

Abstract
Prostate cancer is one of the most commonly diagnosed cancers and a pressing health challenge in men worldwide. Radiation therapy (RT) is widely considered a standard therapy for advanced as well as localized prostate cancer. Although this primary therapy is associated with high cancer control rates, up to one-third of patients undergoing radiation therapy becomes radio-resistant and/or has tumor-relapse/recurrence. Therefore, focus on new molecular targets and pathways is essential to develop novel radio-sensitizing agents for the effective and safe treatment of prostate cancer. Here, we describe functional studies that were performed to investigate the role of structural maintenance of chromosome-1 (SMC1A) in radioresistance of metastatic prostate cancer cells. Short hairpin RNA (shRNA) was used to suppress SMC1A in metastatic castration-resistant prostate cancer cells, DU145 and PC3. Clonogenic survival assays, Western blot, RT-PCR, and γ-H2AX staining were used to assess the effect of SMC1A knockdown on radiation sensitivity of these prostate cancer cells. We demonstrate that SMC1A is overexpressed in human prostate tumors compared to the normal adjacent tissue. SMC1A knockdown limits the clonogenic potential, epithelial-mesenchymal transition (EMT), and cancer stem-like cell (CSC) properties of DU145 and PC3 cells and enhanced efficacy of RT in these cells. Targeted inhibition of SMC1A not only plays a critical role in overcoming radio-resistance in prostate cancer cells, but also suppresses self-renewal and the tumor-propagating potential of x-irradiated cancer cells. We propose that SMC1A could be a potential molecular target for the development of novel radio-sensitizing therapeutic agents for management of radio-resistant metastatic prostate cancer.
Keywords: cancer stem-like cells, DNA-damage repair, epithelial to mesenchymal transition, metastatic castration resistant prostate cancer, radiation-resistance, reactive oxygen species, structural maintenance of chromosome-1
1 |. INTRODUCTION
Prostate cancer is the most common cancer in American men with an estimated 164 690 new cases of prostate cancer expected in 2018. It is also projected that about 29 430 deaths due to the metastatic disease could be recorded.1 While localized prostate cancer can be controlled with surgery and hormonal therapy, the metastatic, castration-resistant (mCRPC) disease remains a major treatment challenge.2,3 Currently available treatment options include administration of androgen receptor inhibitor (enzalutamide, abiraterone), cell cycle disruption (docetaxel, cabazitaxel), immune checkpoint-related therapy (sipuleucel-T), targeting of bone metastasis (radium-223), and radiotherapy.4–8 Radiation therapy (RT) is widely considered a standard care treatment for primary and metastatic prostate cancer and is associated with high cancer control rates, although up to one-third of patients receiving this therapy becomes radio-resistant or manifest tumor-recurrence.8–10 Radiotherapy kills the cancer cells directly by causing DNA damage, but radiation also causes the generation of reactive oxygen species (ROS) which are indirectly involved in DNA damage.11–13 Moreover, RT promotes metastasis and invasion of cancer cells and is involved in epithelial-mesenchymal transition (EMT).14–18 Therefore, attenuating EMT pathways may improve the efficacy of radiotherapy. Unfortunately, currently known anti-metastatic or anti-angiogenic inhibitors have limitations because of their acute toxicity and drug resistance.15–18
Here, we describe that structural maintenance of chromosome-1 (SMC1A), a member of cohesin and one of the major target substrates of ATM kinase is overexpressed in prostate cancer. It is also known to be overexpressed and associated with tumor metastasis in breast, lung, glioma, and colorectal cancers.19–22 Recently, its role has been highlighted in cell proliferation and maintenance of pluripotency.23–25 Interestingly, SMC1A silencing attenuates the activation of Erk1/2 and Akt, which could be related to tumor metastasis and drug resistance.22,26,27 SMC1A binds with breast cancer type 1 susceptibility protein (BRCA1), and together they are involved in the regulation of DNA-damage response and cell cycle checkpoint-mediated DNA-repair in response to x-irradiation and chemotherapeutic agents.28,29 SMC1A phosphorylation is considered as a good prognostic marker for the efficacy of radiotherapy in hematological malignancies.30 However, the role of SMC1A in sensitizing prostate cancer cells toward radiation therapy has not been explored yet.
In this study, we describe a key role of SMC1A in tumor-metastasis and resistance to radiation therapy. We dissected the role of SMC1A in the association of cancer stem-like cells (CSCs), EMT, and DNA-damage response (DDR) pathways. We demonstrate that suppression of SMC1A expression reduces the self-renewal capacity of prostate cancer cells. Our results showed that SMC1A-suppression interferes with the progression of EMT and DDR pathway activation. More significantly, it works synergistically with radiation treatment to kill prostate cancer cells which express an augmented level of SMC1A.
2 |. MATERIALS AND METHODS
2.1 |. Reagents
Details of primary/secondary antibodies, shRNA, and sequence of primers used in this study are listed in Supplementary Tables S1 and S2. Commercially available prostate tissue microarrays (TMAs) were purchased from US Biomax (Rockville, MD). pLKO.1-SMC1A shRNA1 (TRCN0000299515) and pLKO.1-non-silencing shRNA, polybrene, puromycin, and cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) were purchased from Sigma-Aldrich (St. Louis, MO); lentiviral packaging plasmids (pMDLg/pRRE, pRSV-Rev, and pMD2.G) were from Addgene (Cambridge, MA). Direct-zol RNA miniprep kit was from Zymo Research (Irvine, CA). Tetro cDNA synthesis kit was purchased from Bioline (Taunton, MA). GSH-Glo™ glutathione assay kit was purchased from Promega (Madison, WI). Sources of other reagents were the same as previously described.19,31,32
2.2 |. Cell cultures and irradiation
Human prostate cancer cells (DU145 and PC3) were purchased from ATCC (Manassas, VA), authenticated by STR-profiling at the source and passaged for less than 6 months after receipt or resuscitation. Cells were grown at 37°C in a humidified atmosphere of 5% CO2 in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Cells were grown in culture dishes, tissue culture treated multi-well plates, or chamber slides and irradiated with X-RAD SmART system (225 keV) at indicated doses (0.5, 2, 5, and 10 Gy) delivered at a rate of 150 cGy/min (1.5 Gy/min). After irradiation, cells were grown in standard culture conditions further experiments.
2.3 |. SMC1A knockdown
293T cells (purchased from ATCC) were grown in standard culture conditions and transfected by calcium phosphate co-precipitation with either pLKO.1-SMC1A shRNA1 or pLKO.1-non-silencing shRNA and the lentiviral packaging plasmids (pMDLg/pRRE, pRSV-Rev, pMD2.G). The culture medium was replaced with fresh medium after 5 h and the supernatant was collected at 24 and 48 h. Prostate cancer cells (DU145 and PC3) were transduced with the viral vectors in the presence of 4 μg/mL polybrene. The transduced cells were selected with puromycin (1 μg/mL) and used for further experiments.
2.4 |. Quantitative real time RT-PCR and Western blot
The expression of genes involved in EMT and maintenance of CSCs were quantified by real time qRT-PCR using gene-specific primers (Supplementary Table S1). Briefly, total RNA was isolated using the Direct-zol RNA miniprep kit and reverse transcribed using the Tetro cDNA synthesis kit following manufactures directions. qPCR was performed using a CFX96 touch real-time detection system (Bio-Rad, Hercules, CA). Relative mRNA expression was calculated using the comparative method where expression of the target genes in each sample was normalized to GAPDH expression.
The expression of proteins was detected by Western blot analysis. Appropriate amounts of lysates (~20 μg protein) were resolved over Criterion™ TGX precast gels and then transferred onto polyvinylidenedifluoride (PDVF) membrane. The blots were blocked using 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 2 h at room temperature and probed using appropriate primary antibodies in 5% bovine serum albumin (BSA) in TBST buffer for overnight at 4°C. The detail of antibodies is given in Supplementary Table S2. The membranes were then incubated with horseradish peroxidase (HRP) conjugated secondary antibody followed by detection using chemiluminescence ECL kit (Bio-Rad). Densitometry measurements of the bands in Western blot were performed using GS-900 calibrated densitometer (Bio-Rad).
