

Abstract
Rationale
Alteration of human respiratory microbiota had been observed in coronavirus disease (COVID-19). How the microbiota is associated with the prognosis in COVID-19 is unclear.
Objectives
To characterize the feature and dynamics of the respiratory microbiota and its associations with clinical features in patients with COVID-19.
Methods
We conducted metatranscriptome sequencing on 588 longitudinal oropharyngeal swab specimens collected from 192 patients with COVID-19 (including 39 deceased patients) and 95 healthy controls from the same geographic area. Meanwhile, the concentration of 27 cytokines and chemokines in plasma was measured for patients with COVID-19.
Measurements and Main Results
The upper respiratory tract (URT) microbiota in patients with COVID-19 differed from that in healthy controls, whereas deceased patients possessed a more distinct microbiota, both on admission and before discharge/death. The alteration of URT microbiota showed a significant correlation with the concentration of proinflammatory cytokines and mortality. Specifically, Streptococcus-dominated microbiota was enriched in recovered patients, and showed high temporal stability and resistance against pathogens. In contrast, the microbiota in deceased patients was more susceptible to secondary infections and became more deviated from the norm after admission. Moreover, the abundance of S. parasanguinis on admission was significantly correlated with prognosis in nonsevere patients (lower vs. higher abundance, odds ratio, 7.80; 95% CI, 1.70–42.05).
Conclusions
URT microbiota dysbiosis is a remarkable manifestation of COVID-19; its association with mortality suggests it may reflect the interplay between pathogens, symbionts, and the host immune status. Whether URT microbiota could be used as a biomarker for diagnosis and prognosis of respiratory diseases merits further investigation.
Keywords: mortality, microbiome, prognosis, COVID-19, risk stratification
At a Glance Commentary
Scientific Knowledge on the Subject
Human microbiota plays a crucial role in individual health; alteration of the microbiota has been observed in various chronic and acute respiratory diseases and tends to be associated with the severity of the disease. How the upper respiratory tract microbiota is associated with prognosis in COVID-19 is unclear.
What This Study Adds to the Field
This study demonstrates that the composition and dynamics of the oropharyngeal microbiota were significantly correlated with mortality in COVID-19. A higher abundance of Streptococcus on admission predicts a better prognosis.
Coronavirus disease (COVID-19) has infected more than 100 million people worldwide. Although most patients showed mild symptoms or were asymptomatic (1), approximately 14% developed severe diseases, 5% were critically ill, and the overall mortality rate is 3.2% (2). Older people and patients with underlying diseases are at increased risk for severe illness from COVID-19 and have a higher mortality rate. Other risk factors include smoking history, pregnancy, male sex, and obesity (3). Genetic variants on Toll-like receptor 7, ABO blood group locus, and 3p21.31 gene cluster were also associated with severe COVID-19 (4, 5).
Human microbiota plays a crucial role in individual health; alteration of the human microbiota has been observed in various chronic and acute diseases (6). In particular, microorganisms residing in the gastrointestinal tract (GIT) and upper respiratory tract (URT) can alter the susceptibility to and outcomes of infectious diseases (7). The underlying mechanisms include colonization resistance and induction of the immune responses in the host (8). In recent studies, the diversity and composition of GIT microbiota showed significant differences between patients with COVID-19 and healthy controls (HCs) and tended to be associated with the severity of the disease (9–13). Bacterial and fungal opportunistic pathogens were enriched in the feces of patients with COVID-19, and a secondary infection was suspected, suggesting that dysbiosis of the gut microbiota is common in patients with COVID-19 (9–12, 14). Respiratory microbiota contributes to the foremost barrier to viral infection (15). How it is altered in patients with COVID-19 is largely unknown (16–18). Moreover, little is known about the association between the microbiota and the mortality risk, which is one of the most crucial questions regarding microbiota’s contribution to the health of patients with COVID-19.
The power and validity of previous studies are limited by the small sample size and overrepresentation of patients with mild symptoms. To obtain a thorough understanding of the association between microbiota and COVID-19 prognosis, especially those related to mortality, a large cohort with deceased patients is needed. LOTUS (Lopinavir–Ritonavir [L–R] Trial for Suppression of SARS-CoV-2 in China) is a clinical trial that aims to evaluate the efficacy and safety of lopinavir–ritonavir treatment for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (19). LOTUS recruited 192 patients with severe COVID-19, with an overall mortality rate of 22.1%. Serial oropharyngeal swab samples were collected. By analyzing metatranscriptome data from 588 COVID-19 samples and 95 HCs, we found that the URT microbiota was significantly different between recovered and deceased patients on admission and afterwards. The abundance of Streptococcus, particularly S. parasanguinis, was strongly associated with the mortality risk.
Methods
Study Design and Sample Collection
Oropharyngeal swab samples were collected on Days 1, 5, 10, 14, 21, and 28 after admission from patients enrolled in LOTUS (ChiCTR2000029308). The swab was placed into virus transport medium (You Kang Healthcare Trade Co.) and stored at −80°C until transported to the molecular lab for RNA extraction and library preparation. All patients were positive for SARS-CoV-2 RNA tested by RT-PCR assay (Shanghai ZJ Bio Tec or Sansure Biotech) before admission. Owing to the emergency situation at the early stage of the pandemic, an unsophisticated seven-category ordinal scale severity score was assigned to each patient at each time point, ranging from 2 (not hospitalized, not requiring supplemental oxygen) to 7 (death) (19). Ninety-five healthy controls were collected from the community in the same geographic region, who reported no pulmonary disease and no use of antibiotics in the preceding 3 months.
The study was approved by the Institutional Review Board of Jin Yin-Tan Hospital (KY2020-02.01). Written informed consent was obtained from all patients or their legal representatives if they were too unwell to provide consent.
Statistical Analysis
Continuous variables are described as medians (interquartile ranges). α-Diversity and β-diversity were computed using R packages, phyloseq and vegan (20, 21). Correlation between categorical metadata was calculated by Cramer’s V test. Permutational multivariate ANOVA (PERMANOVA) and envfit functions were used to determine the host factors associated with the microbial community and evaluate the similarity between microbiotas. For the permutation test, the P value was calculated based on 2,000 permutations. Fisher exact tests and Mann-Whitney U tests were used to compare categorical variables and continuous variables, respectively. Multiple comparisons were corrected by the false discovery rate algorithm, and a cutoff value of 0.05 was applied. All statistical analysis was implemented in RStudio (22).
Additional methods are provided in an online supplement.
