

Abstract
Rationale
Mechanical signaling through cell–matrix interactions plays a major role in progressive vascular remodeling in pulmonary arterial hypertension (PAH). MMP-8 (matrix metalloproteinase-8) is an interstitial collagenase involved in regulating inflammation and fibrosis of the lung and systemic vasculature, but its role in PAH pathogenesis remains unexplored.
Objectives
To evaluate MMP-8 as a modulator of pathogenic mechanical signaling in PAH.
Methods
MMP-8 levels were measured in plasma from patients with pulmonary hypertension (PH) and controls by ELISA. MMP-8 vascular expression was examined in lung tissue from patients with PAH and rodent models of PH. MMP-8–/– and MMP-8+/+ mice were exposed to normobaric hypoxia or normoxia for 4–8 weeks. PH severity was evaluated by right ventricular systolic pressure, echocardiography, pulmonary artery morphometry, and immunostaining. Proliferation, migration, matrix component expression, and mechanical signaling were assessed in MMP-8–/– and MMP-8+/+ pulmonary artery smooth muscle cells (PASMCs).
Measurements and Main Results
MMP-8 expression was significantly increased in plasma and pulmonary arteries of patients with PH compared with controls and induced in the pulmonary vasculature in rodent PH models. Hypoxia-exposed MMP-8–/– mice had significant mortality, increased right ventricular systolic pressure, severe right ventricular dysfunction, and exaggerated vascular remodeling compared with MMP-8+/+ mice. MMP-8–/– PASMCs demonstrated exaggerated proliferation and migration mediated by altered matrix protein expression, elevated integrin-β3 levels, and induction of FAK (focal adhesion kinase) and downstream YAP (Yes-associated protein)/TAZ (transcriptional coactivator with PDZ-binding motif) activity.
Conclusions
MMP-8 is a novel protective factor upregulated in the pulmonary vasculature during PAH pathogenesis. MMP-8 opposes pathologic mechanobiological feedback by altering matrix composition and disrupting integrin-β3/FAK and YAP/TAZ-dependent mechanical signaling in PASMCs.
Keywords: cellular mechanotransduction, pulmonary arterial hypertension, pulmonary artery smooth muscle cells, integrin-β3, YAP/TAZ
At a Glance Commentary
Scientific Knowledge on the Subject
Matrix stiffening occurs early in the course of experimental pulmonary hypertension (PH) and promotes remodeling phenotypes in pulmonary vascular cells. Mechanical signaling through cell–matrix interactions plays a major role in progressive vascular remodeling in pulmonary arterial hypertension (PAH). MMP-8 (matrix metalloproteinase-8) is an interstitial collagenase involved in regulating inflammation and fibrosis of the lung and systemic vasculature, but its role in PAH pathogenesis has not been explored.
What This Study Adds to the Field
This study reports a novel protective role for MMP-8 in the pulmonary vasculature. MMP-8 expression is markedly increased in the plasma and pulmonary arterioles of patients with PAH compared with control subjects. Deficiency of MMP-8 in mice leads to increased vascular remodeling and right ventricular failure after exposure to hypoxia, indicating that MMP-8 upregulation likely represents an adaptive response to pathogenic PH stimuli. Deficiency of MMP-8 in pulmonary artery smooth muscle cells (PASMCs) alters matrix and integrin expression, enhancing proliferation and migration via proremodeling mechanotransduction by FAK and YAP/TAZ signaling. Disrupting this mechanobiological feedback using a FAK inhibitor ameliorates hyperproliferation and enhanced migration of PASMCs in vitro and improves PH and vascular remodeling in vivo. Our data suggest therapeutic potential in enhancing or mimicking MMP-8 activity to disrupt pathologic mechanical signaling during PAH pathogenesis.
Pulmonary hypertension (PH) is caused by the accumulation of hyperproliferative vascular lesions throughout the pulmonary arterial tree, leading to increased pulmonary arterial pressures and right heart failure (1). Recent work has highlighted the role of cell–matrix interactions in driving pulmonary arterial remodeling (2). In particular, matrix stiffening has been shown to occur early in the course of experimental PH (3), promoting remodeling phenotypes in pulmonary vascular cells (4, 5). Altering matrix composition through inhibition of the collagen cross-linking enzyme LOX (lysyl oxidase) or downstream mechanical signaling via inhibition of the mechanotransducers YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif) disrupts this mechanobiological feedback loop, reducing remodeling behaviors (4, 5) and ameliorating experimental PH (4, 6, 7). Despite these promising results, the primary drivers of altered matrix mechanics and composition during PH pathogenesis are complex and remain poorly understood (2).
Turnover of the extracellular matrix (ECM) is regulated by a family of zinc-dependent proteases called MMPs (matrix metalloproteinases) and endogenous inhibitors named TIMPs (tissue inhibitors of MMPs) (8). The type IV collagenases MMP-2 and MMP-9, as well as TIMP-1, are highly expressed in patients with PH (8, 9) and are elevated in the pulmonary vasculature in experimental PH models (10–12). However, modulation of matrix-altering activity through generalized MMP inhibition has generated mixed results (13, 14). These studies demonstrate the complexity of ECM turnover and suggest that further work requires a more selective approach.
MMP-8 is an interstitial collagenase that has been studied in both inflammatory and fibrotic lung disease as well as systemic vascular remodeling. MMP-8–/– mice have increased neutrophil infiltration and pulmonary edema in response to direct lung injury from LPS, but decreased inflammation during high Vt ventilation (15, 16). In bleomycin-induced lung injury, MMP-8 activity is upregulated in leukocytes and fibroblasts, with MMP-8–/– mice showing increased inflammatory but reduced profibrotic activity (17). In the cardiovascular system, MMP-8 is expressed in endothelial cells, vascular smooth muscle cells (VSMCs), and macrophages of atherosclerotic lesions (18), and elevated MMP-8 concentrations have been associated with atherosclerosis, myocardial infarction, and death (19).
Given these effects on matrix turnover, fibrotic activity, and vascular pathology in the systemic circulation, we investigated the expression and functional role of MMP-8 in PH. Our findings demonstrate that MMP-8 expression is markedly increased in plasma and pulmonary arteries (PAs) of patients with PH compared with control subjects. Absence of MMP-8 in mice leads to increased mortality, vascular remodeling, and right ventricular failure after exposure to hypoxia, indicating that MMP-8 upregulation likely represents an adaptive response to pathogenic PH stimuli. Deficiency of MMP-8 in primary murine PASMCs alters matrix and integrin expression, enhancing proliferation and migration via proremodeling mechanotransduction by FAK (focal adhesion kinase) and YAP/TAZ signaling.
Some of the results of these studies have been previously reported in the form of abstracts (20–24).
Methods
Human Samples
Blood samples were collected and plasma was isolated after informed consent was obtained from patients at the Pulmonary Hypertension Center at Brigham and Women’s Hospital (BWH) and control subjects as described (25). PAH tissue samples derived from explanted lungs of patients with group 1 PAH who underwent lung transplantation were obtained from the Pulmonary Hypertension Breakthrough Initiative (PHBI). Control lung tissue was obtained from donor lungs not suitable for transplantation from the PHBI or from surgical specimens obtained from the BWH Pathology Department. Control and PAH PASMCs were derived from explanted lungs of patients with group 1 PAH who underwent lung transplantation or from control donor lungs not suitable for transplantation and were obtained from the PHBI (5, 26). Informed consent was obtained from subjects or their legal guardians by BWH investigators or the PHBI prior to study participation. All human samples were obtained under a protocol approved by the Mass General Brigham Institutional Review Board.
Animal Studies
All animal experiments were performed in compliance with the relevant laws and guidelines as set forth by the Institutional Animal Care and Use Committees at BWH and the Lovelace Respiratory Research Institute under Institutional Animal Care and Use Committee–approved protocols.
Monocrotaline (MCT) and Sugen/hypoxia were used to induce PH in adult male Sprague-Dawley rats as described (3). Rats were injected subcutaneously with MCT (50 mg/kg) or vehicle (phosphate-buffered saline [PBS]) and harvested after 4 weeks. Alternatively, rats were injected subcutaneously with SU5416 (20 mg/kg) and exposed to hypoxia (10% O2) for 3 weeks followed by 5 weeks of normoxia. MMP-8–/– mice were generated in the mixed SVeV129 × C57BL/6 strain and backcrossed 10 generations into the pure C57BL/6 strain (17). MMP-8–/– and MMP-8+/+ male mice (8–10 weeks old) were exposed to normobaric hypoxia (10% O2) or normoxia for up to 8 weeks (27); some mice were treated twice daily with PF-562271 (25 mg/kg) during hypoxic exposure. PA banding was performed through the constriction of the PA as described (28). Mice were anesthetized with isoflurane (1–3%) for echocardiography and hemodynamic measurements via internal jugular catheterization (3, 29). Hearts were excised, and the ventricles were dissected and weighed (3, 27). Hearts and lungs were embedded in OCT or fixed in formalin for histologic and morphometric analysis. Details are provided in the online supplement.
Mouse PASMCs and In Vitro Experiments
PASMCs were isolated from adult MMP-8–/– and MMP-8+/+ mice and cultured as described (3, 27). Human control and PAH PASMCs were cultured as described (5). Experiments were performed on PASMCs at passages 6–15. RNA interference, transient transfection, flow cytometry, collagenase assay, quantitative PCR, Western blotting, ELISA, immunostaining, proliferation, and migration assays were performed as described (3, 5, 30, 31). Details are provided in the online supplement.
Statistical Analysis
Data are presented as mean ± SD unless otherwise specified. Statistical significance was determined by the Mann-Whitney U test or Wilcoxon signed-rank test for comparisons between two groups as appropriate. The Friedman test was used for comparisons between more than two matched groups with Dunn’s post hoc test for pairwise comparisons. Two-way ANOVA was used to determine significance of a response to two or more factors; post hoc test individual comparisons were obtained using Sidak’s or Tukey’s tests with adjustment for multiple comparisons. All P values were two tailed, and statistical significance was accepted as P < 0.05.
Results
MMP-8 Expression Is Increased in Plasma and PAs of Patients with PH
We examined MMP-8 expression in patients with group 1 PAH and group 3 PH compared with age-matched control subjects. Demographics, clinical characteristics, and MMP-8 levels of patients with PH and controls are shown (Table 1). Mean plasma MMP-8 levels were 18-fold higher in patients with PAH and 3-fold higher in patients with group 3 PH compared with controls (Figure 1A) and showed modest correlation with hemodynamic measures of PH severity (Table E1 in the online supplement). Immunofluorescence staining demonstrated enhanced MMP-8 expression in the PA smooth muscle and endothelium of patients with severe PAH compared with controls or those with mild PAH (P = 0.0044; post hoc test for linear trend effect size R2 = 0.6139, P = 0.0009) (Figures 1B–1D). Moreover, MMP-8 expression correlated with the degree of vascular remodeling, as assessed by α-SMA (α-smooth muscle actin) immunofluorescence staining (R2 = 0.878, P < 0.0001) (Figures E1A and E1B).
Table 1.
