

Abstract
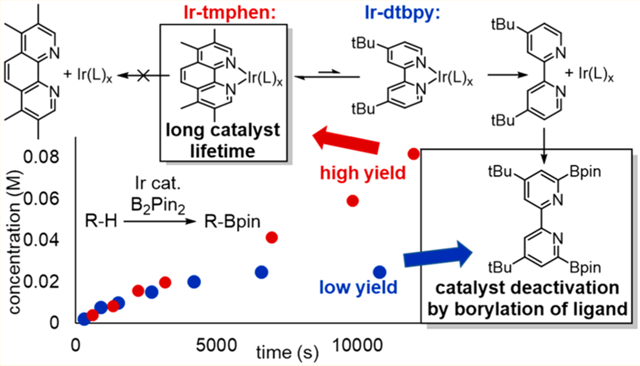
A mechanistic study on the origin of the difference in reactivity between Ir catalysts for C−H borylation reactions is reported. Catalytic reactions of B2pin2 with a series of substrates that require high temperatures and long reaction times were conducted. These reactions catalyzed by the combination of [Ir(COD)(OMe)]2 and 3,4,7,8-tetramethylphenanthroline (tmphen) occur in yields that are substantially higher than those of reactions catalyzed by [Ir(COD)(OMe)]2 and 4,4′-di-tert-butylbipyridine (dtbpy). The electronic properties of Ir catalysts ligated by dtbpy or tmphen and their stoichiometric reactivity were investigated. It was found that a longer lifetime rather than higher reactivity of the catalyst leads to higher yields of reactions catalyzed by Ir-tmphen. The catalyst ligated by dtbpy decomposes principally by dissociation of the ligand and rapid borylation at the positions alpha to nitrogen. Thus, the greater stability of the catalyst containing tmphen results from its greater binding constant.
Graphical Abstract
INTRODUCTION
The borylation of C−H bonds has become a widely used and researched method for the functionalization of C−H bonds (Scheme 1).1,2 These reactions occur without directing groups with regioselectivities that result from steric hindrance and, in some cases, the acidity of the C−H bond. This selectivity complements that of other catalytic and classical functionalizations of aryl C−H bonds.3 The reaction forms organoboronate esters that can be transformed to a variety of C−C and C−heteroatom bonds.4
Scheme 1.
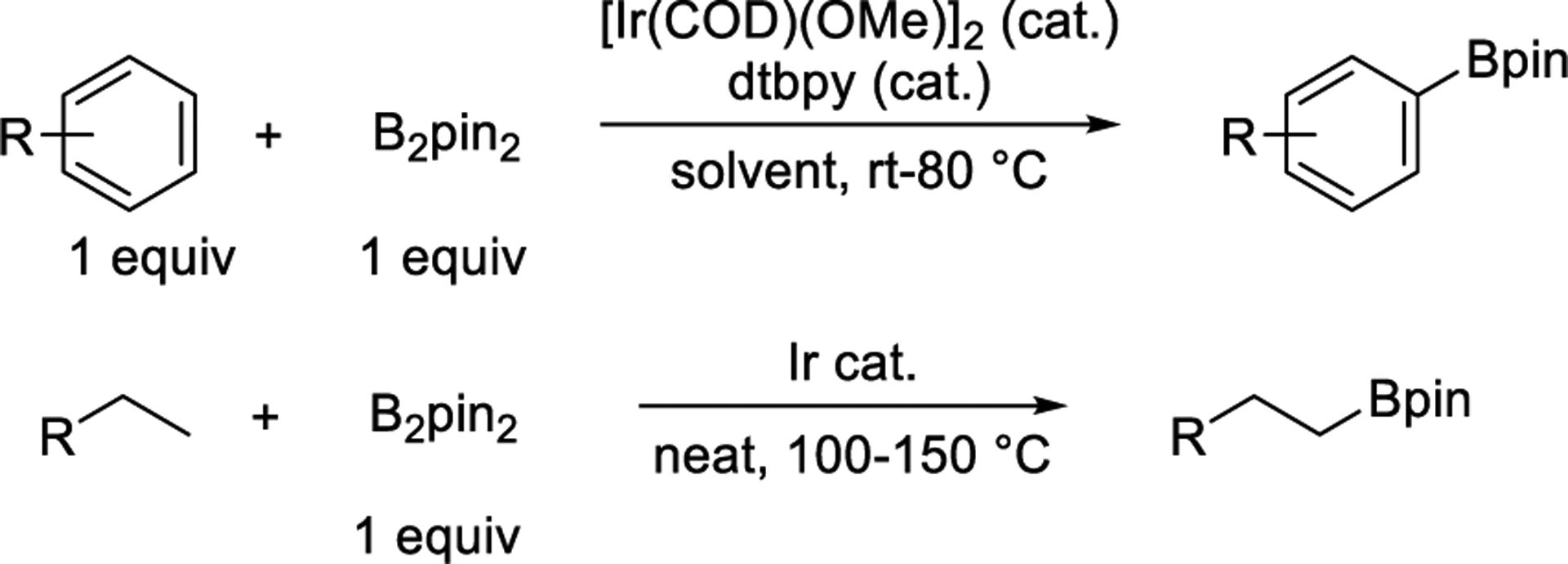
Typical Reaction Conditions for Ir-Catalyzed Borylation of Arenes
The ligands used for these iridium-catalyzed functionalizations are typically based on nitrogen Lewis bases, and the catalyst is often generated from the combination of [Ir(COD)OMe]2 and 4,4′-di-tert-butylbipyridine (dtbpy).5−11 Some of these reactions occur with turnover numbers approaching 20 00011,12 and have been used to produce kilograms of active pharmaceutical ingredients (APIs).13,14 However, the borylation of many types of arenes and heteroarenes catalyzed by [Ir(COD)OMe]2 and dtbpy occur in low yields or require high catalyst loadings (Scheme 2a,b). In these cases, higher yields of the aryl or heteroaryl boronic esters have been achieved by generating the catalyst from [Ir(COD)OMe]2 and 3,4,7,8 tetramethylphenanthroline (tmphen).9,15 This system even catalyzes the borylation of aliphatic C−H bonds (Scheme 2c), including primary and secondary alkyl C−H bonds of amines, ethers,16,17 and silanes18 (Scheme 2d).
Scheme 2.