Results
Microbiota Composition in the Upper Respiratory Tract of Patients with COVID-19
Metatranscriptome sequencing, which targets RNA from both the microbial community and human genes, was conducted on 588 consecutive oropharyngeal swab specimens (OPs) collected from 192 patients with severe COVID-19 (Figure 1A, Figure E1A in the online supplement, and Table 1). For comparison, OPs were collected from 95 healthy individuals without any pulmonary diseases in the community from the same geographic region in April 2020. Deionized water was used as the negative control (NC), which was processed following the same protocol as for clinical samples. Details of the sequencing are described in the online supplement.
Figure 1.

Project design and overview of the upper respiratory tract microbiota. (A) The design of the study. (B) Relative abundance of the 10 most abundant genera in coronavirus disease (COVID-19) and healthy controls (HC). The genera whose abundances in both groups were not significantly higher than negative control (NC) were discarded. The genera were ordered by their abundance in COVID-19. Genera significantly enriched in patients with COVID-19 and HC were labeled in purple and green, respectively. The inverted triangle indicates the genus whose abundance was not significantly higher than that in NC. The boxes represent 25th–75th percentiles, the horizontal lines indicate the median, and the whiskers were drawn from the box to the extremes (values that were lower/greater than first/third quartile minus/plus 1.5 times the interquartile range were regarded as outliers). (C) Principal coordinate analysis plot of samples at the genus level on admission (left) and before discharge/death (right). R2 calculated by permutational multivariate ANOVA when adjusting age and sex is shown in the figure, and all P values were less than 0.001. PCoA = principal coordinate analysis.
Table 1.
Characteristics of the Study Population
| All Patients (N = 192) |
Recovered (N = 153) |
Deceased (N = 39) |
Healthy Controls† (N = 95) |
|
|---|---|---|---|---|
| Age, median (IQR), yr* | 58 (49–68) | 56 (46–65) | 67 (60–74) | 47 (33–61) |
| Sex, M, n (%) | 114 (59) | 87 (57) | 27 (69) | 38 (40) |
| Lopinavir–ritonavir treatment, n (%) | 95 (49) | 80 (52) | 15 (38) | N/A |
| Severity-A, n (%)* | ||||
| 3: no supplemental oxygen | 27 (14) | 22 (14) | 5 (13) | N/A |
| 4: supplemental oxygen | 137 (71) | 122 (80) | 15 (38) | N/A |
| 5: nasal high-flow oxygen/invasive mechanical ventilation | 28 (15) | 9 (6) | 19 (49) | N/A |
| Corticosteroid, n (%)* | 65 (34) | 39 (25) | 26 (67) | N/A |
| Comorbidity, n (%)* | 110 (57) | 80 (52) | 30 (77) | N/A |
| Duration of antibiotic use, median (IQR), d | 11 (7–14) | 11 (6–14) | 10 (7–15) | N/A |
| High-grade antibiotics, n (%)* | 43 (22) | 16 (10) | 27 (69) | N/A |
Definition of abbreviations: IQR = interquartile range; N/A = not available; Severity-A = severity on admission.
Characteristics that significantly differed between recovered patients and deceased patients.
Healthy controls were volunteers from the community in the same city. Only sex and age were collected.
Bacteria accounted for 97.02% of the microbial reads in the OPs of the patients with COVID-19, followed by fungi (2.76%), viruses (0.22%), and archaea (0.001%). Specifically, SARS-CoV-2 accounted for 0.003% of all microbial reads (0.0002–0.05%). The microbial community was mainly composed of Streptococcus, Veillonella, and other known commensal microbes in COVID-19 and HC samples (Figure 1B), which could be assigned into 11 microbial clusters represented by distinct dominant microbes (see details in the online supplement). In contrast, the composition of NC was more homogeneous and consisted of Escherichia, Cutibacterium, and Acinetobacter, which were rarely present in OP samples (Figure E1B).
The overall microbial composition was different between patients with COVID-19 and HCs (R2 = 0.05, P < 0.001 on admission; R2 = 0.04, P < 0.001 before discharge/death; PERMANOVA test) (Figure 1C). Veillonella, Actinomyces, and Rothia were significantly enriched in patients with COVID-19, whereas Streptococcus, Capnocytophaga, and the other eight genera were more abundant in HCs (Figure E1C). However, because of the differences in the age, sex, and disease state between the two groups, the differentially abundant microbes were not necessarily specifically associated with COVID-19. Notably, the microbiota in deceased patients significantly differed from that in recovered patients after adjusting for all covariables, which was more remarkable before discharge/death (R2 = 0.04, P < 0.001 on admission; R2 = 0.10, P < 0.001 before discharge/death). Resampling tests confirmed that the differences in microbiota diversity between different subgroups were not artifacts due to uneven sample sizes (Figure E1D).
Association between the Upper Respiratory Tract Microbiota and Mortality
Samples were then analyzed at two critical time points, admission and discharge/death, which refer to the time that the first sample was collected within the first 2 days after admission and the time that the last sample was collected within 1 week before discharge/death, respectively. Among various demographic and clinical features, mortality was the only feature significantly associated with the microbiota at both time points (Table 2). Besides, the use of high-grade antibiotics (drug list is shown in Table E1) was associated with the microbiota before discharge/death. Moreover, we found that both age and the severity on admission (severity-A) showed an interaction effect with mortality and tended to explain some variation in the microbiota composition before discharge/death (P < 0.05). After taking this interaction effect out, the association between mortality and microbiota was still significant (P < 0.01).
Table 2.
Association between the Upper Respiratory Tract Microbiota Composition and Metadata in Patients with COVID-19
| Admission |
Discharge/Death |
|||
|---|---|---|---|---|
| R 2 | P Value | R 2 | P Value | |
| Mortality | 0.026 | 0.005 | 0.024 | 0.002 |
| Age | 0.011 | 0.219 | 0.008 | 0.168 |
| Sex | 0.006 | 0.593 | 0.004 | 0.571 |
| Corticosteroid | 0.006 | 0.633 | 0.003 | 0.807 |
| Severity-A | 0.015 | 0.508 | 0.017 | 0.100 |
| Lopinavir–ritonavir | 0.009 | 0.314 | 0.002 | 0.957 |
| Comorbidity | 0.004 | 0.828 | 0.004 | 0.539 |
| Antibiotics* | 0.011 | 0.184 | 0.008 | 0.166 |
| High-grade antibiotics | 0.012 | 0.250 | 0.019 | 0.008 |
Definition of abbreviations: COVID-19 = coronavirus disease; Severity-A = severity on admission.
