

Abstract
PURPOSE
Comprehensive genomic profiling has defined key oncogenic drivers and distinct molecular subtypes in esophagogastric cancer; however, the number of clinically actionable alterations remains limited. To establish preclinical models for testing genomically driven therapeutic strategies, we generated and characterized a large collection of esophagogastric cancer patient–derived xenografts (PDXs).
MATERIALS AND METHODS
We established a biobank of 98 esophagogastric cancer PDX models derived from primary tumors and metastases. Clinicopathologic features of each PDX and the corresponding patient sample were annotated, including stage at diagnosis, treatment history, histology, and biomarker profile. To identify oncogenic DNA alterations, we analyzed and compared targeted sequencing performed on PDX and parent tumor pairs. We conducted xenotrials in genomically defined models with oncogenic drivers.
RESULTS
From April 2010 to June 2019, we implanted 276 patient tumors, of which 98 successfully engrafted (35.5%). This collection is enriched for PDXs derived from patients with human epidermal growth factor receptor 2–positive esophagogastric adenocarcinoma (62 models, 63%), the majority of which were refractory to standard therapies including trastuzumab. Factors positively correlating with engraftment included advanced stage, metastatic origin, intestinal-type histology, and human epidermal growth factor receptor 2–positivity. Mutations in TP53 and alterations in receptor tyrosine kinases (ERBB2 and EGFR), RAS/PI3K pathway genes, cell-cycle mediators (CDKN2A and CCNE1), and CDH1 were the predominant oncogenic drivers, recapitulating clinical tumor sequencing. We observed antitumor activity with rational combination strategies in models established from treatment-refractory disease.
CONCLUSION
The Memorial Sloan Kettering Cancer Center PDX collection recapitulates the heterogeneity of esophagogastric cancer and is a powerful resource to investigate mechanisms driving tumor progression, identify predictive biomarkers, and develop therapeutic strategies for molecularly defined subsets of esophagogastric cancer.
INTRODUCTION
Comprehensive genomic analysis has yielded important insights into the molecular complexity of esophagogastric (EG) cancer.1-5 The Cancer Genome Atlas identified four gastric cancer subtypes: Epstein-Barr virus (EBV)-positive, microsatellite-instable (MSI), genomically stable (GS), and chromosomal-instable (CIN).2 Despite this greater understanding of EG cancer biology, genomically targeted therapeutics benefit a limited fraction of patients.4 For patients with human epidermal growth factor receptor 2 (HER2/ERBB2)-positive tumors (approximately 25%), the anti-HER2 antibody trastuzumab and the antibody-drug conjugate trastuzumab-deruxtecan are approved.6,7 Recent data demonstrate synergy between trastuzumab and immunotherapy such as pembrolizumab.8,9 Although studies have suggested potential roles for targeting alterations in other receptor tyrosine kinases (RTKs) such as EGFR and MET, responses tend to be short-lived, and trials in large populations have failed to show a benefit.10-13 One of the main challenges is tumoral heterogeneity (both spatial and temporal), leading to therapeutic resistance such as through loss of the targeted biomarker or co-occurring alterations.14-16 Recent studies highlight that clinical benefit can be attained with individually optimizing chemotherapy, biomarker profiling, and matching of targeting therapies at baseline and over time for EG cancer.17,18 Moreover, correlative analysis suggests that dual targeted inhibition may be advantageous, warranting preclinical studies to optimize these combinatorial strategies.
CONTEXT
Key Objective
Esophagogastric (EG) cancer remains the third leading cause of cancer death globally, and existing targeted therapies are limited. We established a large collection of EG cancer patient–derived xenografts (PDXs) derived from primary tumors and metastases. The objective of this study was to clinically and genomically annotate these PDXs to define features that affect engraftment and develop preclinical models that harbor the genomic alterations and heterogeneity found in patients.
Knowledge Generated
We confirmed factors that correlate with successful PDX engraftment including advanced stage, metastatic origin, intestinal-type histology, and human epidermal growth factor receptor 2–positivity. EG PDXs retain the key genomic features found in parental tumors and can be used in preclinical studies to test rationally designed combinations targeting genomic alterations.
Relevance
This molecularly annotated collection of EG cancer PDXs serves as a valuable resource to understand tumor biology, investigate mechanisms of drug resistance, and evaluate genomically targeted combination strategies.
One preclinical model that has gained traction is patient-derived xenografts (PDXs), in which patient tumor fragments are directly transplanted into immunodeficient mice for propagation.19 Compared to traditional cell lines, PDXs retain the architecture and heterogeneity of patient tumors and can therefore more accurately model human cancer biology.20,21 Importantly, preclinical xenotrials validate that PDXs recapitulate the sensitivities to chemotherapy or targeted therapies of the corresponding patient tumor.22-24
Here, we present a comprehensive collection of EG cancer PDXs that spans histologic and genomic subtypes and uniquely includes many PDXs from patients with treatment-refractory HER2-positive tumors. We clinically annotated and genomically characterized the models using next-generation sequencing (NGS), with the goal of developing this collection as a resource for identifying new drug targets and understanding critical disease mechanisms.
MATERIALS AND METHODS
Generation and Maintenance of PDXs
Patients with esophageal, gastroesophageal junction, and gastric cancer undergoing surgery or biopsy at The Memorial Sloan Kettering Cancer Center (MSK) were consented to an institutional review board–approved protocol for prospective tumor engraftment and genomic profiling. Patients treated on clinical trials and patients with specific genomic alterations such as MSI-H, CDH1, or HER2-positive tumors were enriched in this cohort. The studies were conducted in accordance with the Declaration of Helsinki.
All tumors were prospectively reviewed to confirm histology, Lauren classification, and tumor purity. Samples were cut into small pieces and implanted into 6-week-old NOD SCID gamma mice either subcutaneously or orthotopically into the gastric wall. Tumors reaching a volume of 500-1,000 mm3 (or with evident signs of disease for orthotopic models) were expanded into additional mice by serial transplantation. Harvested tumors were stored in liquid nitrogen for future reimplantation and flash-frozen and formalin-fixed for downstream analysis. Representative hematoxylin and eosin and HER2 immunohistochemistry slides were reviewed by a board-certified pathologist to assess tumor content, histology, differentiation, absence of lymphoma, and HER2 expression.