Demographics, Clinical Characteristics, and MMP-8 Concentrations of Patients with Pulmonary Hypertension and Controls
| Subject | Age (yr) | Sex | Race | PH Classification | Associated Condition | mPAP (mm Hg) |
PVR (dyn ⋅ s ⋅ cm−5) |
Plasma MMP-8 (pg/ml) |
|---|---|---|---|---|---|---|---|---|
| 1 | 60 | M | White | Group 1–IPAH | — | 23 | 134 | 6,394 |
| 2 | 78 | F | White | Group 1–IPAH | — | 32 | 378 | 3,367 |
| 3 | 48 | F | White | Group 1–IPAH | — | 26 | 154 | 42,032 |
| 4 | 71 | F | White | Group 1–IPAH | — | 37 | 452 | 3,742 |
| 5 | 40 | M | White | Group 1–APAH | HIV | 27 | 297 | 1,665 |
| 6 | 63 | F | White | Group 1–APAH | RA | 34 | 263 | 6,306 |
| 7 | 41 | F | White | Group 1–IPAH | — | 48 | 640 | 2,704 |
| 8 | 57 | F | Unknown | Group 1–APAH | Scleroderma | 44 | 457 | 4,329 |
| 9 | 87 | F | White | Group 1–IPAH | — | 46 | 631 | 2,124 |
| 10 | 67 | F | White | Group 1–APAH | Portal hypertension | 44 | 256 | 78,291 |
| 11 | 78 | M | White | Group 1–IPAH | — | 45 | 740 | 16,835 |
| 12 | 81 | F | White | Group 1–IPAH | — | 46 | 736 | 17,380 |
| 13 | 67 | F | White | Group 1–APAH | Portal hypertension | 46 | 333 | 419 |
| 14 | 68 | M | White | Group 1–APAH | Scleroderma | 57 | 571 | 4,837 |
| 15 | 68 | F | White | Group 1–IPAH | — | 55 | 546 | 4,710 |
| 16 | 46 | F | White | Group 1–IPAH | — | 55 | 656 | 3,761 |
| 17 | 68 | F | White | Group 1–APAH | Scleroderma | 59 | 756 | 5,522 |
| 18 | 67 | F | White | Group 2 | — | 56 | 890 | 7,601 |
| 19 | 43 | F | White | Group 4 | CTEPH | 44 | 318 | 7,967 |
| 20 | 73 | F | White | Group 4 | CTEPH | 63 | 856 | 8,336 |
| 21 | 71 | M | White | Group 3 | COPD | 29 | 273 | 2,970 |
| 22 | 64 | F | White | Group 3 | COPD | 28 | 832 | 1,955 |
| 23 | 63 | M | Unknown | Group 3 | COPD | 36 | 499 | 1,873 |
| 24 | 68 | F | White | Group 3 | COPD | 41 | 636 | 2,412 |
| 25 | 64 | M | White | Group 3 | COPD | 38 | 541 | 1,663 |
| 26 | 66 | F | White | Group 3 | COPD | 47 | 577 | 703 |
| 27 | 63 | M | White | Group 3 | COPD | 29 | 367 | 484 |
| 28 | 62 | F | White | Group 3 | COPD | 33 | 553 | 1,188 |
| 29 | 66 | F | White | Group 3 | COPD | 32 | 382 | 3,436 |
| 30 | 63 | M | White | Group 3 | COPD | 37 | 370 | 274 |
| 31 | 46 | F | White | Group 3 | IPF | 33 | 307 | 1,535 |
| 32 | 53 | F | White | Group 3 | IPF | 32 | 226 | 813 |
| 33 | 67 | F | White | Group 3 | IPF | 26 | 391 | 621 |
| 34 | 79 | F | White | Group 3 | IPF | 34 | 502 | 1,206 |
| 35 | 70 | M | White | Group 3 | IPF | 56 | 1,285 | 1,672 |
| 36 | 64 | M | White | Group 3 | IPF | 39 | 330 | 2,668 |
| 37 | 65 | M | White | Group 3 | IPF | 26 | 251 | 539 |
| 38 | 84 | F | White | Group 3 | IPF | 30 | 297 | 5,071 |
| 39 | 56 | M | White | Group 3 | IPF | 30 | 371 | 3,463 |
| 40 | 68 | M | White | Control | — | — | — | 332 |
| 41 | 59 | M | White | Control | — | — | — | 1,795 |
| 42 | 69 | M | White | Control | — | — | — | 772 |
| 43 | 57 | Unknown | Unknown | Control | — | — | — | 249 |
| 44 | 56 | M | White | Control | — | — | — | 1,649 |
| 45 | 72 | M | Black | Control | — | — | — | 254 |
| 46 | 78 | M | White | Control | — | — | — | 120 |
| 47 | 76 | M | White | Control | — | — | — | 1,067 |
| 48 | 56 | M | White | Control | — | — | — | 185 |
| 49 | 60 | Unknown | Unknown | Control | — | — | — | 484 |
| 50 | 69 | Unknown | Unknown | Control | — | — | — | 357 |
| 51 | 77 | F | White | Control | — | — | — | 543 |
Definition of abbreviations: APAH = associated with pulmonary arterial hypertension; COPD = chronic obstructive pulmonary disease; CTEPH = chronic thromboembolic pulmonary hypertension; IPAH = idiopathic pulmonary arterial hypertension; IPF = idiopathic pulmonary fibrosis; MMP-8 = matrix metalloproteinase-8; mPAP = mean pulmonary arterial pressure; PH = pulmonary hypertension; PVR = pulmonary vascular resistance; RA = rheumatoid arthritis.
Figure 1.
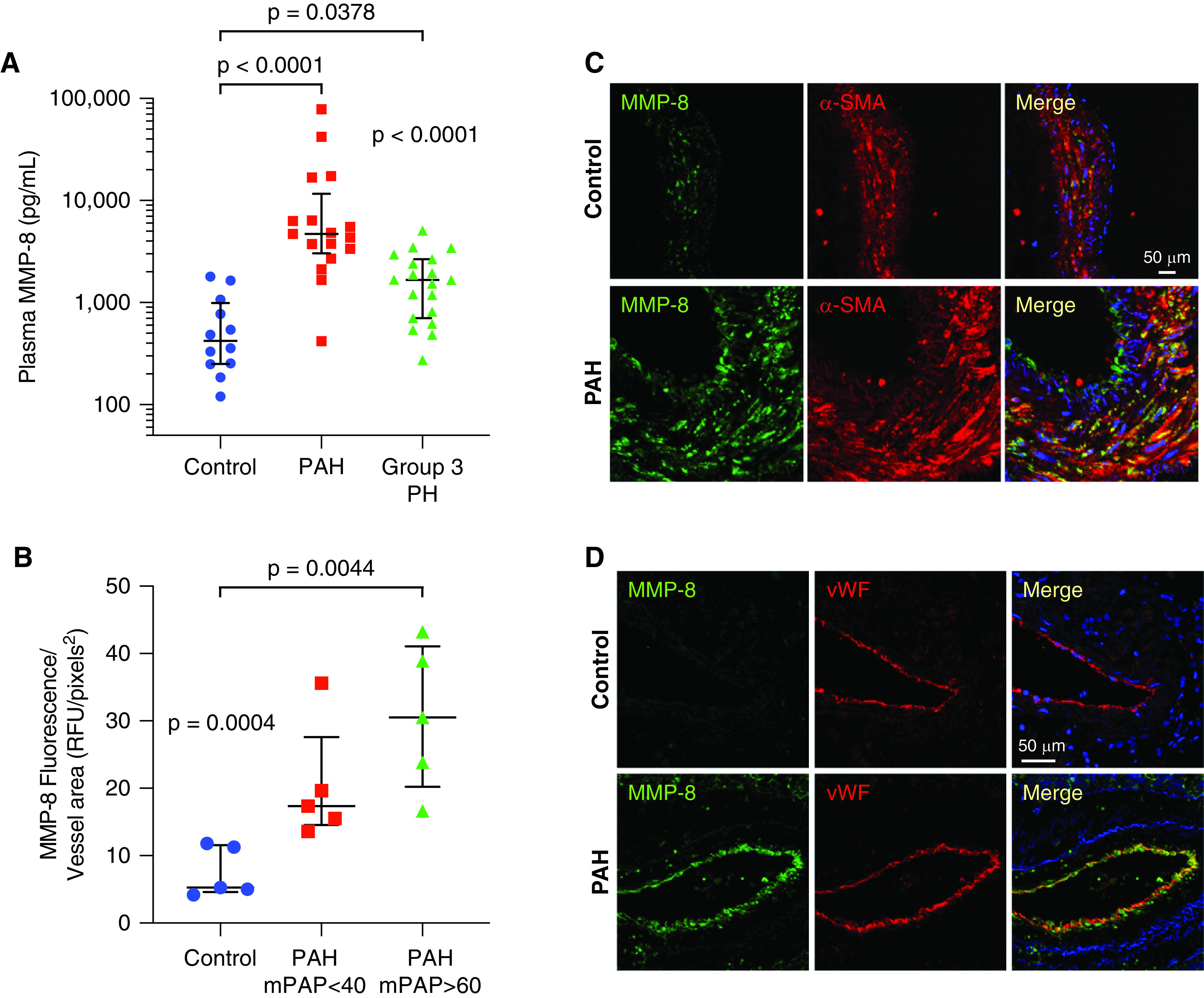
MMP-8 (matrix metalloproteinase-8) expression is increased in plasma and pulmonary arteries (PAs) of patients with pulmonary arterial hypertension (PAH). (A) MMP-8 levels were measured in plasma from patients with group 1 PAH (n = 17, 64 ± 14 yr), patients with group 3 pulmonary hypertension (PH) (n = 19, 65 ± 8 yr), and age-matched controls without PH (n = 12, 66 ± 8 yr) by ELISA. Data represent median and interquartile range (IQR). Statistical significance was determined by Kruskal-Wallis one-way ANOVA (P < 0.0001) followed by Dunn’s multiple comparisons test (P < 0.0001 group 1 PAH vs. controls, P = 0.0378 group 3 PH vs. controls). (B–D) Immunofluorescence staining for MMP-8, α-SMA (α-smooth muscle actin), and vWF (von Willebrand factor) was performed in explanted lungs from patients with group 1 PAH (mPAP < 40 mm Hg, mPAP > 60 mm Hg) who underwent lung transplantation and control lungs (n = 5 per group). (B) MMP-8 fluorescence intensity was measured in 10 PAs (50–150 μm) per patient and normalized per vessel area using Metamorph software. The mean MMP-8 fluorescence intensity per vessel area for each patient is shown with median and IQR per group. Statistical significance was determined by Kruskal-Wallis one-way ANOVA (P = 0.0004) followed by Dunn’s multiple comparisons test (P = 0.0044 control vs. PAH with mPAP > 60 mm Hg). (C and D) Representative confocal images are shown for MMP-8 (green) and α-SMA (red) immunostaining (C) and MMP-8 (green) and vWF (red) immunostaining (D). Merged images are shown on the right of each panel. Scale bars, 50 μm. mPAP = mean pulmonary arterial pressure; RFU = relative fluorescence units.
MMP-8 Expression Is Induced in the Pulmonary Vasculature in Experimental Models of PH
Sprague-Dawley rats demonstrated significantly increased expression of MMP-8 in the pulmonary vasculature 4 weeks after MCT (twofold increase, P = 0.0079) compared with PBS-treated controls (Figures 2A, 2B, and E2A–E2C). Sugen/hypoxia-exposed rats showed similar findings with robust increases in MMP-8 after 8 weeks compared with vehicle/normoxia controls (fivefold increase, P = 0.0079) (Figures 2C, 2D, and E2D–E2F). Wild-type mice exposed to hypoxia also showed significant upregulation of MMP-8 in the pulmonary vasculature at 4 and 8 weeks of hypoxia (P = 0.0013) compared with normoxic controls (Figures 2E and 2F). As in patients with PAH, MMP-8 expression correlated with pulmonary arterial muscularization in mice exposed to chronic hypoxia (R2 = 0.527, P < 0.0001) (Figures E1C and E1D).
Figure 2.
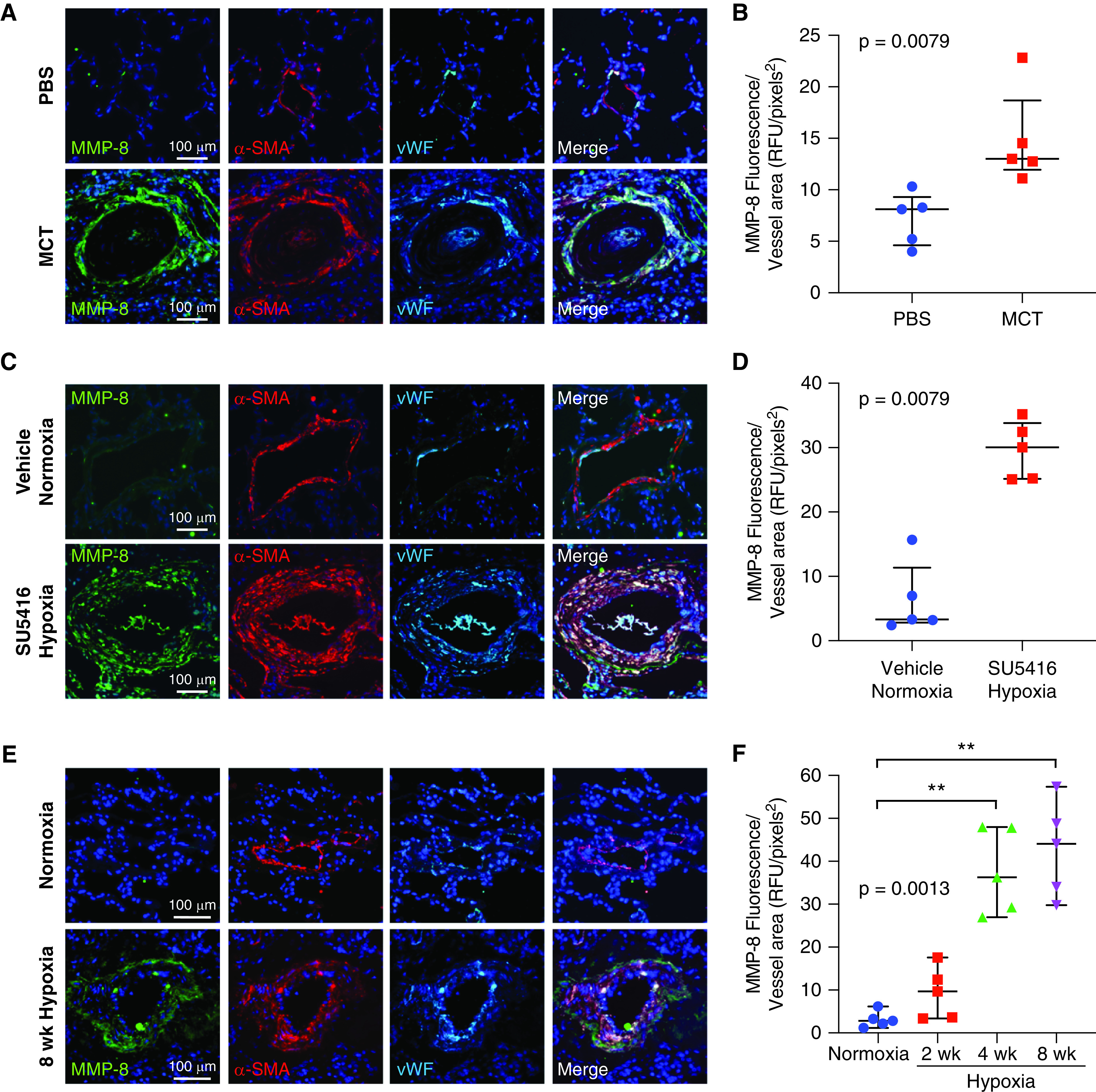
MMP-8 (matrix metalloproteinase-8) expression is induced in the pulmonary vasculature in experimental models of pulmonary hypertension. (A and B) Sprague-Dawley rats were treated with monocrotaline (MCT) or phosphate-buffered saline (PBS; n = 5 per group), and lungs were harvested after 4 weeks. (C and D) Sprague-Dawley rats were treated with SU5416 or vehicle (n = 5 per group), exposed to hypoxia or normoxia for 3 weeks, and then returned to normoxia for an additional 5 weeks. Lungs were triple immunostained for MMP-8, α-SMA (α-smooth muscle actin), and vWF (von Willebrand factor). (A and C) Representative confocal images are shown for MMP-8 (green), α-SMA (red), and vWF (cyan) immunostaining. Merged images are shown to the right of each row. Scale bars, 100 μm. (B and D) MMP-8 fluorescence intensity was measured in 10 pulmonary arteries (PAs) (50–150 μm) per rat and normalized per vessel area using Metamorph software. The mean MMP-8 fluorescence intensity per vessel area for each rat is shown with median and interquartile range (IQR) per group. Statistical significance was determined by the Mann-Whitney U test. (E and F) C57BL/6 wild-type mice were exposed to normoxia or hypoxia for 2, 4, or 8 weeks (n = 5 per group). Lungs were triple immunostained for MMP-8, α-SMA, and vWF. Representative confocal images are shown for MMP-8 (green), α-SMA (red), and vWF (cyan) immunostaining for normoxia and 8 weeks of hypoxia. Merged images are shown to the right of each row. Scale bars, 100 μm. (F) MMP-8 fluorescence intensity was measured in 10 PAs (50–150 μm) per mouse and normalized per vessel area using Metamorph software. The mean MMP-8 fluorescence intensity per vessel area for each mouse is shown with median and IQR per group. Statistical significance was determined by Kruskal-Wallis one-way ANOVA (P = 0.0013) followed by Dunn’s multiple comparisons test (**P ⩽ 0.01). RFU = relative fluorescence units.
