

ABSTRACT
A liver-specific microRNA, miR-122, anneals to the hepatitis C virus (HCV) genomic 5′ terminus and is essential for virus replication in cell culture. However, bicistronic HCV replicons and full-length RNAs with specific mutations in the 5′ untranslated region (UTR) can replicate, albeit to low levels, without miR-122. In this study, we have identified that HCV RNAs lacking the structural gene region or having encephalomyocarditis virus internal ribosomal entry site (EMCV IRES)-regulated translation had reduced requirements for miR-122. In addition, we found that a smaller proportion of cells supported miR-122-independent replication compared a population of cells supporting miR-122-dependent replication, while viral protein levels per positive cell were similar. Further, the proportion of cells supporting miR-122-independent replication increased with the amount of viral RNA delivered, suggesting that establishment of miR-122-independent replication in a cell is affected by the amount of viral RNA delivered. HCV RNAs replicating independently of miR-122 were not affected by supplementation with miR-122, suggesting that miR-122 is not essential for maintenance of an miR-122-independent HCV infection. However, miR-122 supplementation had a small positive impact on miR-122-dependent replication, suggesting a minor role in enhancing ongoing virus RNA accumulation. We suggest that miR-122 functions primarily to initiate an HCV infection but has a minor influence on its maintenance, and we present a model in which miR-122 is required for replication complex formation at the beginning of an infection and also supports new replication complex formation during ongoing infection and after infected cell division.
IMPORTANCE The mechanism by which miR-122 promotes the HCV life cycle is not well understood, and a role in directly promoting genome amplification is still debated. In this study, we have shown that miR-122 increases the rate of viral RNA accumulation and promotes the establishment of an HCV infection in a greater number of cells than in the absence of miR-122. However, we also confirm a minor role in promoting ongoing virus replication and propose a role in the initiation of new replication complexes throughout a virus infection. This study has implications for the use of anti-miR-122 as a potential HCV therapy.
KEYWORDS: hepatitis C virus, miR-122, miR-122-independent replication, 5′ untranslated region, microRNA-122, replication, translation
INTRODUCTION
Hepatitis C virus (HCV) is a positive-strand RNA flavivirus that primarily infects the liver and is carried by over 71 million people worldwide (1). Around 70% of infections cause chronic hepatitis C disease, which can lead to complications such as liver cirrhosis, hepatocellular carcinoma, and decompensated liver disease (2). Chronic HCV was the primary cause of liver transplantations in the United States, Europe, and Japan for many years, but can now be cured using highly effective direct-acting antiviral (DAA) treatments (3).
The positive-strand RNA genome of HCV is approximately 9.6 kb long and contains a single open reading frame (ORF) that encodes a viral polyprotein. The ORF is flanked by 5′ and 3′ untranslated regions (UTR) that have secondary structures essential for viral translation and replication (4, 5). The uncapped HCV 5′ UTR bears an internal ribosomal entry site (IRES) that directs cap-independent synthesis of the viral polyprotein, which is subsequently cleaved by viral and cellular proteases into three structural (core, E1, and E2) and seven nonstructural (p7, NS2, NS3, NS4a and 4b and NS5a and 5b) viral proteins. The nonstructural proteins NS3 to NS5b form the replicase complex and are the minimum viral proteins required for genome replication (6, 7).
Micro RNAs (miRNA) are small noncoding RNAs (approximately 22 nucleotides) that regulate gene expression by suppressing mRNA translation and promoting mRNA decay (8). However, contrary to the conventional suppressive role to miRNAs, the liver-specific miRNA, miR-122, anneals to the HCV genome and is essential for viral propagation (9). miR-122, in association with human Argonaute proteins (Ago 1 to 4) (hAgo:miR-122 complex), binds to two sites on the extreme 5′ UTR of the viral genome and promotes viral RNA accumulation (Fig. 1) (9–11). While the precise mechanism of HCV replication promotion remains incompletely understood, different roles of miR-122 have been identified, including translation stimulation (12–14), genome stabilization (15–17), and a direct role in genome amplification (18). Recent studies have also predicted that miR-122 annealing to the viral 5′ UTR shapes the structure of the viral 5′-UTR RNA and promotes the formation of an “active” or a translation-favorable viral IRES structure (14, 19–21); however, biophysical analyses are still required to support this hypothesis.
FIG 1.

miR-122 annealing to the HCV genome. An Ago:miR-122 complex binds to two sites (Site-1 and Site-2) on the 5′UTR of the HCV genome and promote viral propagation. The miR-122 binding pattern and sequence of nucleotides 1 to 46 of the 5′UTR of hepatitis C virus genome and miR122 annealing to it are shown.
Since miR-122 is an important host factor for HCV replication, miR-122 antagonists have shown promising results in a prolonged reduction of viral load in both preclinical and clinical trials (22–24), but the recent emergence of resistant HCV mutants (25) suggests that the virus can adapt to grow in the absence of miR-122. In addition, lab-derived HCV mutants capable of low-level miR-122-independent replication have been identified. These viruses are either bicistronic, with virus protein synthesis mediated by an encephalomyocarditis virus (EMCV) IRES, or have specific mutations in the miR-122 binding sites of the 5′ UTR (19, 20, 25–30). In contrast, wild-type genome replication is undetectable in the absence of miR-122, even with the use of sensitive reporter genomes. Since miR-122 expression is specific to the liver and composes almost 70% of the total small RNA of the liver (31), it likely plays a role in regulating HCV’s liver tropism. However, there have been reports of extrahepatic HCV RNA and replication (2, 32, 33), and chronic HCV is associated with a broad range of extrahepatic conditions such as cryoglobulinemic vasculitis, diabetes mellitus, and B-cell lymphoma, suggesting a possible role for miR-122-indepenedent HCV replication in these manifestations (2, 34). Further, others have shown selection of cells supporting stable miR-122-independent replication of HCV replicon RNA that suggested a role of miR-122 for establishment, but not maintenance of, replication (25).
In this study, we aimed to compare transient and stable miR-122-dependent versus miR-122-independent HCV replication to identify HCV genetic elements that modulate HCV’s dependency on miR-122 and determine the roles of miR-122 in different stages of the HCV life cycle. We have found that the presence of an EMCV IRES and a smaller genome size allows HCV to replicate independently from miR-122, suggesting roles for modified translation regulation and smaller genome sizes in facilitating miR-122-independent replication. Using several models of miR-122-independent HCV replication, we have also found that miR-122-independent replication appears efficient within individual cells but is established in only a small number of cells in the population. Further, we also found that miR-122 has a dramatic impact during the early stages and the establishment of an HCV infection but a less potent supportive role for ongoing genome amplification. From this, we propose a mechanistic model where miR-122 promotes translation and genomic RNA stability early in the virus life cycle to “jump-start” a virus infection but also has a lesser role in maintaining an ongoing infection, which we propose is to initiate new virus replication complexes as the infection amplifies within a cell and as liver cells divide.
RESULTS
EMCV IRES-mediated synthesis of the HCV nonstructural genes promotes miR-122-independent HCV replication.
In previous works, we have identified that the HCV subgenomic replicon (SGR) JFH-1 Fluc was capable of genome amplification independent of miR-122 in Hep3B cells (26). This construct lacks the structural proteins core, E1, E2, and p2, as well as nonstructural protein NS2; while synthesis of the Fluc reporter protein is driven by the HCV endogenous IRES, synthesis of the nonstructural polyprotein (NS3 through NS5b) is driven by an EMCV IRES. These data suggested that one or all of (i) the addition of an EMCV IRES to regulate viral nonstructural gene synthesis, (ii) the removal of the structural proteins, or (iii) the reduced genome length permitted HCV SGR RNA replication independent of miR-122. In this study, we aimed to determine the independent influence of the EMCV IRES, the structural gene region, and the size of the RNA genome on miR-122-independent replication.
To determine if the addition of an EMCV IRES facilitated miR-122-independent replication, we assessed transient miR-122-dependent and miR-122-independent replication of full-length HCV RNAs in which viral protein synthesis was regulated by an EMCV IRES (Fig. 2A), i.e., FGR JFH-1 Fluc, which has a firefly luciferase reporter gene, or J6/JFH-1 Neo (p7Rluc2a), which has a Renilla luciferase reporter gene and a neomycin selection marker (here referred to as J6/JFH-1 Neo Rluc). To assess miR-122-independent and miR-122-dependent replication, the viral RNAs were coelectroporated into Huh 7.5 miR-122 knockout (KO) cells with either control microRNA (miControl) to assess miR-122-independent replication or synthetic miR-122 to assess miR-122-dependent replication (Fig. 2B to E). HCV genomic RNA amplification was assessed based on luciferase expression as a proxy for virus replication, and SGR JFH-1 Fluc RNA was included as a positive control for miR-122-independent replication (26). All of the viral RNAs replicated efficiently when miR-122 was added (Fig. 2B to E). In the absence of miR-122, we and others have consistently observed that the J6/JFH-1 (p7Rluc2a) full-length wild-type construct (here referred to as J6/JFH-1 Rluc) does not replicate; Renilla luciferase levels decrease between 2 h and 1 day to background levels similar to that seen for a nonreplicating control RNA (GNN) (Fig. 2B). This reflects the established roles for miR-122 in stabilizing the viral genomic RNA and stimulating translation early in the infection cycle. However, both the bicistronic full-length genomic replicon expressing a firefly luciferase reporter gene (FGR JFH-1 Fluc) and a bicistronic full-length genomic replicon that expresses both a neomycin selection marker and a Renilla luciferase reporter gene (J6/JFH-1 Neo Rluc) (Fig. 2A) showed miR-122-independent replication since they demonstrated measurable luciferase levels above their corresponding GNN or GND negative controls (Fig. 2B to F). These results suggest that protein synthesis facilitation by the EMCV IRES allows both full-length and subgenomic HCV to replicate in the absence of miR-122. However, miR-122-independent replication of these RNAs still showed a decrease in luciferase expression between 2 h and 1 day, and at later days were lower than in the presence of miR-122, further highlighting an important role for miR-122 early in the infection cycle that can be partially compensated for by translation regulation by the EMCV IRES. This experiment additionally rules out the contribution of a given reporter gene, Fluc or Rluc, to miR-122-independent replication (Fig. 2C to E). Thus, HCV translation regulation altered by the addition of the EMCV IRES facilitates virus replication in the absence of miR-122. However, miR-122-independent replication of SGR JFH-1 Fluc, which lacks the structural gene region, is more efficient than that of FGR JFH-1 Fluc, in which the structural gene region is intact (Fig. 2F), suggesting that the structural gene region may inhibit, or shorter genome length enhance, miR-122-independent replication of HCV.
FIG 2.
