

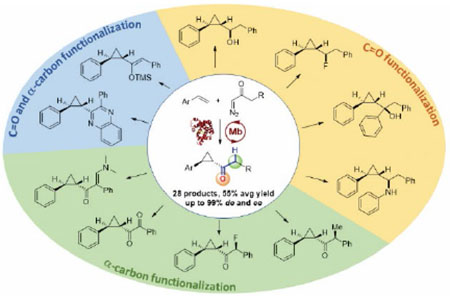
Abstract
Chiral cyclopropane rings are key pharmacophores in pharmaceuticals and bioactive natural products, making libraries of these building blocks a valuable resource for drug discovery and development campaigns. Here, we report the development of a chemoenzymatic strategy for the stereoselective assembly and structural diversification of cyclopropyl ketones, a highly versatile yet underexploited class of functionalized cyclopropanes. An engineered variant of sperm whale myoglobin is shown to enable the highly diastereo- and enantioselective construction of these molecules via olefin cyclopropanation in the presence of a diazoketone carbene donor reagent. This biocatalyst offers a remarkably broad substrate scope, catalyzing this reaction with high stereoselectivity across a variety of vinylarene substrates as well as a range of different α-aryl and α-alkyl diazoketone derivatives. Chemical transformation of these enzymatic products enables further diversification of these molecules to yield a collection of structurally diverse cyclopropane-containing scaffolds in enantiopure form, including core motifs found in drugs and natural products as well as novel structures. This work illustrates the power of combining abiological biocatalysis with chemoenzymatic synthesis for generating collections of optically active scaffolds of high value for medicinal chemistry and drug discovery.
Graphical Abstract
Introduction
Methods for the synthesis and structural diversification of pharmacophores and ‘privileged scaffolds’ represent key assets in medicinal chemistry efforts directed at the discovery of new bioactive molecules and drugs.1–2 Complementing purely synthetic approaches,3–4 chemoenzymatic strategies have recently emerged for this purpose, whereby a selective enzymatic transformation is coupled to a chemical diversification step for functional elaboration of a target molecular scaffold. For example, regio- and stereoselective C—H hydroxylation by means of engineered and natural cytochrome P450s has been integrated with alcohol functionalization/interconversion chemistries for the chemoenzymatic diversification of natural product scaffolds.5–9 As another example, halogenase enzymes have been exploited in combination with Pd-catalyzed cross-coupling chemistry for the diversification of aromatic compounds.10–12 Other approaches have involved other classes of natural enzymes.13–14 Despite this progress, these chemoenzymatic strategies for lead/scaffold diversification has thus far largely relied on enzyme-catalyzed transformations (e.g., hydroxylation, halogenation, glycosylation) that occur in nature.
Over the past few years, engineered hemoproteins (myoglobin, cytochrome P450s, cytochrome c)15–27 as well as artificial metalloenzymes28–35 have been introduced for promoting abiological carbene transfer reactions. In particular, our group has reported that engineered variants of the oxygen-storage hemoprotein myoglobin (Mb) are capable of catalyzing the intermolecular cyclopropanation of olefins with ethyl α-diazoacetate (EDA) with high catalytic activity and high stereoselectivity.16,36 The reaction scope of these myoglobin-based biocatalysts was later extended to intramolecular cyclopropanation reactions37–38 as well as to intermolecular cyclopropanations involving other types of carbene donor precursors such as trifluorodiazoethane and diazoacetonitrile.39–40 More recently, biocatalytic strategies for olefin cyclopropanations involving other non-diazoester reagents have also emerged.41
Given the relevance of optically active cyclopropane rings as pharmacophores in drug molecules and biologically active natural products,42 we have recently begun to explore the potential of combining enzyme-catalyzed cyclopropanation with post-cyclization chemistries for diversification of the enzymatic cyclopropane product, i.e. through the interconversion of nitrile-substituted cyclopropanes (Figure 1a).37,40 Expanding upon this concept, we envisioned diazoketones could represent a promising class of carbene donor reagents for this purpose, offering multiple opportunities for diversification of the cyclopropane product thanks to the versatile reactivity of carbonyls43 along with the unique reactivity imparted by this functional group to the adjacent bond and C—H site (Figure 1b).44–46 Notably, diazoketones have remained largely underexploited in transition metal catalyzed carbene transfer reactions,47–51 with only one isolated example of a highly enantioselective cyclopropanation involving an acceptor-only diazoketone reagent.52 In addition, chemocatalytic protocols for asymmetric cyclopropanations with diazoketone typically require expensive and toxic metals (Ru, Rh) and synthetically complex ligands.47–49,52 Here, we report the development of a biocatalytic strategy for the highly diastereo- and enantioselective synthesis of cyclopropyl ketones using an engineered iron-containing protein. These enzymatic products can be readily diversified by chemical means to provide efficient access to a variety of structurally diverse cyclopropane scaffolds as valuable chiral building blocks for medicinal chemistry and/or natural product synthesis.
Figure 1.
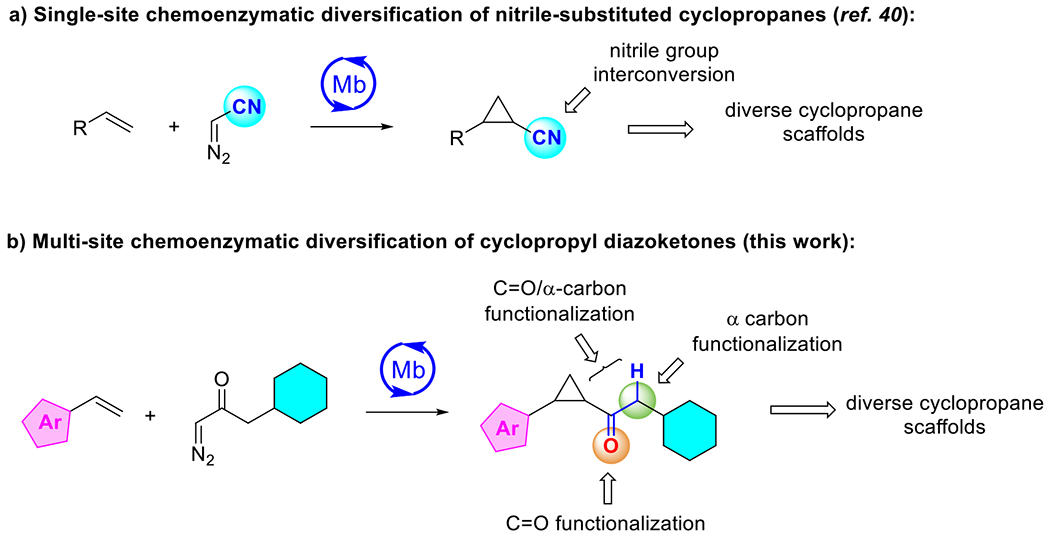
Strategies for chemoenzymatic assembly and diversification of cyclopropanes.
Results and Discussion
Olefin Cyclopropanation with Benzyl Diazoketone.
In initial efforts toward developing this httpology, we targeted the cyclopropanation of styrene (1) in the presence of 1-diazo-3-phenylpropan-2-one (2) (referred here as ‘benzyl diazoketone’) to give the keto-functionalized cyclopropane 3 (Table 1). We envisioned this reaction would offer multiple opportunities for chemoenzymatic diversification of the cyclopropanation product, namely through variation of both the carbene donor and carbene acceptor reagent in the cyclopropanation reaction as well as through the functionalization of the carbonyl group and the alpha C(sp3)—H site in the resulting cyclopropyl ketone product. To our knowledge, this reaction has never been reported using synthetic or biological carbene transfer catalysts. In addition, subjecting these reagents to commonly adopted organometallic catalysts for carbene transfer reactions such as Rh2(OAc)453 and Co(TPP)54 yielded no product or only trace amounts of 3 (Table S1). Similarly, various heme-containing proteins and enzymes, including wild type sperm whale myoglobin (Mb), various cytochromes P450 (P450BM3, P450XplA, P450BezE), and cytochromes c, showed no detectable activity toward this reaction (Table S1). Despite the lack of activity of wild type Mb, we previously found that mutations of the ‘distal’ histidine residue (His64), such as H64V, can improve the reactivity of this metalloprotein toward non-native carbene transfer reactions,16 an effect that has been attributed to an increased accessibility of the metal active center to the carbene donor reagents.55 Promisingly, Mb(H64V) was found to exhibit low but detectable activity in the cyclopropanation reaction, producing 3 in 6% yield in sodium borate buffer at pH 9.0 (Table 1, Entry 5). A sharp decrease in catalytic activity was evident at pH values below or above 9 (Table 1, Entries 2–6). Furthermore, the Mb(H64V) variant was found to catalyze this reaction with excellent degrees of diastereo- and enantioselectivity (>99% de and ee). The latter results are in contrast with the modest enantioselectivity exhibited by this variant in the cyclopropanation of styrene in the presence of ethyl α-diazoacetate as the carbene donor (10% ee).36
Table 1.
