

Abstract
Biofilms, surface-attached communities of bacterial cells, are a concern in health and in industrial operations because of persistent infections, clogging of flows, and surface fouling. Extracellular matrices provide mechanical protection to biofilm-dwelling cells as well as protection from chemical insults, including antibiotics. Understanding how biofilm material properties arise from constituent matrix components and how these properties change in different environments is crucial for designing biofilm removal strategies. Here, using rheological characterization and surface analyses of Vibrio cholerae biofilms, we discover how extracellular polysaccharides, proteins, and cells function together to define biofilm mechanical and interfacial properties. Using insight gained from our measurements, we develop a facile capillary peeling technology to remove biofilms from surfaces or to transfer intact biofilms from one surface to another. We show that our findings are applicable to other biofilm-forming bacterial species and to multiple surfaces. Thus, our technology and the understanding we have developed could potentially be employed to characterize and/or treat biofilm-related infections and industrial biofouling problems.
Keywords: Biofilms, fracture, adhesion energy, rheology, antibiotics
Graphical Abstract
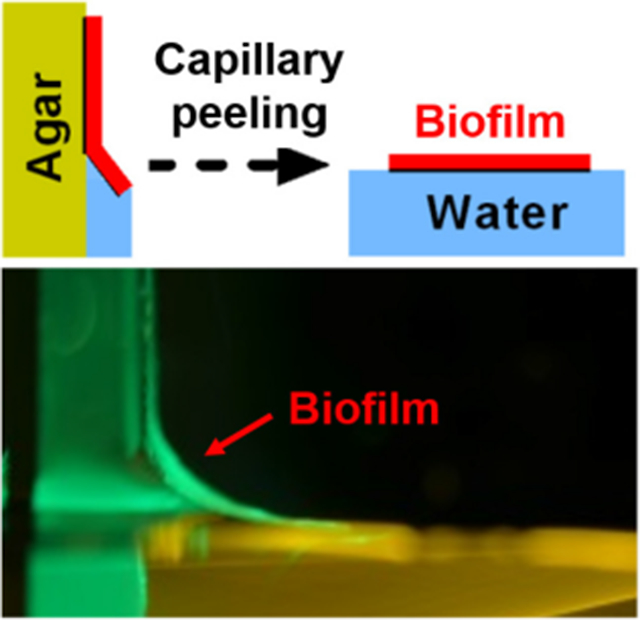
The table of contents entry:
Bacterial biofilms cause serious problems in health and in industrial operations. Using rheological characterization and surface analyses of Vibrio cholerae biofilms, we discover how extracellular polysaccharides, proteins, and cells function together to define biofilm mechanical and interfacial properties. We further develop a facile capillary peeling technology to remove biofilms from surfaces or to transfer intact biofilms from one surface to another.
Biofilms are surface-attached communities of bacteria that cause problems including medical infections, fouling, and clogging in industrial applications.[1] By contrast, beneficial biofilms are crucial in applications including waste-water treatment and microbial fuel cells.[2] To solve biofilm-related problems and to realize the potential of biofilm-promoting technologies, understanding of both the biological and physical properties of biofilms and how these properties interrelate,[3] is necessary. Compared to the extensively studied biological components and regulatory networks responsible for biofilm formation,[4] how the material properties of biofilms are derived from the constituent biocomponents, and how biofilm mechanics change in response to environmental fluctuations are not understood.[5] Similar to synthetic hydrogels, biofilms are composed primarily of water (90%), yet they possess structural integrity and they protect biofilm-dwelling cells from external perturbations such as biocides, shear flows, phagocytosis, and invasion by other bacterial species.[5a, 6] Biofilm matrices are composed of extracellular polysaccharides, accessory proteins, and in some cases, extracellular DNA. Recently, mechanical properties of biofilms have been measured using rheological tools.[5c, 7] However, physical interpretation of biofilm rheological data is not straightforward due to the structural complexity of biofilms,[8] impeding the establishment of general mechanical principles for understanding, disrupting, or, more forward-looking, designing biofilm materials.[9]
The majority of investigations of biofilm material characteristics have focused on bulk biofilm properties, not on their surfaces. However, biofilm interfacial properties are crucial in driving their overall material responses,[10] particularly with respect to how biofilms will interact with underlying substrates. Understanding biofilm interfacial properties could lead to insights relevant to biofilm-related problems. For example, in the context of biofilm removal via mechanical disruption, which is often the first strategy for treating biofilm infections in wounds and in some industrial applications,[5c, 11] to avoid leaving residual biofilm patches, it could be more desirable to detach entire biofilms from the substrate rather than to disrupt biofilms at the wound site or at the contaminated surface. In the language of mechanics, in some cases, inducing interfacial fracture between a biofilm and the substrate could be superior to inducing cohesive fracture within a biofilm. However, to successfully induce interfacial fracture, a deeper understanding of biofilm surface properties is needed.
Here, we use the model biofilm forming bacterium Vibrio cholerae, the causative agent of the pandemic disease cholera,[12] to investigate how biofilm material properties are determined by their components. Our measurements reveal V. cholerae biofilms to be soft viscoelastic solids similar to hydrogels, with surface polarity/hydrophobicity that depends on particular matrix components. We develop a capillary peeling method as a practical and efficient technique to remove biofilms from various surfaces via interfacial fracture. The capillary peeling technique also provides a means to measure the adhesion energy between biofilms and substrates, and, moreover, enables the easy transfer of intact biofilms from one surface to another without destroying their internal structures.
To examine biofilm mechanics, we use a commonly studied immotile V. cholerae mutant that constitutively forms biofilms.[13] This strain is denoted wild type (WT). We grew V. cholerae biofilms on a soft nutritious substrate (LB medium solidified with different concentrations of agar, Figure 1a). To measure biofilm mechanical properties, we transferred the biofilm biomass to a shear rheometer (Figure 1a).[14] Oscillatory shear stresses were applied, and by measuring the resulting strains, we deduced the storage modulus G' (representative of the elastic, solid-like properties) and the loss modulus G" (representative of the viscous, fluid-like properties) of the biofilm as a function of the amplitude of the oscillatory strain ε.[15] Figure 1b shows that biofilms are viscoelastic solids, similar to hydrogels:[15-16] the biofilm possesses a shear modulus G' that is ~10 times larger than G". Indeed, the measured G' value is close to the value previously derived using classical hydrogel theory.[13b] The G'(ε) curve features an initial plateau region in which the biofilm deforms elastically with increasing external stresses, which allows the definition of the plateau modulus of a biofilm, G'p. Above a critical strain, referred to as the yield strain εY, the biofilm starts to yield with a dramatic decrease in G'. Concomitantly, G" displays a peak signifying an increase in energy dissipation during the yielding process. All of these mechanical behaviors are analogous to those of hydrogels.[7a, 15-16] Consistent with this analogy, both G' and G" of a biofilm show only slight frequency dependence in the linear viscoelastic regime (Figure S1).