Interestingly, human PASMCs derived from patients with PAH demonstrated similar degrees of MMP-8 expression and type I collagenase activity compared with control PASMCs (Figures E3A, E3B, and E3D), with no evidence of MMP-8 induction after short-term hypoxia exposure (Figures E3E and E3F). Secretion of MMP-8 was mildly reduced in PAH PASMCs in normoxic (Figure E3C) but not hypoxic (Figure E3G) conditions. These findings, supported by lack of MMP-8 induction in the vasculature after 2 weeks of hypoxia in mice (Figures 2F, E1C, and E1D), suggest that in vivo induction of MMP-8 requires a chronic pathogenic stimulus.
MMP-8 Deficiency Increases Mortality and PH Severity, Exaggerates Vascular Remodeling, and Impairs Right Ventricular Function
To determine the in vivo impact of MMP-8 on PH pathogenesis, MMP-8–/– and MMP-8+/+ mice were exposed to 8 weeks of normobaric hypoxia or normoxia. MMP-8–/– mice had significantly increased mortality compared with MMP-8+/+ mice (P = 0.0014) (Figure 3A). Surviving MMP-8–/– mice demonstrated more severe PH with significantly higher right ventricular systolic pressure (RVSP) after 8 weeks of hypoxia compared with MMP-8+/+ mice (P ⩽ 0.05) and normoxic controls (P ⩽ 0.0001) (Figure 3B). Similarly, MMP-8–/– mice developed more severe PH compared with MMP-8+/+ mice after 2 weeks of hypoxia (Figure E4). Histologic analysis of small and medium-sized PAs in hypoxia-exposed MMP-8–/– mice demonstrated significantly increased wall thickness by morphometry (Figures 3C and 3D) and increased muscularization by α-SMA immunostaining (Figures 3E and 3F).
Figure 3.
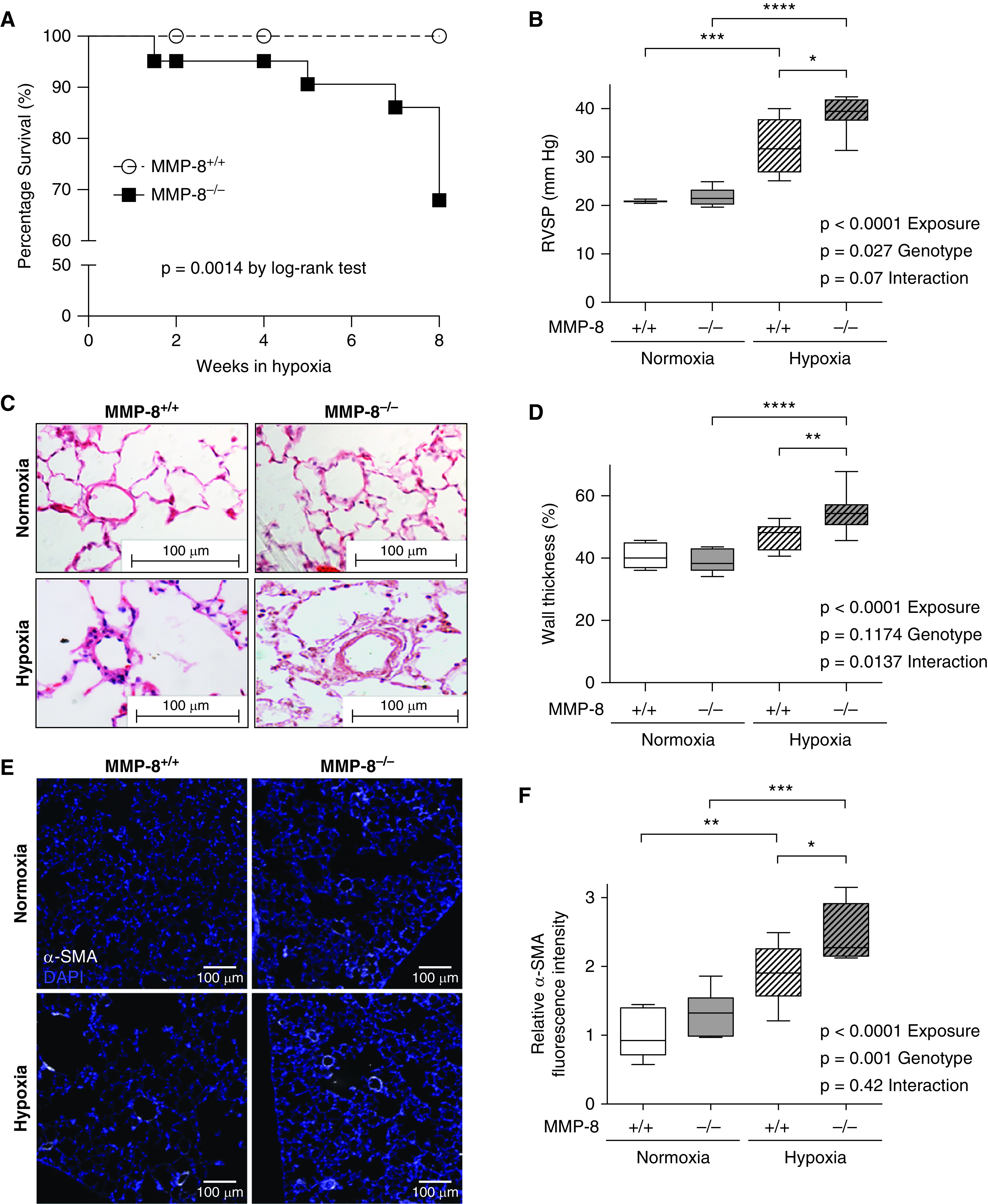
MMP-8 (matrix metalloproteinase-8) deficiency increases mortality, exaggerates vascular remodeling, and increases severity of hypoxia-induced pulmonary hypertension. (A) MMP-8+/+ and MMP-8–/– mice were exposed to normobaric hypoxia (10%) or normoxia (n = 10–15 per group), and survival was monitored over 8 weeks. Data are expressed as percentage of mice alive at each time point (P = 0.0014 by log-rank test). (B) Right ventricular systolic pressure (RVSP) was measured via jugular catheterization after 8 weeks of hypoxia or normoxia (n = 5–8 per group). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (*P ⩽ 0.05, ***P ⩽ 0.001, and ****P ⩽ 0.0001). (C) Representative hematoxylin and eosin staining and (E) α-SMA (α-smooth muscle actin) immunostaining in lungs from MMP-8+/+ and MMP-8–/– mice after normoxia and hypoxia. Scale bar, 100 μm. (D and F) Quantification of percent wall thickness (n = 5–7 per group, 10 vessels per mouse) (D) and relative α-SMA mean fluorescence intensity (F) of pulmonary arterioles <100 μm (n = 6 per group, 5 vessels per mouse). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (*P ⩽ 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001).
Pathologic examination of the right ventricle (RV) revealed significant right ventricular hypertrophy (RVH) and dramatic RV dilation in MMP-8–/– mice after hypoxia compared with MMP-8+/+ controls, indicative of worsening RV failure (Figures 4A–4C). Mild left ventricular hypertrophy also occurred (Figure E5A), but was less pronounced than the RV findings, and total body weight was not different between hypoxic MMP-8–/– and MMP-8+/+ mice (Figure E5B). Echocardiography demonstrated increased RV internal diameter (Figure 4D) and significantly reduced tricuspid annular plane systolic excursion (Figure 4E) in MMP-8–/– animals compared with MMP-8+/+ mice after hypoxia, indicating impaired RV function. LV ejection fraction was preserved and similar between groups (Figure 4F). In addition, MMP-8–/– mice did not develop systemic hypertension or severe diastolic dysfunction, as mean arterial pressures and left ventricular pressures were similar between MMP-8–/– and MMP-8+/+ mice after hypoxia (Figures E5C–E5E). Furthermore, MMP-8 did not have a direct protective effect on the RV, as both MMP-8–/– and MMP-8+/+ mice developed similar increases in RVSP, RVH, and RV dysfunction after PA banding (Figure 5). Taken together, these findings strongly suggest a protective role for MMP-8 in the pulmonary vasculature during the pathogenesis of hypoxia-induced PH.
Figure 4.
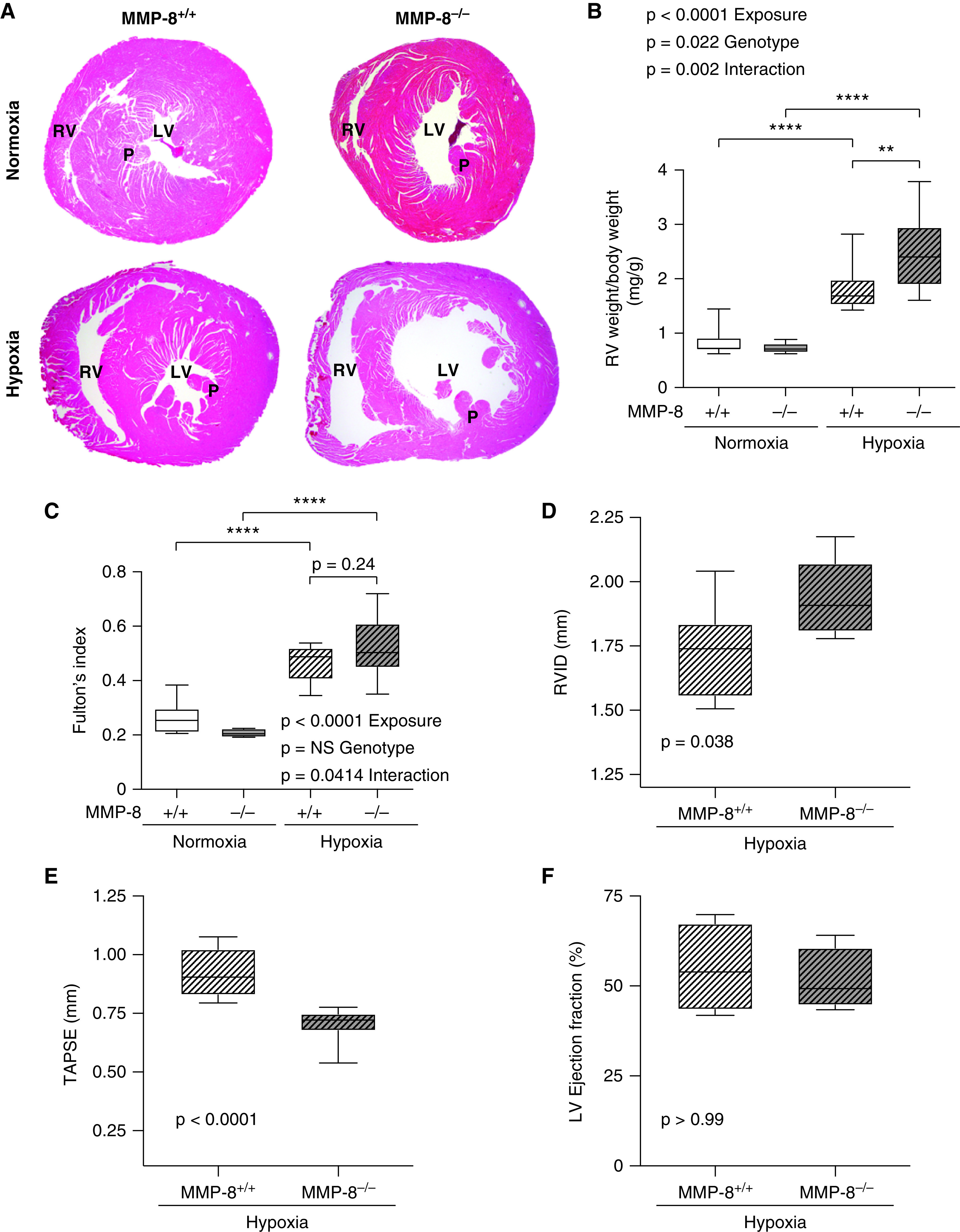
MMP-8 (matrix metalloproteinase-8) deficiency leads to RV hypertrophy and impairs RV function after chronic hypoxia. MMP-8+/+ and MMP-8–/– mice were exposed to 8 weeks of normobaric hypoxia or normoxia (n = 10–15 per group). (A) Representative cross-section images of hematoxylin and eosin–stained hearts at the level of the papillary muscles (P). (B and C) RV hypertrophy was assessed by normalizing RV weight (in milligrams) to total body weight (in grams) (B) and by assessing Fulton’s index (C). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (**P ⩽ 0.01 and ****P ⩽ 0.0001). (D–F) Echocardiography was performed, and measurements of RV internal diameter (RVID), tricuspid annulus plane systolic excursion (TAPSE), and left ventricular ejection fraction were made in MMP-8+/+ and MMP-8–/– mice after 8 weeks of hypoxia (n = 8 per group). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by the Mann-Whitney U test. There was no significant difference in left ventricular ejection fraction between MMP-8+/+ and MMP-8–/– mice after hypoxia. LV = left ventricle; NS = not significant; RV = right ventricle.