2.5 |. Expression and cellular localization of SMC1A in prostate cancer cells
Cellular localization of SMC1A was performed on prostate cancer cells by method described previously with slight modifications.19,32 Briefly, cells (~20 000 cells/well) were plated in polymer 15 μ-slides with 8 well (ibidi) and grown in standard culture conditions and after 24 h, cells were fixed with ice-cold methanol and acetic acid (7:1), permeabilized by incubating for 10 min in phosphate-buffered saline (PBS) containing 0.25% Triton-X100 followed by washing three times with PBS containing 0.1% Tween 20 (PBST). Nonspecific antibody interactions were minimized by incubating cells with 2% bovine serum albumin (BSA) in PBST for 60 min at room temperature. Subsequently, cells were incubated with anti-SMC1A IgG raised in rabbit (1:500 dilution in PBS containing 1% BSA, from Bethyl Labs) overnight at 4°C in a humidified chamber. After washing with PBST five times, the cells were incubated in DyLight® 550-conjugated goat anti-rabbit IgG (1:500 dilution in PBST containing 1% BSA) for 2 h at room temperature in a humidified chamber, followed by washing with PBS five times. DAPI (4′,6-Diamidino-2-phenylindole) was used as a nuclear counter-stain. Slides were analyzed by fluorescence microscope (Zeiss observer II) and the images were acquired at 40× magnification.
2.6 |. Immunohistochemistry and tissue array analysis
Commercially available prostate tissue microarrays (TMAs) were purchased from US Biomax (Rockville, MD). TMAs consisted of cores of formalin-fixed, paraffin embedded prostatectomy cores in duplicate from each prostate. Cores were arrayed in a rectangular fashion and were 1.0–1.5 mm in diameter and 5 μm in thickness. A total of 97 cases of which 81 cases have tumor and matched normal adjacent tissue (NAT), 18 cases have tumor and additional five normal tissues were examined. All the tissue samples were provided with the pathology type, age, grade, and Gleason scores. Immunohistochemical staining of prostate TMAs was performed using standard IHC technique.32,33 Briefly, slides were deparaffinized using a sequential method of rehydration followed by antigen retrieval in citrate buffer with heating and then quenching in 3% hydrogen peroxide. Slides were blocked for 5 min using protein block (from Agilent Dako) and incubated with anti-SMC1A antibody (from Bethyl Labs; 1:500 dilution) overnight at 4°C. Slides were run on Dako autostainer with a rabbit polymer for detection of primary antibodies. After washes in Dako wash buffer, slides were incubated with the chromogen diaminobenzidine tetrahydrochloride (DAB), counterstained with hematoxylin, mounted, and examined under microscope (Zeiss observer II) at 40× magnification. Images were analyzed using ImagePro premier software. Immunoreactivity assessment was based on intensity of staining in epithelial cells relative to any nonspecific stromal reactivity.
2.7 |. Cellular localization of CD44 by flow cytometry (FACS)
Surface localization of CD44 was performed on prostate cancer cells using direct flow cytometry following manufacturer’s directions. Briefly, NT and SMC1A-shRNA expressing DU145 and PC3 cells were seeded in six-well plate and after 24 h, were irradiated on X-RAD SmART system (225keV) at 2 Gy delivered at a rate of 150 cGy/min (1.5 Gy/min) and grown in standard culture conditions. Cells were harvested after 24 h and washed with PBS and suspended in approximately 1 × 106 cells/mL in ice cold PBS, containing 10% FBS and 3% BSA. The cells were stained with FITC-conjugated human CD44 antibodies following manufactures instructions. Cells were washed four times with PBS, resuspended in ice-cold PBS containing 3% BSA and analyzed by flow cytometry (BD Fortessa) at analytical cytometry core, City of Hope).
2.8 |. Quantification of γH2AX foci formation after irradiation
NT and SMC1A shRNA expressing DU145 and PC3 cells were subjected to immunocytochemistry (ICC) to quantify γH2AX foci (DNA DSB marker) as described previously.19,32 Briefly, 20 000 cells/well were grown in 1.5 polymer tissue culture treated μ-slides with 8-well (Ibidi) and after 24 h, exposed to 2 Gy radiation. At specific time points (pre-radiation, 0.5, 2, 12, and 24 h after 2 Gy), medium was aspirated and cells were fixed with ice-cold methanol, acetic acid (7:1), permeabilized with 0.25% Triton X-100, and incubated with 2% BSA in phosphate-buffered saline, 0.05% Tween 20 (PBST) for 1 h at room temperature. Cells were incubated with rabbit γH2AX (phosphor-Ser139) antibody (from Cell Signaling 1:1000 dilution in 1% BSA in PBST) overnight at 4°C, followed by incubation with goat anti-rabbit DyLight®550-conjugate (from Bethyl Labs, 1:500 dilution) for 1 h at 37°C in a humidified chamber. Cells were washed with PBS and cell nuclei were stained with DAPI and visualized by fluorescence microscopy (Zeiss observer II). For each data point, γH2AX foci were counted in at least 20 cells to yield average foci/cell and plotted.
2.9 |. Colony formation assay after irradiation
DU145 and PC3 and their corresponding SMC1A knockdown (SMC1A shRNA) cells were seeded in tissue culture treated 24-well plates with different cell numbers for each dose group (100–1000). After 24 h, cells were subjected to single dose of radiation (0.5, 2, 5, or 10 Gy) and then incubated for a period of 3 weeks in complete medium to permit colony growth. The media of all cultures was renewed every 3 days. After 3 weeks, colonies were fixed with 60% methanol and stained with 0.5% crystal violet. The colonies, defined as groups of >50 cells, were scored manually with the aid of microscope (Zeiss observer II). Data from irradiated cells were normalized against the untreated cells (scored as 100% colony forming ability). Plating efficiency and survival fractions were calculated to obtain survival parameters and to plot cell survival curves. The radiation survival curves were fitted according to the linear quadratic (LQ) model.34 The colony sensitizing enhancement ratio (SER), defined as SER = mean inactivation dose of irradiation on NT shRNA/mean inactivation dose SMC1A shRNA expressing DU145 and PC3 cells was calculated from the survival curves.
2.10 |. Prostate sphere culture and assay
Prostate cancer cells (DU145 and PC3, NT) and their corresponding SMC1A knockdown (SMC1A shRNA) cells were irradiated (5 Gy), seeded on 24-well ultra-low attachment plates (Thermo Scientific) at a density of ~1000 cells/well and grown in stem cell culture medium (StemCell, Vancouver, Canada). Sphere formation was evaluated every 24 h for up to 5 days and quantitated at day 5 by counting spherical cell clusters (>60 μm) in 10 random fields. Images were captured by fluorescence microscope (Zeiss observer II) at 10× magnification.
2.11 |. ROS measurement
Cellular ROS was measured using 2′,7′-dichlorofluorescein diacetate (DCF-DA), a fluorogenic dye that measures hydroxyl, peroxyl, and other ROS activity within the cell as described.35 Briefly, NT and SMC1A shRNA expressing DU145 and PC3 cells (20 000 cells/well) were plated in 96-well plate with clear flat bottom and black sides and grown overnight in standard culture conditions. Cells were treated with DCF-DA (a final concentration of 5 μM) in HSSB buffer and incubated for 30 min at 37°C, washed three times with PBS and fresh media was added and irradiated at 2 Gy and grown in standard culture conditions for 2 h. DCF-DA is deacetylated by cellular esterases to a non-fluorescent compound, which is oxidized by ROS into fluorescent compound 2′,7′-dichlorofluorescin (DCF).35 Fluorescence was detected by micro-plate reader (Tecan) with maximum excitation and emission spectra of 495 and 529 nm, respectively.
2.12 |. Cellular glutathione measurement
Cellular concentrations of glutathione (GSH) were measured using GSH-Glo™ assay kit following manufacturer’s directions. Briefly, DU145 and PC3 and their corresponding SMC1A knockdown (SMC1A-shRNA) cells were seeded in 96-well plates (10 000 cells/well) and grown in standard culture conditions overnight and irradiated on X-RAD SmART X-ray machine (225keV) at 2 Gy as described above. In parallel, equal number of cells were plated and grown as a control (no radiation treatment). Two hours after radiation, culture medium was removed and 100 μL of 1× GSH-Glo™ reagent was added to each well and were incubated at room temperature for 30 min. Reconstituted luciferin detection reagent (100 μL) was added to each well, mixed on a plate shaker and incubated for 15 min. Luminescence was measured on microplate luminometer (Turner Biosystems). The ratio of cellular GSH concentration was calculated by control untreated versus radiation treated cells at different time points.
