

Abstract
PURPOSE
PIK3CA mutations frequently contribute to oncogenesis in solid tumors. Taselisib, a potent and selective inhibitor of phosphoinositide 3-kinase, has demonstrated clinical activity in PIK3CA-mutant breast cancer. Whether PIK3CA mutations predict sensitivity to taselisib in other cancer types is unknown. National Cancer Institute–Molecular Analysis for Therapy Choice Arm EAY131-I is a single-arm, phase II study of the safety and efficacy of taselisib in patients with advanced cancers.
METHODS
Eligible patients had tumors with an activating PIK3CA mutation. Patients with breast or squamous cell lung carcinoma, or whose cancer had KRAS or PTEN mutations, were excluded. Patients received taselisib 4 mg, orally once daily continuously, until disease progression or unacceptable toxicity. The primary end point was objective response rate. Secondary end points included progression-free survival (PFS), 6-month PFS, overall survival (OS), and identification of predictive biomarkers.
RESULTS
Seventy patients were enrolled, and 61 were eligible and initiated protocol therapy. Types of PIK3CA mutations included helical 41 of 61 (67%), kinase 11 of 61 (18%), and other 9 of 61 (15%). With a median follow-up of 35.7 months, there were no complete or partial responses. Six-month PFS was 19.9% (90% CI, 12.0 to 29.3) and median PFS was 3.1 months (90% CI, 1.8 to 3.7). Six-month OS was 60.7% (90% CI, 49.6 to 70.0) and median OS was 7.2 months (90% CI, 5.9 to 10.0). Individual comutations were too heterogeneous to correlate with clinical outcome. Fatigue, diarrhea, nausea, and hyperglycemia were the most common toxicities, and most were grade 1 and 2.
CONCLUSION
In this study, taselisib monotherapy had very limited activity in a heterogeneous cohort of heavily pretreated cancer patients with PIK3CA-mutated tumors; the presence of a PIK3CA mutation alone does not appear to be a sufficient predictor of taselisib activity.
BACKGROUND
Phosphoinositide 3-kinase (PI3K) is a lipid kinase with central roles in cell proliferation, survival, and migration. Upon activation by growth factor receptors or integrins, PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate to generate phosphatidylinositol-3,4,5-trisphosphate, a second messenger involved in the activation of the kinase Akt and associated proteins in the Akt and mammalian target of rapamycin (mTOR) pathway.1,2 Class I PI3Ks comprise a family of proteins using one of four catalytic subunit isoforms: α, β, γ, and δ. Activating and transforming mutations of the gene encoding the p110α catalytic subunit of PI3K (PIK3CA) are among the most frequent oncogenic events in human cancers.1,3-5 For example, PIK3CA mutations were found in 12% of 104,000 tumors in the AACR GENIE database Version 9.0.6 Approximately 80% of PIK3CA mutations occur in three hotspots, E542X, E545X, and H1047X, although other, less common recurrent mutations have been demonstrated to be oncogenic.7 In addition, the PI3K pathway is activated in many PIK3CA wild-type cancers by receptor tyrosine kinase (RTK) signaling, mutation or loss of the PTEN, or mutations in KRAS. In nonclinical studies, cell lines with activating PIK3CA mutations show greater sensitivity to pan-class I PI3K inhibitors and α isoform selective PI3K inhibitors, whereas cell lines with concurrent KRAS mutations are more resistant to these inhibitors.8,9 Several nonclinical studies have shown that PTEN-deficient tumors preferentially depend on the β isoform of PI3K for survival and respond to β-specific, but not α-specific, inhibitors.10-12 A clinical study demonstrated that PTEN mutations can emerge as an acquired resistance mechanism to an α-specific PI3K inhibitor.13
Taselisib is a potent, selective inhibitor of Class I PI3K α, δ, and γ isoforms, with approximately 30-fold less inhibition of the p110β isoform. In vitro, taselisib is approximately 2- to 3-fold more potent against helical domain and catalytic domain mutant forms of p110α than the wild-type kinase, and more potently inhibits proliferation of cell lines with a PIK3CA mutation than those without a mutation.14 In addition, taselisib treatment led to selective downregulation of mutant p110α protein in a PIK3CA H1047R-mutant breast cancer cell line.15 In a phase I study, taselisib pharmacokinetics were dose-proportional, with frequent dose-dependent treatment-related adverse events (AEs), including GI toxicity, rash, and hyperglycemia. Notably, monotherapy activity in PIK3CA-mutant cancers was observed, with most responses observed in PIK3CA-mutant breast cancers and no responses in cancers without detectable PIK3CA mutations.16
At the time this current study was conceived, several PI3K inhibitors, including taselisib, were in clinical development. Phase III trials of taselisib and the α-selective PI3K inhibitor alpelisib evaluated these agents in combination with fulvestrant for hormone receptor–positive (HR+) breast cancer and have since demonstrated that both inhibitors improve progression-free survival (PFS) versus fulvestrant alone.17,18 On the basis of these data, alpelisib was recently approved by the US Food and Drug Administration in combination with fulvestrant for the treatment of HR+ metastatic breast cancer. By contrast, despite taselisib's positive phase III trial result in breast cancer,18 it is not being developed further because the benefit to risk ratio in PIK3CA-mutant breast cancer was deemed not sufficiently favorable. The class I PI3K inhibitor copanlisib has been approved for relapsed follicular lymphoma,19 and the δ-specific inhibitor idelalisib20 and the δ/γ isoform inhibitor duvelisib21 have been approved for the treatment of chronic lymphocytic leukemia. Interestingly, these hematologic indications do not require the presence of an activating PI3K mutation for drug activity. Despite these approvals, important questions remain regarding the extent to which PIK3CA mutations predict for sensitivity to PI3K inhibitors across other less-studied tumor types, and whether the type of PIK3CA mutation, or other comutated genes affect tumor sensitivity to PI3K inhibitor therapy. To address these questions, we evaluated taselisib in the National Cancer Institute-Molecular Analysis for Therapy Choice (NCI-MATCH) trial (NCT0246506).
The NCI-MATCH study, one of the largest precision medicine trials to date evaluating targeted therapy selected on the basis of molecular alterations, was developed by the ECOG-ACRIN Cancer Research Group (ECOG-ACRIN) and the NCI. It is a multiarm molecular profile–driven, nonrandomized, open-label, phase II study in patients with advanced cancers.22 It aims to identify signals of efficacy for treatments targeted to actionable molecular alterations irrespective of tumor histologic type. Briefly, this national effort required subjects to have tumor analyzed by a next-generation sequencing (NGS) assay and immunohistochemistry (IHC) for select proteins (eg, PTEN and mismatch repair proteins). Patients with actionable mutations were assigned to an available treatment arm through an informatics algorithm (MATCHBOX). Herein, we report the results of the NCI-MATCH EAY131-Arm I evaluating taselisib.
