

Pulmonary hypertension (PH) is a progressive cardiopulmonary disorder defined as a mean pulmonary artery (PA) pressure ⩾20 mm Hg at rest, leading to right heart failure and premature death (1). Despite the progress of past decades, pulmonary arterial hypertension (PAH) (group I PH) remains a life-threatening disease with a high mortality rate and a lack of curative options. The mechanisms of development and progression of other groups of PH are also hugely understudied. The key pathophysiologic feature of PAH is a hyperproliferation of resident cells in small PAs, resulting in remodeling, lumen obliteration, and an unresolved increase of PA pressure (2).
The remodeling and stiffening of extracellular matrix (ECM) in PAH PAs promote the proliferative response of PA smooth muscle cells (PASMCs), PA endothelial cells (PAECs), and PA adventitial fibroblasts, making cell–ECM interaction a desirable antiremodeling target. To date, several pathologic mechanisms by which defective cell–matrix interactions drive pulmonary vascular proliferation and PH have been identified, including upregulation of the proproliferative transcriptional coactivators YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif) (3–5), dysregulation of the aldosterone–NEDD9 pathway (6), BMPR2 mutation–driven endothelial-to-mesenchymal transition (7), and disbalance between MMPs (matrix metalloproteinases) and their inhibitors, TIMPs (tissue inhibitors of metalloproteinases) (7).
MMPs are proteolytic enzymes that degrade structural ECM proteins and regulatory molecules and are involved in the pathogenesis of proliferative, fibrotic, and vascular diseases, including PH (7, 8). Elevated plasma amounts of several MMPs and TIMPs have been associated with worse clinical outcomes, PA stiffness, remodeling, and right ventricular dysfunction in adult and pediatric PAH, suggesting their potential attractiveness as clinical biomarkers (7, 9, 10). Preclinical studies, however, report both pro- and antiremodeling roles for different MMPs, cautioning against the use of pan-MMP inhibitors and calling for careful investigation of individual MMPs as candidate targets for antiremodeling therapeutics in PH (8).
In the current issue of the Journal, Dieffenbach and colleagues (pp. 1433–1451) performed comprehensive analysis of previously understudied member of the MMP family, MMP-8 (collagenase 2 or neutrophil collagenase), in PH (11). The authors found that MMP-8 concentrations are significantly higher in plasma from patients with PAH and group III PH, as well as in small PAs from subjects with PAH and three rodent models of PH. Importantly, the authors detected a modest association between MMP-8 plasma concentrations and PH severity and detected a strong correlation between pulmonary vascular MMP-8 amount and PA muscularization and remodeling, strongly suggesting involvement of MMP-8 in PASMC remodeling and PH. Intriguingly, mice with MMP-8 germline knockout were not protected from hypoxia-induced PH but developed exaggerated PASMC proliferation, pulmonary vascular remodeling, and PH upon hypoxia exposure (11). Further confirming the antiremodeling role of MMP-8 in PASMCs, MMP-8 deficiency induced PASMC hyperproliferation via activating FAK (focal adhesion kinase)–YAP/TAZ signaling (Figure 1).
Figure 1.