R2, which represents the proportion of variance explained by the factor, and the P value were calculated by permutational multivariate ANOVA analysis. P values below 0.05 are in bold.
Duration of antibiotic use (0 if no antibiotics were taken).
Generalized additive model for location, scale, and shape (GAMLSS) regression analysis identified Streptococcus as the most enriched microbe in the recovered group on admission (Figure 2A and Figure E2A), which was also true before discharge/death but was ranked behind a low abundant genus Atopobium (0.0014 [0.0001–0.0054]) (Figure 2B and Figure E2B). The enrichment of Streptococcus had been further confirmed by linear discriminant analysis effect size (LEfSe) analysis (Figures E2C and E2D) but cannot be verified by multiple linear regression when controlling for all covariates on admission (adjusted P value [Padj] = 0.14 on admission; Padj < 0.05 before discharge/death), suggesting possible confounding effects.
Figure 2.
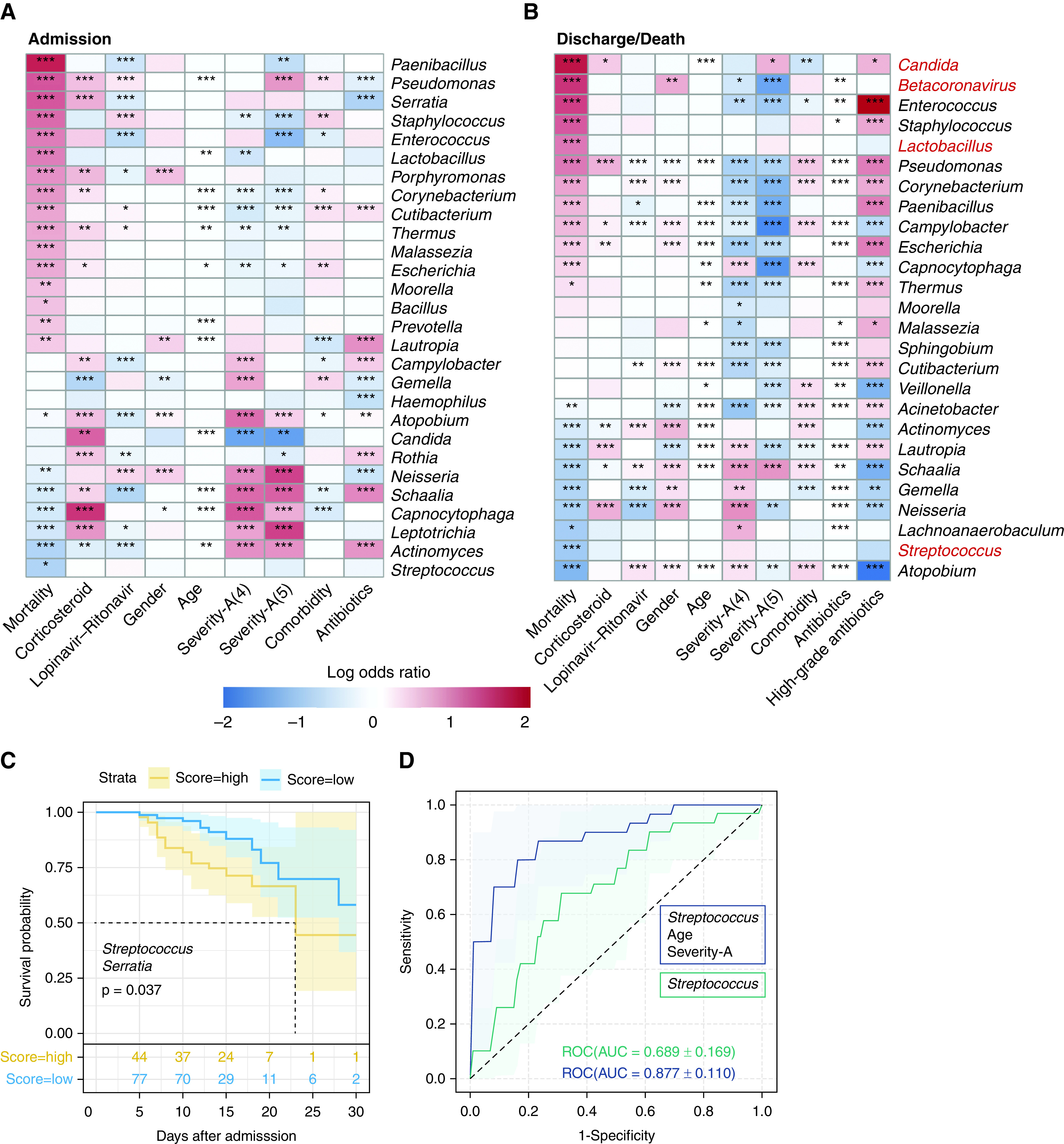
The associations between the upper respiratory tract microbiota and mortality. Genera that are associated with different metadata identified by generalized additive model for location, scale, and shape analysis are shown in (A) (on admission) and (B) (before discharge/death) (adjusted P value < 0.05). Genera were ordered by the regression coefficients with mortality. Mortality-associated genera identified by the other two methods (multivariate linear regression and linear discriminant analysis effect size) were marked in red. The color in the heat map represents the regression coefficients (log odds ratio) estimated by generalized additive model for location, scale, and shape. *P < 0.05, **P < 0.01, and ***P < 0.001. (C) Kaplan-Meier survival curves for the two groups classified by two genera selected by Cox regression, which included Streptococcus and Serratia. The risk score was calculated as the sum of all variables (the abundance of the genus) weighted by their multivariate Cox regression coefficients. The cutoff to classify the patients as the high-score group and the low-score group was selected as the risk score that resulted in the highest area under the curve (AUC) (see details in the online supplement). Log-rank P value is shown in the figure. (D) Receiver operator characteristic (ROC) curves for the mortality classifier based on both host factors and the microbiota composition (blue line), and merely on microbiota composition (green line). Severity-A = severity on admission.
To further investigate microbes associated with disease progression, Cox regression was conducted on mortality-associated genera selected by GAMLSS on admission together with all demographic and clinical covariates. Finally, three variables were selected as best predictors, including age and the abundance of two genera (Streptococcus and Serratia) (Figure E2E). Kaplan-Meier survival analysis confirmed that the abundances of these two bacteria were significantly associated with the mortality risk (Figure 2C). Moreover, we built a classifier based on L1 regularized logistic regression model to distinguish the recovered and deceased patients on admission. The best performance was achieved with three variables (Figure 2D). Streptococcus was the only microbe with a nonzero coefficient besides age and severity-A (area under the curve [AUC] = 0.877) (see more performance metrics in Table E2). The classifier based merely on Streptococcus had an AUC of 0.689 (Table E2), indicating that the abundance of Streptococcus on admission was correlated with the clinical outcome.
