

Abstract
Rationale
Mesenchymal stem/stromal cell (MSC)–small extracellular vesicle (MEx) treatment has shown promise in experimental models of neonatal lung injury. The molecular mechanisms by which MEx afford beneficial effects remain incompletely understood.
Objectives
To investigate the therapeutic mechanism of action through assessment of MEx biodistribution and impact on immune cell phenotypic heterogeneity.
Methods
MEx were isolated from the conditioned medium of human umbilical cord Wharton’s jelly–derived MSCs. Newborn mice were exposed to hyperoxia (HYRX, 75% O2) from birth and returned to room air at Postnatal Day 14 (PN14). Mice received either a bolus intravenous MEx dose at PN4 or bone marrow–derived myeloid cells (BMDMy) pretreated with MEx. Animals were killed at PN4, PN7, PN14, or PN28 to characterize MEx biodistribution or for assessment of pulmonary parameters. The therapeutic role of MEx-educated BMDMy was determined in vitro and in vivo.
Measurements and Main Results
MEx therapy ameliorated core histological features of HYRX-induced neonatal lung injury. Biodistribution and mass cytometry studies demonstrated that MEx localize in the lung and interact with myeloid cells. MEx restored the apportion of alveolar macrophages in the HYRX-injured lung and concomitantly suppressed inflammatory cytokine production. In vitro and ex vivo studies revealed that MEx promoted an immunosuppressive BMDMy phenotype. Functional assays demonstrated that the immunosuppressive actions of BMDMy are driven by phenotypically and epigenetically reprogrammed monocytes. Adoptive transfer of MEx-educated BMDMy, but not naive BMDMy, restored alveolar architecture, blunted fibrosis and pulmonary vascular remodeling, and improved exercise capacity.
Conclusions
MEx ameliorate hyperoxia-induced neonatal lung injury though epigenetic and phenotypic reprogramming of myeloid cells.
Keywords: bronchopulmonary dysplasia, exosome, mesenchymal stem cells, macrophage, monocyte
At a Glance Commentary
Scientific Knowledge on the Subject
Mesenchymal stem/stromal cell (MSC)–small extracellular vesicles (MEx) bestow considerable therapeutic benefits in various experimental models of disease, including bronchopulmonary dysplasia. However, little is known about the in vivo pharmacokinetics of MEx and the target cells they interact with, most importantly the cell type(s) representing the mediators of MEx therapeutic functionality.
What This Study Adds to the Field
We used the murine model of bronchopulmonary dysplasia to analyze MEx–target cell interaction at a single-cell level and demonstrated that MEx interact with lung myeloid cells, restore them to normal levels, and redirect them to a noninflammatory cell phenotype. In addition, we report that adoptive transfer of MEx-educated myeloid cells protects from hyperoxic lung injury and normalizes lung development. These findings provide new mechanistic insights of MEx–myeloid cell interactions that can be harnessed for the further development of MEx-based therapeutics for diseases of the lung.
Bronchopulmonary dysplasia (BPD) is a multifactorial chronic lung disease that occurs almost exclusively in infants born at early gestational age who require mechanical ventilation and/or supplemental oxygen (1). It is characterized by reduced formation of alveoli coupled with perturbed matrix remodeling, immature pulmonary vasculature, impaired pulmonary function, and, in a subset of patients with moderate-to-severe BPD, secondary pulmonary hypertension (PH) (2–5). The antecedents of BPD, whether oxygen toxicity, reactive oxygen species, mechanical ventilation, infection, or fetal and postnatal inflammation, inflict their destructive effects via common inflammatory pathways (6, 7).
Mesenchymal stem/stromal cell (MSC)-based therapies have shown promise in numerous models of neonatal cardiorespiratory disease (8–15). It is widely recognized that the main therapeutic modality of MSCs resides in their secretome (16–19), and recent studies have shown that the chief therapeutic vector therein is represented by “small” extracellular vesicles (sEVs), an EV subset that includes exosomes (20–22). Notably, we (20–23) and others (24–26) have demonstrated that treatment with purified human MSC-derived–sEV (MEx) ameliorates and even reverses core histological and functional outcomes in experimental lung injury models including PH (20), pulmonary fibrosis (22), and BPD (21, 23). Indeed, MEx-based interventions have promoted tissue repair and regeneration through modulation of the immune response in numerous preclinical models where pulmonary dysfunction and inflammation occurs (8, 21, 22, 27). MEx are now recognized as potent immunomodulators that have the capacity to regulate the macrophage (Mϕ) fulcrum and “instruct” monocyte/Mϕ-mediated immune responses (21, 22, 28–30). It has been therefore suggested that myeloid cells may serve as a cellular mediator of MEx biologic activity.
Despite the encouraging transition of MEx-based therapeutics toward clinical applications, the molecular mechanisms by which MEx afford beneficial effects are incompletely understood. Key issues for effective clinical translation of any biologic medicine include characterization of the in vivo biodistribution plus identification of cellular targets and downstream signaling. Here, using a model of hyperoxia-induced neonatal lung injury, we sought to investigate the therapeutic mechanism(s) of action of MEx through detailed assessment of biodistribution and impact on immune cell phenotypic heterogeneity.
Methods
An extended description of our experimental BPD model and analytical methods are described in the online supplement.
Study Approval
Animal experiments were approved by the Boston Children’s Hospital Animal Care and Use Committee.
Hyperoxia induced-neonatal lung injury model
Newborn Friend leukemia virus B (FVB) mice were exposed to hyperoxia (HYRX, 75% O2) from birth until Postnatal Day 14 (PN14). After HYRX exposure, mice were returned to room air/normoxia (NRMX). Mice that were exposed to HYRX were compared with animals that remained in NRMX for the study duration. A bolus intravenous MEx treatment was administered at PN4 (Figure 1A). Additional experimental groups include HYRX-exposed animals that received either MEx-educated or naive bone marrow derived–myeloid cells (BMDMy). Control groups included NRMX animals that received MEx and HYRX-exposed animals that received appropriate vehicle controls. Notably, separate sets of independent in vivo experiments were performed to assess the therapeutic capacity of MEx, BMDMy, or BMDMy + MEx accordingly. Furthermore, different MEx preparations were used in each independent in vivo experiment, as outlined in the online supplement.
Figure 1.
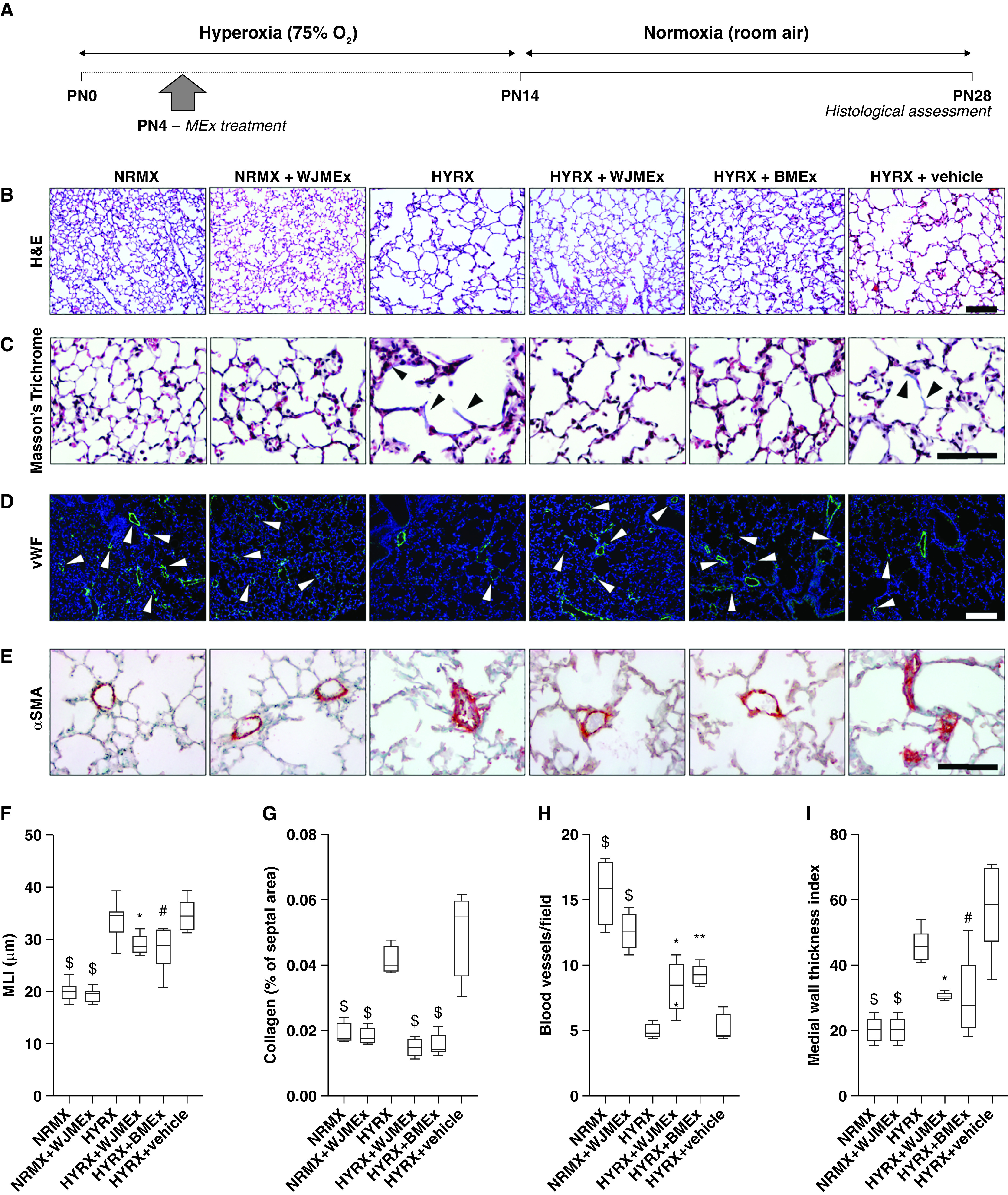
A single mesenchymal stromal/stem cell (MSC)–small extracellular vesicle (MEx) treatment improves core features of hyperoxia (HYRX)-induced neonatal lung injury. (A) Schematic of experimental model. Briefly, newborn Friend leukemia virus B mice were exposed to HYRX (75% O2) for 14 days. HYRX-exposed mice were compared with mice that remained at room air (normoxia [NRMX]). MEx treatments (both Wharton’s jelly MSC-EVs [WJMEx] and bone marrow MSC-EVs [BMEx]) were delivered intravenously at PN4. Animals were assessed at PN28. (B–E) Harvested lung sections were stained for hematoxylin and eosin (H&E) to assess lung architecture (B), Masson’s trichrome to evaluate septal collagen deposition (C), and vWF (von Willebrand factor) (D) or α-SMA (α-smooth muscle actin) (E) to assess the effect of HYRX on peripheral pulmonary blood vessel loss and pulmonary vascular remodeling, respectively. As additional experimental controls, NRMX animals received WJMEx (NRMX + WJMEx), and HYRX-exposed animals also received vehicle control (HYRX + vehicle). (F) Quantification of mean linear intercept (MLI, μm) represents a surrogate of average air space diameter. (G) Collagen deposition was used as a surrogate of fibrosis and was reported as percent of septal area. Arrowheads in C highlight collagen deposition. (H) vWF-positive vessels <500 μm in outer diameter were counted at 100× magnification in 8–12 random views. Arrowheads in D highlight vWF-stained pulmonary vessels, and values are expressed as the average blood vessel count per field. (I) Medial thickness index = 100 × (area[ext] – area[int])/area[ext]. Area[ext] and area[int] denote the areas within the external and internal boundaries of the α-SMA layer, respectively. Data represent results from three individual studies. n = 6–12 per group; *P < 0.05, **P < 0.01, #P < 0.001, and $P < 0.0001 versus HYRX. Scale bars: H&E and vWF, 100 μm; Masson’s trichrome and α-SMA, 50 μm. PN = Postnatal Day.
