journal.pone.0063812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Campbell KA, Colacino JA, Bakulski KM. Cell types in environmental epigenetic studies: biological and epidemiological frameworks. Curr Environ Heal Reports. 2020;7(3):185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Montaño CM, Irizarry RA, Kaufmann WE, et al. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 2013;14(8):R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zheng SC, Breeze CE, Beck S, et al. Identification of differentially methylated cell types in epigenome-wide association studies. Nat Methods. 2018;15(12):1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bakulski KM, Feinberg JI, Andrews SV, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11(5):354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zheng SC, Webster AP, Dong D, et al. A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics. 2018;10(7):925–940. [DOI] [PubMed] [Google Scholar]
- [14].Langie SAS, Szarc Vel Szic K, Declerck K, et al. Whole-genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS One. 2016;11(3):e0151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hearn NL, Coleman AS, Ho V, et al. Comparing DNA methylation profiles in saliva and intestinal mucosa. BMC Genomics. 2019;20(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bruinsma FJ, Joo JE, Wong EM, et al. The utility of DNA extracted from saliva for genome-wide molecular research platforms. BMC Res Notes. 2018;11(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Walker D. Oral mucosal immunology: an overview. Ann Acad Med. 2004;33(4):27S–30S. [PubMed] [Google Scholar]
- [18].Vidović A, Vidović Juras D, Vučićević Boras V, et al. Determination of leucocyte subsets in human saliva by flow cytometry. Arch Oral Biol. 2012;57(5):577–583. [DOI] [PubMed] [Google Scholar]
- [19].Groeger S, Meyle J. Oral mucosal epithelial cells. Front Immunol. 2019;10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Eckhart L, Lippens S, Tschachler E, et al. Cell death by cornification. Biochim Biophys Acta Mol Cell Res. 2013;1833(12):3471–3480. [DOI] [PubMed] [Google Scholar]
- [21].Aps JKM, Van Den Maagdenberg K, Delanghe JR, et al. Flow cytometry as a new method to quantify the cellular content of human saliva and its relation to gingivitis. Clin Chim Acta. 2002;321(1–2):35–41. [DOI] [PubMed] [Google Scholar]
- [22].Langie SAS, Moisse M, Declerck K, et al. Salivary DNA methylation profiling: aspects to consider for biomarker identification. Basic Clin Pharmacol Toxicol. 2017;121(Suppl Suppl 3):93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Titus AJ, Gallimore RM, Salas LA, et al. Cell-type deconvolution from DNA methylation: a review of recent applications. Hum Mol Genet. 2017;26(R2):R216–R224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].DNA Genotek Inc . Laboratory protocol for manual purification of DNA from 0.5 ML of sample; 2018. [cited 2020 Apr 16]. Available from: www.dnagenotek.com
- [25].Just AC, Heiss JA. ewastools: EWAS Tools; 2018. Available from: https://github.com/hhhh5/ewastools/blob/master/DESCRIPTION
- [26].Triche TJ, Weisenberger DJ, Van Den Berg D, et al. Low-level processing of Illumina Infinium DNA Methylation Beadarrays. Nucleic Acids Res. 2013;41(7):e90-e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hansen KD IlluminaHumanMethylationEPICanno.ilm10b2.hg19: annotation for Illumina’s EPIC methylation arrays. 2016. R package version 0.6.0.
- [30].Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2015;32(2):286–288. [DOI] [PubMed] [Google Scholar]
- [31].Heiss JA, Just AC. Improved filtering of DNA methylation microarray data by detection p values and its impact on downstream analyses. Clin Epigenetics. 2019;11(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heiss JA, Just AC. Identifying mislabeled and contaminated DNA methylation microarray data: an extended quality control toolset with examples from GEO. Clin Epigenetics. 2018;10(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Davis CA, Hitz BC, Sloan CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46(D1):D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Reinius LE, Acevedo N, Joerink M, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7(7):e41361. Ting AH, ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Reinius LE, Acevedo N, Joerink M, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7(7):e41361–e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McEwen LM, O’Donnell KJ, McGill MG, et al. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc Natl Acad Sci U S A. 2020;117(38):23329–23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mayo Clinic Laboratories . Complete blood count normal pediatric values. [cited April 9 2020]. Available from: http://a1.mayomedicallaboratories.com/webjc/attachments/110/30a2131-complete-blood-count-normal-pediatric-values.pdf
- [39].Comans-Bitter WM, De Groot R, Van Den Beemd R, et al. Immunophenotyping of blood lymphocytes in childhood reference values for lymphocyte subpopulations. J Pediatr. 1997;130(3):388–393. [DOI] [PubMed] [Google Scholar]
- [40].Shearer WT, Rosenblatt HM, Gelman RS, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the pediatric AIDS clinical trials group P1009 study. J Allergy Clin Immunol. 2003;112(5):973–980. [DOI] [PubMed] [Google Scholar]
- [41].Duchamp M, Sterlin D, Diabate A, et al. B-cell subpopulations in children: national reference values. Immun Inflamm Dis. 2014;2(3):131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pediatric Center - Penn State Hershey Medical Center . Blood differential test; Published January 29, 2019; [cited 2020 Apr 9]. Available from: http://pennstatehershey.adam.com/content.aspx?productId=112&pid=1&gid=003657
- [43].LabCorp . T- and B-lymphocyte and natural killer cell profile; [cited 2020 April 9]. Available from: https://www.labcorp.com/tests/505370/t-and-b-lymphocyte-and-natural-killer-cell-profile
- [44].Fisher JD, Middleton LYM, Bakulski KM (2020). BeadSorted.Saliva.EPIC: Illumina DNA methylation data on sorted saliva cell populations. R package version 0.0.900. [Google Scholar]
- [45].Theda C, Hwang SH, Czajko A, et al. Quantitation of the cellular content of saliva and buccal swab samples. Sci Rep. 2018;8(1):6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Thom JT, Weber TC, Walton SM, et al. The salivary gland acts as a sink for tissue-resident memory CD8+ T cells, facilitating protection from local cytomegalovirus infection. Cell Rep. 2015;13(6):1125–1136. [DOI] [PubMed] [Google Scholar]
- [47].Nunes AP, Oliveira IO, Santos BR, et al. Quality of DNA extracted from saliva samples collected with the OrageneTM DNA self-collection kit. BMC Med Res Methodol. 2012;12(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Farré P, Jones MJ, Meaney MJ, et al. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin. 2015;8(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Maestre-Batlle D, Pena OM, Hirota JA, et al. Novel flow cytometry approach to identify bronchial epithelial cells from healthy human airways. Sci Rep. 2017;7(1):42214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Trzpis M, McLaughlin PMJ, De Leij LMFH, et al. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol. 2007;171(2):386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schmelzer E, Reid L. EpCAM expression in normal, non-pathological tissues. Front Biosci. 2008;13(13):3096–3100. [DOI] [PubMed] [Google Scholar]
- [52].Maden SK, Thompson RF, Hansen KD, et al. Human methylome variation across Infinium 450K data on the Gene Expression Omnibus. bioRxiv. 2020. January :2020.11.17.387548. DOI: 10.1101/2020.11.17.387548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2). DOI: 10.1186/gb-2014-15-2-r31 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.