Genomic Analysis
Targeted sequencing was performed on DNA extracted from PDX tissue using MSK-IMPACT, a cancer-associated gene-bait capture, NGS assay.25 Additional samples from formalin-fixed paraffin-embedded patient tumors and matched normal blood were sequenced. For PDX and patient sequences run on the same MSK-IMPACT pipeline, the corresponding matched normal was used for mutation calling; otherwise, a pooled normal was used. Genomic alterations were filtered for oncogenic variants using OncoKB,26 and molecular subtypes (MSI-H, GS, and CIN) were assigned as described previously.4 EBV testing was not performed.
In Vivo Animal Experiments
Animal work was approved by the MSK Institutional Animal Care and Use Committee. For drug efficacy experiments, tumors were implanted subcutaneously into mice and treatment was initiated when the tumor volumes reached 100 mm3. Xenografts were randomly assigned and mice were dosed with vehicle, afatinib 25 mg/kg PO once daily, rapamycin 20 mg/kg IP three times weekly, AZD8055 75 mg/kg PO three times weekly, or duligotuzumab 10 mg/kg IP twice weekly. Tumor volume was measured twice weekly using the formula (π/6) × length × width.2
Statistical Analysis
Statistical analysis was performed using GraphPad Prism Version 9.0.2 or R version 4.0.0. Two-sided Student's t-tests were used to evaluate significant differences in tumor volumes. Associations between successful PDX engraftment and clinical features were analyzed using a two-sided Fisher's exact test or chi square test. The Kaplan-Meier methodology and log-rank test were used to compare survival outcomes in patients with successful or failed PDX generation. Recurrence-free survival (RFS) was defined as the time from surgery to date of recurrence or death. Overall survival was defined as the time from date of diagnosis of metastatic disease to time of death. Patients without a known event were censored at the date of last follow-up. Baseline patient characteristics were summarized using descriptive statistics.
RESULTS
Patient Characteristics
Between April 2010 and June 2019, we collected tumor samples from 225 patients with EG cancer for PDX generation. Baseline patient characteristics are summarized in the Data Supplement. The median age at initial collection was 63 years (range, 23-92 years), and 94 patients (41%) had metastatic disease. Most patients had tumors of adenocarcinoma histology (95%), including five patients (2%) with MSI-H tumors. Notably, 88 patients (39%) had HER2-positive tumors, as many PDXs were generated from patients on clinical studies testing HER2-directed therapies.
Generation of a Comprehensive Collection of Esophagogastric Cancer PDXs
In total, 276 tumor samples were implanted either subcutaneously or orthotopically into the gastric wall of mice (Fig 1A). Of the 117 xenografts that engrafted, we excluded 19 models because of histology consistent with possible lymphoproliferative disease, which is known to develop in xenografts implanted into immunodeficient mice and is typically driven by EBV-transformed lymphocytes.27 Consequently, we established 98 EG PDXs, correlating with a success rate of 35.5%. The median time to first passage of successfully generated PDXs was 16.1 weeks (range, 4.6-67.6 weeks).
FIG 1.
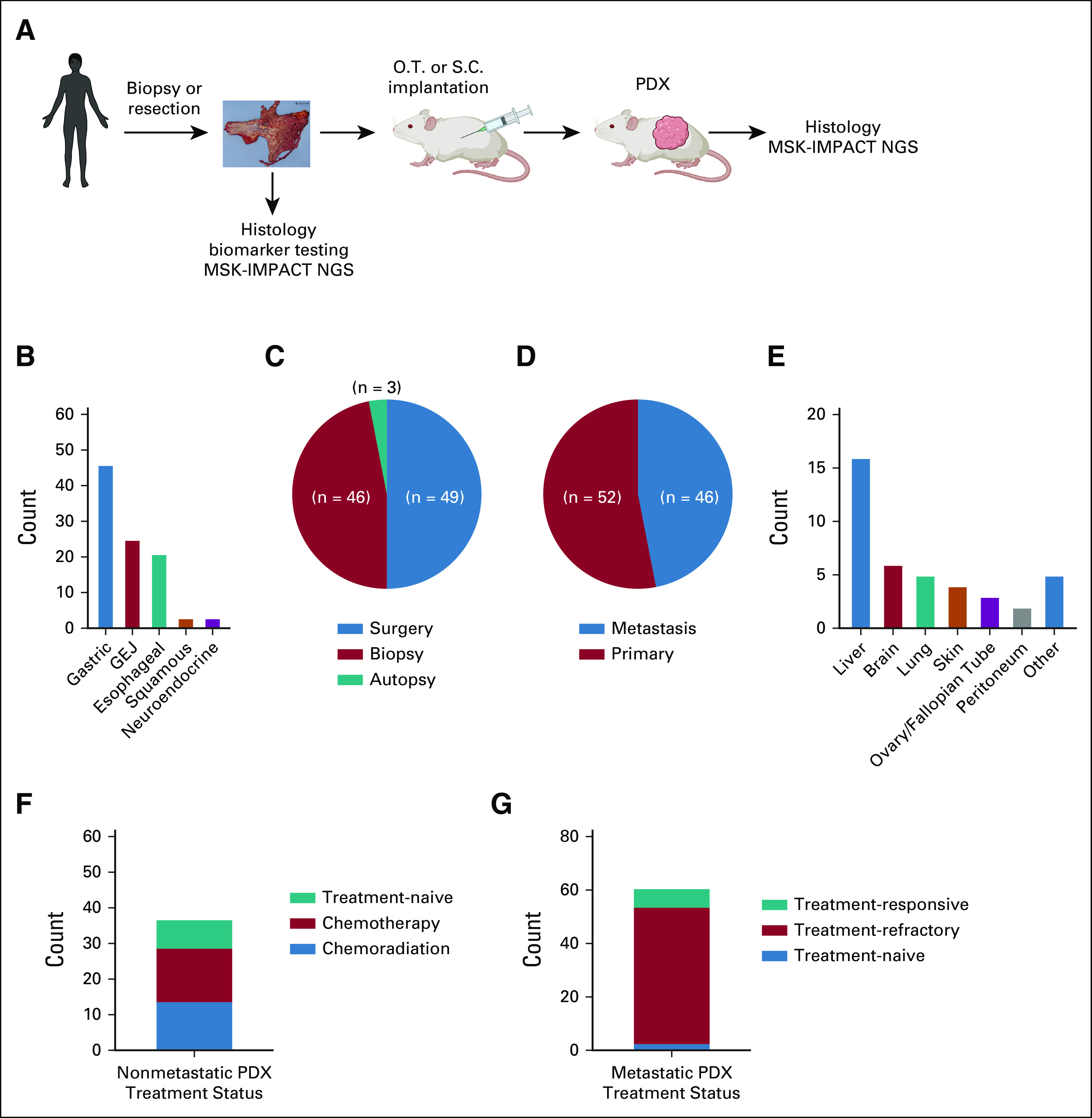
Generation of a comprehensive collection of esophagogastric cancer PDXs. (A) Schematic overview of esophagogastric cancer PDX pipeline. (B) Numbers of successfully established PDX by disease subtype. (C) Distribution of procedures from which PDXs were generated. (D) Distribution of PDXs generated from primary tumors versus metastatic sites. (E) Numbers of PDXs generated by metastatic site of origin. (F) Treatment status of PDXs from patients treated for early-stage (I-III) disease. (G) Treatment status of PDXs from patients treated for metastatic disease. PDX, patient-derived xenograft.