2.13 |. Statistical analyses
All data were presented as the mean ± SD. We evaluated significance of differences between control and treatment groups using a two-tailed unpaired Student’s t-test. Differences were considered statistically significant when the P-value was less than 0.05. All statistical analysis was carried out in the freely downloadable software R (http://cran.r-project.org/).36
3 |. RESULTS
3.1 |. Differential overexpression of SMC1A in mCRPC cells and tumors
To validate and explore the function of SMC1A in prostate cancer, we determined the expression of SMC1A in prostate cancer cell lines and in tissue arrays containing prostate tumor of various grades and their adjacent normal tissues (NAT) or normal prostate tissue sections. SMC1A expression, determined by Western blot shows that it is significantly overexpressed in androgen-independent prostate cancer cell lines (DU145 and PC3) compared with the androgen-sensitive cell line (LNCaP). Androgen receptor (AR) was expressed in LNCaP cells while DU145 and PC3 cells were AR negative and SMC1A expression was negatively correlated with the expression of androgen receptor (Figure 1A). Immunocytochemistry showed that cellular localization of SMC1A occurs in multiple compartments including nucleus, cell membrane, and cytoplasm in all three prostate cancer cell lines (Figure 1B). These observations are consistent with our previous report showing the extra-nuclear localization and function of SMC1A in triple negative breast cancer (TNBC).19 Considering that SMC1A expression was much higher in androgen-independent prostate derived human cancer cells DU145 and PC3, these cells were used for further defining its role in prostate cancer. Expression of SMC1A was also tested in TMAs by immunohistochemistry. SMC1A was found to be significantly upregulated in prostate tumor compared to normal or adjacent normal tissues (NAT) with positive correlation with stage and Gleason scores (Figure 1C). A Kruskal-Wallis test showed that the three groups namely non-tumor adjacent tissue (NAT), histological sections from patients tumors with Gleason score <8, and tumor sections with Gleason score ≥8, come from different distribution (P < 0.00001). In addition, pair-wise comparisons of means using a t-test indicate each pair is significantly different (all P-values <0.001) (Figure 1D).
FIGURE 1.
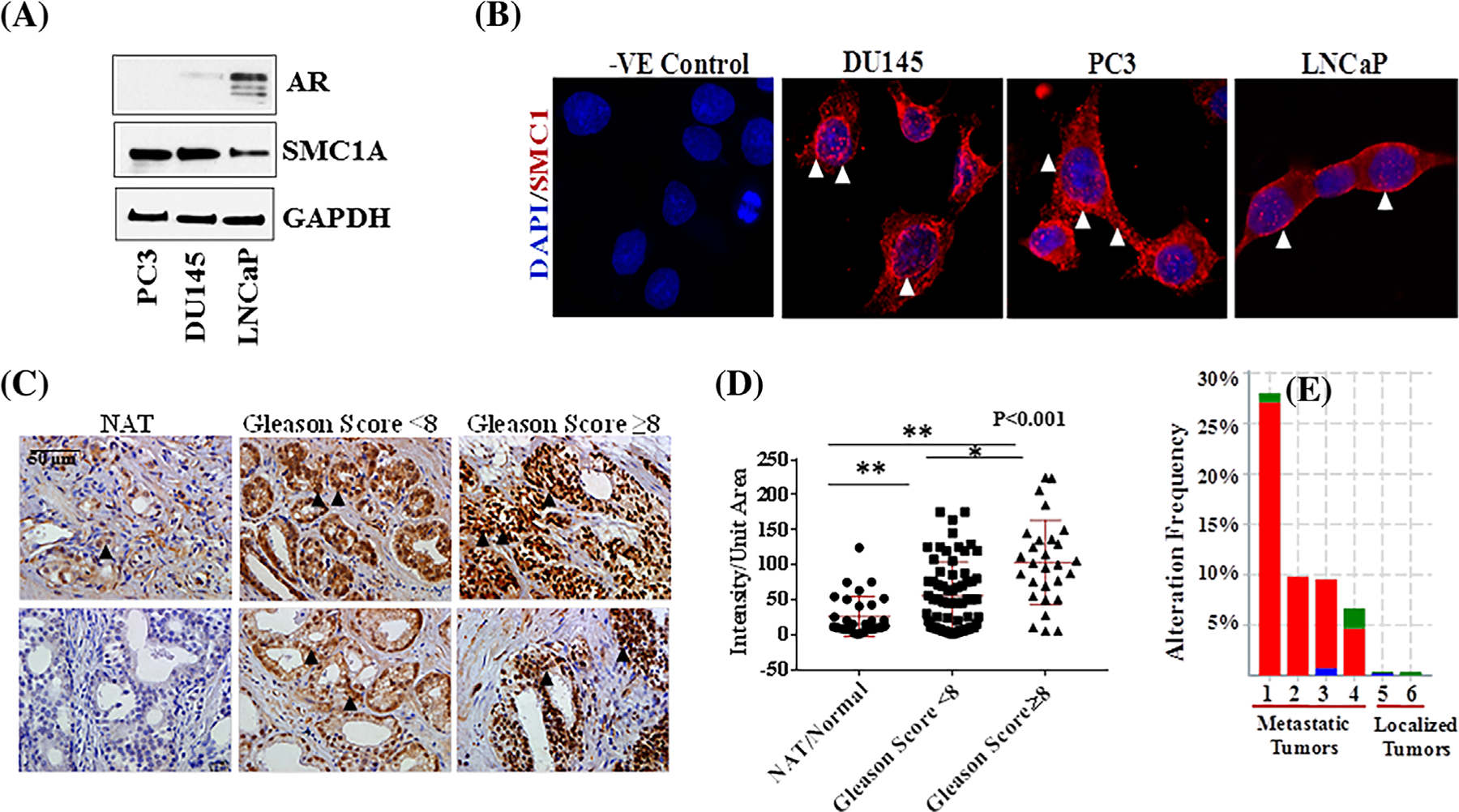
Expression and cellular localization of SMC1A in prostate cancer. A, Expression of SMC1A was tested in prostate cancer cells by Western blot as detailed in section 2. The expression of androgen receptor (AR) was validated in hormone responsive (LNCaP) and resistant (DU145 and PC3) cells by Western blot. B, Cellular localization of SMC1A was tested in prostate cancer cells by immuno-cytochemistry (ICC) as described.19 Slides were analyzed by fluorescence microscope (Zeiss Observer II) at 40× magnification. C and D, The expression of SMC1A was tested in prostate cancer tissue array (from US Biomax) by immunohistochemistry using our established protocol32,33 and images were quantified by ImagePro9.2. A Kruskal-Wallis test showed that the three groups come from different distribution (P < 0.00001). In addition, pair-wise comparisons of means using a t-test indicate each pair is significantly different (all P-values < 0.001). E, Amplification of SMC1A and its clinical significance in prostate cancer patients utilizing data mining of publicly available datasets from CBioPortal, a platform which provides large-scale cancer genomics data sets. 1–4 represents samples primarily from metastatic tumors while 5 and 6 represents primary localized tumors
3.2 |. SMC1A up-regulation is associated with recurrence and metastasis in prostate cancer patients
To illustrate the expression level of SMC1A and its clinical significance in prostate cancer patients, we performed the data mining of publicly available datasets CBioPortal, which provides large-scale cancer genomics data sets.37,38 Data include prostate adenocarcinoma from cancer genome atlas (TCGA, Cell 2015, 492 samples),39 prostate adenocarcinoma (MSKCC/DFCI, Nature Genetics 2018, 1013 cases),40 primary localized tumors and the samples from patient cohort including primarily metastatic tumor from SU2C/PCF Dream Team (Robinson et al, Cell 2015, 150 samples),41 prostate adenocarcinoma, metastatic (Michigan, Nature 2012, 61 cases),42 prostate adenocarcinoma (Fred Hutchinson CRC, Nat Med 2016, 136 cases),43 and neuroendocrine prostate cancer (Trento/Cornell/Broad 2016, 107 samples) databases.44 SMC1A was found to be significantly upregulated in prostate tumors and there was a positive correlation with clinical stage, Gleason score, and progression of tumors. The expression level of SMC1A was significantly higher in prostate tumors compared to normal adjacent tissues (Figure 1E; P < 0.05).