Figure 5.
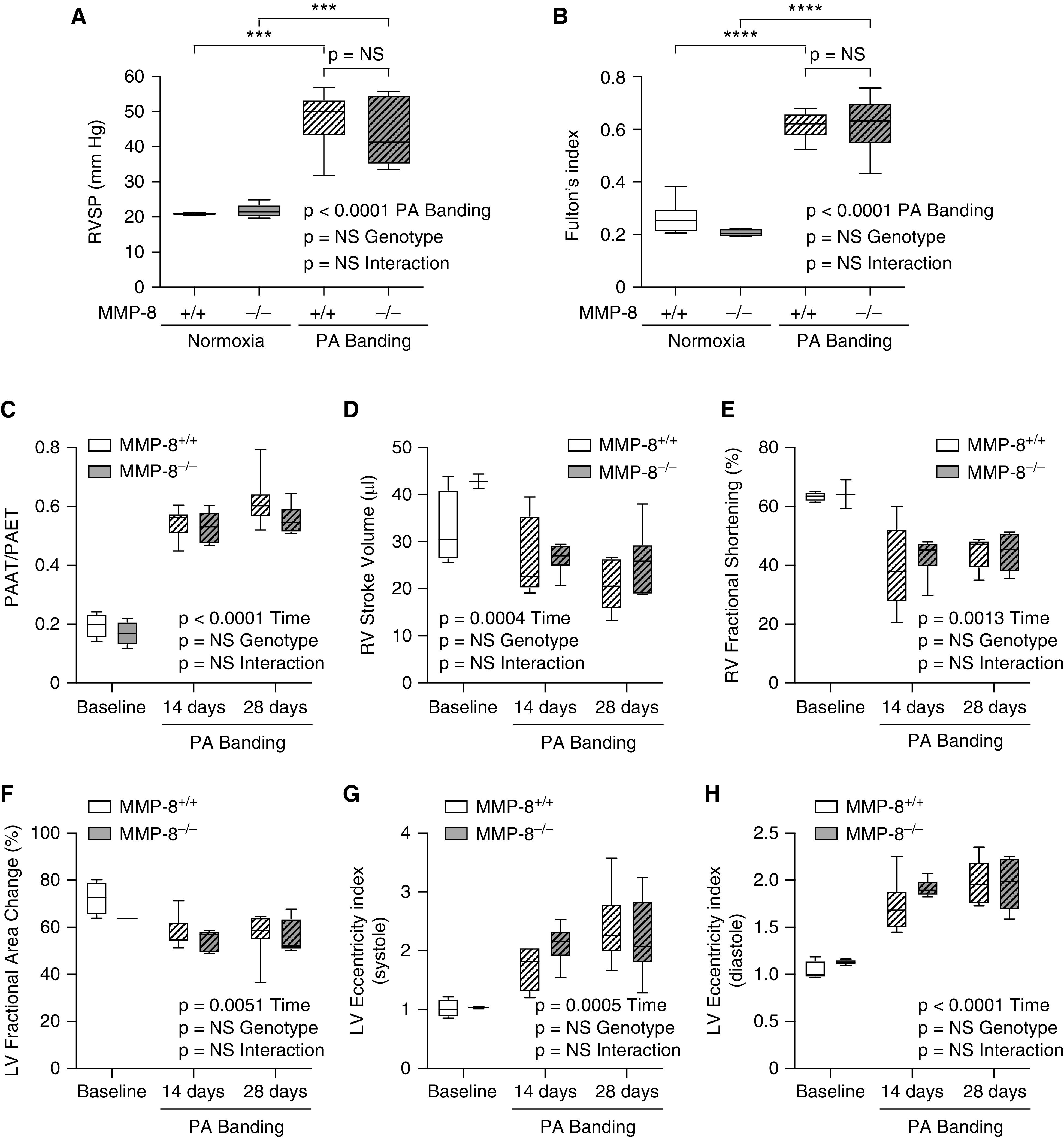
MMP-8 (matrix metalloproteinase-8) deficiency does not directly alter right ventricular (RV) function independent of its effects on the pulmonary vasculature. MMP-8+/+ and MMP-8–/– mice (n = 6) were anesthetized and subjected to pulmonary artery (PA) banding surgery using a 24-gauge needle to tie the PA ligature. (A) Right ventricular systolic pressure (RVSP) was measured via jugular catheterization after 28 days. (B) RV hypertrophy was assessed by Fulton’s index at 28 days after PA banding surgery. Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (***P ⩽ 0.001 and ****P ⩽ 0.0001). (C–H) Echocardiography was performed, and measurements of pulmonary arterial acceleration time/pulmonary arterial ejection time (PAAT/PAET) (C), RV stroke volume (D), RV fractional shortening (E), left ventricular (LV) fractional area change (F), and LV eccentricity indices (G and H) were made in MMP-8+/+ and MMP-8–/– mice at baseline, 14 days, and 28 days after PA banding surgery. Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by a repeated measures mixed-effects model followed by Holm-Šídák’s post hoc test. NS = not significant.
MMP-8 Deficiency Leads to Enhanced Proliferation and Migration of PASMCs
Immunofluorescence staining of the pulmonary vasculature in MMP-8–/– and MMP-8+/+ mice revealed a significant increase in Ki67+/α-SMA+ cells in MMP-8–/– mice after hypoxia compared with MMP-8+/+ and normoxic controls, suggesting that increased PASMC proliferation contributes to enhanced vascular remodeling and exaggerated PH in MMP-8–/– mice (Figures 6A and 6B).
Figure 6.
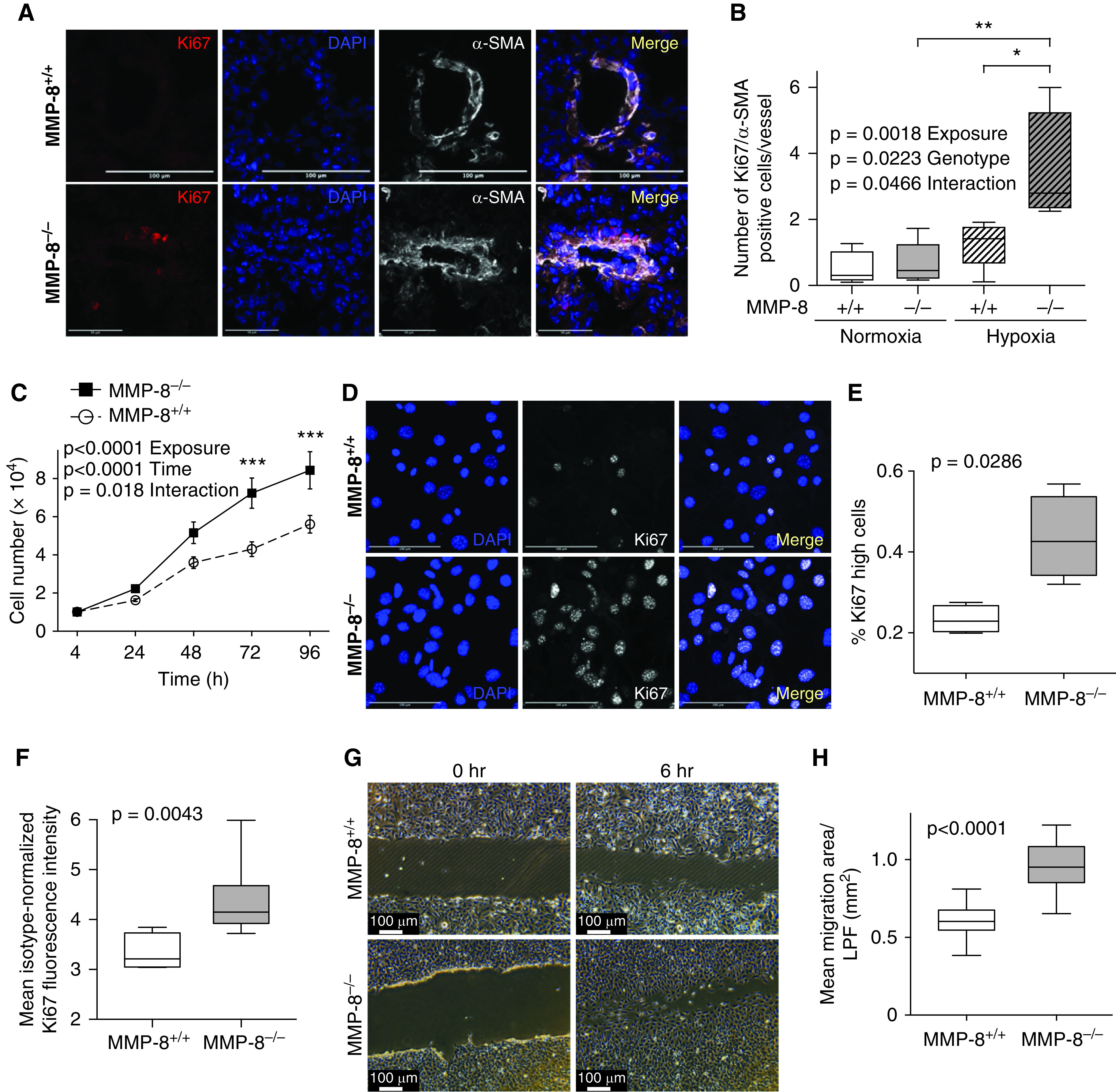
MMP-8 (matrix metalloproteinase-8) deficiency leads to enhanced proliferation and migration of pulmonary artery smooth muscle cells (PASMCs). (A and B) MMP-8+/+ and MMP-8–/– mice were exposed to 4 weeks of normobaric hypoxia or normoxia (n = 4–5 per group). Lungs were immunostained for Ki67 and α-SMA (α-smooth muscle actin), and counterstained with DAPI. (A) Representative Ki67 (red) and α-SMA (white) immunostaining in lungs from MMP-8+/+ and MMP-8–/– mice exposed to hypoxia. Merged images are shown on the right of each panel. Scale bars, 100 μm. (B) The number of Ki67+/α-SMA+ cells per vessel (<100 μm) was quantified (average of 14 vessels per mouse). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (*P ⩽ 0.05 and **P ⩽ 0.01). (C) PASMCs isolated from MMP-8+/+ and MMP-8–/– mice were serum starved for 24 hours, seeded at 10,000 cells/cm2, and subsequently grown in full serum conditions over 96 hours. Cell number was quantified by CyQUANT assay (n = 5 independent experiments). Data represent the mean ± SEM. Statistical significance was determined by two-way ANOVA followed by Sidak’s post hoc test (***P ⩽ 0.001). (D) MMP-8+/+ and MMP-8–/– PASMCs were seeded on glass slides and grown in low serum conditions (1% fetal bovine serum) for 24 hours. Cells were fixed and permeabilized, and immunostaining was performed for Ki67. Counterstaining was performed with DAPI. Representative Ki67 (white) and DAPI (blue) images of MMP-8+/+ and MMP-8–/– PASMCs. Merged images are shown on the right of each panel. Scale bars, 100 μm. (E) Quantification of the percentage of Ki67 high cells using CellProfiler in MMP-8+/+ and MMP-8–/– PASMCs (n = 4 independent experiments). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by the Mann-Whitney U test (P = 0.0286). (F) MMP-8+/+ and MMP-8–/– PASMCs were cultured in low serum conditions for 24 hours, detached using Accutase (StemPro), fixed, permeabilized, stained with anti-Ki67 fluorescein isothiocyanate, and analyzed via flow cytometry. Quantification of mean isotype-normalized Ki67 fluorescence intensity in MMP-8+/+ and MMP-8–/– PASMCs by flow cytometry (n = 5 independent experiments). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by the Mann-Whitney U test (P = 0.0043). (G and H) MMP-8+/+ and MMP-8–/– PASMCs were seeded to confluency in 6- or 12-well plates and wounded using a 1,000-μl pipette tip. Wound healing was evaluated by measuring the total area over which the cells migrated in 6 hours per low-power field (LPF; n = 18 independent experiments). Statistical significance was determined by the Mann-Whitney U test (P ⩽ 0.0001).
Based on these in vivo findings, we hypothesized that MMP-8–/– PASMCs (Figure E6) would be biased toward vascular remodeling behaviors in vitro. Under growth conditions, MMP-8–/– PASMCs demonstrated increased cell numbers over time compared with MMP-8+/+ PASMCs (sevenfold vs. fourfold at 72 h, P < 0.001) (Figure 6C). To confirm a hyperproliferative state under more stringent conditions, we assessed Ki67 immunostaining in low serum conditions. MMP-8–/– PASMCs demonstrated a marked increase in Ki67 positivity compared with control cells by immunofluorescence staining (Figures 6D, 6E, and E7A), and a skewed distribution of highly positive Ki67 cells when assessed via flow cytometry (Figures 6F, E7B, and E7C). MMP-8–/– PASMCs also demonstrated enhanced migration over 6 hours when assessed via scratch assay (P < 0.0001) (Figures 6G and 6H).