METHODS
Study Design
Patients were eligible for NCI-MATCH through a two-step registration process, requiring written consent before each step. In Step 0, patients underwent a research tumor biopsy or submitted archival tissue collected < 6 months before. Tumor profiling of the biopsy specimen was accomplished as described.22,23 Briefly, actionable mutations were assessed using a next-generation sequencing panel including single-nucleotide variants, indels, amplifications and selected fusions, and immunohistochemistry assays for PTEN, MLH1, and MSH2 expression. Patients were assigned to a study arm using a prospectively defined NCI-designed informatics rules algorithm (MATCHBOX), as previously described.22
Study Population
Patients must have been eligible for the NCI-MATCH master protocol to be considered potentially eligible for the EAY131-Arm I subprotocol. NCI-MATCH master protocol eligibility has been described elsewhere.22,23 Key eligibility for the master protocol Step 0 include age 18 years or older with histologically confirmed solid tumors, lymphoma, or multiple myeloma. Patients must have had disease for which no standard treatment exists that had been shown to prolong overall survival (OS), or had progressed on at least one line of standard systemic therapy. All patients had measurable disease (specified criteria appropriate to underlying disease), Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, no other concurrent investigational or chemotherapy agents or radiation, no uncontrolled intercurrent illness, ability to swallow, and adequate hematologic and organ function. Patients were permitted with well controlled HIV, well-controlled brain metastases, no evidence of QTc prolongation, and not taking QT-prolonging medications or steroids (patients with glioblastoma on stable dose of steroids were allowed). Patients with prior cancers who were disease-free for 5 years, or adequately treated basal or squamous cell skin cancer, in situ cervical cancer, or adequately treated other stage I or II cancer in complete remission were permitted.
Patients determined to have an actionable mutation via Step 0 were eligible to be screened for Step 1. Key Step 1 EAY131-Arm I eligibility included presence of an activating PIK3CA mutation (Data Supplement) and absence of a KRAS mutation and/or PTEN mutation or loss (Data Supplement), identified by the NCI-MATCH screening assessment. Patients were excluded if they had clinically significant ECG abnormalities, left ventricular ejection fraction ≤ 50%, fasting blood glucose > 125 mg/dL or known type 1 or 2 diabetes requiring antihyperglycemic medication, resting dyspnea or supplemental oxygen requirement, inflammatory bowel disease, known hypersensitivity to taselisib, or prior therapy with a PI3K, dual PI3K/mTOR, or Akt inhibitor. Prior mTOR inhibitors were permitted. Patients with breast cancer and patients with squamous cell carcinoma of the lung who were eligible for and had access to the Lung-MAP trial (NCT02154490) were excluded. All relevant institutional review boards approved the protocol, in compliance with the recommendations of the Helsinki Declaration.
Study Treatment
In EAY131-Arm I, taselisib was administered at 4 mg orally, once daily, on a continuous 28-day cycle, until disease progression or unacceptable toxicity. Toxicity was evaluated using CTCAEv4. Doses were permitted to be held for up to 28 days because of treatment-related toxicities or unanticipated medical events. Dose modifications were as follows: first reduction to 2 mg daily, second reduction to 2 mg every other day, then discontinuation if further indication for dose reduction. Dose re-escalation was not permitted. Because of the 40-hour half-life of taselisib, investigators were advised to consider holding taselisib for certain grade 2 toxicities (stomatitis, mucositis, rash, colitis, and pneumonitis) until resolved to grade 1 or less, and to carefully monitor for delayed toxicities up to 4 weeks after holding or stopping taselisib. Arm I-specific expedited reporting beyond the master Comprehensive Adverse Events and Potential Risks (CAEPR) list included greater than or equal to grade 2 colitis, diarrhea, nausea, vomiting, fatigue, anorexia, and greater than or equal to grade 3 mucositis, rash, and hyperglycemia. Safety was monitored by the NCI-MATCH steering committee, as well as continuous monitoring by the safety chairs and monthly reviews by the study team.
Evaluation of Response
Response was evaluated every two cycles (56 days) for the first 26 cycles, and every three cycles thereafter using criteria for solid tumors (using RECIST guideline version 1.124), lymphoma, glioblastoma multiforme, or multiple myeloma as appropriate.
Statistics
The overall NCI-MATCH statistics have been described elsewhere.22,23 Total accrual goal for Step 0 screening was more than 6,000 patients. The initial accrual goal in each arm was 35 patients (31 eligible), providing 92% power to distinguish an objective response rate (ORR) of 25% from a null of 5% with a one-sided type 1 error of 1.8%. The regimen would be declared promising and worthy of further study if ≥ 5 of 31 patients achieve a response. In December 2016, the taselisib arm accrual goal was expanded to 70 patients, using the same null hypothesis (5% ORR) with a Type 1 error of 1.8%. This expansion was done on the basis of protocol criteria allowing up to 6 months of additional accrual and a maximum of 35 additional patients, as well as to provide more data on the individual PIK3CA mutation types. The primary end point for this arm, as with others, was ORR as defined in the protocol for respective diseases. Secondary end points included PFS, PFS at 6 months, and the identification of biomarkers predictive of response.
RESULTS
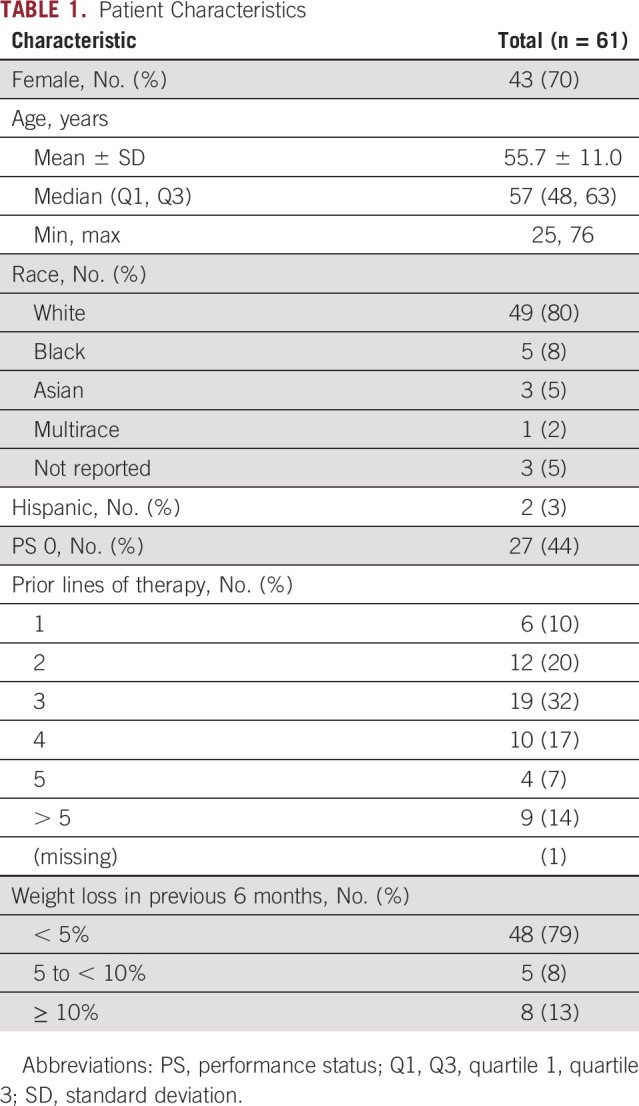
Patient Characteristics
Between March 18, 2016, and April 20, 2017, 70 patients were enrolled. Four patients did not initiate protocol therapy and five patients initiated therapy but were subsequently found to be ineligible by central review. Thus, 61 patients initiated therapy and were analyzable for the primary end point (Data Supplement). The database was locked on November 10, 2020. Patient demographics are summarized in Table 1. Notably, 43 of 61 (70%) were female, and the majority were heavily pretreated (70% with ≥ 3 prior lines of therapy). Consistent with the tumor agnostic NCI-MATCH trial platform and the Arm I-specific exclusion of breast and squamous cell lung cancer, patients with 37 distinct tumor histologies were enrolled. The most common cancers included gynecologic, anogenital, and colorectal cancers (Data Supplement). Types of PIK3CA mutations included helical 41 of 61 (67%), kinase 10 of 61 (16%), and other 10 of 61 (16%; Data Supplement). The distribution of PIK3CA mutation types was similar to their expected prevalence, given the tumor type and comutation restrictions on eligibility. Comutations included alterations in the TP53 or MDM2, RTKs or mitogen-activated protein kinase (MAPK), Wnt, cell cycle, FBXW7, IDH1 or IDH2 pathways, and other PI3K pathway mutations. Approximately one third of patients had no comutations detected (Data Supplement).