In contrast, Enterococcus and Candida were recognized as the most enriched genera in deceased patients by GAMLSS and LEfSe analysis before discharge/death, but not on admission (Figures 2A, 2B, and E2A–E2D). Analysis at the species level confirmed Candida albicans and Enterococcus faecium as the only species in the aforementioned two genera that enriched in deceased patients (Figures E3A and E3B).
Association between the Abundance of Streptococcus Parasanguinis and Mortality
The abundance of Streptococcus was higher in the recovered group at all time points except Day 21 (P < 0.01) (Figure 3A) and remained stable during the hospitalization (admission vs. discharge/death, P = 0.19). In contrast, the abundance of Streptococcus was marginally decreased over time in the deceased group (P = 0.095). Also, the case mortality rate (CMR) declined dramatically with the increased abundance of Streptococcus, and this trend was more evident before discharge/death (Figure 3B).
Figure 3.

Association between the abundance of Streptococcus and mortality in patients with coronavirus disease (COVID-19). (A) The abundance of Streptococcus in recovered and deceased patients at different time points. (B) The accumulative mortality rate for individuals with different Streptococcus abundances. (C) The Kaplan-Meier curves for two groups classified by the abundance of S. parasanguinis. The cutoff to classify the patients as the high-score group and the low-score group was the median of the abundance of S. parasanguinis. The P value was calculated by log-rank test. (D) The abundance of S. parasanguinis in different severity groups. The boxes represent 25th–75th percentiles, the horizontal lines indicate the median, and the whiskers were drawn from the box to the extremes (values that were lower/greater than first/third quartile minus/plus 1.5 times the interquartile range were regarded as outliers). **P < 0.01 and ***P < 0.001. ns = not significant.
Among all species belonging to Streptococcus, S. parasanguinis showed the greatest fold change between recovered and deceased patients (14.7-fold on admission and 229-fold before discharge/death) (Figures E3C and E3D) and assigned with the lowest odds ratio by GAMLSS analysis (Figure E3A). Meanwhile, the L1 regularized logistic regression method identified S. parasanguinis abundance as the primary variable associated with CMR, followed by Streptococcus sp. oral taxon 431 (Figure E3E and Table E2). Kaplan-Meier survival analysis confirmed S. parasanguinis as a candidate marker for prognosis prediction but not for Streptococcus sp. oral taxon 431 (Figure 3C).
We then examined whether any confounding factors were involved in the association between the abundance of S. parasanguinis on admission and mortality. First, we found that mortality was the only factor associated with the abundance of S. parasanguinis among all variables (Padj < 0.05). Second, multivariate logistic regression analysis confirmed that the abundance of S. parasanguinis as well as age and severity-A were independently correlated with mortality (Table 3). However, considering that there were many possible species associated with mortality (31 species as revealed by GAMLSS analysis), the adjusted P value became insignificant after multiple testing corrections despite the fact that the P value for S. parasanguinis was the lowest among all species (Padj = 0.060).
Table 3.
Logistic Regression Analysis of Variables Associated with Mortality
| Univariate OR (95% CI) | P Value | Multivariate OR (95% CI) | P Value | |
|---|---|---|---|---|
| S. parasanguinis, relative abundance | 0.30 (0.13–0.66) | 0.004 | 0.09 (0.02–0.38) | 0.002 |
| Sex (M vs. F) | 1.70 (0.79–3.84) | 0.185 | 1.93 (0.53–7.93) | 0.333 |
| Age | 1.08 (1.04–1.12) | <0.001 | 1.12 (1.06–1.21) | 0.001 |
| Lopinavir–ritonavir (yes vs. no) | 0.50 (0.23–1.05) | 0.071 | 0.59 (0.16–2.02) | 0.399 |
| Severity-A (4 vs. 3) | 0.56 (0.20–1.87) | 0.312 | 0.56 (0.11–3.14) | 0.485 |
| Severity-A (5 vs. 3) | 11.73 (3.26–49.99) | <0.001 | 19.56 (2.38–243.84) | 0.010 |
| Antibiotics* | 0.79 (0.31–1.93) | 0.608 | 0.98 (0.26–3.65) | 0.980 |
| Corticosteroid (yes vs. no) | 5.78 (2.68–12.92) | <0.001 | 1.8 (0.3–10.05) | 0.504 |
| Comorbidity (with vs. without) | 3.09 (01.37–7.68) | 0.009 | 0.97 (0.21–4.31) | 0.964 |
Definition of abbreviations: CI = confidence interval; OR = odds ratio; S. parasanguinis = Streptococcus parasanguinis; severity-A = severity on admission.
P values below 0.05 are in bold.
Duration of antibiotic use. High-grade antibiotics were not included in the analysis as only one patient took high-grade antibiotics before sampling.
Intriguingly, the degree of correlation between the abundance of S. parasanguinis and mortality was greater in patients with less severe symptoms on admission (P = 0.080 and 0.010 in patients with a severity score of 3 and 4, respectively) (Figure 3D). Specifically, 97.3% (36/37) of patients with a high abundance of S. parasanguinis (>10%) and 93.75% (60/64) of patients with an intermediate abundance of S. parasanguinis (>1%) on admission in the less severe group recovered, resulting in a CMR of 6.25%. In contrast, patients with negligible S. parasanguinis (<0.1%) suffered a higher CMR of 35% (7/20) (odds ratio, 7.8; 95% CI, 1.7–42.0; P < 0.01). Consequently, the predictive power of S. parasanguinis was stronger in the less severely ill patients compared with that in all patients (AUC, 0.754 vs. 713) (Table E2). Of note, patients with a severity score of 5 on admission showed the highest CMR (76.9%), which agreed with the observation that a higher severity score on admission itself was a risk factor for mortality (Table 3).
Codetection of Pathogens in the Upper Respiratory Tract of Patients with COVID-19
We then screened all samples for 58 common respiratory pathogens (Table E3). Overall, 21.9% of patients had at least one pathogen besides SARS-CoV-2, and the proportion of patients with pathogens was higher in deceased patients than that in recovered patients (48.7% vs. 15.0%, P < 0.001). Also, the number of pathogens was significantly higher in deceased patients than in recovered patients (0 [0–1] vs. 0 [0–0], P < 0.001). Specifically, Candida albicans and Enterococcus faecium were more prevalent in deceased patients than in recovered patients and HCs (Figure 4A). Notably, these two species’ abundances on admission were relatively low and similar between recovered and deceased patients but remarkably increased during hospitalization in deceased patients (Figures 4B and 4C), suggesting secondary infections.