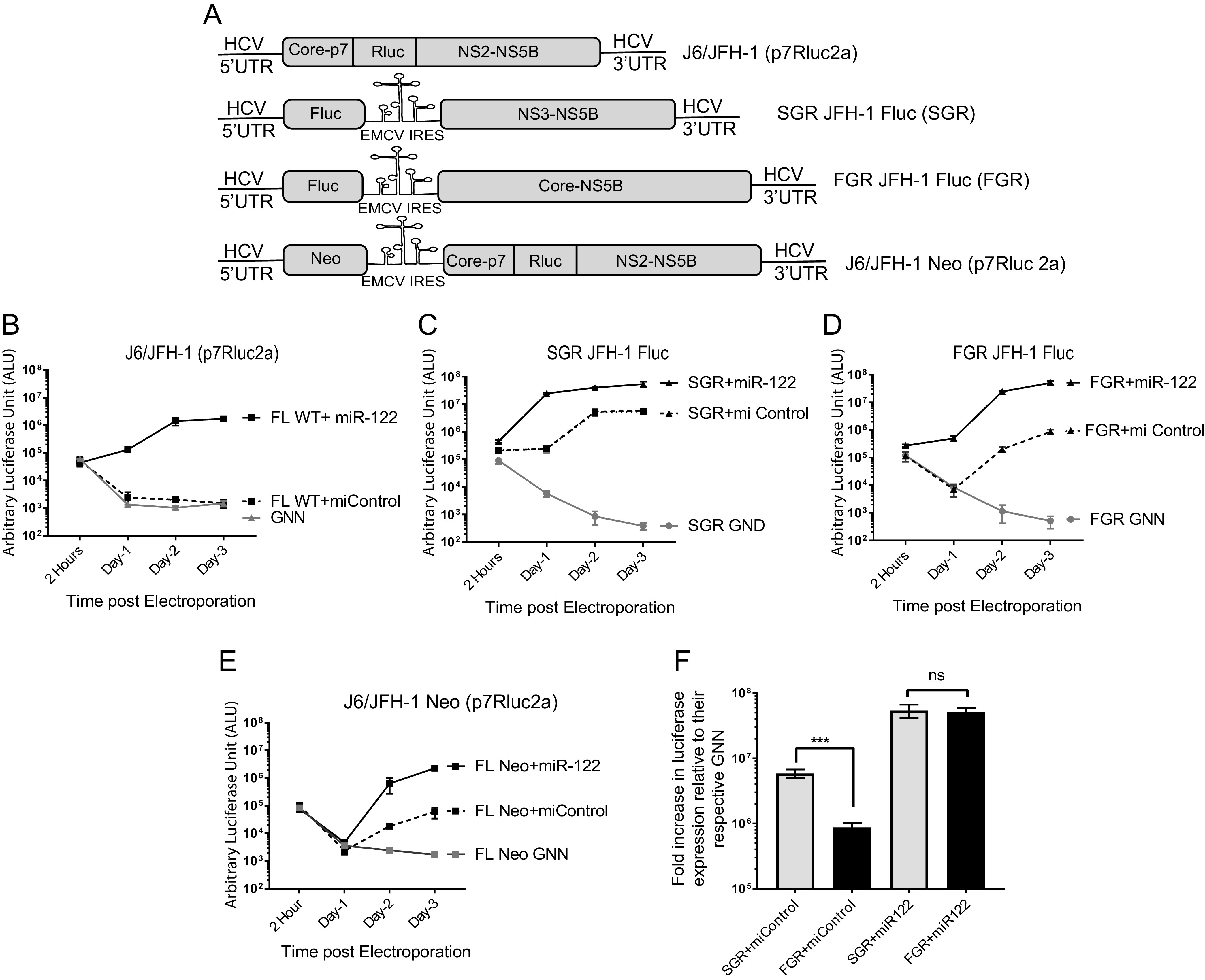
HCV RNAs containing an EMCV IRES replicate independently from miR-122. (A) Schematic diagram of the HCV viral genomes. (Top to bottom) Monocistronic full-length wild-type J6/JFH-1 (p7Rluc2a) with a Renilla luciferase reporter, bicistronic subgenomic replicon (SGR) JFH-1 with a Firefly luciferase reporter SGR JFH-1 Fluc, bicistronic full-length genomic replicon (FGR) or FGR JFH-1Fluc with a firefly luciferase reporter, and bicistronic full-length wild-type J6/JFH-1 Neo (p7Rluc2a) with a Renilla luciferase reporter and a neomycin selection marker. (B to F) Transient replication of HCV viral genomes shown in panel A in the presence of a control miRNA (miControl) or miR-122 in Huh 7.5 miR-122 knockout cells measured by luciferase reporter expression. (B) J6/JFH-1 (p7Rluc2a), (C) SGR JFH-1 Fluc, (D) FGR JFH-1 Fluc, and (E) J6/JFH-1 Neo (p7Rluc2a). The replication-defective versions of each genome, GNN or GND, are negative controls. (F) Fold change in luciferase expression versus nonreplicative GNN controls of a full-length (FGR JFH-1 Fluc) and a subgenomic (SGR JFH-1 Fluc) HCV adapted from data shown in panels B and C. All data shown are the average of three or more independent experiments, and error bars indicate the standard deviation of the mean. The significance was determined by using Student’s t test (ns, not significant, ***, P < 0.001).
Shorter genomic RNAs support miR-122-independent HCV replication.
To determine the contribution of the structural gene region and the size of the RNA genome to miR-122-independent replication, we assessed transient miR-122-dependent and miR-122-independent replication of HCV RNAs in which various regions or all of the structural gene region was removed. For this we designed several monocistronic constructs derived from J6/JFH-1 (p7Rluc2a) (lacking the EMCV IRES), where we have deleted part, or all, of the structural gene region (Fig. 3A). The deletion mutants were designed such that they did not adversely affect polyprotein processing, and all were replication competent in the presence of miR-122 (Fig. 3B to F). However, only two mutants in which the entire structural gene region was removed, SGR JFH-1 (Rluc2a) NS2 and SGR JFH-1 (Rluc2a) NS3, were capable of replicating in the absence of miR-122 (Fig. 4A to C). Thus, we concluded that the absence of the structural genes in their entirety, rather than a specific protein-coding region, modulated the ability of HCV RNA to replicate independently from miR-122, and we speculate that may be due to a general increase in rate of genomic RNA replication based on a smaller genome size but could also be caused by increased efficiency of synthesis by T7 polymerase during in vitro transcription.
FIG 3.
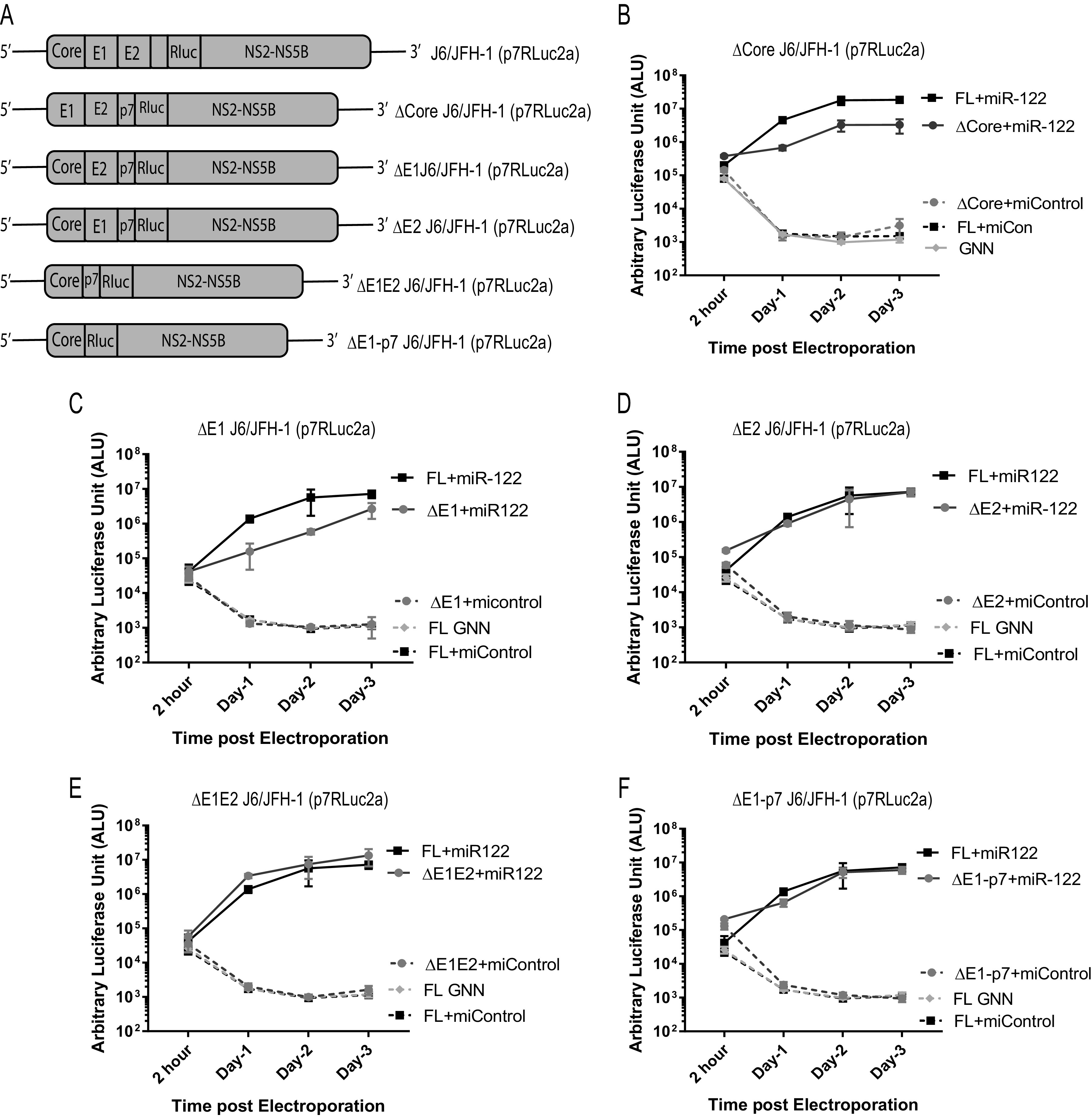
Deletion of the portions of the structural gene region does not facilitate miR-122-independent replication of HCV. (A) Schematic diagrams of the structural gene deletion mutant HCV constructs used, from top to bottom: full-length WT, J6/JFH-1(p7Rluc2a); J6/JFH-1 with deletion of core region, Δcore J6/JFH-1 (p7Rluc2a); J6/JFH-1 with deletion of E1 region, ΔE1 J6/JFH-1 (p7Rluc2a), J6/JFH-1 with deletion of E2 region, ΔE2 J6/JFH-1 (p7Rluc2a), J6/JFH-1 with deletion of E1E2 region, ΔE1E2 J6/JFH-1 (p7Rluc2a); and J6/JFH-1 with deletion of E1-p7 region, ΔE1-p7 J6/JFH-1 (Rluc2a). Transient replication of structural gene deletion HCV RNAs in Huh 7.5 miR-122 KO cells in the presence of either control microRNA (miControl) or miR-122. (B) WT J6/JFH-1 Rluc HCV RNA and Δcore J6/JFH-1 (p7Rluc2a) RNA; (C) ΔE1 J6/JFH-1 (p7Rluc2a) RNA; (D) ΔE2 J6/JFH-1 (p7Rluc2a) RNA; (E) ΔE1E2 J6/JFH-1 (p7Rluc2a) RNA; and (F) ΔE1-p7 J6/JFH-1 (Rluc2a). A replication-defective GNN mutant was included for each RNA as a negative control. All the experiments are a representation of 3 or more replicates. The significance was determined by using Student’s t test (ns, not significant; *, P < 0.033).
FIG 4.
