

Abstract
PURPOSE
IDH mutations occur in about 30% of patients with cholangiocarcinoma. Analysis of mutations in circulating tumor DNA (ctDNA) can be performed by droplet digital polymerase chain reaction (ddPCR). The analysis of ctDNA is a feasible approach to detect IDH mutations.
METHODS
We isolated ctDNA from the blood of patients with IDH-mutated advanced cholangiocarcinoma collected at baseline, on therapy, and at progression to isocitrate dehydrogenase (IDH) inhibitors.
RESULTS
Of 31 patients with IDH1R132 (n = 26) or IDH2R172 mutations (n = 5) in the tumor, IDH mutations were detected in 84% of ctDNA samples analyzed by ddPCR and in 83% of ctDNA samples analyzed by next-generation sequencing (NGS). Patients with a low variant allele frequency of ctDNA detected by NGS at baseline had a longer median time to treatment failure compared to patients with high variant allele frequency of ctDNA (3.6 v 1.5 months; P = .008). Patients with a decrease in IDH-mutated ctDNA on therapy by ddPCR compared with no change/increase had a trend to a longer median survival (P = .07). Most frequent emergent alterations in ctDNA by NGS at progression were ARID1A (n = 3) and TP53 mutations (n = 3).
CONCLUSION
Detection of IDH mutations in ctDNA in patients with advanced cholangiocarcinoma is feasible, and dynamic changes in ctDNA can correspond with the clinical course and clonal evolution.
INTRODUCTION
Oncogenic mutations in the isocitrate dehydrogenase 1 (IDH1) and IDH2 genes, coding for an essential enzyme for cellular respiration in the tricarboxylic acid cycle,1 are present in up to 30% of patients with cholangiocarcinoma and offer molecular targets for cancer therapy.2,3 The IDH1 inhibitor ivosidenib and IDH2 inhibitor enasidenib are approved for the treatment of acute myeloid leukemia with respective IDH mutations.4,5 In addition, ivosidenib is approved for previously treated IDH1-mutated advanced cholangiocarcinoma and other IDH inhibitors are in the late stage of clinical development in cholangiocarcinoma and in various stages of clinical development in other solid tumors such as glioma or chondrosarcoma.6-8 Treatment with IDH inhibitors is usually well tolerated; however, objective responses are infrequent, and similar to other targeted therapies, acquired resistance evolves in nearly all patients who initially responded.9
CONTEXT
Key Objective
IDH mutations are prevalent in cholangiocarcinoma, and the isocitrate dehydrogenase 1 inhibitor ivosidenib has been recently approved for patients with advanced cholangiocarcinoma previously treated with chemotherapy. However, therapeutic responses are relatively infrequent, and nearly all patients ultimately develop acquired therapeutic resistance. The objective of this study was to investigate whether analysis of plasma-derived circulating tumor DNA (ctDNA) corresponds with molecular testing of tumor tissue, and whether monitoring of dynamic changes and clonal evolution can be performed using ctDNA analysis.
Knowledge Generated
We used two different orthogonal methods, droplet digital polymerase chain reaction and targeted digital next-generation sequencing, to detect ctDNA. Our data demonstrate that IDH mutation status in ctDNA is concordant with tumor tissue and that low ctDNA levels are associated with longer time to treatment failure. Emerging alterations with predicted oncogenic potential were detected in ctDNA at the time of progression.
Relevance
We demonstrated that detection of IDH mutations in ctDNA in patients with advanced cholangiocarcinoma is feasible. Therefore, ctDNA analysis can be potentially used to select patients for treatment with isocitrate dehydrogenase inhibitors, and to monitor the emergence of molecular alteration–associated therapeutic resistance.
Circulating tumor DNA (ctDNA) is secreted into the circulation by apoptotic and necrotic cells originating from the primary and/or metastatic cancer lesions and can be used for molecular testing in lieu of cancer tissue.10 Unlike tumor biopsies, collection of blood samples for isolation of ctDNA is minimally invasive, can be repeated multiple times during therapy and, therefore, can be used to study clonal evolution and mechanisms of adaptive resistance. The purpose of our study was to investigate whether the results of molecular testing of plasma-derived ctDNA using two orthogonal methods (comprehensive targeted next-generation sequencing [NGS] and droplet digital polymerase chain reaction [ddPCR]) each correspond with the results of molecular testing of tumor tissue, and whether serial collection of ctDNA can be used for monitoring dynamic changes in ctDNA and clonal evolution studies in patients with advanced cholangiocarcinoma and IDH1/2 mutation treated with IDH inhibitors.
METHODS
Patients
The study enrolled patients with advanced cholangiocarcinoma and known IDH1 or IDH2 mutations detected by NGS in archival formalin-fixed paraffin-embedded tumor samples as a part of clinical care referred to MD Anderson Cancer Center for treatment with experimental IDH inhibitors between March 2015 and October 2017 and who consented to the optional collection of blood samples for retrospective analysis of ctDNA. The study was conducted in accordance with institutional review board guidelines.
Blood Collection and Processing
Blood samples were collected before starting treatment and then serially on therapy with IDH inhibitors (approximately every 3-4 weeks). Whole blood was collected in EDTA tubes and centrifuged and spun twice within 2 hours to yield plasma. The QIAamp Circulating Nucleic Acid kit (Qiagen, Valencia, CA) was used to isolate cell-free DNA (cfDNA) according to the manufacturer's instructions. Quantitation of cfDNA was done with Quant-iT PicoGreen dsDNA Reagent and Kits (Invitrogen, Carlsbad, CA).
Mutation Detection in cfDNA by ddPCR
Mutation-specific assays were used to distinguish the wild-type allele from hotspot mutations in the IDH1 (R132C, R132G, R132F, R132L, and R132S), IDH2 (R172W, R172K, R172M, and R172G), KRAS (G12V and G12D), and PIK3CA (H1047R) genes present in the tumor tissue of the patients using the QX200 Droplet Digital PCR platform (Bio-Rad, Hercules, CA) according to the manufacturer's standard protocol. A total of 16 ng of cfDNA (if available; median, 16 ng; range, 1-55 ng) was used as input in duplicate reactions. The investigator who performed the mutation analysis of the cfDNA samples was blinded to the results of testing of archival tumor tissue, and appropriate positive and negative controls were used. The lower limit of detection was approximately < 0.1% variant allele frequency (VAF) per single well for the mutation-specific assays.