Figure 4.

Codetection of other pathogens in patients with coronavirus disease (COVID-19). (A) The incidence of potential pathogens in different groups. The bar plot shows the incidence of each potential pathogen (left y-axis), and the triangle shows the median abundance of the pathogen in positive samples (right y-axis). The potential pathogens enriched in deceased patients with COVID-19 were labeled in bold (Fisher’s exact test). (B and C) The abundance of Candida albicans (B) and Enterococcus faecium (C) on admission and before discharge/death in paired samples from the same patient. Samples from the same patient were connected by a solid line. The boxes represent 25th–75th percentiles, the horizontal lines indicate the median, and the whiskers were drawn from the box to the extremes (values that were lower/greater than first/third quartile minus/plus 1.5 times the interquartile range were regarded as outliers). *P < 0.05, ***P < 0.001, and ****P < 0.0001. A. baumannii = Acinetobacter baumannii; K. pneumoniae = Klebsiella pneumoniae; P. aeruginosa = Pseudomonas aeruginosa; S. aureus = Staphylococcus aureus; S. pneumoniae = Streptococcus pneumoniae.
Virus coinfection was rare. Human alphaherpesvirus 1 and rhinovirus A were detected with more than 50 reads in 10 samples from three deceased patients and five recovered patients, and 1 sample from a recovered patient, respectively. Other possible viral coinfections included influenza, enterovirus, etc., which were revealed by less than 50 reads (Table E3).
Dynamics of the Upper Respiratory Tract Microbiota in Patients with COVID-19
The magnitude of microbiota change in deceased patients was higher than that in recovered patients and tended to increase over time (P = 0.10) (Figure 5A). A similar trend was observed when using the Bray-Curtis distance to HC (Figure 5B). Strikingly, the distance to HC increased in deceased patients over time but tended to decline in the recovered group (Figure 5B). Specifically, Streptococcus and Schaalia were the two genera whose abundances were significantly approaching the level in HC during hospitalization (Figures E4A and E4B). Moreover, multivariate linear regression results indicated that multiple variables were correlated with the microbiota change besides mortality (Table E4).
Figure 5.

Dynamics of the upper respiratory tract microbiota and its association with mortality risk. (A) Microbiota change (measured by Bray-Curtis distance to the sample of the first day after admission) for each patient at different time points. (B) Bray-Curtis distance to the healthy controls at different time points. The distances within healthy controls are shown for comparison. The boxes represent 25th–75th percentiles, the horizontal lines indicate the median, and the whiskers were drawn from the box to the extremes (values that were lower/greater than first/third quartile minus/plus 1.5 times the interquartile range were regarded as outliers). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Of note, the distance from the recovered group to HC before discharge was still significantly greater than that within HC (Figure 5B), indicating that the URT microbiota was not restored at discharge. The dynamics of the URT microbiota had also been analyzed as transitions between different CSs. The results are shown in the online supplement.
Association between Immune Response and the Microbiota
The interaction between human microbiota and host immune homeostasis has been extensively investigated (23, 24). To explore the correlation between OP microbiota and human immune status in patients with COVID-19, we have quantified the level of 27 cytokines and chemokines using the plasma of patients with COVID-19. Meanwhile, the concentration of white blood cells (WBCs), neutrophils (NEUs), lymphocytes (LYMs), and procalcitonin and erythrocyte sedimentation rate were retrieved from clinical records. First, we found that the concentration of multiple cytokines was positively correlated with mortality on admission and before discharge/death (Figures 6A and 6B), indicating enhanced immune response in deceased patients. Furthermore, the concentration of WBCs, NEUs, and IL-6 significantly increased after admission in deceased patients (Figure 6C), suggesting that exacerbated and prolonged inflammation might result in poor outcomes in patients with COVID-19. Meanwhile, decreased LYM proportion was observed in deceased patients, supporting it as a possible biomarker for predicting the disease severity of COVID-19 (25).
Figure 6.
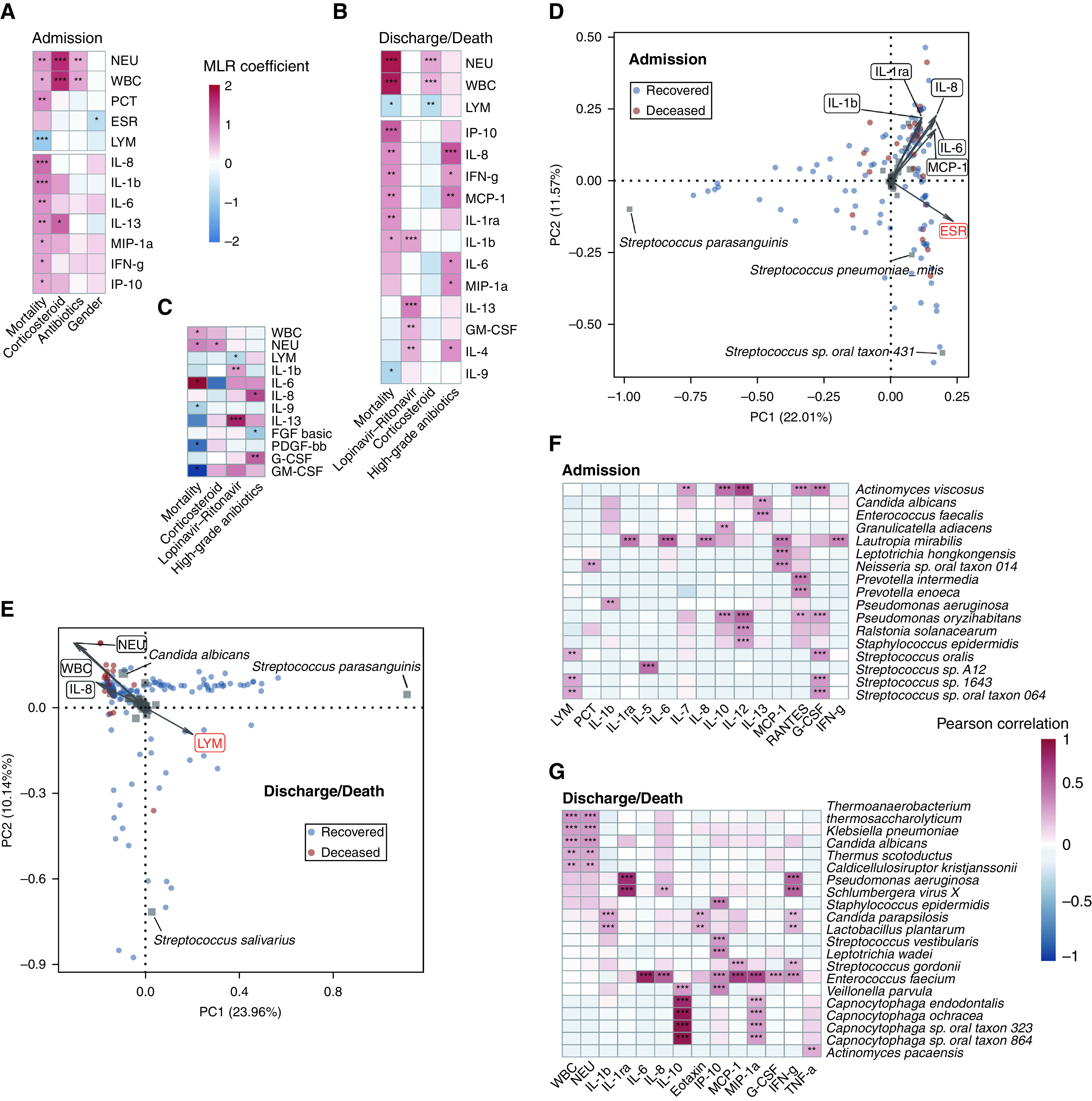