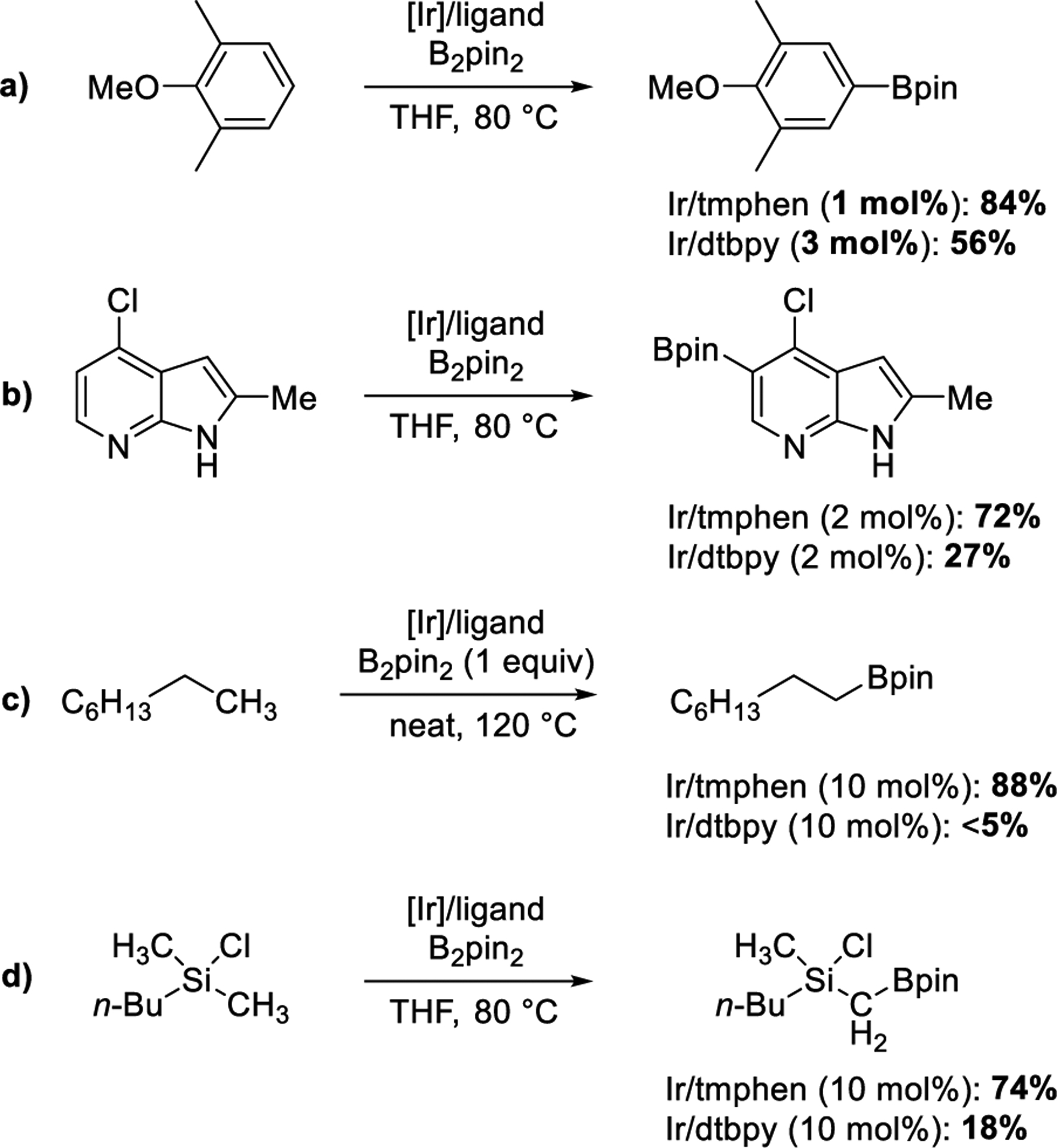
Published Examples of the Borylations of C−H Bonds Showing the Different Yields for Reactions Catalyzed by Ir-tmphen and Ir-dtbpy
Many catalysts for reactions involving organometallic intermediates of noble metals contain phosphines as dative ligands,19−21 and many studies have revealed the effects of the properties of phosphines on such catalytic reactions.22−24 Far fewer catalysts of such metals contain dative ligands bearing only Lewis-basic nitrogen donors.25−29 Few studies on the origins of the activity of such metal−ligand complexes have been published, and most of these studies focus on the polymerization of alkenes.30−32 Thus, the origins of the differences in reactivity of complexes of bipyridine and phenanthroline ligands on the borylation of C−H bonds are difficult to glean from prior published literature.
To reveal the origin of the difference in reactivity between the combination of [Ir(COD)OMe]2 and dtbpy or tmphen as ligand for the borylation of C−H bonds, we studied the relative rates for reaction of the boryl intermediates with various substrates, the relative rates for catalyst decomposition, and the origin of catalyst decomposition. We show that the rates of the reactions catalyzed by the two types of complexes are similar to each other, but the lifetime of the catalyst containing tmphen is much longer than that containing dtbpy, and this longer lifetime, rather than higher reactivity of the catalytic boryl intermediate, leads to the higher yields of reactions catalyzed by tmphen than of those catalyzed by dtbpy. The catalyst lifetime is limited by the borylation of the free nitrogen-based ligand; the higher binding constant of phenanthroline ligands and thereby lower concentration of free ligand, not slower modification of free, phenanthrolines, accounts for the difference in lifetime.
RESULTS AND DISCUSSION
To determine the origins of the difference in reactivity between catalysts containing 4,4′-di-tert-butylbipyridine (dtbpy) and 3,4,7,8-tetramethylphenanthroline (tmphen), we first monitored the rates of reactions of a series of substrates of varying reactivity with the iridium-trisboryl complexes [Ir(Bpin)3(L2)-(alkene)] (1 and 2) in which L2 is dtbpy or tmphen and the alkene is derived from the catalyst precursor. Such tris-boryl complexes are known to be the resting state of the catalyst during the borylation of arenes,33 and the reaction of such complexes with the C−H bond of the substrate is known to be rate limiting for reactions of arenes catalyzed by complexes of both ligands. These complexes react with the substrate after dissociation of the alkene.33
The rates of the stoichiometric reactions of tris-boryl complexes 1 and 2 with 1,3-dimethoxybenzene (3), an arene that forms the product of borylation in high yield with both catalysts, was studied first to assess differences in the rate of reaction of aryl C−H bonds with the two complexes containing the different ligands. These data are provided in Figure 1 and show that the rate of the reaction with dtbpy complex 1 is similar to that with tmphen complex 2. Thus, an inherently higher reactivity of the complex ligated by tmphen with aryl C−H bonds than of the complex ligated by dtbpy does not explain the difference in yields of reactions catalyzed by complexes of the two ligands. Analogous reactions of 1 and 2 with benzene and 2-methoxy-1,3-dimethylbenzene also showed that the rates of the reactions of complex 1 are similar to those of complex 2 with the same arene (see Supporting Information).
Figure 1.
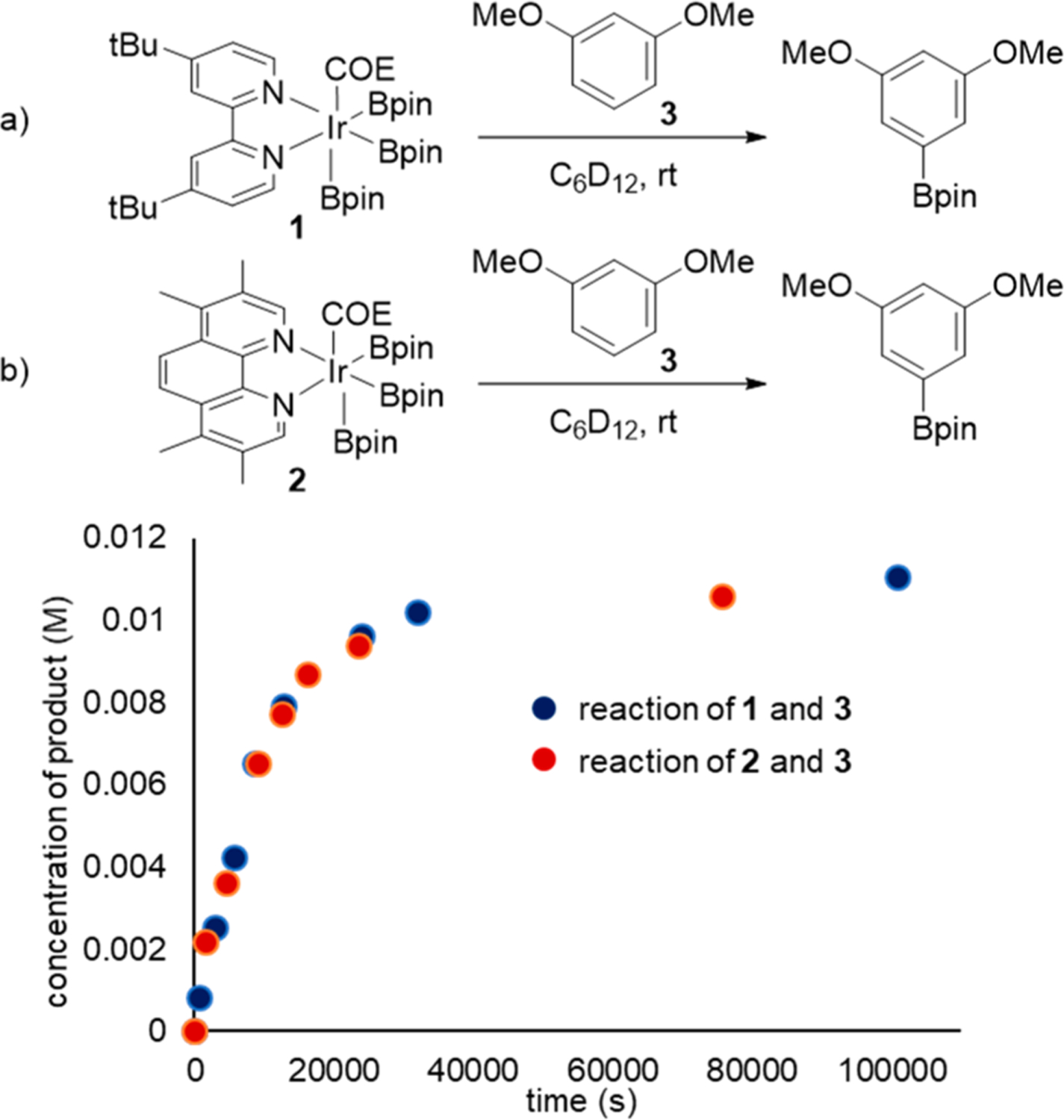
Reaction profile of stoichiometric borylation of m-dimethoxybenzene (3) with Ir complexes (10.5 μM) 1 (blue) and 2 (red).