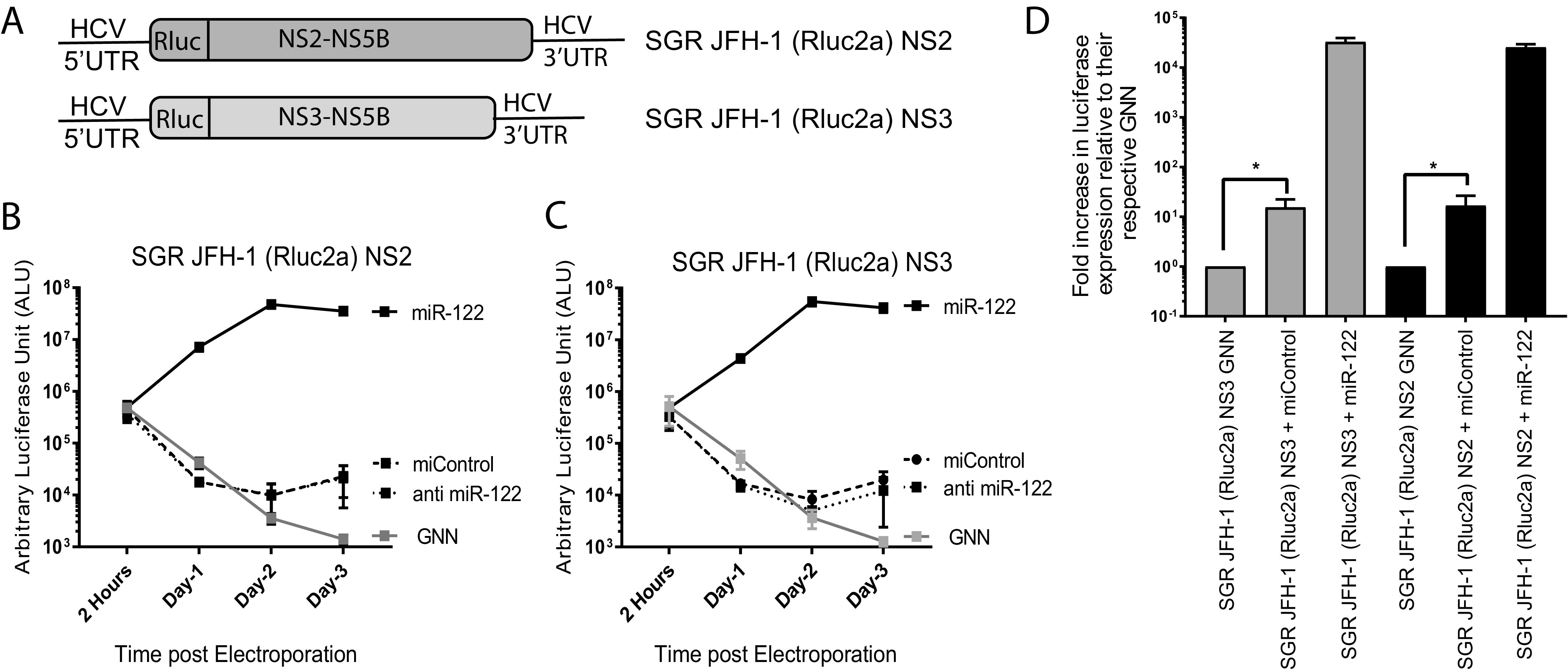
Deletion of the entire structural gene region facilitates miR-122-independent replication of HCV. (A) Schematic diagrams of the structural gene deletion mutants missing the entire structural gene region. SGR JFH-1 (Rluc2a) NS2 has a deletion of the structural gene region and retains either NS2 to NS5b nonstructural genes, and SGR JFH-1 (Rluc2a) NS3 has a complete deletion of structural genes and NS2 and retains NS3 to NS5b nonstructural genes. (B and C) Transient replication assay of (B) SGR JFH-1 (Rluc2a) NS2 and (C) SGR JFH-1 (Rluc2a) NS3 HCV RNA in the presence of a control miRNA (miControl) or miR-122 in Huh 7.5 miR-122 knockout cells or anti-miR-122 LNA to verify miR-122-independent replication. GNN denotes replication assays using the respective replication defective mutants. (D) Fold increase in luciferase expression versus nonreplicative GNN controls for SGR JFH-1 (Rluc2a) NS2 and (C) SGR JFH-1 (Rluc2a) NS3 HCV RNA adapted from data shown in panels B and C. All data shown are the average of three or more independent experiments. Error bars indicate the standard deviation of the mean, and asterisks indicate significant differences. The significance was determined by using Student’s t test (ns, not significant; *, P < 0.033).
HCV genomes capable of miR-122-independent replication replicate efficiently but in a small number of cells.
Next, we wanted to characterize and compare cells supporting transient miR-122-dependent and miR-122-independent HCV replication. We hypothesized that low-level transient miR-122-independent HCV replication will be displayed as either a large number of cells that support low levels of HCV replication or, alternatively, as a small number of cells that support efficient HCV replication. To test this hypothesis, we used immunofluorescence to detect the viral protein NS5a in infected cells and observed that a small number of cells support miR-122-independent replication compared to a large number of cells that support miR-122-dependent replication (Fig. 5A). We used flow cytometry to compare the intensity and number of cells stained with NS5a during miR-122-dependent versus miR-122-independent replication of HCV SGR JFH-1 Fluc RNA (∼2 to 6% and ∼45 to 60%, respectively), U4C/G28A/C37U J6/JFH-1 (p7Rluc2a), a full-length viral RNA carrying three point mutations in the 5′ UTR previously reported to promote miR-122-independent replication of full-length genomic HCV RNA (∼2 to 6% versus ∼25 to 30%), (27), and J6/JFH-1 Rluc wild-type RNA, a viral RNA that does not support miR-122-independent replication (∼0.2 to 1.5 versus ∼55 to 70%) (Fig. 5B and C). In addition, the staining intensity appeared similar in individual cells supporting either miR-122-dependent or miR-122-independent replication (Fig. 5A), and the intensity of viral protein staining in NS5a-positive cells from miR-122-independent replication was equal to, or higher than in miR-122-dependent replication on day 3 post electroporation (Fig. 5B and D). Thus, HCV RNAs capable of miR-122-independent HCV replication establish replication in a smaller proportion of cells than miR-122-dependent systems, but once established, viral protein levels appear similar. This suggests that miR-122 functions to enhance the establishment of HCV replication in a cell but appears dispensable to maintain efficient viral protein expression during the infection.
FIG 5.
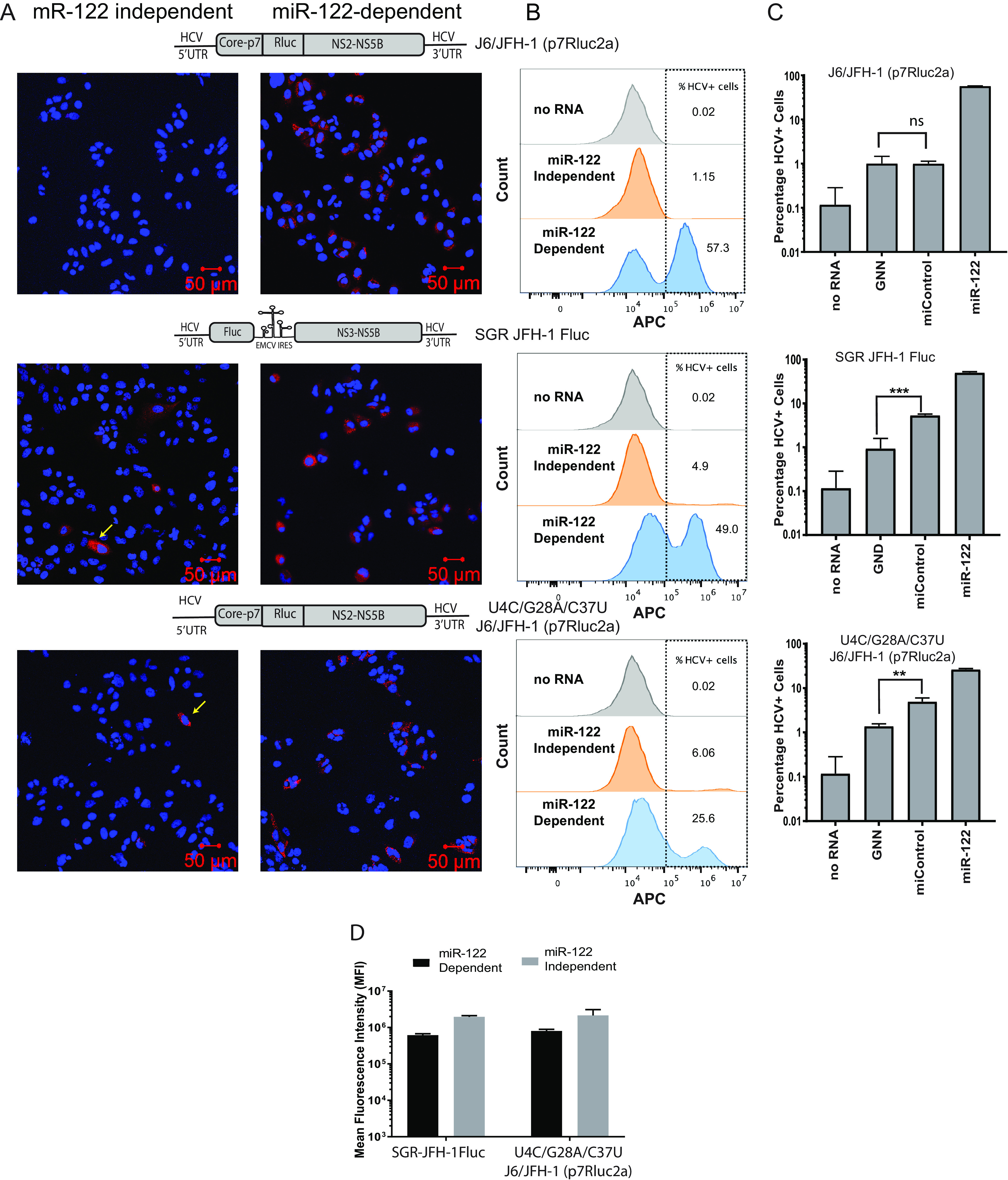
miR-122-independent replication is supported by a few cells within the population. (A) Immunostaining and confocal microscopy of Huh 7.5 miR-122 knockout cells coelectroporated with wild-type J6/JFH-1 (p7Rluc2a), SGR JFH-1 Fluc, and U4C/G28A/C37U J6/JFH-1 (p7Rluc2a) HCV RNA with control microRNA (miControl, left panel) or miR-122 (right panel). Cells were coelectroporated with viral RNA and microRNA and immunostained to detect NS5a on day 3. HCV NS5a is stained red with Alexa Fluor-594 secondary antibody, and the cells were counterstained with DAPI to identify the nucleus. The RNA constructs are shown above the panel. Cells supporting miR-122-independent replication are indicated with yellow arrows. (B) Representative flow cytometry plots of Huh 7.5 miR-122 KO cells coelectroporated with J6/JFH-1 (p7Rluc2a), SGR JFH-1 Fluc, or U4C/G28A/C37U J6/JFH-1(p7Rluc2a) HCV RNA and control microRNA or miR-122. Cells were collected on day 3 postelectroporation, stained for HCV NS5a with 9E10 anti-NS5a and APC-conjugated goat anti-mouse secondary antibody. The y axis indicates the count of the cells, and the x axis indicates the intensity of the APC signal. Data are represented as a histogram overlay of no RNA and miR-122-independent and miR-122-dependent replication. Gray histogram indicates the “no-RNA” control, whereas orange and blue histograms indicate the miR-122-independent and miR-122-dependent replication. The no-RNA control was used to gate HCV-positive cells. (C) Percentage of Huh 7.5 miR-122 KO HCV-positive cells electroporated with viral RNAs indicated (wild-type J6/JFH-1 [p7Rluc2a], SGR JFH-1 Fluc, or U4C/G28A/C37U J6/JFH-1 [p7Rluc2a]) and control microRNA or miR-122. Cells electroporated with no RNA and GNN are used as negative controls. (D) The mean fluorescence intensity (MFI) of cells supporting HCV replication (panel B) was calculated with FlowJo software v10.6. The MFI was measured as a proxy for viral NS5a protein accumulation in the cells. All data shown are the average of three or more independent experiments. Error bars indicate the standard deviation of the mean, and asterisks indicate significant differences. The significance was determined by using Student’s t test (**, P < 0.002; ***, P < 0.001).