Figure 1.

Rheological characterization of V. cholerae biofilms. a) Representative images of WT biofilms grown for two days on LB medium solidified with 0.6% agar (left) and following transfer to the lower plate of a rheometer (right). Scale bars: 1 cm. b) Representative storage modulus G' and loss modulus G" curves as a function of the amplitude of oscillatory shear strain ε measured for the biofilm sample in (a). From the G' curves, plateau storage modulus G'p and yield strain εY (indicated by the vertical dotted line) were extracted for each biofilm. c) Measured G'p, εY, and yield stress σY values, from left to right, for biofilms containing cells only (ΔrbmAΔbap1ΔrbmCΔvpsL, white), cells and the vibrio polysaccharide (VPS) (ΔrbmAΔbap1ΔrbmC, red), and with RbmA (Δbap1ΔrbmC, green), and with RbmC and Bap1 (ΔrbmA, blue), and with all the matrix components (WT, gray), respectively. All biofilms were grown for two days on LB medium solidified with 0.6% agar. Unpaired t-tests with Welch’s correction were performed for statistical analyses. NS denotes not significant; * denotes P < 0.05, ** denotes P < 0.01, **** denotes P < 0.0001. Error bars correspond to standard deviations with n = 3.
To define the structural elements responsible for biofilm mechanics, we generated V. cholerae mutants lacking particular matrix components.[17] In V. cholerae, the vibrio polysaccharide (VPS), a hydrophilic polymer composed primarily of glucose and galactose and other minor building blocks,[18] serves as the main scaffold for the extracellular matrix.[19] The accessory protein RbmA connects neighboring cells by dimerizing and interacting with VPS through extensive surface binding.[20] Two other proteins with high homology, Bap1 and RbmC, perform partially redundant functions in which Bap1 more than RbmC is responsible for cell-surface adhesion and RbmC more than Bap1 provides additional crosslinks with the VPS.[21] We generated single, double, triple, and quadruple mutants to systematically investigate the distinct contribution of each matrix component to biofilm mechanics.
We first present data for biofilms grown on the softest agar we tested (0.6%). Figure 1c shows the measured plateau modulus G'p (left) and yield strain εY (middle) for each bacterial strain. The quadruple mutant ΔrbmAΔbap1ΔrbmCΔvpsL (white) exhibits a low basal G'p (~ 120 Pa) and εY (~ 0.1). Upon introduction of an unstructured extracellular matrix (i.e., +VPS), the ΔrbmAΔbap1ΔrbmC (red) strain displays a similar G'p to the quadruple mutant but possesses a significantly increased yield strain εY (~ 0.4). When the cell-cell linkage protein RbmA is present (Δbap1ΔrbmC strain, green), the biofilm shows increased elasticity (G'p ~ 760 Pa), although at a cost of reduced εY (~ 0.1). Similarly, in the ΔrbmA strain (blue), the presence of RbmC/Bap1 causes G'p to increase to ~ 590 Pa. Finally, the WT strain (gray) exhibits the highest G'p, around 1 kPa with a small εY (~ 0.1). We also plotted the yield stress σY (the product of G'p and εY), which indicates the maximum shear stress a biofilm can sustain before it begins to yield, for the WT and mutant biofilms (Figure 1c, right). A monotonic increase in σY occurred as components are incorporated into the biofilm matrix, from ~ 10 Pa for the quadruple mutant biofilm to ~ 100 Pa for the WT biofilm. In nature, V. cholerae biofilms grow on sinking marine particles or on swimming organisms and so they are subjected to flow-induced shear stress.[22] Our results suggest that the flow environment could impose a selective pressure on V. cholerae to evolve multiple matrix components that enable the biofilms to withstand shear stress.
We propose a dual network model to understand the mechanical properties of V. cholerae biofilms (Figure 2a). The key is the presence of two interacting networks, one composed of interconnected, stiff bacterial cells, and the other composed of soft secreted polymers.[23] In the quadruple mutant, cells are in direct contact with one another, and we propose that neighboring cells interact weakly through their surface lipopolysaccharides (LPS) or other surface appendages such as pili.[24] Similar to colloidal gels with short-range attractions,[25] this cellular network yields at a relatively low strain, εY ~ 0.1. In the ΔrbmAΔbap1ΔrbmC biofilm that lacks accessory proteins but possesses the VPS polysaccharidethe, VPS fills the spaces between neighboring cells and increases εY to ~ 0.4 (Figure 1c, red). Cells interact indirectly through the hydrogel network of the VPS, which can sustain larger deformations than the naked cells. However, our measurements show that the unstructured VPS does not increase the biofilm storage modulus, presumably because the hydrogen-bond based interactions between individual polysaccharide chains are similar to those between individual LPS units.[26] To strengthen the biofilm, two strategies are employed. First, the polymer network is reinforced by cross-linking via RbmC/Bap1 (Figure 1c, blue).[21a, 27] Second, the cellular network is strengthened by enhancing the weak cell-cell interactions via crosslinking by RbmA.[20, 28] In the latter case, a stronger colloidal gel with short-range attractions forms,[25] and hence εY once again declines while G'p increases (Figure 1c, green). Note that the strengthening effect of RbmA depends on VPS and RbmC/Bap1 (Figure S2), suggesting that the two networks function together to fortify the V. cholerae biofilm. Indeed, the WT biofilm possesses both the cell-cell and polymeric networks so it exhibits the highest G'p (~ 1 kPa), despite a small εY due to the short-range nature of RbmA-mediated cell-cell interactions. Future experiments investigating the rheology of combinations of purified matrix components will be necessary to provide a molecular-level understanding of how the precise interactions between particular matrix components and between matrix components and cells drive macroscopic biofilm mechanics.
Figure 2.