3.3 |. SMC1A down-regulation increases the radiosensitivity of prostate cancer cells
SMC1A is a target substrate of ATM kinase and is required for efficient and accurate repair of a variety of DSBs and DNA lesions in response to radiation and chemotherapy.28,29,45 This is the first report showing that suppression of SMC1A sensitizes prostate cancer cells toward x-irradiation. To investigate the role of SMC1A in radioresistance, we first stably knocked down SMC1A by transducing DU145 and PC3 cells with lentiviral vectors expressing SMC1A shRNA or non-targeted shRNA (NT shRNA) as a control. SMC1A expression, tested by qRT-PCR and Western blot was found to be reduced to ~20% and 30% in DU145 and PC3 cells (DU145-SMC1A shRNA and PC3-SMC1A shRNA) respectively and remained largely unaffected in control (DU145-NT shRNA and PC3-NT shRNA) cells compared to their respective parental cells (Figures 2A and 2B). Radiation did not significantly alter the expression of SMC1A in control and SMC1A knockdown cells (Figure 2A).
FIGURE 2.
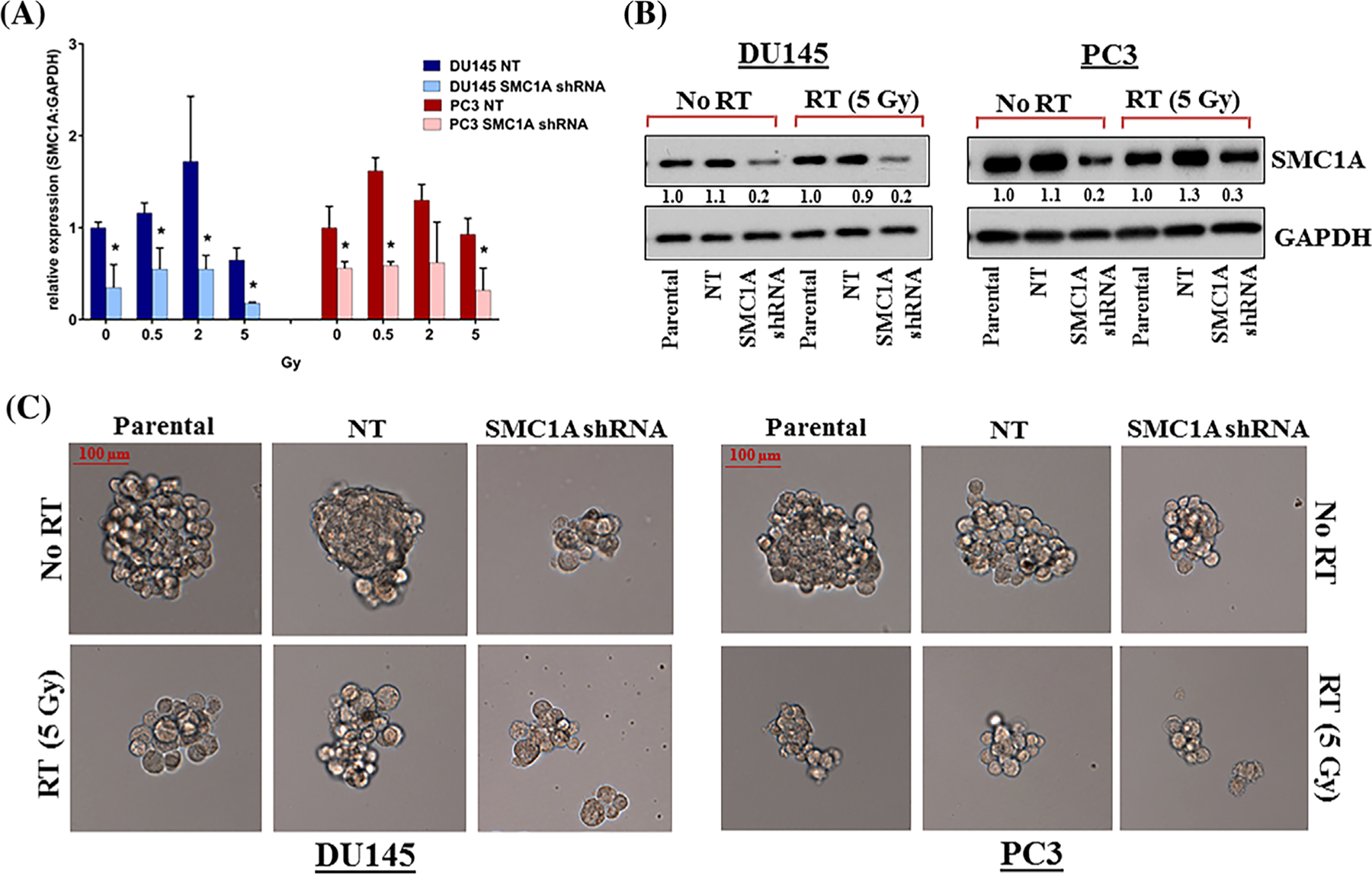
SMC1A knockdown attenuates sphere formation and sensitizes prostate cancer cells toward x-irradiation. A, SMC1A expression was detected by 48 h after irradiation by qRT-PCR in NT shRNA and SMC1A shRNA expressing DU145 and PC3 cells. SMC1A expression was significantly reduced in both DU145-SMC1A shRNA and PC3-SMC1A shRNA cells, compared to their corresponding NT shRNA expressing cells (*P < 0.05). B, Protein expression in parental, NT shRNA and SMC1A shRNA expressing DU145 and PC3 cells was analyzed by Western blot with GAPDH as a loading control. The expression of SMC1A was reduced to ~20% and 30% in DU145-SMC1A shRNA and PC3-SMC1A shRNA cells respectively compared to their NT shRNA controls. C, Ability of prostate cancer cells to form spheres was tested in x-irradiated and non-irradiated parental, NT and SMC1A shRNA expressing cells. Sphere formation was evaluated every 24 h for up to 5 days and quantitated at day 5 by counting spherical cell clusters (>60 μm) in 10 random fields. Representative images captured by fluorescence microscope (Zeiss Observer II) at 10× exposure are shown
As radiation affect the sphere formation capability of prostate cancer cells, we tested the ability of SMC1A shRNA expressing DU145 and PC3 cells to form spheres in control (no radiation treatment) and cells irradiated with 5 Gy. Cells were grown in 24-well ultra-low attachment plates (Thermo Scientific) at a density of ~1000 cells/well in stem cell culture medium (StemCell, Vancouver) for 5 days. DU145 and PC3 cells expressing NT shRNA were also irradiated and used as positive control. Our result shows that suppression of SMC1A attenuated the sphere forming ability of DU145 and PC3 cells as lower number of prostate spheres formed in cells having reduced SMC1A expression (Supplementary Table S3). In addition, sphere sizes were smaller in SMC1A shRNA expressing DU145 and PC3 cells (Figure 2C). Irradiating these cells further suppressed their ability to form spheres in both DU145-SMC1A shRNA and PC3-SMC1A shRNA cells, suggesting that SMC1A down-regulation enhanced the efficacy of radiotherapy in prostate cancer.
3.4 |. SMC1A down-regulation enhances the efficacy of radiotherapy by decreasing the colony formation ability of prostate cancer cells
To further delineate the role of SMC1A in radioresistance, SMC1A shRNA and NT shRNA expressing DU145 and PC3 cells were irradiated (0–10 Gy) and grown in 24-well tissue culture treated plate in standard culture conditions for 3 weeks and their colony formation ability was tested. Our results show that suppression of SMC1A radiosensitized both DU145 (Figures 3A and 3B) and PC3 (Figures 3C and 3D) cells more than twofold, as determined by colony formation units. However, SMC1A suppression has no significant effect on the plating efficiency of control (non-irradiated) cells. Our data support that SMC1A protein is important for cell survival after x-irradiation. Parameters including surviving fraction and plating efficiency generated from linear quadratic model are listed in Supplementary Table S4. Survival fraction was significantly reduced in x-irradiated SMC1A shRNA expressing DU145 and PC3 cells compared to NT shRNA expressing cells, indicating a radiosensitizing effect of SMC1A suppression (Figure 3).
FIGURE 3.