HCV variants supporting miR-independent replication do not show evidence of adaptation to growth without miR-122.
An alternative explanation for efficient ongoing miR-122-independent replication of HCV is molecular adaptation of the viral genome to replication without miR-122. To identify potential adaptation to growth independent from miR-122, we used a biological assay that assessed the replication ability of viral RNA derived from cells supporting transient miR-122-independent replication. We used a biological method instead of sequencing as a rapid and sensitive way to detect if adaptation was a major influence in the observed miR-122-independent replication. We hypothesized that if viral RNA replicating in Huh 7.5 miR-122 KO cells had mutated to adapt to miR-122-independent replication, the viral RNA derived from these cells would have an enhanced ability to replicate when electroporated into new Huh 7.5 miR-122 KO cells, and we would observe an increase in replication efficiency with repeated RNA passage through Huh 7.5 miR-122 KO cells. To test this, we harvested total cellular RNA from cells supporting miR-122-independent replication of SGR JFH-1 Fluc HCV RNA and electroporated 5 μg of the total RNA into new Huh 7.5 miR-122 KO cells with and without miR-122 supplementation. miR-122-independent virus replication was measured based on Fluc expression in miR-122 KO cells electroporated with total RNA + miControl, and miR-122-dependent RNA replication was measured in miR-122 KO cells supplemented with total RNA + miR-122. The ratio of Rluc signal in miControl/miR-122 represented the relative efficiency of the viral RNA present in the total RNA sample to grow independently from miR-122 RNA (Fig. 6A). We also harvested and passaged RNA from cells supporting miR-122-dependent replication as a control. Based on miControl/miR-122 ratios over 3 RNA passages, we did observe an increase in ratios between RNA passage 1 and passage 2, but the ratios did not differ from those of control RNA from cells harboring miR-122-dependent replication. In addition, miR-122-independent replication was undetectable in the total RNA after 2 RNA passages (Fig. 6B). Thus, we concluded that there was no detectible evolution of viral genomes with enhanced miR-122-independent replication.
FIG 6.
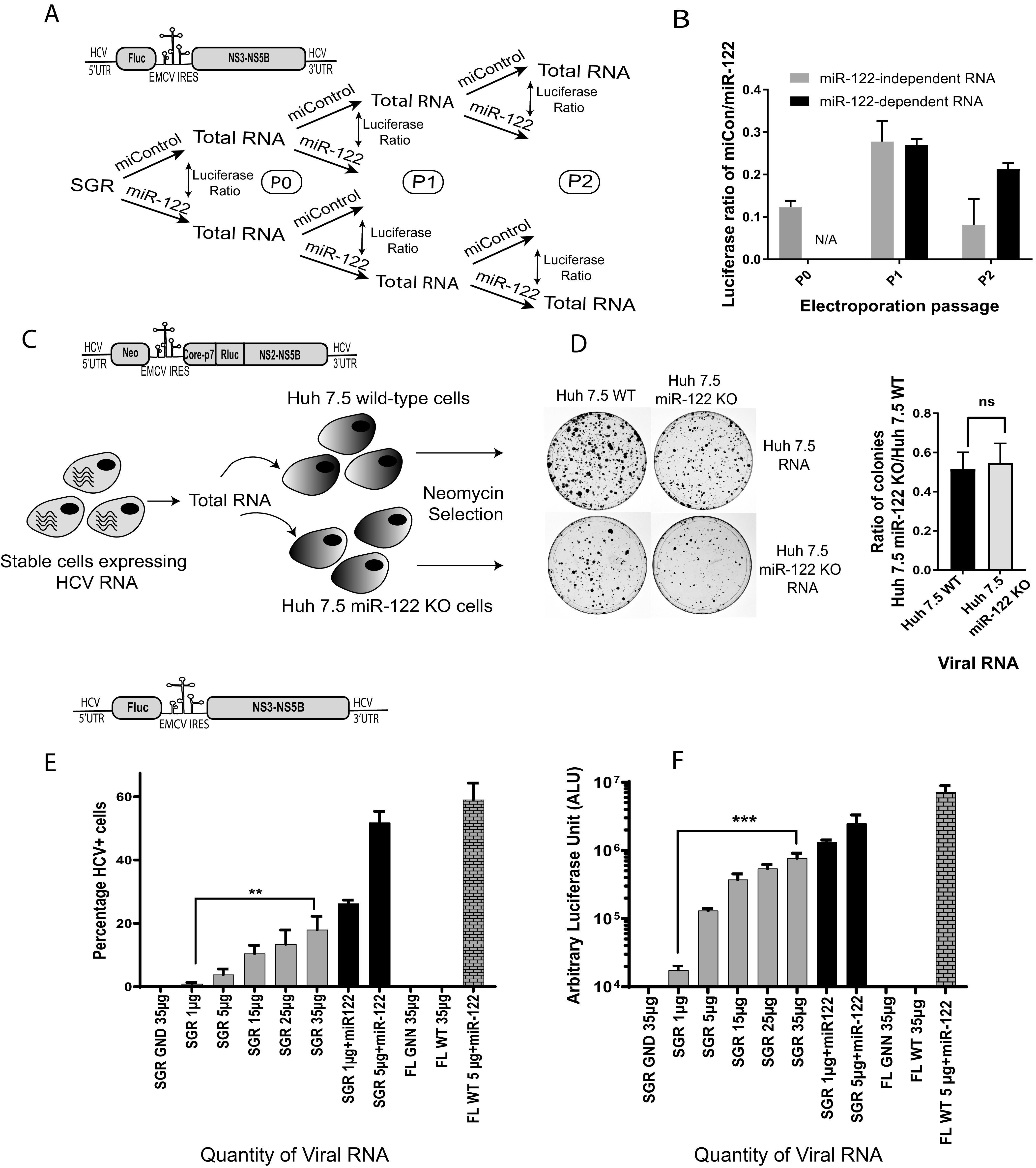
There was no evidence of genome adaptation to the absence of miR-122 in HCV genomes passaged under miR-122-independent conditions. (A) Schematic diagram of the experiment to assess HCV adaptation to growth without miR-122. Total RNA was isolated from cells supporting transient miR-122-independent (miControl) or miR-122-dependent (miR-122) replication of HCV SGR JFH-1 Fluc and then reelectroporated into miR-122 KO Huh 7.5 cells with either miControl or miR-122. This was done for 3 RNA passages as shown. (B) The relative luciferase expression when RNA was electroporated with miControl was calculated versus the same RNA electroporated with miR-122 to determine the relative miR-122-independent replication ability of each RNA and plotted. (C) A schematic diagram of the experiment to test for HCV adaptation to replication without miR-122 in cells supporting stable miR-122-independent HCV replication. Huh 7.5 cells stably supporting miR-122-independent and miR-122-dependent replication of HCV J6/JFH-1 Neo (p7Rluc2a) were generated by electroporation of Huh 7.5 miR-122 KO or WT cells and selection with G418. (D) Total RNA from Huh 7.5 cells stably supporting miR-122-independent or miR-122-dependent replication was isolated and electroporated into new WT or miR-122 KO Huh 7.5 cells, and the ability to initiate miR-122-independent HCV replication was assessed based on colony formation and the ratio of colonies formed in Huh 7.5 miR-122 KO/WT cells. The constructs used for each experiment are shown above each diagram. (E and F) Transient replication assays showing the impact of the quantity of SGR JFH-1 Fluc RNA on the number of HCV + cells (E) and Fluc expression (F). In panel E the percentage of HCV positive cells was determined by using a fluorescence-activated cell sorter (FACS) and in panel F luciferase activity was assessed. SGR JFH-1 GND and J6/JFH-1 (p7Rluc2a) GNN (replication-defective mutants) were used as the negative controls. All data shown are the average of three or more independent experiments. Error bars indicate the standard deviation of the mean, and asterisks indicate significant differences. The significance was determined by using Student’s t test (ns, not significant; **, P < 0.002; ***, P < 0.001).
We next tested for HCV adaptation to miR-122-independent replication in cells stably supporting miR-122-independent replication. To test for adaptation, we assessed the ability of viral RNA extracted from Huh 7.5 miR-122 KO cells stably harboring J6/JFH-1 Neo Rluc RNA to form colonies when reelectroporated into naive miR-122 KO cells. If adaptation had occurred, we expected a higher ratio of colonies formed after electroporation of total RNA derived from stable cells supporting miR-122-independent J6/JFH-1 Neo Rluc RNA replication compared with total RNA from cells supporting miR-122-dependent J6/JFH-1 Neo Rluc RNA replication (Fig. 6C). Similar to our transient adaptation assay, we did not observe a significant difference in the ratio of colonies formed following electroporation of total RNA derived from stable miR-122 KO versus stable miR-122 wild-type cell lines (Fig. 6D). Thus, we concluded that transient and stable miR-122-independent HCV replication was not due to observable adaptation of the viral RNA.
The quantity of viral RNA affects the percentage of cells supporting miR-122-independent replication.
Since we did not detect viral RNA adaptation to miR-122-independent replication, we wondered what mechanism led to some cells supporting efficient miR-122-independent replication and some cells remaining uninfected. Based on a role for miR-122 in stabilizing the HCV genome, we hypothesized that the cells supporting miR-122-independent replication were ones that had stochastically received sufficient viral RNA to initiate an infection. If this was the case, then we surmised that electroporation of larger quantities of HCV RNA would increase the numbers of cells that received enough viral RNA to initiate an infection, and thus the numbers of cells supporting miR-122-independent replication should increase. Alternatively, if the cells supporting miR-122-independent replication had a specific phenotype that allowed for miR-122-independent HCV replication, then greater amounts of viral RNA would not enhance the numbers of cells supporting miR-122-independent replication. To test this, we electroporated increasing amounts of SGR JFH-1 Fluc HCV RNA into miR-122-knockout cells (5 μg, 10 μg, 15 μg, 25 μg, and 35 μg) and measured the numbers of cells supporting miR-122-independent replication by flow cytometry. We observed an increase in the proportion of cells supporting HCV replication with an increased amount of viral RNA. A negative-control sample electroporated with 35 μg of nonreplicative SGR JFH-1 Fluc GND RNA did not show any fluorescence signal. These data indicate that the amount of RNA affects the numbers of cells supporting miR-122-independent replication and suggests that the cells supporting miR-122-independent replication do not have a specific phenotype (Fig. 6E). Similar results were obtained in an experiment in which luciferase activity was used to assess viral replication and confirmed greater luciferase expression following larger amounts of virus RNA electroporation (Fig. 6F). Further, this experiment also showed that using larger amounts of J6/JFH-1 wild-type (WT) viral RNA, which is incapable of miR-122-independent replication, did not enhance luciferase expression in miR-122 knockout cells. Thus, electroporation of larger amounts of viral RNA enhances miR-122-independent replication but did not enhance replication of wild-type genomes. This suggests that larger amounts of viral RNA can partially compensate for the lack of miR-122 and that the selection of cells that support miR-122-independent replication is likely stochastic. Since we have shown in our previous experiment that miR-122-independent HCV replication levels appear similar to miR-122-dependent replication, but in fewer cells, then we propose that cells supporting miR-122-independent replication had randomly received sufficient RNA to reach a threshold quantity required to initiate genome amplification. That electroporation of larger amounts of viral RNA can compensate for the lack of miR-122 is consistent with the proposed role for miR-122 in genome stabilization. However, because wild-type HCV RNA does not exhibit miR-122-independent replication, a high RNA concentration cannot compensate for the lack of miR-122. Thus, it appears that genetic modifications that alter virus translation regulation, such as an EMCV IRES or 5′ UTR mutations, are also necessary (35). This supports our and others’ findings that stabilization alone is not sufficient to promote miR-122-independent replication and that both stabilization and altered translation regulation are required (14, 20, 36).
miR-122 has a small but significant impact on the maintenance of an established HCV infection.