Osmotic pressure affects the biofilm plateau modulus and the effect depends on particular matrix components. a) Schematic of the V. cholerae biofilm matrix components contributing to its mechanical properties. Yellow cylinders denote cells. Black curvy lines (left) denote cell surface LPS. Red wavy lines (right) denote VPS filling interstitial spaces. Blue dots (right bottom) denote the RbmC and Bap1 proteins that crosslink the VPS network. The green symbols (bottom left) denote RbmA proteins that connect neighboring cells. b) Shown from left to right are the plateau storage moduli G'p for biofilms containing cells only (ΔrbmAΔbap1ΔrbmCΔvpsL, white), cells and VPS (ΔrbmAΔbap1ΔrbmC, red), and with RbmA (Δbap1ΔrbmC, green), and with RbmC and Bap1 (ΔrbmA, blue), and with all matrix components (WT, gray), grown on medium solidified with the designated concentrations of agar, respectively. Error bars correspond to standard deviations with n = 3.
Biofilm mechanical properties often depend on physicochemical conditions.[7c, 14, 29] This feature has often been attributed to biological responses to the environment, directed by gene expression changes. However, we hypothesized that the biofilm matrix, as a hydrogel, could respond physically to environmental changes, and hence alter biofilm mechanics, for example by swelling or deswelling in response to external osmotic pressure. Recently, we and others showed that the concentration of agar on which biofilms are grown controls biofilm growth primarily by dictating the osmotic pressure of the agar substrate.[13b, 30] Indeed, the biofilm matrix will either take up or lose water depending on the osmotic contrast between the biofilm and the agar, potentially altering biofilm mechanics. Consistent with this reasoning, we demonstrate here a strong dependence of the biofilm modulus on the concentration of agar in the substrate (Figure 2b, Figure S3, and Table S1). Specifically, Figure 2b shows that the rheological properties of the quadruple mutant biofilm (white) are significantly affected by the agar concentration: the value of G'p declines appreciably with decreasing agar concentration (i.e., with higher osmotic contrast across the biofilm-agar interface,[13b, 30] see Figure S4). This trend holds for the ΔrbmAΔbap1ΔrbmC mutant (red) that possesses an unstructured, easily swollen VPS network lacking proteins that make crosslinks. Introducing RbmA-mediated cell-cell linkages (the Δbap1ΔrbmC strain, green) raises the overall G'p for all agar concentrations but does not mitigate the decrease in G'p that occurs upon swelling. By contrast, crosslinking the VPS network with RbmC/Bap1 eliminates the strong dependence of G'p on agar concentration as shown by the ΔrbmA biofilm (blue). Nonetheless, the overall G'p remains low (~ 400-700 Pa), suggesting that cell-cell connections are more effective in raising the biofilm modulus than are VPS crosslinks. Again, the WT biofilm, containing the entire complement of matrix components (gray), displays properties of both strengthened networks: the overall G'p value remains high (~ 1 kPa) for all agar concentrations, and thus, is nearly independent of how the agar affects the osmotic pressure contrast. Such a doubly strengthened network structure is reminiscent qualitatively of recently developed tough synthetic gels.[7b, 31] From an evolutionary perspective, a robust mechanical response to the environment could be beneficial to V. cholerae, which transitions from sea water to fresh water to the human intestine.[12] The possibility exists that other physicochemical changes occur in the substrate (such as changes in stiffness or friction) as the agar concentration is changed that could also contribute to changes in biofilm mechanics.
Our above analyses of biofilm bulk material characteristics reveal that the biofilm has hydrogel-like properties. This finding suggested to us a rationale that could underlie some of the difficulties encountered in thorough biofilm removal via mechanical perturbation. Specifically, hydrogels have low cohesiveness,[31] and hence they easily break into pieces upon mechanical perturbation. That understanding inspired us to imagine alternative strategies for biofilm removal that would enable biofilms to be detached, intact, from substrates. However, to achieve such interfacial fracture requires information about biofilm interfacial properties, and such data are generally lacking. To overcome this issue, we systematically characterized interfacial energies of V. cholerae biofilms with respect to air, liquids, and solid substrates.
We first characterized the surface energy γf of a biofilm in air using the Owens-Wendt method (Figure 3a, Table S2, Supplementary Methods).[32] Surprisingly, despite a high water content, the surface of the WT biofilm is highly nonpolar () with a negligible polar component (). We confirmed the magnitude of the nonpolar energy using the Zisman method,[33] which measures the contact angles of nonpolar fluids of different surface tensions on a surface, in this case the biofilm surface (Figure 3b). The quantified values are comparable between the two methods ( for the Zisman method), and independent of the substrate agar concentration. Removal of Bap1 makes the biofilm surface become predominantly polar (), identical to that of the surface of the ΔvpsL mutant. Indeed, an earlier report noted that V. cholerae biofilms are hydrophobic but become hydrophilic upon deletion of the bap1 gene.[34] Additional deletion of rbmC in the Δbap1 strain further increases the polar nature of the biofilm surface (). A vivid demonstration of biofilm polarity is shown in Figure 3c and Figure S5, in which a WT biofilm and a Δbap1ΔrbmC biofilm show differences in affinity for polar (water) and nonpolar (CH2I2) liquids. Hence, Bap1, and to a minor extent, RbmC, behave similarly to surfactant molecules that alter the surface energy of a material. Using the classic Young equation, the interfacial energy between a WT biofilm and water γfw was determined to be ~ 50-60 mJ m−2 (Figure 3d). γfw depends minimally on the agar concentration (Figure 3d), suggesting that interfacial energies are not influenced by the growth conditions of the biofilm. Finally, we note that deleting rbmA did not alter these measured values implying that cell-cell connections do not contribute to biofilm surface properties.
Figure 3.

Characterization of biofilm surface energies. a) Measured values of the polar () and dispersion () components of the surface energy (γf) for the designated V. cholera biofilms grown for two days on 1.5% agar using the Owens-Wendt method.[39] b) Measurement of the surface energies of WT biofilms grown on medium containing the designated concentrations of agar, using the Zisman method.[40] The contact angles θ for nonpolar liquids (CH2I2, CH2Br2, 1-methylnaphthalene, and 1-bromonaphthalene) on biofilms were measured and plotted against the surface energies of the liquids (γ1) for different concentrations of agar. Linear extrapolation to cosθ = 1 gives the critical surface tension for wetting γc corresponding to the nonpolar component of the surface energy. The symbols designate measurements made for biofilms grown on LB medium solidified with the designated agar concentrations. c) Representative images showing preferential wetting of the WT biofilm by a polar (water) liquid and the Δbap1ΔrbmC mutant biofilm by a nonpolar (CH2I2) liquid, when both liquids were present on top of each biofilm. The green lines denote the interfaces between water and CH2I2. Scale bar: 1 mm. d) Measured biofilm-water interfacial energies γfw for biofilms grown on medium solidified with the designated concentrations of agar. Unpaired t-tests with Welch’s correction were performed for statistical analyses. NS stands for not significant. Error bars correspond to standard deviations with n = 3.