Statistics
Data was reported as mean ± SEM unless stated otherwise. Differences between experimental groups were compared by ANOVA coupled with an ad hoc Tukey’s test using GraphPad Prism (v6.0; GraphPad). Supervised flow cytometry and mass cytometry analysis was performed using FlowJo software v10.2 (TreeStar). Unsupervised mass cytometry analysis was performed using Cytobank (Beckman Coulter). Investigators were blinded to experimental groups for histological analysis and physiological measurements. Significance was considered at P < 0.05. For in vivo studies, we based our sample size calculations on previous work (17, 21), suggesting that detection of a 15% improvement in lung architecture (assessed by mean linear intercept [MLI]), with >90% power at the 5% α level, requires a minimum of five animals per group.
Results
MEx: Nomenclature, Isolation, and Characterization
MEx were comprehensively characterized in accordance with the 2018 Minimal Information for Studies of Extracellular Vesicles (31), as detailed in the online supplement (see Figure E1).
A Single MEx Treatment Improves Core Features of HYRX-induced Neonatal Lung Injury Lung
To assess the effect of HYRX on alveolar simplification, a hallmark of BPD, we assessed the MLI value across all experimental groups. As anticipated, 14 days of HYRX-exposure resulted in a BPD phenotype that resembles the clinical BPD presentation witnessed in the modern-day neonatal intensive care unit. Compared with NRMX subjects, HYRX-control mice presented with a greater MLI value (P < 0.0001), an elevated amount of septal collagen deposition (P < 0.0001), diminished peripheral pulmonary blood vessel number (50- to 350-μm diameter, P < 0.0001), and enhanced peripheral pulmonary vascular muscularization (P < 0.0001). HYRX-exposed animals treated with MEx (either human Wharton’s jelly MSC-EVs [WJMEx] or human bone marrow MSC-EVs [BMEx]) presented with dramatically improved alveolarization (P < 0.001). Furthermore, WJMEx or BMEx treatments blunted pulmonary fibrosis (P < 0.0001 and P < 0.0001, respectively), improved blood vessel count (P < 0.05 and P < 0.01, respectively), and dramatically decreased the degree of HYRX-induced pulmonary muscularization (P < 0.05, P < 0.001 respectively, Figure 1). In our previous studies, on both BPD and fibrosis models, we used human dermal fibroblast EVs as a biological control and demonstrated that the observed therapeutic actions are specific to MEx (21, 22). Thus, in this study only the appropriate vehicle controls were employed. Importantly, compared with their respective controls, across all analyses no parenchymal changes were found in NRMX animals treated with MEx or HYRX-exposed animals that received vehicle (P > 0.05, Figure 1). Here, to aid interstudy comparison and reproducibility, BMEx served as an alternate MEx source. Herein, all experiments that employ MEx refers to WJMEx.
HYRX-induced Apoptosis Is Inhibited by MEx
Given the well-known cytotoxic actions of HYRX, we next explored the antiapoptotic effect of MEx in the lung parenchyma. Here, terminal deoxynucleotidyl transferase–mediated dUTP nick end label (TUNEL) staining in whole-lung sections revealed that HYRX-exposed mice presented with increased number of apoptotic cells compared with NRMX animals (P < 0.01) and that MEx treatment efficiently reduced the degree of apoptosis (P < 0.05, Figures E2A and E2B). Similar trends were recapitulated on assessment of whole-lung Casp1 and Bax mRNA assessment (P < 0.05, Figures E2C and E2D).
MEx Biodistribution
Despite MEx therapy providing considerable therapeutic benefits in various experimental models, little is known about the in vivo pharmacokinetics, arguably as a consequence of limitations in current technology. Here, we labeled MEx with a near-infrared (NIR) membrane dye before intravenous administration (Figure 2A). Animals (PN4 pups) were assessed at 0 minutes, 5 minutes, 30 minutes, 1 hour, 3 hours, and 24 hours after injection. Immediately after injection of NIR-MEx, live (isoflurane-sedated) animal imaging detected the NIR-MEx transition from the injection site to the mouse torso, before declining in intensity at 24 hours after injection (Figures 2B and E3). Corresponding ex vivo analysis of harvested organs revealed the lung and liver are the major sites of NIR signal. Notably, over the 24-hour time course, the proportion of NIR signal was reduced in the lung and concomitantly accumulated in the liver (Figures 2C and E3).
Figure 2.