Consistent with the similar rates for reactions of complexes of the two ligands, the degree of electron density at iridium in the trisboryl complex containing dtbpy was similar to that containing tmphen. The infrared stretching frequencies of the Ir-bound carbonyl23 ligands in trisboryl complexes [Ir(Bpin)3(L2)(CO)] (L2 = dtbpy, 4;34 and L2 = tmphen, 5) ligated by CO in place of the alkene (Scheme 3) were 1971 and 1972 cm−1, respectively. These data, together with the kinetic data for stoichiometric reactions, show that the origin of the difference in yields of reactions catalyzed by the two iridium-ligand combinations is more likely due to factors besides a difference in reactivity resulting from variations in the electronic properties of the two trisboryl complexes.
Scheme 3.
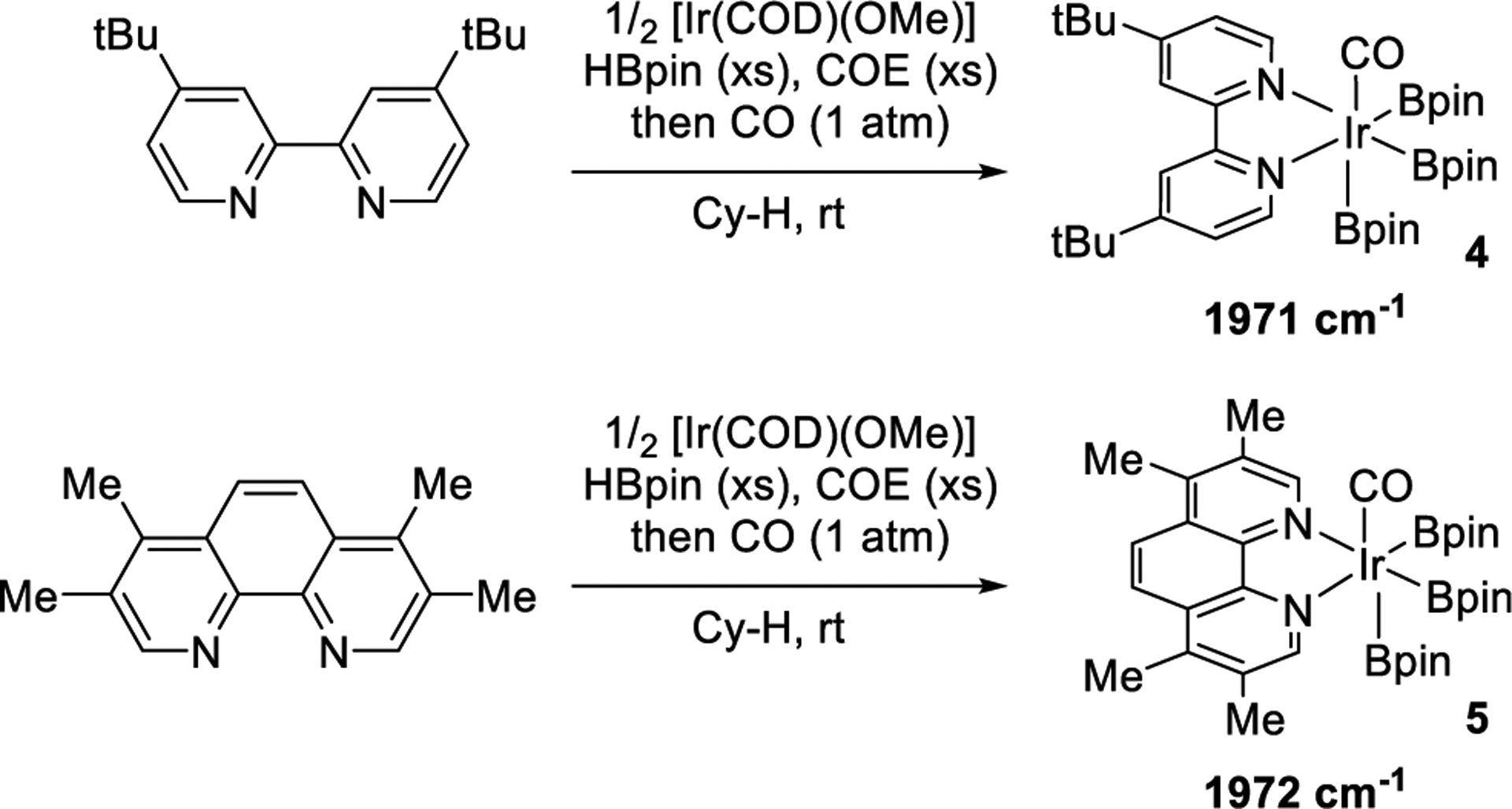
C−O Stretching Frequencies of Iridium-Trisboryl Carbonyl Complexes 4 and 5
To point toward alternative origins of the differences in yields of reactions catalyzed by complexes of the two ligands, we conducted catalytic reactions of B2pin2 with THF and with two aromatic compounds that react in higher yield with the catalyst ligated by tmphen than with that ligated by dtbpy. (Mes)Ir(Bpin)3 was used as catalyst precursor instead of [Ir(COD)(OMe)]2, which is more typically used for synthetic purposes, to eliminate the induction period sometimes observed with [Ir(COD)(OMe]2.33 The profiles of the borylation of with the two catalysts are shown in Figure 2. The reactions of these arenes with the trisboryl complex ligated by tmphen formed the products in high yield (7, 103%; 9, 91%; 11, 88%), whereas the reactions with the trisboryl complex ligated by dtbpy formed the product in much lower yield (7, 5%; 9, 31%; 11, 30%; Table 1).
Figure 2.