Features of the EG PDX collection are detailed in Figure 1. Of the 98 PDXs, there are 46 gastric adenocarcinomas (47%), 25 gastroesophageal junction adenocarcinomas (26%), 21 esophageal adenocarcinomas (32%), three squamous cell carcinomas (3%), and three neuroendocrine tumors (3%; Fig 1B). Forty-nine PDXs were generated from surgical resections, predominantly from primary tumors but also a few from metastasectomies (ie, ovary and lung), and 46 PDXs were from biopsies (Fig 1C). In addition, three PDXs were generated from participants in a rapid autopsy program. The 46 PDXs generated from metastases were collected from a range of sites, most commonly liver (Figs 1D and 1E).
Treatment history of the parental patient tumors is critical for understanding selective pressures, which may affect the biology of each model, as well for correlating PDX and patient treatment response. Of the 37 PDXs generated from localized EG cancer, 29 tumors had been treated with preoperative chemotherapy or chemoradiation (Fig 1F). PDXs from patients with metastatic disease were primarily generated from patients who had progressed following treatment with one or more lines of systemic therapy (Fig 1G).
Factors Associated with Successful PDX Establishment
To define factors affecting PDX generation, we compared clinicopathologic features of tumors that failed to versus successfully engrafted (Table 1). Interestingly, EG PDXs were significantly more likely to be established from metastases than primary tumors (Fig 2A), with 52% of metastatic tumors resulting in established PDXs versus 28% of primary tumors. Consistent with metastatic tumors having enhanced growth capacity, PDXs were more likely to engraft from tumor samples harvested from patients with stage IV disease (Fig 2B). Histology also affected PDX establishment, as gastric tumors of intestinal-type histology were more likely to form PDXs compared with diffuse or mixed-type tumors (Fig 2C). In addition, HER2 expression positively correlated with successful engraftment (Fig 2D).
TABLE 1.
Factors Associated With Successful Patient-Derived Xenograft Generation
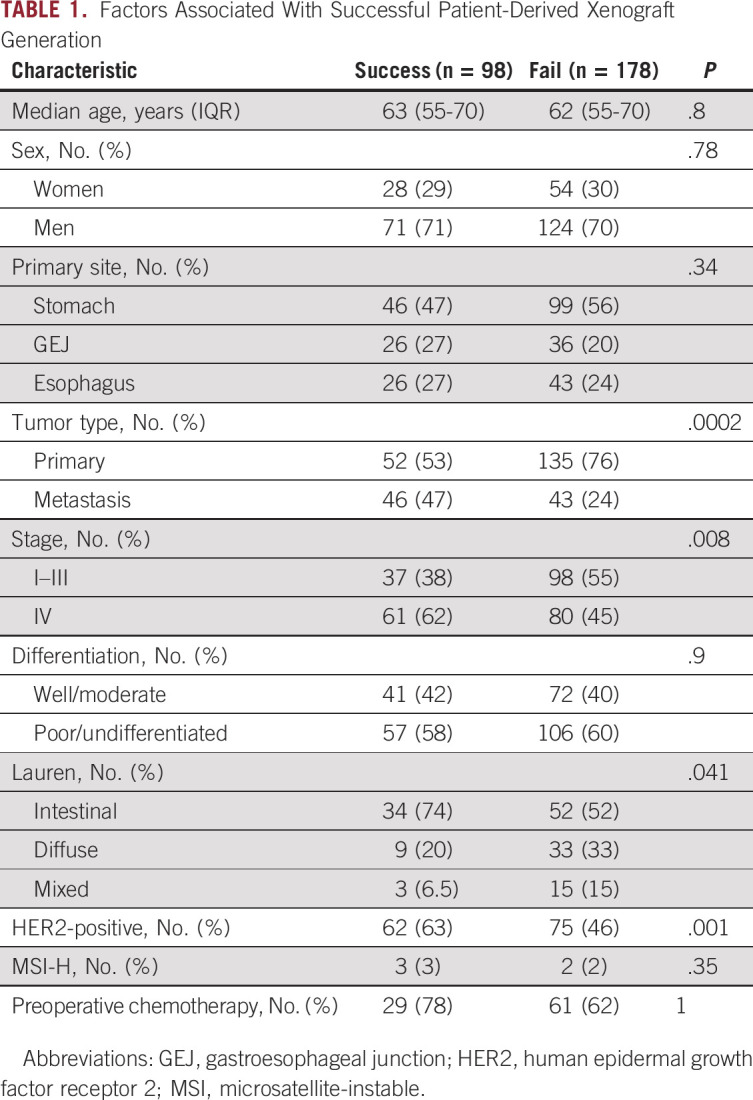
FIG 2.