Catalytic activity and stereoselectivity of Mb variants for the cyclopropanation of styrene with 1-diazo-3-phenyl-2-propanone (2).a
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Entry | Catalyst | Catalyst loading | [1] (mM) | [2] (mM) | Buffer | Yield (SFC) | TON | d.e. | e.e. |
| 1 | Mb | 0.1 mol% | 20 | 20 | KPi pH 7 | trace | n.d. | n.d. | n.d. |
| 2 | Mb(H64V) | 0.1 mol% | 20 | 20 | KPi pH 7 | 1% | 10 | n.d. | n.d. |
| 3 | Mb(H64V) | 0.1 mol% | 20 | 20 | KPi pH 6 | 1% | 10 | n.d. | n.d. |
| 4 | Mb(H64V) | 0.1 mol% | 20 | 20 | KPi pH 8 | 2% | 20 | n.d. | n.d. |
| 5 | Mb(H64V) | 0.1 mol% | 20 | 20 | NaBB pH 9 | 6% | 60 | >99% | >99% |
| 6 | Mb(H64V) | 0.1 mol% | 20 | 20 | NaBB pH 10 | n.a. | - | - | - |
|
| |||||||||
| 7 | Mb(L29F,H64V) | 0.1 mol% | 20 | 20 | NaBB pH 9 | n.a. | - | - | - |
| 8 | Mb(L29S,H64V) | 0.1 mol% | 20 | 20 | NaBB pH 9 | n.a. | - | - | - |
| 9 | Mb(F43S,H64V) | 0.1 mol% | 20 | 20 | NaBB pH 9 | 1% | 10 | n.d. | n.d. |
| 10 | Mb(F43I,H64V) | 0.1 mol% | 20 | 20 | NaBB pH 9 | n.a. | - | - | - |
| 11 | Mb(H64V,V68F) | 0.1 mol% | 20 | 20 | NaBB pH 9 | n.a. | - | - | - |
| 12 | Mb(H64V,V68A) | 0.1 mol% | 20 | 20 | NaBB pH 9 | 8% | 80 | >99% | >99% |
| 13 | Mb(H64V,I107S) | 0.1 mol% | 20 | 20 | NaBB pH 9 | 1% | 10 | n.d. | n.d. |
| 14 | Mb(H64V,I107Y) | 0.1 mol% | 20 | 20 | NaBB pH 9 | n.a. | - | - | - |
| 15 | Mb(H64A,V68A) | 0.1 mol% | 20 | 20 | NaBB pH 9 | 12% | 120 | >99% | >99% |
| 16 | Mb(H64G,V68A) | 0.1 mol% | 20 | 20 | NaBB pH 9 | 20% | 200 | >99% | >99% |
|
| |||||||||
| 17 | Mb(H64G,V68A) | 0.2 mol% | 10 | 10 | NaBB pH 9 | 33% | 160 | >99% | >99% |
| 18 | Mb(H64G,V68A) | 0.4 mol% | 5 | 5 | NaBB pH 9 | 50% | 120 | >99% | >99% |
| 19 | Mb(H64G,V68A) | 0.4 mol% | 10 | 5 | NaBB pH 9 | 80% | 200 | >99% | >99% |
| 20 | Mb(H64G,V68A) | 0.4 mol% | 20 | 5 | NaBB pH 9 | >99% | >250 | >99% | >99% |
| 21 | Mb(H64G,V68A) | 0.04 mol% | 20 | 5 | NaBB pH 9 | 40% | 995 | >99% | >99% |
| 22b | Mb(H64G,V68A) | 0.4 mol% | 10 | 2.5 | NaBB pH 9 | >99% | >250 | >99% | >99% |
The reactions were carried using purified protein at the indicated reagents concentration in 50 mM potassium phosphate buffer (KPi) or 50 mM sodium borate buffer (NaBB) containing 10 mM sodium dithionite, overnight, under anaerobic conditions. Product yield and % de and ee as determined by chiral supercritical fluid chromatography (SFC) analysis. Reported values are mean values from n ≥ 2 experiments (SE <15%). N.a. = not active.
Reaction time: 10 min (see Figure S1a for complete time-course reaction profile).
Using Mb(H64V) as background, we then tested a small library of Mb variants in which the shape of the distal heme cavity is systematically varied by mutating each of the ‘active site’ residues (i.e., Leu29, Phe43, Val68, and Ile107; Figure 2) with an apolar amino acid of significantly different size (i.e. Leu29 → Ala/Phe: was mutated to Ala or Phe; Phe43 was mutated to Ile or Ser). This strategy has previously proven effective toward improving the efficiency of other Mb-catalyzed carbene transfer reactions such as the C—H functionalization of indoles.24 While most of these mutations resulted in loss of catalytic activity (Table 1, Entries1 7–14), Mb(H64V,V68A) showed a slight improvement in activity (80 vs. 60 turnovers or TON), while maintaining excellent diastereo- and enantioselectivity (>99% de and ee) (Table 1, Entry 12). This variant was previously found to exhibit high activity and stereoselectivity for the cyclopropanation of styrene with EDA16 and other small-sized acceptor-only diazo compounds such as trifluorodiazoethane39 and diazoacetonitrile.40 Considering the significantly bulkier diazoketone reagent utilized in the present reaction, we envisioned that the latter could benefit from increasing the accessibility of the heme pocket through substitution of the ‘gating’ residue at position 64 (Figure 2) with a small amino acid residue. Gratifyingly, a progressive enhancement in catalytic activity (8% (Val) → 12% (Ala) → 20% (Gly)) was observed upon reducing steric hindrance at this position through substitution of Val64 with alanine and glycine to give Mb(H64A,V68A) and Mb(H64G,V68A), respectively (Table 1, Entries 15–16). Altogether, this structure-activity data showed a clear correlation between cyclopropanation activity with benzyl diazoketone 2 (TON) and the decreasing size of the amino acid at the ‘proximal’ site (Gly > Ala > Val >> His). Upon optimization of the reaction conditions, the Mb(H64G,V68A)-catalyzed cyclopropanation reaction of styrene with 2 could be further optimized to produce 3a in quantitative yield and with excellent stereocontrol (>99% de and ee; Table 1, Entries 20). To further deconvolute the functional role of the mutations in Mb(H64G,V68A), the activity of a series of single mutants was investigated under these optimized reaction conditions (Figure S2). While confirming the beneficial effect of space-creating mutations at the level of the gating residue 64 (Gly > Ala = Val), these results also revealed a synergistic effect of the H64G and V68A mutations toward enhancing biocatalyst’s activity in this transformation, as judged based on the significantly higher TON exhibited by Mb(H64G,V68A) compared to Mb(H64G) and Mb(V68A) (>250 vs. 24–32 TON; Figure S2). As noted for Mb(H64V) (Table 1, Entries 3–6), the efficiency of the Mb(H64G,V68A)-catalyzed cyclopropanation reaction with diazoketone 2 was found to be pH dependent, showing an optimum at pH 9 (Table S3). This trend differs from that observed for other Mb-catalyzed carbene transfer reactions investigated previously, which showed either no pH dependence (e.g., indole C—H functionalization)24 or optimal performance at neutral pH (e.g., benzofuran cyclopropanation).26 Thus, the mildly alkaline conditions seem to be specifically beneficial for Mb-catalyzed olefin cyclopropanation with diazoketones. Using X-ray crystallography (vide infra), the absolute configuration of the cyclopropanation product was determined to be (1S,2S). The high trans-(1S,2S) stereoselectivity exhibited by Mb(H64G,V68A) and Mb(H64V,V68A) in this reaction mirrors that of Mb(H64V,V68A) for the cyclopropanation of styrene with EDA,16 suggesting a similar mechanism of catalyst-induced stereoinduction.55 Kinetic experiments showed that the Mb(H64G,V68A)-catalyzed reaction proceeds with an initial rate of 196 turnovers per minute (Figure S2) and it reaches completion within 10 minutes (Table 1, Entry 22; Figure S1). Furthermore, the biocatalyst supports up to ~1,000 total turnovers (TTN) under catalyst-limited conditions (Table 1, Entry 21).
Figure 2.