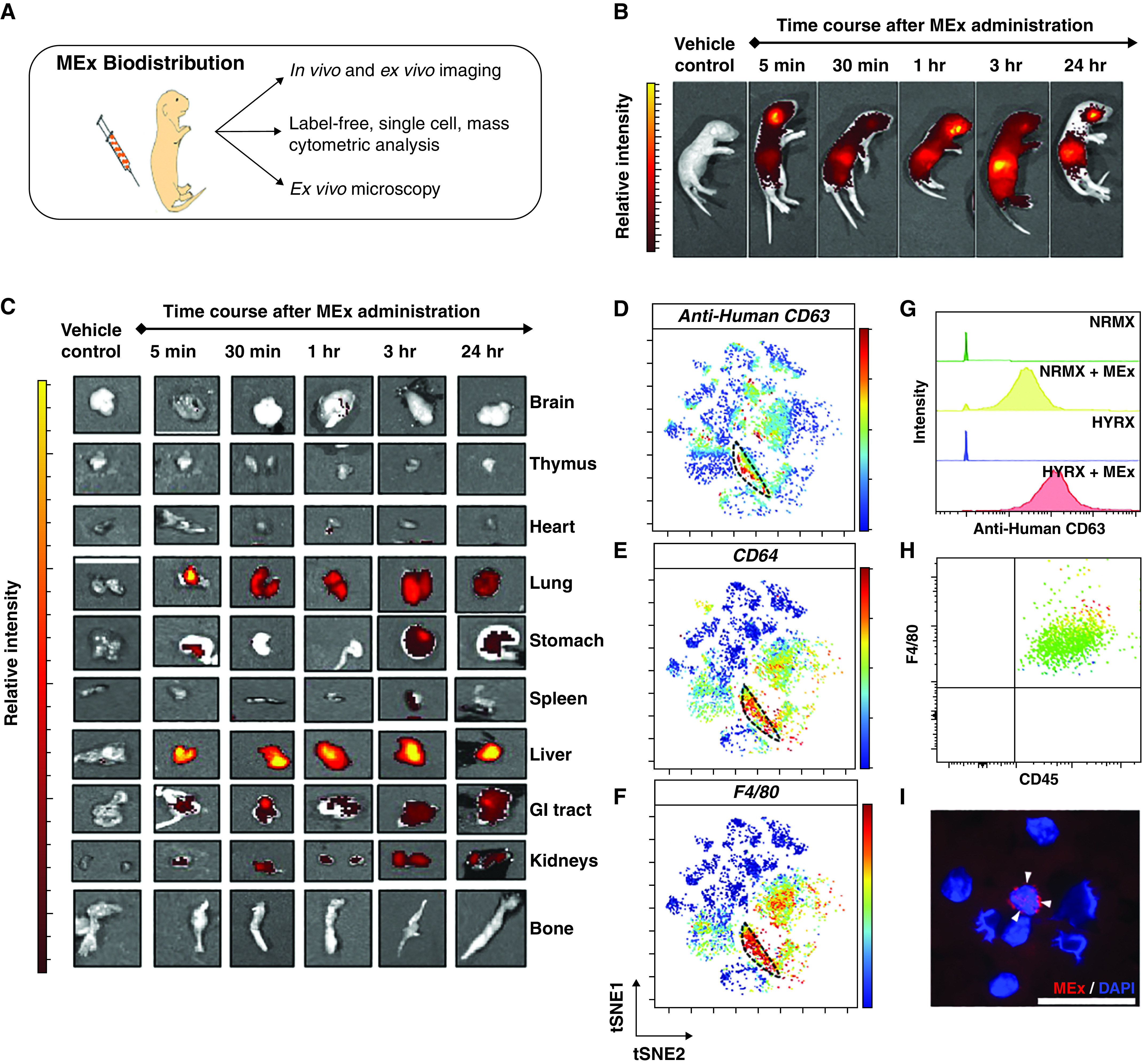
Mesenchymal stem/stromal cell–small extracellular vesicle (MEx) biodistribution: MEx localize to the lung and interact with myeloid cells. (A) Schematic of the comprehensive trimodal approach to assess MEx biodistribution. (B) Representative live animal images of Friend leukemia virus B mice (PN4). Mice received MEx labeled with a membrane-bound near-infrared (NIR) dye, as described in Methods. Animals were assessed at 5 minutes, 30 minutes, 1 hour, 3 hours, and 24 hours after NIR-MEx administration. (C) Ex vivo imaging was undertaken to detect MEx biodistribution at an organ level. Vehicle control represents the dye-free supernatant from the dye removal wash step. Mass cytometry was used as a novel “dye-free” approach to assess MEx–target cell interaction at a single-cell level. (D) Representative t-distributed stochastic neighbor embedding (tSNE) analysis, depicting relative human anti-CD63 intensity across CD45+ cells. (E and F) In addition, tSNE analysis depicting the relative intensity for F4/80 and CD64 reveals that the greatest degree of human CD63 intensity appears to colocalize, albeit not exclusively, with cells that express CD64 (E) and F4/80 (F). The dashed black line highlights colocation of human anti-CD63, F4/80, and CD64. Scale bars represent relative intensity (blue to red through green/yellow, reflects low to high intensity). (G) Anti-human CD63 histograms after conventional biaxial gating demonstrated MEx was associated with CD45+, CD11b+, CD64+, F4/80+ cells in both normoxia (NRMX)- and hyperoxia (HYRX)-exposed mice. Minimal background was seen in respective control animals. Histograms: green, NRMX; yellow, NRMX + MEx; blue, HYRX; red, HYRX + MEx. (H) Flow cytometric analysis confirmed CD45+, F4/80+ cell enrichment in BAL fluid of PN4 pups. MEx were labeled with ExoGlow-membrane dye (red) and were administered to PN4 pups as described in Methods. (I) Immunofluorescence imaging confirming MEx interact with CD45+, F4/80+ cells (stained with DAPI). White arrowheads highlight labeled MEx. Scale bar, 50 μm. GI = gastrointestinal; PN = Postnatal Day.
Mass Cytometry Detection of MEx–Myeloid Cell Interaction in Whole Lung
MEx appears to localize to the lung; however, it remains unclear which cells MEx interact with, and as such, which cells are likely the mediators for orchestrating their beneficial actions. Thus, we next employed a novel mass cytometry–based approach to achieve “label-free” assessment of MEx–target cell interaction at a single-cell level. We used mass cytometry to gain a hierarchal overview of pulmonary immune cell populations and included an anti-human CD63 antibody, an abundant tetraspanin that is readily accessible on the surface of MEx (Figure E1), to assess MEx-target cell interaction.
After MEx administration, unsupervised (whole-lung) analysis on CD45+ cells revealed that anti-human CD63 was detected in the mouse lung, and albeit not exclusively, appeared to co-localize with F4/80+, CD64+ myeloid cells (Figures 2D–2F). Moreover, this was confirmed using a supervised biaxial gating strategy in NRMX and HYRX animals (Figure 2G). Importantly, minimal cross-reactivity with endogenous mouse CD63 was detected in control NRMX or HYRX animals that did not receive MEx (Figure 2G).
MEx–target cell interaction was confirmed by immunofluorescence microscopy. Using PN4 pups, we confirmed F4/80+ cells were enriched in BAL fluid (BALF) (Figure 2H and E4A). We obtained and processed BALF F4/80+ cells at 3 hours and 24 hours after ExoGlow-labeled MEx delivery (Figure E4). Immunofluorescence assessment of cytocentrifuged BALF-derived cells revealed MEx were associated with F4/80+ myeloid cells at both 3 hours (Figures 2I and E4B) and 24 hours (Figure E4B) after administration.
MEx Treatment Restores the Normal Levels of Pulmonary Myeloid Cells Disrupted by HYRX and Suppresses Inflammatory Cell Phenotype
Considering the beneficial histological findings after a single MEx dose, and the interaction of MEx-myeloid cells, we next wanted to generate a hierarchical perspective of whole-lung cellular heterogeneity. Using a mass cytometry approach, we queried 22 cell surface markers and 5 intracellular cytokine markers to assess activation and inflammatory state in whole-lung cell suspensions (Table E1 and Figure E5). At PN7, we found that, compared with NRMX animals, HYRX-exposed animals presented with a striking reduction in CD45+ cells within the lung (P < 0.0001), and this was fully restored by MEx therapy (P < 0.0001, Figure 3A). By PN14, levels of CD45+ cells were normalized across all experimental groups (P > 0.05, Figure 3B).
Figure 3.

Mesenchymal stem/stromal cell–small extracellular vesicles (MEx) restore pulmonary myeloid cell apportion. Mass cytometry was undertaken on whole-lung samples across all experimental groups at PN7 and PN14. (A and B) The proportion of CD45+ cells at PN7 (A) and PN14 (B). (C and D) The cell proportions of alveolar macrophage (Mϕ), interstitial Mϕ, Ly6Chigh, and Ly6Clow monocyte populations at PN7 (C) and PN14 (D). Gating strategy is shown in Figure E5. Data expressed as percentage of CD45+ population. n = 4–8 per group; *P < 0.05 and $P < 0.0001 versus HYRX. HYRX = hyperoxia; NRMX = normoxia; PN = Postnatal Day.
At PN7, HYRX-exposed mice presented with a markedly reduced proportion of alveolar Mϕs compared with NRMX animals (P < 0.0001). MEx ameliorated the HYRX-induced reduction in AMϕs (P < 0.05, Figure 3C). Albeit to lesser magnitude, a similar trend was also found at PN14 (P < 0.05, Figure 3D). No differences were found in interstitial Mϕ apportion across all experimental groups (P > 0.05, Figures 3C and 3D). Levels of Ly6Chigh monocytes at PN7 were either absent in HYRX animals or present in very low numbers in the MEx-treated and NRMX animals (Figure 3C). HYRX-exposed animals that received MEx presented with an increased apportion of Ly6Clow monocytes (P < 0.05), while no differences were detected in HYRX and NRMX counterparts (P > 0.05). At PN14, compared with NRMX mice, HYRX-exposed animals displayed a reduced apportion of Ly6Clow monocytes (P < 0.0001), and this was normalized by MEx treatment (P < 0.0001, Figure 3D). There was little evidence to suggest that numbers of T lymphocytes, NK cells, or B lymphocytes were impacted by MEx treatment at either PN7 or PN14. The apportion of whole-lung immune cells for PN7 and PN14 are detailed in Tables E2 and E3, respectively.
HYRX Promotes a Proinflammatory Monocyte/Macrophage Phenotype
To assess the inflammatory state of lung myeloid cells, we analyzed intracellular levels of TNFα, IL4, IL6, IFNγ, and IL17a. We found that MEx treatment was able to blunt the HYRX-induced elevation in intracellular AMϕ levels of TNFα, IL4, and IL6 at PN7 (P < 0.05, Table E4). At PN14, interstitial Mϕs displayed a pronounced “proinflammatory” activation state, characterized by increased levels of TNFα, IL4, IL6, IL17a, and IFNγ, which was blunted by MEx treatment (P < 0.05, Table E5). Notably, Ly6Chigh monocytes populations were not detected in the HYRX-exposed mice lung, arguably a downstream consequence of overall CD45+ depletion.