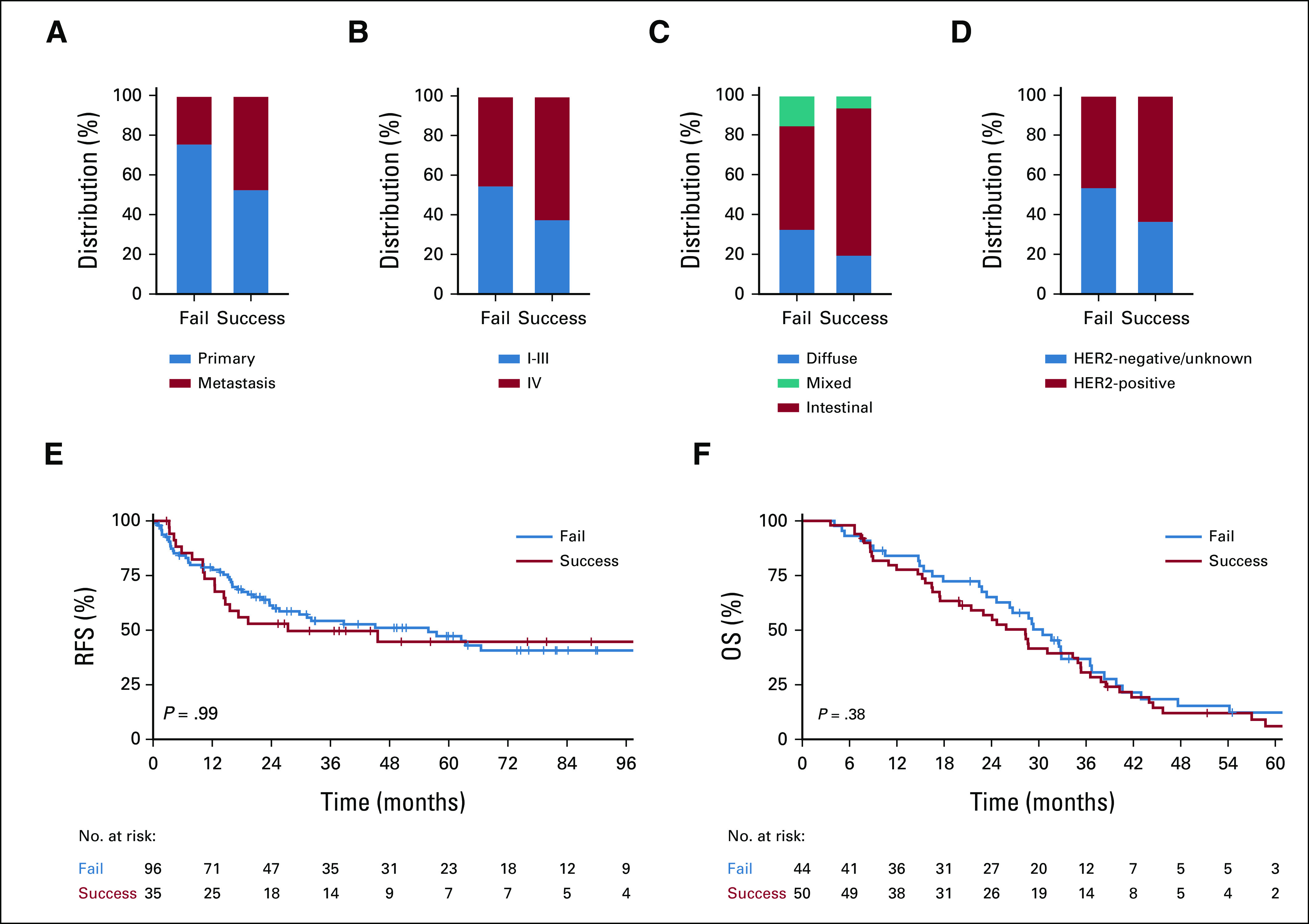
Factors associated with successful PDX establishment. (A) Distribution of tumor site among successful and failed PDX attempts. (B) Distribution of cancer stage for the corresponding patient among successful and failed PDX attempts. (C) Distribution of tumor histology by Lauren classification among successful and failed PDX attempts. (D) Distribution of HER2-negative/unknown and HER2-positive tumors among successful and failed PDX attempts. (E) Recurrence-free survival of patients with early-stage disease and successful or failed PDX generation. (F) Overall survival of patients with metastatic disease and successful or failed PDX generation. Select patients had multiple PDX tumor samples harvested for PDX generation, and patients were considered in the success cohort if any PDX attempt resulted in successful engraftment. HER2, human epidermal growth factor receptor 2; OS, overall survival; PDX, patient-derived xenograft; RFS, recurrence-free survival.
In other tumor types such as pancreatic cancer28 and colorectal cancer,29 studies have found that patients with successful PDX engraftment have worse survival outcomes, suggesting that PDX engraftment can be prognostic. We assessed RFS of patients who underwent primary tumor resection for localized disease and found no difference in RFS between patients with failed and successful PDX generation (Fig 2E). Similarly, there was no difference in overall survival in patients with stage IV disease on the basis of PDX engraftment (Fig 2F). Thus, in this cohort, successful PDX establishment was not prognostic of patient outcome.
EG PDXs Retain Genomic Features of Patient Tumors
To determine whether key genomic alterations were retained in PDXs, we performed NGS using MSK-IMPACT25 and compared PDX sequencing to clinical sequencing from the corresponding patient (Fig 3A, Data Supplement). Mutations in TP53 were the most commonly identified alterations in PDXs (80%), similar to prior clinical sequencing cohorts of EG cancer.1,2,4 On the basis of the high representation of HER2-positive tumors and increased likelihood of PDX generation, a large percentage of sequenced PDXs showed alterations in ERBB2, primarily copy-number amplifications (57%). We also observed frequent alterations in KRAS; RTKs such as EGFR, MET, and FGFR2; PI3K pathway genes such as PIK3CA and PTEN; cell-cycle regulators including CDKN2A and CCNE1; and CDH1, which is commonly altered in diffuse-type and GS tumors. Importantly, genomic alterations found in patient tumor sequencing by clinical MSK-IMPACT were highly concordant with those identified by PDX sequencing: 234 mutation events observed in the clinical samples were also identified in the PDXs (58%), with 76 driver mutations in the clinical samples also being identified in the PDXs (60%). Among the microsatellite-stable samples, 202 mutation events observed in the clinical samples were also identified in the PDXs (83%), with 66 driver mutations in the clinical samples also being identified in the PDXs (85%). The concordance rate of ERBB2 amplification was 89% between PDX and clinical samples. We also sequenced 34 additional PDXs lacking corresponding patient sequencing (Data Supplement).
FIG 3.

Genomic characterization of esophagogastric PDXs. (A) Relevant clinical features (tumor type, sample type of metastasis v primary tumor, patient HER2 status, and prior trastuzumab treatment) and key genomic alterations (mutations, structural alteration, and The Cancer Genome Atlas molecular subtype) in sequenced PDX and patient tumor samples. The percent alteration for each gene for PDX (top) and patient tumors (bottom) is provided. PDX sequencing was analyzed using either matched normal or a pooled normal as noted. Asterisk (*) denotes mutations present in clinical samples at low read counts below normal thresholds for mutation calling. (B) Representative examples of genomic alterations observed in two PDXs generated from the same patient. The site of origin (primary tumor or metastasis) is indicated. PDXs were generated at different time points unless indicated. CIN, chromosomal-instable; GEJ, gastroesophageal junction; GS, genomically stable; HER2, human epidermal growth factor receptor 2; MSI, microsatellite-instable; NA, not available; NE, neuroendocrine; PDX, patient-derived xenograft; SCC, squamous cell carcinoma; VUS, variants of unknown significance.