As alluded to above, being able to completely remove biofilms has been a challenging goal in medicine and industry. Knowing the hydrophobic nature of V. cholerae biofilms, coupled with the above description of their hydrogel-like bulk properties, put us in a position to exploit a simple technique, previously developed by our group, to peel thin hydrophobic films off of hydrophilic substrates using capillary forces.[35] When the carrier substrate of a thin hydrophobic film is slowly dipped into water, the air-water contact line, pinned at the edge of the film on the substrate, initiates a crack and leads to the peeling of the film, provided that penetration of water into the crack is energetically favorable. Applying this technique to a WT V. cholerae biofilm allowed us to peel the biofilm off the agar substrate (Figure 4a, Figure S6, Video S1). Moreover, during the peeling process, a constant peeling angle θp developed at the triple contact point between the biofilm, the agar substrate, and the water (Figure 4b). An energy balance leads to Γ = γfw (1 − cos θp), in which Γ is the minimum effective mechanical energy (per unit area) required to separate the biofilm from the substrate if no other dissipation mechanism exists.[35-36] Here, we minimized the energy dissipation through viscous effects by performing the peeling at very low speeds.[35] Therefore, capillary peeling, beyond removing the biofilm, provides a way to determine Γ by measuring θp. We determined θp experimentally for each agar concentration, and we found a small positive correlation (Figure 4c). This method yields Γ values on the order of 4-7 mJ m−2 that depend only modestly on agar concentration (Figure 4d).
Figure 4.

Measurement of the adhesion energy between a biofilm and a substrate (agar) using capillary peeling. a) Representative time-lapse images of the capillary peeling process. A green laser line illuminates the biofilm and the dye in the water. As the water level rises at a speed of 0.035 mm s−1, the biofilm is peeled off the agar substrate (agar = 0.6%). Scale bar: 3 mm. b) Schematic of the capillary peeling process. The interfacial tension γfw between the water and the biofilm causes peeling of the biofilm from the substrate, with a constant peeling angle θP. Γ denotes the adhesion energy (energy area−1) between the biofilm and the agar substrate. c,d) Peeling angle θP and Γ, respectively, as a function of agar concentration. e) Successful peeling as a function of peeling velocity (Vpeel). Fourteen biofilms were tested at each peeling velocity. The success rate is defined as the ratio between the number of completely removed biofilms and the total number of biofilms. f) Biofilm morphology before (upper) and after (lower) capillary peeling and transfer. Scale bar: 5 mm.
To demonstrate the thoroughness of biofilm removal using capillary peeling, we imaged live and dead cells in the biofilm before and after peeling (Figure S7). Most of the live cells were removed from the agar substrate, while some dead cells remained, primarily at the location that harbored the original central core of the biofilm where significant cell death occurs during biofilm growth.[13b, 37] Therefore, we suggest that the capillary peeling process could fulfill an unmet need for biofilm removal because it causes interfacial fracture at the biofilm-substrate interface rather than cohesive fracture within the biofilm. To establish optimum conditions for capillary peeling, we scanned different peeling velocities Vpeel and found that the success rate of peeling decreases with increasing Vpeel above a threshold value around 0.1 mm s−1 (Figure 4e). This observation confirms the quasi-equilibrium nature of the apillary peeling process. If Vpeel is too high, water passes over the edge of the biofilm and traps the system in a high-energy configuration.
This simple capillary peeling technique can be applied to other biofilm-forming bacterial species and other surfaces. As a proof of concept, we successfully peeled Pseudomonas aeruginosa biofilms from agar substrates (Figure S8). Moreover, we used the capillary peeling procedure to remove biofilms grown on a variety of surfaces, ranging from paper, to semi-permeable membranes, to metal (Figure S9).
Due to the gentle nature of the capillary peeling process, the peeled biofilm remains intact and floats on the liquid used for peeling (Figure 4a, Video S1). Therefore, in situations in which biofilms need to be grown on one surface and transferred to another, the capillary peeling method provides a convenient option. Indeed, using a substrate with a lower surface energy (untreated glass in this case), the floating biofilm can be picked up without changing its morphology apart from modest swelling (Figure 4f).
We provide two examples to demonstrate the utility of the biofilm transfer technique. First, we imaged the internal structure of a living, peeled biofilm grown at an air-solid interface with single-cell resolution (Figure 5a,b), a useful but heretofore inaccessible operation in biofilm research. Moreover, by reversing the dipping direction of the receiving glass substrate, that is by picking up the floating biofilm from each side, we can image the top and bottom of the biofilm, respectively (Figure 5a,b). The bottom layer possesses patches of cells with low fluorescence intensities. In the top layer, by contrast, cells possess strong fluorescence and pack densely. The contrast in the internal structures of the top and bottom of the biofilm likely reflects the oxygen gradient present in the original biofilm.[38]
Figure 5.

Application of the biofilm transfer technique. a) Schematic of biofilm transfer procedures. Yellow denotes agar. Red denotes the biofilm. Blue denotes water. Gray denotes the glass receiving surface. The black lines highlight the bottoms of the biofilms. The solid arrows indicate the direction of motion of the glass, which is the receiving surface. b) High-resolution imaging of the top (upper) and bottom (lower) of the WT V. cholerae biofilm architecture imaged through the glass coverslips used for pick-up. Cells constitutively express mKate2 from the chromosome. Arrows indicate the direction of the corresponding glass motion during the picking-up step in (a). Scale bar: 5 μm. c) Percent survival of cells grown planktonically, either to stationary phase (S) or to exponential phase (E), or of cells grown in biofilms, peeled off the surface, and subsequently subjected to 1 h treatment with 50 μg mL−1 tetracycline in three different configurations: floated, submerged, or submerged as disrupted pieces. NS stands for not significant; * denotes P < 0.05, ** denotes P < 0.01, *** denotes P < 0.001. Error bars correspond to standard deviations with n = 4. An agar concentration of 0.6% was used for all experiments in this figure.