MEx Modulates BMDMy Phenotype and Promotes an Immunosuppressive Phenotype in vitro
The above data show that MEx interact with myeloid cells and promote a prohomeostatic shift in immune cell proportion and phenotype akin to baseline NRMX. We next assessed the direct impact of MEx on myeloid cells in vitro. Compared with naive BMDMy, MEx-“educated” BMDMy presented with increased levels of CD11b, F4/80, CX3CR1, and Ly6C/G, while CCR2 expression was dramatically blunted in MEx-treated BMDMy (Figure E6). In accordance, a targeted gating strategy confirmed that MEx promoted a Ly6C/G+, CX3CR1+, and CCR2− phenotype (P < 0.001, Figures 4B and 4C). To explore if such trends occurred in vivo, we undertook expedient real-time qRT-PCR assays on whole-lung samples. MEx treatment reduced the HYRX-induced elevation in whole-lung Ccr2 mRNA levels (P < 0.01, Figure E7).
Figure 4.

Mesenchymal stem/stromal cell–small extracellular vesicle (MEx)-educated bone marrow-derived myeloid cells (BMDMy) favor an immunosuppressive phenotype. (A) Schematic representation of workflow and BMDMy culture. (B) Representative t-distributed stochastic neighbor embedding (tSNE) analysis highlighted CCR2 intensity across BMDMy that were treated with/without MEx. tSNE plots are accompanied by conventional biaxial gating on CD45+, CX3CR1+, and CCR2+/− populations. Scale bars represent relative intensity (blue to red through green/yellow, reflects low to high intensity). (C) Quantification of CD45+, CX3CR1+, and CCR2+/− phenotypes are expressed as percentage of CD45+ cells. The immunosuppressive capacity of MEx-educated BMDMy was tested using cocultures of carboxyfluorescein succinimidyl ester (CFSE)-labeled splenocytes with BMDMy (or BMDMy + MEx). After a 5-day coculture, cells were harvested and analyzed for CFSE dilution via flow cytometry. Splenocytes were cultured in media alone (unstimulated) and in the presence of anti-CD3/CD28 activation beads, which were used as negative and positive controls, respectively. (D) Gating on CD3+ cells, histograms depict proliferation of splenocytes, conceptualized by dilution of CFSE. Purple histogram: CFSE-labeled splenocytes. Dotted histogram: unstained control. (E) Quantification of CFSE dilution across experimental groups. n = 4 per group; *P < 0.05, **P < 0.01 #P < 0.001, and $P < 0.0001. FITC = fluorescein isothiocyanate.
To test the functional significance of such phenotypic changes, we next evaluated their immunosuppressive capacity using cocultures of single-cell suspensions of carboxyfluorescein succinimidyl ester (CFSE)-labeled splenocytes with BMDMy (or BMDMy + MEx). Splenocytes showed negligible proliferation in the presence of media alone and high-proliferative capacity (>85%) in the presence of anti-CD3/CD28 beads. Interestingly, naive BMDMy modestly inhibited CD3+ cell proliferation. Compared with the BMDMy, this effect was significantly exaggerated in the MEx-educated BMDMy (P < 0.05, Figures 4D and 4E). There was non-indication of autoreactivity across any experimental group cocultures without anti-CD3/CD28 bead stimulation. MEx alone had no impact on CD3+ cell proliferation (P > 0.05).
Transcriptomic and Epigenetic Reprogramming of Monocytes (BMDMono) Drive the Immunosuppressive Actions of MEx-educated BMDMy
MEx treatment modulates BMDMy phenotype and function, promoting a CCR2− monocyte phenotype within the BMDMy population. To confirm if monocytes are the cell population responsible for the immune-modulatory actions in vitro, we isolated monocytes (BMDMono) from BMDMy by magnetic bead enrichment (Figure E8).
To generate an unbiased global overview of MEx impact, we undertook bulk RNA sequencing on the purified monocytes (BMDMono) that were primed with or without MEx. We found that 208 genes were differentially expressed at a magnitude >1 log2 (P < 0.05). Of these, MEx upregulated 130 and downregulated 78 genes (Figure 5, Tables E6–E7). Ingenuity pathway analysis revealed that the Ccr2–Ccl2 axis was significantly modulated by MEx treatment and involved in the infiltration and recruitment of monocyte-derived suppressive cell (MDSC)-like cells (Figure E9D). In support, MEx treatment elevated Arg1 and Il-10 mRNA levels, key characteristics of MDSCs (P < 0.05, Figure 5). Upstream regulators that govern the differentially expressed genes were explored using ingenuity pathway analysis. Ccr2 was found to be a key upstream regulator of MEx-mediated gene expression. Of note, most upstream regulators were transcription factors that were involved in binding and remodeling chromatin (Table E8).
Figure 5.
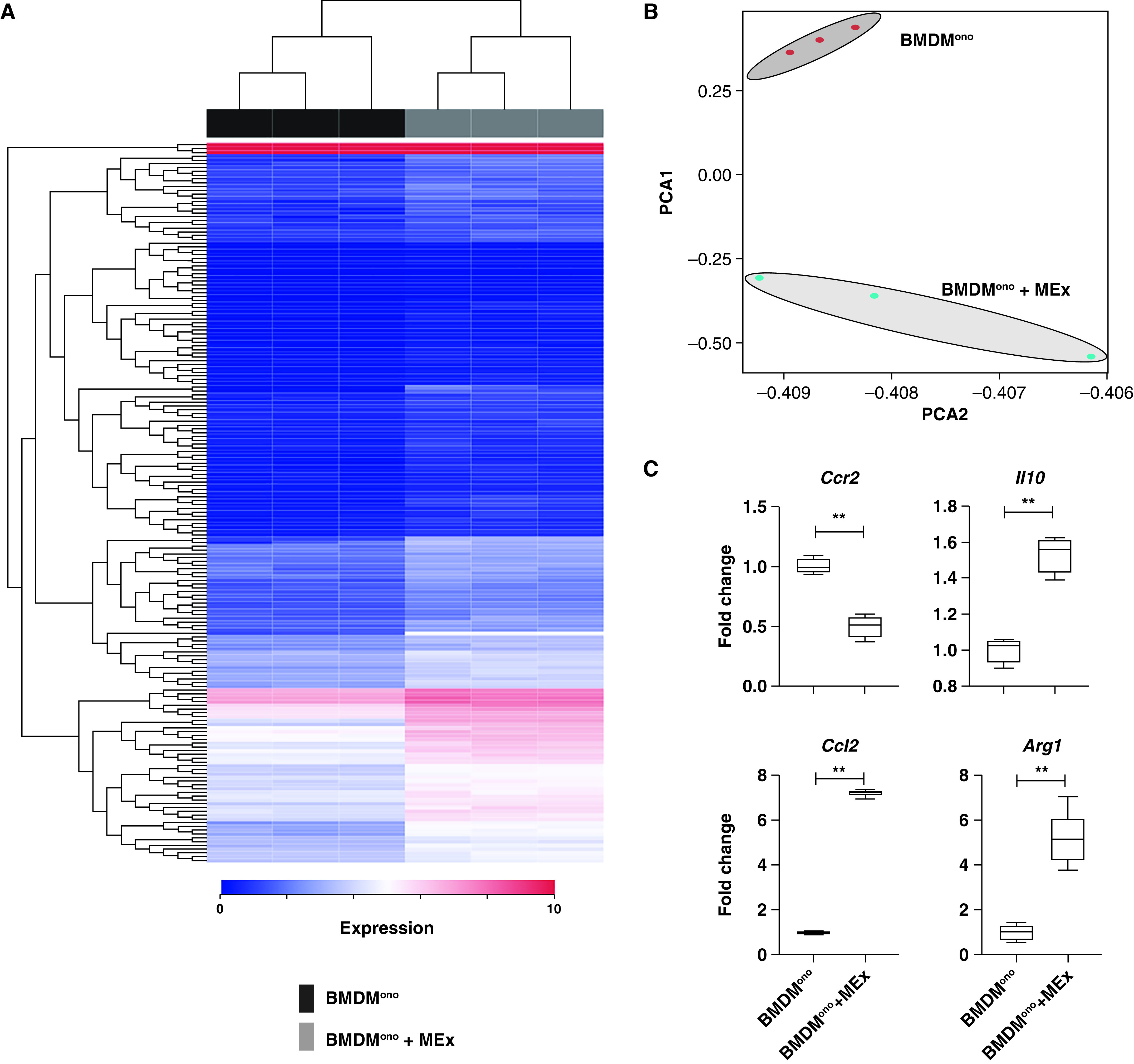
Transcriptomic reprogramming by mesenchymal stem/stromal cell–small extracellular vesicles (MEx) promote an immunosuppressive monocytic phenotype. (A) Poly-A capture RNA sequencing of isolated bone marrow–derived monocytes (BMDMono) was performed on control (BMDMono) and MEx-educated (BMDMono + MEx) groups, n = 3 per group. The heat map shows the differentially expressed genes (blue [0] to red [10] through white) ranked according to adjusted P value. (B) Principal component analysis (PCA) between control (BMDMono) and MEx-educated (BMDMono + MEx) groups. (C) Validation of candidate RNA seqencing markers was assessed by qRT-PCR in independent (separate) experiments. Data are shown as median (minimum–maximum). n = 3–6 per group; **P < 0.01.