For a few patients, we generated PDXs from multiple tumor sites at the same time point (synchronous) or at different time points (ie, preprogression and postprogression; Fig 3B). In some cases, we observed the same genomic alterations across sites. For example, two PDXs generated from pleural fluid and ascites both harbored a TP53 mutation and amplifications in CCNE1, ERBB2, and KRAS, consistent with these being early oncogenic driver alterations. By contrast, we observed intralesional and temporal heterogeneity in other cases, such as a PDX from an ovarian metastasis carrying an acquired MET amplification compared with a PDX from the primary tumor.
Several studies have identified mechanisms of intrinsic and acquired resistance to HER2-targeted therapy.4 Therefore, we evaluated the presence of known resistance mechanisms in HER2-positive PDXs. One mechanism of resistance is coalteration of RTK, RAS, or PI3K pathway genes. Among the 24 ERBB2-amplified PDXs, three had amplifications in KRAS, three had amplifications in RTK genes (ie, EGFR, FGFR2, or MET), and five had oncogenic driver alterations in PIK3CA or PTEN. Similar rates of coalterations were found in PDXs from metastases (41%) and primary tumors (43%). In addition to several trastuzumab-refractory PDX and clinical samples with no ERBB2 amplification identified, we also observed three PDXs with discordance in ERBB2 amplification between PDX and clinical sequencing, consistent with either heterogeneity in HER2 expression or loss of HER2 expression following trastuzumab.4 Taken together, this genomic analysis demonstrates that EG PDXs retain key molecular features relevant to human tumor biology.
Utilization of EG PDXs for Investigation of Genomically Driven Therapeutic Strategies
PDXs have been used to characterize resistance mechanisms to targeted therapies, such as mutations affecting drug binding or activation of bypass pathways. For example, we previously reported a phase II study of afatinib, an irreversible pan-HER kinase inhibitor, in patients with HER2-positive EG cancer refractory to trastuzumab.30 Investigation of a PDX from a patient with an acquired MET amplification after progression on afatinib demonstrated that the PDX was sensitive to the combination of afatinib and an MET inhibitor, implicating MET amplification as the biologic basis for afatinib resistance. Less is known about the role of PDXs in understanding variants of unknown significance (VUS) detected in clinical sequencing. To that end, we examined additional PDXs from patients with progression on afatinib to evaluate whether shared alterations between PDXs and resistant patient tumors could be successfully targeted.
One PDX was established from an umbilical nodule from a patient with metastatic HER2-positive gastric adenocarcinoma following progression on second-line afatinib. From clinical sequencing, we observed a TP53 mutation and ERBB2 amplification that was found in a pretreatment liver biopsy as well as the before and after afatinib biopsies. Interestingly, however, we identified a VUS in MTOR (D1105N) detected only in samples collected after progression on first-line therapy and retained in the PDX (Fig 4A). We treated mice bearing this PDX with afatinib, the mTORC1 inhibitor rapamycin, the selective mTORC1/2 inhibitor AZD8055, and the combination of afatinib/rapamycin. Both rapamycin and AZD8055 slowed tumor growth, and notably, the combination of rapamycin and afatinib most potently induced tumor regression (Fig 4B). These results provide further justification for additional biologic studies to assess the oncogenicity of the D1105N MTOR VUS.
FIG 4.
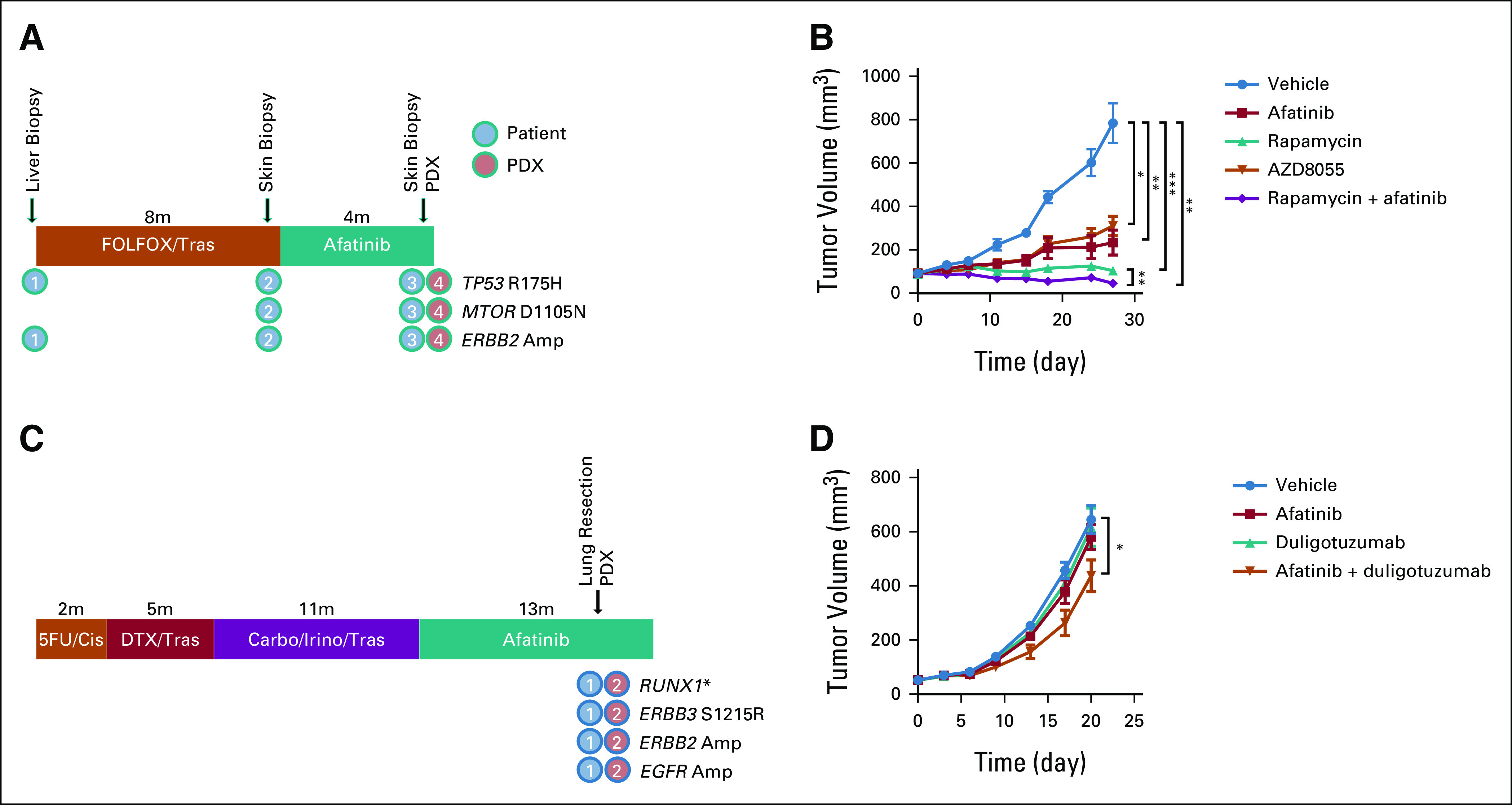
Utilization of esophagogastric PDXs for investigation of genomically driven therapeutic strategies. (A) A PDX was generated from a patient with HER2-positive gastric cancer after progression on first-line trastuzumab and second-line afatinib. Next-generation sequencing revealed a mutation in MTOR (VUS) in clinical sequencing that was retained in the PDX. (B) Efficacy of afatinib and MTOR inhibitors in an ERBB2-amplified/MTOR-altered PDX. (C) A PDX was generated from a patient with a VUS in ERBB3 after progression on afatinib. (D) Efficacy of afatinib or a dual EGFR/HER3 inhibitor (duligotuzumab) as monotherapy or in combination. *P < .05, **P < .01, ***P < .001; Student's t-test. 5-FU, fluorouracil; Carbo, carboplatin; Cis, cisplatin; DTX, docetaxel; FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; HER2, human epidermal growth factor receptor 2; Irino, irinotecan; PDX, patient-derived xenograft; Tras, trastuzumab.