MMP-8–/– PASMCs Demonstrate Altered Matrix Protein Expression Leading to Enhanced Integrin Signaling via FAK
To assess the possible impact of MMP-8 deficiency on the contents of the extracellular matrix, we screened a panel of matrix proteins using quantitative PCR. Although MMP-8–/– PASMCs showed modest increases in expression of the structural components of the matrix, such as type I collagen and fibronectin, there was a much more profound alteration in matricellular proteins such as thrombospondin-1, tenascin-C, and osteopontin in MMP-8–/– PASMCs compared with MMP-8+/+ PASMCs (Figure 7A). We verified significant upregulation of a selection of these matrix components in MMP-8–/– PASMCs using Western blotting (Figures 7B–7D) and immunofluorescence staining (Figures 7E and 7F). In addition, we observed enhanced membrane vinculin accumulation in MMP-8–/– compared with MMP-8+/+ PASMCs (Figure 7F), suggesting activation of mechanotransduction signaling pathways in MMP-8–/– PASMCs. We next examined the expression profile of integrins, ECM receptors that facilitate mechanical signaling by coupling the actin cytoskeleton with matrix proteins. Our results showed no difference in integrin-β1 expression between MMP-8–/– and MMP-8+/+ PASMCs (Figure E8); however, we found a robust increase in surface expression (Figures 7G–7I) and total protein expression (Figures 7J and 7K) of integrin-β3 in MMP-8–/– cells. Given increased integrin-β3 expression and enhanced vinculin membrane expression in MMP-8–/– PASMCs, we next assessed FAK activity (phospho-FAKTyr397/total FAK expression) as a surrogate marker of integrin-mediated mechanotransduction. Phospho-FAKTyr397 was highly upregulated in MMP-8–/– PASMCs (Figures 7L and 7M), indicating enhanced integrin-β3 mechanosignaling in MMP-8–/– PASMCs compared with MMP-8+/+ cells. In vitro exposure of MMP-8–/– PASMCs to hypoxia did not accentuate activation of this mechanosensitive pathway or differentially activate other hypoxia-inducible pathways (PHD2, PDK1, or VEGFA) (Figure E9).
Figure 7.
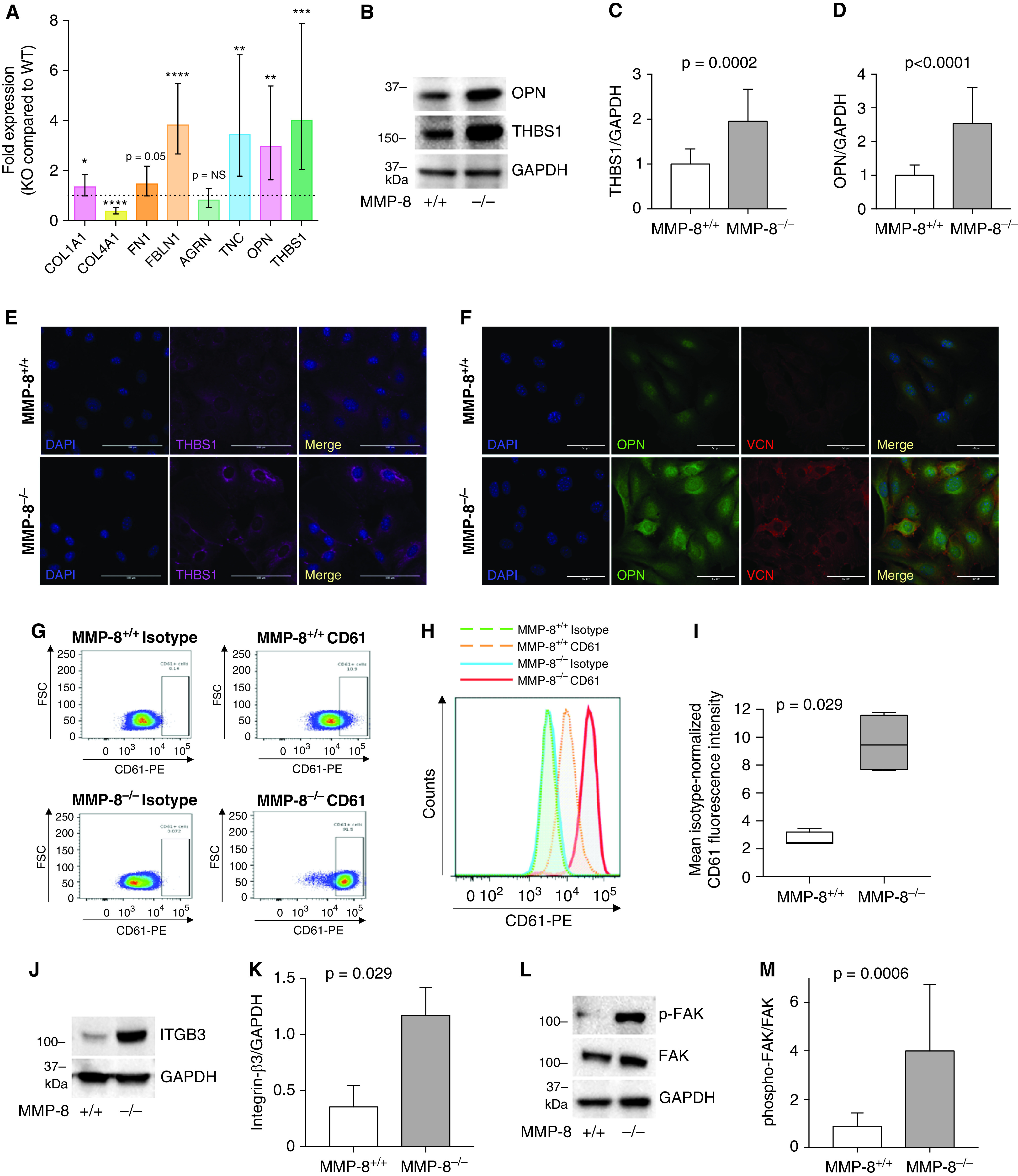
MMP-8 (matrix metalloproteinase-8)–deficient pulmonary artery smooth muscle cells (PASMCs) demonstrate altered matrix protein expression leading to enhanced integrin signaling via FAK (focal adhesion kinase). (A) RNA was isolated from MMP-8+/+ and MMP-8–/– PASMCs and quantitative PCR was performed for COL1A1, COL4A1, FN1, FBLN1, AGRN, TNC, OPN, and THBS1. Data represent mean expression ± SD in MMP-8–/– (knockout [KO]) PASMCs normalized to MMP-8+/+ (wild-type [WT]) PASMC expression (n = 8 independent experiments). Statistical significance was determined by the Wilcoxon signed-rank test using ΔΔCT values (*P ⩽ 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001). (B–D) Protein was isolated from MMP-8+/+ and MMP-8–/– PASMCs, and Western blot was performed using anti-OPN (osteopontin), anti-THBS1 (thrombospondin-1) and anti-GAPDH antibodies. Representative blots are shown. Quantification of (C) THBS1 and (D) OPN normalized to GAPDH protein expression (n = 8 independent experiments). Statistical significance was determined by the Mann-Whitney U test. (E and F) Immunofluorescence staining was performed for (E) THBS1 and (F) OPN and VCN (vinculin) in MMP-8+/+ and MMP-8–/– PASMCs, and counterstaining was performed with DAPI. Representative THBS1 (magenta), OPN (green), VCN (red), and DAPI (blue) images of MMP-8+/+ and MMP-8–/– PASMCs. Merged images are shown to the right of each row. Scale bars, 100 μm. (G–I) MMP-8+/+ and MMP-8–/– PASMCs were stained with anti-CD61-PE (integrin-β3), fixed, and analyzed via flow cytometry. (G and H) Representative dot and histogram plots for CD61 in MMP-8+/+ and MMP-8–/– PASMCs compared with isotype controls. (I) Mean isotype-normalized CD61 fluorescence intensity in MMP-8+/+ and MMP-8–/– PASMCs (n = 4 independent experiments). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by the Mann-Whitney U test (P = 0.029). (J–M) Protein was isolated from MMP-8+/+ and MMP-8–/– PASMCs, and Western blot was performed using anti–ITGB3 (integrin-β3), anti-phospho-FAK (Tyr397) (p-FAK), FAK, and anti-GAPDH antibodies. Representative blots are shown. Quantification of (K) integrin-β3 (n = 4) normalized to GAPDH protein expression and (M) p-FAK normalized to FAK (n = 7) protein expression. Statistical significance was determined by the Mann-Whitney U test. FSC = forward scatter; NS = not significant; PE = phycoerythrin.
Enhanced Proliferation and Migration in MMP-8–/– PASMCs Are Driven by Increased FAK Activity
We next assessed functional consequences of increased mechanical signaling in MMP-8–/– PASMCs. Treatment with the FAK inhibitor PF-562271 at 1 μM demonstrated abrogation of the hyperproliferative phenotype in MMP-8–/– PASMCs but had minimal effect on proliferation in MMP-8+/+ PASMCs (Figure 8A). Assessment of Ki67 by flow cytometry (Figures 8B, E7D, and E7E) and immunofluorescence staining (Figures 8C–8E and E7F) revealed profound attenuation of the hyperproliferative phenotype in MMP-8–/– PASMCs after low-dose FAK inhibition. Low-dose FAK inhibition also significantly reduced migration in MMP-8–/– PASMCs with minimal effect in MMP-8+/+ cells (Figure 8F). These effects were specific to activation of integrin-mediated FAK signaling and its downstream effectors, as inhibition of TGF-β (transforming growth factor-β) signaling, which has been linked to MMP-8 deficiency (32), did not attenuate proliferation in MMP-8–/– PASMCs despite significantly reducing proliferation in MMP-8+/+ PASMCs (Figures 8G and 8H). Moreover, in vivo treatment with PF-562271 attenuated hypoxia-induced PH, with significant reduction in RVSP, vascular remodeling, and trend toward reduced RVH in MMP-8–/–, but not MMP-8+/+, mice (Figures 8I–8K).
Figure 8.