Thus, to complement the RNA sequencing, we next undertook assay for transposase-accessible chromatin (ATAC) sequencing on BMDMono and BMDMono+MEx to assess the impact of MEx on the epigenetic landscape of their target cells and explore any epigenetic signatures that may be responsible for the long-term benefits. Importantly, ATAC sequencing found that MEx did not alter monocytic genomic fragment distribution (P > 0.05, Figure E10). Unbiased quantitative analysis of annotated peaks mapped to the mouse genome revealed that MEx promoted accessible chromatin regions in 65 genes and closed 19 genomic locations (P < 0.05). Upstream promoter regions of candidate markers were identified through UCSC genome browser (upstream promoter region defined as 50 kbp). Interestingly, in accordance with our RNA sequencing and quantitative PCR data, ATAC sequencing demonstrated that the upstream promoters for CCL2, IL10, and ARG1 had enhanced number of peak calls (P < 0.05, Figures 6A–6D). CCR2 upstream promotor chromatin accessibility was not significantly impacted by MEx (P > 0.05, Figures 6A–6D).
Figure 6.
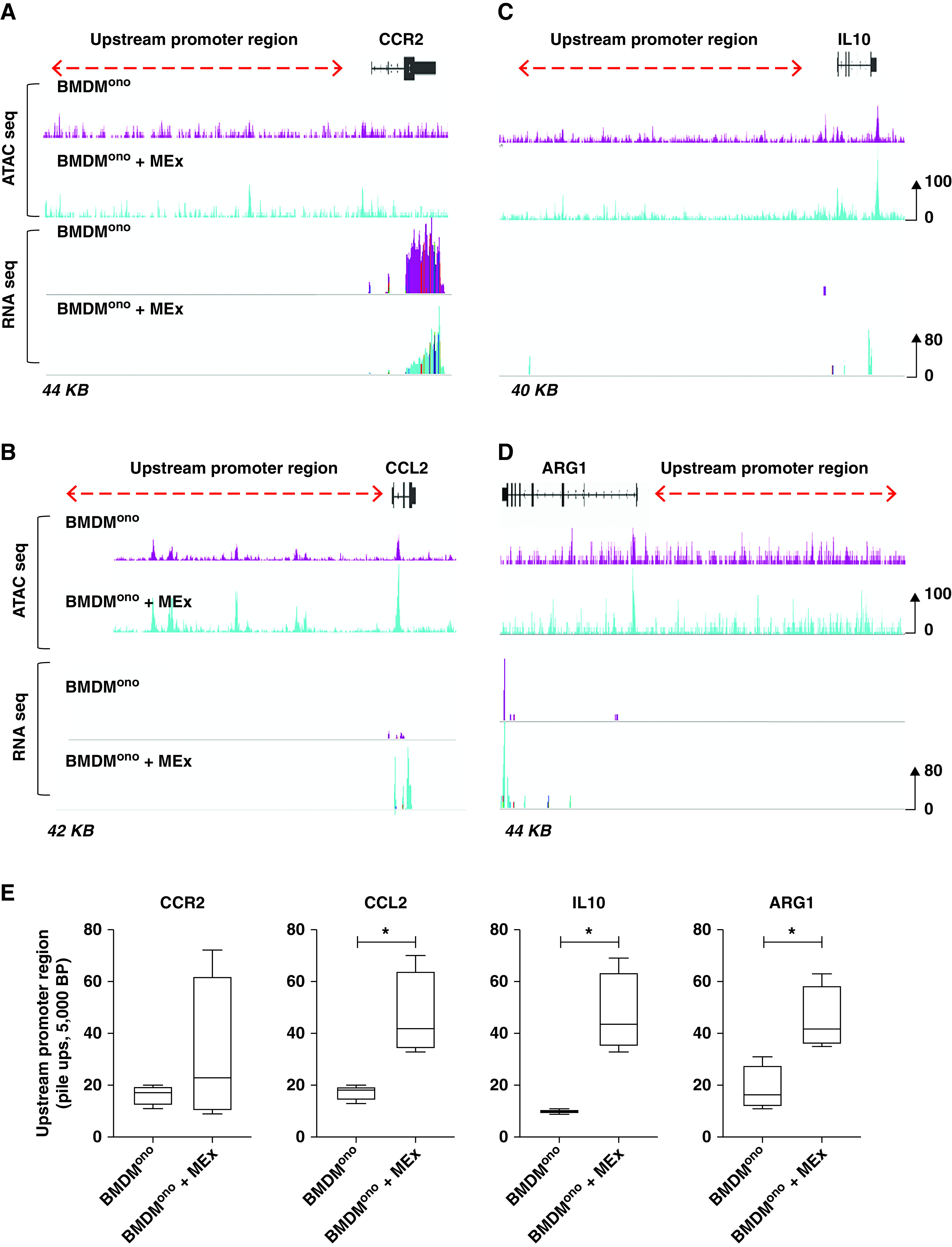
Mesenchymal stem/stromal cell–small extracellular vesicles (MEx) modulate the epigenetic landscape of bone marrow–derived monocytes (BMDMono). BMDMono were isolated and characterized as described in text. Transposase-accessible chromatin with high-throughput sequencing (ATAC seq) is a method for mapping genome-wide chromatin accessibility. Upstream promoter regions for candidate markers were obtained using the University of California Santa Cruz genome browser, with a search radius of 50,000 bp upstream. IGV software was used to overlay and visualize ATAC seq peak calls and representative RNA sequencing (RNA seq) expression for (A) CCR2, (B) CCL2, (C) IL10, and (D) ARG1. Purple peaks = BMDMono; turquoise peaks = MEx-educated BMDMono. Peak heights were set at 100 for ATAC seq and 80 for RNA seq. (E) Respective quantification of upstream promoter region peak calls. n = 3 per group; *P < 0.05.
To determine the relevance of such transcriptomic changes, we evaluated the immunosuppressive capacity using cocultures of single-cell suspensions of CFSE-labeled splenocytes with BMDMono or BMDMono+MEx. Here, BMDMono cells exhibited subtle immunosuppressive properties, modestly inhibiting CD3+ proliferation; however, this effect was exaggerated in the MEx-educated BMDMono cells (P < 0.05, Figure E11).
Adoptive Transfer of MEx-educated BMDMy Increases Alveolarization, Reduces Pulmonary Fibrosis, and Improves HYRX-induced Vascular Muscularization
Employing our established experimental BPD model, we sought to determine whether MEx-educated BMDMy could alleviate core features of neonatal HYRX-induced lung injury. Again, we compared animals that were exposed to HYRX to mice that remained at room air for the experimental duration. As anticipated, compared with NRMX animals, HYRX-control mice presented with severe alveolar simplification, elevated septal collagen deposition, diminished peripheral blood vessel count, and increased pulmonary vascular remodeling (P < 0.0001, P < 0.05, P < 0.01, respectively, Figure 7).
Figure 7.
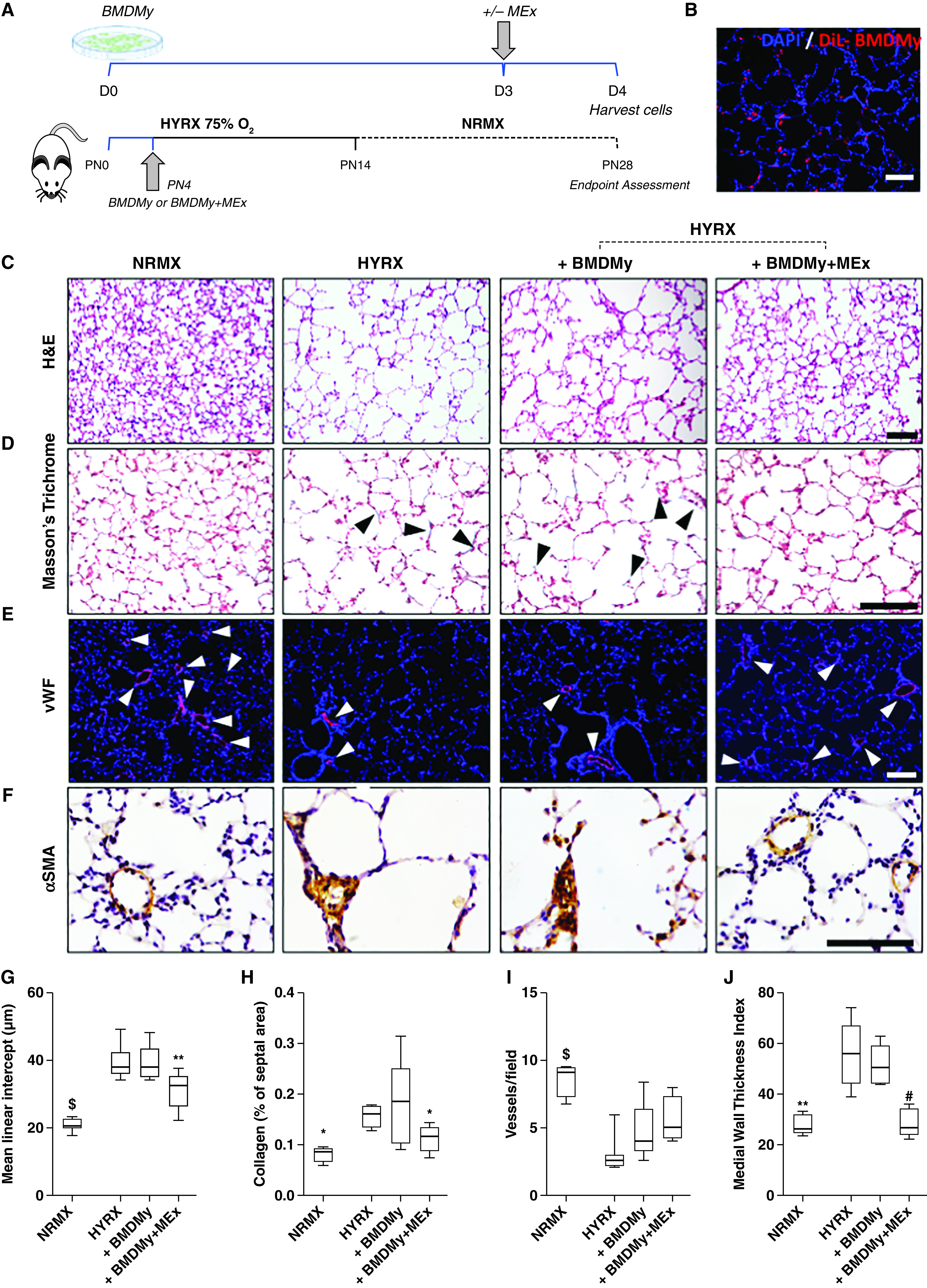
Adoptive transfer of mesenchymal stem/stromal cell–small extracellular vesicle (MEx)-educated bone marrow-derived myeloid cells (BMDMy) increases alveolarization and reduces pulmonary fibrosis and hyperoxia (HYRX)-induced vascular muscularization. (A) Schematic representation of adoptive transfer workflow. Briefly, newborn Friend leukemia virus B mice were exposed to HYRX (75% O2) for 14 days. HYRX-exposed mice were compared with mice that remained at room air (normoxia [NRMX]). BMDMy or MEx-educated BMDMy were delivered intravenously at PN4. Animals were assessed at PN28. (B) Proof-of-concept studies showed that dialkylcarbocyanine (DiL)-labeled BMDMy were detected in the lung at PN14. Blue = DAPI. (C–F) Harvested lung sections were stained for hematoxylin and eosin (H&E) to assess lung architecture (C), Masson’s trichrome to evaluate septal collagen deposition (D), and vWF (von Willebrand factor) (E) or α-SMA (α-smooth muscle actin) (F) to assess the effect of HYRX on peripheral pulmonary blood vessel loss and pulmonary vascular remodeling, respectively. (G–J) Quantification of mean linear intercept (μm) (G), collagen deposition (reported as % of septal area) (H), blood vessel count (positive <500-μm outer diameter) (I), and medial thickness index (J). Black arrowheads highlight collagen deposition. White arrowheads highlight vWF-stained pulmonary vessels. Data represent results from three individual studies. n = 6–12 per group; *P < 0.05, **P < 0.01, #P < 0.001, and $P < 0.0001 versus HYRX. Scale bars: H&E and vWF, 100 μm; Masson’s trichrome and α-SMA, 50 μm. PN = Postnatal Day.