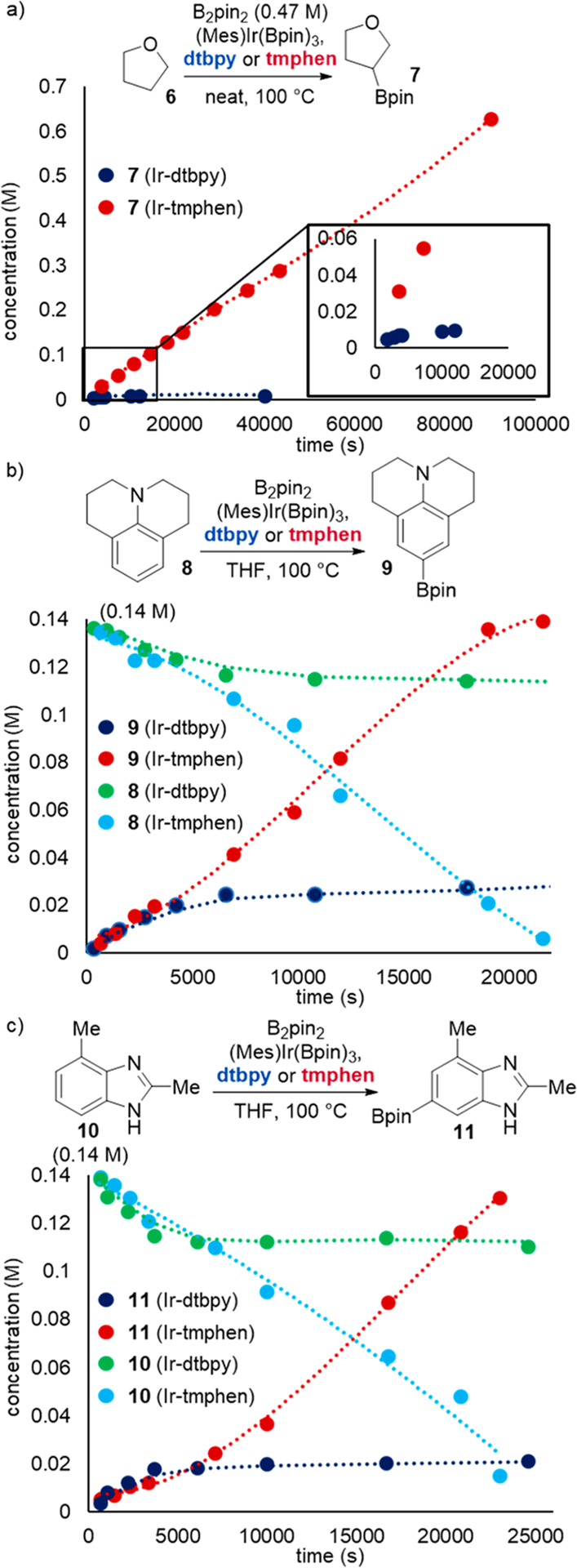
Catalytic borylation of (a) THF (6), (b) Julolidine (8), and c) 2,4-dimethylbenzimidazole (10) with (Mes)Ir(Bpin)3 (5 mol%). Initial rates: 6, 5.7 × 10−6 M s−1 (Ir-tmphen) and 2.7 × 10−6 M s−1 (Ir-dtbpy); 8, 7.1 × 10−6 M s−1 (Ir-tmphen) and 6.6 × 10−6 M s−1 (Ir-dtbpy); 10, 3.7 × 10−6 M s−1 (Ir-tmphen) and 4.9 × 10−4 M s−1 (Ir-dtbpy).
Table 1.
Yields of Borylated Product after 24 h, Initial Rates of the Borylation Reactions, and Percentage of Borylated Dtbpy Ligand for the Catalytic Reactions of THF (6), Julolidine (8), and 2,4-Dimethylbenzimidazole (10)
| product | |||
|---|---|---|---|
| 7 | 9 | 11 | |
| yield with 5 mol% Ir-dtbpy (%) | 5 | 31 | 30 |
| yield with 5 mol% Ir-tmphen (%) | 103 | 91 | 88 |
| initial rates with 5 mol% Ir-dtbpy (M s−1) | 2.7 × 10−6 | 6.6 × 10−6 | 4.9 × 10−6 |
| initial rates with 5 mol% Ir-tmphen (M s−1) | 5.7 × 10−6 | 7.1 × 10−6 | 3.7 × 10−6 |
| percentage of dtbpy ligand borylated after 24 h | 80 | 60 | 45 |
The formation of product over time began to reveal the origin of these differences in yields. These plots showed that the initial rates (Figure 2 and Table 1) of the reactions catalyzed by Ir-tmphen are similar to those catalyzed by Ir-dtbpy. However, the reactions catalyzed by Ir-tmphen occurred to high conversion and high yield, whereas the reactions catalyzed by Ir-dtbpy stopped after partial conversion. The plots show that the remaining material in these reactions was starting substrate, and no significant accumulation of side products was observed (see Supporting Information). These results show that the lower yields from the borylations of alkanes, electron-rich arenes, and heteroarenes catalyzed by Ir-dtbpy than those from the borylations catalyzed by Ir-tmphen is due to catalyst lifetime, not due to a difference in inherent reactivity of the trisboryl intermediate with the substrate or formation of side products from reactions of the catalyst bearing dtbpy.
To reveal the origin of the difference in catalyst lifetime, the borylation of THF catalyzed by Ir-dtbpy was analyzed by 1H NMR spectroscopy after 12 h at 100 °C. We observed the formation of a new species corresponding to signals at 8.70 and 7.82 ppm. A comparison of the 1HNMR spectra of independently synthesized 2,2′-(pinB)2-4,4′-(tBu)2 bpy (13) matched that of the new species observed during the reaction (see Supporting Information for solid-state structure by X-ray diffraction).35 To gauge the rate of the borylation of dtbpy versus that of the borylation of the substrate, the reactions of equal concentrations of dtbpy and ο-xylene side by side were monitored. The plot in Figure 3a shows that the rate of the borylation of dtbpy is comparable to that of the borylation of o-xylene. To determine if the borylation of dtbpy is competitive with the borylation of o-xylene in the same reaction solution, we conducted the reaction of B2pin2 with 1 equiv of ο-xylene and 1 equiv of dtbpy catalyzed by the Ir-dtbpy system (Scheme 3b). Monoborylated dtbpy (46%), diborylated dtbpy (8%), and monoborylated o-xylene (58%) all formed. These experiments show that the rate of the borylation of dtbpy is similar to that of the borylation of arenes and that it is plausible that dtbpy undergoes borylation during the catalytic processes, particularly during catalytic reactions of substrates, such as THF, containing less reactive C−H bonds.
Figure 3.