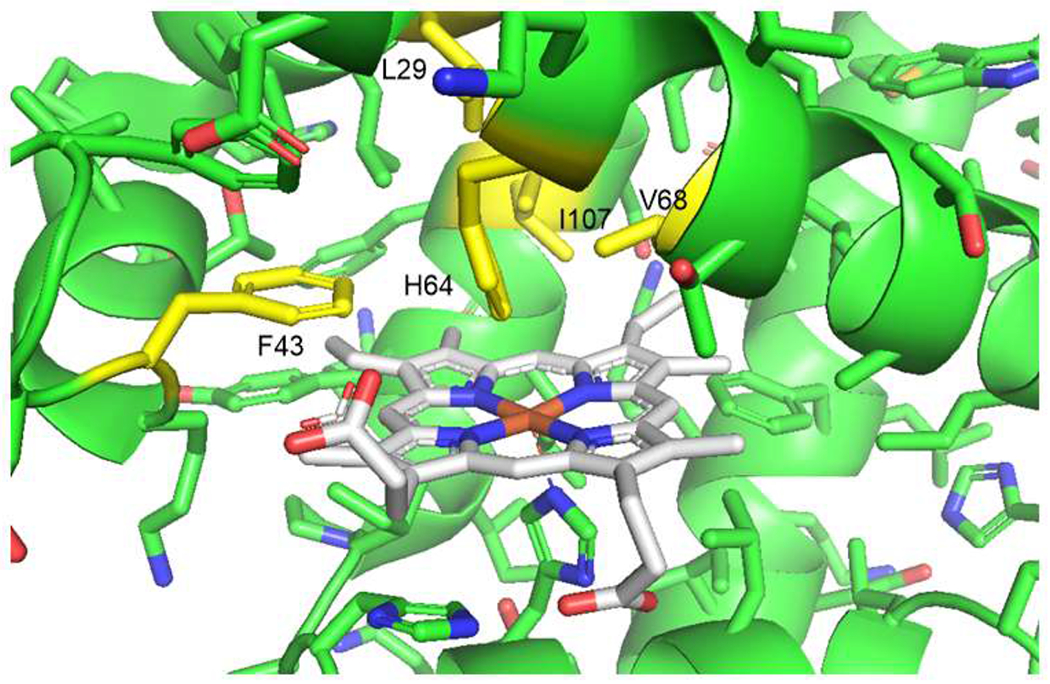
Distal heme pocket of sperm whale myoglobin (pdb 1VXA). The heme cofactor (grey) and neighbouring residues are shown as stick models. The active site residues targeted for mutagenesis are labeled and highlighted in yellow.
Analysis of Substrate Scope Across Olefin Substrates
To assess the substrate tolerance of the Mb(H64G,V68A) catalyst across the olefin reagent, the Mb variant was tested against a diverse set of styrene derivatives and other vinylarenes in the cyclopropanation reaction with diazoketone 2. As summarized in Figure 3, these experiments showed that this variant is able to accept a range of para-substituted styrene derivatives (4a-4e) yielding the corresponding cyclopropyl ketones in enantiopure form (>99% de and ee for 5a,b,d,e) or highly enantioenriched form (95% ee for 5c). Both electrondonating and electrowithdrawing groups were accepted with generally higher yields for the electronrich styrenes (5a-b) compared to the electronpoor ones (5c-e). This trend is generally consistent with the electrophilic reactivity of the heme-carbene intermediate expected to mediate this reaction as determined for α-diazoacetates.55–56 Kinetic analysis of these reactions (Figure S2) showed indeed that electronrich styrenes (176-190 TON min−1 for 5a-b) are cyclopropanated at faster rates than electropoor ones (123-132 TON min−1 5c-d). At the same time, the cyclopropanation rate of electronrich styrenes was comparable to that of styrene (Figure S2), indicating that while electronic effects are dominating, steric effects also play a role in influencing catalyst’s reactivity toward these substrates. The results with 5f and 5g indicated that the catalyst exhibits good activity and excellent stereoselectivity (99% de and ee) also toward the cyclopropanation of meta- and ortho- substituted styrenes (Figure 3). The results across the 5a, 5f, and 5c series indicated that substitutions at the para position are generally better tolerated than ortho and meta substitutions in terms of catalytic activity, while excellent stereoselectivity is retained in all cases. The Mb(H64G,V68A)-catalyzed cyclopropanation of electrodeficient pentafluorostyrene (4i) was also successful, producing 5i in good yield and with excellent stereoselectivity (>99% de and ee). Notably, other challenging substrates such as the disubstituted α-methyl-styrene (4h) and thionyl- and pyridine-containing vinylarenes 4j and 4k, respectively, could be processed with good efficiency (60–65% SFC yields) to afford the corresponding trisubstituted cyclopropanes 5h, 5j, and 5k, respectively, in high diastereomeric and enantiomeric excess (>99% de and 97–99% ee; Figure 3). Of note, substrates such as 4j and 4k are notoriously challenging substrates for transition-metal catalyzed cyclopropanations due to the propensity of N- and S-containing heterocycles to bind and thus inhibit the activity of metal centers. Conversely, aliphatic olefins such as 1-octene and internal olefins such as cyclohexene did not participate in the reaction, defining the boundaries of the biocatalyst’s scope with respect to the olefin substrate.
Figure 3.
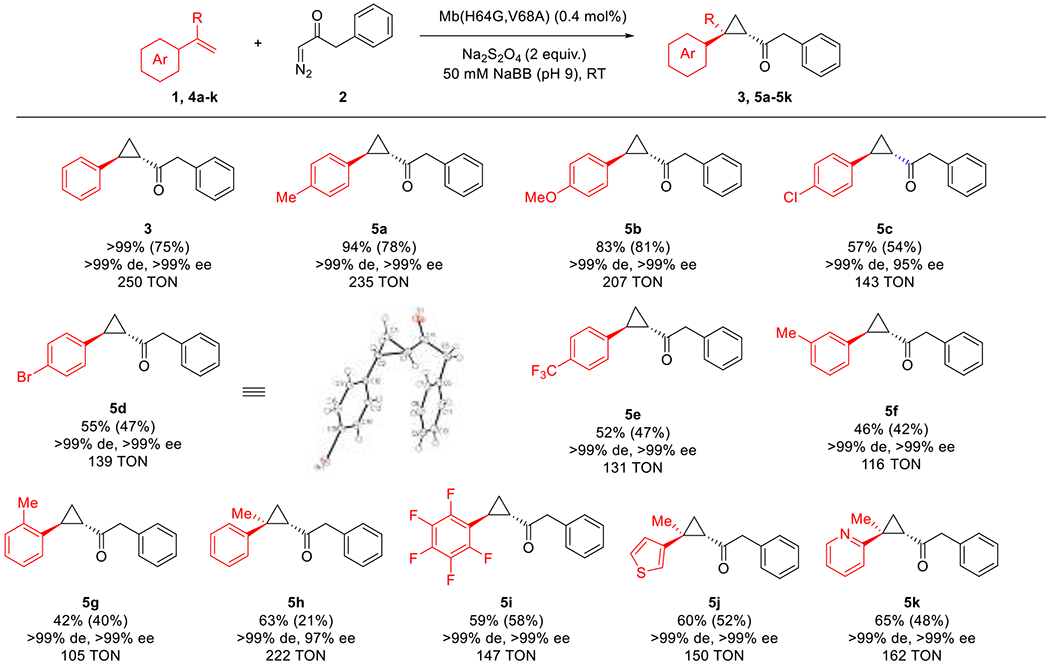
Substrate scope of Mb(H64G,V68A)-catalyzed cyclopropanation with diazoketone 2 in the presence of different olefin substrates.a
a Reaction Conditions: 20 mM olefin, 5 mM diazo compound, 10 mM Na2S2O4, 20 μM Mb(H64G,V68A) in sodium borate buffer (50 mM, pH 9) containing 10% ethanol, room temperature, 12 hours, anaerobic conditions. Product conversion and stereoselectivity was determined by chiral SFC using calibration curves with authentic standards and yields of isolated products are reported in parentheses.
Crystallographic analysis of 5d confirmed the (1S,2S) configuration of the cyclopropanation product (Figure 3, Table S3 and Figure S4), as it was anticipated based on the results with Mb(H64V,V68A) and its previously established stereopreference in the cyclopropanation of vinylarenes in the presence of other acceptor-only diazo compounds.16,39–40 Using 5d as reference, chiral chromatographic analysis of the other cyclopropane products indicated an identical enantioselectivity across all the other cyclopropanation products, denoting the generality and predicable stereoselectivity of the Mb(H64G,V68A) catalyst. All of the aforementioned reactions could be performed at the semi-preparative scale (0.2 mol) enabling the synthesis of the desired cyclopropyl ketone products with an average isolated yield of 54%. Altogether, these results demonstrated the broad tolerance of the Mb(H64G,V68A) catalyst toward accepting a variety of vinylarene substrates.