TABLE 1.
Patient Characteristics
Efficacy
After a median follow-up of 35.7 months, there were no complete or partial responses observed. A best response of stable disease occurred in 32 of 61 patients (52%), whereas progressive disease occurred in 21 of 61 patients (34%). The remaining eight patients were not evaluable (13%; Table 2). PFS at 6 months was 19.9% (90% CI, 12.0 to 29.3) and the median PFS was 3.1 months (90% CI, 1.8 to 3.7; Fig 1A). OS at 6 months was 60.7% (90% CI, 49.6 to 70.0) and the median OS was 7.2 months (90% CI, 5.9 to 10.0; Fig 1B).
TABLE 2.
Best Confirmed Response
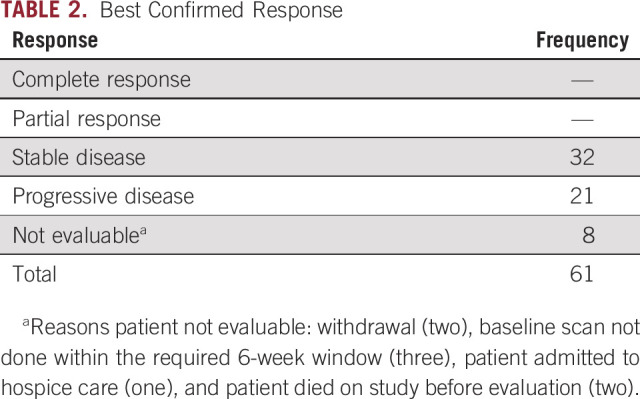
FIG 1.
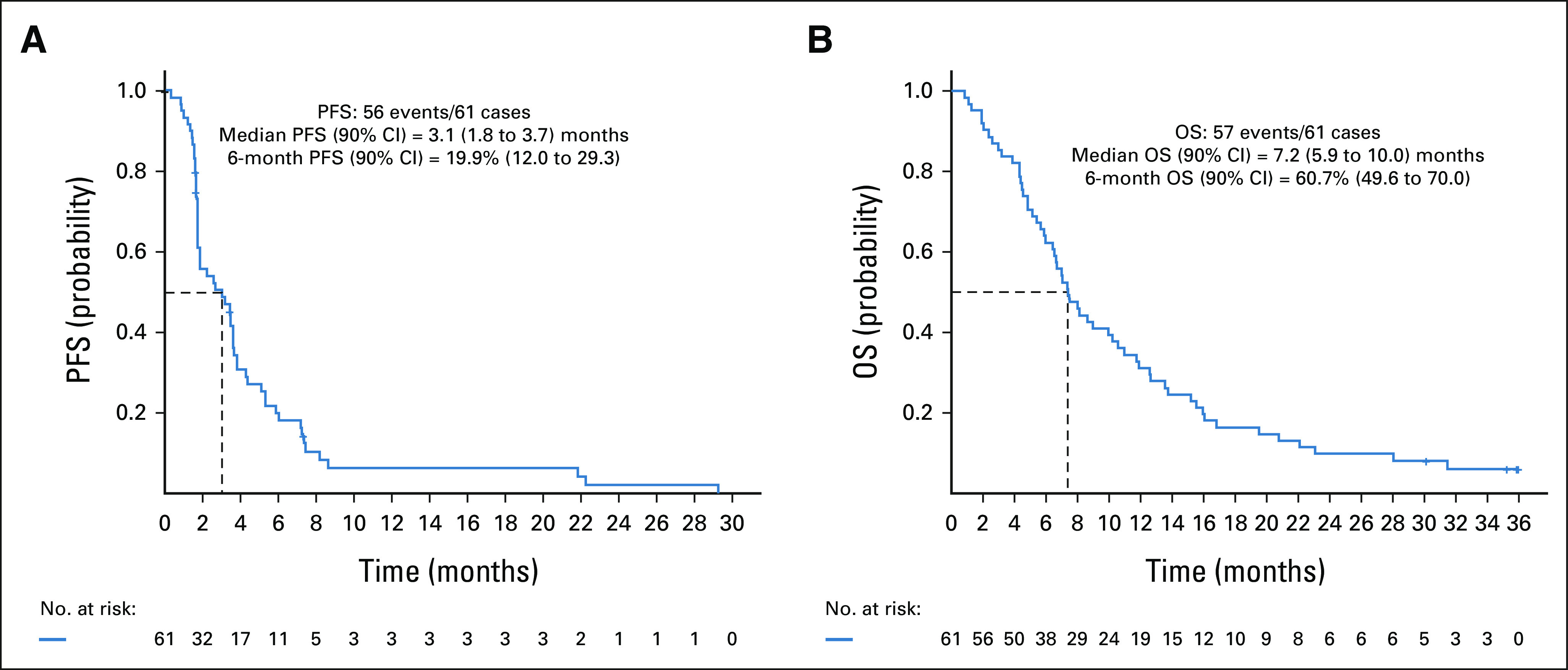
Kaplan-Meier curves of PFS (A) and OS (B) for patients treated with taselisib. The dashed line indicates the median duration. OS, overall survival; PFS, progression-free survival.
Impact of Mutation Type and Comutations on Outcome
The median PFS was 3.7 months for patients with PIK3CA kinase domain mutations, 3.2 months for helical domain mutations, and 1.8 months for mutations in other domains (Data Supplement). These differences did not reach statistical significance. Two subjects with kinase domain mutations remained on study for more than 24 months (Data Supplement). Best change in tumor target lesion diameters by mutation type is shown in the waterfall plots (Fig 2).
FIG 2.