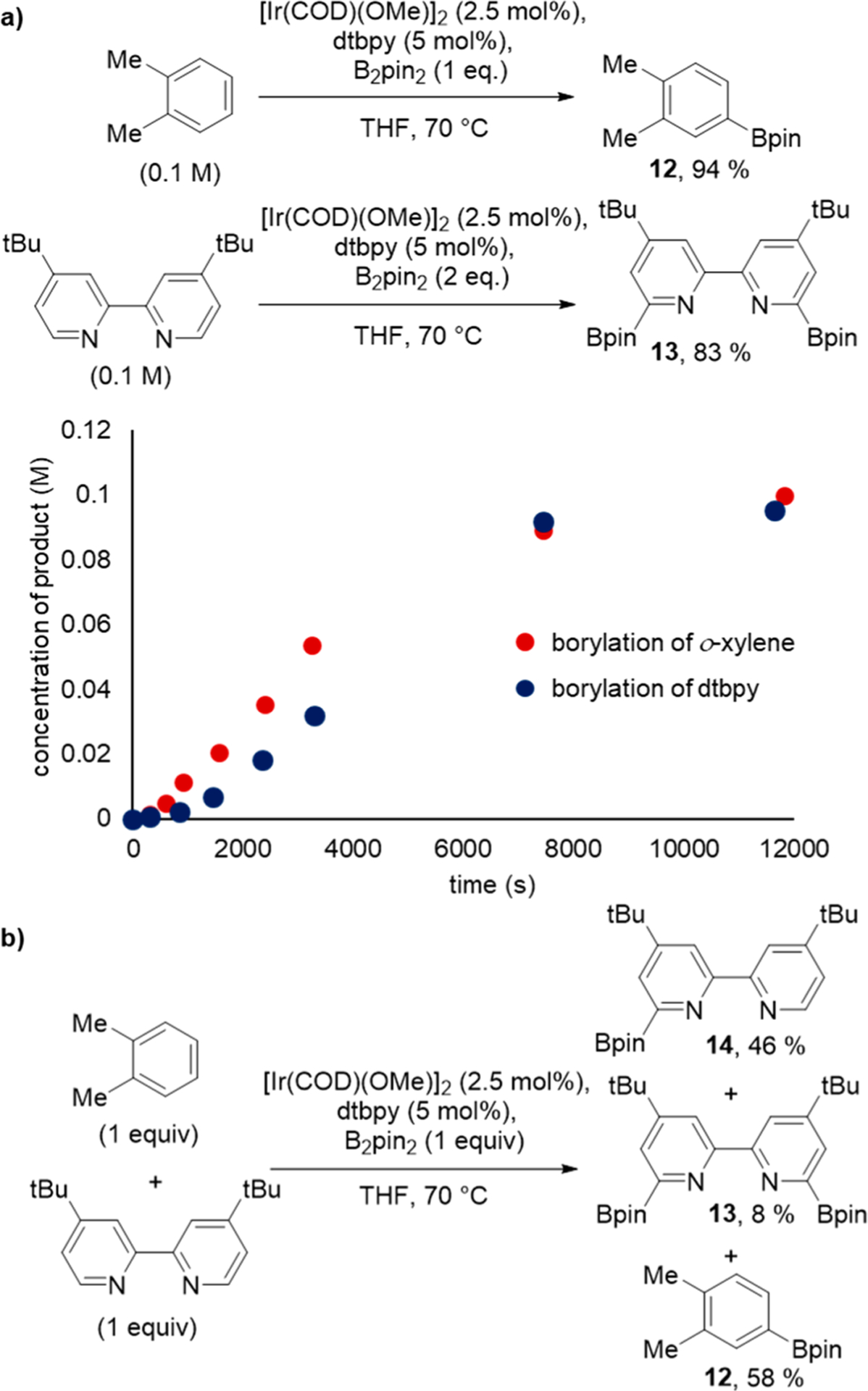
(a) Catalytic borylation of o-xylene and tert-butylbipyridine with [Ir(COD)(OMe)]2 (2.5 mol%). (b) Reaction of 1 equiv o-xylene, 1 equiv tert-butylbipyridine, and 1 equiv B2pin2.
To assess whether dtbpy underwent borylation during reactions of substrates that form borylated products in higher yields when catalyzed by Ir-tmphen than when catalyzed by Ir-dtbpy, we obtained 1H NMR spectra of crude reactions of THF, aminoarene 8, and heteroarene 10 after 12 h of reaction time at 80 °C. These spectra showed that 80%, 60%, and 45% of the dtbpy ligand had converted to the borylated dtbpy 13 for the reactions of THF, aminoarene 8 and heteroarene 10, respectively (Table 1). These data are consistent with the conclusion that the ligand undergoes borylation in competition with the substrate and that this borylation leads to catalyst deactivation.
To investigate whether the borylation of the ligand inactivates the catalyst, we tested the activity of Ir catalysts containing diborylated ligand 13 for the borylation of Julolidine (8) and 2,4-dimethylbenzimidazole (10). Consistent with the borylation of dtbpy leading to catalyst deactivation, the borylation of 8 and 10 catalyzed by the combination of (Mes)Ir(Bpin)3 and modified ligand 13 (Scheme 4) gave only traces of product (<5%).
Scheme 4.
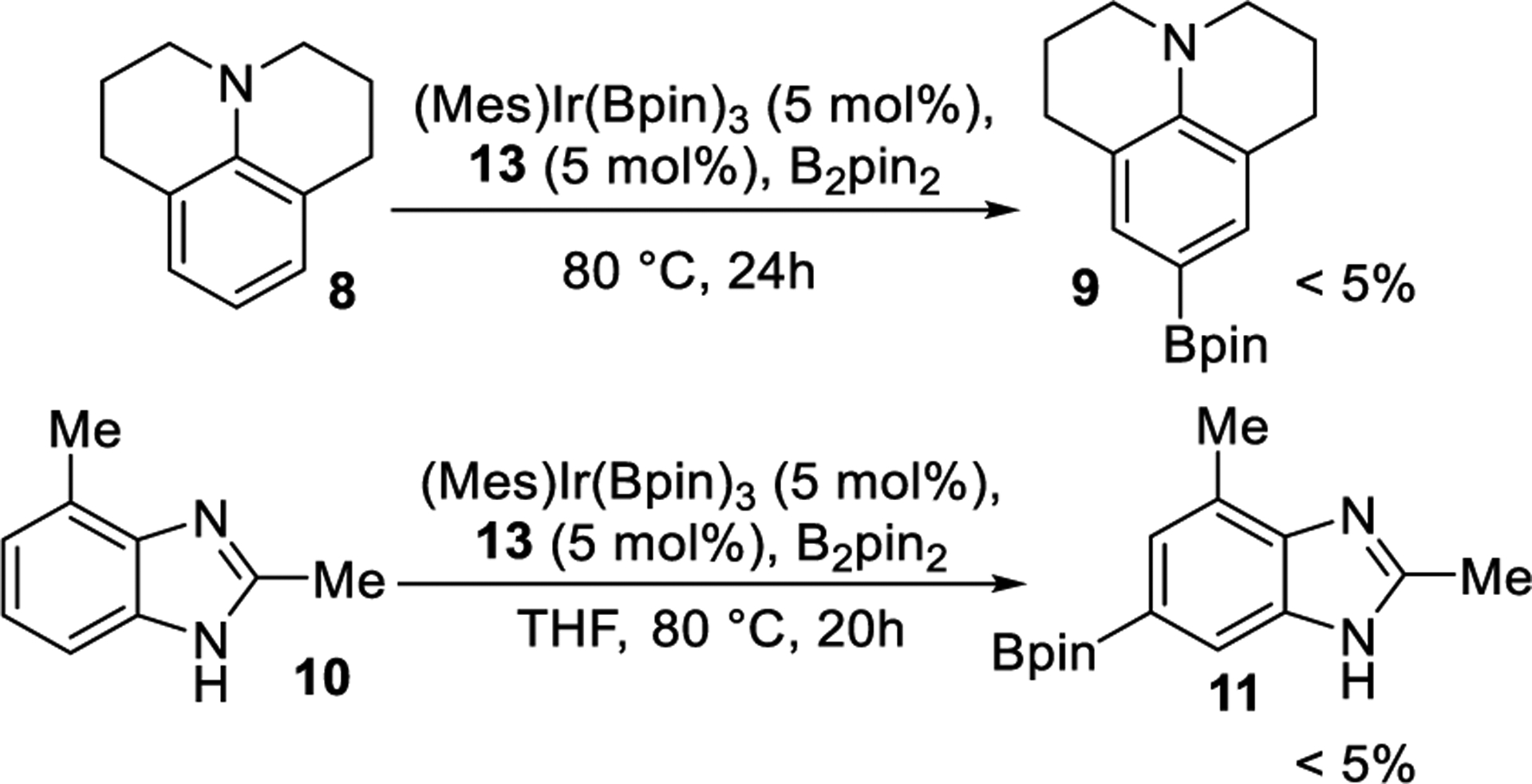
Reactions with Bpin2-tbpy 13 as Ligand
To test whether the longer lifetime of the catalyst containing tmphen is due simply to a difference in rate of the borylation of the ligand, which could be retarded by the presence of methyl groups in the 3 and 8 positions of the phenanthroline, we conducted the reaction of 1 equiv of tmphen with 2 equiv of B2pin2 under the same conditions that led to the borylation of dtbpy. In contrast to the slower rate of decomposition of the catalyst containing tmphen than of that containing dtbpy, the reaction of free tmphen with B2pin2 was faster than that of dtbpy (Figure 4). This reaction of tmphen formed a complex mixture of products. After oxidation of the borylation products with H2O2 and NaOH, the mixture was analyzed by HPLC/MS. Several species corresponding to mono and dihydroxy-lated phenanthrolines were detected, suggesting that tmphen is borylated at both aromatic and benzylic positions (see Supporting Information).