Mutation Detection in cfDNA by Targeted Digital NGS
Targeted digital NGS of cfDNA was done using the 74 genes assay Guardant360, which is Clinical Laboratory Improvement Amendments–certified, College of American Pathologist–accredited, and performed in the New York State Department of Health–approved laboratory (Guardant Health, Redwood City, CA).11-13 In our study, we used a median of 36 ng (range, 1.8-1,106 ng) of cfDNA, which was exposed to barcoding to be processed and sequenced into a digital library as described earlier (Data Supplement).11,12
Statistical Analysis
The Mann-Whitney U test was used to compare groups, and the Wilcoxon signed-rank test was used to compare paired samples collected before therapy and at progression. Time to treatment failure (TTF) was defined as the time from the initiation of treatment with IDH inhibitor to the date of treatment discontinuation. Survival was defined as the time from the initiation of treatment with IDH inhibitor to the date of death or last follow-up. The Kaplan-Meier method was used to estimate TTF and survival, and a log-rank test was used to compare differences among subgroups. All tests were two-sided, and P values < .05 were considered statistically significant. All statistical analyses were performed with the SPSS 24 (SPSS, Chicago, IL) software.
RESULTS
Patient Characteristics
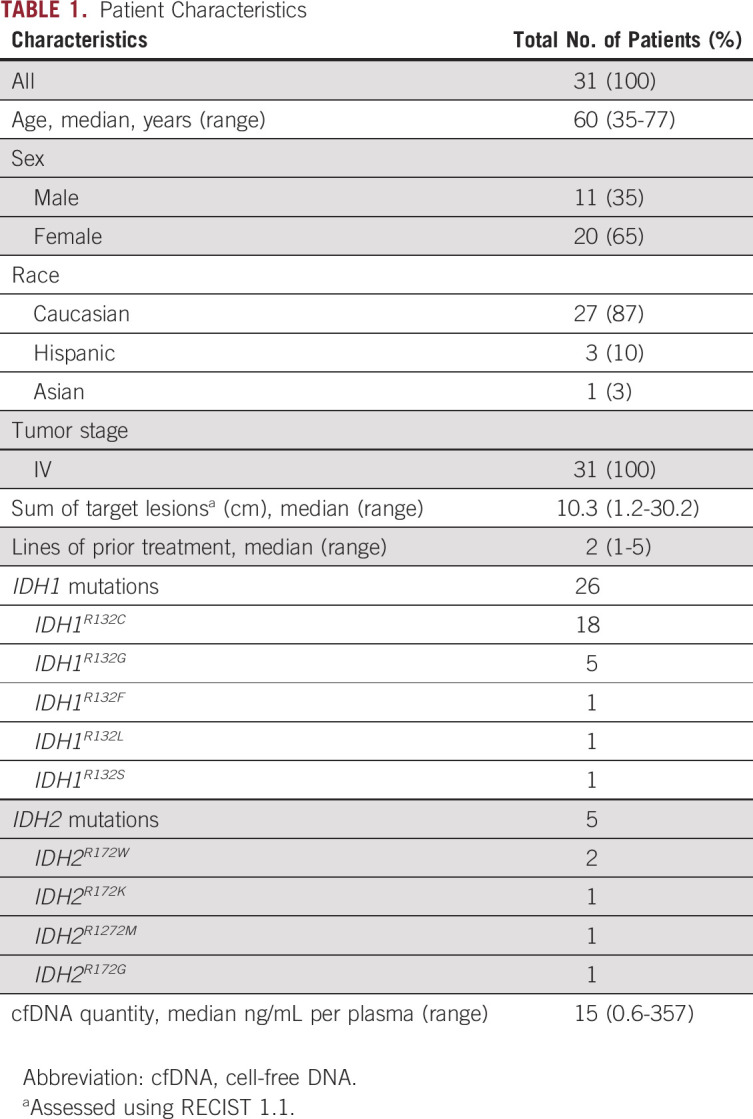
The study enrolled 31 patients with advanced cholangiocarcinoma and mutations in the IDH1 or IDH2 genes determined by analysis of archival formalin-fixed paraffin-embedded tumor tissue, who were dispositioned to start on treatment with IDH1 or IDH2 inhibitors (Table 1). The median age was 60 years (range, 35-77 years). Most patients were White (27; 87%) and female (21; 65%). All patients had stage IV disease, a median tumor burden measured by the sum of longest diameters of target lesions (per RECIST 1.1) was 10.3 cm, and patients received a median of two prior therapies. The most common IDH mutations were IDH1R132C mutation (18; 58%), followed by IDH1R132G mutation (five; 16%). The median amount of cfDNA isolated per 1 mL of plasma was 15 ng (range, 0.6-357 ng).
TABLE 1.
Patient Characteristics
Concordance Between Tumor Tissue and ctDNA
All 31 patients included in the study had known IDH1R132 (26; 84%) or IDH2R172 (5; 16%) mutations in the tumor tissue. First, we analyzed the presence of IDH mutations in ctDNA from available blood samples from 31 patients collected before starting therapy with IDH inhibitors with ddPCR using 16 ng of cfDNA as input. IDH mutations were detected in ctDNA from 26 of 31 patients yielding a sensitivity and observed agreement of 84% (95% CI, 66 to 95). The sensitivity for IDH1 mutations was 85% (95% CI, 65 to 96) and for IDH2 mutations was 80% (95% CI, 28 to 99). Observed VAFs ranged from 0% to 16.2%, with a median of 1.4%.
Then, we analyzed the presence of IDH mutations in ctDNA from available blood samples from 29 of 31 patients (samples from two patients failed quality control) collected before starting therapy with IDH inhibitors with targeted NGS using a median 36 ng (2.6 ng-175.7 ng) of cfDNA as input. IDH mutations were detected in ctDNA from 24 of 29 patients yielding a sensitivity and observed agreement of 83% (95% CI, 64 to 94). The sensitivity for IDH1 mutations was 83% (95% CI, 63 to 95) and for IDH2 mutations was 80% (95% CI, 28 to 99). Observed VAFs ranged from 0% to 12.9%, with a median 2.2%.
Finally, we analyzed agreement between 29 patients, who had available blood samples collected before therapy, analyzed by both ddPCR and digital NGS. Of these patients, 23 (79%: 95% CI, 60 to 92) had concordant results. Moreover, only two (7%) patients were negative for IDH mutations by both ddPCR and digital targeted NGS (Table 2). There was a correlation for reported VAF between ddPCR and NGS (0.8; P < .001; Fig 1A).
TABLE 2.