Substrate Scope Across Diazoketone Substrates
Next, we assessed the performance of Mb(H64G,V68A) in the cyclopropanation reaction with styrene in the presence of different diazoketone reagents, as we envisioned this approach could provide an additional means for diversification of the cyclopropyl ketone products (Figure 4). To this end, a diverse set of diazoketones was prepared according to the Arndt-Eistert method57–58 through reaction of the corresponding acyl chloride precursors with diazomethane generated in situ from diazald. This procedure yielded the desired carbene donor reagents in up to ~80% yield over two steps from readily available carboxylic acid precursors. Notably, a variety of benzyl diazoketone analogues containing para-, meta-, and ortho-substitutions (6a-6g) could be processed by the Mb(H64G,V68A) catalyst to afford the corresponding cyclopropanes 7a-7g in up to 90% conversion, excellent trans-selectivity (>99% de) and very good to excellent enantioselectivity (83 to >99% ee) (Figure 4a). Interestingly, higher conversions and enantioselectivity were generally observed for the reactions involving the ortho- or meta-substituted diazo compounds compared to the para-substituted counterparts (e.g., 7d vs.7b and 7g vs 7f; Figure 4a) and regardless of the electronic effect of these substituents, suggesting a stronger influence of steric over electronic effects on the activity and selectivity of the biocatalyst upon variation of the diazo reagent. Of note, a bulky carbene donor such as the benzodioxolane derivative 6h was also readily converted by the hemoprotein to give 7h in good yield and with excellent enantioselectivity (>99% de, 98% ee). Finally, the results with 7i showed that the biocatalyst is also able to tolerate alpha substitutions in the diazo reagent. Using enantiopure (S)-α-methyl-β-phenyl diazoketone 6i, the desired cyclopropyl ketone 7i was produced in 50% yield and with a (1S,2S,3S):(1R,2R,3S) diastereomeric ratio of 96:4 (Figure 4a), the latter result indicating that the Mb(H64G,V68A)-catalyzed cyclopropanation reaction has retained high stereoselectivity also in the presence of the α-substituted diazo reagent. Interestingly, the same reaction in the presence of (R)-α-methyl-β-phenyl diazoketone 6j yielded no product, indicating that the Mb variant is enantioselective toward the chiral diazo reagent. Consistent with these results, a reaction with racemic (±)-α-methyl-β-phenyl diazoketone produced 7i in 28% yield and with a d.r. of 96:4, further demonstrating that the enzyme is indeed able to resolve the racemic diazo reagent by engaging only the (S)-enantiomer. As observed for the reaction with styrene (Table 1) and other vinylarenes (Figure 2), the Mb(H64G,V68A)-catalyzed reactions involving the different diazo compounds occur cleanly with no signs of carbene dimerization and with the desired product and unreacted substrate accounting for total mass balance. In some cases, trace amounts (<1%) of a reduced by-product resulting from diazotization of the diazo reagent was observed. These results indicate that catalyst inactivation is the primary factor limiting product conversion under suboptimal reaction conditions (e.g. Entry 15, Table 1) or in the presence of less favorable substrates (e.g., 5h, 7f).
Figure 4.
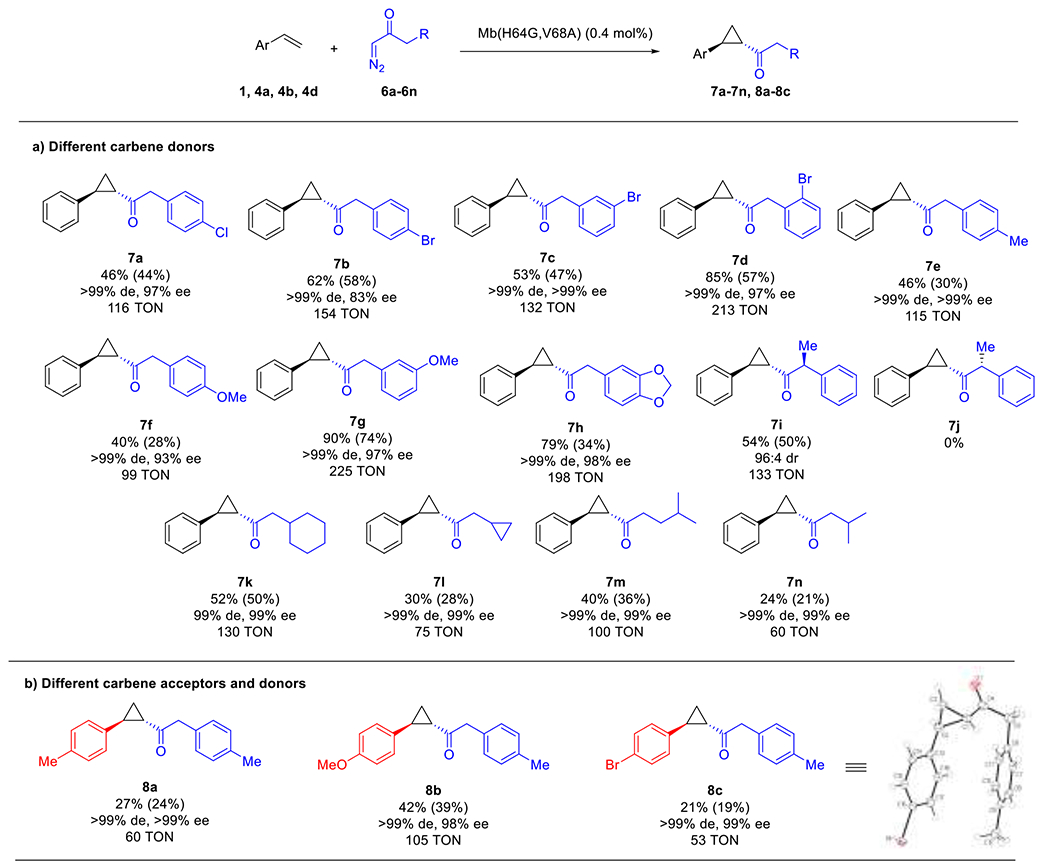
Substrate scope of Mb(H64G,V68A)-catalyzed cyclopropanation with styrene (a) and styrene derivatives (b) in the presence of different diazoketone reagents.a
a Reaction Conditions: 20 mM olefin, 5 mM diazo compound, 10 mM Na2S2O4, 20 μM Mb(H64G,V68A) in sodium borate buffer (50 mM, pH 9) containing 10% ethanol, room temperature, 12 hours, anaerobic conditions. Product conversion and stereoselectivity was determined by chiral SFC (or chiral HPLC) using calibration curves with authentic standards and yields of isolated products are reported in parentheses.
To further explore the substrate scope of the reaction, a panel of different α-alkyl-substituted diazoketones (6k-6n) was investigated. Notably, all of these diazoketone reagents were accepted by the Mb(H64G,V68A) catalyst, enabling the synthesis of the desired cyclopropanation products 7k-7n in 21–50% isolated yields and with excellent diastereo- and enantioselectivity (99% de and ee; Figure 4a). Finally, we tested the ability of Mb(H64G,V68A) to accept combinations of substituted styrenes and substituted benzyl diazoketone reagents (Figure 4b). Albeit in moderate yields (21–40%), the Mb(H64G,V68A)-catalyzed cyclopropanation of various styrene derivatives in the presence of 6e produced the desired disubstituted cyclopropyl ketones 8a-8c with excellent degrees of diastereo- and enantioselectivity (>99% de, 98–99% ee; Figure 4b). Crystallographic analysis of 8c confirmed the 1S,2S absolute configuration of the cyclopropane ring (Figure 4b and S5, Table S4), further highlighting the predictable and highly conserved stereoselectivity of this biocatalyst in these transformations. Together with those discussed earlier (Figure 3), these results demonstrated the remarkable tolerance of the Mb(H64G,V68A) biocatalyst toward accepting various diazoketone reagents in order to generate a diverse set of cyclopropyl ketones with high stereoselectivity.
Diverse Cyclopropane Scaffolds via Chemoenzymatic Diversification of Cyclopropyl Ketones
While remaining largely underexploited as pharmacophores, the optically active cyclopropyl ketones made accessible using the present methodology are expected to constitute valuable building blocks for medicinal chemistry as α-cyclopropyl ketone motifs are found in a number of bioactive molecules including the natural product bisgersolanolide59 and approved drugs such as the larotaxel (anticancer)60 and prasugrel (anticoagulant)61. In addition to that, we envisioned that another key advantage of this biocatalytic strategy would derive from the opportunity to diversify the cyclopropanation product via chemoenzymatic means in order to obtain a variety of valuable chiral cyclopropane-based scaffolds (Figure 1b). To illustrate this point, enantiopure 3 produced from the Mb-catalyzed reaction was converted, through different chemistries, into a panel of structurally diverse cyclopropane-containing scaffolds, including α-cyclopropyl alcohols, amines, and fluorides, α-substituted cyclopropyl ketones, and diketones (Scheme 1). These compounds encompass both structural motifs found in medicinally active molecules as well as entirely new structures. As shown in Scheme 1, α-cyclopropyl alcohol 9 could be readily obtained via reduction of the carbonyl group in 3 with sodium borohydride (84% yield, 2.5:1 d.r., >99% ee), whereas a nucleophilic addition reaction with phenylmagnesium bromide afforded the tertiary α-cyclopropyl alcohol 10 (45% yield, 2.4:1 d.r., >99% ee,; Scheme 1). Similar α-cyclopropyl alcohol motifs are found in various members of eicosanoid-like families of natural products such as constanolactones, solandelactones, and halicholactones 62–63 as well as in synthetic drugs such as the triple re-uptake inhibitor GSK1360707F.64 Deoxofluorination of 9 in the presence of the nucleophilic fluorinating reagent XtalFluor-E afforded α-cyclopropyl fluoride 11 (66% yield over two steps), whereas α-cyclopropyl amine 12 could be obtained in good yield (63%) via direct reductive amination of cyclopropyl ketone 3 in the presence of aniline (Scheme 1). An α-cyclopropyl fluoride moiety is found in the anti-HIV agent lenacapavir,65 whereas α-cyclopropyl amines are common pharmacophore in a variety of drugs, including the serotonin reuptake inhibitors milnacipram66 and BMS-505130.67 For all the aforementioned reactions, transformation of the carbonyl group introduces a third stereogenic center which could be formed with a significant (and comparable) degree of diastereoselectivity (~ 2.5 : 1 d.r.) as influenced by the chirality of the enzymatically assembled cyclopropane ring. Furthermore, in each case the two diastereomers could be separated and isolated as enantiopure entities (>99% ee).