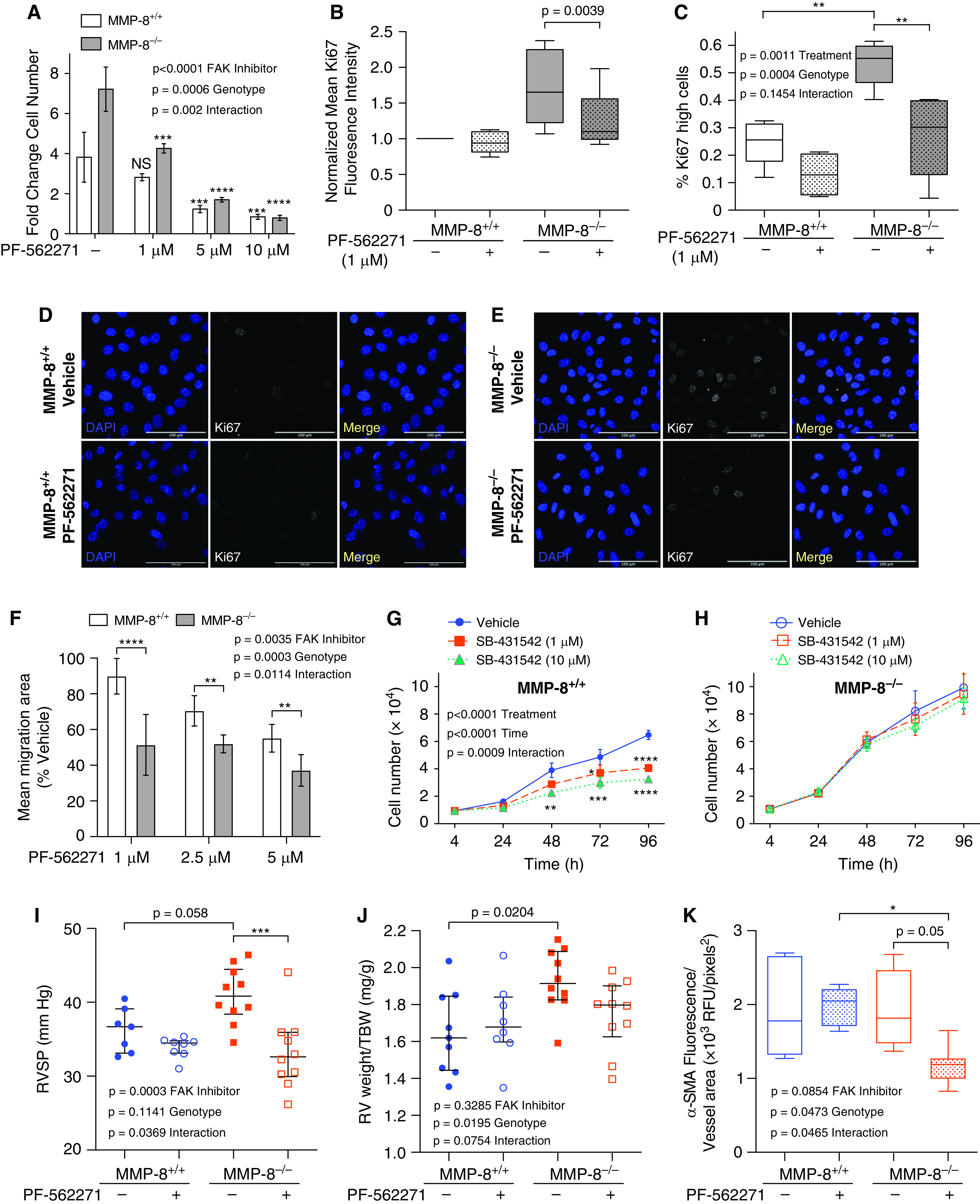
Inhibition of FAK (focal adhesion kinase) activity decreases vascular remodeling behaviors in MMP-8 (matrix metalloproteinase-8)–deficient pulmonary artery smooth muscle cells (PASMCs) and attenuates hypoxia-induced pulmonary hypertension in MMP-8–deficient mice. (A) MMP-8+/+ and MMP-8–/– PASMCs were serum starved for 24 hours followed by treatment with vehicle or PF-562271 (1, 5, and 10 μM) in full serum conditions for 48 hours. Cell number was quantified by CyQUANT assay (n = 2–4 independent experiments). Data represent the mean fold change ± SD. Statistical significance was determined by two-way ANOVA followed by Sidak’s post hoc test (***P ⩽ 0.001 and ****P ⩽ 0.0001 compared with vehicle-treated respective controls). (B–E) MMP-8+/+ and MMP-8–/– PASMCs were serum starved for 12 hours followed by treatment with vehicle or PF-562271 (1 μM) in low serum conditions for 12 hours. (B) Cells were fixed and permeabilized, stained with anti-Ki67 fluorescein isothiocyanate, and analyzed via flow cytometry. Quantification of mean isotype-normalized Ki67 fluorescence intensity in vehicle-treated and PF-562271–treated MMP-8+/+ and MMP-8–/– PASMCs (n = 9 independent experiments) by flow cytometry. Data are normalized to vehicle-treated MMP-8+/+ PASMCs and represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by the Wilcoxon matched-pairs signed-rank test (P = 0.0039). (C–E) Cells were fixed and permeabilized, and immunostaining was performed for Ki67. (C) Quantification of the percentage of Ki67 high cells using CellProfiler in vehicle-treated and PF-562271–treated MMP-8+/+ and MMP-8–/– PASMCs (n = 4–5 independent experiments). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (**P ⩽ 0.01). (D and E) Representative Ki67 immunostaining (white) and DAPI (blue) images of vehicle-treated and PF-562271–treated (D) MMP-8+/+ and (E) MMP-8–/– PASMCs. Merged images are shown to the right of each row. Scale bars, 100 μm. (F) MMP-8+/+ and MMP-8–/– PASMCs were grown to confluency, wounded using a 1,000-μl pipette tip, and treated with vehicle or PF-562271 (1, 2.5, and 5 μM) under full serum conditions. Wound healing was evaluated by measuring the total area over which the cells migrated in 6 hours per low-power field (n = 9 independent experiments) and normalized to vehicle-treated control cells. Statistical significance was determined by two-way ANOVA followed by Sidak’s post hoc test (**P ⩽ 0.01 and ****P ⩽ 0.0001). (G) MMP-8+/+ and (H) MMP-8–/– PASMCs were serum starved for 24 hours followed by treatment with vehicle or SB-431542 (1 and 10 μM) in full serum conditions for 96 hours. Cell number was quantified by CyQUANT assay (n = 3 independent experiments). Data represent the mean ± SEM. Statistical significance was determined by two-way ANOVA followed by Sidak’s post hoc test (*P ⩽ 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001). There was no significant difference in proliferation in SB-431542–treated MMP-8–/– PASMCs compared with vehicle-treated MMP-8–/– PASMCs. (I–K) MMP-8+/+ and MMP-8–/– mice were treated with vehicle or PF-562271 (25 mg/kg) by oral gavage twice daily and exposed to normobaric hypoxia (10%) (n = 8–10 per group) for 5 weeks. (I) Right ventricular systolic pressure (RVSP) was measured via jugular catheterization. Data represent median and interquartile range (IQR). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (***P ⩽ 0.001). (J) Right ventricular (RV) hypertrophy was assessed by normalizing RV weight (mg) to total body weight (TBW; g). Data represent median and IQR. Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test. (K) Immunostaining was performed for α-SMA (α-smooth muscle actin) in lungs from vehicle-treated and PF-562271–treated MMP-8+/+ and MMP-8–/– hypoxia-exposed mice, and α-SMA mean fluorescence intensity was quantified in pulmonary arterioles less than 100 μm (average 10 vessels per mouse). Data represent 25th–75th percentiles (box), median (line), and minimum and maximum values (whiskers). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (*P ⩽ 0.05). NS = not significant; RFU = relative fluorescence units.
YAP/TAZ Activation Downstream of FAK Is Required for Enhanced Proliferation and Migration of PASMCs in the Absence of MMP-8 Activity
We have previously demonstrated that activation of the transcriptional regulators YAP and TAZ is required for matrix stiffness–induced mechanical signaling and proremodeling phenotypes in PASMCs (5). As YAP/TAZ are known to be activated downstream of FAK (33, 34), we assessed YAP/TAZ transcriptional activity in MMP-8–/– PASMCs. YAP/TAZ activity was markedly increased (approximately fivefold, P < 0.0001) in MMP-8–/– versus MMP-8+/+ PASMCs when assessed via TEA domain (TEAD) promoter assay (Figure 9A), a finding corroborated by increased YAP/TAZ nuclear translocation (Figures 9B–9E), and the transcription of multiple genes (ANKRD1, CTGF, Cyr61, and PAI-1) known to be regulated by YAP/TAZ activation (Figure 9F). Interestingly, hypoxia did not further enhance activation of the YAP/TAZ pathway in MMP-8–/– PASMCs (Figure E9C). At 1–2.5 μM concentrations, PF-562271 dramatically reduced YAP/TAZ-dependent gene activation (Figure 9G), as well as YAP/TAZ nuclear translocation in MMP-8–/– PASMCs but not MMP-8+/+ cells (Figures 9H and E10A), indicating YAP/TAZ activation is FAK-dependent in the absence of MMP-8. Moreover, YAP/TAZ activation in MMP-8–/– PASMCs did not appear to be dependent on other upstream activators, including AMPK and mTORC1, p38 MAPK, AKT, and ERK, as these pathways were similarly expressed in MMP-8–/– and MMP-8+/+ PASMCs (Figure E11).
Figure 9.
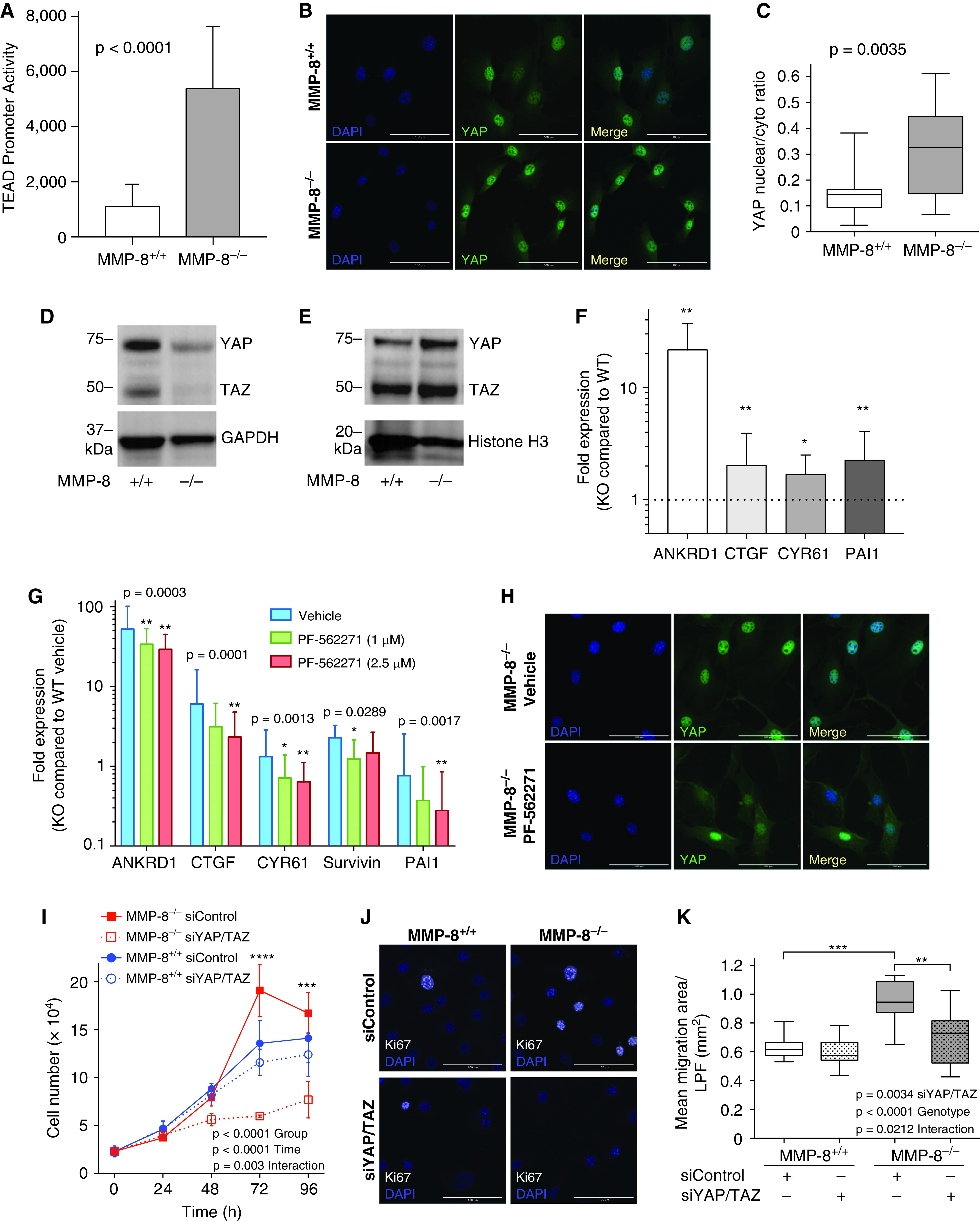
YAP (Yes-associated protein)/TAZ (transcriptional coactivator with PDZ-binding motif) activation downstream of FAK (focal adhesion kinase) is required for enhanced proliferation and migration of pulmonary artery smooth muscle cells (PASMCs) in the absence of MMP-8 (matrix metalloproteinase-8) activity. (A) MMP-8+/+ and MMP-8–/– PASMCs were cotransfected with a YAP/TAZ-responsive TEAD promoter reporter driving luciferase expression and a β-galactosidase–expressing reporter construct. TEAD promoter activity is expressed as the ratio of luciferase luminescence and β-galactosidase absorbance. Data represent mean and SD (n = 9 independent experiments). Statistical significance was determined by the Mann-Whitney U test (P ⩽ 0.0001). (B) Immunofluorescence staining was performed for YAP in MMP-8+/+ and MMP-8–/– PASMCs, and counterstaining was performed with DAPI. Representative YAP (green) immunostaining and DAPI (blue) images of MMP-8+/+ and MMP-8–/– PASMCs. Merged images are shown on the right of each panel. Scale bars, 100 μm. (C) Quantification of the ratio of YAP nuclear/cytoplasmic expression in MMP-8+/+ and MMP-8–/– PASMCs was performed using CellProfiler (4 high-power fields per experiment, n = 5 independent experiments). Statistical significance was determined by the Mann-Whitney U test (P = 0.0035). (D) Cytoplasmic and (E) nuclear protein extracts were isolated from MMP-8+/+ and MMP-8–/– PASMCs, and Western blot was performed using anti-YAP/TAZ, anti-GAPDH, and anti-histone H3 antibodies. Representative blots are shown. (F) RNA was isolated from MMP-8+/+ and MMP-8–/– PASMCs, and quantitative PCR was performed for YAP/TAZ target genes ANKRD1, CTGF, CYR61, and PAI-1. Data represent mean expression ± SD in MMP-8–/– (knockout [KO]) PASMCs and are normalized to MMP-8+/+ (wild-type [WT]) PASMC expression (n = 9–11 independent experiments). Statistical significance was determined by the Wilcoxon signed-rank test on ΔΔCT values (*P ⩽ 0.05 and **P ⩽ 0.01). (G and H) MMP-8+/+ and MMP-8–/– PASMCs were treated with vehicle or PF-562271 (1 and 2.5 μM) for 12 hours. (G) RNA was isolated, and quantitative PCR was performed for YAP/TAZ target genes ANKRD1, CTGF, CYR61, survivin, and PAI-1. Data represent mean expression ± SD in MMP-8–/– (KO) PASMCs and are normalized to vehicle-treated MMP-8+/+ (WT) PASMCs (n = 5 independent experiments). Statistical significance was determined by the Friedman test with Dunn’s post hoc test on ΔΔCT values (*P ⩽ 0.05 and **P ⩽ 0.01 compared with vehicle-treated MMP-8–/– PASMCs). (H) Immunofluorescence staining was performed for YAP in vehicle-treated and PF-562271–treated MMP-8+/+ and MMP-8–/– PASMCs. Counterstaining was performed with DAPI. Representative YAP (green) immunostaining and DAPI (blue) images in vehicle-treated and PF-562271–treated MMP-8–/– PASMCs. Merged images are shown on the right of each panel. Scale bars, 100 μm. (I–K) MMP-8+/+ and MMP-8–/– PASMCs were serum starved for 24 hours, transfected with control siRNA (siControl) or siRNA to YAP and TAZ (siYAP/TAZ), and subsequently grown in full serum conditions over 96 hours. (I) Cell number was quantified by CyQUANT assay (n = 3 independent experiments). Data represent the mean ± SEM. Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (***P ⩽ 0.001 and ****P ⩽ 0.0001 in MMP-8–/– PASMCs siControl vs. siYAP/TAZ). (J) Immunofluorescence staining for Ki67 and DAPI counterstaining was performed in siControl- and siYAP/TAZ-transfected MMP-8+/+ and MMP-8–/– PASMCs 48 hours after transfection. Representative merged images of Ki67 (white) and DAPI (blue) staining in siControl- and siYAP/TAZ-transfected MMP-8+/+ and MMP-8–/– PASMCs. Scale bars, 100 μm. (K) siControl- and siYAP/TAZ-transfected MMP-8+/+ and MMP-8–/– PASMCs were wounded using a 1,000-μl pipette tip 72 hours after transfection. Wound healing was evaluated by measuring the total area over which the cells migrated in 6 hours per low-power field (LPF; n = 9 independent experiments). Statistical significance was determined by two-way ANOVA followed by Tukey’s post hoc test (**P ⩽ 0.01 and ***P ⩽ 0.001).