Treatment groups received a single intravenous dose of either BMDMy or BMDMy + MEx at PN4. HYRX-exposed animals that received a bolus BMDMy + MEx dose presented with an improved MLI, reflecting enhanced alveolarization (P < 0.01, Figures 7C and 7G). BMDMy treatment alone had no impact on alveolarization (P > 0.05). A similar trend was recapitulated on assessment of lung fibrosis. Here, mice that received BMDMy + MEx, but not BMDMy, presented with levels of septal collagen deposition levels that were akin to their NRMX counterparts (P < 0.05, Figures 7D and 7H). We undertook vWF staining on the lung sections to assess for peripheral pulmonary blood vessel loss. Although BMDMy + MEx–treated animals displayed a trend toward an increased pulmonary blood vessel count, this did not reach significance (P > 0.05, Figures 7E and 7I). Moreover, α-SMA staining revealed BMDMy + MEx–treated animals, but not BMDMy-treated mice, had significantly reduced levels of pulmonary vascular muscularization (P < 0.001, Figures 7F and 7J). These beneficial effects were dose dependent and observed in mice that received a dose of 5 × 106 cells, whereas 0.25–1 × 106 cell doses did not provide therapeutic benefits (P > 0.05, Figure E12).
MEx-educated BMDMy Improves Functional Exercise Capacity
To determine the functional significance of MEx-educated BMDMy, we undertook exercise capacity testing in our experimental groups at PN28. Compared with NRMX-animals, the HYRX-exposed mice presented with diminished exercise capacity (P < 0.01). Akin to their NRMX counterparts, HYRX-exposed animals that received MEx were able to complete the exercise challenge. BMDMy + MEx treatment improved the exercise capacity (P < 0.01). BMDMy treatment had no impact on exercise capacity (P > 0.05, Figure E13).
Discussion
In this study we demonstrate that a single MEx intervention improved lung architecture, decreased pulmonary fibrosis, increased peripheral pulmonary blood vessel loss, and ameliorated pulmonary vascular muscularization in experimental BPD. Furthermore, using in vivo and ex vivo imaging coupled with a novel “label-free” mass cytometric approach, we extend our observations to show that MEx localize to the lung and interact with myeloid cells, promoting a prohomeostatic shift in myeloid cell apportion coupled with a concomitant suppression in cellular inflammation. T-cell proliferation assays coupled with expedient in vitro and in vivo phenotypic characterization showed that MEx promoted an immunosuppressive, CCR2−-associated myeloid cell phenotype. Notably, we are the first to demonstrate that adoptive transfer of MEx-educated BMDMy, but not naive BMDMy, ameliorated the lung architecture, blunted fibrosis and pulmonary vascular remodeling, and improved exercise capacity in an experimental model of “severe” BPD. Furthermore, we extend our observations to show that the immunosuppressive actions are driven by phenotypically and epigenetically reprogrammed monocytes.
The considerable promise of MEx-based therapies has been demonstrated in a diverse array of preclinical pathologies. However, more detailed information on the in vivo trafficking of MEx is required to support effective clinical translation. Few studies have focused on the biodistribution of MEx, arguably because of the inadequate tools for in vivo EV tracking (32–34). In this study, we demonstrate that after administration, the lung and liver are the major, albeit not exclusive, sites for early MEx accumulation. Our findings are in accordance with other animal models that have demonstrated that the liver and lungs are preferential target organs for systemically administered EVs (35, 36). Notably, it remains unclear if the observed accumulation in the liver is related to NIR-dye leakage and/or MEx clearance. Of interest, the liver is enriched in Kupffer cells that likely engulf EVs and may serve as a site for systemic immunomodulation. Ultimately, the impact of MEx on the lung–liver axis and systemic cytokine/inflammatory levels is intriguing, understudied, and warrants further investigation. Here, we were able to extend our observations and assess MEx-target cell interaction at a single level, demonstrating that within the lung, MEx readily interact with CD45+, CD11b+, CD64+, F4/80+ myeloid cells. Growing evidence confirms that MEx directly interact with monocytes and Mϕs in vivo. Lankford and colleagues showed that intravenously delivered “MSC-EVs” target “M2-like” Mϕs in an experimental model of spinal cord injury (37). Interestingly, recent studies have demonstrated that monocytes readily uptake EVs in vivo and mediate homing of intravenously administered EVs to the pulmonary vasculature during low-grade systemic inflammation (38).
Clinical reports show that the arrest in lung development is associated with altered myeloid cell dynamics (39–41). In this study we employed mass cytometry to generate a hierarchal perspective of immune cell heterogeneity. We found that ∼50% of pulmonary CD45+ cells were absent in the HYRX-exposed animals at PN7. Moreover, Mϕ levels were dramatically reduced in animals exposed to HYRX, and MEx treatment restored CD45+ cells and replenished Mϕ levels. Our findings are in accordance with Nold and coworkers who demonstrated that HYRX-exposure depletes the overall murine lung immune cell population by a third (6). On a parallel vein, Kalymbetova and colleagues demonstrated that HYRX exposure diminishes resident alveolar Mϕ populations, in turn disrupting lung structural development (42). Furthermore, through antibody or via transgenic death receptor–based approaches to deplete or prevent lung recruitment of myeloid cells, they demonstrated that neither neutrophils nor exudate Mϕs (that may include interstitial Mϕs) contribute to the structural perturbations of HYRX exposure; however, cells of the monocyte/Mϕ lineage were the likely mediators of arrested lung development (42). Coupled with the aforementioned limitations in assessing MEx biodistribution, it remains unknown if MEx manipulate the bone marrow directly, and it remains to be characterized if the therapeutic effect is a consequence of bone marrow mobilization or of an impact on the circulating myeloid cells. To assess this, using appropriate BPD models, future work would require carefully designed lineage tracing studies. It also remains unclear whether the initial HYRX-induced reduction in CD45+ cells and Mϕs is explained solely through cell death, or through differentiation into a detrimental phenotype, or a delay of the normal developmental process of Mϕ maturation that is restored by MEx. Collectively, our findings suggest that the early interaction and subsequent restoration of pulmonary myeloid populations is associated with the beneficial effects of MEx.
It is noteworthy that other studies have found that HYRX exposure leads to elevated numbers of CD45+ cells or a steady-state increase in lung Mϕs (43, 44). The reasons for conflicting findings are not immediately clear. It is convenient to suggest that subtle differences in mice strain and in O2 concentration and duration are responsible for such findings. However, studies often assess different pulmonary compartments, for example whole-lung analysis versus BALF, and often assess the mice at different developmental stages (arguably at a single study endpoint). It is well-known that Mϕ influx, plasticity, and diversity rapidly change in the developing neonatal lung (45). Furthermore, studies seldom use comparable cell surface markers to distinguish different monocyte and Mϕ lineages, hampering a full appreciation of the immune response.