Scheme 1.
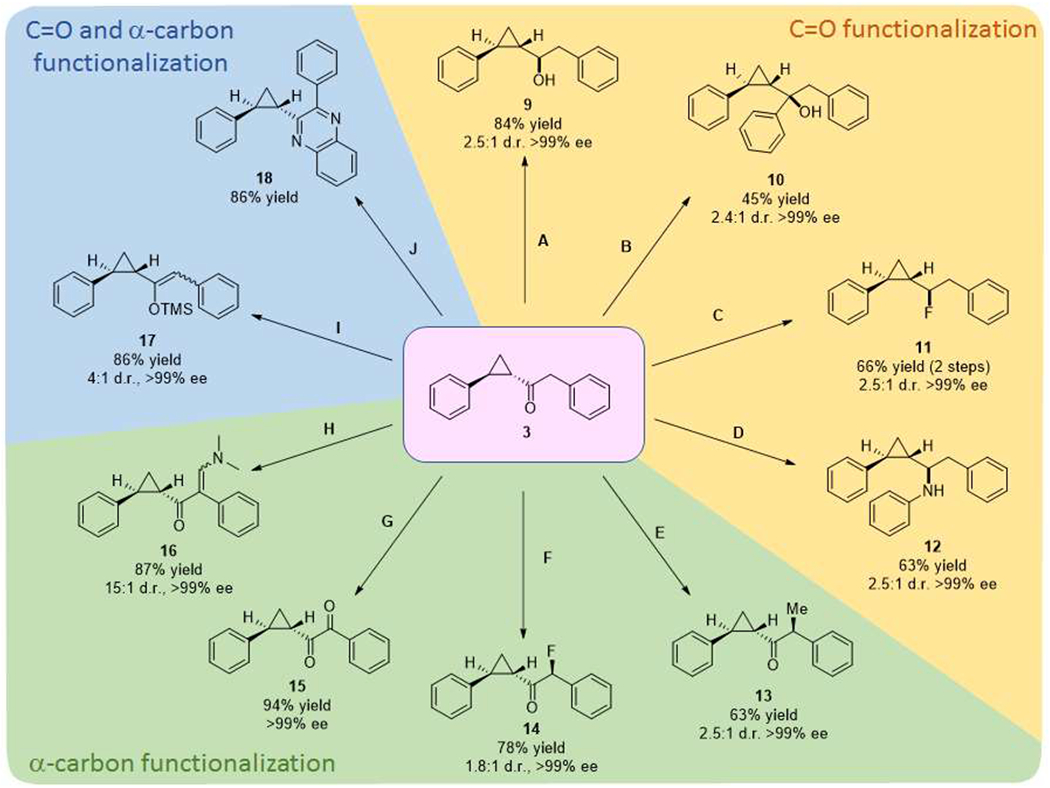
Diverse cyclopropane scaffolds via diversification of cyclopropyl ketones.
Reaction conditions: A: sodium borohydride (1.5 equiv.) MeOH, r.t., 3 hrs. B: Phenylmagnesium bromide (3 equiv.), CsCl2 (3 equiv.), THF, 0 °C, 2 hrs. C: 1) NaBH4 (1.5 equiv.) MeOH, r.t., 3 hr, 2) XtalFluor-E (1.5 equiv.), DBU (1.5 equiv.), DCM, 0 °C to r.t., overnight. D: Aniline (2 equiv.), sodium borohydride (NaBH4) (3 equiv.), THF, r.t., overnight. E: Lithium diisopropylamide, ., MeI, THF, 0 °C to r.t., overnight. F: Sodium dodecyl benzenesulfonate, SelectF, DCM, r.t., overnight. G: CuBr2 (20 mol%), thiomorpholine (3 equiv.), aerobic, DMSO, r.t., overnight. H: N,N-dimethylformamide dimethyl acetal, neat, 90 °C, overnight. I: Lithium diisopropylamide (1.1 equiv.), trimethylsilyl chloride (1.2 equiv.), Et2O, 0 °C to r.t., overnight. J: 1) copper (II) bromide (CuBr2) (20 mol%), thiomorpholine (3 equiv.), aerobic, DMSO, r.t., overnight. 2) o-phenylenediamine (1 equiv.), potassium tert-butoxide (4 mol%), MeOH, r.t., overnight.
We further posited that functionalization of the alpha C—H site in the enzymatic product 3 could furnish a viable route to gain access to other types of functionalized cyclopropane scaffolds. Accordingly, base-catalyzed α-alkylation of 3 in the presence of methyl iodide could be applied to obtain cyclopropyl ketone 13 (63% yield; 2.5:1 d.r.; >99% ee; Scheme 1), whereas its α-fluorination in the presence of electrophilic reagent Selectfluor in an emulsion68 successfully afforded fluorinated cyclopropyl ketone 14 (78% yield; 1.8:1 d.r.; >99% ee; Scheme 1). Both of products were obtained as a separable mixture of two diastereomers in enantiopure form (>99% ee). The configuration of the newly generated stereogenic center in 13 was assigned by direct comparison with the enzymatic product 7j and that in 14 by analogy with 13, since the two reactions are expected to exhibit similar facial selectivity during electrophilic attack of the enolate intermediate to generate the α-functionalized ketone. Aerobic oxidation of 3 in the presence of copper bromide and thiomorpholine produced cyclopropyl-1,2-diketone 15 in high yield as a single stereoisomer (94% yield, >99% ee). While cyclopropane scaffolds 14 and 15 have never been obtained via cyclopropanation protocols before, both α-fluoroketones and 1,2-diketones have represented key pharmacophores for the design of potent inhibitors of proteases,69–71 due to the ability of these moieties to mimic the tetrahedral intermediate and/or inactivate the nucleophilic residue mediating the cleavage of peptide bonds in these enzymes.69–71 Subjecting 3 to an excess of dimethylformamide dimethyl acetal produced the α,β-unsaturated ketone 16 in high yield (86%) as a mixture of enantiopure Z and E isomers (Scheme 1). Whereas 16 represents a novel cyclopropane-based scaffold per se, α,β-unsaturated carbonyl derivatives have found a growing number of applications in the development of drugs acting as covalent protein/enzyme inhibitors.72–73 Finally, other novel cyclopropane-based scaffolds could be obtained through functionalization of both the carbonyl group and α position. Enolization of 3 with lithium diisopropylamine (LDA) followed by enolate trapping with trimethylsilyl chloride generated the cyclopropane silyl ether 17 in 84% yield. On the other hand, subjecting diketone 15 to a double condensation reaction in the presence of phenylenediamine74 readily afforded the cyclopropane-containing quinoxaline 18 in 66% yield over two steps from 3. The latter chemoenzymatic route can provide access to diaryl-substituted cyclopropanes which are not readily accessible via transition metal-catalyzed carbene transfer.
Conclusion
In summary, we have reported the development of a highly diastereo- and enantioselective biocatalytic strategy for the synthesis of keto-functionalized cyclopropanes. This represents the first example of a highly stereoselective protocol for olefin cyclopropanation with diazoketones involving a biocatalyst and iron-based catalyst. Notable features of the present strategy include a broad substrate scope and a consistently high stereoselectivity toward promoting this reaction in the presence of different vinylarenes and diazoketone reagents, as demonstrated through the synthesis of 28 different cyclopropyl ketone products by means of a single Mb-based catalyst. These compounds could be readily assembled and diversified by chemoenzymatic means to produce a collection of structurally diverse cyclopropane-based scaffolds (Scheme 1) useful for medicinal chemistry and other applications. This work paves the way to the application of diazoketones in other biocatalytic carbene transfer reactions and it illustrates the power of combining abiological biocatalysis with chemoenzymatic synthesis for generating collections of optically active synthons of potentially high value for the synthesis of new bioactive molecules.
EXPERIMENTAL DETAILS
General Information.