Concordance for IDH1 and IDH2 Mutations Among Tumor Tissue, and Blood ctDNA Analyzed by ddPCR or Targeted Digital NGS
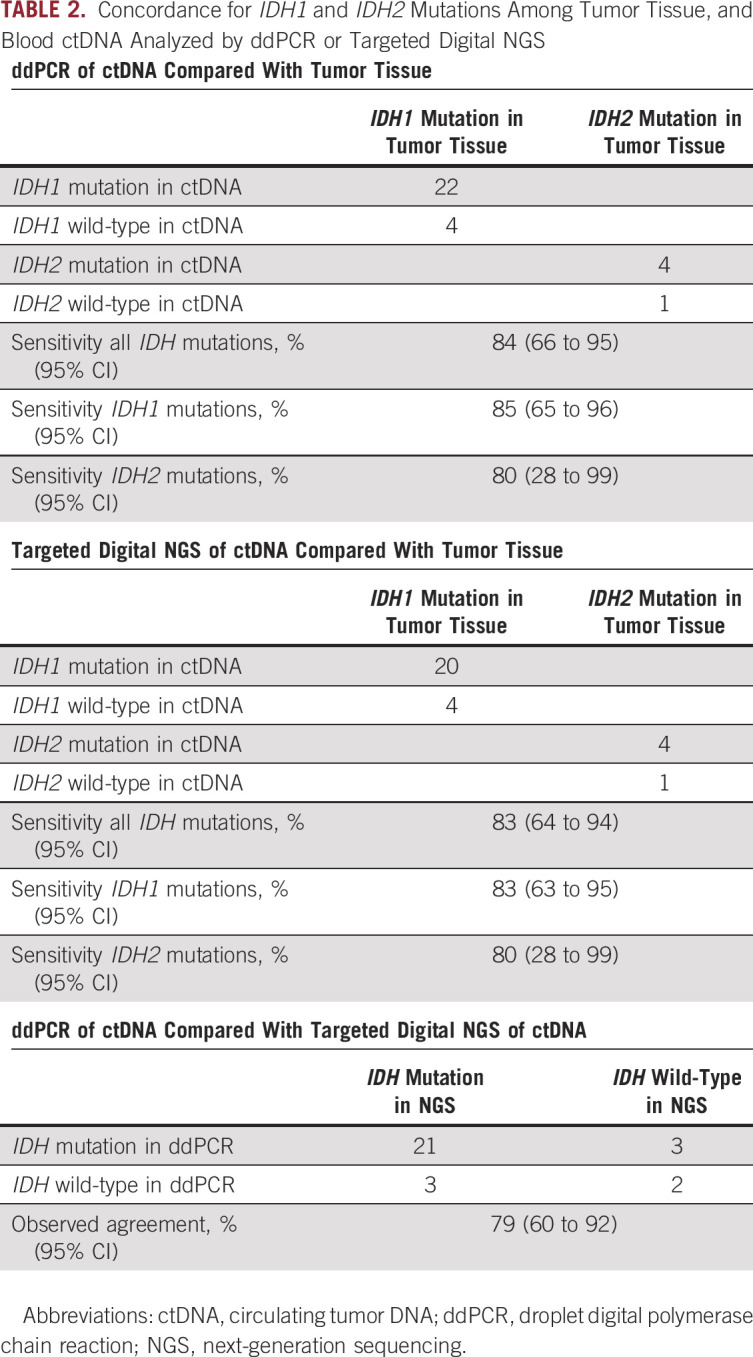
FIG 1.
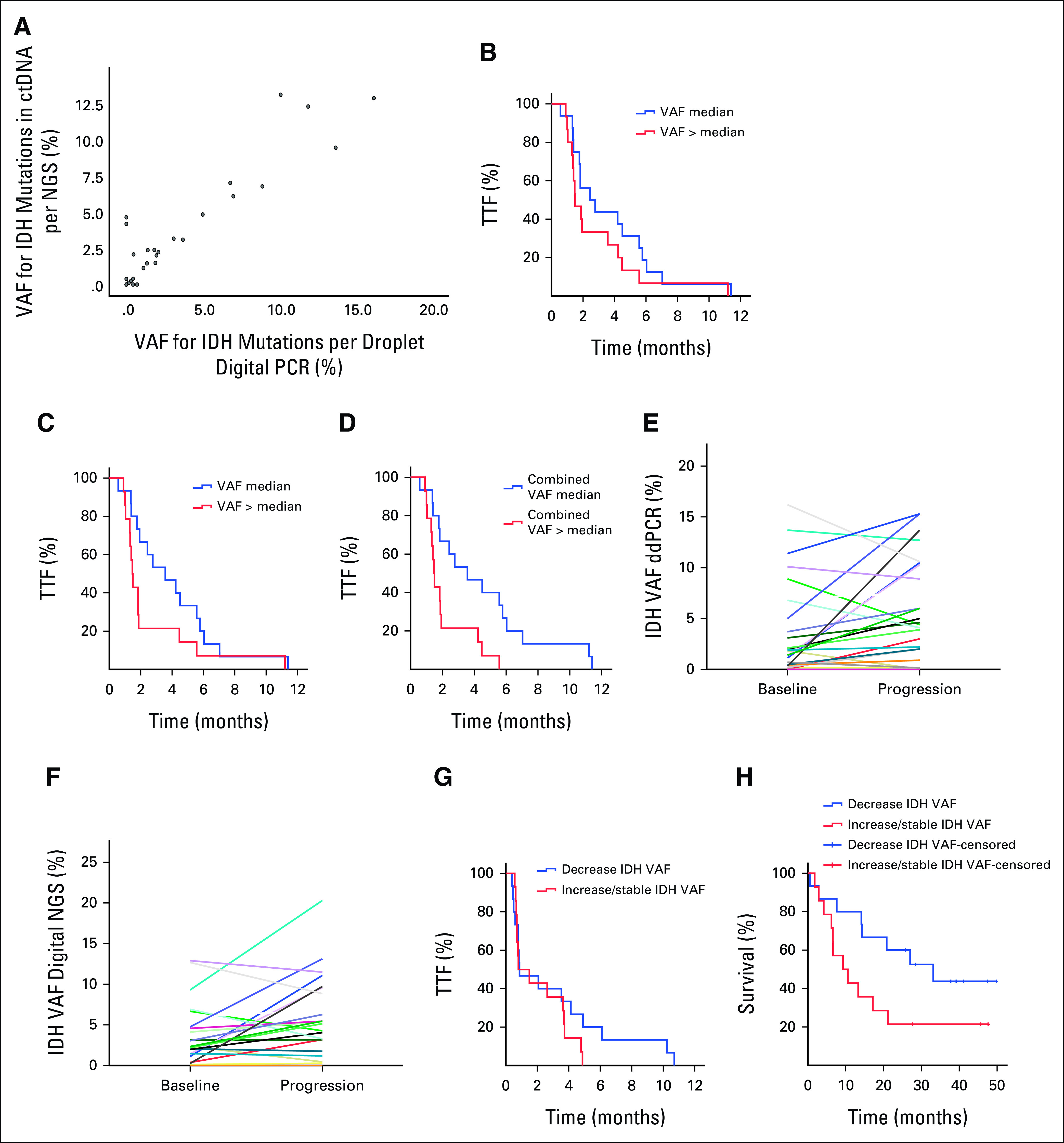
(A) There was a correlation (0.8, P < .001) between ddPCR and digital NGS for reported VAF in ctDNA. (B) Kaplan-Meier curves for TTF analyses per baseline quantity of ctDNA (≤ median v > median) in blood samples for IDH mutations detected by ddPCR (VAF ≤ 1.4% v VAF > 1.4 %; P = .21), (C) IDH mutations detected by digital NGS (VAF ≤ 2.2% v VAF > 2.2%; P = .09), (D) and the combined VAF for all alterations detected by digital NGS (VAF ≤ 4.6% v VAF > 4.6%; P = .008). (E) Comparisons of ctDNA samples for the levels of IDH mutation detected by ddPCR in baseline and progression samples from the same patient (P = .049) and (F) the levels of IDH mutation detected by digital NGS in baseline and in progression samples from the same patient (P = .06). (G) Kaplan-Meier curves of changes in VAF of IDH-mutated ctDNA during therapy demonstrated no difference in the median TTF between patients with decrease in IDH-mutated ctDNA on therapy compared with increase or no change (0.9 months; 95% CI, 0 to 2.5 v 0.8 months; 95% CI, 0 to 2.1; P = .29), (H) but there was a trend toward a longer median survival in patients with decrease in quantity of IDH-mutated ctDNA compared with patients with no change or increase in IDH-mutated ctDNA (33.1 months; 95% CI, 13 to 53.2 v 9.3 months; 95% CI, 2.1 to 16.4 months; P = .07). ctDNA, circulating tumor DNA; ddPCR, droplet digital polymerase chain reaction; IDH, isocitrate dehydrogenase; NGS, next-generation sequencing; VAF, variant allele frequency; TTF, time to treatment failure.