The functional significance of such changes was confirmed using adoptive transfer studies, where administration of MEx-educated BMDMy ameliorated core histological and functional features of experimental BPD. Naive BMDMy and subsequently BMDMono exhibited subtle immunosuppressive actions in vitro, arguably as an apportion of such cells occupy a ‘regulatory’ CCR2−-associated phenotype. However, naive BMDMy had no impact in our in vivo BPD model. Importantly, these beneficial effects appear dose dependent, as they were only observed in mice that received a dose of 5 × 106 cells. Animals that received 0.25–1 × 106 cell doses did not manifest therapeutic benefits. Findings from this study support and expand upon our recent work that demonstrated that MEx preconditioned BMDMy provide antifibrotic actions in a murine model of bleomycin-induced pulmonary fibrosis (22). Others have shown that “MSC-EV” educated Mϕs promote early Achilles tendon healing (46) and afford antiinflammatory actions (47).
In support, using a neonatal hyperoxia-induced BPD model, we recently demonstrated MEx therapy is able to restore HYRX-damaged thymic medullary architecture, which results in a restoration of thymocyte counts, FoxP3+ regulatory T cell generation, and prevented T cell autoreactivity (48). Notably, in this body of work our biodistribution and ex vivo studies provided no evidence that MEx directly localize/interact with the thymus, nor do they provide any direct immunosuppressive actions. Collectively, this body of work supports the growing consensus that MEx afford their immunomodulatory actions via myeloid cells.
In this study, we demonstrated that MEx modulate BMDMono phenotype, promoting an Arg1+ and Il10+ state. Interestingly, we and others have previously observed that MEx promotes a “pro-resolving” M2-like Mϕ phenotype that is characterized by the super-induction of Arg1 (21, 49, 50). Here, the enhanced immunosuppressive actions of MEx-educated BMDMono, coupled with the Arg1+ and Il10+ gene expression pattern, suggest that MEx promote an MDSC-like phenotype. Supporting the notion that MEx modulate the transcriptome, this is the first study to demonstrate that MEx reprogram the epigenetic landscape, increasing open chromatin regions, thus regulating gene expression and subsequent function. Considering the long-lasting actions of MEx in vivo, our findings demonstrates that MEx alter the epigenetic landscape of target cells.
The results of this investigation should be judged in the context of the experimental model used, and we acknowledge several limitations of this work. First, we administered MEx intravenously. Other studies have demonstrated therapeutic actions of MEx through intraperitoneal and intratracheal administration. Future studies should investigate how different administration routes impact MEx biodistribution and efficacy. Elucidating the biodistribution and metabolic fate of MEx is critical to supporting smooth clinical translation. However, current biodistribution technologies are often inadequate. In addition to dye dilution via target cell fusion, a potential limitation of using dyes to track MEx is the disparity in half-life between MEx and the dye. Furthermore, dyes may influence the size and charge of MEx, in turn modifying the natural tropism and efficacy (13). At present, with each labeling method harboring advantages and limitations, a multimodal approach should be encouraged in this area. Finally, several animal models are routinely employed to recapitulate the modern-day clinical presentation of BPD. It is commonly modeled in mice because of their short gestation times and because neonatal mice are born in the saccular stage of murine lung development resembling that of a human preterm neonate (∼24 and 28 wk gestation) (51). However, every preclinical model of BPD is subject to its own advantages and limitations. Notably, adequate blood and bone marrow/tissue sampling is often not feasible in the initial postnatal period of a neonatal pup. Thus, future studies may consider larger rodent/animal models of BPD to further explore the impact of MEx on the bone marrow and on circulating immune cell populations.
Conclusions
The therapeutic actions of MEx are recapitulated, at least in part, through adoptive transfer of MEx-educated BMDMy. Our findings provide novel mechanistic insights into MEx–monocytic interaction that can be harnessed for the further development of MEx-based therapeutics for diseases of the lung as well as a diverse array of immune-mediated pathologies.
Acknowledgments
Acknowledgment
The authors thank Dr. Eleni Delavogia (Boston Children’s Hospital, Harvard Medical School, Boston, MA) and Mr. John Cortinas and Mrs. Janhavi Nadkarni (Boston Children’s Hospital) for their scientific discussions and blinded analysis. The authors also thank Dr. Nick Andrew (Boston Children’s Hospital Neurodevelopment Behavioral Core) for providing training and assistance with the murine treadmill experiments. The Boston Children’s Hospital Neurodevelopment Behavioral Core receives support from the Children’s Hospital Intellectual and Developmental Disabilities Research Center (CHB IDDRC 1U54HD090255).
Footnotes
Supported in part by NHLBI grants R01HL146128 (S.K.) and K99HL146986 (G.R.W.), National Institute of Allergy and Infectious Diseases grant R21AI134025 (S.K.), and Eunice Kennedy Shriver National Institute of Child Health and Human Development T32HD098061 Neonatal Research Training Program (E.S.T.), Charles H. Hood Foundation Major Grants Initiative to Advance Child Health (S.K.), a United Therapeutics Corporation Sponsored Research Agreement (S.K. and S.A.M.), American Thoracic Society Unrestricted Foundation Grant (G.R.W.), and The Little Giraffe Foundation Research Grant (G.R.W.).
Author Contributions: G.R.W. participated in conception and design, collection and/or assembly of data, data analysis and interpretation, financial support, and manuscript writing; M.R., A.F.-G., E.S.T., and V.Y. participated in conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing; A.H.G. provided data analysis and interpretation; X.L., M.E., and E.H. provided collection and/or assembly of data; and S.A.M. and S.K. contributed to conception and design, data analysis and interpretation, financial support, manuscript writing, and final approval.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202102-0329OC on October 26, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Kinsella JP, Greenough A, Abman SH. Bronchopulmonary dysplasia. Lancet . 2006;367:1421–1431. doi: 10.1016/S0140-6736(06)68615-7. [DOI] [PubMed] [Google Scholar]
- 2. Al-Ghanem G, Shah P, Thomas S, Banfield L, El Helou S, Fusch C, et al. Bronchopulmonary dysplasia and pulmonary hypertension: a meta-analysis. J Perinatol . 2017;37:414–419. doi: 10.1038/jp.2016.250. [DOI] [PubMed] [Google Scholar]
- 3. Mourani PM, Abman SH. Pulmonary hypertension and vascular abnormalities in bronchopulmonary dysplasia. Clin Perinatol . 2015;42:839–855. doi: 10.1016/j.clp.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol . 2005;67:623–661. doi: 10.1146/annurev.physiol.67.040403.102229. [DOI] [PubMed] [Google Scholar]
- 5. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med . 2007;357:1946–1955. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- 6. Nold MF, Mangan NE, Rudloff I, Cho SX, Shariatian N, Samarasinghe TD, et al. Interleukin-1 receptor antagonist prevents murine bronchopulmonary dysplasia induced by perinatal inflammation and hyperoxia. Proc Natl Acad Sci USA . 2013;110:14384–14389. doi: 10.1073/pnas.1306859110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willis GR, Kourembanas S, Mitsialis SA. Therapeutic applications of extracellular vesicles: perspectives from newborn medicine. Methods Mol Biol . 2017;1660:409–432. doi: 10.1007/978-1-4939-7253-1_34. [DOI] [PubMed] [Google Scholar]
- 8. Mitsialis SA, Kourembanas S. Stem cell-based therapies for the newborn lung and brain: possibilities and challenges. Semin Perinatol . 2016;40:138–151. doi: 10.1053/j.semperi.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matthay MA, Anversa P, Bhattacharya J, Burnett BK, Chapman HA, Hare JM, et al. Cell therapy for lung diseases. Report from an NIH-NHLBI workshop, November 13-14, 2012. Am J Respir Crit Care Med . 2013;188:370–375. doi: 10.1164/rccm.201303-0522WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guillot M, Offringa M, Lacaze-Masmonteil T, Thébaud B. Cell-based therapy for bronchopulmonary dysplasia in preterm infants. Can J Physiol Pharmacol . 2019;97:232–234. doi: 10.1139/cjpp-2018-0342. [DOI] [PubMed] [Google Scholar]
- 11. Álvarez-Fuente M, Moreno L, Mitchell JA, Reiss IK, Lopez P, Elorza D, et al. Preventing bronchopulmonary dysplasia: new tools for an old challenge. Pediatr Res . 2019;85:432–441. doi: 10.1038/s41390-018-0228-0. [DOI] [PubMed] [Google Scholar]
- 12. Kang M, Thébaud B. Stem cell biology and regenerative medicine for neonatal lung diseases. Pediatr Res . 2018;83:291–297. doi: 10.1038/pr.2017.232. [DOI] [PubMed] [Google Scholar]
- 13. Willis GR, Mitsialis SA, Kourembanas S. “Good things come in small packages”: application of exosome-based therapeutics in neonatal lung injury. Pediatr Res . 2018;83:298–307. doi: 10.1038/pr.2017.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Augustine S, Avey MT, Harrison B, Locke T, Ghannad M, Moher D, et al. Mesenchymal stromal cell therapy in bronchopulmonary dysplasia: systematic review and meta-analysis of preclinical studies. Stem Cells Transl Med . 2017;6:2079–2093. doi: 10.1002/sctm.17-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeung V, Willis GR, Taglauer E, Mitsialis SA, Kourembanas S.Paving the road for mesenchymal stem cell-derived exosome therapy in bronchopulmonary dysplasia and pulmonary hypertension. In: Burgess JK, Heijink IH, editors. Stem cell-based therapy for lung disease. Springer Nature Switzerland 2019. pp. 131–152. [Google Scholar]