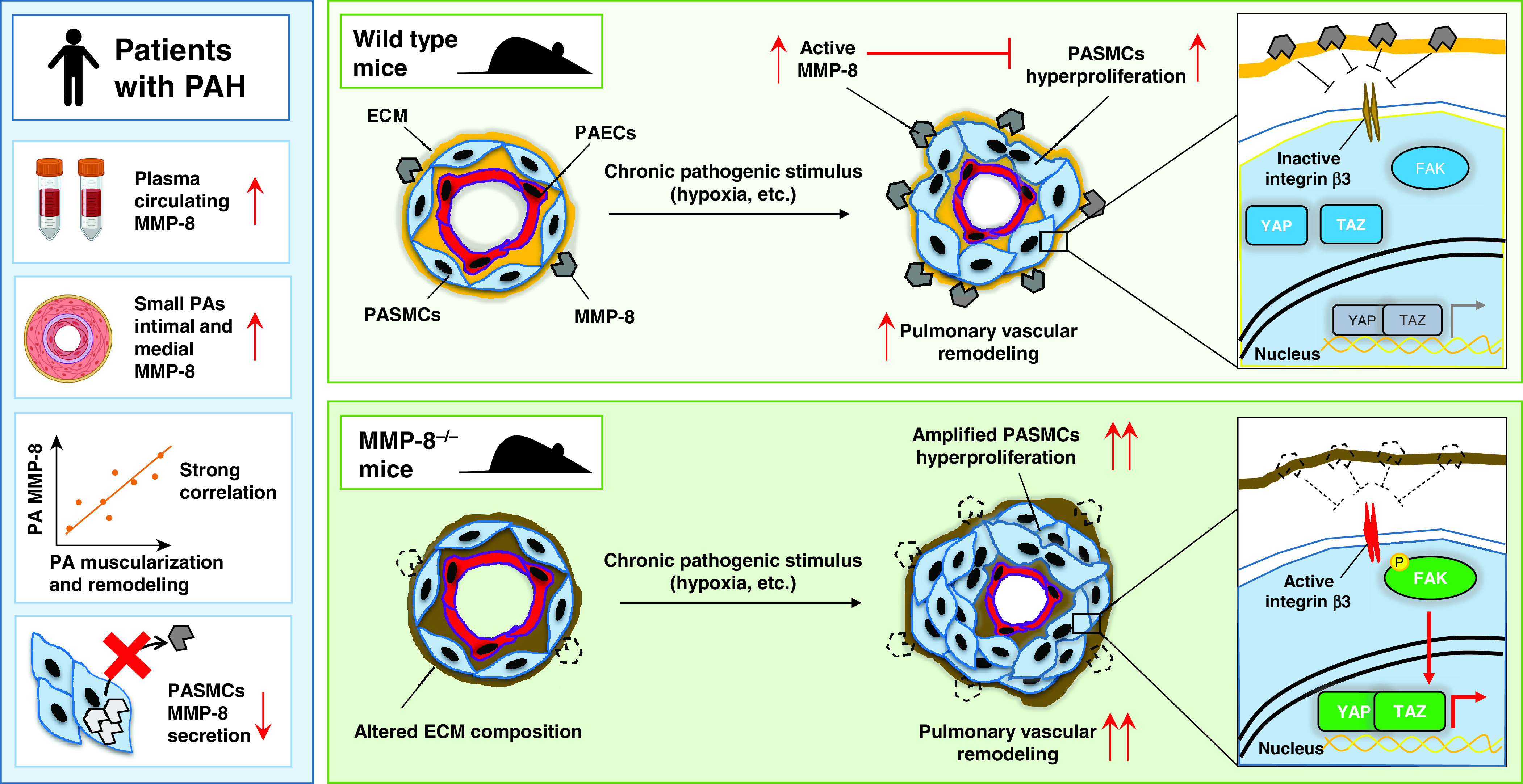
Protective role of MMP-8 (matrix metalloproteinase-8) in vascular remodeling and pulmonary hypertension (PH) as shown by the study of Dieffenbach and colleagues (11). Left: Dysregulation of MMP-8 in plasma and pulmonary vasculature from patients with PAH. Right: MMP-8 loss augments hypoxia-induced PH in mice via FAK (focal adhesion kinase)–YAP (Yes-associated protein)/TAZ (transcriptional coactivator with PDZ-binding motif) signaling. Chronic pathogenic stimuli, such as prolonged hypoxia exposure, induce a drastic upregulation of MMP-8 in the intimal and medial layers of pulmonary arteries (PAs), which in turn prevents activation of the FAK–YAP/TAZ axis via integrin β3 signaling (top panel). Hypoxia exposure in MMP-8–deficient PAs results in exacerbated PASMCs hyperproliferation and exaggerated vascular remodeling, leading to worse PH outcomes. Red arrows indicate increase (↑) or decrease (↓) compared with the nondiseased state. Double red arrows indicate amplified response. ECM = extracellular matrix; P = phosphorylation; PAEC = PA endothelial cell; PAH = pulmonary arterial hypertension; PASMC = PA smooth muscle cell.