All the chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, Alfa Aesar, ACS Scientific, Acros) and used without any further purification, unless otherwise stated. All dry reactions were carried out under argon or nitrogen gas in flame-dried glassware with magnetic stirring using standard gas-tight syringes, cannula and septa. 1H and 13C NMR spectra were measured on a Bruker DPX-500 spectrometer (operating at 400 MHz for 1H and 100 MHz for 13C) or Bruker DPX-400 (operating at 500 MHz for 1H and 125 MHz for 13C). 19F was measured on Bruker DPX-400 (operating at 375 MHz). TMS was used as the internal standard (0 ppm) for 1H NMR, CDCl3 was used as the internal standard (77.0 ppm) for 13C NMR, and trifluorotoluene served as the internal standard (−63 ppm) for 13F NMR. Silica gel chromatography purifications were carried out using AMD Silica Gel 60 230-400 mesh. Preparative thin layer chromatography was performed on TLC plates (1 mm thickness, Sigma Aldrich).
Protein Expression and Purification.
Wild-type sperm whale myoglobin and the engineered Mb variants were cloned and expressed in E. coli C41(DE3) cells as described previously.36 Briefly, cells were grown in terrific broth (TB) medium (ampicillin, 100 mg L−1) at 37 °C (200 rpm) until OD600 reached 1.0–1.2. Cells were then induced with 0.25 mM β-d-1-thiogalactopyranoside (IPTG) and 0.3 mM δ-aminolevulinic acid (ALA). After induction, cultures were shaken at 180 rpm and 27 °C and harvested after 18–20 h by centrifugation at 4000 rpm at 4 °C. After cell lysis by sonication, the proteins were purified by Ni-affinity chromatography. The lysate was transferred to a Ni-NTA column equilibrated with Ni-NTA Lysis Buffer (50 mM KPi, 250 mM, NaCl, 10 mM imidazole, pH 8.0). The resin was washed with 50 mL of Ni-NTA Lysis Buffer and then 50 mL of Ni-NTA Wash Buffer (50 mM KPi, 250 mM, NaCl, 20 mM imidazole, pH 8.0). Proteins were eluted with Ni-NTA Elution Buffer (50 mM KPi, 250 mM, NaCl, 250 mM histidine, pH 7.0). After elution, the proteins were buffer exchanged against 50 mM KPi buffer (pH 7.0) using 10 kDa Centricon filters. Protein concentration in ferric form was determined using ε410 = 156 mM−1 cm−1 as the extinction coefficient.
Enzymatic reactions with Purified Protein.
Substrate screening reactions were carried out at a 400 μL scale using 20 μM myoglobin, 20 mM styrene, 5 mM diazoketone, and 10 mM sodium dithionite inside an anaerobic chamber. In a typical procedure, a buffered solution containing the myoglobin variant was carefully charged to a vial inside an anaerobic chamber. After diluting the catalyst solution with sodium borate buffer (50 mM, pH 9.0), 20 μL of sodium dithionite solution (200 mM stock solution in 50 mM pH 9 sodium borate buffer) was added. Reactions were initiated by addition of 20 μL of styrene (from 400 mM stock solution in ethanol), followed by the addition of 20 μL of diazoketone (from 100 mM stock solution in ethanol). The reaction mixture was left under magnetic stirring for 16 hours at room temperature inside an anaerobic chamber. The reaction mixtures were added with 20 μL of internal standard (50 mM benzodioxole in ethanol), followed by extraction with 400 μL of dichloromethane. For determination of product yield, diastereomeric excess, and enantiomeric excess, the extraction solutions were analyzed by chiral SFC, chiral GC or chiral HPLC as described in the Supporting Information (see Analytical Methods in SI and Figures S3 and S4). Time-course experiments (Figure S1) and kinetic experiments (Figure S2) were performed in a similar manner using 10 μM enzyme, 10 mM styrene 1, 2.5 mM diazoketone 2, 10 mM Na2S2O4 in 50 mM sodium borate buffer (pH 9.0) with 10% EtOH, room temperature, anaerobic atmosphere. The reactions were quenched with 3 M HCl at the different time points, followed by extraction and chiral SFC analysis as described above.
Reactions with Whole Cells.
Whole cell experiments were carried out at a 400 μL scale using 370 μL of E. coli cells expressing Mb(H64V,V68A), 20 mM styrene derivative, and 5 mM diazoketone inside an anaerobic chamber. In a typical procedure, cells suspended in potassium phosphate buffer (50 mM, pH 7.2) with 10% glycerol were charged to a vial inside an anaerobic chamber. After diluting the catalyst solution with potassium phosphate buffer (50 mM, pH 7.2) with 10% glycerol, reactions were initiated by addition of 20 μL of styrene (from 400 mM stock solution in ethanol), followed by the addition of 20 μL of diazoketone (from 100 mM stock solution in ethanol). The reaction mixture was stirred for 16 hours at room temperature inside an anaerobic chamber. The reactions were prepared for chiral SFC analysis by adding 20 μL of internal standard (50 mM benzodioxole in ethanol) to the reaction mixture, followed by extraction with 400 μL of dichloromethane. The TON for the whole-cell reactions were calculated based on Mb concentration in the reaction mixture as measured via UV-vis spectroscopy (ε410 = 156 mM−1 cm−1) after cell lysis.
General procedure for synthesis of diazoketones 2, 6a-6n (Procedure A).
The diazoketone reagents were prepared from the corresponding carboxylic acids according to the following procedure. To a flame dried round bottom flask, carboxylic acid (1.0 mmol) was dissolved in DCM (1.0 mL) under argon with a drop of dimethylformamide as catalyst. After dropwise addition of thionyl chloride (1.5 equiv.), the reaction was stirred for 2 hours at room temperature. Solvent was removed in vacuo and used in the next step without further purification. Synthesis of diazoketones from acetyl chlorides was performed in a diazomethane generator (Aldrich). CAUTION: diazomethane is a toxic and explosive gas and it must be handled with great caution at all times! In the outer tube of diazomethane-generator, acetyl chloride (0.3 mmol) was dissolved in Et2O (3.0 mL) and in the inner tube, diazald (5.7 equiv.) was suspended in carbitol (1.0 mL). Once the reaction mixture was immersed in an ice bath, aqueous KOH (37%, 1.5 mL) was injected dropwise via syringe into the inner tube. After stirring for 3 hours at 0 °C, silicic acid (0.15 g) was added to the inner tube to quench any unreacted diazomethane. Solvent in the outer tube was removed and the reaction mixture was subjected to column chromatography to yield diazoketones 2 and 6a-6n. See Supporting information for compound characterization data.
General procedure for preparative-scale enzymatic synthesis of cyclopropanation products 3, 5a-5k, 7a-7n and 8a-8c (Procedure B).
To a round bottom flask, 18 mL of 20 μM myoglobin in argon-purged sodium borate buffer (50 mM, pH 9.0) and 70 mg of sodium dithionite was added under argon. Reaction was initiated by addition of 1 mL of 400 mM alkene solution in ethanol (20 mM final conc.), followed by addition 1 mL of 100 mM diazoketone solution in ethanol (5 mM final conc.). The reaction was left under magnetic stirring for 16 hours at room temperature under argon. After extraction with DCM (3 x 50 mL), organic layers were combined and dried over NaSO4. Solvent was removed in vacuo and the reaction mixture was subjected to column chromatography to yield cyclopropanes 3, 5a-5k, 7a-7n and 8a-8c. See Supporting information for compound characterization data.
Racemic standards.
Analytical amounts of racemic products were prepared from 400 μL-scale cyclopropanation reactions with 1 mM Fe(TPP)Cl (10 mol%), 20 mM olefin, 10 mM diazoketone and 10 mM sodium dithionite in toluene:H2O:EtOH (8:1:1). In a typical procedure, a mixture containing 40 μL sodium dithionite (100 mM stock solution in distilled H2O) and 300 μL toluene was degassed by sparging with argon for 5 min in a sealed vial. A separate vial containing 20 μL Fe(TPP)Cl (20 mM stock solution in toluene) was carefully degassed in a similar manner. The two solutions were then mixed together via cannula. Reactions were initiated by addition of 20 μL olefin (400 mM stock solution in ethanol), followed by the addition of 20 μL diazoketone (200mM stock solution in ethanol) with a syringe, and the reaction mixture was stirred for 16 hours at room temperature under positive argon pressure. The product was isolated via preparative TLC (hexanes:EtOAc 9:1), extracted with dichloromethane, and analysed by chiral SFC, GC, or HPLC as described in SI.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institute of Health grant GM098628. The authors are grateful to Dr. William Brennessel for assistance with crystallographic analyses. MS and X-ray instrumentation are supported by U.S. National Science Foundation grants CHE-0946653 and CHE-1725028.
Footnotes
Supporting Information
Supporting information includes supplementary Tables and Figures, chiral GC and SFC chromatograms, synthetic procedures, compound characterization data, NMR spectra.