Given increased YAP/TAZ signaling in MMP-8–/– PASMCs, we further assessed the role of YAP/TAZ in remodeling phenotypes using siRNA knockdown (Figure E12). YAP/TAZ knockdown significantly inhibited proliferation of MMP-8–/– PASMCs while having minimal effect in MMP-8+/+ cells (Figure 9I), indicating that YAP/TAZ signaling mediates PASMC proliferation in the absence of MMP-8. Similarly, YAP/TAZ knockdown reduced the number of strongly Ki67 positive MMP-8–/– PASMCs, while having minimal effect on Ki67 levels in MMP-8+/+ cells (Figures 9J and E10B). Genetic silencing of YAP/TAZ activity also significantly attenuated migration in MMP-8–/–, but not MMP-8+/+, PASMCs (Figure 9K).
In summary, MMP-8–/– PASMCs demonstrate robust activation of YAP/TAZ signaling downstream of integrin-mediated FAK signaling. This mechanical signaling axis acts as a primary driver of a hyperproliferative phenotype, which may explain increased vascular remodeling and exaggerated PH development in the setting of MMP-8 deficiency in response to hypoxia in vivo.
Discussion
This study identifies a novel protective role for MMP-8 in the pathobiology of PH. MMP-8 is an intriguing target based not only on its function in inflammation, ECM turnover, and fibrosis (15–17), but also for its role as a marker in cardiopulmonary disease (19, 35). MMP-8 has been shown to be upregulated in the lung after MCT (36); however, its role in PH has otherwise not been well defined. Here, we demonstrate that MMP-8 is markedly upregulated in both the plasma and PAs of patients with severe PAH, as well as in three different rodent PH models. Pulmonary vascular expression of MMP-8 correlates with disease severity in patients with PAH, PH development in mice exposed to hypoxia, and measures of vascular remodeling in both.
MMP-8 deficiency in mice leads to decreased survival and severe PH in response to chronic hypoxia, with exaggerated vascular remodeling and RV dysfunction. These findings strongly implicate upregulation of MMP-8 as an adaptive response during PH pathogenesis. Upregulation of adaptive processes during disease pathogenesis is a common phenomenon exemplified by other cardiovascular proteins including HO-1 in vascular disease, natriuretic peptides in heart failure, and LC3B in PH (37–39). The mortality observed in MMP-8–/– mice in chronic hypoxia is somewhat out of proportion to their RVSP, particularly given the pressures achieved during PA banding. Although survivorship bias may partly explain this, we cannot exclude the possibility that hypoxemia, arrhythmia, or cardiac ischemia may be contributing to mortality in this setting. Further investigation of the interplay between hypoxemia, MMP-8 deficiency, and cardiac remodeling may be revealing.
Our study also demonstrates that absence of MMP-8 in PASMCs leads to marked hyperproliferation and enhanced migration, increased expression of several integrin-interacting matrix components, and robust expression of integrin-β3. Downstream of matrix–integrin interactions, we show that hyperproliferation and hypermotility in MMP-8–/– PASMCs are dependent on mechanical signaling through FAK-YAP/TAZ activation (Figure E13).
Cell–matrix interactions provide critical cues for regulation of proliferation, migration, and contractile forces in vascular cells (2). We have demonstrated that these remodeling phenotypes are modulated via mechanotransduction after alteration in the stiffness of the underlying substrate (3, 5). Prior studies have shown that elastases (40) and MMPs (41) can also induce PASMC mitogenic activity directly through release of mitogens, exposure of hidden integrin binding sites, or mitogen receptor activation (40, 41). Additionally, MMP activity can reveal hidden integrin-β3 binding sites, triggering a feedforward loop of integrin-β3 activation via upregulation of tenascin-C expression and deposition (41).
Multiple integrin-interacting matricellular proteins such as tenascin-C are linked to PH development, including osteopontin and thrombospondin-1. Elevated osteopontin expression is a disease marker with prognostic value in PH (42, 43), and has mitogenic and promigratory effects on PASMCs (44) and adventitial fibroblasts (45). Thombospondin-1 is elevated in human PH (46–48), correlates with PH severity and cardiac output (47), and is highly expressed in distal PAs in end-stage disease (49). The potential pathogenic roles of thrombospondin-1 in PH are complex (50) but include induction of PASMC migration and proliferation via integrin-β3 interactions (51, 52). Our finding that absence of MMP-8 drives upregulation of multiple matricellular components important in PH pathogenesis highlights the intricacy of cell–matrix crosstalk in PH. Undoubtedly, these interactions set into motion multiple pathogenic mechanisms, but here we focus on altered integrin signaling and its downstream consequences.
Regulation of integrin signaling by MMPs has been well established. Gross inhibition of MMP activity in cancer cell lines decreases traction forces, cell rounding, and cell softening via perturbed integrin-β1 levels and localization (53). This may be due to absence of exposed integrin binding sites generated by collagen cleavage, as observed in a study of MMP-mediated integrin-β3 activation (41). In contrast, TIMP-1 has been found to activate integrin-β1 via CD63 (54, 55), and MMP-8 has been shown to negatively regulate integrin-β1 signaling in cancer cell lines (56). These data are more consistent with our finding that MMP-8 deficiency enhances integrin signaling, although we demonstrate this effect is not mediated by integrin-β1, but rather by integrin-β3. Integrin-β3 has a well-known role in SMC migration, particularly chemotaxis (57), and recent work shows that integrin-β3 is more greatly associated with actin stress fibers than integrin-β1 and is critical for transduction of shape signals in SMCs (33). These signals are transmitted in part through interaction with Merlin, an important regulator of YAP/TAZ activity (58).
Recently, there has been much interest in regulation of YAP/TAZ activity downstream of integrin and FAK signaling. Mechanotransduction via this pathway is likely due to both canonical Hippo pathway suppression via Merlin and a RhoA, tension, and actin-stress fiber-dependent mechanism that likely acts simultaneously and synergistically (34). In fact, YAP nuclear localization has been found to be highly dependent on intact cytoskeletal-mediated stress and strain transmission to the nucleus, which allows nuclear flattening and increased pore exposure for YAP/TAZ transport (59). As is typical in these systems, there is significant feedforward regulation, with YAP activation as a critical driver of focal adhesion protein transcription and required for focal adhesion assembly (33). Through these overlapping and synergistic mechanisms, enhanced integrin activity has been found to be a critical driver of YAP/TAZ activation. Here we find that a change in matrix composition driven by MMP-8 deficiency is sufficient to enhance integrin and FAK signaling upstream of YAP/TAZ activity. As we have previously established (5), YAP/TAZ activation is a critical driver of PASMC remodeling phenotypes in vitro and is likely a key mechanoregulator in PH pathogenesis.
Our findings differ in some respects from studies of MMP-8 in different contexts. Astrom and colleagues found increased TGF-β1 expression and activity in MMP-8–/– fibroblasts (32), whereas we found that hyperproliferation in MMP-8–/– PASMCs is TGF-β independent. We suspect that this phenotype may be cell-type specific, as the effect of MMP-8 in collagen breakdown may predominate in fibroblasts given their high collagen expression and secretion compared with SMCs. Our results also differ from those of Xiao and colleagues, who showed that MMP-8 deficiency decreased proliferation and migratory capacity in ApoE–/– VSMCs (60). This may be due to absence of ApoE, which can affect proliferation and migratory capacity (61).
Limitations of this study include the possibility that the mechanism underlying the MMP-8–deficient PH phenotype is indirect. Matrix composition is controlled in part by the balance between multiple MMPs and their endogenous inhibitors (8), thus MMP-8 deficiency is likely to have significant second-order effects. Indeed, our results suggest that a complex alteration in multiple matricellular components may be responsible for enhanced integrin-β3 activity in MMP-8–/– PASMCs. Despite these second-order effects, the loss of MMP-8 is a specific driver in the pulmonary vasculature, as MMP-9–/– mice do not have a similar PH phenotype (data not shown). It is also possible that additional pulmonary vascular cell types may play a key role driving the severe PH phenotype in MMP-8–/– mice. Although PASMC proliferation was striking in these mice after hypoxia exposure, MMP-8 expression was induced by hypoxia in the endothelium as well, warranting further study.
Future extensions of this work include additional characterization of upstream matrix alterations induced by MMP-8 deficiency in vitro and in vivo, such as soluble matrisome analysis. Future studies may also focus on enhancing the protective effects of MMP-8 by increasing substrate availability (for example through collagen cross-linking inhibitors) or altering downstream signaling (such as integrin-αvβ3 inhibition). Development of more potent YAP/TAZ inhibitors, along with appropriate lung targeting, may also allow interruption of mechanobiological feedback and have ameliorative effects on vascular remodeling in PH.
Conclusions
MMP-8 is a novel protective factor induced during PH development that acts in opposition to pathologic mechanobiological feedback by altering matrix composition and suppressing mechanical signaling by integrin-β3, FAK, and YAP/TAZ. Our data suggest therapeutic potential in enhancing or mimicking MMP-8 activity to disrupt pathologic mechanical signaling during PH pathogenesis.
Acknowledgments
Acknowledgement
The authors thank Dr. Daniel Tschumperlin and Dr. Mark Perrella for helpful discussions. They also thank Dr. Xaralabos (Bob) Varelas for helpful discussions and for providing the TEAD promoter reporter construct. They thank Dr. Stephen Walsh for biostatistical assistance, as well as Laura Aspelund and Dr. Dezhao Liu for technical assistance. They also thank Dr. Suzana Zorca for assistance with the echocardiogram analyses. They thank Dr. Sudeshna Fisch and Souen Ngoy from the MGB Cardiovascular Physiology Core for helpful discussions and assistance with echocardiograms and cardiovascular procedures.
Footnotes
Supported by funding from NIH/NHLBI, including T32HL007633, F32HL131228, K08HL143197, R03HL115106, R01HL114839, R01HL137366, and R01HL147059, as well as funding from the American Heart Association (17GRNT33660449). The Pulmonary Hypertension Breakthrough Initiative is supported by NHLBI (R24HL123767) and the Cardiovascular Medical Research and Education Fund.