Of note, four patients also had five simultaneous mutations in KRASG12 (n = 4) and PIK3CAH1047 (n = 1) in the tumor tissue. Detection rates in ctDNA from blood samples collected before therapy were three of five mutations (60%) for ddPCR and three of three mutations (100%, one sample failed quality control) for digital NGS.
Mutation Detection in ctDNA Before Therapy and Treatment Outcomes
To determine whether the quantity of IDH-mutated ctDNA from blood samples collected before therapy with IDH inhibitors determined by VAF was associated with outcomes, we divided patients into two groups separated by median (low VAF ≤ 1.4% v high VAF > 1.4% for ddPCR and low VAF ≤ 2.2% v high VAF > 2.2% for targeted digital NGS). For ddPCR, there was no significant difference in a median TTF between patients with low IDH-mutated ctDNA compared with high IDH-mutated ctDNA (2.4 months; 95% CI, 0.6 to 4.3 v 1.5 months; 95% CI, 0.9 to 2.1; P = .21; Fig 1B). For targeted digital NGS, there was a trend toward a longer median TTF in patients with low IDH-mutated ctDNA compared with high IDH-mutated ctDNA (3.6 months; 95% CI, 1.3 to 5.8 v 1.5 months; 95% CI, 1.3 to 1.7; P = .09; Fig 1C). Finally, we analyzed association between the quantity of ctDNA from blood samples collected before therapy with IDH inhibitors determined by the aggregate total VAF for all somatic mutations detected by targeted digital NGS excluding variants with VAF below 0.25% to prevent inclusion of low-frequent sequencing errors and variants likely associated with clonal hematopoiesis of indeterminate potential. Patients were again divided into two groups per median quantity of total ctDNA (VAF ≤ 4.6% v VAF > 4.6%). Patients with low quantity of total ctDNA had a longer median TTF compared with patients with high total ctDNA (3.6 months; 95% CI, 1 to 6.2 v 1.5 months; 95% CI, 1.3 to 1.7; P = .008; Fig 1D).
There were no significant associations with median survival for IDH-mutated ctDNA detected by ddPCR (17.6 months v 11.3 months; P = .86), IDH-mutated ctDNA detected by targeted digital NGS (16.2 months v 14.1 months; P = .36), or the total ctDNA detected by targeted digital NGS (22 months v 10.2 months; P = .53).
Dynamic Changes in Quantity of IDH-Mutated ctDNA
Of the 31 patients, 28 had available ctDNA from blood samples collected before therapy and at progression on IDH inhibitors, whereas three patients had only ctDNA samples collected before therapy. For ddPCR, the paired analysis of the 28 patients with available ctDNA obtained before therapy and at progression demonstrated an increase in IDH-mutant ctDNA at progression (P = .049; Fig 1E). Similarly, for NGS, the paired analysis of 26 patients with available ctDNA obtained before therapy and at progression demonstrated a strong trend toward an increase in IDH-mutant ctDNA at progression (P = .06; Fig 1F).
Next, we assessed dynamic changes in quantity of IDH-mutated ctDNA detected by ddPCR in blood samples serially collected during therapy (approximately every 3-4 weeks [median, 26 days; range, 7-57]) with IDH inhibitors (Fig 2A). Patients with a decrease in IDH-mutated ctDNA on therapy with IDH inhibitors had a similar median TTF as patients with no change or increase in IDH-mutated ctDNA (0.9 months; 95% CI, 0 to 2.5 v 0.8 months; 95% CI, 0 to 2.1; P = .29; Fig 1G). Nevertheless, there was a trend toward a longer median survival in patients with a decrease in quantity of IDH-mutated ctDNA compared to patients with no change or increase in IDH-mutated ctDNA (33.1 months; 95% CI, 13 to 53.2 v 9.3 months; 95% CI, 2.1 to 16.4 months; P = .07; Fig 1H).
FIG 2.