References
- (1).Welsch ME; Snyder SA; Stockwell BR Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol 2010, 14, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Barnes EC; Kumar R; Davis RA The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep 2016, 33, 372. [DOI] [PubMed] [Google Scholar]
- (3).Wencel-Delord J; Glorius F C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem 2013, 5, 369. [DOI] [PubMed] [Google Scholar]
- (4).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 2016, 45, 546. [DOI] [PubMed] [Google Scholar]
- (5).Zhang K; Shafer BM; Demars MD 2nd; Stern HA; Fasan R Controlled oxidation of remote sp3 C-H bonds in artemisinin via P450 catalysts with fine-tuned regio- and stereoselectivity. J. Am. Chem. Soc 2012, 134, 18695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kolev JN; O’Dwyer KM; Jordan CT; Fasan R Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C-H Functionalization. ACS Chem. Biol 2014, 9, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tyagi V; Alwaseem H; O’Dwyer KM; Ponder J; Li QY; Jordan CT; Fasan R Chemoenzymatic synthesis and antileukemic activity of novel C9-and C14-functionalized parthenolide analogs. Bioorg. Med. Chem 2016, 24, 3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lowell AN; DeMars MD; Slocum ST; Yu FA; Anand K; Chemler JA; Korakavi N; Priessnitz JK; Park SR; Koch AA; Schultz PJ; Sherman DH Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C-H Functionalization. J. Am. Chem. Soc 2017, 139, 7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schmidt JJ; Khatri Y; Brody SI; Zhu C; Pietraszkiewicz H; Valeriote FA; Sherman DH A Versatile Chemoenzymatic Synthesis for the Discovery of Potent Cryptophycin Analogs. ACS Chem. Biol 2020, 15, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Roy AD; Gruschow S; Cairns N; Goss RJM Gene Expression Enabling Synthetic Diversification of Natural Products: Chemogenetic Generation of Pacidamycin Analogs. J. Am. Chem. Soc 2010, 132, 12243. [DOI] [PubMed] [Google Scholar]
- (11).Runguphan W; O’Connor SE Diversification of Monoterpene Indole Alkaloid Analogs through Cross-Coupling. Org. Lett 2013, 15, 2850. [DOI] [PubMed] [Google Scholar]
- (12).Durak LJ; Payne JT; Lewis JC Late-Stage Diversification of Biologically Active Molecules via Chemoenzymatic C-H Functionalization. ACS Catal. 2016, 6, 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Langenhan JM; Griffith BR; Thorson JS Neoglycorandomization and chemoenzymatic glycorandomization: Two complementary tools for natural product diversification. J. Nat. Prod 2005, 68, 1696. [DOI] [PubMed] [Google Scholar]
- (14).Tailhades J; Zhao YW; Ho YTC; Greule A; Ahmed I; Schoppet M; Kulkarni K; Goode RJA; Schittenhelm RB; De Voss JJ; Cryle MJ A Chemoenzymatic Approach to the Synthesis of Glycopeptide Antibiotic Analogues. Angew. Chem. Int. Ed 2020, 59, 10899. [DOI] [PubMed] [Google Scholar]
- (15).Coelho PS; Brustad EM; Kannan A; Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307. [DOI] [PubMed] [Google Scholar]
- (16).Bordeaux M; Tyagi V; Fasan R Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew. Chem. Int. Ed 2015, 54, 1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang ZJ; Peck NE; Renata H; Arnold FH Cytochrome P450-catalyzed insertion of carbenoids into N-H bonds. Chem. Sci 2014, 5, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tyagi V; Bonn RB; Fasan R Intermolecular carbene S-H insertion catalysed by engineered myoglobin-based catalysts. Chem. Sci 2015, 6, 2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tyagi V; Fasan R Myoglobin-Catalyzed Olefination of Aldehydes. Angew. Chem. Int. Ed 2016, 55, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kan SBJ; Lewis RD; Chen K; Arnold FH Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life. Science 2016, 354, 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tyagi V; Sreenilayam G; Bajaj P; Tinoco A; Fasan R Biocatalytic Synthesis of Allylic and Allenyl Sulfides through a Myoglobin-Catalyzed Doyle-Kirmse Reaction. Angew. Chem. Int. Ed 2016, 55, 13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Weissenborn MJ; Low SA; Borlinghaus N; Kuhn M; Kummer S; Rami F; Plietker B; Hauer B Enzyme-Catalyzed Carbonyl Olefination by the E. coli Protein YfeX in the Absence of Phosphines. Chemcatchem 2016, 8, 1636. [Google Scholar]
- (23).Kan SBJ; Huang X; Gumulya Y; Chen K; Arnold FH Genetically programmed chiral organoborane synthesis. Nature 2017, 552, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Vargas DA; Tinoco A; Tyagi V; Fasan R Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles. Angew. Chem. Int. Ed 2018, 57, 9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen K; Huang XY; Kan SBJ; Zhang RK; Arnold FH Enzymatic construction of highly strained carbocycles. Science 2018, 360, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vargas D; Khade R; Zhang Y; Fasan R Biocatalytic strategy for highly diastereo- and enantioselective synthesis of 2,3-dihydrobenzofuran based tricyclic scaffolds. Angew. Chem. Int. Ed 2019, 58, 10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic assembly of carbon-carbon bonds via iron-catalysed sp(3) C-H functionalization. Nature 2019, 565, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Srivastava P; Yang H; Ellis-Guardiola K; Lewis JC Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat. Commun 2015, 6, 7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dydio P; Key HM; Nazarenko A; Rha JY; Seyedkazemi V; Clark DS; Hartwig JF An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sreenilayam G; Moore EJ; Steck V; Fasan R Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts. Adv. Synth. Cat 2017, 359, 2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sreenilayam G; Moore EJ; Steck V; Fasan R Stereoselective olefin cyclopropanation under aerobic conditions with an artificial enzyme incorporating an iron-chlorin e6 cofactor. ACS Catal 2017, 7, 7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Moore EJ; Steck V; Bajaj P; Fasan R Chemoselective Cyclopropanation over Carbene Y-H Insertion Catalyzed by an Engineered Carbene Transferase. J. Org. Chem 2018, 83, 7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ohora K; Meichin H; Zhao LM; Wolf MW; Nakayama A; Hasegawa J; Lehnert N; Hayashi T Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species. J. Am. Chem. Soc 2017, 139, 17265. [DOI] [PubMed] [Google Scholar]
- (34).Villarino L; Splan KE; Reddem E; Alonso-Cotchico L; de Souza CG; Lledos A; Marechal JD; Thunnissen AMWH; Roelfes G An Artificial Heme Enzyme for Cyclopropanation Reactions. Angew. Chem. Int. Ed 2018, 57, 7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Carminati DM; Fasan R Stereoselective Cyclopropanation of Electron-Deficient Olefins with a Cofactor Redesigned Carbene Transferase Featuring Radical Reactivity. ACS Catal 2019, 9, 9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bajaj P; Sreenilayam G; Tyagi V; Fasan R Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed 2016, 55, 16110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chandgude AL; Ren X; Fasan R Stereodivergent Intramolecular Cyclopropanation Enabled by Engineered Carbene Transferases. J. Am. Chem. Soc 2019, 141, 9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ren X; Chandgude AL; Fasan R Highly Stereoselective Synthesis of Fused Cyclopropane-γ-Lactams via Biocatalytic Iron-Catalyzed Intramolecular Cyclopropanation. ACS Catal. 2020, 10, 2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tinoco A; Steck V; Tyagi V; Fasan R Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene. J. Am. Chem. Soc 2017, 139, 5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chandgude AL; Fasan R Highly Diastereo- and Enantioselective Synthesis of Nitrile-Substituted Cyclopropanes by Myoglobin-Mediated Carbene Transfer Catalysis. Angew. Chem. Int. Ed 2018, 57, 15852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wittmann BJ; Knight AM; Hofstra JL; Reisman SE; Kan SBJ; Arnold FH Diversity-Oriented Enzymatic Synthesis of Cyclopropane Building Blocks. ACS Catal. 2020, 10, 7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Talele TT The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem 2016, 59, 8712. [DOI] [PubMed] [Google Scholar]