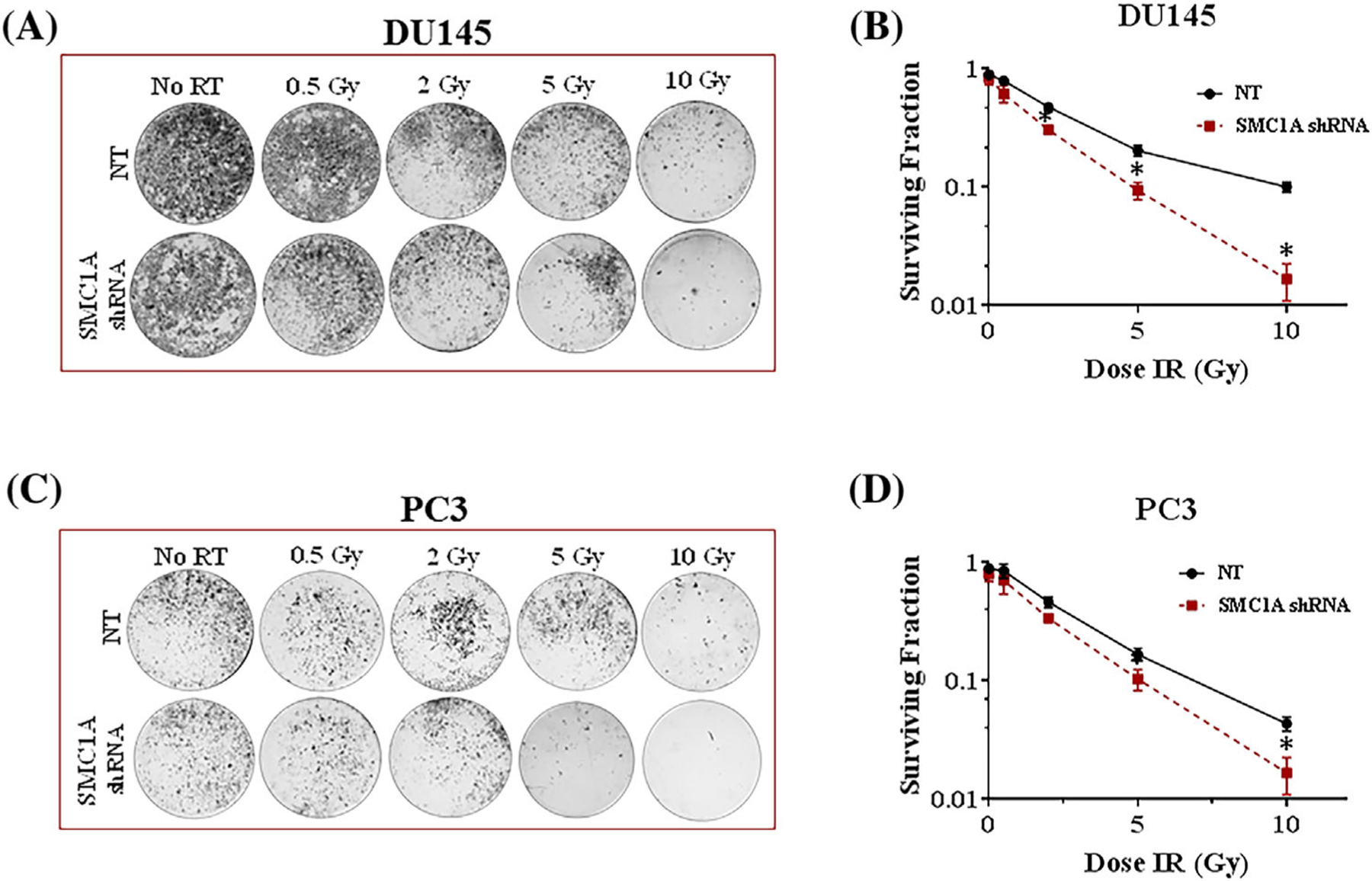
SMC1A knockdown radiosensitizes prostate cancer cells and limits their colony survival potential. (A, B) Clonogenic survival of SMC1A shRNA expressing DU145 and (C, D) PC3 cells was reduced compared to their corresponding NT shRNA expressing cells after a single dose radiation (0, 0.5, 2, 5, and 10 Gy) with the X-RAD SmART system (225 keV) delivered at a rate of 150 cGy/min (1.5 Gy/min). Data were fitted according the linear quadratic model. All results were from three independent experiments (Mean ± SD, n = 3)
3.5 |. SMC1A down-regulation enhances efficacy of radiotherapy by regulating the EMT in prostate cancer cells
Coordinated lost and gain, respectively, of epithelial (E-cadherin) and mesenchymal (Vimentin, N-cadherin) markers are known to enhance migratory and invasive abilities and increased radiation resistance in cells. SMC1A has shown to be involved in metastasis and regulation of EMT in cancer cells including breast, lung, and colorectal cancer19–22; we determined the effect of radiation treatment on the expression of EMT markers in prostate cancer cells. We analyzed mRNA levels of NT shRNA and SMC1A shRNA expressing x-irradiated (0–5 Gy) and non-irradiated DU145 and PC3 cells for the expression of EMT biomarkers, VIM (Vimentin), and CDH1 (E-cadherin). The qPCR analysis of total RNA isolated from control (no radiation treatment) and irradiated cells showed that the expression of EMT markers were significantly altered in SMC1A shRNA expressing DU145 and PC3 cells in control and irradiated cells (Figures 4A and 4B). Expression of VIM was significantly decreased in both DU145-SMC1A shRNA and PC3-SMC1A shRNA cells compared to their corresponding NT shRNA expressing control and irradiated cells, indicating reduction of the mesenchymal phenotype (Figure 4A). The expression of CDH1 was higher in PC3 shRNA expressing cells in both irradiated and non-irradiated cells (Figure 4B). We analyzed mRNA level for EMT-inducer, SNAI1 (Snail) in x-irradiated and non-irradiated cells. Consistent changes were observed for Snail expression, a transcription factor known to regulate stress resistance, stem cell-like phenotype, and EMT in various cancer types. Snail was induced by x-irradiation in both PC3 and DU145 cells but the expression of Snail was lowered in shRNA SMC1A transduced cells as compared to their corresponding NT shRNA expressing cells at mRNA (Figure 4C) and protein (Figures 4D) levels. Our results are in agreement with literature showing that the loss of cell adhesion depends on Snail, and knockdown of Snail reverted the enhanced loss of adhesion in irradiated cells, while having no effect on control cells.14 In addition, SMC1A expression correlated with the protein levels of N-cadherin, E-cadherin, Claudin-1 and Vimentin in SMC1A shRNA expressing DU145 and PC3 cells (Figure 4D). The expression of these proteins increases in response to x-irradiation but to a lower degree in SMC1A shRNA expressing cells (Figure 4D). Although there was a decrease in the expression of E-cadherin in SMC1A shRNA expressing DU145 and PC3 cells, the ratio of E-cadherin to N-cadherin is higher which support the role of SMC1A in regulation of EMT phenomenon of coordinated gain and loss of epithelial (E-cadherin) and mesenchymal (Vimentin, N-cadherin) markers in prostate cancer cells.
FIGURE 4.