Author Contributions: P.B.D., C.M.H., and R.R. contributed to the experimental conception and design of the work; acquisition, analysis, and interpretation of the data; and writing of the manuscript. A.M.C., F.P., A.M.F.C., S.K.V., I.C., S.H.V., and R.F.P. contributed to the acquisition, analysis, and/or interpretation of the data. A.B.W. and L.M.S. provided human plasma and tissue samples. D.L. and C.A.O. contributed to the experimental conception and design of the work and interpretation of the data. L.E.F. supervised the work and contributed to the experimental conception and design of the work; analysis and interpretation of the data; and writing of the manuscript. All authors contributed to drafting the work or revising it critically for important intellectual content and gave final approval of the version to be published.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202108-1863OC on September 22, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Malenfant S, Neyron AS, Paulin R, Potus F, Meloche J, Provencher S, et al. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ . 2013;3:278–293. doi: 10.4103/2045-8932.114752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dieffenbach PB, Maracle M, Tschumperlin DJ, Fredenburgh LE. Mechanobiological feedback in pulmonary vascular disease. Front Physiol . 2018;9:951. doi: 10.3389/fphys.2018.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu F, Haeger CM, Dieffenbach PB, Sicard D, Chrobak I, Coronata AM, et al. Distal vessel stiffening is an early and pivotal mechanobiological regulator of vascular remodeling and pulmonary hypertension. JCI Insight . 2016;1:e86987. doi: 10.1172/jci.insight.86987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep . 2015;13:1016–1032. doi: 10.1016/j.celrep.2015.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dieffenbach PB, Haeger CM, Coronata AMF, Choi KM, Varelas X, Tschumperlin DJ, et al. Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ-mediated repression of cyclooxygenase-2. Am J Physiol Lung Cell Mol Physiol . 2017;313:L628–L647. doi: 10.1152/ajplung.00173.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerr JS, Ruppert CL, Tozzi CA, Neubauer JA, Frankel HM, Yu SY, et al. Reduction of chronic hypoxic pulmonary hypertension in the rat by an inhibitor of collagen production. Am Rev Respir Dis . 1987;135:300–306. doi: 10.1164/arrd.1987.135.2.300. [DOI] [PubMed] [Google Scholar]
- 7. Nave AH, Mižíková I, Niess G, Steenbock H, Reichenberger F, Talavera ML, et al. Lysyl oxidases play a causal role in vascular remodeling in clinical and experimental pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol . 2014;34:1446–1458. doi: 10.1161/ATVBAHA.114.303534. [DOI] [PubMed] [Google Scholar]
- 8. Chelladurai P, Seeger W, Pullamsetti SS. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur Respir J . 2012;40:766–782. doi: 10.1183/09031936.00209911. [DOI] [PubMed] [Google Scholar]
- 9. Benisty JI, Folkman J, Zurakowski D, Louis G, Rich S, Langleben D, et al. Matrix metalloproteinases in the urine of patients with pulmonary arterial hypertension. Chest . 2005;128:572S. doi: 10.1378/chest.128.6_suppl.572S. [DOI] [PubMed] [Google Scholar]
- 10. Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, et al. Role of the TGF-β/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med . 2008;177:896–905. doi: 10.1164/rccm.200707-1083OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frisdal E, Gest V, Vieillard-Baron A, Levame M, Lepetit H, Eddahibi S, et al. Gelatinase expression in pulmonary arteries during experimental pulmonary hypertension. Eur Respir J . 2001;18:838–845. doi: 10.1183/09031936.01.00084601. [DOI] [PubMed] [Google Scholar]
- 12. Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest . 2000;105:21–34. doi: 10.1172/JCI6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vieillard-Baron A, Frisdal E, Eddahibi S, Deprez I, Baker AH, Newby AC, et al. Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer or doxycycline aggravates pulmonary hypertension in rats. Circ Res . 2000;87:418–425. doi: 10.1161/01.res.87.5.418. [DOI] [PubMed] [Google Scholar]
- 14. Vieillard-Baron A, Frisdal E, Raffestin B, Baker AH, Eddahibi S, Adnot S, et al. Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer limits monocrotaline-induced pulmonary vascular remodeling in rats. Hum Gene Ther . 2003;14:861–869. doi: 10.1089/104303403765701150. [DOI] [PubMed] [Google Scholar]
- 15. Albaiceta GM, Gutierrez-Fernández A, García-Prieto E, Puente XS, Parra D, Astudillo A, et al. Absence or inhibition of matrix metalloproteinase-8 decreases ventilator-induced lung injury. Am J Respir Cell Mol Biol . 2010;43:555–563. doi: 10.1165/rcmb.2009-0034OC. [DOI] [PubMed] [Google Scholar]
- 16. Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 α to reduce acute lung inflammation and injury in mice. J Immunol . 2010;184:1575–1588. doi: 10.4049/jimmunol.0900290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Craig VJ, Quintero PA, Fyfe SE, Patel AS, Knolle MD, Kobzik L, et al. Profibrotic activities for matrix metalloproteinase-8 during bleomycin-mediated lung injury. J Immunol . 2013;190:4283–4296. doi: 10.4049/jimmunol.1201043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation . 2001;104:1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 19. Kormi I, Nieminen MT, Havulinna AS, Zeller T, Blankenberg S, Tervahartiala T, et al. Matrix metalloproteinase-8 and tissue inhibitor of matrix metalloproteinase-1 predict incident cardiovascular disease events and all-cause mortality in a population-based cohort. Eur J Prev Cardiol . 2017;24:1136–1144. doi: 10.1177/2047487317706585. [DOI] [PubMed] [Google Scholar]
- 20. Mallarino Haeger C, Coronata AMF, Chrobak I, Polverino F, Vitali SH, Padera RF, et al. Deficiency of MMP-8 exacerbates hypoxia-induced pulmonary hypertension [abstract] Am J Respir Crit Care Med . 2015;191:A1925. [Google Scholar]
- 21. Dieffenbach PB, Mallarino Haeger C, Coronata AMF, Polverino F, Vitali SH, Padera RF, et al. MMP-8 inhibits vascular remodeling and is protective in hypoxia-induced pulmonary hypertension. Presented at the Grover Conference. September 6, 2017, Sedalia, CO. Abstract 5, p. 8.
- 22. Dieffenbach PB, Mallarino Haeger C, Coronata AMF, Polverino F, Vitali SH, Padera RF, et al. MMP-8 inhibits vascular remodeling and is protective in hypoxia-induced pulmonary hypertension [abstract] Am J Respir Crit Care Med . 2018;197:A7397. [Google Scholar]
- 23. Dieffenbach PB, Rehman R, Corcoran AM, Mallarino Haeger C, Coronata AMF, Fredenburgh LE. MMP-8 deficiency promotes vascular remodeling phenotypes in pulmonary artery smooth muscle cells via FAK-dependent induction of YAP/TAZ signaling [abstract] Am J Respir Crit Care Med . 2019;199:A3448. [Google Scholar]
- 24. Dieffenbach PB, Rehman R, Mallarino Haeger C, Corcoran AM, Coronata AMF, Polverino F, et al. MMP-8 deficiency promotes vascular remodeling through enhanced integrin-β3 signaling. Presented at the 62nd Annual Thomas L Petty Aspen Lung Conference. June 5, 2019, Aspen, CO.
- 25. Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail . 2013;15:277–283. doi: 10.1093/eurjhf/hfs173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, et al. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation . 2014;129:864–874. doi: 10.1161/CIRCULATIONAHA.113.004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, et al. Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary hypertension and enhances contractility of vascular smooth muscle cells. Circulation . 2008;117:2114–2122. doi: 10.1161/CIRCULATIONAHA.107.716241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S, Izumo S. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol Genomics . 2004;16:349–360. doi: 10.1152/physiolgenomics.00041.2003. [DOI] [PubMed] [Google Scholar]
- 29. Samokhin AO, Stephens T, Wertheim BM, Wang RS, Vargas SO, Yung LM, et al. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med . 2018;10:eaap7294. doi: 10.1126/scitranslmed.aap7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fredenburgh LE, Velandia MM, Ma J, Olszak T, Cernadas M, Englert JA, et al. Cyclooxygenase-2 deficiency leads to intestinal barrier dysfunction and increased mortality during polymicrobial sepsis. J Immunol . 2011;187:5255–5267. doi: 10.4049/jimmunol.1101186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang X, Zhang D, Fucci QA, Dollery CM, Owen CA. Surface-bound matrix metalloproteinase-8 on macrophages: contributions to macrophage pericellular proteolysis and migration through tissue barriers. Physiol Rep . 2021;9:e14778. doi: 10.14814/phy2.14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Åström P, Pirilä E, Lithovius R, Heikkola H, Korpi JT, Hernández M, et al. Matrix metalloproteinase-8 regulates transforming growth factor-β1 levels in mouse tongue wounds and fibroblasts in vitro. Exp Cell Res . 2014;328:217–227. doi: 10.1016/j.yexcr.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 33. Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skládal P, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun . 2017;8:15321. doi: 10.1038/ncomms15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakraborty S, Njah K, Pobbati AV, Lim YB, Raju A, Lakshmanan M, et al. Agrin as a mechanotransduction signal regulating YAP through the Hippo pathway. Cell Rep . 2017;18:2464–2479. doi: 10.1016/j.celrep.2017.02.041. [DOI] [PubMed] [Google Scholar]
- 35. Sng JJ, Prazakova S, Thomas PS, Herbert C. MMP-8, MMP-9 and neutrophil elastase in peripheral blood and exhaled breath condensate in COPD. COPD . 2017;14:238–244. doi: 10.1080/15412555.2016.1249790. [DOI] [PubMed] [Google Scholar]
- 36. Pullamsetti S, Krick S, Yilmaz H, Ghofrani HA, Schudt C, Weissmann N, et al. Inhaled tolafentrine reverses pulmonary vascular remodeling via inhibition of smooth muscle cell migration. Respir Res . 2005;6:128. doi: 10.1186/1465-9921-6-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Araujo JA, Zhang M, Yin F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front Pharmacol . 2012;3:119. doi: 10.3389/fphar.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goetze JP, Bruneau BG, Ramos HR, Ogawa T, de Bold MK, de Bold AJ. Cardiac natriuretic peptides. Nat Rev Cardiol . 2020;17:698–717. doi: 10.1038/s41569-020-0381-0. [DOI] [PubMed] [Google Scholar]
- 39. Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med . 2011;183:649–658. doi: 10.1164/rccm.201005-0746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular-matrix bound basic fibroblast growth factor. J Cell Physiol . 1996;166:495–505. doi: 10.1002/(SICI)1097-4652(199603)166:3<495::AID-JCP4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 41. Jones PL, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the α v β 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol . 1997;139:279–293. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosenberg M, Meyer FJ, Gruenig E, Lutz M, Lossnitzer D, Wipplinger R, et al. Osteopontin predicts adverse right ventricular remodelling and dysfunction in pulmonary hypertension. Eur J Clin Invest . 2012;42:933–942. doi: 10.1111/j.1365-2362.2012.02671.x. [DOI] [PubMed] [Google Scholar]
- 43. Mura M, Cecchini MJ, Joseph M, Granton JT. Osteopontin lung gene expression is a marker of disease severity in pulmonary arterial hypertension. Respirology . 2019;24:1104–1110. doi: 10.1111/resp.13557. [DOI] [PubMed] [Google Scholar]
- 44. Saker M, Lipskaia L, Marcos E, Abid S, Parpaleix A, Houssaini A, et al. Osteopontin, a key mediator expressed by senescent pulmonary vascular cells in pulmonary hypertension. Arterioscler Thromb Vasc Biol . 2016;36:1879–1890. doi: 10.1161/ATVBAHA.116.307839. [DOI] [PubMed] [Google Scholar]
- 45. Anwar A, Li M, Frid MG, Kumar B, Gerasimovskaya EV, Riddle SR, et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol . 2012;303:L1–L11. doi: 10.1152/ajplung.00050.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat Commun . 2017;8:15494. doi: 10.1038/ncomms15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaiser R, Frantz C, Bals R, Wilkens H. The role of circulating thrombospondin-1 in patients with precapillary pulmonary hypertension. Respir Res . 2016;17:96. doi: 10.1186/s12931-016-0412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rogers NM, Yao M, Sembrat J, George MP, Knupp H, Ross M, et al. Cellular, pharmacological, and biophysical evaluation of explanted lungs from a patient with sickle cell disease and severe pulmonary arterial hypertension. Pulm Circ . 2013;3:936–951. doi: 10.1086/674754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rogers NM, Sharifi-Sanjani M, Yao M, Ghimire K, Bienes-Martinez R, Mutchler SM, et al. TSP1-CD47 signaling is upregulated in clinical pulmonary hypertension and contributes to pulmonary arterial vasculopathy and dysfunction. Cardiovasc Res . 2017;113:15–29. doi: 10.1093/cvr/cvw218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rogers NM, Ghimire K, Calzada MJ, Isenberg JS. Matricellular protein thrombospondin-1 in pulmonary hypertension: multiple pathways to disease. Cardiovasc Res . 2017;113:858–868. doi: 10.1093/cvr/cvx094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao AG, Lindberg FP, Dimitry JM, Brown EJ, Frazier WA. Thrombospondin modulates α v β 3 function through integrin-associated protein. J Cell Biol . 1996;135:533–544. doi: 10.1083/jcb.135.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stouffer GA, Hu Z, Sajid M, Li H, Jin G, Nakada MT, et al. β3 integrins are upregulated after vascular injury and modulate thrombospondin- and thrombin-induced proliferation of cultured smooth muscle cells. Circulation . 1998;97:907–915. doi: 10.1161/01.cir.97.9.907. [DOI] [PubMed] [Google Scholar]
- 53. Das A, Monteiro M, Barai A, Kumar S, Sen S. MMP proteolytic activity regulates cancer invasiveness by modulating integrins. Sci Rep . 2017;7:14219. doi: 10.1038/s41598-017-14340-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jung KK, Liu XW, Chirco R, Fridman R, Kim HR. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J . 2006;25:3934–3942. doi: 10.1038/sj.emboj.7601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ando T, Charindra D, Shrestha M, Umehara H, Ogawa I, Miyauchi M, et al. Tissue inhibitor of metalloproteinase-1 promotes cell proliferation through YAP/TAZ activation in cancer. Oncogene . 2018;37:263–270. doi: 10.1038/onc.2017.321. [DOI] [PubMed] [Google Scholar]
- 56. Pellinen T, Rantala JK, Arjonen A, Mpindi JP, Kallioniemi O, Ivaska J. A functional genetic screen reveals new regulators of β1-integrin activity. J Cell Sci . 2012;125:649–661. doi: 10.1242/jcs.090704. [DOI] [PubMed] [Google Scholar]
- 57. Varadarajulu J, Laser M, Hupp M, Wu R, Hauck CR. Targeting of α(v) integrins interferes with FAK activation and smooth muscle cell migration and invasion. Biochem Biophys Res Commun . 2005;331:404–412. doi: 10.1016/j.bbrc.2005.03.175. [DOI] [PubMed] [Google Scholar]
- 58. Ron A, Azeloglu EU, Calizo RC, Hu M, Bhattacharya S, Chen Y, et al. Cell shape information is transduced through tension-independent mechanisms. Nat Commun . 2017;8:2145. doi: 10.1038/s41467-017-02218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell . 2017;171:1397–1410.e14. doi: 10.1016/j.cell.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 60. Xiao Q, Zhang F, Grassia G, Hu Y, Zhang Z, Xing Q, et al. Matrix metalloproteinase-8 promotes vascular smooth muscle cell proliferation and neointima formation. Arterioscler Thromb Vasc Biol . 2014;34:90–98. doi: 10.1161/ATVBAHA.113.301418. [DOI] [PubMed] [Google Scholar]
- 61. Ishigami M, Swertfeger DK, Hui MS, Granholm NA, Hui DY. Apolipoprotein E inhibition of vascular smooth muscle cell proliferation but not the inhibition of migration is mediated through activation of inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol . 2000;20:1020–1026. doi: 10.1161/01.atv.20.4.1020. [DOI] [PubMed] [Google Scholar]