In another PDX model, we evaluated the potential role of targeting an ERBB3 coalteration (Fig 4C). The corresponding patient with trastuzumab-refractory HER2-positive gastric adenocarcinoma received afatinib for 10 months before developing a lung metastasis, which was resected and used for PDX generation. Intriguingly, both the PDX and corresponding tumor harbored a VUS in ERBB3 (S1215R), as well as amplifications in ERBB2 and EGFR. Although afatinib is a pan-HER kinase inhibitor, it has strongest activity against HER2, EGFR, and HER4, with only indirect activity on HER3/ERBB3.31 We treated mice bearing this PDX with afatinib, a dual EGFR/HER3 antibody duligotuzumab,32 combined afatinib/duligotuzumab, or vehicle. Consistent with the clinical history, the PDX was resistant to afatinib monotherapy, as well as duligotuzumab; however, we observed a modest decrease in tumor growth rate with the combination (Fig 4D), suggesting that HER3 may partially mediate resistance, although other mechanisms likely contribute. Collectively, these studies highlight the potential utility of EG PDXs in evaluating therapeutic targets on the basis of genomic data.
DISCUSSION
PDXs have emerged as powerful preclinical models for interrogating clinically relevant disease biology as they retain the three-dimensional architecture, molecular complexity, and heterogeneity of human cancers. Here, we have established a large collection of clinically and genomically annotated EG PDXs, including many PDXs from metastatic tumors that are refractory to standard therapies.
A few collections of gastric cancer PDXs have been reported. For example, in one collection from Yonsei University, 15 PDXs were established from 62 gastric cancers (24.2% success rate).33 This study found that diffuse-type tumors and low tumor cell percentages were associated with poor engraftment. Another study attempted to generate PDXs from 100 gastric tumors, successfully generating 27 PDXs, which were more likely intestinal histology, CIN subtype, and MSI-H.34 Finally, a collection of 100 gastric cancer PDXs generated from 349 tumors was recently reported (28.7% success rate).35 Again, histologic type and MSI-H status were associated with engraftment, as well as advanced stage and high RTK/RAS copy-number variation. In our study, we generated 98 PDXs from 276 tumors (35.5% success rate). This rate is slightly higher than previous studies, although it may reflect the clinicopathologic features of tumors rather than procedural differences, as our population had a higher proportion of patients with metastatic disease and HER2-positive tumors, which we found as being positively correlated with PDX success. Of note, PDX engraftment is highly variable by tumor type, with some cancers such as colorectal cancer showing engraftment rates of over 89%,36 whereas breast cancer PDXs have a lower take rate of 10%-25%.37 The lower engraftment rate for EG PDXs is likely related to tumor-intrinsic factors. For example, diffuse-type gastric tumors are characterized by single tumor cells or small clusters embedded within dense fibrous stroma. Moreover, the PDX versus patient tumor microenvironment may affect engraftment as well as treatment response because of differences in tissue stroma (including immune cells), local cytokines, and growth factor milieu.
Importantly, NGS revealed that EG PDXs maintained key genomic alterations found in the corresponding patients, suggesting that they may be suitable models to investigate potentially targetable alterations. Indeed, we and others have shown that EG PDXs can mimic responses to standard therapy and investigational agents, and that PDXs with coalterations in HER2 and RTK pathways can be used to validate putative resistance mechanisms.20,30 In this study, we found that VUS identified in PDXs can be evaluated to prioritize genomic alterations such as those in MTOR and ERBB3 of unknown biologic and clinical significance for further studies. These studies are hypothesis-generating, and further mechanistic studies would be needed to define the oncogenicity of these VUS. Of note, we validated that these alterations were also found in clinical sequencing; in the absence of matched clinical sequencing or normal tissue, it is difficult to ascertain whether variants outside of known driver mutations represent true somatic tumor alterations or polymorphisms.
Interestingly, several academic and industry pipelines are interrogating PDXs as avatar models in coclinical trials to guide clinical decision making.20 In our study, the median time to first passage was 16 weeks, and therefore, it takes several months before a PDX model is ready for efficacy studies. Given that the median survival of patients with metastatic EG cancer is approximately a year, it is unlikely that these studies can be reliably performed on a timeframe that is relevant to most patient donors. Thus, other models such as patient-derived organoids may be more relevant for predicting drug response and guiding treatment as avatars for individual patients.38 Nevertheless, we believe that PDXs still serve an important role as a clinically relevant model for testing genomically driven concepts that can affect drug development and clinical trial design.