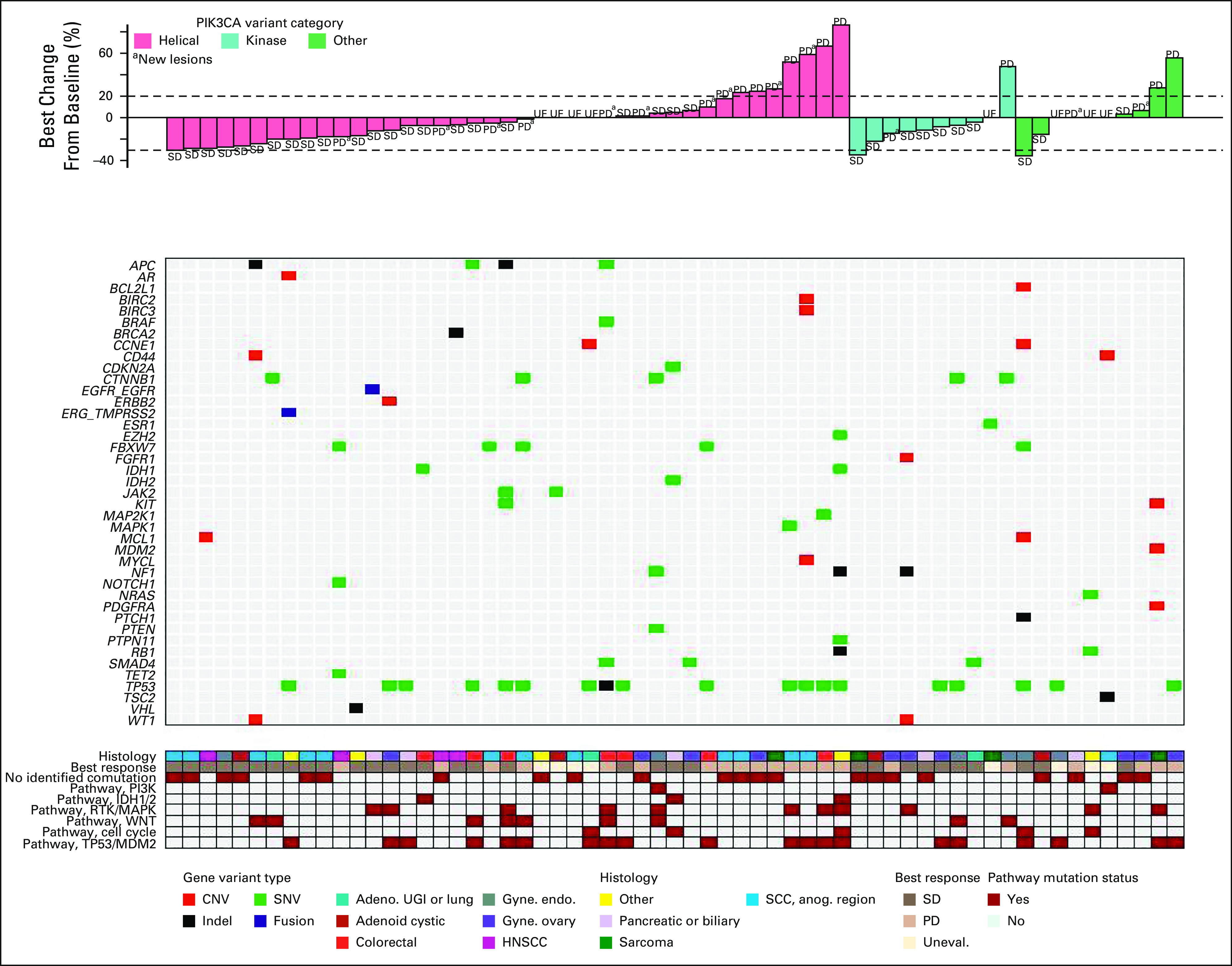
Best change in tumor diameter according to PIK3CA mutation domain and comutations. Investigator-assessed best change in tumor diameter is shown. Patients are grouped by PIK3CA mutation type (top). Co-occurring mutations or CNVs are shown and color-coded by variant type (middle). Comutations categorized by pathway, best overall response, and tumor histology are indicated (bottom). Adeno, adenocarcinoma; CNV, copy-number variant; Gyne. endo., endometrial carcinoma; Gyne ovary, ovarian carcinoma; HNSCC, head and neck squamous cell carcinoma; Indel, insertion or deletion; PD, progressive disease; RTK, receptor tyrosine kinase; SCC Anog., squamous cell carcinoma of the anogenital region; SD, stable disease; SNV, single-nucleotide variant; UE or Uneval, unevaluable; UGI, upper GI.
Individual comutations were too heterogeneous to correlate with clinical outcome. When grouped into pathways, the largest groups of coaltered genes included TP53 or MDM2 (34%), RTKs or MAPK signaling (19%), Wnt signaling (14%), cell cycle (11%), FBXW7 (7%), IDH1 or IDH2 (6%), and other PI3K pathway genes (3%). None of these gene groups or pathways was significantly correlated with either time on treatment (Data Supplement) or change in the sum of target lesion diameters (Fig 2). The absence of another detectable genomic alteration was also not associated with outcomes.
Impact of Disease Type on Outcome
Tumor histologies were very heterogeneous. Several tumor types, including adenoid cystic carcinoma and squamous cell carcinomas of the head and neck, showed some evidence of tumor size reductions in all or most patients, although the sample sizes were small for each group (Fig 3).
FIG 3.
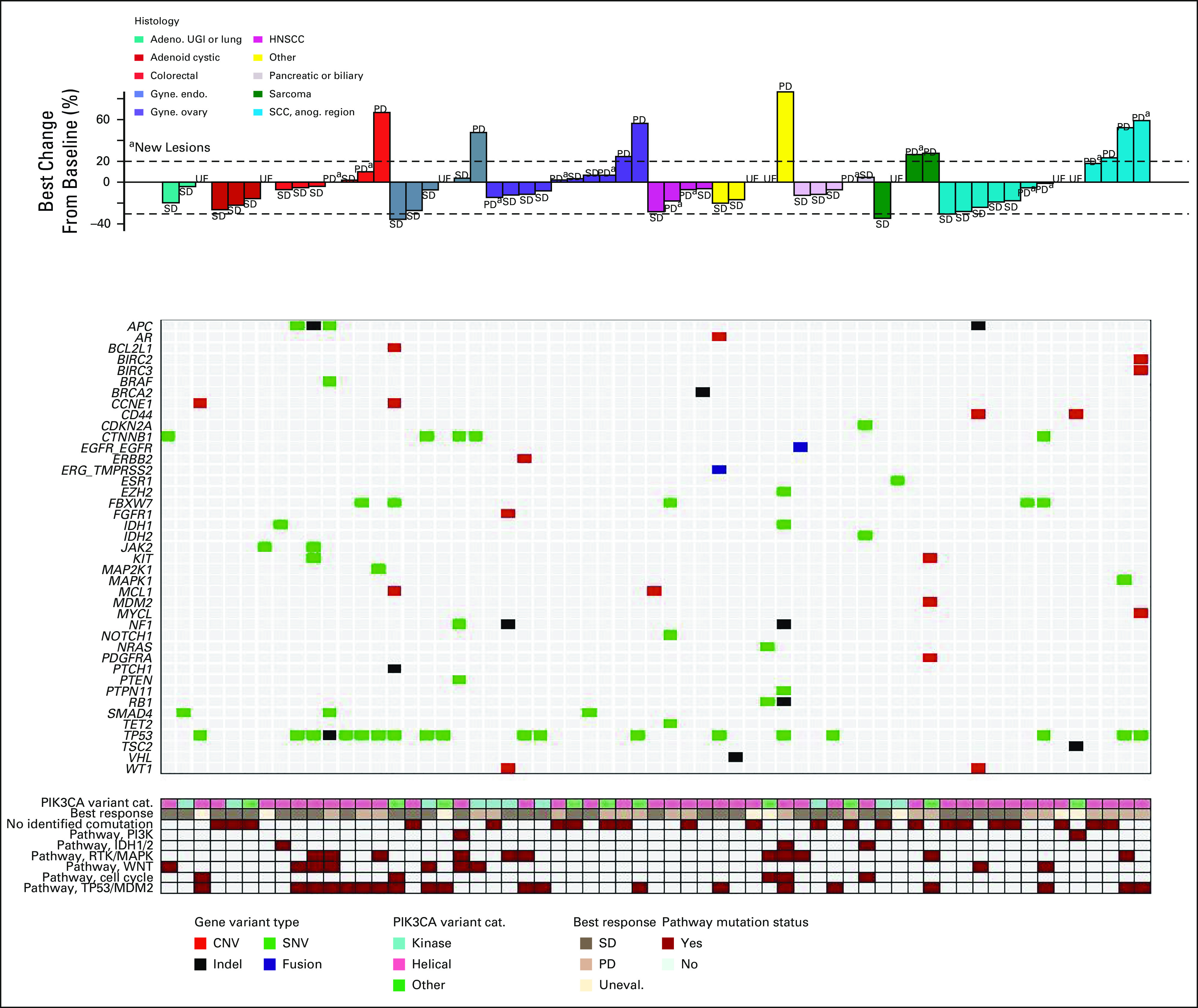
Best change in tumor diameter according to histology and co-occurring mutations. Investigator-assessed best change in tumor diameter is shown. Patients are grouped by PIK3CA mutation type (top). Co-occurring mutations or CNVs are shown and color-coded by variant type (middle). PIK3CA mutation domain, best overall response, and comutations categorized by pathway are indicated (bottom). Adeno, adenocarcinoma; CNV, copy-number variant; Gyne. endo., endometrial carcinoma; Gyne ovary, ovarian carcinoma; HNSCC, head and neck squamous cell carcinoma; Indel, insertion or deletion; PD, progressive disease; RTK, receptor tyrosine kinase; SCC Anog., squamous cell carcinoma of the anogenital region; SD, stable disease; SNV, single nucleotide variant; UE or Uneval, unevaluable; UGI, upper GI.