As a second example of the applicability of the capillary peeling method, we peeled biofilms off of agar substrates and floated them on liquid medium containing 50 μg mL−1 tetracycline, an antibiotic used to treat clinical V. cholerae infections.[39] We compared survival of the floating biofilm cells after 1 h of antibiotic treatment to that of isogenic cells that were grown planktonically to exponential or stationary phase (Figure 5c). Cells in the floating biofilm were significantly more resistant to antibiotic treatment (percent survival Sbiofilm ~ 80%) than planktonically-grown cells (Sexponential phase = 6%, Sstationary phase = 21%). To understand whether the increased antibiotic tolerance is due to a biofilm-specific physiological state or due to the spatial structure of the biofilm, we disrupted the floating biofilm immediately prior to antibiotic treatment and repeated the assay (Figure S10). This treatment caused Sbiofilm to decrease over three-fold (to 24%) to nearly the level of the stationary phase planktonic cells. This result indicates that the biofilm spatial structure, rather than some biofilm-specific cellular state,[40] is responsible for increased tolerance to antibiotics. Indeed, the effectiveness of antibiotic treatment tracked with the amount of surface area the biofilm had in contact with the antibiotic-containing medium: if the floated biofilm was submerged in the antibiotic-containing medium so that tetracyline could diffuse in from both sides of the biofilm, Sbiofilm declines to an intermediate level (~ 57%). Consistent with this observation, using live-dead staining, we observed that cells in the biofilm interior showed higher survival than cells close to the antibiotic-containing medium (Figure S10). Future experiments including visualization of the distribution of antibiotic molecules throughout the biofilm could reveal the underlying mechanism of the increased antibiotic resistance of biofilm cells.[41]
Currently the capillary peeling method is limited to biofilms that have been grown statically at an air-solid interface, which provides the necessary means for an external liquid to come into contact with the biofilms and initiate the peeling process. Going forward, we envision two potential strategies to remove biofilms grown under liquids. First, if submerged biofilms could be dried out, the same capillary peeling method should apply. Second, the capillary peeling method hints at the general possibility of exploiting interfacial energies to remove biofilms from surfaces. Hence, we expect that introducing air bubbles into the medium could potentially generate similar triple contact points and thus remove biofilms that have been grown under liquid.[42]
We envision a variety of applications for the capillary peeling technique. For example, in biofilm infections in wounds, applying appropriate fluids to slowly detach the infectious biofilm could possibly be used instead of the common practice of mechanical debridement.[5c] This technique could also facilitate basic research investigations. For example, it is difficult to study biofilm-host interactions in vitro due to problems encountered in growing host cells and biofilms together under laboratory conditions.[43] The capillary peeling technique could enable scientists to grow the bacterial and host cells separately and bring them into contact at the time of choosing. Also in terms of fundamental biofilm studies, the capillary peeling process yields a convenient measurement of the adhesion energy Γ between a biofilm and a substrate. An alternative method for measurement of bulk biofilm adhesion energy is a micromanipulation method that involves scraping biofilms off substrates and integrating the forces measured during scraping.[11, 44] However, such measurements are confounded by the significant energy dissipation caused by friction and deformation, rather than detachment of the biofilm. Therefore, such measures suffer from overestimation of the adhesion energy. Indeed, our measured Γ value of ~ 4-7 mJ m−2 is an order of magnitude lower than values reported with the scraping method. We argue that we are measuring the actual, close-to-equilibrium work of adhesion between a biofilm and a substrate.
To conclude, we used V. cholerae as a model biofilm former to systematically reveal connections between bulk rheological and interfacial biofilm properties, the biocomponents that make up the biofilm material, and relevant external conditions. We discovered V. cholerae biofilms to be soft, hydrophobic materials possessing a dual-network internal structure with surface polarity that can be tuned by altering the matrix composition. The discovery of a dual-network structure might allow biofilms to be used as materials for bioengineering, as the strength and chemistry of both the cellular and the polymer networks can be independently manipulated. The understanding gained here also points to new design approaches to disrupt or enhance biofilms by targeting specific components in the biofilm matrix. Finally, we demonstrated capillary peeling as a simple method to measure the adhesion energy and to remove and/or transfer intact biofilms, steps which are otherwise difficult. The capillary peeling technique can be integrated into current or future treatments of biofilm-related infections and industrial processes.
Experimental Section
Strains and media:
All V. cholerae strains used in this study are derivatives of the wild-type Vibrio cholerae O1 biovar El Tor strain C6706, harboring a missense mutation in the vpvC gene (VpvC W240R).[13a] This mutation causes V. cholerae cells to have elevated c-di-GMP level and to constitutively produce biofilms. Additional mutations were engineered into this strain using Escherichia coli S17 λpir carrying pKAS32. All P. aeruginosa strains used in this study are derivatives of the wild-type PA14 strain. Mutants were constructed using established procedures.[45] A strain list is provided in Table S3. All strains were grown in lysogeny both (LB) medium at 37°C with shaking. When designated, fresh LB medium solidified with different percentages of agar was used. To measure the osmotic pressure of agar substrates, dextran solutions were prepared by adding dextran powder (1500-2800 kDa, Sigma-Aldrich) to LB medium followed by shaking at 30 °C overnight.
Microscopy:
Transmission images were taken with a home-built setup consisting of an LED illumination pad (Huion) and a Nikon D3300 camera equipped with a macrolens (Sigma). Fluorescence images were obtained using a Leica stereoscope with GFP and mCherry filter sets or with no filter (reflection mode). High-resolution confocal images were acquired with a Yokogawa CSU-X1 confocal spinning disk unit mounted on a Nikon Ti-E inverted microscope, using a 60× water objective with a numerical aperture of 1.2 plus a 1.5× post-magnification lens and an Andor iXon 897 EMCCD camera. A 591 nm laser (OEM DPSS) was used to excite cells expressing mKate2 as well as to visualize dead cells stained with propidium iodide (BacLight Bacterial Viability Kit, ThermoFisher). A 488 nm laser was used to excite live cells stained with Syto9 (BacLight Bacterial Viability Kit, ThermoFisher).