In summary, we have generated a comprehensive clinically and molecularly annotated collection of EG PDXs that will provide an invaluable resource for interrogating EG cancer biology and genomic targets to drive discoveries that can be translated to the clinic. Future efforts should focus on using PDXs to dissect drug resistance mechanisms through pharmacologic and genetic studies, as well as optimizing methods to generate these models more efficiently by integrating transcriptional, proteomic, and other molecular studies to identify additional factors that regulate engraftment.
APPENDIX
FIG A1.

Genomic profiling of esophagogastric cancer patient–derived xenografts.
Daniela Molena
Honoraria: Bristol Myers Squibb/Pfizer, Merck
Consulting or Advisory Role: Johnson & Johnson, UroGen pharma, Boston Scientific, AstraZeneca/MedImmune
Steven B. Maron
Stock and Other Ownership Interests: Calithera Biosciences
Consulting or Advisory Role: Natera, Basilea, Daichi Sankyo, Bicara Therapeutics, Novartis
Research Funding: Roche/Genentech (Inst), Guardant Health (Inst)
Travel, Accommodations, Expenses: Bayer
David R. Jones
Consulting or Advisory Role: Merck, AstraZeneca
Jaclyn F. Hechtman
Employment: NeoGenomics Laboratories
Stock and Other Ownership Interests: NeoGenomics Laboratories
Honoraria: WebMD, Illumina, Bayer
Consulting or Advisory Role: Cor2Ed, Axiom Healthcare Strategies, Bayer
Research Funding: Bayer, Lilly, Boehringer Ingelheim
David B. Solit
This author is a member of the JCO Precision Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Stock and Other Ownership Interests: Scorpion Therapeutics, Vividion Therapeutics, Fore Biotherapeutics
Consulting or Advisory Role: Pfizer, Lilly, BridgeBio Pharma, Scorpion Therapeutics, Vividion Therapeutics, Syros Pharmaceuticals
Yelena Y. Janjigian
Stock and Other Ownership Interests: Rgenix
Consulting or Advisory Role: Pfizer, Merck, Bristol Myers Squibb, Merck Serono, Daiichi Sankyo, Rgenix, Bayer, Imugene, AstraZeneca, Lilly, Zymeworks, Basilea Pharmaceutical, Michael J. Hennessy Associates, Paradigm, Seattle Genetics
Research Funding: Bayer (Inst), Rgenix (Inst), Bristol Myers Squibb (Inst), Merck (Inst), Lilly (Inst), NCI (Inst), Department of Defense (Inst), Cycle for Survival (Inst), Fred's Team (Inst), Genentech/Roche (Inst)
Other Relationship: Clinical Care Options, Axis Medical Education, Research to Practice
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part as a poster at the virtual ASCO Gastrointestinal Cancers Symposium, January 15-17, 2021.
SUPPORT
Supported by grants from the Department of Defense Congressionally Directed Medical Research Program (CA150646 and CA150647P1; YYJ); Marie-Josée and Henry R. Kravis Center for Molecular Oncology, Conquer Cancer Foundation of the American Society of Clinical Oncology (Y.Y.J., R.H.M.), the Society of MSK (Y.Y.J.), NIH grant K12CA184746 (S.B.M.), NIH grant U54OD020355 (E.d.S.), and an NIH/NCI Cancer Center Support Grant P30CA008748. R.H.M. was supported by NIH training grant T32CA009207 and is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG 121-20).
AUTHOR CONTRIBUTIONS
Conception and design: Ryan H. Moy, Vivian E. Strong, Daniel G. Coit, David R. Jones, Yelena Y. Janjigian
Financial support: David B. Solit, Yelena Y. Janjigian
Administrative support: Daniel G. Coit, David B. Solit, Yelena Y. Janjigian
Provision of study materials or patients: Daniela Molena, Laura H. Tang, Steven B. Maron, Daniel G. Coit, David B. Solit, Yelena Y. Janjigian
Collection and assembly of data: Ryan H. Moy, Henry S. Walch, Marissa Mattar, Laura H. Tang, Daniel G. Coit, Jaclyn F. Hechtman, David B. Solit, Elisa de Stanchina, Yelena Y. Janjigian
Data analysis and interpretation: Ryan H. Moy, Henry S. Walch, Marissa Mattar, Walid K. Chatila, Daniela Molena, Vivian E. Strong, Laura H. Tang, Steven B. Maron, Daniel G. Coit, Jaclyn F. Hechtman, David B. Solit, Nikolaus Schultz, Elisa de Stanchina, Yelena Y. Janjigian
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Ryan H. Moy, Henry S. Walch, Marissa Mattar, Walid K. Chatila, Daniela Molena, Vivian E. Strong, Laura H. Tang, Steven B. Maron, Daniel G. Coit, David R. Jones, Jaclyn F. Hechtman, David B. Solit, Nikolaus Schultz, Elisa de Stanchina, David B. Solit, Yelena Y. Janjigian
Daniela Molena
Honoraria: Bristol Myers Squibb/Pfizer, Merck
Consulting or Advisory Role: Johnson & Johnson, UroGen pharma, Boston Scientific, AstraZeneca/MedImmune
Steven B. Maron
Stock and Other Ownership Interests: Calithera Biosciences
Consulting or Advisory Role: Natera, Basilea, Daichi Sankyo, Bicara Therapeutics, Novartis
Research Funding: Roche/Genentech (Inst), Guardant Health (Inst)
Travel, Accommodations, Expenses: Bayer
David R. Jones
Consulting or Advisory Role: Merck, AstraZeneca
Jaclyn F. Hechtman
Employment: NeoGenomics Laboratories
Stock and Other Ownership Interests: NeoGenomics Laboratories
Honoraria: WebMD, Illumina, Bayer
Consulting or Advisory Role: Cor2Ed, Axiom Healthcare Strategies, Bayer
Research Funding: Bayer, Lilly, Boehringer Ingelheim
David B. Solit
This author is a member of the JCO Precision Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Stock and Other Ownership Interests: Scorpion Therapeutics, Vividion Therapeutics, Fore Biotherapeutics
Consulting or Advisory Role: Pfizer, Lilly, BridgeBio Pharma, Scorpion Therapeutics, Vividion Therapeutics, Syros Pharmaceuticals
Yelena Y. Janjigian
Stock and Other Ownership Interests: Rgenix
Consulting or Advisory Role: Pfizer, Merck, Bristol Myers Squibb, Merck Serono, Daiichi Sankyo, Rgenix, Bayer, Imugene, AstraZeneca, Lilly, Zymeworks, Basilea Pharmaceutical, Michael J. Hennessy Associates, Paradigm, Seattle Genetics
Research Funding: Bayer (Inst), Rgenix (Inst), Bristol Myers Squibb (Inst), Merck (Inst), Lilly (Inst), NCI (Inst), Department of Defense (Inst), Cycle for Survival (Inst), Fred's Team (Inst), Genentech/Roche (Inst)
Other Relationship: Clinical Care Options, Axis Medical Education, Research to Practice
No other potential conflicts of interest were reported.