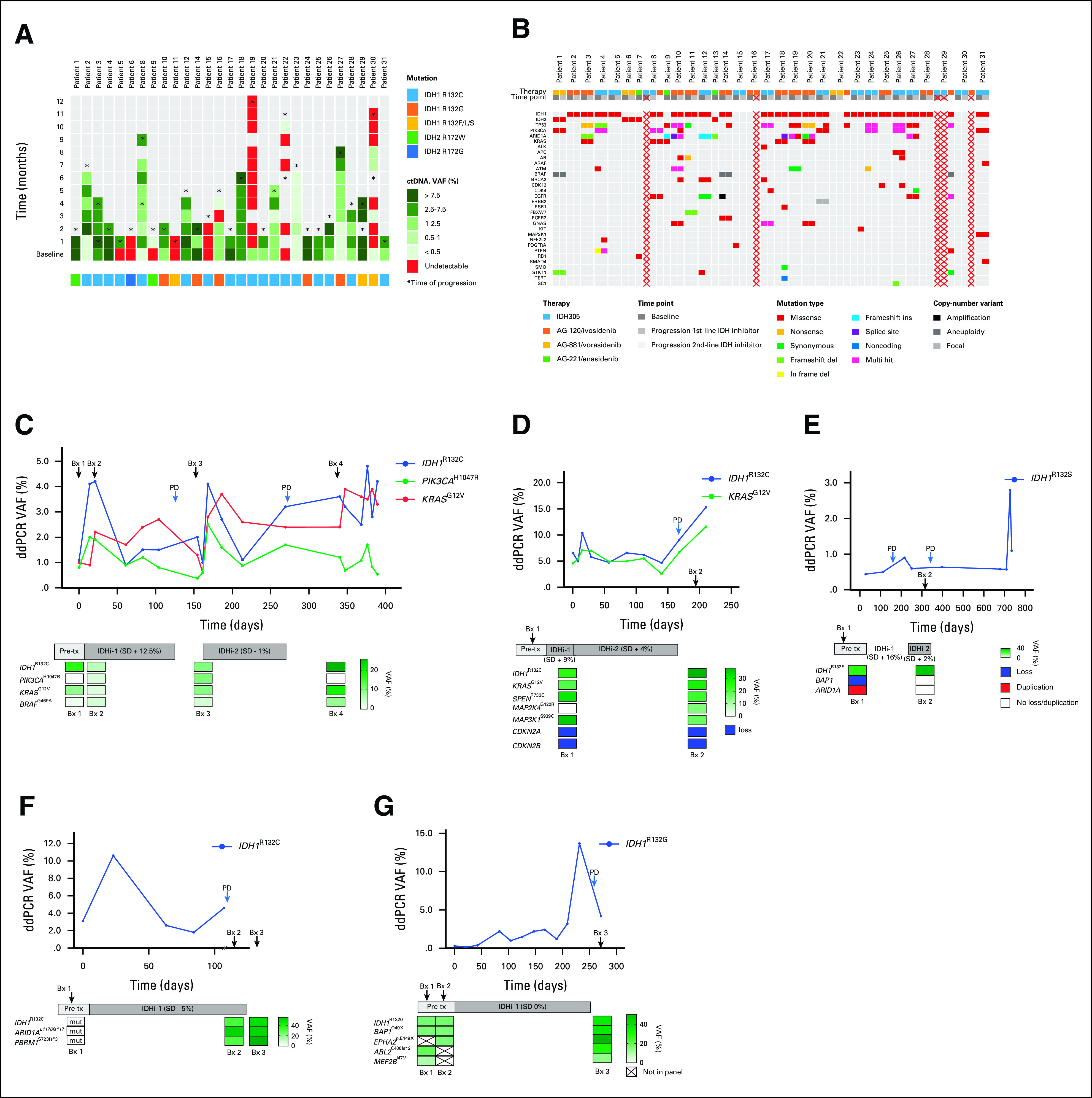
(A) IDH-mutated ctDNA by ddPCR in serially collected plasma during therapy with IDH inhibitors. (B) Heatmap of mutations detected by digital NGS in ctDNA from patients with IDH-mutated cholangiocarcinoma treated with IDH inhibitors at baseline and progression. Red crosses mark samples that failed quality control. (C-G) Dynamic tracking of VAF for detected mutations in ctDNA (ddPCR) from serial plasma samples or tumor tissue (NGS sequencing) from serial biopsies in patients treated with IDH inhibitors. Bx, biopsy; ctDNA, circulating tumor DNA; ddPCR, droplet digital polymerase chain reaction; IDH, isocitrate dehydrogenase; IDHi-1, first IDH1 inhibitor; IDHi-2, second IDH inhibitor; mut, mutant without reported VAF; NGS, next-generation sequencing; PD, progressive disease; Pre-tx, pretreatment; SD, stable disease; VAF, variant allele frequency.
Emerging Mutations in ctDNA Detected at Progression and in Serial Tumor Biopsies
We also analyzed with targeted digital NGS, ctDNA from blood samples collected before therapy and at progression on IDH inhibitors. Of, note four of 31 patients also had ctDNA obtained at progression on a second IDH inhibitor (Fig 2B). In ctDNA obtained before therapy, we detected a total of 96 alterations (mean 3.7 alterations per patient sample; range, 1-9) and three patients had no detectable alterations. The most frequently co-occurring alterations with IDH mutations were TP53 mutations (18 in 11 patients), ARID1A mutations (10 in 8 patients), PIK3CA mutations (10 in 6 patients), and KRAS mutations (four in four patients). Of interest, KRAS mutations were detected only in patients with IDH1 mutations (Fig 2B).
At the time of progression on IDH inhibitors, we detected 118 alterations in 27 ctDNA samples (mean 4.4 alterations per patient sample; range, 1-13). Three samples had no detectable alterations. The most frequently co-occurring alterations with IDH mutations were TP53 mutations (22 in 13 patients), ARID1A mutations (nine in seven patients), PIK3CA mutations (15 in 9 patients) and KRAS mutations (five in five patients, only present in patients with IDH1 mutations).
There were 15 new emerging alterations with predicted oncogenic potential in eight patients (range, 1-3 per patient) detected in ctDNA from blood samples collected at progression. The most frequent alterations were ARID1A mutations (n = 3) and TP53 mutations (n = 3; Data Supplement). Of note, one patient had no alteration in ctDNA before therapy; however, ctDNA sample obtained at progression showed TP53I255F mutation (not detected in archival tumor tissue) and IDH1R132C mutation (present in archival tumor tissue). Of interest, previously reported isoform switching from IDH1 to IDH2 mutation has not been observed.9
Finally, five patients had tumor tissue samples from serial biopsies collected before therapy, on therapy, and at progression, which were analyzed with targeted NGS or whole-exome sequencing. The first patient with an IDH1R132C mutation demonstrated emergence of a PIK3CAH1047R mutation in the tumor tissue obtained after 3 weeks of experimental therapy with the first IDH1 inhibitor and persisted in the tumor during therapy with the second IDH1 inhibitor. Of interest, the PIK3CAH1047R mutation disappeared from the tumor tissue 2 months after discontinuation of the second IDH1 inhibitor; however, the mutation has been consistently detectable in all ctDNA samples from the time before the first IDH1 inhibitor until 2 months after the second IDH1 inhibitor with VAF ranging from 0.38% to 1.70% (Fig 2C). The second patient with an IDH1R132C mutation demonstrated the emergence of a MAP2K4G122R mutation in the tumor tissue obtained 4 weeks after experimental therapy (Fig 2D). This mutation was not included in our ctDNA assays. The third patient with an IDH1R132S mutation demonstrated disappearance of BAP1 loss and ARID1A duplication during treatment with the second IDH1 inhibitor (Fig 2E). Unfortunately, corresponding ctDNA samples failed quality control. The fourth patient with an IDH1R132C mutation and the fifth patient with an IDH1R132G mutation showed no changes in molecular profile of the serially collected tumor tissue (Figs 2F and 2G).