Figure 4.
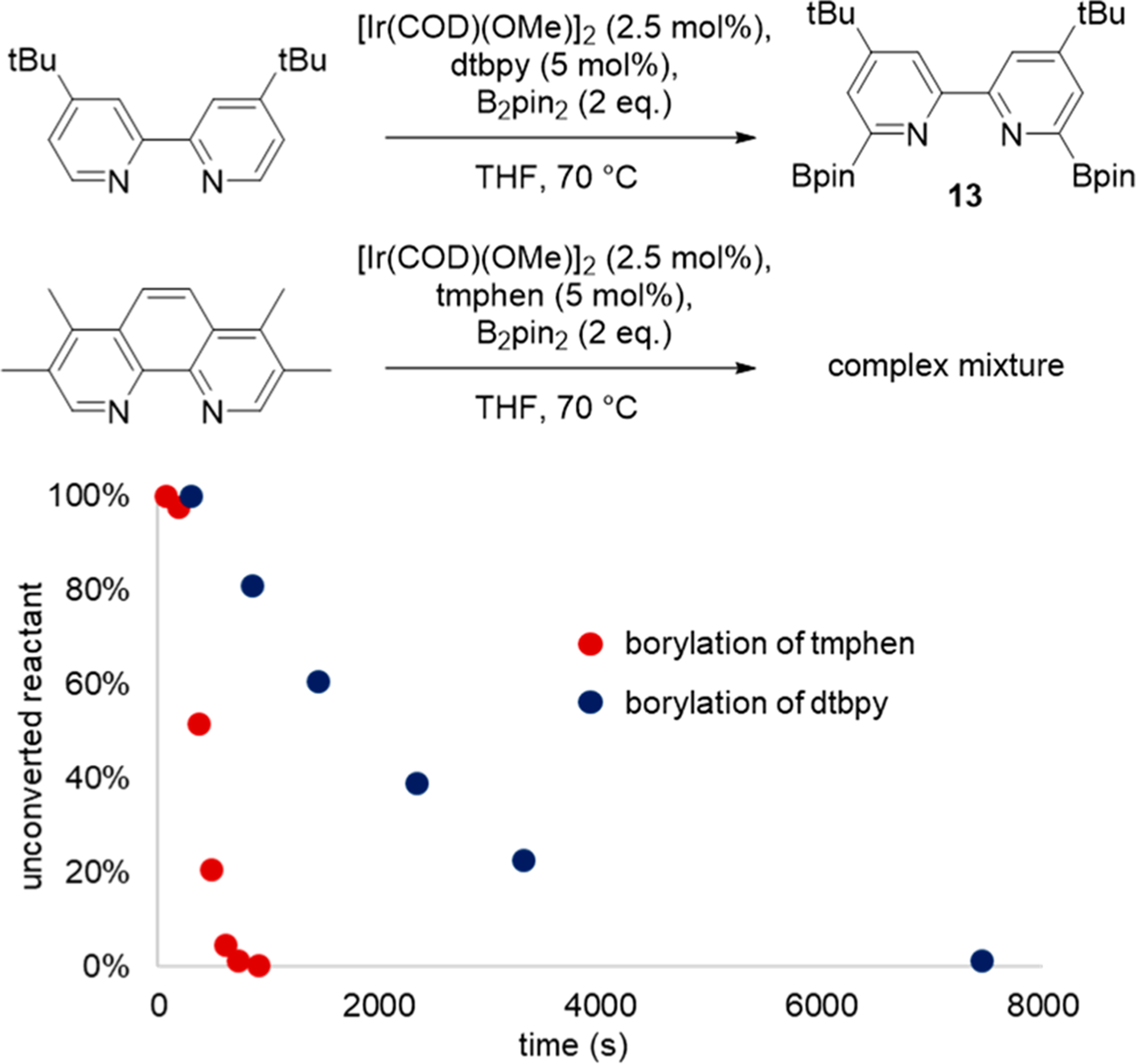
Consumption of dtbpy (blue) and tmphen (red) over time.
Because the faster catalytic borylation of free tmphen than of free dtbpy runs counter to the relative catalyst lifetimes, we considered that the differences in binding of the ligand to iridium and effect of binding on the borylation process could account for the difference in catalyst stability. To determine the relative binding of dtbpy and tmphen to the trisboryl-iridium fragment and qualitative rate of ligand exchange, we conducted ligand substitution reactions (Scheme 5). The reaction of 1 equiv of (dtbpy)Ir(Bpin)3(COE) (1) with 1 equiv of tmphen in THF-d8 was monitored by 1H NMR spectroscopy. Free tmphen fully replaced bound dtbpy within 5 min, showing that tmphen binds substantially more strongly to Ir than does dtbpy.36 The higher binding constants of phenanthroline ligands than of bipyridine ligands are due to the more favorable orientation of the nitrogen atoms for binding the metal in free phenanthrolines than in free bipyridines. In contrast to the orientation of the nitrogen atoms in free phenanthroline, the two nitrogen atoms in the most stable conformation of free dtbpy are distal from each other, and the dipoles are aligned more favorably than they are in the required orientation of the nitrogen atoms for binding of bpy ligands.37,38
Scheme 5.