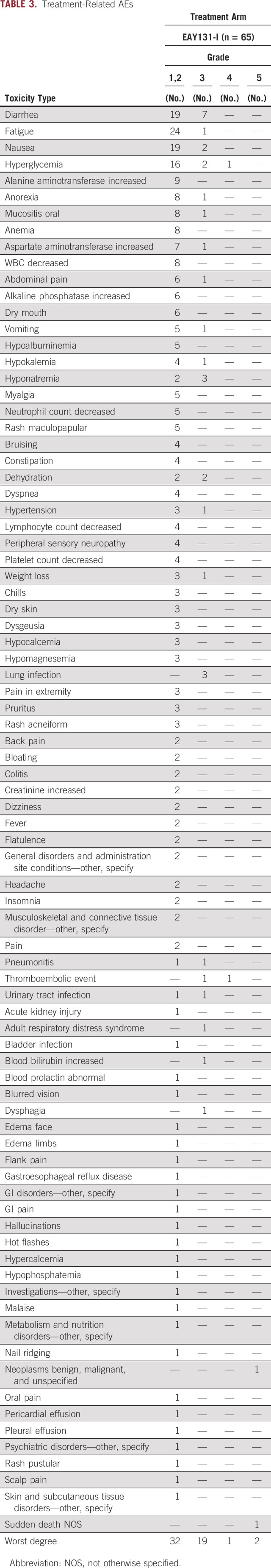
Safety
The AEs observed in this study were typical of the class effects of PI3K inhibitors and were consistent with prior experience with taselisib (Table 3). AEs that were considered at least possibly related to taselisib occurred in 54 of 66 (82%) patients. The most common toxicities were fatigue, diarrhea, nausea, and hyperglycemia. Most AEs were grade 1 and 2, although 19 of 66 (29%) patients experienced grade 3 AEs, one patient (2%) experienced grade 4 hyperglycemia, and two patients had grade 5 AEs (sudden death and neoplasm).
TABLE 3.
Treatment-Related AEs
The median time on treatment was 83 days. In total, six unique patients (10%, 6 of 61) had a total of seven dose reductions (one patient had dose modification twice during cycles 5 and 7), and 5 (8%, 5 of 61) because of AEs (Data Supplement). Fifteen unique patients (25%, 15 of 61) discontinued taselisib, nine of which were because of AEs. Approximately half (51%, 31 of 61) of the participants discontinued protocol therapy by the end of cycle 3, mostly because of disease progression (61%, 19 of 31). Overall, 44 of 61 participants (72%) discontinued treatment because of disease progression (Data Supplement).
DISCUSSION
In the NCI-MATCH EAY131 Arm I, taselisib monotherapy had very limited activity in a heterogeneous cohort of heavily pretreated patients with tumors harboring PIK3CA mutations. Although approximately 20% of patients experienced PFS of at least 6 months, the study failed to meet the primary end point of ORR. No objective responses were observed in this study and given the heterogeneous population, some of the prolonged stable disease may be in part related to indolent cancers.
Exploratory analyses of the impact of PIK3CA mutation type, comutations (considered singly or grouped by pathway), or tumor type on other clinical outcomes including change in tumor size or PFS did not identify any statistically significant associations with these variables. However, these analyses are limited by the study's lack of a control arm, the heterogeneity of tumor types and mutations, the lack of responses generally, and the small sample size.
A similar study of taselisib in 166 patients with diverse tumor types harboring activating PIK3CA mutations reported by Jhaveri et al showed only marginally greater clinical activity than that seen in the current study. In that study, the ORR was 9% (95% CI, 5.1 to 14.5) and the median PFS was 3.6 months (95% CI, 3.1 to 4.2), with a trend toward greater tumor shrinkage and response rates in tumors with helical domain mutations versus kinase or other domain mutations and variability in clinical activity across tumor types.25 The patient selection criteria were similar to those in the current study and are unlikely to account for the slight differences in observed clinical activity. However, most (73%) subjects in the study reported by Jhaveri et al received taselisib at a higher dose of 6 mg. Although some responses were observed at 4 mg, there was a trend toward higher rates of tumor regression and response in subjects receiving 6 mg versus 4 mg. However, the higher dose of taselisib was characterized by higher rates of AEs, grade 3 AEs, serious AEs, and dose reduction or treatment discontinuation. It is likely that the different dose intensity between the two studies contributed to the clinical activity observed in the study by Jhaveri et al. If true, these results highlight the challenge of achieving the necessary dose intensity to achieve clinical responses with taselisib monotherapy in subjects whose tumors harbor sensitizing PIK3CA mutations. Whether this problem could be overcome using a more specific PI3Kα inhibitor such as alpelisib, which appears to have a better therapeutic index, is unknown.
There are several other potential explanations for the lack of objective responses in this study. It is possible that comutated genes or other activated pathways contributed to resistance to the targeted inhibition of PI3K. Because of substantial preclinical and limited clinical data showing that comutation of KRAS leads to resistance to taselisib or other PI3K inhibitors, subjects were excluded if their tumors harbored KRAS mutations; however, other MAPK pathway–activating mutations in BRAF, NRAS, MAP2K1, and other genes were not excluded. It is possible that these comutations contributed to resistance to taselisib in some cases. Most subjects on the study, however, did not have an identifiable comutation known or hypothesized to cause resistance to targeting PIK3CA.