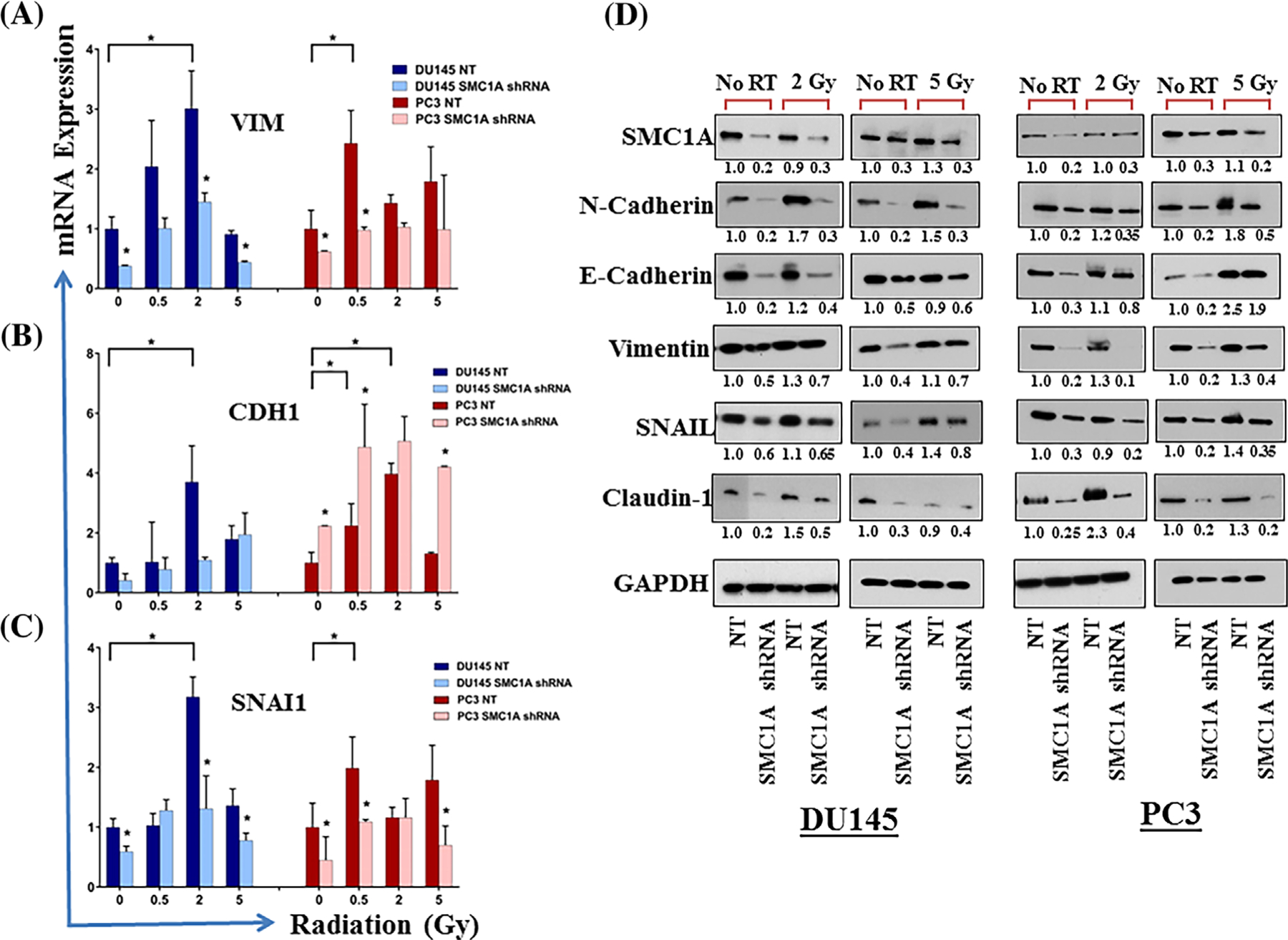
SMC1A regulates EMT in control and irradiated prostate cancer cells. A, Expression of genes involved in EMT was tested by qRT-PCR in NT shRNA and SMC1A shRNA expressing DU145 and PC3 cells irradiated (0–5 Gy) and grown in standard culture conditions for 48 h. Our data show lower levels of VIM, CDH1 and SNAI1 in cells transduced with SMC1A shRNA compared to their respective NT shRNA expressing cells. These EMT genes were overexpressed in x-irradiated cells, but their expression was significantly lower in SMC1A knockdown cells. Asterisk indicate statistically significant differences (P < 0.05). B, The expression of EMT marker proteins (N-cadherin, E-cadherin, Vimentin, Snail, and Claudin-1) was quantified by Western blot. Asterisk indicate statistically significant difference (P < 0.05)
3.6 |. SMC1A down-regulation enhances the efficacy of radiotherapy by regulating the CSC population in prostate cancer cells
SMC1A, a member of cohesin complex is known to be involved in stem cell maintenance and renewal.23–25 The transition of cells to a CSC phenotype is a critical event that enables redifferentiation to allow growth and colonization at distant metastatic sites.14,15,23 To expand the role of SMC1A in response to x-irradiation on the interplay between the EMT and CSCs, we examined the impact of SMC1A suppression on the regulation of the stem-like cell properties in DU145 and PC3 cells.14,15 Expression of genes involved in CSCs were tested in both (DU145-SMC1A shRNA and PC3-SMC1A shRNA) cells that were irradiated (0–5 Gy) and grown in standard culture conditions for 48 h by qRT-PCR and Western blot (2 Gy). We selected prostate cancer stem cell-specific biomarker genes: CD44, LEF-1, and POU5F1 (OCT4). The qPCR analysis of total RNA isolated from control and irradiated cells showed alterations in the expression of these stem-like cell related genes (Figure 5A). A significantly lower expression of CD44, LEF-1, and POU5F1 was observed in SMC1A shRNA expressing DU145 and PC3 cells compared with control cells (Figure 5A–C). Radiation increases the expression of these CSC marker genes in clinically relevant doses of ionizing radiation (≤2 Gy) but the expression was significantly attenuated in x-irradiated SMC1A shRNA transduced prostate cancer cells. In addition, SMC1A expression correlated with the expression of CSC markers including CD44, OCT4, and ALDH1 and in both DU145-SMC1A shRNA and PC3-SMC1A shRNA control and x-irradiated cells determined by and flow cytometry (CD44) and Western blot analysis (Figures 5D and 5E). These results suggest that silencing of SMC1A may attenuate the CSC-like populations in control and x-irradiated prostate cancer cells.
FIGURE 5.
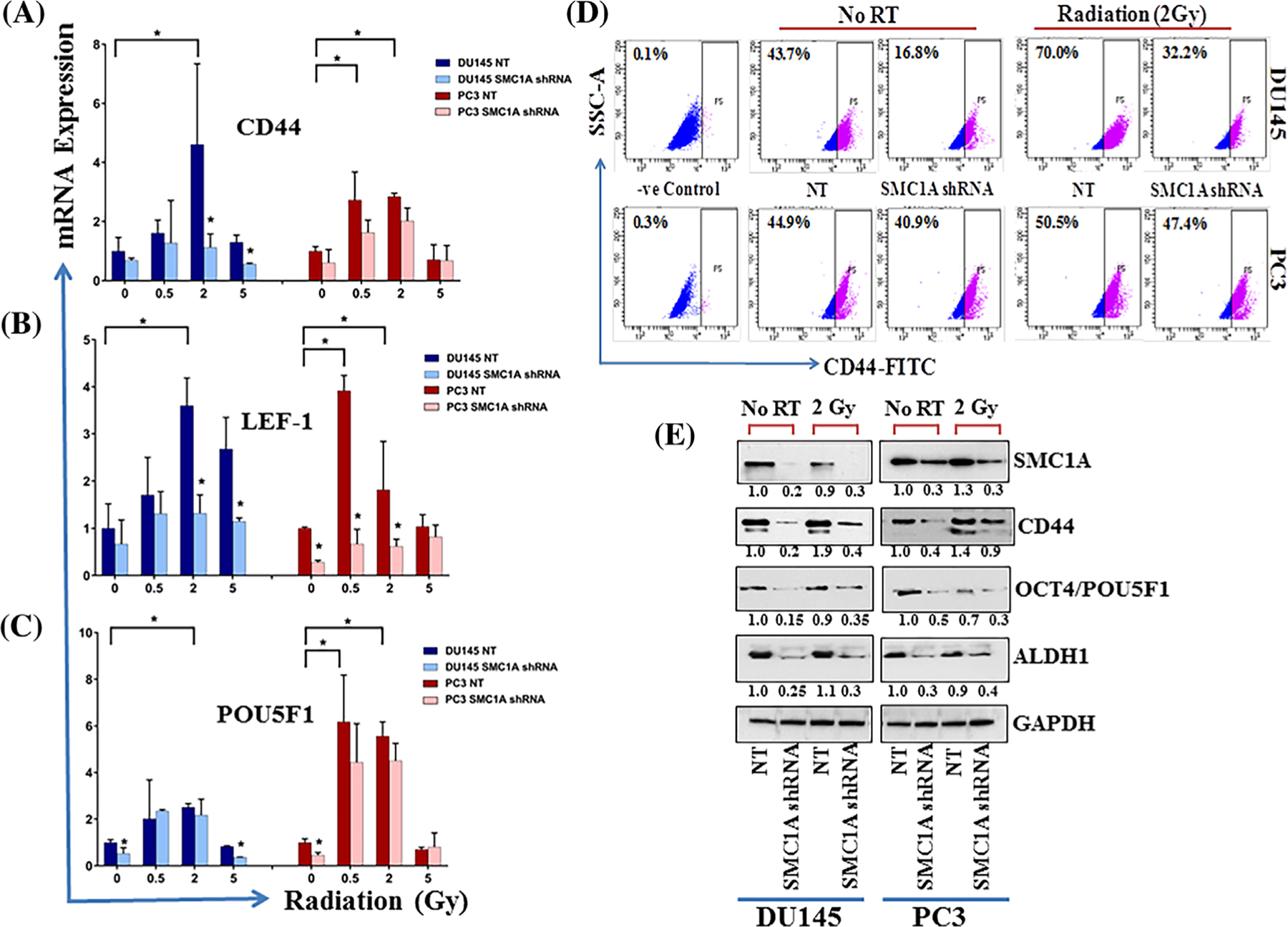
SMC1A regulates CSCs in control and x-irradiated prostate cancer cells. A-C, Expression of stem cell markers, CD44, LEF1, and POU5F1 was checked in DU145-SMC1A shRNA and PC3-SMC1A shRNA cells irradiated (0–5 Gy) and grown in standard culture conditions for 48 h, by qRT-PCR. D, Expression of CD44 was further quantified by flow cytometry in control and SMC1A expressing DU145 and PC3 cells x-irradiated with clinically relevant dose (2 Gy). E, Expression of stem-like cell markers, POU5F1, CD44, and ALDH1 was also checked in control and x-irradiated (2 Gy) NT and SMC1A shRNA expressing DU145 and PC3 cells by Western blot. Our data show that SMC1A regulates the expression of stem-like cells in x-irradiated and control cells
3.7 |. Suppression of SMC1A enhances the efficacy of radiation therapy by altering DNA-repair capacity and ROS