The association between host factors and the upper respiratory tract microbiota. Host factors whose concentrations were associated with metadata identified by multivariate linear regression are shown in A (on admission) and B (before discharge/death) (adjusted P value < 0.05). The host factors whose concentrations changed during hospitalization were associated with metadata as shown in C. The color in the heat map represents regression coefficients estimated by multivariate linear regression. Principal component analysis plots of samples are shown in D (on admission) and E (before discharge/death). Host factors significantly associated with the upper respiratory tract microbiome were identified by envfit analysis and are shown with arrows. The length of the arrow represents the variance of the variables, and the angle among arrows represents the degree of correlation between individual variables. Positively correlated variables are shown as arrows pointing in the same direction, whereas negatively correlated variables point in opposite directions. The factors that were also revealed by permutational multivariate ANOVA are marked in red. Red dots represent deceased patients, and blue dots represent recovered patients. Species with relative abundance greater than 1% in at least one sample are shown as gray squares in the plot, and the names of species with the largest loadings on the two axes are labeled. Species whose abundances were correlated with the concentration of host factors are shown in F (on admission) and G (before discharge/death) (adjusted P value < 0.01, Pearson’s correlation test). The color in the heat map represents correlation coefficients. *P < 0.05, **P < 0.01, and ***P < 0.001. ESR = erythrocyte sedimentation rate; LYM = lymphocytes; NEU = neutrophils; PC = principal component; PCT = procalcitonin; WBC = white blood cells.
Multiple host factors were associated with the diversity of OP microbiota. The concentration of IL-1β, IL-6, IL-8, MCP-1, and IL-1ra showed significant correlations with the microbiota on admission (Figure 6D), and most of these cytokines were upregulated in deceased patients. Meanwhile, the concentration of NEUs, WBCs, IL-8, and LYMs was significantly correlated with the microbiota before discharge/death (Figure 6E). Notably, the former three factors were inversely correlated with LYMs regarding their correlations with the microbiota.
We then looked further into the correlation between host factors and individual microbes. A positive correlation was observed between the abundance of Strepcococcus species and LYMs on admission (Figure 6F), whereas the concentration of WBCs and NEUs increased in patients with higher abundances of Klebsiella pneumonia and Candida albicans before discharge/death (Figure 6G). Additionally, the abundance of Enterococcus faecium was positively correlated with the concentration of multiple cytokines. Collectively, we found that the immune response varied between deceased and recovered patients at different stages of the hospitalization. The correlation between the concentration of immune cells and cytokines and OP microbiota suggested possible host–microbe interactions. Hence, the association between microbiota and mortality might be mediated by the host immune response.
Additional results of validation of S. parasanguinis, virome, clustering, microbial network, and microbial functionality analysis are included in the online supplement.
Discussion
In this study, we have demonstrated the features and dynamics of the URT microbiota and its association with mortality in patients with COVID-19. The metatranscriptomic analysis was conducted, which provided a real-time snapshot of the microbiota. Moreover, it allows the analysis of RNA viruses and host transcriptome (26).
The URT microbiota of COVID-19 was partially distinguishable from the HC (Figure 1C), indicating disrupted URT microbiota in some patients with COVID-19 (17, 18). However, the dispersive distribution of URT microbiota in COVID-19 suggested no definitive alteration associated with SARS-COV-2 infection, which follows the “Anna Karenina principle” that dysbiotic microbiotas vary in different forms and are more diverse than that in healthy individuals (27). The URT microbiota of deceased patients showed a distinct composition compared with that of the HC and recovered patients, and the discrepancy was more significant when the collection time was closer to discharge/death, suggesting that the extent of the microbiota dysbiosis might be correlated with the severity of the disease. However, this may also reflect that deceased patients were administered with more broad-spectrum antibiotics and/or invasive procedures, which can greatly affect the microbiota (28).
The pharyngeal swab is an easily accessible sample that is routinely collected for the diagnosis of respiratory infection. Besides the use for the detection of pathogens (29), URT microbiota has been associated with the susceptibility to respiratory infections and bronchiolitis, as well as symptom severity and clinical outcome (30–34). Our results showed a remarkable correlation between URT microbiota on admission and mortality in patients with COVID-19, indicating its potential as a prognostic biomarker and/or an index to screen for patients who are more likely to become critically ill. Notably, instead of microbial diversity, various metrics revealed Streptococcus as the only candidate genus associated with prognosis.