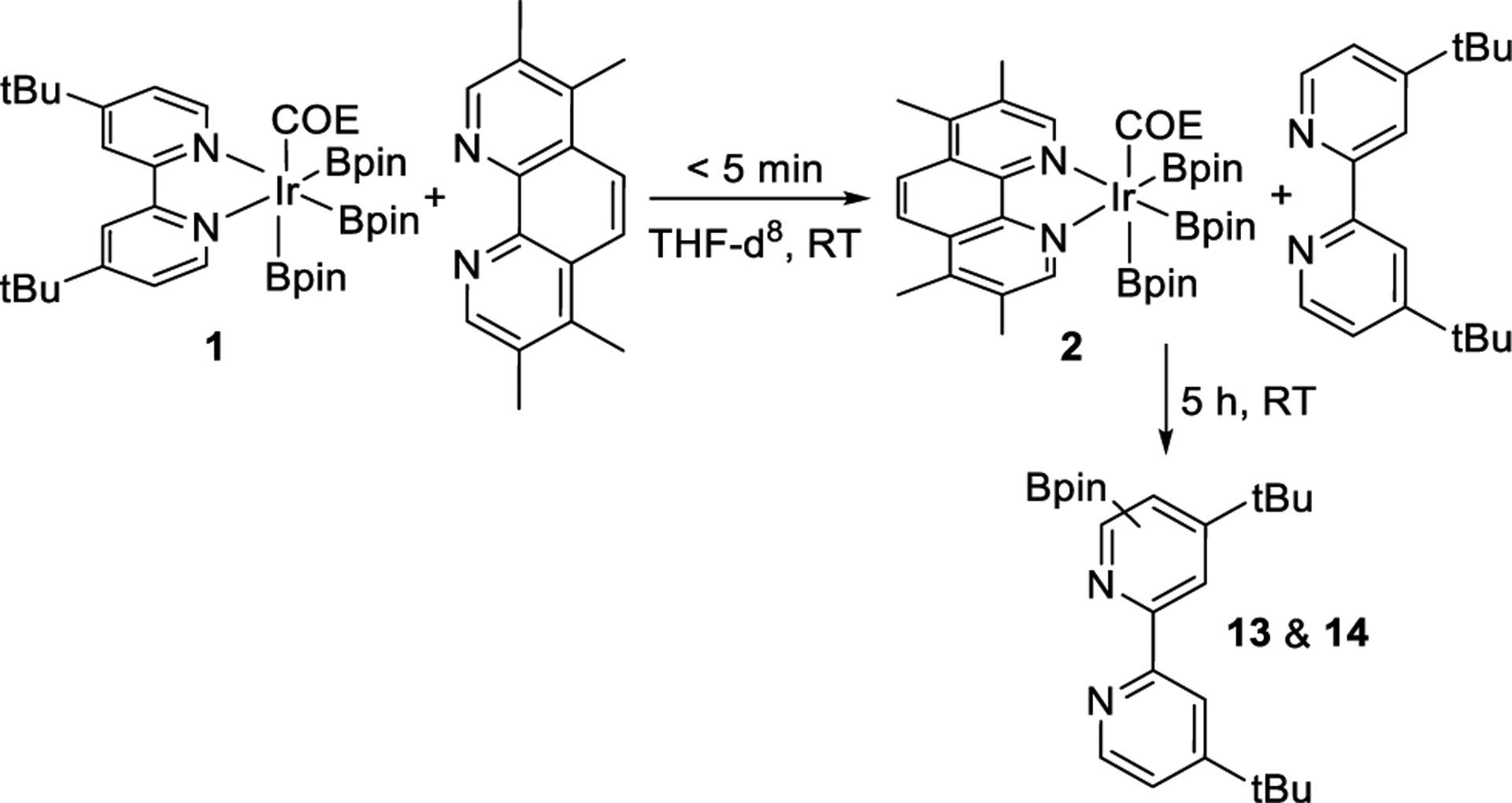
Ligand Exchange Followed by Borylation of dtbpy
After this solution was allowed to sit for 5 h at room temperature, borylated dtbpy was observed by 1H NMR spectroscopy (ca. 40%), but no signals corresponding to borylated tmphen were observed. On the other hand, a solution of 1 equiv of (tmphen)Ir(Bpin)3(COE) (2) and 1 equiv of dtbpy was unchanged after 5 min, and only borylated dtbpy was observed after 5 h. These results, together with the faster borylation of free tmphen than of free dtbpy, imply that dtbpy and tmphen are borylated when they are unbound from the metal. Thus, the stronger binding of tmphen than of dtbpy leads to a lower concentration of free ligand in reactions catalyzed by Ir-tmphen than in those catalyzed by Ir-dtbpy. This lower concentration of free ligand leads to slower borylation of tmphen and accompanying slower catalyst decomposition.
The borylation of free bipyridine-based structures has been observed previously but has not been connected to the lifetime of the catalyst. For example, the diborylation of dtbpy catalyzed by Ir-dtbpy was reported by Marder et al. in a study on C−H borylations of pyridines.35 In related work, the borylation of a pincer ligand on cobalt was reported by Chirik et al., and, in this case, the borylation of ligand was shown to decrease activity of cobalt catalysts for the borylation of arenes.39 However, the ligand on cobalt is a strongly bound pincer. Thus, borylation of the pincer ligand presumably occurs directly on the bound ligand, and borylation process likely imparts unfavorable changes to the electronic properties of the catalyst that decreases its reactivity.
CONCLUSIONS
A series of experiments on reaction rates, catalyst lifetime and pathways to catalyst decomposition shows that the activity of the iridium catalysts ligated by tmphen and dtbpy are similar but the yields for reactions catalyzed by Ir-dtbpy are lower than those catalyzed by Ir-tmphen in many cases because of catalyst deactivation. Table 1 summarizes our data on reaction yields, initial rates, and amount of borylated dtbpy formed in reactions of an aliphatic compound, an electron-rich arene, and a heteroarene: THF (6), Julolidine (8), and 2,4-dimethylbenzimidazole (10). The electronic properties of the two catalysts are similar to each other, and the rates of the stoichiometric reactions of arenes with the trisboryl complexes ligated by the two ligands are similar to each other. However, the catalyst containing dtbpy decomposes during borylations that require higher temperatures and longer reaction times, such as the borylation of Csp3−H bonds, electron-rich arenes, and heteroarenes, and this catalyst decomposition limits reaction yields.
The major decomposition pathway occurs by borylation of the nitrogen-based ligand when it is unbound from the metal. Counterintuitively, tmphen, which leads to the more stable catalyst, reacts in its free form with B2pin2 faster than free dtbpy reacts with B2pin2. Thus, the longer lifetime of the Ir-tmphen catalyst than of the Ir-dtbpy catalyst is due to stronger binding of tmphen to the metal than of dtbpy, as a result of its more rigid structure. Stronger binding leads to a lower concentration of free ligand and, thereby, slower borylation of the ligand and catalyst decomposition. These data predict that catalysts containing other rigid bidentate ligands should resist catalyst deactivation during the borylation of C−H bonds, and these findings will guide our future studies on the development of new catalysts for the borylation of C−H bonds.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the NIH R35GM130387. R.J.O. is a Swiss National Science Foundation postdoctoral fellow. AB thanks the Royal Society of Chemistry for a Researcher Mobility Grant and EPSRC and CRITICAT CDT (Ph.D. studentship to A.B.; No. EP/L016419/1). R.J.O. thanks Caleb Karmel for helpful discussions and assistance in the preparation of the manuscript.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08920.