Radiation therapy kills the cells directly by damaging the DNA (double or single-stranded breaks) or indirectly producing free radicals including hydroxyl radical, hydrogen peroxide, and superoxide which cause damage to biomolecules.10–13 Since cohesin has shown to involve in DSB repair through sister-chromatid homologous recombination (HR) and non-homologous end joining (NHEJ)30,46–48 the two major pathways of repairing DSBs, we dissected the effect of SMC1A suppression in altering these repair pathways. Our data showed that expression of Ku70 and Ku80, two regulatory subunit of NHEJ pathway was unaffected in SMC1A shRNA expressed DU145 and PC3 cells and radiation activated both Ku70 and Ku80 in control (NT shRNA) cells but not in SMC1A shRNA expressing cells (Figure 6A). These results support that SMC1A suppression sensitized the cells toward x-irradiation by attenuating the NHEJ pathway (Figure 6A). The effect of SMC1A suppression was also explored on HR pathway by testing the expression of BRCA1, BRCA2, and Rad51, the crucial member of HR pathway by Western blot. Our results showed that SMC1A regulated the expression of Rad51 in control and x-irradiated cells (Figure 6A). BRCA2 expression was not significantly altered in control and x-irradiated cells but BRCA1 expression was activated in response to radiation in control (NT shRNA expressing cells) but not in SMC1A shRNA expressing DU145 and PC3 cells (Figure 6A).
FIGURE 6.
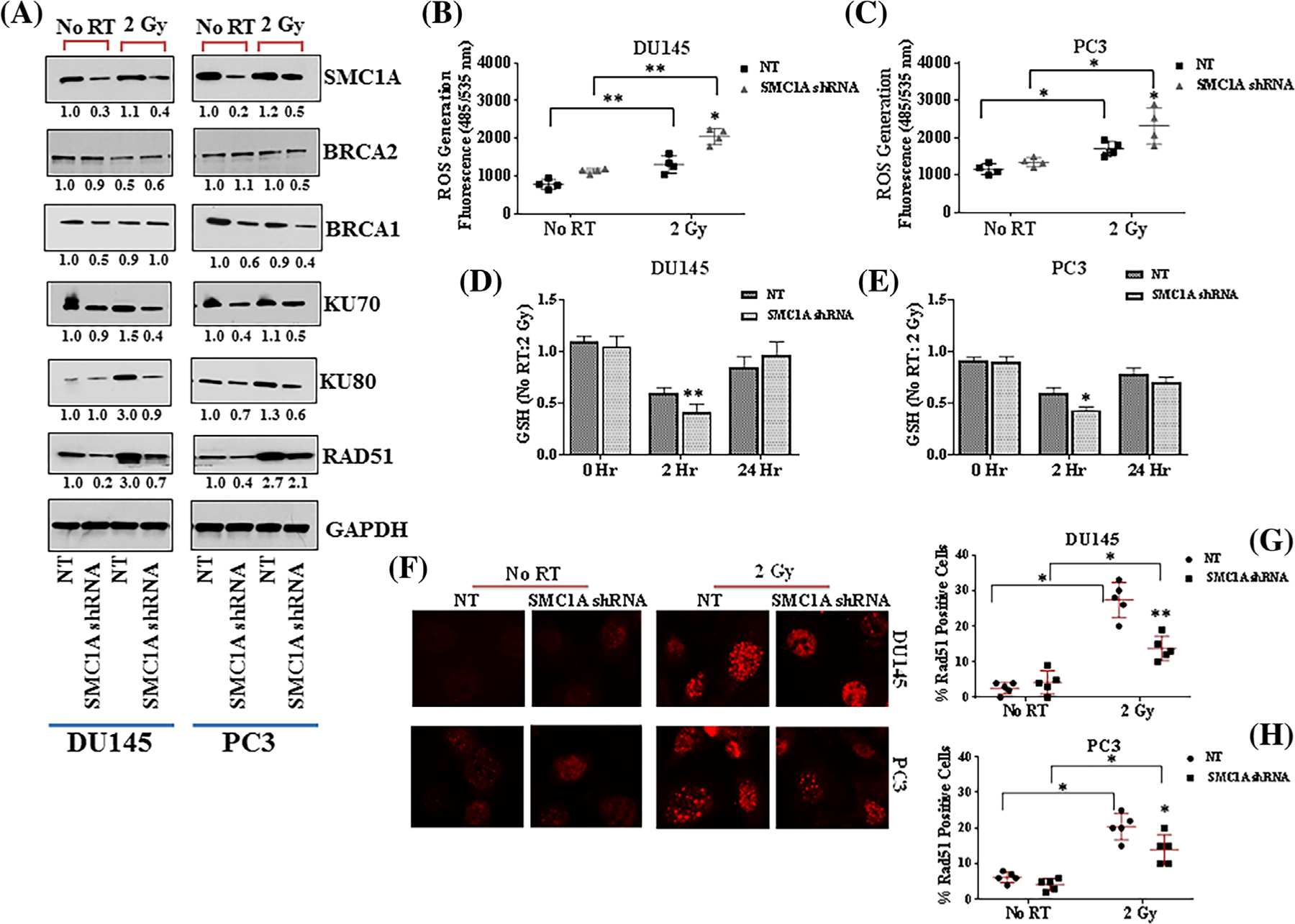
SMC1A regulates DDR and oxidative stress in control and irradiated prostate cancer cells. A, The expression of proteins involved in homologous recombination (BRCA1, BRCA2, Rad51) and NHEJ (Ku70, Ku80) were tested in SMC1A shRNA expressing DU145 and PC3 cells by Western blot. Our results showed that expression of Ku70 and Ku80, two regulatory subunit of NHEJ pathway was suppressed in SMC1A shRNA expressing DU145 and PC3 cells. Radiation activated both Ku70 and Ku80 in control cells but not in SMC1A shRNA expressing cells. SMC1A regulated the expression of Rad51 in control and x-irradiated cells. BRCA1 expression was activated in response to radiation in control cells but not in SMC1A shRNA expressing cells. BRCA2 expression was not significantly altered in control and irradiated cells. B and C, Effect of SMC1A suppression on ROS (DCF-DA) and cellular glutathione (D, E) in control and x-irradiated DU145 and PC3 cells transduced with NT or SMC1A shRNA. Our results showed that SMC1A shRNA expressing cells have significantly higher intracellular levels of ROS and reduced level of glutathione in x-irradiated cells. F, Rad51 foci formation induced by x-irradiation was significantly higher in NT shRNA expressing cells compared to SMC1A shRNA expressing DU145 and PC3 cells. G and H, The percentage of cells with persisting Rad51 foci was counted and it was higher in NT shRNA compared to SMC1A shRNA expressing cells
Intracellular antioxidants including glutathione (GSH), glutathione S-transferases (GSTs), thioredoxin reductase, glutathione peroxidase, catalase, and superoxide dismutase form the first line of defense against ROS induced oxidative stress in the cells.49 ROS can alter the cellular GSH level and promote oxidative stress. We tested the effect of SMC1A suppression on oxidative stress by measuring the ROS (DCF-DA) and GSH levels in control and x-irradiated DU145 and PC3 cells transduced with NT or SMC1A shRNA. Our results showed that SMC1A suppression did not alter the intracellular level of ROS in normal conditions (no radiation treatment) but SMC1A shRNA expressing cells have significantly higher intracellular levels of ROS and lower glutathione levels in x-irradiated cells after 2 h in culture (Figure 6B–E). These results supported the role of SMC1A in sensitizing the cells toward x-irradiation by elevating the intracellular level of ROS. Rad51 foci formation induced by x-irradiation were significantly higher in NT shRNA compared to SMC1A shRNA expressing DU145 and PC3 cells (Figure 6F–H). The relative fraction of cells with persisting Rad51 foci was higher in NT shRNA expressing DU145 cells compared to PC3 cells while lower in SMC1A shRNA expressing cells.