DISCUSSION
Our study demonstrated that molecular testing for IDH mutations in ctDNA samples from patients with advanced cholangiocarcinoma using two orthogonal methods has high concordance (84% for ddPCR and 83% for digital NGS) with IDH mutation testing of tumor tissue. To our knowledge, this study is the first to focus on assessing agreement rates between ctDNA and tumor tissue for IDH mutations in cholangiocarcinoma, which implemented both ddPCR and NGS. Aguado et al14 presented in an abstract form the data from ClarIDHy study (ivosidenib v placebo in advanced previously treated IDH1-mutated cholangiocarcinoma) using the BEAMing digital PCR, which demonstrated concordance with tumor tissue of 92%. Overall, previously presented studies in advanced cancers demonstrated agreement in rates between ctDNA and tumor tissue between 68% and 90%.15,16 Ettrich et al16 used NGS to detect molecular alterations in 15 frequently mutated genes in 24 patients with cholangiocarcinoma and reported an overall concordance of 74%. Okamura et al15 used digital targeted NGS to detect molecular alterations in 68-73 genes and reported concordance of 68% for TP53 alterations, 80% for KRAS alterations, and 90% for PIK3CA alterations. For IDH1 mutations, the detection rate in ctDNA was 75%.
We also noticed that high VAF of mutated ctDNA determined by NGS before starting on therapy was associated with a shorter median TTF compared with low VAF of mutated ctDNA (P = .008). This is not unexpected as similar observations have been made in other cancers and in preliminary reports for the ClarIDHy study of IDH1 inhibitor ivosidenib or placebo in IDH1-mutated cholangiocarcinoma.14,17,18 For instance, in one of our previous studies, we noticed that in patients with BRAFV600-mutated advanced cancers per tumor tissue analysis, patients with detectable BRAFV600 mutations in ctDNA had an inferior median TTF on BRAF/MEK inhibitors compared to patients without BRAFV600 mutations.17 Similarly, in patients with BRAFV600-mutated advanced melanoma per tumor tissue analysis, patients with detectable BRAFV600 mutations in ctDNA had an inferior median progression-free survival on BRAF/MEK inhibitors compared to patients without BRAFV600 mutations.18
We also demonstrated that the quantity of IDH1-mutated ctDNA at progression defined by VAF is higher compared with pretreatment levels, and we noticed a trend toward longer median TTF in patients with a decrease in IDH1-mutated ctDNA compared to patients with no change or an increase. Although there are no comparable data in cholangiocarcinoma, similar observations have been made with ddPCR and NGS in other advanced cancers.19,20
Molecular testing of ctDNA is minimally invasive and can be repeated at multiple time points to investigate clonal evolution and mechanisms of innate and adaptive resistance.10 Anecdotal experience in patients with acute myeloid leukemia, myelodysplastic syndrome, and cholangiocarcinoma described isoform switching from IDH1 to IDH2 mutations and vice versa as a possible mechanism of resistance to IDH1 or IDH2 inhibitors.9 In our study, we noticed 15 new emerging alterations with predicted oncogenic potential in patients with blood samples collected at progression. ARID1A mutations and TP53 mutations were most frequent, and no IDH1 or IDH2 mutations switching were noted. ARID1A mutations belong to a broad group of DNA damage response (DDR) alterations.21 In addition, DDR targeting of IDH-mutated cancers with poly (ADP-ribose) polymerase inhibitors demonstrated preclinical efficacy in some studies and is currently assessed in early-stage clinical testing.22 Therefore, development of IDH inhibitors in combination with DDR targeting agents would be of special interest, although better understanding of the nature of possible synergistic activity is needed as neomorphic IDH mutations were also suggested to be associated with BRCAness phenotype.22
Our study had several limitations. The study sample size was relatively small. In addition, patients evaluated in our study received four different IDH inhibitors (ivosidenib, enasidenib, vorasidenib, or IDH305), which were in early-phase clinical development at the time of study enrollment, and some patients received more than one IDH inhibitor. Therefore, our results require further confirmation in larger prospective studies. Despite these limitations, our study demonstrated that detection of IDH mutations in ctDNA in patients with advanced cholangiocarcinoma is feasible and can be potentially used to select patients for treatment with IDH inhibitors. We also demonstrated that dynamic changes in ctDNA can correspond with the clinical course and clonal evolution.
ACKNOWLEDGMENT
The authors would like to acknowledge Mr Joseph Munch from the Department of Scientific Publications, the University of Texas MD Anderson Cancer Center, for his editorial and grammar assistance.