Based on our data, miR-122 is required early to establish an infection in a particular cell, but thus far appears dispensable for ongoing maintenance of viral replication. To further investigate the impact of miR-122 on the maintenance of a viral infection, we assayed the impact of miR-122 supplementation or depletion during later stages of the virus life cycle. We first assessed the impact of miR-122 supplementation or antagonization on the maintenance of transient miR-122-dependent HCV replication. For this assay we analyzed miR-122-dependent replication of J6/JFH-1 Rluc wild-type RNA, an RNA that cannot replicate without miR-122, in wild-type Huh 7.5 cells. J6/JFH-1 Rluc FL WT reached a maximum replication level at 3 days postelectroporation, and we supplemented or antagonized miR-122 at that time point and measured the impact on RNA replication by measuring Rluc expression 2 days later. After supplementation with miR-122, we saw a small but significant increase in luciferase expression from J6/JFH-1 Rluc FL WT (Fig. 7A). Similarly, antagonization of miR-122 caused a small but significant decrease in luciferase expression (Fig. 7A). We observed similar results using ΔE2 J6/JFH-1 Rluc RNA, a mutant that cannot produce infectious particles and thus eliminates the potential impact of cell to cell spread to initiate new infections. This suggests that miR-122 has a significant impact in supporting ongoing miR-122-dependent HCV replication separate from its effect during the establishment of an infection. Similarly, we tested the influence of miR-122 supplementation on the maintenance of transient miR-122-independent HCV replication. To test this, we used miR-122 KO cells supporting transient replication of a packaging-defective HCV mutant capable of miR-122-independent replication (U4C/G28A/C37U ΔE2 J6/JFH-1 Rluc HCV), and like in the previous experiment, we supplemented the construct with miR-122 3 days after viral RNA electroporation and measured the impact on Rluc expression 2 days later (Fig. 7B). In this case miR-122 supplementation did not change luciferase levels compared to miControl-supplemented cells during maintenance of replication. These data suggest that miR-122 does not have a significant additional impact on the maintenance of viral RNAs capable of miR-122-independent replication after establishment of replication (Fig. 7B). Reports suggest that the mutations present on the 5′ end of this viral RNA promote viral RNA stability (U4C), favor translation (G28A), and promote viral genomic RNA synthesis by altering the structure of the negative strand (C37U) (19, 30, 35), and this may allow ongoing replication of this viral RNA in the absence of miR-122.
FIG 7.
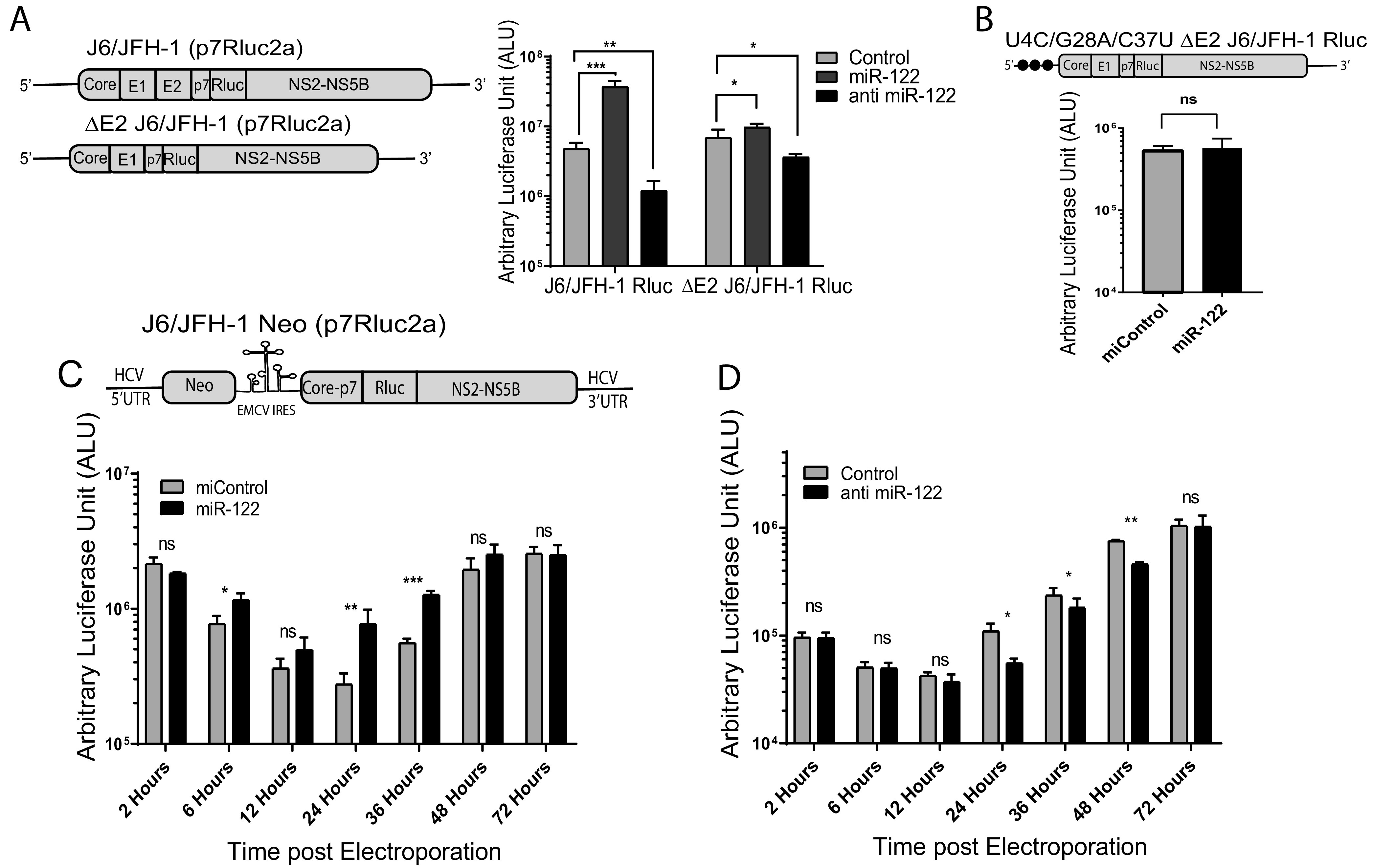
miR-122 has a small but significant influence on the maintenance of HCV replication. (A) To test for the influence of miR-122 on ongoing HCV, replication cells were supplemented with miR-122 or an miR-122 antagonist after the establishment of an infection. MiR-122-dependent replication was established by electroporating Huh 7.5 miR-122 KO cells with wild-type J6/JFH-1 (p7Rluc2a) or ΔE2 J6/JFH-1 (p7Rluc2a) RNA and miR-122. Then, 3 days later the cells were transfected with miR-122, anti-miR-122, or miControl to assess the influence of miR-122 supplementation and antagonization, and 2 days later luciferase activity was measured as an indication of viral RNA amplification. (B) To assess the impact of miR-122 supplementation on HCV RNA replicating independently of miR-122, replication of U4C/G28A/C37U ΔE2 J6/JFH-1 (p7Rluc2a) HCV infection was established in Huh 7.5 miR-122 KO cells without miR-122 and on day 3 was supplemented by transfection with either miR-122 or control microRNA. Luciferase activity was measured on day 2 posttransfection as an indication of viral propagation. (C) To assess the impact of miR-122 supplementation on cells stably supporting miR-122-independent HCV RNA, we used Huh 7.5 miR-122 KO cells electroporated with J6/JFH-1 Neo (p7Rluc2a) HCV and selected with G418. The cells were then electroporated with either control microRNA or miR-122, and luciferase activity was measured at different time points posttransfection. (D) To assess the impact of miR-122 antagonization on miR-122-dependent HCV replication, we electroporated Huh 7.5 wild-type (WT) cells with J6/JFH-1 Neo (p7Rluc2a) HCV RNA and selected with G418. Stable cells were then transfected with either control LNA or anti-miR-122 LNA, and luciferase activity was measured at different time points posttransfection. All data shown are the average of three or more independent experiments. Error bars indicate the standard deviation of the mean, and asterisks indicate significant differences versus the control microRNA/LNA. The significance was determined by using Student’s t test (ns, not significant; *, P < 0.033; **, P < 0.002; ***, P < 0.001).
To further assess the impact of miR-122 on ongoing HCV replication, we also analyzed the impact of miR-122 supplementation and antagonization on cells supporting stable miR-122-dependent and -independent replication of J6/JFH-1 Neo Rluc HCV RNA (Fig. 2A and E). When cells supporting stable miR-122-independent replication were supplemented with miR-122, there was a significant stimulation of luciferase expression observed at 24 and 36 h postsupplementation, but by 72 h this effect was lost (Fig. 7C). In cells supporting miR-122-dependent HCV replication, electroporation of an miR-122 antagonist significantly reduced luciferase expression at 24, 36, and 48 h postelectroporation, and again no difference was observed after 72 h (Fig. 7D). These experiments suggest that miR-122 has a small but measurable influence on the maintenance of both miR-122-dependent and miR-122-independent HCV replication but also suggests the influence is temporary.
Altogether, our data show that miR-122 exerts its strongest effect by promoting the establishment of an infection in a cell by stimulating viral translation and stabilizing the viral genome and thus increases the chance that the virus will synthesize sufficient viral proteins to initiate an active infection before the RNA genome is degraded. In the absence of miR-122, the effect can be partially compensated for by using an alternative IRES or 5′ UTR mutations that increase the efficiency of viral protein synthesis (20, 27, 29) or by shortening the viral genome length, which may simply decrease the time to complete genome replication. Delivery of increasing amounts of viral RNA can also increase the chance of establishing miR-122-independent replication in a cell, likely by compensating for the function of miR-122 in genome stabilization. Finally, our data suggest a small but significant positive influence on the maintenance of ongoing viral RNA replication. Based on these findings, we suggest a model (Fig. 8) in which viral RNAs entering a cell are translated while being subject to degradation by host defense and RNA degradation pathways but in which miR-122 promotes translation and stabilizes the genome and thus increases the chances that enough viral proteins will be produced to initiate genome replication and establishment of virus replication complexes. In addition, we propose that miR-122 is required for ongoing HCV infections for similar reasons—to stabilize viral RNAs that exit the replication complex to initiate new replication complexes as the infection proceeds or when the infected cell divides.
FIG 8.