Biofilm growth on agar plates:
V. cholerae strains were streaked on LB plates containing 1.5% agar and grown at 37°C overnight. Individual colonies were inoculated into 3 mL of LB liquid medium containing glass beads, and the cultures were grown with shaking at 37°C to mid-exponential phase (5-6 h). Subsequently, the cells in the cultures were mixed by vortex, OD600 was measured, and the cultures were back diluted to an OD600 of 0.5. 1 μL of this inoculum was spotted onto pre-warmed agar plates (100 mm) solidified with different percentages of agar. For contact angle measurements, 50 μL of this inoculum was applied to an agar plate and spread with glass beads to enable growth of a biofilm covering the entire plate. Plates were incubated at 37°C for two days. For capillary peeling experiments, two to four colonies were grown per plate. For rheological measurements, seven colonies were grown per plate for agar concentrations of 0.8% or lower, and sixteen colonies were grown per plate for agar concentrations of 1.0% or 1.5%. Fifteen plates were prepared for each rheological measurement. For P. aeruginosa biofilms, 2 μL of overnight cultures of WT P. aeruginosa PA14 and mutant strains were spotted onto 60 x 15 mm Petri plates containing 10 mL of 1% TB medium fortified with 40 mg/L Congo red and 20 mg/L Coomassie brilliant blue dyes and solidified with 1% agar. P. aeruginosa biofilms were grown at 25°C and images were acquired after 120 h.
Biofilm growth on non-agar substrates:
In the case of biofilms grown on paper or on stainlesss steel, two pieces of 3×3 cm paper (Whatman) or sheets of 304 stainless steel mesh (McMaster, # 500 × 500) were pre-sterilized with ethanol and placed on top of an agar plate (0.6%). 1 μL of inoculant prepared according to the method described above was spotted onto the paper/stainless steel mesh, and the plate was incubated at 37°C overnight. In the case of biofilms grown on a semi-permeable membrane, the inoculant was spotted onto EMD Millipore, VSWP04700 semi-permeable membranes that had been placed on top of the agar, and the plate was incubated at 37°C for 2 days.
Rheology:
All rheological measurements were performed with a stress-controlled Anton Paar Physica MCR 301 rheometer at 37°C. For each measurement, 100-240 colonies were collected with a pipette tip or a razor blade and transferred onto the lower plate of the rheometer. After sandwiching the biofilms between the upper and lower plates with a gap size of 0.5 mm, silicone oil (5 cSt at 25°C, Sigma Aldrich) was applied to surround the biofilm to avoid evaporation. We confirmed that the silicone oil did not affect the measurement result. Sandblasted surfaces were used for both the upper and lower plates to avoid slippage at the boundary. Oscillatory shear tests were performed. During amplitude sweeps, a strain range of 0.01 – 2000% was scanned at a fixed frequency of 6.28 rad s−1. During frequency sweeps, a range of 0.1 to 200 rad s−1 was scanned at a fixed strain of 1% (in the linear viscoelastic regime). Segmented linear fittings were applied to G'(ε) curves on a log-log scale. The strain value at which the two linear lines intersect is defined as εY. G' varies minimally in the plateau region. We used the fitted G' value at ε = 1% as G'p.[14] Yield stress σY is defined as the stress measured at εY. Three biological replicates were performed for each bacterial strain under each osmotic condition.
Contact angle and surface energy measurements:
Side views of biofilm-liquid interfaces were recorded with a Nikon camera (D3300) equipped with a macrolens (Sigma). For all nonpolar liquids, the static sessile drop method was used, in which a 1 μL droplet was gently deposited onto the biofilm and subsequently imaged. For water, to overcome uptake of water by the underlying biofilm/agar, we used a dynamic sessile drop method. Water was slowly added to the surface by a syringe pump, and the advancing contact angle was measured to approximate the equilibrium contact angle. The contact angle θ was extracted using the Droplet_Analysis plugin in ImageJ. We used two methods to calculate the biofilm-air surface energy γf. In the Owens-Wendt method,[32] the contact angles of water and CH2I2 on biofilms were measured and, by knowing the dispersion (nonpolar) and polar components of the surface energies of the liquids, two linear equations can be written to solve for the polar and dispersion portions of the total biofilm surface energy γf. In the Zisman method,[33] we measured the contact angles θ of four different nonpolar liquids (CH2I2, CH2Br2, 1-methylnaphthalene, and 1-bromonaphthalene, all from Sigma Aldrich) on the biofilm. Plotting their surface tensions γ1 versus cosθ (Figure 3b) resulted in a straight line. Linear extrapolation to cosθ = 1 yielded the critical surface tension of wetting γc as an approximation for . Once γf was determined, γfw was calculated using the Young equation γf − γfw = γw cosθ.
Peeling angle measurements:
A piece of agar (~ 2 cm × 2 cm) on which a biofilm was grown for two days was transferred onto a 1×3 inch glass, and secured with a clamp. The setup was attached vertically to a stationary stand. Underneath the biofilm, a water bath containing Rhodamine B was placed on a translational stage (Thorlabs) and moved upward with a controlled velocity to initiate peeling. A cylindrical lens (Thorlabs) was mounted on a 543 nm laser (BioRay) to generate a laser sheet that illuminated the biofilm from the side. The peeling process was recorded with a Nikon camera at a 90° angle. Peeling angles were measured manually using the Nikon Element software at five time points and averaged. Three biological replicates were performed for each condition.
Biofilm removal and transfer:
For biofilms grown directly on agar plates, the entire agar plate was slowly dipped into water. For biofilms grown on other surfaces, each piece of paper/membrane/stainless steel mesh was individually dipped into water. The biofilm was peeled off of the substrate and floated at the water-air interface. Subsequently, an untreated coverslip was dipped into water to pick up the floated biofilm. Before and immediately after transfer, the biofilm and the original substrate were imaged with a Leica Stereoscope or with a Nikon camera. SytoX green nucleic acid stain (ThermoFisher) was added to the agar at 5 μg mL−1 to label dead cells. Live cells constitutively express mKate2.