Streptococcus is one of the most abundant genera inhabiting the respiratory tract in a healthy population (26, 35, 36). The depletion of Streptococcus might represent a dysbiosis state of the URT microbiota, which may result from the introduction and overgrowth of competing microbes, enhanced immune response to SARS-CoV-2 infection, or use of antibiotics (37, 38). The immune imbalance scenario was supported by the increased level of cytokines in deceased patients and its significant correlation with microbiota variation (Figure 6). The heightened immune responses induced by the viral infection could potentially alter respiratory microbiota, whereas the latter could further modulate the local immune response (39–41). Meanwhile, although no significant difference in the abundance of potential pathogens was observed between deceased patients and recovered patients on admission (Figures 4B and 4C), the concentration of procalcitonin was significantly higher in deceased patients (0.13 [0.08–0.33] vs. 0.05 [0.05–0.05] ng/ml; P < 0.001) (Figure 6A), suggesting possible coinfections or secondary infections in deceased patients on admission. Furthermore, no correlation was observed between the use of antibiotics and microbiota on admission (Table 2), which may be because of the short duration of antibiotic use. In contrast, the use of antibiotics (including high-grade antibiotics) was associated with the alteration of the URT microbiota during hospitalization (Table E4). However, we did not find a significant correlation between the abundance of Streptococccus and the use of antibiotics after adjusting for other covariates. Nevertheless, a dysbiotic microbiota may diminish the colonization resistance against pathogens and predispose a patient to a secondary infection, thus leading to poor prognosis, which agrees with our observation that microbiota with a high abundance of Streptococcus were more stable and resistant to coinfection or secondary infection and had a lower mortality rate.
There are some limitations to our study. First, although pharyngeal microbiota can potentially serve as a proxy for the lower respiratory tract (LRT) microbiota (42), an investigation of the lung microbiota would be more valuable to reveal the host–microbiome interactions in COVID-19. Unfortunately, it is difficult to collect serial LRT samples owing to practicalities and safety concerns (43). Second, many confounding factors related to the severity of the disease could potentially influence the URT microbiota, including the use of antibiotics, diet, and intubation. Although multivariate regression analysis had been applied, we cannot fully control for the confounding effect because of the limited sample size and unavailability of some data. However, this should have little impact on the analysis of the microbiota on admission as the medical intervention was limited before being admitted to the hospital. Third, the cohort only includes patients with severe COVID-19, whereas the mild and asymptomatic patients may display distinct features. Fourth, our study is a single-center study. It is unclear whether the findings could be generalized to other populations, particularly considering that URT microbiota could be influenced by geography, host genetics, and medication (36, 44). Finally, the time from symptom onset to the recruitment of the patients was 10 days; thus, it is unknown when the initial change of URT microbiota occurred.
Conclusions
Our study has characterized the features and dynamics of the URT microbiota and its association with mortality in COVID-19, suggesting that URT microbiota on admission was significantly associated with the prognosis. Notably, no causality can be inferred from the associations we observed, and some of the associations might be related to unmeasured confounders that need to be explored further. Additional studies are needed to unveil the underlying mechanisms responsible for the associations and validate the findings in other cohorts.
Acknowledgments
Acknowledgment
The authors thank the patients, clinicians, and healthy volunteers who participated in this study.
Footnotes
Supported by the National Major Science and Technology Project for Control and Prevention of Major Infectious Diseases in China (2017ZX10103004), National Natural Science Foundation of China (82161148009), the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS 2020-I2M-2-013, 2018-I2M-1-003, and 2020-I2M-CoV19-005), National Key R&D Program of China (2020YFC0848900), Beijing Municipal Science and Technology Commission (Z191100006619100, Z201100005320016, and Z201100007920017), Beijing Advanced Innovation Center for Genomics, and Beijing Advanced Innovation Center for Structural Biology.
Author Contributions: L.R., B.C., Jianwei Wang, and M.L. conceived of and designed the project; Yeming Wang, X.L., G.F., L.G., H.L., and X.Z. contributed to patient recruitment and management and collection of specimens and clinical data; L.R., Y.X., Jie Li, L.G., Q.L., Jizhou Li, L.D., C.W., Ying Wang, J.R., X.W., Jianbin Wang, and Y.H. conducted the laboratory work and metatranscriptome sequencing; J.Z., J.Y., Z.S., L.K., L.S., and L.Z. conducted the bioinformatics and statistical analysis. M.L., Jianwei Wang, and B.C. wrote the manuscript. All authors contributed to the interpretation of the data and revision of the manuscript and approved the final version. M.L., L.R., and J.Z. had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202103-0814OC on September 17, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Pollán M, Pérez-Gómez B, Pastor-Barriuso R, Oteo J, Hernán MA, Pérez-Olmeda M, et al. ENE-COVID Study Group. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): a nationwide, population-based seroepidemiological study. Lancet . 2020;396:535–544. doi: 10.1016/S0140-6736(20)31483-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Clinical management of severe acute respiratory infection when novel coronavirus (2019-nCoV) infection is suspected: interim guidance 28 January 2020 World Health Organization; 2020. [accessed 2021 Nov 16]. Available from: https://apps.who.int/iris/handle/10665/330893. [Google Scholar]
- 3. Wolff D, Nee S, Hickey NS, Marschollek M. Risk factors for Covid-19 severity and fatality: a structured literature review. Infection . 2021;49:15–28. doi: 10.1007/s15010-020-01509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Severe Covid-19 GWAS Group. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med . 2020;383:1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA . 2020;324:663–673. doi: 10.1001/jama.2020.13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ . 2017;356:j831. doi: 10.1136/bmj.j831. [DOI] [PubMed] [Google Scholar]
- 7. de Steenhuijsen Piters WAA, Binkowska J, Bogaert D. Early life microbiota and respiratory tract infections. Cell Host Microbe . 2020;28:223–232. doi: 10.1016/j.chom.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 8. Libertucci J, Young VB. The role of the microbiota in infectious diseases. Nat Microbiol . 2019;4:35–45. doi: 10.1038/s41564-018-0278-4. [DOI] [PubMed] [Google Scholar]
- 9. Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L, et al. Alterations of the gut microbiota in patients with coronavirus disease 2019 or H1N1 influenza. Clin Infect Dis . 2020;71:2669–2678. doi: 10.1093/cid/ciaa709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology . 2020;159:944–955.e8. doi: 10.1053/j.gastro.2020.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo T, Zhan H, Zhang F, Liu Q, Tso EYK, Lui GCY, et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology. 2020;159:1302–1310.e5. doi: 10.1053/j.gastro.2020.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zuo T, Liu Q, Zhang F, Lui GCY, Tso EYK, Yeoh YK, et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut . 2021;70:276–284. doi: 10.1136/gutjnl-2020-322294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yeoh YK, Zuo T, Lui GC-Y, Zhang F, Liu Q, Li AY, et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut . 2021;70:698–706. doi: 10.1136/gutjnl-2020-323020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang H, Ai JW, Yang W, Zhou X, He F, Xie S, et al. Metatranscriptomic characterization of coronavirus disease 2019 identified a host transcriptional classifier associated with immune signaling. Clin Infect Dis . 2021;73:376–385. doi: 10.1093/cid/ciaa663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaul D, Rathnasinghe R, Ferres M, Tan GS, Barrera A, Pickett BE, et al. Microbiome disturbance and resilience dynamics of the upper respiratory tract during influenza A virus infection. Nat Commun . 2020;11:2537. doi: 10.1038/s41467-020-16429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L, et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin Infect Dis . 2020;71:713–720. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu R, Liu P, Zhang T, Wu Q, Zeng M, Ma Y, et al. Progressive worsening of the respiratory and gut microbiome in children during the first two months of COVID-19 [preprint] medRxiv 2020. [accessed 2021 Nov 16]. Available from: https://www.medrxiv.org/content/10.1101/2020.07.13.20152181v1.