Milind Javle
Honoraria: QED Therapeutics, Incyte, TransThera Biosciences, Merck, EMD Serono/Merck, AstraZeneca/MedImmune
Consulting or Advisory Role: QED Therapeutics, OncoSil, Incyte, Mundipharma EDO GmbH, AstraZeneca, Merck, EMD Serono, Derazantinib
Other Relationship: Rafael Pharmaceuticals, Incyte, Pieris Pharmaceuticals, Merck, Merck Serono, Novartis, Seattle Genetics, BeiGene, QED Therapeutics, Bayer
Rachna T. Shroff
Consulting or Advisory Role: Exelixis, Merck, QED Therapeutics, Incyte, AstraZeneca, Taiho Pharmaceutical, Boehringer Ingelheim, Servier, Genentech, Basilea, Helsinn Therapeutics
Speakers' Bureau: Servier, Helsinn Therapeutics
Research Funding: Pieris Pharmaceuticals, Taiho Pharmaceutical, Merck, Exelixis, QED Therapeutics, Rafael Pharmaceuticals, Bristol Myers Squibb, Bayer, Immunovaccine, Seattle Genetics, Novocure, Nucana, Loxo/Lilly
Shubham Pant
Honoraria: 4D Pharma
Consulting or Advisory Role: Xencor, Zymeworks, Ipsen
Research Funding: Mirati Therapeutics (Inst), Lilly (Inst), RedHill Biopharma (Inst), Xencor (Inst), Five Prime Therapeutics (Inst), Novartis (Inst), Rgenix (Inst), Sanofi/Aventis (Inst), ArQule (Inst), Bristol Myers Squibb (Inst), Onco Response (Inst), GlaxoSmithKline (Inst), Ipsen (Inst), Astellas Pharma (Inst), Purple Biotech (Inst), 4D Pharma (Inst), Boehringer Ingelheim (Inst), NGM Biopharmaceuticals (Inst), Janssen (Inst), Arcus Biosciences (Inst), Elicio Therapeutics (Inst)
S. Greg Call
Employment: Tempus
Richard B. Lanman
Employment: Guardant Health
Leadership: Guardant Health, Biolase, Circulogene Theranostics
Stock and Other Ownership Interests: Guardant Health, Biolase, Forward, Circulogene, Teiko Bio, Inc, Glympse Bio
Consulting or Advisory Role: Forward, Guardant Health, Glympse Bio, Teiko Bio, Inc
Research Funding: Guardant Health
Funda Meric-Bernstam
Employment: MD Anderson Cancer Center
Honoraria: Rutgers Cancer Institute of New Jersey
Consulting or Advisory Role: Samsung Bioepis, Xencor, Debiopharm Group, Silverback Therapeutics, IBM Watson Health, Roche, PACT Pharma, eFFECTOR Therapeutics, Kolon Life Sciences, Tyra Biosciences, Zymeworks, Puma Biotechnology, Zentalis, Alkermes, Infinity Pharmaceuticals, AbbVie, Black Diamond Therapeutics, Eisai, OnCusp Therapeutics, Lengo Therapeutics, Tallac Therapeutics, Karyopharm Therapeutics, Biovica
Speakers' Bureau: Chugai Pharma
Research Funding: Novartis (Inst), AstraZeneca (Inst), Taiho Pharmaceutical (Inst), Genentech (Inst), Calithera Biosciences (Inst), Debiopharm Group (Inst), Bayer (Inst), Aileron Therapeutics (Inst), PUMA Biotechnology (Inst), CytomX Therapeutics (Inst), Jounce Therapeutics (Inst), Zymeworks (Inst), Curis (Inst), Pfizer (Inst), eFFECTOR Therapeutics (Inst), AbbVie (Inst), Boehringer Ingelheim (I), Guardant Health (Inst), Daiichi Sankyo (Inst), GlaxoSmithKline (Inst), Seattle Genetics (Inst), Klus Pharma (Inst), Takeda (Inst)
Travel, Accommodations, Expenses: Beth Israel Deaconess Medical Center
Victoria M. Raymond
Employment: Guardant Health
Stock and Other Ownership Interests: Guardant Health, Trovagene
Lawrence N. Kwong
Stock and Other Ownership Interests: Sarepta Therapeutics
Research Funding: Array BioPharma
Filip Janku
Stock and Other Ownership Interests: Cardiff Oncology
Consulting or Advisory Role: Deciphera, Novartis, Sequenom, Foundation Medicine, Guardant Health, Synlogic, Valeant/Dendreon, IFM Therapeutics, Sotio, PureTech, Jazz Pharmaceuticals, Immunomet, IDEAYA Biosciences, Cardiff Oncology
Research Funding: Novartis (Inst), BioMed Valley Discoveries (Inst), Roche (Inst), Agios (Inst), Astellas Pharma (Inst), Deciphera (Inst), Plexxikon (Inst), Piqur (Inst), Fujifilm (Inst), Symphogen (Inst), Bristol Myers Squibb (Inst), Asana Biosciences (Inst), Astex Pharmaceuticals (Inst), Genentech (Inst), Proximagen (Inst)
Other Relationship: Bio-Rad
No other potential conflicts of interest were reported.
SUPPORT
Supported by grants from the Sheikh Khalifa Al Nahyan Bin Zayed Institute for Personalized Cancer Therapy (Filip Janku) and the Sabin Family Foundation (F.J.), MD Anderson Cancer Center Support grant (NIH/NCI P30 CA016672), Clinical Translational Science Award (NIH/HHS 1UL1 TR003167) and Cancer Prevention Research Institute of Texas (CPRIT) Precision Oncology Decision Support Core (RP150535), and Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy. Targeted digital next-generation sequencing of blood cell-free DNA was supported by Guardant Health.
AUTHOR CONTRIBUTIONS
Conception and design: Morten Lapin, Helen J. Huang, Milind Javle, Shubham Pant, Richard B. Lanman, Lawrence N. Kwong, Filip Janku
Financial support: Filip Janku
Administrative support: Funda Meric-Bernstam, Filip Janku
Provision of study materials or patients: Veronica R. Holley, Funda Meric-Bernstam, Victoria M. Raymond, Filip Janku
Collection and assembly of data: Morten Lapin, Helen J. Huang, Sharmeen Chagani, Rachna T. Shroff, Mohamed A. Gouda, Anjali Raina, Kiran Madwani, Veronica R. Holley, S. Greg Call, Richard B. Lanman, Funda Meric-Bernstam, Victoria M. Raymond, Lawrence N. Kwong, Filip Janku
Data analysis and interpretation: Morten Lapin, Helen J. Huang, Sharmeen Chagani, Milind Javle, Rachna T. Shroff, Kiran Madwani, S. Greg Call, Derek J. Dustin, Richard B. Lanman, Funda Meric-Bernstam, Victoria M. Raymond, Lawrence N. Kwong, Filip Janku
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Milind Javle
Honoraria: QED Therapeutics, Incyte, TransThera Biosciences, Merck, EMD Serono/Merck, AstraZeneca/MedImmune
Consulting or Advisory Role: QED Therapeutics, OncoSil, Incyte, Mundipharma EDO GmbH, AstraZeneca, Merck, EMD Serono, Derazantinib
Other Relationship: Rafael Pharmaceuticals, Incyte, Pieris Pharmaceuticals, Merck, Merck Serono, Novartis, Seattle Genetics, BeiGene, QED Therapeutics, Bayer
Rachna T. Shroff
Consulting or Advisory Role: Exelixis, Merck, QED Therapeutics, Incyte, AstraZeneca, Taiho Pharmaceutical, Boehringer Ingelheim, Servier, Genentech, Basilea, Helsinn Therapeutics
Speakers' Bureau: Servier, Helsinn Therapeutics
Research Funding: Pieris Pharmaceuticals, Taiho Pharmaceutical, Merck, Exelixis, QED Therapeutics, Rafael Pharmaceuticals, Bristol Myers Squibb, Bayer, Immunovaccine, Seattle Genetics, Novocure, Nucana, Loxo/Lilly
Shubham Pant
Honoraria: 4D Pharma
Consulting or Advisory Role: Xencor, Zymeworks, Ipsen