Proposed model of miR-122’s function in the establishment of HCV infection. We propose that binding of miR-122 to the incoming viral RNA promotes viral translation and genome stability to allow the viral RNA to generate a threshold amount of viral protein required to initiate genome replication and the formation of virus replication complexes. After the establishment of replication complexes, miR-122 may support ongoing virus replication by promoting the establishment of new replication complexes inside an infected cell or after an infected cell divides.
DISCUSSION
While miR-122 is required for efficient and detectable HCV replication in cell culture, we and others have demonstrated that in certain contexts HCV can replicate independently from miR-122 (20, 25–27, 29, 37). Previously, we have identified that bicistronic HCV subgenomic replicons, and HCV genomes with point mutations within the miR-122 binding region of the 5′ UTR, promote miR-122-independent replication (17, 20, 26). In this paper, we have shown that miR-122-independent replication is facilitated by the addition of an EMCV IRES (Fig. 2) and by a smaller viral genome with the deletion of structural genes (Fig. 3). Since viral IRES elements regulate cap-independent translation, we believe that the presence of the EMCV IRES enhances the overall efficiency of the viral polyprotein synthesis and thus substitutes for the function of miR-122 in stimulating translation. However, the efficiency of miR-122-independent HCV replication is significantly lower than the miR-122-dependent replication, and thus the presence of an EMCV IRES cannot completely rescue the viral replication in the absence of miR-122. Our data have also suggested that shorter viral genomes with the deletion of whole structural gene regions can replicate to low but detectable levels in the absence of miR-122 (Fig. 4), even in viral genomes that lack an EMCV IRES or 5′-UTR point mutations. We speculate that the small genome size resulting from the removal of the structural gene region allows for more rapid virus protein synthesis and RNA replication simply based on the size of the protein product and genome, but we cannot omit the possibility that the structural gene region somehow inhibits viral propagation in the absence of miR-122. Thus, 5′-UTR point mutations, an EMCV IRES, and removal of the structural protein region each facilitate detectable HCV replication in the absence of miR-122.
We also observed that miR-122-independent replication is manifested as efficient genome replication within a small percentage of cells of a population, rather than inefficient genome replication in a large proportion of the cells (Fig. 5). This suggests that miR-122-independent RNA replication is efficient but is initiated in only a small number of cells. We propose that the few cells that support miR-122-independent replication stochastically received enough RNA to initiate an infection and do not have a cellular phenotype that promotes initiation, as evidenced by increasing amounts of viral RNA resulting in increasing numbers of cells supporting miR-122-independent replication (Fig. 6E and F). In this context, we propose that the EMCV IRES and the small genome size facilitates viral protein synthesis and genome replication such that the threshold for establishment of an active HCV replication complex is lower. These findings also support the notion that miR-122 promotes the virus life cycle by stabilizing the genome and stimulating its translation, influencing the ability of the virus to establish an infection within a particular cell. Studies of HCV capable of replicating independently of miR-122 showed that enhanced viral protein synthesis and genome stability can completely rescue HCV replication in the absence of miR-122 (35, 36), further confirming miR-122’s role in viral translation and genome stability in early stages of viral infection. Thus, we propose that miR-122 stabilizes the genome to provide sufficient viral genomes and protein synthesis to jump-start the early stages of an infection.
Microscopy and flow cytometry data also suggested that once the infection is established in the absence of miR-122, viral protein levels are equivalent to infection established in the presence of miR-122. This finding supports the data published by the Matsuura group indicating that miR-122 is essential for the initial stage of viral infection but is dispensable for the maintenance of infection (25). In addition, Villanueva et al. showed that the addition of miR-122 or anti-miR-122 has no effect on HCV RNA synthesis ex vivo in membrane-bound replicase complexes isolated from HCV-infected cells. They also observed no significant detectable quantities of miR-122 associated with replicase complexes in vivo, suggesting no significant role of miR-122 on the elongation phase of viral RNA synthesis (38). In contrast, we have found that miR-122 has a small effect on the maintenance of viral replication. This supports work published by Masaki et al. (18), who proposed a direct role for miR-122 in promoting genome amplification. However, based on our data, we propose an alternative hypothetical model that miR-122 promotes the establishment of new replication complexes within an already-infected cell, an extension of its effect at infection initiation. This is supported by our experiments showing a requirement for miR-122 from 24 to 36 h post-miR-122-supplementation in stably infected cells, but not after 72 h (Fig. 7). We speculate that this is the time when there is active cell growth, and by 72 h the cells have become confluent, and we propose that miR-122 enhances the rate at which new replication complexes form as infected cells divide. This could be through a direct role of miR-122 in promoting genome amplification as proposed by Masaki et al. but is also consistent with the roles for miR-122 in stabilizing and stimulating protein synthesis from newly synthesized viral RNAs as they are released from one replication complex to promote their initiation of new replication complexes. Indeed, miRNAs and Ago have been implicated in liquid-liquid phase separation and sequestration of mRNAs into protein complexes, and miR-122 could also function to directly stimulate replication complex formation (39). This would explain why Vilanueva et al. did not see any effect of miR-122 supplementation or antagonization on HCV propagation in isolated membrane-bound replicase complexes, as they were assayed ex vivo without provision for the formation of new replication complexes. Interestingly, however, miR-122 supplementation did not affect the ongoing replication of the mutant HCV (U4C/G28A/C37U) capable of replicating independently of miR-122. A recent report suggests that these mutations substitute for miR-122 by stabilizing the viral genome (U4C), promoting viral translation (G28) and promoting the synthesis of positive-strand RNA (C37U) (30), and this could explain why miR-122 supplementation had no influence on the ongoing replication of the virus. Overall, our model (Fig. 8) posits that miR-122 promotes genome stabilization and translation to initiate replication complex formation to initiate an infection but may similarly affect newly synthesized viral RNAs to promote the development of new replication complexes within an infected cell and in daughter cells following cell division.
Initiation of an HCV infection in the liver is likely more dependent on miR-122 than in tissue culture cells since the HCV RNA copy number varies from less than 1 to 8 per hepatocyte under the in vivo conditions (40), 100-fold lower than in tissue culture cells. Thus, despite the abundance of miR-122 in hepatocytes, infection with a single or a low-copy-number of viral genomes may require multiple rounds of viral replication to establish an infection and could explain the importance of miR-122 in both the establishment and ongoing viral replication in the liver. In addition, recent findings reported that other RNA viruses, such as enterovirus, norovirus, and rotavirus, transmit among hosts in vesicle-cloaked viral clusters that are found to be more infectious than the single free viral particles (41, 42). Similarly, miR-122 may also enhance the viral genome quantity by slowing RNA degradation. Further, although there are no reports of HCV virion clusters, exosomes containing replication-competent HCV RNAs in complex with miR-122, Ago-2, and HSP90 can transmit HCV infection, which could increase the copy number and stability of infecting HCV genomes (43).
The use of anti-miR-122 locked nucleic acid (miravirsen, SPC3649) has shown promising results as a therapeutic agent, as its use in infected patients decreased HCV levels to undetectable levels (44–46). Our findings suggest that anti-miR-122 exerts its effect primarily during the establishment of HCV infection and suggest that infections in the liver must be highly dynamic such that new cells are continually being infected, and antagonization of miR-122 might be restricting new cell infection and decrease the overall viral burden of the liver. This also aligns with the hypothesis proposed by Stiffler et al. (40), which suggests that HCV infection might be a transient infection in vivo, and the chronic condition of the virus is maintained by a continuous cycle of viral infection and clearance. However, the low HCV genome copy number in infected cells in vivo could also make HCV infections more reliant on the supportive role of miR-122 during ongoing infections; thus, miR-122 antagonism may also decrease HCV replication levels within infected liver cells.
Since miravirsen targets a host factor, it has a high barrier to resistance and is effective against all HCV genotypes (44, 47). However, our work and that of others cautions that HCV can replicate independently from miR-122 and can develop resistance to miR-122 targeting therapy. Resistance-associated substitutions (RAS) variants G28A (guanine is replaced by adenine at position 28 of the viral genome) and C3U (cytosine is replaced by uracil at position 3 of the viral genome) were identified in a patient treated with miravirsen and are capable of replicating at a low abundance in the absence of miR-122 (23, 48). In addition, several studies have identified mutants capable of replicating independently of miR-122 in cell culture (20, 27). Thus, the application of anti-miR-122 should be done with care. The addition of Miravirsen to the DAA treatment has been shown to be synergistic in suppressing HCV replication, and thus, rather than being a stand-alone therapeutic, miravirsen may instead be best used as a part of the treatment cocktail for patients who are nonresponsive to first-line DAAs (49).
Overall, in this study, we show that the influence of miR-122 can be partially compensated for by using an alternative IRES element to drive viral polyprotein synthesis or 5′-UTR mutations that increase the efficiency of viral protein synthesis or by shortening the viral genome length, and we speculate that this increases the efficiency with which the viral genome can be translated and replicated. Next, we showed that miR-122-independent replication is displayed as efficient replication in a smaller number of cells compared to miR-122-dependent replication. This suggested that miR-122 is required to initiate an infection in each cell, but we have found that miR-122 does have a small but measurable impact on ongoing replication. Based on these findings, we propose a model (Fig. 8) in which miR-122 stabilizes the viral genome and stimulates translation, thus increasing the chance that the virus will produce sufficient viral proteins to initiate an active infection before the RNA genome is degraded. In addition, we propose that miR-122 is required for ongoing HCV infections for similar reasons—to promote the generation of new replication complexes as the infection proceeds or as the infected cell divides. Thus, we propose that a successful viral infection is an interplay between viral protein synthesis, RNA replication, and RNA degradation and that miR-122 favors the successful formation of the replication complex and the establishment of an infection.
MATERIALS AND METHODS
Viral cDNA plasmids.
Plasmids pSGR JFH-1 Fluc WT and pSGR JFH-1 Fluc GND contain bicistronic JFH-1-derived subgenomic replicon (SGR) cDNAs with a firefly luciferase reporter and an encephalomyocarditis virus (EMCV) IRES and were provided by T. Wakita (50). pSGR GND is the control nonreplicative version containing a mutation to the viral polymerase-active site, GDD, to an inactive GND. Plasmids encoding a full-length HCV genome expressing a Renilla luciferase (Rluc) gene, pJ6/JFH-1 RLuc (p7RLuc2A) (51), and a full-length bicistronic HCV cDNA expressing neomycin from the HCV IRES and Rluc within the full-length HCV polyprotein, pFLneo-J6/JFH-1 (p7-Rluc2a), were provided by C. M. Rice (here called pJ6/JFH-1 Rluc and pJ6/JFH-1 Neo Rluc, respectively) (51). A GDD to GNN mutation rendering the viral polymerase nonfunctional in the negative-control pJ6/JFH-1 Rluc GNN was also provided by C. M. Rice (51). Plasmids pFGR Fluc JFH-1 WT and pFGR Fluc JFH-1 GNN bear full-genomic bicistronic replicons of JFH-1 and a firefly luciferase reporter (52). Plasmid pJ6/JFH1 Rluc, encoding triple mutations in the 5′ UTR of the virus genome (U4C/G28A/C37U), was made by site-directed mutagenesis of a smaller plasmid (pBSKS+) encoding EcoRI to KpnI fragment of J6/JFH-1 Rluc. Once the mutations were confirmed in pBSKS+, the EcoRI to KpnI fragment from pBSKS+ was cloned into pJ6/JFH-1 to obtain U4C/G28A/C37U pJ6/JFH-1 Rluc. HCV structural gene deletion mutants were constructed in pJ6/JFH-1 Rluc plasmid using 5′ phosphorylated primers to perform inverse PCR. Unmodified template was then removed by DpnI digestion, and PCR products were then circularized with T4 DNA ligase. All the deletion mutants were sequenced and confirmed for deletions.