Antibiotic killing assay:
The killing assay is adapted and modified from Ref. [19] and Ref.[41a]. First, pieces of agar (~ 2 cm × 2 cm) on which biofilms were grown for two days were transferred onto a 1×3 inch glass. These setups were vertically dipped into 50 mL Corning conical tubes containing 25 mL of liquid LB medium and glass beads. The biofilms were gently peeled off of the agar substrates and floated at the air-liquid interfaces. The LB medium either contained or lacked 50 μg mL−1 of tetracycline. The conical tubes were left at 37° for 1 h, and subsequently, vigorously mixed by vortex for 1 min. 1 mL of the suspensions were transferred to Eppendorf tubes containing small glass beads (acid-washed, 425–500 μm, Sigma), mixed by vortex for 1 min, and serially diluted onto LB plates. The LB plates were incubated overnight in 37° and, subsequently, assessed for colony forming units (CFU). The ratios between CFUs with and without tetracycline treatment were defined as the percent survival. The identical procedure was used for experiments with submerged biofilms except that, immediately after the biofilm was peeled off of the agar substrate, the biofilm was gently pushed completely into the antibiotic-containing medium. For experiments using disrupted biofilms, the conical tube containing the peeled biofilm was agitated by vigorous vortex for 1 min prior to antibiotic treatment. In experiments involving planktonic cells, the cells were grown to mid-exponential (5 h) or stationary (12 h) phase in liquid LB medium at 37°. Subsequently, the cultures were diluted 100-fold into LB medium with or without tetracycline. The final culture densities were adjusted to make it so that planktonically-grown and biofilm-grown cell densities were comparable.
Supplementary Material
Acknowledgements
This work was supported by the Howard Hughes Medical Institute, NSF MCB-1713731 and the Max Planck Society-Alexander von Humboldt Foundation (B.L.B.) and MCB-1344191 (to N.S.W., B.L.B., and H.A.S.). Jing Yan and Alexis Moreau contributed equally to this work. J.Y. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. S.M. holds a Life Sciences Research Foundation Postdoctoral Fellowship through the Gordon and Betty Moore Foundation through Grant GBMF2550.06. We thank Chari D. Smith, Wei Yu, Hongbin Zhang, Zhong Zheng, Ruobing Bai, and Shmuel Rubinstein for helpful discussion.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jing Yan, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Alexis Moreau, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Sepideh Khodaparast, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Antonio Perazzo, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Jie Feng, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Chenyi Fei, Department of Molecular Biology, Princeton University, 329 Lewis Thomas Laboratory, Princeton, NJ, 08544, United States.
Sheng Mao, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Sampriti Mukherjee, Department of Molecular Biology, Princeton University, 329 Lewis Thomas Laboratory, Princeton, NJ, 08544, United States.
Andrej Košmrlj, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States.
Ned S. Wingreen, Department of Molecular Biology, Princeton University, 329 Lewis Thomas Laboratory, Princeton, NJ, 08544, United States
Bonnie L. Bassler, Department of Molecular Biology, Princeton University, 329 Lewis Thomas Laboratory, Princeton, NJ, 08544, United States; Howard Hughes Medical Institute, Chevy Chase, MD, 20815, USA
Howard A. Stone, Department of Mechanical and Aerospace Engineering, Princeton University, D328 E-Quad, Olden St., Princeton, NJ, 08544, United States
References
- [1].Ghannoum M, Parsek M, Whiteley M, Mukherjee P, Microbial Biofilms, ASM Press, Washington, DC: 2015. [Google Scholar]
- [2].a) Nerenberg R, Curr. Opin. Biotechnol 2016, 38, 131. [DOI] [PubMed] [Google Scholar]; b) Franks AE, Malvankar N, Nevin KP, Biofuels 2010, 1, 589. [Google Scholar]
- [3].Mazza MG, J. Phys. D 2016, 49, 203001. [Google Scholar]
- [4].Hobley L, Harkins C, Macphee CE, Stanley-Wall NR, FEMS Microbiol. Rev 2015, 39, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Flemming H-C, Wingender J, Nat. Rev. Microbiol 2010, 8, 623; [DOI] [PubMed] [Google Scholar]; b) Peterson BW, He Y, Ren Y, Zerdoum A, Libera MR, Sharma PK, van Winkelhoff A-J, Neut D, Stoodley P, van der Mei HC, Busscher HJ, FEMS Microbiol. Rev 2015, 39, 234; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gordon VD, Davis-Fields M, Kovach K, Rodesney CA, J. Phys. D: Appl. Phys 2017, 50, 223002; [Google Scholar]; d) Wilking JN, Angelini TE, Seminara A, Brenner MP, Weitz DA, MRS Bull. 2011, 36, 385. [Google Scholar]
- [6].Dragoš A, Kovács ÁT, Trends Microbiol. 2017, 25, 257. [DOI] [PubMed] [Google Scholar]
- [7].a) Billings N, Birjiniuk A, Samad TS, Doyle PS, Ribbeck K, Rep. Prog. Phys 2015, 78, 036601. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Even C, Marlière C, Ghigo J-M, Allain J-M, Marcellan A, Raspaud E, Adv. Colloid Interface Sci 2017, 247, 573; [DOI] [PubMed] [Google Scholar]; c) Pavlovsky L, Younger JG, Solomon MJ, Soft Matter 2013, 9, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lieleg O, Caldara M, Baumgartel R, Ribbeck K, Soft Matter 2011, 7, 3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Klapper I, Rupp CJ, Cargo R, Purvedorj B, Stoodley P, Biotechnol. Bioeng 2002, 80, 289. [DOI] [PubMed] [Google Scholar]
- [9].a) Chen AY, Deng Z, Billings AN, Seker UOS, Lu Michelle Y., Citorik RJ, Zakeri B, Lu TK, Nat. Mater 2014, 13, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jin X, Riedel-Kruse IH, Proc. Natl. Acad. Sci. USA 2018, 115, 3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Epstein AK, Pokroy B, Seminara A, Aizenberg J, Proc. Natl. Acad. Sci. USA 2011, 108, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shivapooja P, Wang Q, Orihuela B, Rittschof D, López GP, Zhao X, Adv. Mater 2013, 25, 1430. [DOI] [PubMed] [Google Scholar]