MMP production could be induced by multiple stimuli, including growth factors, cytokines, and cell–cell and cell–ECM interactions, all of which are involved in PAH pathogenesis (7, 8). The majority of MMPs, however, are generated as inactive proenzymes and become activated only after secretion, which appeared to be suppressed in PAH PASMCs compared with controls. Furthermore, MMPs were shown to directly cleave and inactivate FAK (12), supporting the potential existence of negative cross-talk between MMP-8 and FAK in PAH PASMCs. In line with this, the findings by Dieffenbach and colleagues (11) implicate potential defects in MMP-8 secretion, which can lead to both maladaptive overaccumulation of MMP-8 in PAH PASMCs and deficiency of active MMP-8 in the extracellular space, resulting in constitutive activation of the pro-remodeling FAK–YAP/TAZ axis. However, better understanding of spatiotemporal regulation of MMP-8 expression and secretion as well as the cellular origin of MMP-8 in the PAH context are required to gain comprehensive knowledge on the role of MMP-8 in PAH.
To overcome the limitations of a mouse model with global MMP-8 deficiency, the authors elegantly excluded the direct effects of MMP-8 loss on the right ventricle by using a PA banding approach, which confirmed the importance of MMP-8 loss, specifically in the pulmonary vasculature (11). However, in addition to playing a role in the heart, MMP-8 plays a role in multiple cell types. This not only could interfere with pulmonary vasculature–focused studies but could also make systemic targeting of MMP-8 challenging because of potential off-target effects. As an example, MMP-8 plays a pro-proliferative role in systemic smooth muscle cells and is expressed by several inflammatory cell types, making MMP-8–deficient mice protected from the remodeling of systemic arteries but prone to an exacerbated inflammatory response and delayed wound healing (13, 14).
In the pulmonary vasculature, the striking increase of MMP-8 in the intimal layer of PAH PAs suggests a potential role for MMP-8 in PAH PAECs. Furthermore, MMP-8 overaccumulation, seen in whole PAs, could not be detected in isolated PAH PASMCs, implying a complex scenario in which MMP-8 overproduction by PASMCs requires a PAH-specific microenvironment and, potentially, a PAEC–PASMC interaction. Although enabling total ablation of extracellular MMP-8, the use of mice with germline MMP-8 knockout lacks resolution for identifying cell-specific roles of MMP-8 in PAH. In this sense, use of conditional knockout models to uncover the specific functions of MMP-8 in different inflammatory and pulmonary vascular cell populations in PAH is warranted. In addition, a detailed analysis of ECM composition and specific matrix components altered by MMP-8 to provide insights on adaptive versus maladaptive ECM composition in the PAH setting is required. Previous studies have reported alterations in ECM composition upon the onset of MMP-8 deficiency, as well as differential expression of important regulators of mechanosensitive pathways (ADAM10, N-cadherin, β-catenin) (13). Thus, it would be also interesting to investigate which events that are mediated by a lack of MMP-8 precede and potentially cause the exaggerated phenotype that arises upon chronic pathologic stimulation.
Interestingly, the authors clearly demonstrate that plasma MMP-8 concentrations were significantly higher in patients with PH than in those without the disease. This may have practical implications for both diagnosis and therapy in PH. As the neutrophils are an important source of MMP-8 (15), this has to be correlated with neutrophil subsets as well as with the markers of systemic inflammation to improve the prognostic and diagnostic significance of MMP-8 in PH. Furthermore, genetic association studies on the role of specific MMP variants in PH are warranted, as SNPs of MMP-8 in atherosclerosis have been shown to influence its gene expression (16).
In conclusion, the study by Dieffenbach and colleagues (11) uncovers a novel role and mechanism of action for MMP-8 in PH. Importantly, the pharmacologic attractiveness of targeting the MMP-8 downstream effectors FAK and YAP/TAZ has already proven promising as an antiproliferative, antiremodeling therapy in experimental PH. However, an additional endeavor would be required to identify crucial targetable nodes of MMP-8 signaling and develop safe and efficient cell-specific therapeutic strategies to target MMP-8 in PH.
Footnotes
Supported by NIH/NHLBI grants R01 HL113178 and HL130261 (E.A.G.), Deutsche Forschungsgemeinschaft project number 268555672– Sonderforschungsbereich (SFB) 1213 (projects A01 and A05) (S.S.P.), and European Research Council Consolidator grant 866051 (S.S.P.).