The following primers were used: ΔCore, 5′Phos-CCTTTTTCTTTGAGGTTTAGGATTTG3′-F and 5′Phos-TGCTCCTTTTCTATCTTCTTG3′-R; ΔE1, 5′Phos-GGAGCAACCGGGTAAGTTCC3′-F and 5′Phos-AAAGTCGTTGTCATCCTTCTG3′-R; ΔE2, 5′Phos-CTTCTGTTGGCCGCCGGGGTGGAC3′-F and 5′Phos-CAGGCCGAAGCAGCACTAGAGAAGC3′-R; ΔE1E2, 5′Phos-GCATTGCCCCAACAGGCTTATGC3′-F and 5′Phos-GCCGGTACTGATGTTCTTCACTTC3′-R.
J6/JFH-1 Rluc with deletion of envelope protein 2 (ΔE2) was used to construct U4C/G28A/C37U ΔE2 pJ6/JFH-1 Rluc HCV where the EcoRI to AgeI fragment of ΔE2 pJ6/JFH-1 Rluc plasmid was replaced with the EcoRI to AgeI fragment from U4C/G28A/C37U pJ6/JFH-1 Rluc. pΔE1E2 J6/JFH-1 Rluc plasmid was obtained from Ralf Bartenschlager (52).
Construction of monocistronic and bicistronic subgenomic constructs.
Monocistronic pSGR JFH-1Rluc NS2 was made from J6/JFH-1 (p7Rluc2a) by generating a short intermediate PCR product using primers SGR NS2 F and R1 and then using this product as a forward megaprimer in tandem with SGR NS2-R2; the region from the core to p7 was removed.
The following primers were used: SGR NS2 5′CGACGGCCAGTGAATTCTAATAC3′-F, 5′CTGGATCATAAACTTTCGAAGTCATAGGCCGGCCGGTTTTTCTTTGAGGTTTAGGATTTG3′-R1, and 5′CGGCCCATATGATGCCATCG3′-R2.
Once confirmed through sequencing, the EcoRI to NotI region of the plasmid was swapped in to the original J6/JFH-1 (p7Rluc2a) plasmid to create pSGR JFH-1Rluc NS2. pSGR JFH-1Rluc NS3 was generated with the same approach using primers SGR NS3 F, R1, and R2 to remove the NS2 region from the pSGR J6/JFH-1Rluc NS2, and then the EcoRI to SpeI fragment was inserted into the original JFH-1 Rluc to create pSGR JFH-1Rluc NS3.
The primers used were SGR NS3 5′GCCTCGTGAAATCCCGTTAG3′-F, 5′GCTGGGCATAAGCAGTGATGGGAGCGGGCCCTGGGTTGGACTCGACGTC3′-R1, and 5′TCCACACTTGCACGGCTCCAAAGAC3′-R2.
Replication -defective mutant controls were generated by cloning the GNN fragment from pJ6/JFH-1 (p7RLuc2a) GNN with SnaBI and XbaI into the monocistronic and bicistronic HCV plasmids. All modifications were confirmed by sequencing.
In vitro RNA transcription.
Viral RNAs were transcribed in vitro using the MEGAScript T7 high-yield in vitro transcription kit (Life Technologies, Burlington, ON, Canada). HCV cDNA plasmids were linearized with XbaI and made blunt with mung bean nuclease and transcribed as suggested in our previous studies (17).
Cell culture.
Huh 7.5 (WT) cells, Huh 7.5 miR-122 KO cells, and Huh 7.5 cells stably expressing J6/JFH-1 Neo Rluc were grown and maintained as described by Amador-Cañizares et al. (20).
Transient HCV replication assays.
First, 6.0 × 106 cells suspended in 400 μL 1× phosphate-buffered saline (PBS) were coelectroporated with 5 μg of HCV genomic RNA (unless otherwise indicated) and 3 μL miR-122/miControl/anti-miR-122/anti-miR-124 at 10 mM concentration and resuspended in 4 mL cell culture medium (Dulbecco’s modified Eagle’s medium [DMEM] + 10% fetal bovine serum [FBS] + 1 nM nonessential amino acid + 100 μg/mL Pen/Strep). A total of 500 μL of the electroporated cells were plated onto each well of a 6-well plate in culture medium and incubated at 37°C for the indicated lengths of time. A control luciferase mRNA was also coelectroporated with the viral RNA and microRNAs and measured at the 2-h time point. Experiments with more than a 10% difference in control luciferase expression, indicating differences in electroporation efficiencies among samples, were discarded.
Analysis of the effects of miR-122 supplementation and antagonization on transient HCV replication.
To analyze the impact of miR-122 supplementation or antagonization on transient HCV replication assays, cells were coelectroporated with HCV RNA and miR-122 as described above, and a total of 250 μL of resuspended cells was allowed to grow on 6-well plates for 3 days. On day 3 the cells were transfected with 60 pMol of either miR-122 or control microRNA (miR-122 p2-8) or anti-miR-122 LNA or control LNA (anti-miR-124) with 5 μL of Lipofectamine 2000 (Invitrogen). Cells were harvested 2 days posttransfection, and luciferase activity was measured.
Establishment of miR-122 KO stable cell line expressing bicistronic pJ6/JFH-1 Neo Rluc.
Huh 7.5 miR-122 KO cells were electroporated with 10 μg of pJ6/JFH-1 Neo Rluc RNA, and all the cells were plated on a 15-cm tissue culture plate with DMEM + 800 μg/mL G418 sulfate. Cells were selected for more than 60 days, and flow cytometry was used to compare the cells with the already-established Huh 7.5 cells stably expressing pJ6/JFH-1 Neo Rluc (data not shown).
Analysis for adaptation to replication without miR-122 during transient replication assays.
Initially, 5 μg of SGR (JFH-1) was coelectroporated with miR-122 or miControl into 6.0 × 106 Huh 7.5 miR-122 KO cells and then resuspended into 4 mL of cell culture medium. Then, 500 μL of cells was plated in 6-well plates for luciferase as well as for total RNA. Total RNA was extracted at each passage, and a total of 10 μg of total RNA was again coelectroporated with either miR-122 or miControl, and the process was repeated for 3 passages.
Analysis for adaptation to replication without miR-122 in stable HCV cells.
Total RNA was isolated from Huh 7.5/Huh 7.5 miR-122 KO stable cell lines, and 10 μg of total RNA was electroporated into approximately 6.0 × 106 Huh 7.5 and Huh 7.5 miR-122 KO cells. After electroporation, cells were resuspended into 4 mL of cell culture medium. A total of 1 or 2 mL of cells was plated on a 10-cm tissue culture dish containing DMEM + 800 μg/mL G418 sulfate and incubated at 37°C for approximately 2 weeks until the colonies developed. The plates were then washed with 1× PBS, fixed with ice-cold methanol, and stained with crystal violet. Plates were scanned and colony numbers were quantified using ImageJ.
Analysis of the effects of miR-122 supplementation and antagonization on stable HCV replication.
Huh 7.5 WT/Huh 7.5 miR-122 KO cells expressing pJ6/JFH-1 Neo Rluc were electroporated with miR-122 or control microRNA. Cells were plated on a 6-well plate and harvested at different time points according to above-mentioned protocol for luciferase analysis. For transfection, approximately 5.0 × 105 cells were plated on a 6-well plate. Then, 60 pmol of miR-122/control microRNA was transfected to the cells the next day using 5 μL Lipofectamine 2000. Cells were harvested 48 h posttransfection, and luciferase activity was measured.
Luciferase assays.
Cells were washed with 1× Dulbecco’s PBS and harvested with 100 μL of 1× passive lysis buffer diluted in double-distilled water (ddH2O; Promega, Madison WI, USA). Luciferase activity in cell lysates was measured by using either Firefly or Renilla luciferase kits (Promega), and light emission was measured in arbitrary light units on a Glomax 20/20 luminometer (Promega). The luciferase assays were performed as suggested by the manufacturer’s protocols.
Total cellular RNA extraction.
Following the suggested manufacturer’s protocol (Life Technologies, Burlington, ON, Canada), 1 mL of TRIzol was used to harvest the total cellular RNA.
Immunostaining for confocal microscopy.
A total of 150 μL of electroporated cells were placed on a (18 mM diameter, 1.5 thickness) coverslip and placed in a 12-well plate with culture medium. On day 3 cells were washed with PBS and fixed with 4% paraformaldehyde. After washing three times with PBS, cells were permeabilized and blocked with 0.1 to 0.2% Triton X-100 + 3% bovine serum albumin (BSA) in PBS for 1 h. Mouse anti-NS5a 1° antibody (Ab; 9E10, provided by Charles Rice [53]) was incubated on the cells at a 1:5,000 dilution in the blocking buffer overnight at 4°C. Cells were further washed three times with washing buffer (0.02 to 0.01% Triton X-100 + 0.3% BSA in PBS) and incubated with 2° Ab at a 1:1,000 dilution (goat Ab-AF 594; Life Technologies, A11005) for 1 h at room temperature. After washing the cells three times with washing buffer, 1 μg/mL DAPI in PBS was used to stain the nucleus for 10 min. Cells were further washed with PBS and ddH2O to remove the residual salts, and the coverslip was mounted on a glass slide using SlowFade Gold antifade mounting solution (Life Technologies). Images were captured using a Zeiss confocal microscope (LSM 700), and images were processed using Zen 3.1 (Blue edition) and ImageJ software.
Immunostaining for flow cytometry.
On day 3 of electroporation cells were harvested from 6-well plates, and flow cytometry was done as described by Kannan et al. (54). Primary Ab against HCV NS5a was used at a dilution of 1:2,500 (9E10, mouse anti-NS5a), and secondary antibody allophycocyanin (APC):(F ab′) 2 goat anti-mouse IgG APC (eBioscience; catalog [cat.] no 17-4010-82) was used as suggested by the manufacturer. Samples were suspended in 300 μL of 1× PBS and acquired in a Beckman Coulter CytoFlex flow cytometer. Data were analyzed using FlowJo v10.6.
Statistical analysis.
All data are displayed as the mean of three or more independent experiments, and error bars indicate the standard deviation. Statistical analysis was performed using Graph Pad Prism v7, and the statistical tests are indicated in the figures.
ACKNOWLEDGMENTS
We acknowledge Charlie Rice (The Rockefeller University) for providing the J6/JFH-1(p7Rluc2A), FL-Neo J6/JFH-1(p7Rluc2A), and Huh-7.5 wild-type cells. We also acknowledge Matthew Evans (Icahn School of Medicine at Mount Sinai) for Huh-7.5 miR-122 knockout cells. We also thank Saurav Saswat Rout for his help with flow cytometry data analysis.
M.P., P.A.T., and J.A.W. designed and performed the experiments and analyzed the data; M.P. and J.A.W. wrote the manuscript.
This work was supported by grants from the Canadian Institutes of Health Research (MOP-133458), the Canada Foundation for Innovation (18622), and the University of Saskatchewan (CoMBRIDGE) to J.A.W. M.P. was funded by a University of Saskatchewan Graduate Teaching Fellowship. P.A.T. was funded by the National CIHR Research Training Program in hepatitis C (NCRTP-HepC).