- [12].Nelson EJ, Harris JB, Morris JGJ, Calderwood SB, Camilli A, Nat. Rev. Microbiol 2009, 7, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Beyhan S, Yildiz FH, Mol. Microbiol 2007, 63, 995. [DOI] [PubMed] [Google Scholar]; b) Yan J, Nadell CD, Stone HA, Wingreen NS, Bassler BL, Nat. Commun 2017, 8, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kovach K, Davis-Fields M, Irie Y, Jain K, Doorwar S, Vuong K, Dhamani N, Mohanty K, Touhami A, Gordon VD, npj Biofilms Microbiomes 2017, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rubinstein M, Colby RH, Polymer Physics, Oxford University Press, 2003. [Google Scholar]
- [16].Wloka M, Rehage H, Flemming HC, Wingender J, Colloid Polym. Sci 2004, 282, 1067. [Google Scholar]
- [17].Teschler JK, Zamorano-Sanchez D, Utada AS, Warner CJA, Wong GCL, Linington RG, Yildiz FH, Nat. Rev. Microbiol 2015, 13, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yildiz F, Fong J, Sadovskaya I, Grard T, Vinogradov E, PLoS ONE 2014, 9, e86751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yildiz FH, Schoolnik GK, Proc. Natl. Acad. Sci. USA 1999, 96, 4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fong JCN, Rogers A, Michael AK, Parsley NC, Cornell W-C, Lin Y-C, Singh PK, Hartmann R, Drescher K, Vinogradov E, Dietrich LEP, Partch CL, Yildiz FH, eLife 2017, 6, e26163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Berk V, Fong JCN, Dempsey GT, Develioglu ON, Zhuang XW, Liphardt J, Yildiz FH, Chu S, Science 2012, 337, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fong JCN, Yildiz FH, J. Bacteriol 2007, 189, 2319. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Absalon C, Van Dellen K, Watnick PI, PLoS Pathog. 2011, 7, e1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Drescher K, Nadell CD, Stone HA, Wingreen NS, Bassler BL, Curr. Biol 2014, 24, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Azam F, Malfatti F, Nat. Rev. Microbiol 2007, 5, 782. [DOI] [PubMed] [Google Scholar]
- [23].a) Reichert WF, Göritz D, Duschl EJ, Polymer 1993, 34, 1216 [Google Scholar]; b) Baeza GP, Dessi C, Costanzo S, Zhao D, Gong S, Alegria A, Colby RH, Rubinstein M, Vlassopoulos D, Kumar SK, Nat. Commun 2016, 7, 11368. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Filippone G, Salzano de Luna M, Macromolecules 2012, 45, 8853. [Google Scholar]
- [24].Wang H, Wilksch JJ, Lithgow T, Strugnell RA, Gee ML, Soft Matter 2013, 9, 7560. [Google Scholar]
- [25].Lu PJ, Zaccarelli E, Ciulla F, Schofield AB, Sciortino F, Weitz DA, Nature 2008, 453, 499. [DOI] [PubMed] [Google Scholar]
- [26].Ganesan M, Knier S, Younger JG, Solomon MJ, Macromolecules 2016, 49, 8313. [Google Scholar]
- [27].Yan J, Sharo AG, Stone HA, Wingreen NS, Bassler BL, Proc. Natl. Acad. Sci. USA 2016, 113, E5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Giglio KM, Fong JC, Yildiz FH, Sondermann H, J. Bacteriol 2013, 195, 3277. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Maestre-Reyna M, Wu W-J, Wang AHJ, PLoS ONE 2013, 8, e82458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Sutherland IW, Trends Microbiol. 2001, 9, 222. [DOI] [PubMed] [Google Scholar]; b) Tallawi M, Opitz M, Lieleg O, Biomater. Sci 2017, 5, 887. [DOI] [PubMed] [Google Scholar]; c) Chew SC, Kundukad B, Seviour T, van der Maarel JRC, Yang L, Rice SA, Doyle P, Kjelleberg S, mBio 2014, 5, e01536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Seminara A, Angelini TE, Wilking JN, Vlamakis H, Ebrahim S, Kolter R, Weitz DA, Brenner MP, Proc. Natl. Acad. Sci. USA 2012, 109, 1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Gong JP, Katsuyama Y, Kurokawa T, Osada Y, Adv. Mater 2003, 15, 1155 [Google Scholar]; b) Sun J-Y, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z, Nature 2012, 489, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Owens DK, Wendt RC, J. Appl. Polym. Sci 1969, 13, 1741. [Google Scholar]
- [33].a) Weisberg DS, Dworkin M, Appl. Environ. Microbiol 1983, 45, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zisman WA, in Contact Angle, Wettability, and Adhesion, Vol. 43 (Ed: Fowkes FM), Americal Chemical Society, 1964, pp. 1. [Google Scholar]
- [34].Hollenbeck EC, Fong JCN, Lim JY, Yildiz FH, Fuller GG, Cegelski L, Biophys. J 2014, 107, 2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Khodaparast S, Boulogne F, Poulard C, Stone HA, Phys. Rev. Lett 2017, 119, 154502. [DOI] [PubMed] [Google Scholar]
- [36].Kendall K, J. Phys. D 1971, 4, 1186. [Google Scholar]
- [37].Asally M, Kittisopikul M, Rué P, Du Y, Hu Z, Çağatay T, Robinson AB, Lu H, Garcia-Ojalvo J, Süel GM, Proc. Natl. Acad. Sci. USA 2012, 109, 18891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dietrich LEP, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, Newman DK, J. Bacteriol 2013, 195, 1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kitaoka M, Miyata ST, Unterweger D, Pukatzki S, J. Med. Microbiol 2011, 60, 397. [DOI] [PubMed] [Google Scholar]
- [40].Costerton JW, Stewart PS, Greenberg EP, Science 1999, 284, 1318. [DOI] [PubMed] [Google Scholar]
- [41].a) Stone G, Wood P, Dixon L, Keyhan M, Matin A, Antimicrob. Agents Chemother 2002, 46, 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tseng BS, Zhang W, Harrison JJ, Quach TP, Song JL, Penterman J, Singh PK, Chopp DL, Packman AI, Parsek MR, Environ. Microbiol 2013, 15, 2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].a) Khodaparast S, Kim MK, Silpe JE, Stone HA, Environ. Sci. Technol 2017, 51, 1340. [DOI] [PubMed] [Google Scholar]; b) Pitt WG, McBride MO, Barton AJ, Sagers RD, Biomaterials 1993, 14, 605. [DOI] [PubMed] [Google Scholar]
- [43].Stewart PS, Pathog. Dis 2014, 70, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen MJ, Zhang Z, Bott TR, Biotechnol. Tech 1998, 12, 875. [Google Scholar]
- [45].a) Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, Siehnel RJ, Howell PL, Singh PK, Tolker-Nielsen T, Parsek MR, Schweizer HP, Harrison JJ, Nat. Protoc 2015, 10, 1820. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mukheijee S, Bree AC, Liu J, Patrick JE, Chien P, Kearns DB, Proc. Natl. Acad. Sci. USA 2015, 112, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.