Maintenance of PI3K pathway signaling via other PI3K isoforms could also potentially explain the overall low clinical activity of taselisib. PTEN loss-of-function mutations or protein loss has been shown to mediate acquired resistance to the α-selective PI3K inhibitor alpelisib via preferential activation of the p110β isoform of PI3K.13 Taselisib is less selective than alpelisib, although it is approximately 30-fold less potent against the p110β isoform than p110α. Subjects in this study were excluded if their tumor had an inactivating PTEN mutation or PTEN protein loss, but we cannot rule out the acquisition of such mutations on treatment, or other potential drivers of p110β signaling. Planned correlative studies using circulating tumor DNA samples from subjects in this study may identify these or other mechanisms contributing to the low clinical activity of taselisib monotherapy.
In addition to PI3K inhibitors, there are novel inhibitors of other nodes within the PI3K pathway in late-stage clinical development. Akt inhibitors have shown some promise in solid tumors. Capivasertib is a selective pan-Akt inhibitor with preclinical activity against multiple solid tumor models with PIK3CA, AKT, or PTEN alterations.26 In the phase II FAKTION trial, the addition of capivasertib to fulvestrant in postmenopausal patients with HR+ metastatic breast cancer improved PFS.27 In the NCI-MATCH subgroup EAY131-Y, 35 patients with an AKT1 E17K-mutated metastatic tumor received capivasertib. Breast (51%) and gynecologic (31%) cancers were most common. The ORR was 28.6% (95% CI, 15 to 46) and the 6-month PFS was 50% (95% CI, 35 to 71).28 Ipatasertib is another selective pan-Akt inhibitor that has displayed antitumor activity against multiple human tumor cell lines and xenografts.29 In the phase III IPATential150 trial, the addition of ipatasertib to abiraterone and prednisone improved radiographic PFS in patients with metastatic castration-resistant prostate cancer with evidence of PTEN loss.30
In summary, although taselisib has shown clinical activity in PIK3CA-mutant HR+ breast cancer as monotherapy and when added to antiestrogen therapy,16,18,31 in this study, very little activity was observed in other cancer types with PIK3CA mutations. At this time, there is insufficient evidence to conclude that breast cancer is a special case among PIK3CA-mutated cancer types in terms of sensitivity to PI3K inhibition. However, the results of this study, along with those of the study by Jhaveri et al,25 suggest that PIK3CA mutation alone does not appear to be a sufficient predictor of taselisib activity. Further research is needed to determine the degree to which tumor histology, PIK3CA mutation type, or comutations influence clinical outcomes with PI3K pathway inhibitors. In addition, evaluation of PI3K inhibitors with an improved therapeutic index or other pathway-targeting agents such as Akt inhibitors may potentially demonstrate meaningful activity in a histology-agnostic setting for cancers harboring activating PIK3CA mutations.
ACKNOWLEDGMENTS
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Peter J. O'Dwyer, MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs).
Ian E. Krop
Employment: Freeline Therapeutics (I), PureTech (I), AMAG Pharmaceuticals (I)
Leadership: AMAG Pharmaceuticals (I), Freeline Therapeutics (I), PureTech (I)
Stock and Other Ownership Interests: AMAG Pharmaceuticals (I), Freeline Therapeutics (I), PureTech (I)
Honoraria: Genentech/Roche, AstraZeneca, Celltrion
Consulting or Advisory Role: Genentech/Roche, Seattle Genetics, Daiichi Sankyo, Macrogenics, Novartis, Merck, Bristol Myers Squibb, AstraZeneca
Research Funding: Genentech (Inst), Pfizer (Inst)
Juneko E. Grilley-Olson
Consulting or Advisory Role: Bayer, Chimerix, Kura Oncology, SpringWorks Therapeutics
Research Funding: NanoCarrier (Inst), Genentech (Inst), Seattle Genetics (Inst), Pfizer (Inst), Loxo (Inst), Astellas Pharma (Inst), Iovance Biotherapeutics (Inst)
Josh D. Lauring
Employment: Janssen Research & Development
Stock and Other Ownership Interests: Johnson & Johnson
Consulting or Advisory Role: Galderma (I), Regeneron (I), Novartis (I)
Speakers' Bureau: Galderma (I), AbbVie (I), Pfizer (I)
Patents, Royalties, Other Intellectual Property: I intermittently receive royalty payments for cell lines created in my laboratory, which are licensed for commercial sale to Horizon Discovery, Ltd by Johns Hopkins University
Travel, Accommodations, Expenses: Galderma (I), AbbVie (I)
Edith P. Mitchell
Leadership: Corvus Pharmaceuticals
Honoraria: Sanofi, Exelixis
Consulting or Advisory Role: Genentech, Novartis, Merck, Bristol Myers Squib
Speakers' Bureau: Ipsen
Research Funding: Genentech (Inst), sanofi (Inst)
Robert J. Gray
Research Funding: Agios, Amgen, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Genentech/Roche, Genomic Health, Genzyme, GlaxoSmithKline, Janssen-Ortho, Onyx, Pfizer, Sequenta, Syndax, Novartis, Takeda, AbbVie, Sanofi, Merck Sharp & Dohme
P. Mickey Williams
Research Funding: Illumina (Inst)
Patents, Royalties, Other Intellectual Property: I was a coinventor of the DLBCL cell of origin patent recently filed by the NIH
Stanley R. Hamilton
Research Funding: Minerva Biotechnologies, Intima
James M. Ford
This author is the Editor-in-Chief for JCO Precision Oncology. Journal policy recused the author from having any role in the peer review of this manuscript.
Research Funding: Genentech (Inst), AstraZeneca (Inst), Puma Biotechnology (Inst), Pfizer (Inst), Merus (Inst), Bayer (Inst), Incyte (Inst)
Agustin A. Garcia
Consulting or Advisory Role: Biotheranostics, GlaxoSmithKline
Research Funding: Advenchen Laboratories (Inst), Seattle Genetics (Inst), Merck (Inst), Iovance Pharm (Inst)
Xingwei D. Sui
Consulting or Advisory Role: Novartis
Robert D. Siegel
Research Funding: Merck (Inst), Mirati Therapeutics (Inst), GRAIL (Inst), Altor BioScience (Inst), Galera Therapeutics (Inst), Apollomics (Inst), Strata Oncology (Inst), Arcus Biosciences (Inst), Bristol Myers Squibb (Inst), Cancer Insight (Inst), Puma Biotechnology (Inst), Conjupro Biotherapeutics (Inst), Razor Genomics (Inst), Sanofi (Inst), Seattle Genetics (Inst)
Other Relationship: American Board of Internal Medicine (ABIM)
Brian M. Slomovitz
Consulting or Advisory Role: Clovis Oncology, AstraZeneca, Genentech, Incyte, Agenus, GlaxoSmithKline, GOG Foundation, Myriad Genetics, Merck, Eisai
Carlos L. Arteaga
Stock and Other Ownership Interests: Provista Diagnostics
Consulting or Advisory Role: Novartis, Lilly, Sanofi, Radius Health, Taiho Pharmaceutical, Puma Biotechnology, Merck, Origimed, Immunomedics, Daiichi Sankyo, Athenex, Astrazeneca, Arvinas
Research Funding: Pfizer, Lilly, Takeda
Other Relationship: Susan G. Komen for the Cure
Lyndsay N. Harris
Patents, Royalties, Other Intellectual Property: Philips Healthcare
Peter J. O'Dwyer
Consulting or Advisory Role: Genentech
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), Novartis (Inst), Genentech (Inst), Mirati Therapeutics (Inst), Celgene (Inst), GlaxoSmithKline (Inst), BBI Healthcare (Inst), Pharmacyclics (Inst), Five Prime Therapeutics (Inst), Forty Seven (Inst), Amgen (Inst), H3 Biomedicine (Inst), Taiho Pharmaceutical (Inst), Array BioPharma (Inst), Lilly/ImClone (Inst), Syndax (Inst), Syndax (Inst), Syndax (Inst), Syndax (Inst), syndax (Inst), Minneamrita Therapeutics (Inst)
Expert Testimony: Lilly, Dai-ichi Sankyo
Alice P. Chen
(OPTIONAL) Uncompensated Relationships: Frontiers in Medicine
Keith T. Flaherty
Stock and Other Ownership Interests: Clovis Oncology, Loxo, X4 Pharma, Strata Oncology, PIC Therapeutics, Shattuck Labs, Apricity Health, Oncoceutics, FOGPharma, Tvardi Therapeutics, Checkmate Pharmaceuticals, Kinnate Biopharma, Scorpion Therapeutics, ALX Oncology, xCures, Monopteros Therapeutics, Vibliome Therapeutics, Transcode Therapeutics, Soley Therapeutics
Consulting or Advisory Role: Novartis, Lilly, Oncoceutics, Tvardi Therapeutics, Takeda, Boston Biomedical, Debiopharm Group, FOGPharma
No other potential conflicts of interest were reported.