Contributor Information
Joyce A. Wilson, Email: joyce.wilson@usask.ca.
J.-H. James Ou, University of Southern California
REFERENCES
- 1.World Health Organization. 2017. Global hepatitis report, 2017. https://www.who.int/publications/i/item/global-hepatitis-report-2017.
- 2.Blackard JT, Kemmer N, Sherman KE. 2006. Extrahepatic replication of HCV: insights into clinical manifestations and biological consequences. Hepatology 44:15–22. 10.1002/hep.21283. [DOI] [PubMed] [Google Scholar]
- 3.Tsoulfas G, Goulis I, Giakoustidis D, Akriviadis E, Agorastou P, Imvrios G, Papanikolaou V. 2009. Hepatitis C and liver transplantation. Hippokratia 13:211–215. [PMC free article] [PubMed] [Google Scholar]
- 4.Friebe P, Bartenschlager R. 2009. Role of RNA structures in genome terminal sequences of the hepatitis C virus for replication and assembly. J Virol 83:11989–11995. 10.1128/JVI.01508-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sagan SM, Chahal J, Sarnow P. 2015. Cis-acting RNA elements in the hepatitis C virus RNA genome. Virus Res 206:90–98. 10.1016/j.virusres.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Frese M, Pietschmann T. 2004. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res 63:71–180. 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 7.Lindenbach BD, Rice CM. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11:688–700. 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammond SM. 2015. An overview of microRNAs. Adv Drug Deliv Rev 87:3–14. 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 10.Jopling CL, Schütz S, Sarnow P. 2008. Position-dependent function for a tandem MicroRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4:77–85. 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson JA, Zhang C, Huys A, Richardson CD. 2011. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol 85:2342–2350. 10.1128/JVI.02046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jangra RK, Yi M, Lemon SM. 2010. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 84:6615–6625. 10.1128/JVI.00417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts APE, Lewis AP, Jopling CL. 2011. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 39:7716–7729. 10.1093/nar/gkr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunden RD, Ghezelbash S, Khan JQ, Wilson JA. 2020. Location specific annealing of miR-122 and other small RNAs defines an hepatitis C virus 5′ UTR regulatory element with distinct impacts on virus translation and genome stability. Nucleic Acids Res 48:9235–9249. 10.1093/nar/gkaa664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. 2012. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci USA 109:941–946. 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Yamane D, Lemon SM. 2015. Dissecting the roles of the 5′ exoribonucleases Xrn1 and Xrn2 in restricting hepatitis C virus replication. J Virol 89:4857–4865. 10.1128/JVI.03692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thibault PA, Huys A, Amador-Cañizares Y, Gailius JE, Pinel DE, Wilson JA. 2015. Regulation of hepatitis C virus genome replication by Xrn1 and MicroRNA-122 binding to individual sites in the 5′ untranslated region. J Virol 89:6294–6311. 10.1128/JVI.03631-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masaki T, Arend KC, Li Y, Yamane D, McGivern DR, Kato T, Wakita T, Moorman NJ, Lemon SM. 2015. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 17:217–228. 10.1016/j.chom.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schult P, Roth H, Adams RL, Mas C, Imbert L, Orlik C, Ruggieri A, Pyle AM, Lohmann V. 2018. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat Commun 9:2613. 10.1038/s41467-018-05053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amador-Cañizares Y, Panigrahi M, Huys A, Kunden RD, Adams HM, Schinold MJ, Wilson JA. 2018. miR-122, small RNA annealing and sequence mutations alter the predicted structure of the hepatitis C virus 5′ UTR RNA to stabilize and promote viral RNA accumulation. Nucleic Acids Res 46:9776–9792. 10.1093/nar/gky662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chahal J, Gebert LFR, Gan HH, Camacho E, Gunsalus KC, MacRae IJ, Sagan SM. 2019. miR-122 and Ago interactions with the HCV genome alter the structure of the viral 5′ terminus. Nucleic Acids Res 47:5307–5324. 10.1093/nar/gkz194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201. 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ottosen S, Parsley TB, Yang L, Zeh K, van Doorn L-J, van der Veer E, Raney AK, Hodges MR, Patick AK. 2015. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob Agents Chemother 59:599–608. 10.1128/AAC.04220-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thibault PA, Wilson JA. 2013. Targeting miRNAs to treat hepatitis C virus infections and liver pathology: inhibiting the virus and altering the host. Pharmacol Res 75:48–59. 10.1016/j.phrs.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Ono C, Fukuhara T, Motooka D, Nakamura S, Okuzaki D, Yamamoto S, Tamura T, Mori H, Sato A, Uemura K, Fauzyah Y, Kurihara T, Suda T, Nishio A, Hmwe SS, Okamoto T, Tatsumi T, Takehara T, Chayama K, Wakita T, Koike K, Matsuura Y. 2017. Characterization of miR-122-independent propagation of HCV. PLoS Pathog 13:e1006374. 10.1371/journal.ppat.1006374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thibault PA, Huys A, Dhillon P, Wilson JA. 2013. MicroRNA-122-dependent and -independent replication of hepatitis C virus in Hep3B human hepatoma cells. Virology 436:179–190. 10.1016/j.virol.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Hopcraft SE, Azarm KD, Israelow B, Lévêque N, Schwarz MC, Hsu T-H, Chambers MT, Sourisseau M, Semler BL, Evans MJ. 2016. Viral determinants of miR-122-independent hepatitis C virus replication. mSphere 1:e00009-15. 10.1128/mSphere.00009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibault PA, Wilson JA. 2014. Transient replication of hepatitis C virus sub-genomic RNA in murine cell lines is enabled by miR-122 and varies with cell passage. PLoS One 9:e89971. 10.1371/journal.pone.0089971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Israelow B, Mullokandov G, Agudo J, Sourisseau M, Bashir A, Maldonado AY, Dar AC, Brown BD, Evans MJ. 2014. Hepatitis C virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat Commun 5:5408. 10.1038/ncomms6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chahal J, Gebert LFR, Camargo C, MacRae IJ, Sagan SM. 2021. miR-122-based therapies select for three distinct resistance mechanisms based on alterations in RNA structure. Proc Natl Acad Sci USA 118:e2103671118. 10.1073/pnas.2103671118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girard M, Jacquemin E, Munnich A, Lyonnet S, Henrion-Caude A. 2008. miR-122, a paradigm for the role of microRNAs in the liver. J Hepatol 48:648–656. 10.1016/j.jhep.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 32.Chang T, Young K, Yang Y, Lei H, Wu H. 1996. Hepatitis C virus RNA in peripheral blood mononuclear cells: comparing acute and chronic hepatitis C virus infection. Hepatology 23:977–981. 10.1002/hep.510230506. [DOI] [PubMed] [Google Scholar]
- 33.Manzin A, Candela M, Paolucci S, Caniglia ML, Gabrielli A, Clementi M. 1994. Presence of hepatitis C virus (HCV) genomic RNA and viral replicative intermediates in bone marrow and peripheral blood mononuclear cells from HCV-infected patients. Clin Diagn Lab Immunol 1:160–163. 10.1128/cdli.1.2.160-163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherman AC, Sherman KE. 2015. Extrahepatic manifestations of hepatitis C infection: navigating CHASM. Curr Hiv/Aids Rep 12:353–361. 10.1007/s11904-015-0274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panigrahi M, Palmer MA, Wilson JA. 2021. Enhanced virus translation enables miR-122-independent hepatitis C virus propagation. bioRxiv 2021.06.08.447644. [DOI] [PMC free article] [PubMed]
- 36.Amador-Cañizares Y, Bernier A, Wilson JA, Sagan SM. 2018. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res 46:5139–5158. 10.1093/nar/gky273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y-P, Gottwein JM, Scheel TK, Jensen TB, Bukh J. 2011. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci USA 108:4991–4996. 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villanueva RA, Jangra RK, Yi M, Pyles R, Bourne N, Lemon SM. 2010. miR-122 does not modulate the elongation phase of hepatitis C virus RNA synthesis in isolated replicase complexes. Antiviral Res 88:119–123. 10.1016/j.antiviral.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheu-Gruttadauria J, MacRae IJ. 2018. Phase transitions in the assembly and function of human miRISC. Cell 173:946–957.e16. 10.1016/j.cell.2018.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stiffler JD, Nguyen M, Sohn JA, Liu C, Kaplan D, Seeger C. 2009. Focal distribution of hepatitis C virus RNA in infected livers. PLoS One 4:e6661. 10.1371/journal.pone.0006661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y-H, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, Brantner CA, Stempinski ES, Connelly PS, Ma H-C, Jiang P, Wimmer E, Altan-Bonnet G, Altan-Bonnet N. 2015. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 160:619–630. 10.1016/j.cell.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santiana M, Ghosh S, Ho BA, Rajasekaran V, Du W-L, Mutsafi Y, Jésus-Diaz DAD, Sosnovtsev SV, Levenson EA, Parra GI, Takvorian PM, Cali A, Bleck C, Vlasova AN, Saif LJ, Patton JT, Lopalco P, Corcelli A, Green KY, Altan-Bonnet N. 2018. Vesicle-cloaked virus clusters are optimal units for inter-organismal viral transmission. Cell Host Microbe 24:208–220.e8. 10.1016/j.chom.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. 2014. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog 10:e1004424. 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. 2013. Treatment of HCV infection by targeting microRNA. N Engl J Med 368:1685–1694. 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 45.van der Ree MH, van der Meer AJ, de Bruijne J, Maan R, van Vliet A, Welzel TM, Zeuzem S, Lawitz EJ, Rodriguez-Torres M, Kupcova V, Wiercinska-Drapalo A, Hodges MR, Janssen HLA, Reesink HW. 2014. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res 111:53–59. 10.1016/j.antiviral.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 46.Zeisel MB, Baumert TF. 2017. Clinical development of hepatitis C virus host-targeting agents. Lancet 389:674–675. 10.1016/S0140-6736(17)30043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Ree MH, de Vree JM, Stelma F, Willemse S, van der Valk M, Rietdijk S, Molenkamp R, Schinkel J, van Nuenen AC, Beuers U, Hadi S, Harbers M, van der Veer E, Liu K, Grundy J, Patick AK, Pavlicek A, Blem J, Huang M, Grint P, Neben S, Gibson NW, Kootstra NA, Reesink HW. 2017. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet 389:709–717. 10.1016/S0140-6736(16)31715-9. [DOI] [PubMed] [Google Scholar]
- 48.Mata M, Neben S, Majzoub K, Carette J, Ramanathan M, Khavari PA, Sarnow P. 2019. Impact of a patient-derived hepatitis C viral RNA genome with a mutated microRNA binding site. PLoS Pathog 15:e1007467. 10.1371/journal.ppat.1007467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu F, Shimakami T, Murai K, Shirasaki T, Funaki M, Honda M, Murakami S, Yi M, Tang H, Kaneko S. 2016. Efficient suppression of hepatitis C virus replication by combination treatment with miR-122 antagonism and direct-acting antivirals in cell culture systems. Sci Rep 6:30939. 10.1038/srep30939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wakita T, Kato T. 2006. Development of an infectious HCV cell culture system. In Tan S-L (ed), Hepatitis C viruses: genomes and molecular biology. Horizon Bioscience, Norfolk, UK. [PubMed] [Google Scholar]
- 51.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich H-G, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 54.Kannan RP, Hensley LL, Evers LE, Lemon SM, McGivern DR. 2011. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis. J Virol 85:7989–8001. 10.1128/JVI.00280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]