- 16. Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, et al. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med . 2009;180:1122–1130. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hansmann G, Fernandez-Gonzalez A, Aslam M, Vitali SH, Martin T, Mitsialis SA, et al. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm Circ . 2012;2:170–181. doi: 10.4103/2045-8932.97603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Haaften T, Byrne R, Bonnet S, Rochefort GY, Akabutu J, Bouchentouf M, et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med . 2009;180:1131–1142. doi: 10.1164/rccm.200902-0179OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emukah C, Dittmar E, Naqvi R, Martinez J, Corral A, Moreira A, et al. Mesenchymal stromal cell conditioned media for lung disease: a systematic review and meta-analysis of preclinical studies. Respir Res . 2019;20:239. doi: 10.1186/s12931-019-1212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, et al. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation . 2012;126:2601–2611. doi: 10.1161/CIRCULATIONAHA.112.114173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Willis GR, Fernandez-Gonzalez A, Anastas J, Vitali SH, Liu X, Ericsson M, et al. Mesenchymal stromal cell exosomes ameliorate experimental bronchopulmonary dysplasia and restore lung function through macrophage immunomodulation. Am J Respir Crit Care Med . 2018;197:104–116. doi: 10.1164/rccm.201705-0925OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mansouri N, Willis GR, Fernandez-Gonzalez A, Reis M, Nassiri S, Mitsialis SA, et al. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight . 2019;4:e128060. doi: 10.1172/jci.insight.128060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willis GR, Fernandez-Gonzalez A, Reis M, Yeung V, Liu X, Ericsson M, et al. Mesenchymal stromal cell-derived small extracellular vesicles restore lung architecture and improve exercise capacity in a model of neonatal hyperoxia-induced lung injury. J Extracell Vesicles . 2020;9:1790874. doi: 10.1080/20013078.2020.1790874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Braun RK, Chetty C, Balasubramaniam V, Centanni R, Haraldsdottir K, Hematti P, et al. Intraperitoneal injection of MSC-derived exosomes prevent experimental bronchopulmonary dysplasia. Biochem Biophys Res Commun . 2018;503:2653–2658. doi: 10.1016/j.bbrc.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaubey S, Thueson S, Ponnalagu D, Alam MA, Gheorghe CP, Aghai Z, et al. Early gestational mesenchymal stem cell secretome attenuates experimental bronchopulmonary dysplasia in part via exosome-associated factor TSG-6. Stem Cell Res Ther . 2018;9:173–173. doi: 10.1186/s13287-018-0903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Porzionato A, Zaramella P, Dedja A, Guidolin D, Van Wemmel K, Macchi V, et al. Intratracheal administration of clinical-grade mesenchymal stem cell-derived extracellular vesicles reduces lung injury in a rat model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol . 2019;316:L6–L19. doi: 10.1152/ajplung.00109.2018. [DOI] [PubMed] [Google Scholar]
- 27. Monsel A, Zhu YG, Gudapati V, Lim H, Lee JW. Mesenchymal stem cell derived secretome and extracellular vesicles for acute lung injury and other inflammatory lung diseases. Expert Opin Biol Ther . 2016;16:859–871. doi: 10.1517/14712598.2016.1170804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burrello J, Monticone S, Gai C, Gomez Y, Kholia S, Camussi G. Stem cell-derived extracellular vesicles and immune-modulation. Front Cell Dev Biol . 2016;4:83. doi: 10.3389/fcell.2016.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen W, Huang Y, Han J, Yu L, Li Y, Lu Z, et al. Immunomodulatory effects of mesenchymal stromal cells-derived exosome. Immunol Res . 2016;64:831–840. doi: 10.1007/s12026-016-8798-6. [DOI] [PubMed] [Google Scholar]
- 30. Willis GR, Fernandez-Gonzalez A, Reis M, Mitsialis SA, Kourembanas S. Macrophage Immunomodulation: The gatekeeper for mesenchymal stem cell derived-exosomes in pulmonary arterial hypertension? Int J Mol Sci . 2018;19:2534. doi: 10.3390/ijms19092534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles . 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Rocco G, Baldari S, Toietta G. Towards therapeutic delivery of extracellular vesicles: strategies for in vivo tracking and biodistribution analysis. Stem Cells Int . 2016;2016:5029619. doi: 10.1155/2016/5029619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gangadaran P, Hong CM, Ahn B-C. Current perspectives on in vivo noninvasive tracking of extracellular vesicles with molecular imaging. BioMed Res Int . 2017;2017:9158319. doi: 10.1155/2017/9158319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gangadaran P, Hong CM, Ahn B-C. An update on in vivo imaging of extracellular vesicles as drug delivery vehicles. Front Pharmacol . 2018;9:169. doi: 10.3389/fphar.2018.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wiklander OPB, Nordin JZ, O’Loughlin A, Gustafsson Y, Corso G, Mäger I, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles . 2015;4:26316. doi: 10.3402/jev.v4.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Royo F, Cossío U, Ruiz de Angulo A, Llop J, Falcon-Perez JM. Modification of the glycosylation of extracellular vesicles alters their biodistribution in mice. Nanoscale . 2019;11:1531–1537. doi: 10.1039/c8nr03900c. [DOI] [PubMed] [Google Scholar]
- 37. Lankford KL, Arroyo EJ, Nazimek K, Bryniarski K, Askenase PW, Kocsis JD. Intravenously delivered mesenchymal stem cell-derived exosomes target M2-type macrophages in the injured spinal cord. PLoS One . 2018;13:e0190358. doi: 10.1371/journal.pone.0190358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Dea KP, Tan YY, Shah S, V Patel B, C Tatham K, Wilson MR, et al. Monocytes mediate homing of circulating microvesicles to the pulmonary vasculature during low-grade systemic inflammation. J Extracell Vesicles . 2020;9:1706708. doi: 10.1080/20013078.2019.1706708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prince LR, Maxwell NC, Gill SK, Dockrell DH, Sabroe I, McGreal EP, et al. Macrophage phenotype is associated with disease severity in preterm infants with chronic lung disease. PLoS One . 2014;9:e103059. doi: 10.1371/journal.pone.0103059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nupponen I, Pesonen E, Andersson S, Mäkelä A, Turunen R, Kautiainen H, et al. Neutrophil activation in preterm infants who have respiratory distress syndrome. Pediatrics . 2002;110:36–41. doi: 10.1542/peds.110.1.36. [DOI] [PubMed] [Google Scholar]
- 41. Clement A, Chadelat K, Sardet A, Grimfeld A, Tournier G. Alveolar macrophage status in bronchopulmonary dysplasia. Pediatr Res . 1988;23:470–473. doi: 10.1203/00006450-198805000-00007. [DOI] [PubMed] [Google Scholar]
- 42. Kalymbetova TV, Selvakumar B, Rodríguez-Castillo JA, Gunjak M, Malainou C, Heindl MR, et al. Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia. J Pathol . 2018;245:153–159. doi: 10.1002/path.5076. [DOI] [PubMed] [Google Scholar]
- 43. Al-Rubaie A, Wise AF, Sozo F, De Matteo R, Samuel CS, Harding R, et al. The therapeutic effect of mesenchymal stem cells on pulmonary myeloid cells following neonatal hyperoxic lung injury in mice. Respir Res . 2018;19:114. doi: 10.1186/s12931-018-0816-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jankov RP, Luo X, Belcastro R, Copland I, Frndova H, Lye SJ, et al. Gadolinium chloride inhibits pulmonary macrophage influx and prevents O(2)-induced pulmonary hypertension in the neonatal rat. Pediatr Res . 2001;50:172–183. doi: 10.1203/00006450-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 45. Tan SYS, Krasnow MA. Developmental origin of lung macrophage diversity. Development . 2016;143:1318–1327. doi: 10.1242/dev.129122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chamberlain CS, Clements AEB, Kink JA, Choi U, Baer GS, Halanski MA, et al. Extracellular vesicle-educated macrophages promote early achilles tendon healing. Stem Cells . 2019;37:652–662. doi: 10.1002/stem.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, et al. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med . 2017;196:1275–1286. doi: 10.1164/rccm.201701-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reis M, Willis GR, Fernandez-Gonzalez A, Yeung V, Taglauer E, Magaletta M, et al. Mesenchymal stromal cell-derived extracellular vesicles restore thymic architecture and T cell function disrupted by neonatal hyperoxia. Front Immunol . 2021;12:640595. doi: 10.3389/fimmu.2021.640595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heo JS, Choi Y, Kim HO. Adipose-derived mesenchymal stem cells promote M2 macrophage phenotype through exosomes. Stem Cells Int . 2019:7921760. doi: 10.1155/2019/7921760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu H, Liang Z, Wang F, Zhou C, Zheng X, Hu T, et al. Exosomes from mesenchymal stromal cells reduce murine colonic inflammation via a macrophage-dependent mechanism. JCI Insight . 2019;4:e131273. doi: 10.1172/jci.insight.131273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nardiello C, Mižíková I, Morty RE. Looking ahead: where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res . 2017;367:457–468. doi: 10.1007/s00441-016-2534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]