DISCLAIMER
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
PRIOR PRESENTATION
Presented at ASCO Annual Meeting, Chicago, IL, June 1-5, 2018.
SUPPORT
Supported by the National Cancer Institute of the National Institutes of Health under the following award numbers: U10CA180820, U10CA180794, U10CA180888, U10CA180868, UG1CA189821, UG1CA189854, UG1CA189858, UG1CA189953, UG1CA233180, UG1CA233196, UG1CA233302, UG1CA233329, UG1CA233341, and UG1CA233373.
CLINICAL TRIAL INFORMATION
I.E.K., O.A.J., J.E.G.-O., and J.D.L. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Ian E. Krop, Juneko E. Grilley-Olson, Josh D. Lauring, Edith P. Mitchell, Robert J. Gray, Lisa M. McShane, Larry V. Rubinstein, David Patton, P. Mickey Williams, Stanley R. Hamilton, Barbara A. Conley, Carlos L. Arteaga, Peter J. O'Dwyer, Alice P. Chen, Keith T. Flaherty
Financial support: David Patton
Administrative support: David Patton, Carlos L. Arteaga, Lyndsay N. Harris
Collection and assembly of data: Ian E. Krop, Opeyemi A. Jegede, Juneko E. Grilley-Olson, James A. Zwiebel, Robert J. Gray, Victoria Wang, David Patton, P. Mickey Williams, Stanley R. Hamilton, Scott A. Kono, James M. Ford, Agustin A. Garcia, Xingwei D. Sui, Robert D. Siegel, Brian M. Slomovitz, Lyndsay N. Harris
Data analysis and interpretation: Ian E. Krop, Opeyemi A. Jegede, Juneko E. Grilley-Olson, Josh D. Lauring, Robert J. Gray, Victoria Wang, Lisa M. McShane, David Patton, P. Mickey Williams, Stanley R. Hamilton, Agustin A. Garcia, Carlos L. Arteaga, Lyndsay N. Harris, Peter J. O'Dwyer, Alice P. Chen, Keith T. Flaherty
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
Provision of study material or patients: David Patton, Stanley R. Hamilton, James M. Ford, Agustin A. Garcia, Xingwei D. Sui, Robert D. Siegel
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Ian E. Krop
Employment: Freeline Therapeutics (I), PureTech (I), AMAG Pharmaceuticals (I)
Leadership: AMAG Pharmaceuticals (I), Freeline Therapeutics (I), PureTech (I)
Stock and Other Ownership Interests: AMAG Pharmaceuticals (I), Freeline Therapeutics (I), PureTech (I)
Honoraria: Genentech/Roche, AstraZeneca, Celltrion
Consulting or Advisory Role: Genentech/Roche, Seattle Genetics, Daiichi Sankyo, Macrogenics, Novartis, Merck, Bristol Myers Squibb, AstraZeneca
Research Funding: Genentech (Inst), Pfizer (Inst)
Juneko E. Grilley-Olson
Consulting or Advisory Role: Bayer, Chimerix, Kura Oncology, SpringWorks Therapeutics
Research Funding: NanoCarrier (Inst), Genentech (Inst), Seattle Genetics (Inst), Pfizer (Inst), Loxo (Inst), Astellas Pharma (Inst), Iovance Biotherapeutics (Inst)
Josh D. Lauring
Employment: Janssen Research & Development
Stock and Other Ownership Interests: Johnson & Johnson
Consulting or Advisory Role: Galderma (I), Regeneron (I), Novartis (I)
Speakers' Bureau: Galderma (I), AbbVie (I), Pfizer (I)
Patents, Royalties, Other Intellectual Property: I intermittently receive royalty payments for cell lines created in my laboratory, which are licensed for commercial sale to Horizon Discovery, Ltd by Johns Hopkins University
Travel, Accommodations, Expenses: Galderma (I), AbbVie (I)
Edith P. Mitchell
Leadership: Corvus Pharmaceuticals
Honoraria: Sanofi, Exelixis
Consulting or Advisory Role: Genentech, Novartis, Merck, Bristol Myers Squib
Speakers' Bureau: Ipsen
Research Funding: Genentech (Inst), sanofi (Inst)
Robert J. Gray
Research Funding: Agios, Amgen, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Genentech/Roche, Genomic Health, Genzyme, GlaxoSmithKline, Janssen-Ortho, Onyx, Pfizer, Sequenta, Syndax, Novartis, Takeda, AbbVie, Sanofi, Merck Sharp & Dohme
P. Mickey Williams
Research Funding: Illumina (Inst)
Patents, Royalties, Other Intellectual Property: I was a coinventor of the DLBCL cell of origin patent recently filed by the NIH
Stanley R. Hamilton
Research Funding: Minerva Biotechnologies, Intima
James M. Ford
This author is the Editor-in-Chief for JCO Precision Oncology. Journal policy recused the author from having any role in the peer review of this manuscript.