Experimental procedures, characterization of new compounds, and crystallographic information (PDF)
X-ray crystallographic data for 13 (CIF)
REFERENCES
- (1).Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF C-H Activation for the Construction of C-B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- (2).Xu L; Wang G; Zhang S; Wang H; Wang L; Liu L; Jiao J; Li P Recent advances in catalytic C-H borylation reactions. Tetrahedron 2017, 73, 7123–7157. [Google Scholar]
- (3).Hartwig JF Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev 2011, 40, 1992–2002. [DOI] [PubMed] [Google Scholar]
- (4).Hartwig JF; Larsen MA Undirected, Homogeneous C−H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci 2016, 2, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ishiyama T; Miyaura N Iridium-catalyzed borylation of arenes and heteroarenes via C-H activation. Pure Appl. Chem 2006, 78, 1369–1375. [Google Scholar]
- (6).Ishiyama T; Nobuta Y; Hartwig JF; Miyaura N Room temperature borylation of arenes and heteroarenes using stoichiometric amounts of pinacolborane catalyzed by iridium complexes in an inert solvent. Chem. Commun 2003, 2924–2925. [DOI] [PubMed] [Google Scholar]
- (7).Ishiyama T; Takagi J; Yonekawa Y; Hartwig JF; Miyaura N Iridium-Catalyzed Direct Borylation of Five-Membered Heteroarenes by Bis(pinacolato)diboron: Regioselective, Stoichiometric, and Room Temperature Reactions. Adv. Synth. Catal 2003, 345, 1103–1106. [Google Scholar]
- (8).Kallepalli VA; Shi F; Paul S; Onyeozili EN; Maleczka RE; Smith MR Boc Groups as Protectors and Directors for Ir-Catalyzed C-H Borylation of Heterocycles. J. Org. Chem 2009, 74, 9199–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Larsen MA; Hartwig JF Iridium-Catalyzed C−H Borylation of Heteroarenes: Scope, Regioselectivity, Application to Late-Stage Functionalization, and Mechanism. J. Am. Chem. Soc 2014, 136, 4287–4299. [DOI] [PubMed] [Google Scholar]
- (10).Mkhalid IAI; Coventry DN; Albesa-Jove D; Batsanov AS; Howard JAK; Perutz RN; Marder TB Ir-Catalyzed Borylation of C-H Bonds in N-Containing Heterocycles: Regioselectivity in the Synthesis of Heteroaryl Boronate Esters. Angew. Chem., Int. Ed 2006, 45, 489–491. [DOI] [PubMed] [Google Scholar]
- (11).Ishiyama T; Takagi J; Ishida K; Miyaura N; Anastasi NR; Hartwig JF Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc 2002, 124, 390–391. [DOI] [PubMed] [Google Scholar]
- (12).Ishiyama T; Takagi J; Hartwig JF; Miyaura N A Stoichiometric Aromatic C-H Borylation Catalyzed by Iridium(I)/2,2-Bipyridine Complexes at Room Temperature. Angew. Chem., Int. Ed 2002, 41, 3056–3058. [DOI] [PubMed] [Google Scholar]
- (13).Sieser JE; Maloney MT; Chisowa E; Brenek SJ; Monfette S; Salisbury JJ; Do NM; Singer RA Ir-Catalyzed Borylation as an Efficient Route to a Nicotine Hapten. Org. Process Res. Dev 2018, 22, 527–534. [Google Scholar]
- (14).Campeau L-C; Chen Q; Gauvreau D; Girardin M; Belyk K; Maligres P; Zhou G; Gu C; Zhang W; Tan L; O’Shea PD A Robust Kilo-Scale Synthesis of Doravirine. Org. Process Res. Dev 2016, 20, 1476–1481. [Google Scholar]
- (15).Preshlock SM; Ghaffari B; Maligres PE; Krska SW; Maleczka RE; Smith MR High-Throughput Optimization of Ir-Catalyzed C−H Borylation: A Tutorial for Practical Applications. J. Am. Chem. Soc 2013, 135, 7572–7582. [DOI] [PubMed] [Google Scholar]
- (16).Liskey CW; Hartwig JF Iridium-Catalyzed Borylation of Secondary C−H Bonds in Cyclic Ethers. J. Am. Chem. Soc 2012, 134, 12422–12425. [DOI] [PubMed] [Google Scholar]
- (17).Zhong R-L; Sakaki S sp3 C−H Borylation Catalyzed by Iridium(III) Triboryl Complex: Comprehensive Theoretical Study of Reactivity, Regioselectivity, and Prediction of Excellent Ligand. J. Am. Chem. Soc 2019, 141, 9854–9866. [DOI] [PubMed] [Google Scholar]
- (18).Ohmura T; Torigoe T; Suginome M Catalytic Functionalization of Methyl Group on Silicon: Iridium-Catalyzed C(sp3)−H Borylation of Methylchlorosilanes. J. Am. Chem. Soc 2012, 134, 17416–17419. [DOI] [PubMed] [Google Scholar]
- (19).Gillespie JA; Zuidema E; van Leeuwen PW; Kamer PC Phosphorus Ligand Effects in Homogeneous Catalysis and Rational Catalyst Design. In Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; Kamer PC, van Leeuwen PW, Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2012. [Google Scholar]
- (20).Hartwig JF Dative Ligands. Organotransition Metal Chemistry-From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010; Chapter 2. [Google Scholar]
- (21).Jones NL; Ibers JA, Structurally Characterized Transition-Metal Phosphine Complexes of Relevance to Catalytic Reactions. In Homogeneous Catalysis with Metal Phosphine Complexes; Pignolet LH, Ed.; Springer US: Boston, MA, 1983; pp 111–135. [Google Scholar]
- (22).Crabtree RH Carbonyls, Phosphines, and Substitution. In The Organometallic Chemistry of the Transition Metals; John Wiley & Sons, Inc.: Hoboken, NJ, 2014; Chapter 4, pp 98–133. [Google Scholar]
- (23).Tolman CA Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev 1977, 77, 313–348. [Google Scholar]
- (24).Clarke ML; Frew JJR Ligand electronic effects in homogeneous catalysis using transition metal complexes of phosphine ligands. Organometallic Chemistry; The Royal Society of Chemistry: London, 2009; Vol. 35, pp 19–46. [Google Scholar]
- (25).Wang D; Weinstein AB; White PB; Stahl SS Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev 2018, 118, 2636–2679. [DOI] [PubMed] [Google Scholar]
- (26).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Guo J-Y; Minko Y; Santiago CB; Sigman MS Developing Comprehensive Computational Parameter Sets To Describe the Performance of Pyridine-Oxazoline and Related Ligands. ACS Catal. 2017, 7, 4144–4151. [Google Scholar]
- (28).Hashiguchi BG; Bischof SM; Konnick MM; Periana RA Designing Catalysts for Functionalization of Unactivated C−H Bonds Based on the CH Activation Reaction. Acc. Chem. Res 2012, 45, 885–898. [DOI] [PubMed] [Google Scholar]
- (29).McDonald RI; Liu G; Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Johnson LK; Killian CM; Brookhart M New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and alpha-Olefins. J. Am. Chem. Soc 1995, 117, 6414–6415. [Google Scholar]
- (31).Chen C Designing catalysts for olefin polymerization and copolymerization: beyond electronic and steric tuning. Nat. Rev. Chem 2018, 2, 6–14. [Google Scholar]
- (32).Wang F; Chen C A continuing legend: the Brookhart-type α-diimine nickel and palladium catalysts. Polym. Chem 2019, 10, 2354–2369. [Google Scholar]
- (33).Boller TM; Murphy JM; Hapke M; Ishiyama T; Miyaura N; Hartwig JF Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc 2005, 127, 14263–14278. [DOI] [PubMed] [Google Scholar]
- (34).Larsen MA; Wilson CV; Hartwig JF Iridium-Catalyzed Borylation of Primary Benzylic C−H Bonds without a Directing Group: Scope, Mechanism, and Origins of Selectivity. J. Am. Chem. Soc 2015, 137, 8633–8643. [DOI] [PubMed] [Google Scholar]
- (35).Sadler SA; Tajuddin H; Mkhalid IAI; Batsanov AS; Albesa-Jove D; Cheung MS; Maxwell AC; Shukla L; Roberts B; Blakemore DC; Lin Z; Marder TB; Steel PG Iridium-catalyzed C−H borylation of pyridines. Org. Biomol. Chem 2014, 12, 7318–7327. [DOI] [PubMed] [Google Scholar]
- (36).This reaction is likely associative; the dissociation of COE from the trisboryl complex is known to occur on the NMR time scale.
- (37).Alreja P; Kaur N Recent advances in 1,10-phenanthroline ligands for chemosensing of cations and anions. RSC Adv. 2016, 6, 23169–23217. [Google Scholar]
- (38).Bencini A; Lippolis V 1,10-Phenanthroline: A versatile building block for the construction of ligands for various purposes. Coord. Chem. Rev 2010, 254, 2096–2180. [Google Scholar]
- (39).Obligacion JV; Semproni SP; Pappas I; Chirik PJ Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design. J. Am. Chem. Soc 2016, 138, 10645–10653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.