- 18. Xu R, Lu R, Zhang T, Wu Q, Cai W, Han X, et al. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun Biol . 2021;4:240. doi: 10.1038/s42003-021-01796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med . 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McMurdie PJ, Holmes S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One . 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: Community Ecology Package. R package version 2.0-9 2013. [accessed 2021 Jul 9]. Available from: http://cran.r-project.org/package=vegan
- 22.RStudio Team. RStudio: Integrated Development for R Boston, MA: RStudio, Inc; 2016. [accessed 2021 Nov 16]. Available from: http://www.rstudio.com. [Google Scholar]
- 23. Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, et al. Mining the human gut microbiota for immunomodulatory organisms. Cell . 2017;168:928–943.e11. doi: 10.1016/j.cell.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Man WH, de Steenhuijsen Piters WAA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol . 2017;15:259–270. doi: 10.1038/nrmicro.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang Y-Q, et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther . 2020;5:33. doi: 10.1038/s41392-020-0148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ren L, Zhang R, Rao J, Xiao Y, Zhang Z, Yang B, et al. Transcriptionally active lung microbiome and its association with bacterial biomass and host inflammatory status. mSystems . 2018;3:e00199-18. doi: 10.1128/mSystems.00199-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaneveld JR, McMinds R, Vega Thurber R. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat Microbiol . 2017;2:17121. doi: 10.1038/nmicrobiol.2017.121. [DOI] [PubMed] [Google Scholar]
- 28. de Koff EM, Man WH, van Houten MA, Jansen NJG, Arp K, Hasrat R, et al. The respiratory microbiota during and following mechanical ventilation for respiratory infections in children. Eur Respir J . 2021;57:2002652. doi: 10.1183/13993003.02652-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ali M, Han S, Gunst CJ, Lim S, Luinstra K, Smieja M. Throat and nasal swabs for molecular detection of respiratory viruses in acute pharyngitis. Virol J . 2015;12:178. doi: 10.1186/s12985-015-0408-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verhagen LM, Rivera-Olivero IA, Clerc M, Chu MLJN, van Engelsdorp Gastelaars J, Kristensen MI, et al. Nasopharyngeal microbiota profiles in rural Venezuelan children are associated with respiratory and gastrointestinal infections. Clin Infect Dis . 2021;72:212–221. doi: 10.1093/cid/ciaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hasegawa K, Linnemann RW, Mansbach JM, Ajami NJ, Espinola JA, Petrosino JF, et al. Nasal airway microbiota profile and severe bronchiolitis in infants: a case-control study. Pediatr Infect Dis J . 2017;36:1044–1051. doi: 10.1097/INF.0000000000001500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lehtinen MJ, Hibberd AA, Männikkö S, Yeung N, Kauko T, Forssten S, et al. Nasal microbiota clusters associate with inflammatory response, viral load, and symptom severity in experimental rhinovirus challenge. Sci Rep . 2018;8:11411. doi: 10.1038/s41598-018-29793-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitsios GD, Yang H, Yang L, Qin S, Fitch A, Wang XH, et al. Respiratory tract dysbiosis is associated with worse outcomes in mechanically ventilated patients. Am J Respir Crit Care Med . 2020;202:1666–1677. doi: 10.1164/rccm.201912-2441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dickson RP, Schultz MJ, van der Poll T, Schouten LR, Falkowski NR, Luth JE, et al. Biomarker Analysis in Septic ICU Patients (BASIC) Consortium. Lung microbiota predict clinical outcomes in critically ill patients. Am J Respir Crit Care Med . 2020;201:555–563. doi: 10.1164/rccm.201907-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Einarsson GG, Zhao J, LiPuma JJ, Downey DG, Tunney MM, Elborn JS. Community analysis and co-occurrence patterns in airway microbial communities during health and disease. ERJ Open Res. 2019;5:00128–02017. doi: 10.1183/23120541.00128-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dickson RP, Huffnagle GB. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog . 2015;11:e1004923. doi: 10.1371/journal.ppat.1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanada S, Pirzadeh M, Carver KY, Deng JC. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front Immunol . 2018;9:2640. doi: 10.3389/fimmu.2018.02640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dickson RP, Erb-Downward JR, Falkowski NR, Hunter EM, Ashley SL, Huffnagle GB. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Respir Crit Care Med . 2018;198:497–508. doi: 10.1164/rccm.201711-2180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. O’Dwyer DN, Ashley SL, Gurczynski SJ, Xia M, Wilke C, Falkowski NR, et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1127–1138. doi: 10.1164/rccm.201809-1650OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Segal LN, Clemente JC, Tsay J-CJ, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol . 2016;1:16031. doi: 10.1038/nmicrobiol.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Man WH, van Houten MA, Mérelle ME, Vlieger AM, Chu MLJN, Jansen NJG, et al. Bacterial and viral respiratory tract microbiota and host characteristics in children with lower respiratory tract infections: a matched case-control study. Lancet Respir Med . 2019;7:417–426. doi: 10.1016/S2213-2600(18)30449-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sulaiman I, Chung M, Angel L, Tsay J-CJ, Wu BG, Yeung ST, et al. Microbial signatures in the lower airways of mechanically ventilated COVID19 patients associated with poor clinical outcome [preprint] medRxiv 2021. [accessed 2021 Nov 16]. Available from: https://www.medrxiv.org/content/10.1101/2021.02.23.21252221v1. [DOI] [PMC free article] [PubMed]
- 44. Gupta VK, Paul S, Dutta C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol . 2017;8:1162. doi: 10.3389/fmicb.2017.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]