Research Funding: Mirati Therapeutics (Inst), Lilly (Inst), RedHill Biopharma (Inst), Xencor (Inst), Five Prime Therapeutics (Inst), Novartis (Inst), Rgenix (Inst), Sanofi/Aventis (Inst), ArQule (Inst), Bristol Myers Squibb (Inst), Onco Response (Inst), GlaxoSmithKline (Inst), Ipsen (Inst), Astellas Pharma (Inst), Purple Biotech (Inst), 4D Pharma (Inst), Boehringer Ingelheim (Inst), NGM Biopharmaceuticals (Inst), Janssen (Inst), Arcus Biosciences (Inst), Elicio Therapeutics (Inst)
S. Greg Call
Employment: Tempus
Richard B. Lanman
Employment: Guardant Health
Leadership: Guardant Health, Biolase, Circulogene Theranostics
Stock and Other Ownership Interests: Guardant Health, Biolase, Forward, Circulogene, Teiko Bio, Inc, Glympse Bio
Consulting or Advisory Role: Forward, Guardant Health, Glympse Bio, Teiko Bio, Inc
Research Funding: Guardant Health
Funda Meric-Bernstam
Employment: MD Anderson Cancer Center
Honoraria: Rutgers Cancer Institute of New Jersey
Consulting or Advisory Role: Samsung Bioepis, Xencor, Debiopharm Group, Silverback Therapeutics, IBM Watson Health, Roche, PACT Pharma, eFFECTOR Therapeutics, Kolon Life Sciences, Tyra Biosciences, Zymeworks, Puma Biotechnology, Zentalis, Alkermes, Infinity Pharmaceuticals, AbbVie, Black Diamond Therapeutics, Eisai, OnCusp Therapeutics, Lengo Therapeutics, Tallac Therapeutics, Karyopharm Therapeutics, Biovica
Speakers' Bureau: Chugai Pharma
Research Funding: Novartis (Inst), AstraZeneca (Inst), Taiho Pharmaceutical (Inst), Genentech (Inst), Calithera Biosciences (Inst), Debiopharm Group (Inst), Bayer (Inst), Aileron Therapeutics (Inst), PUMA Biotechnology (Inst), CytomX Therapeutics (Inst), Jounce Therapeutics (Inst), Zymeworks (Inst), Curis (Inst), Pfizer (Inst), eFFECTOR Therapeutics (Inst), AbbVie (Inst), Boehringer Ingelheim (I), Guardant Health (Inst), Daiichi Sankyo (Inst), GlaxoSmithKline (Inst), Seattle Genetics (Inst), Klus Pharma (Inst), Takeda (Inst)
Travel, Accommodations, Expenses: Beth Israel Deaconess Medical Center
Victoria M. Raymond
Employment: Guardant Health
Stock and Other Ownership Interests: Guardant Health, Trovagene
Lawrence N. Kwong
Stock and Other Ownership Interests: Sarepta Therapeutics
Research Funding: Array BioPharma
Filip Janku
Stock and Other Ownership Interests: Cardiff Oncology
Consulting or Advisory Role: Deciphera, Novartis, Sequenom, Foundation Medicine, Guardant Health, Synlogic, Valeant/Dendreon, IFM Therapeutics, Sotio, PureTech, Jazz Pharmaceuticals, Immunomet, IDEAYA Biosciences, Cardiff Oncology
Research Funding: Novartis (Inst), BioMed Valley Discoveries (Inst), Roche (Inst), Agios (Inst), Astellas Pharma (Inst), Deciphera (Inst), Plexxikon (Inst), Piqur (Inst), Fujifilm (Inst), Symphogen (Inst), Bristol Myers Squibb (Inst), Asana Biosciences (Inst), Astex Pharmaceuticals (Inst), Genentech (Inst), Proximagen (Inst)
Other Relationship: Bio-Rad
No other potential conflicts of interest were reported.
REFERENCES
- 1.Fujii T, Khawaja MR, DiNardo CD, et al. Targeting isocitrate dehydrogenase (IDH) in cancer Discov Med 21373–3802016 [PubMed] [Google Scholar]
- 2.Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: Utility of next-generation sequencing for clinical management Cancer 1223838–38472016 [DOI] [PubMed] [Google Scholar]
- 3.Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention Clin Cancer Res 244154–41612018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML N Engl J Med 3782386–23982018 [DOI] [PubMed] [Google Scholar]
- 5.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia Blood 130722–7312017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study Lancet Oncol 21796–8072020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mellinghoff IK, Ellingson BM, Touat M, et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma J Clin Oncol 383398–34062020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tap WD, Villalobos VM, Cote GM, et al. Phase I study of the mutant IDH1 inhibitor ivosidenib: Safety and clinical activity in patients with advanced chondrosarcoma J Clin Oncol 381693–17012020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition Cancer Discov 81540–15472018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polivka J, Jr, Pesta M, Janku F.Testing for oncogenic molecular aberrations in cell-free DNA-based liquid biopsies in the clinic: Are we there yet? Expert Rev Mol Diagn 151631–16442015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10:e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies Clin Cancer Res 243539–35492018 [DOI] [PubMed] [Google Scholar]
- 13.Zill OA, Greene C, Sebisanovic D, et al. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas Cancer Discov 51040–10482015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aguado E, Abou-Alfa GK, Zhu AX, et al. IDH1 mutation detection in plasma circulating tumor DNA (ctDNA) and association with clinical response in patients with advanced intrahepatic cholangiocarcinoma (IHC) from the phase III ClarIDHy study. J Clin Oncol. 2020;38 suppl; abstr 4576. [Google Scholar]
- 15.Okamura R, Kurzrock R, Mallory RJ, et al. Comprehensive genomic landscape and precision therapeutic approach in biliary tract cancers Int J Cancer 148702–7122021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ettrich TJ, Schwerdel D, Dolnik A, et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep. 2019;9:13261. doi: 10.1038/s41598-019-49860-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janku F, Huang HJ, Claes B, et al. BRAF mutation testing in cell-free DNA from the plasma of patients with advanced cancers using a rapid, automated molecular diagnostics system Mol Cancer Ther 151397–14042016 [DOI] [PubMed] [Google Scholar]
- 18.Santiago-Walker A, Gagnon R, Mazumdar J, et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials Clin Cancer Res 22567–5742016 [DOI] [PubMed] [Google Scholar]
- 19.Fujii T, Barzi A, Sartore-Bianchi A, et al. Mutation-enrichment next-generation sequencing for quantitative detection of KRAS mutations in urine cell-free DNA from patients with advanced cancers Clin Cancer Res 233657–36662017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janku F, Huang HJ, Fujii T, et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction Ann Oncol 28642–6502017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen J, Peng Y, Wei L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors Cancer Discov 5752–7672015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9:eaal2463. doi: 10.1126/scitranslmed.aal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]