Research Funding: Genentech (Inst), AstraZeneca (Inst), Puma Biotechnology (Inst), Pfizer (Inst), Merus (Inst), Bayer (Inst), Incyte (Inst)
Agustin A. Garcia
Consulting or Advisory Role: Biotheranostics, GlaxoSmithKline
Research Funding: Advenchen Laboratories (Inst), Seattle Genetics (Inst), Merck (Inst), Iovance Pharm (Inst)
Xingwei D. Sui
Consulting or Advisory Role: Novartis
Robert D. Siegel
Research Funding: Merck (Inst), Mirati Therapeutics (Inst), GRAIL (Inst), Altor BioScience (Inst), Galera Therapeutics (Inst), Apollomics (Inst), Strata Oncology (Inst), Arcus Biosciences (Inst), Bristol Myers Squibb (Inst), Cancer Insight (Inst), Puma Biotechnology (Inst), Conjupro Biotherapeutics (Inst), Razor Genomics (Inst), Sanofi (Inst), Seattle Genetics (Inst)
Other Relationship: American Board of Internal Medicine (ABIM)
Brian M. Slomovitz
Consulting or Advisory Role: Clovis Oncology, AstraZeneca, Genentech, Incyte, Agenus, GlaxoSmithKline, GOG Foundation, Myriad Genetics, Merck, Eisai
Carlos L. Arteaga
Stock and Other Ownership Interests: Provista Diagnostics
Consulting or Advisory Role: Novartis, Lilly, Sanofi, Radius Health, Taiho Pharmaceutical, Puma Biotechnology, Merck, Origimed, Immunomedics, Daiichi Sankyo, Athenex, Astrazeneca, Arvinas
Research Funding: Pfizer, Lilly, Takeda
Other Relationship: Susan G. Komen for the Cure
Lyndsay N. Harris
Patents, Royalties, Other Intellectual Property: Philips Healthcare
Peter J. O'Dwyer
Consulting or Advisory Role: Genentech
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), Novartis (Inst), Genentech (Inst), Mirati Therapeutics (Inst), Celgene (Inst), GlaxoSmithKline (Inst), BBI Healthcare (Inst), Pharmacyclics (Inst), Five Prime Therapeutics (Inst), Forty Seven (Inst), Amgen (Inst), H3 Biomedicine (Inst), Taiho Pharmaceutical (Inst), Array BioPharma (Inst), Lilly/ImClone (Inst), Syndax (Inst), Syndax (Inst), Syndax (Inst), Syndax (Inst), syndax (Inst), Minneamrita Therapeutics (Inst)
Expert Testimony: Lilly, Dai-ichi Sankyo
Alice P. Chen
(OPTIONAL) Uncompensated Relationships: Frontiers in Medicine
Keith T. Flaherty
Stock and Other Ownership Interests: Clovis Oncology, Loxo, X4 Pharma, Strata Oncology, PIC Therapeutics, Shattuck Labs, Apricity Health, Oncoceutics, FOGPharma, Tvardi Therapeutics, Checkmate Pharmaceuticals, Kinnate Biopharma, Scorpion Therapeutics, ALX Oncology, xCures, Monopteros Therapeutics, Vibliome Therapeutics, Transcode Therapeutics, Soley Therapeutics
Consulting or Advisory Role: Novartis, Lilly, Oncoceutics, Tvardi Therapeutics, Takeda, Boston Biomedical, Debiopharm Group, FOGPharma
No other potential conflicts of interest were reported.
REFERENCES
- 1.Cantley LC.The phosphoinositide 3-kinase pathway Science 2961655–16572002 [DOI] [PubMed] [Google Scholar]
- 2.Guertin DA, Sabatini DM.Defining the role of mTOR in cancer Cancer Cell 129–222007 [DOI] [PubMed] [Google Scholar]
- 3.Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers Cancer Biol Ther 3772–7752004 [DOI] [PubMed] [Google Scholar]
- 4.Massion PP, Taflan PM, Shyr Y, et al. Early involvement of the phosphatidylinositol 3-kinase/Akt pathway in lung cancer progression Am J Respir Crit Care Med 1701088–10942004 [DOI] [PubMed] [Google Scholar]
- 5.Shayesteh L, Lu Y, Kuo WL, et al. PIK3CA is implicated as an oncogene in ovarian cancer Nat Genet 2199–1021999 [DOI] [PubMed] [Google Scholar]
- 6.AACR Project GENIE Consortium AACR Project GENIE: Powering precision medicine through an International Consortium Cancer Discov 7818–8312017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang S, Bader AG, Vogt PK.Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic Proc Natl Acad Sci USA 102802–8072005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ihle NT, Lemos R, Jr, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance Cancer Res 69143–1502009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.She QB, Halilovic E, Ye Q, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors Cancer Cell 1839–512010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edgar KA, Wallin JJ, Berry M, et al. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors Cancer Res 701164–11722010 [DOI] [PubMed] [Google Scholar]
- 11.Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis Nature 454776–7792008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wee S, Wiederschain D, Maira SM, et al. PTEN-deficient cancers depend on PIK3CB Proc Natl Acad Sci USA 10513057–130622008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juric D, Castel P, Griffith M, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor Nature 518240–2442015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore HM, Savage HM, O'Brien C, et al. Predictive and pharmacodynamic biomarkers of response to the phosphatidylinositol 3-kinase inhibitor taselisib in breast cancer preclinical models Mol Cancer Ther 19292–3032020 [DOI] [PubMed] [Google Scholar]
- 15. Song KW, Edgar KA, Kirkpatrick DS, et al. The PI3K inhibitor, taselisib, has a unique mechanism of action that leads to enhanced potency in PIK3CA mutant models. Cancer Res. 2017;77 suppl; abstr 146. [Google Scholar]
- 16.Juric D, Krop I, Ramanathan RK, et al. Phase I dose-escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors Cancer Discov 7704–7152017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andre F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer N Engl J Med 3801929–19402019 [DOI] [PubMed] [Google Scholar]
- 18.Dent S, Cortes J, Im YH, et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: The SANDPIPER trial Ann Oncol 32197–2072021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreyling M, Santoro A, Mollica L, et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma J Clin Oncol 353898–39052017 [DOI] [PubMed] [Google Scholar]
- 20.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia N Engl J Med 370997–10072014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flinn IW, Hillmen P, Montillo M, et al. The phase 3 DUO trial: Duvelisib vs ofatumumab in relapsed and refractory CLL/SLL Blood 1322446–24552018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flaherty KT, Gray R, Chen A, et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) trial: Lessons for genomic trial design J Natl Cancer Inst 1121021–10292020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flaherty KT, Gray RJ, Chen AP, et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH) J Clin Oncol 383883–38942020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer 45228–2472009 [DOI] [PubMed] [Google Scholar]
- 25.Jhaveri K, Chang MT, Juric D, et al. Phase I Basket study of taselisib, an isoform-selective PI3K inhibitor, in patients with PIK3CA-mutant cancers Clin Cancer Res 27447–4592021 [DOI] [PubMed] [Google Scholar]
- 26.Davies BR, Greenwood H, Dudley P, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background Mol Cancer Ther 11873–8872012 [DOI] [PubMed] [Google Scholar]
- 27.Jones RH, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): A multicentre, randomised, controlled, phase 2 trial Lancet Oncol 21345–3572020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalinsky K, Hong F, McCourt CK, et al. Effect of capivasertib in patients with an AKT1 E17K-mutated tumor: NCI-MATCH subprotocol EAY131-Y nonrandomized trial JAMA Oncol 7271–2782021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin J, Sampath D, Nannini MA, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models Clin Cancer Res 191760–17722013 [DOI] [PubMed] [Google Scholar]
- 30.de Bono JS, Bracarda B, Sternberg CN, et al. IPATential150: Phase III Study of Ipatasertib (Ipat) Plus Abiraterone (Abi) vs Placebo (Pbo) Plus Abi in Metastatic Castration-Resistant Prostate Cancer (mCRPC) ESMO Virtual Congress, 2020 (abstr)
- 31.Dickler MN, Saura C, Richards DA, et al. Phase II study of taselisib (GDC-0032) in combination with fulvestrant in patients with HER2-negative, hormone receptor-positive advanced breast cancer Clin Cancer Res 244380–43872018 [DOI] [PMC free article] [PubMed] [Google Scholar]