Originally Published in Press as DOI: 10.1164/rccm.202109-2144ED on October 13, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J . 2019;53:1801913. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res . 2017;367:643–649. doi: 10.1007/s00441-016-2539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep . 2015;13:1016–1032. doi: 10.1016/j.celrep.2015.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kudryashova TV, Goncharov DA, Pena A, Kelly N, Vanderpool R, Baust J, et al. HIPPO-integrin-linked kinase cross-talk controls self-sustaining proliferation and survival in pulmonary hypertension. Am J Respir Crit Care Med . 2016;194:866–877. doi: 10.1164/rccm.201510-2003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dieffenbach PB, Haeger CM, Coronata AMF, Choi KM, Varelas X, Tschumperlin DJ, et al. Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ-mediated repression of cyclooxygenase-2. Am J Physiol Lung Cell Mol Physiol . 2017;313:L628–L647. doi: 10.1152/ajplung.00173.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Samokhin AO, Stephens T, Wertheim BM, Wang RS, Vargas SO, Yung LM, et al. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med . 2018;10:eaap7294. doi: 10.1126/scitranslmed.aap7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol . 2018;315:H1322–H1331. doi: 10.1152/ajpheart.00136.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chelladurai P, Seeger W, Pullamsetti SS. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur Respir J . 2012;40:766–782. doi: 10.1183/09031936.00209911. [DOI] [PubMed] [Google Scholar]
- 9. Schäfer M, Ivy DD, Nguyen K, Boncella K, Frank BS, Morgan GJ, et al. Metalloproteinases and their inhibitors are associated with pulmonary arterial stiffness and ventricular function in pediatric pulmonary hypertension. Am J Physiol Heart Circ Physiol . 2021;321:H242–H252. doi: 10.1152/ajpheart.00750.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arvidsson M, Ahmed A, Bouzina H, Rådegran G. Matrix metalloproteinase 7 in diagnosis and differentiation of pulmonary arterial hypertension. Pulm Circ . 2019;9:2045894019895414. doi: 10.1177/2045894019895414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dieffenbach PB, Mallarino Haeger C, Rehman R, Corcoran AM, Coronata AMF, Vellarikkal SK, et al. A novel protective role for matrix metalloproteinase-8 in the pulmonary vasculature. Am J Respir Crit Care Med . doi: 10.1164/rccm.202108-1863OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shofuda T, Shofuda K, Ferri N, Kenagy RD, Raines EW, Clowes AW. Cleavage of focal adhesion kinase in vascular smooth muscle cells overexpressing membrane-type matrix metalloproteinases. Arterioscler Thromb Vasc Biol . 2004;24:839–844. doi: 10.1161/01.ATV.0000126680.78500.4c. [DOI] [PubMed] [Google Scholar]
- 13. Xiao Q, Zhang F, Grassia G, Hu Y, Zhang Z, Xing Q, et al. Matrix metalloproteinase-8 promotes vascular smooth muscle cell proliferation and neointima formation. Arterioscler Thromb Vasc Biol . 2014;34:90–98. doi: 10.1161/ATVBAHA.113.301418. [DOI] [PubMed] [Google Scholar]
- 14. Gutiérrez-Fernández A, Inada M, Balbín M, Fueyo A, Pitiot AS, Astudillo A, et al. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8) FASEB J . 2007;21:2580–2591. doi: 10.1096/fj.06-7860com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ong CWM, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-Derived MMP-8 Drives AMPK-Dependent Matrix Destruction in Human Pulmonary Tuberculosis. PLoS Pathog . 2015;11:e1004917. doi: 10.1371/journal.ppat.1004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salminen A, Vlachopoulou E, Havulinna AS, Tervahartiala T, Sattler W, Lokki ML, et al. Genetic variants contributing to circulating matrix metalloproteinase 8 levels and their association with cardiovascular diseases: a genome-wide analysis. Circ Cardiovasc Genet . 2017;10:e001731. doi: 10.1161/CIRCGENETICS.117.001731. [DOI] [PubMed] [Google Scholar]