REFERENCES
- 1.Cancer Genome Atlas Research Network, Analysis Working Group, Asan University, BC Cancer Agency. et al. Integrated genomic characterization of oesophageal carcinoma Nature 541169–1752017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma Nature 513202–2092014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes Nat Med 21449–4562015 [DOI] [PubMed] [Google Scholar]
- 4.Janjigian YY, Sanchez-Vega F, Jonsson P, et al. Genetic predictors of response to systemic therapy in esophagogastric cancer Cancer Discov 849–582018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J Clin 71209–2492021 [DOI] [PubMed] [Google Scholar]
- 6.Bang Y-J, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial Lancet 376687–6972010 [DOI] [PubMed] [Google Scholar]
- 7.Shitara K, Bang Y-J, Iwasa S, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer N Engl J Med 3822419–24302020 [DOI] [PubMed] [Google Scholar]
- 8.Janjigian YY, Maron SB, Chatila WK, et al. First-line pembrolizumab and trastuzumab in HER2-positive oesophageal, gastric, or gastro-oesophageal junction cancer: An open-label, single-arm, phase 2 trial Lancet Oncol 21821–8312020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janjigian YY, Kawazoe A, Yanez PE, et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2+ metastatic gastric or gastroesophageal junction (G/GEJ) cancer: Initial findings of the global phase 3 KEYNOTE-811 study. J Clin Oncol. 2021;39 suppl; abstr 4013. [Google Scholar]
- 10.Lordick F, Kang Y-K, Chung H-C, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomised, open-label phase 3 trial Lancet Oncol 14490–4992013 [DOI] [PubMed] [Google Scholar]
- 11.Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial Lancet Oncol 14481–4892013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial Lancet Oncol 181467–14822017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah MA, Bang Y-J, Lordick F, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: The METGastric randomized clinical trial JAMA Oncol 3620–6272017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pectasides E, Stachler MD, Derks S, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma Cancer Discov 837–482018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maron SB, Alpert L, Kwak HA, et al. Targeted therapies for targeted populations: Anti-EGFR treatment for EGFR-amplified gastroesophageal adenocarcinoma Cancer Discov 8696–7132018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura Y, Kawazoe A, Lordick F, et al. Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: An emerging paradigm Nat Rev Clin Oncol 18473–4872021 [DOI] [PubMed] [Google Scholar]
- 17.Catenacci DVT, Moya S, Lomnicki S, et al. Personalized antibodies for gastroesophageal adenocarcinoma (PANGEA): A phase II study evaluating an individualized treatment strategy for metastatic disease Cancer Discov 11308–3252021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Kim ST, Kim K, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial Cancer Discov 91388–14052019 [DOI] [PubMed] [Google Scholar]
- 19. Mattar M, McCarthy CR, Kulick AR, et al. Establishing and maintaining an extensive library of patient-derived xenograft models. Front Oncol. 2018;8:18. doi: 10.3389/fonc.2018.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hidalgo M, Amant F, Biankin AV, et al. Patient-derived xenograft models: An emerging platform for translational cancer research Cancer Discov 4998–10132014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tentler JJ, Tan AC, Weekes CD, et al. Patient-derived tumour xenografts as models for oncology drug development Nat Rev Clin Oncol 9338–3502012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Misale S, Bozic I, Tong J, et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun. 2015;6:8305. doi: 10.1038/ncomms9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drapkin BJ, George J, Christensen CL, et al. Genomic and functional fidelity of small cell lung cancer patient-derived xenografts Cancer Discov 8600–6152018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izumchenko E, Paz K, Ciznadija D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors Ann Oncol 282595–26052017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients Nat Med 23703–7132017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: A precision oncology knowledge base JCO Precis Oncol 11–162017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen K, Ahmed S, Adeyi O, et al. Human solid tumor xenografts in immunodeficient mice are vulnerable to lymphomagenesis associated with Epstein-Barr virus. PLoS One. 2012;7:e39294. doi: 10.1371/journal.pone.0039294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pergolini I, Morales-Oyarvide V, Mino-Kenudson M, et al. Tumor engraftment in patient-derived xenografts of pancreatic ductal adenocarcinoma is associated with adverse clinicopathological features and poor survival. PLoS One. 2017;12:e0182855. doi: 10.1371/journal.pone.0182855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh BY, Lee WY, Jung S, et al. Correlation between tumor engraftment in patient-derived xenograft models and clinical outcomes in colorectal cancer patients Oncotarget 616059–160682015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Vega F, Hechtman JF, Castel P, et al. EGFR and MET amplifications determine response to HER2 inhibition in ERBB2-amplified esophagogastric cancer Cancer Discov 9199–2092019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker J Pharmacol Exp Ther 343342–3502012 [DOI] [PubMed] [Google Scholar]
- 32.Schaefer G, Haber L, Crocker LM, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies Cancer Cell 20472–4862011 [DOI] [PubMed] [Google Scholar]
- 33. Choi YY, Lee JE, Kim H, et al. Establishment and characterisation of patient-derived xenografts as paraclinical models for gastric cancer. Sci Rep. 2016;6:22172. doi: 10.1038/srep22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peille A-L, Vuaroqueaux V, Wong S-S, et al. Evaluation of molecular subtypes and clonal selection during establishment of patient-derived tumor xenografts from gastric adenocarcinoma. Commun Biol. 2020;3:367. doi: 10.1038/s42003-020-1077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corso S, Isella C, Bellomo SE, et al. A comprehensive PDX gastric cancer collection captures cancer cell-intrinsic transcriptional MSI traits Cancer Res 795884–58962019 [DOI] [PubMed] [Google Scholar]
- 36.Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer Cancer Discov 1508–5232011 [DOI] [PubMed] [Google Scholar]
- 37. Murayama T, Gotoh N. Patient-derived xenograft models of breast cancer and their application. Cells. 2019;8:621. doi: 10.3390/cells8060621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers Science 359920–9262018 [DOI] [PMC free article] [PubMed] [Google Scholar]