- (43).Otera J Modern Carbonyl Chemistry; Wiley-CH, 2000. [Google Scholar]
- (44).Mukherjee S; Yang JW; Hoffmann S; List B Asymmetric enamine catalysis. Chem. Rev 2007, 107, 5471. [DOI] [PubMed] [Google Scholar]
- (45).Culkin DA; Hartwig JF Palladium-catalyzed alpha-arylation of carbonyl compounds and nitriles. Acc. Chem. Res 2003, 36, 234. [DOI] [PubMed] [Google Scholar]
- (46).Huang ZX; Lim HN; Mo FY; Young MC; Dong GB Transition metal-catalyzed ketone-directed or mediated C-H functionalization. Chem. Soc. Rev 2015, 44, 7764. [DOI] [PubMed] [Google Scholar]
- (47).Lindsay VNG; Nicolas C; Charette AB Asymmetric Rh(II)-Catalyzed Cyclopropanation of Alkenes with Diacceptor Diazo Compounds: p-Methoxyphenyl Ketone as a General Stereoselectivity Controlling Group. J. Am. Chem. Soc 2011, 133, 8972. [DOI] [PubMed] [Google Scholar]
- (48).Denton JR; Davies HM Enantioselective reactions of donor/acceptor carbenoids derived from alpha-aryl-alpha-diazoketones. Org. Lett 2009, 11, 787. [DOI] [PubMed] [Google Scholar]
- (49).Nicolas I; Le Maux P; Simonneaux G Intermolecular asymmetric cyclopropanation with diazoketones catalyzed by chiral ruthenium porphyrins. Tetrahedron Lett. 2008, 49, 2111. [Google Scholar]
- (50).Nicolas I; Roisnel T; Le Maux P; Simonneaux G Asymmetric intermolecular cyclopropanation of alkenes by diazoketones catalyzed by Halterman iron porphyrins. Tetrahedron Lett. 2009, 50, 5149. [Google Scholar]
- (51).Kim T; Kassim AM; Botejue A; Zhang C; Forte J; Rozzell D; Huffman MA; Devine PN; McIntosh JA Hemoprotein-Catalyzed Cyclopropanation En Route to the Chiral Cyclopropanol Fragment of Grazoprevir. Chembiochem 2019, 20, 1129. [DOI] [PubMed] [Google Scholar]
- (52).Chi LL; Suharto A; Da HL; Chanthamath S; Shibatomi K; Iwasa S Catalytic Asymmetric Intermolecular Cyclopropanation of a Ketone Carbene Precursor by a Ruthenium(II)-Pheox Complex. Adv. Synth. Cat 2019, 361, 951. [Google Scholar]
- (53).Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev 1998, 98, 911. [DOI] [PubMed] [Google Scholar]
- (54).Dzik WI; Xu X; Zhang XP; Reek JNH; de Bruin B ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc 2010, 132, 10891. [DOI] [PubMed] [Google Scholar]
- (55).Tinoco A; Wei Y; Bacik J-P; Carminati DM; Moore EJ; Ando N; Zhang Y; Fasan R Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase. ACS Catal. 2019, 9 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wei Y; Tinoco A; Steck V; Fasan R; Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc 2018, 140, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Mckervey MA; Tuladhar SM; Twohig MF Efficient Synthesis of Bicyclo[5.3.0]Decatrienones and of 2-Tetralones Via Rhodium(Ii) Acetate-Catalysed Cyclization of Alpha-Diazoketones Derived from 3-Arylpropionic Acids. J. Chem. Soc. Chem. Comm 1984, 129. [Google Scholar]
- (58).Arndt F; Eistert B Ein Verfahren zur Überführung von Carbonsäuren in ihre höheren Homologen bzw. deren Derivate. Ber. Dtsch. Chem. Ges 1935, 68, 200. [Google Scholar]
- (59).Rodriguez AD; Shi JG Isolation, structure elucidation, and synthesis of bisgersolanolide, a novel heptacyclic bis-diterpenoid from the gorgonian octocoral Pseudopterogorgia bipinnata. Org. Lett 1999, 1, 337. [DOI] [PubMed] [Google Scholar]
- (60).Metzger-Filho O; Moulin C; de Azambuja E; Ahmad A Larotaxel: broadening the road with new taxanes. Expert Opin. Inv. Drug 2009, 18, 1183. [DOI] [PubMed] [Google Scholar]
- (61).Baker WL; White CM Role of Prasugrel, a Novel P2Y(12) Receptor Antagonist, in the Management of Acute Coronary Syndromes. Am. J. Cardiovasc. Drug 2009, 9, 213. [DOI] [PubMed] [Google Scholar]
- (62).Nagle DG; Gerwick WH Isolation and Structure of Constanolactone A and Constanolactone B, New Cyclopropyl Hydroxyeicosanoids from the Temperate Red Alga Constantinea-Simplex. Tetrahedron Lett. 1990, 31, 2995. [Google Scholar]
- (63).Proteau PJ; Rossi JV; Gerwick WH Absolute Stereochemistry of Neohalicholactone from the Brown Alga Laminaria-Sinclairii. J. Nat. Prod 1994, 57, 1717. [DOI] [PubMed] [Google Scholar]
- (64).Micheli F; Cavanni P; Andreotti D; Arban R; Benedetti R; Bertani B; Bettati M; Bettelini L; Bonanomi G; Braggio S; Carletti R; Checchia A; Corsi M; Fazzolari E; Fontana S; Marchioro C; Merlo-Pich E; Negri M; Oliosi B; Ratti E; Read KD; Roscic M; Sartori I; Spada S; Tedesco G; Tarsi L; Terreni S; Visentini F; Zocchi A; Zonzini L; Di Fabio R 6-(3,4-Dichlorophenyl)-1-[(Methyloxy)methyl]-3-azabicyclo[4.1.0]heptane: A New Potent and Selective Triple Reuptake Inhibitor. J. Med. Chem 2010, 53, 4989. [DOI] [PubMed] [Google Scholar]
- (65).Link JO; Rhee MS; Tse WC; Zheng J; Somoza JR; Rowe W; Begley R; Chiu A; Mulato A; Hansen D; Singer E; Tsai LK; Bam RA; Chou CH; Canales E; Brizgys G; Zhang JR; Li JY; Graupe M; Morganelli P; Liu Q; Wu QY; Halcomb RL; Saito RD; Schroeder SD; Lazerwith SE; Bondy S; Jin DB; Hung M; Novikov N; Liu XH; Villasenor AG; Cannizzaro CE; Hu EY; Anderson RL; Appleby TC; Lu B; Mwangi J; Liclican A; Niedziela-Majka A; Papalia GA; Wong MH; Leavitt SA; Xu YL; Koditek D; Stepan GJ; Yu H; Pagratis N; Clancy S; Ahmadyar S; Cai TZ; Sellers S; Wolckenhauer SA; Ling J; Callebaut C; Margot N; Ram RR; Liu YP; Hyland R; Sinclair GI; Ruane PJ; Crofoot GE; McDonald CK; Brainard DM; Lad L; Swaminathan S; Sundquist WI; Sakowicz R; Chester AE; Lee WE; Daar ES; Yant SR; Cihlar T Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, pages, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Rizvi SMD; Shaikh S; Khan M; Biswas D; Hameed N; Shakil S Fetzima (levomilnacipran), a Drug for Major Depressive Disorder as a Dual Inhibitor for Human Serotonin Transporters and Beta-Site Amyloid Precursor Protein Cleaving Enzyme-1. CNS Neurol. Disord 2014, 13, 1427. [DOI] [PubMed] [Google Scholar]
- (67).Taber MT; Wright RN; Molski TF; Clarke WJ; Brassil PJ; Denhart DJ; Mattson RJ; Lodge NJ Neurochemical, pharmacokinetic, and behavioral effects of the novel selective serotonin reuptake inhibitor BMS-505130. Pharmacol. Biochem. Be 2005, 80, 521. [DOI] [PubMed] [Google Scholar]
- (68).Stavber G; Zupan M; Stavber S Micellar-System-Mediated Direct Fluorination of Ketones in Water. Synlett 2009, 589. [Google Scholar]
- (69).Gelb MH; Svaren JP; Abeles RH Fluoro Ketone Inhibitors of Hydrolytic Enzymes. Biochemistry 1985, 24, 1813. [DOI] [PubMed] [Google Scholar]
- (70).Angelastro MR; Mehdi S; Burkhart JP; Peet NP; Bey P Alpha-Diketone and Alpha-Keto Ester Derivatives of N-Protected Amino-Acids and Peptides as Novel Inhibitors of Cysteine and Serine Proteinases. J. Med. Chem 1990, 33, 11. [DOI] [PubMed] [Google Scholar]
- (71).Hu LY; Abeles RH Inhibition of Cathepsin-B and Papain by Peptidyl, Alpha-Keto Esters, Alpha-Keto Amides, Alpha-Diketones, and Alpha-Keto Acids. Arch. Biochem. Biophys 1990, 281, 271. [DOI] [PubMed] [Google Scholar]
- (72).Singh J; Petter RC; Baillie TA; Whitty A The resurgence of covalent drugs. Nat. Rev. Drug Discov 2011, 10, 307. [DOI] [PubMed] [Google Scholar]
- (73).Dalton SE; Campos S Covalent Small Molecules as Enabling Platforms for Drug Discovery. Chembiochem 2020, 21, 1080. [DOI] [PubMed] [Google Scholar]
- (74).Ghosh P; Mandal A Greener approach toward one pot route to pyrazine synthesis. Green Chem. Lett. Rev 2012, 5, 127. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.