Phosphorylation of histone H2AX (γH2AX) occurs in response to DNA double-strand breaks produced by ionizing radiation and is a sensitive biomarker for the detection of radiation sensitivity.50,51 The change of γH2AX foci after x-irradiation (2 Gy) is shown in Figure 7. Positive γH2AX foci appeared in DU145 and PC3 cells at 2 h after irradiation (2 Gy) in both SMC1A shRNA and NT shRNA cells without any significant difference in control versus SMC1A knockdown cells (Figure 7A–D. However, the average foci number in NT shRNA cells gradually decreased at the later time points while that of the SMC1A shRNA cells increased. PC3 cells have the lower decay of γ-H2AX foci at 24 h than DU145 cells which is in agreement with the literature50,51 (Figures 7C and 7D). Foci of γ-H2AX can also be induced by factors other than ionizing radiation, such as during the process of replication and therefore not all cells have similar numbers of γ-H2AX foci after equal radiation doses.51 The effect of SMC1A suppression was more pronounced in DU145 cells (Figures 7A and 7B). This is also corroborated by our observation that higher surviving fractions after irradiation correlated with a lower number of residual γ-H2AX foci in the prostate cancer cells and DSB repair was impaired in SMC1A shRNA cells, compared with NT shRNA cells.
FIGURE 7.
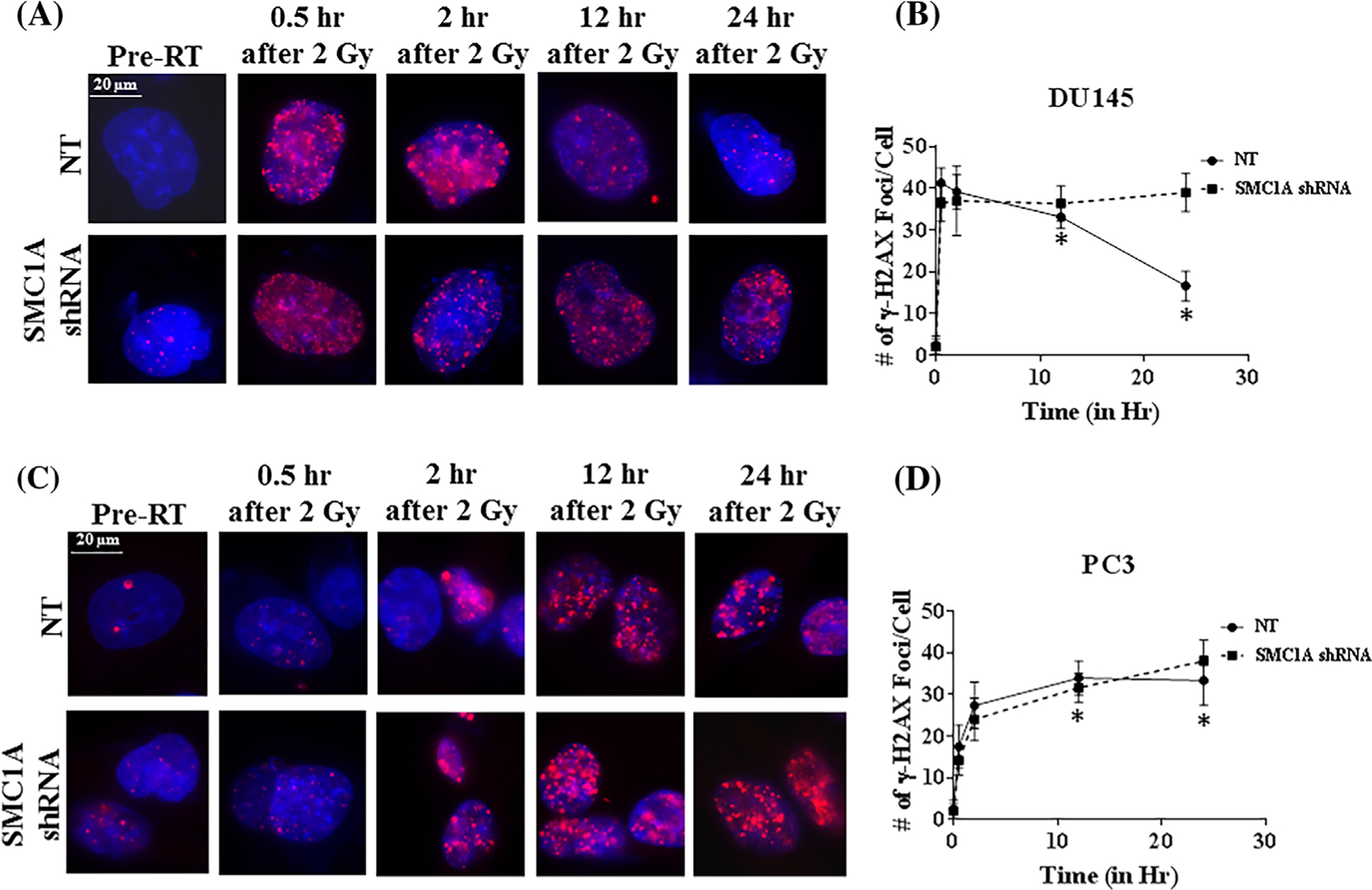
Quantification of DNA DSB marker, γH2AX foci after irradiation in SMC1A shRNA expressing DU145 and PC3 cells. A, Representative images of γH2AX staining at the specific time points in NT and SMC1A-shRNA expressing cells after irradiation (2 Gy). Red fluorescence staining indicates positive while blue stain from DAPI indicates nuclei (magnification 40×). B, Number of γH2AX foci in SMC1A shRNA expressing DU145 and PC3 cells (Mean ± SD, n = 20) were counted and plotted. At 12 and 24 h after radiation (asterisks), significant difference was found between SMC1A shRNA expressing DU145 cells. The difference was more pronounced in DU145 cells compared to PC3 cells showing these cells are more radio-resistant. SMC1A suppression sensitized both DU145 and PC3 cells (P < 0.05; SMC1A shRNA vs NT shRNA expressing DU145 and PC3 cells, no radiation treatment and irradiated with single dose of 2 Gy)
Our data support that radiation promotes a phenotypic switch toward EMT and stem-like traits in prostate cancer cells which may cause relapse and resistance after radiation therapy. SMC1A is involved in overcoming the resistance of cancer cells to radiation treatment. Thus, SMC1A represents an attractive novel target for radio-sensitizing prostate cancer cells. Development of novel therapeutic agents that target SMC1A for the treatment of this highly aggressive neoplasm could be a novel strategy to manage radio-resistant, metastatic prostate cancer cells.
4 |. DISCUSSION
Radiation therapy remains an important modality for primary as well as locally advanced prostate cancer patients.7–13 However, radioresistance and relapse/recurrence of tumor remains a major clinical challenge.8–10 Therefore, it is critical to identify tumor-specific targets and explore their linked signaling pathways associated with radioresistance. Emerging evidence suggests that aberrant activation of DNA-damage repair (DDR) signaling pathways play important roles in prostate cancer metastasis and progression and are involved in radio-resistance.10,11,46–48 Targeting these pathways by employing multiple approaches involving gene therapy, specific inhibitors, or immunotherapy has the potential to enhance the radiosensitivity of prostate cancer cells and thereby improve the survival of prostate cancer patients.
The cohesin protein structural maintenance of chromosome-1 (SMC1A) is one of the target substrate of ataxia-telangiectasia mutated (ATM) kinase, the key sensor of the DNA damage response along with p53, Nibrin (NBS1), and breast cancer 1 (BRCA1).28,29,46–48 In response to ionizing radiation, ATM kinase is activated and phosphorylates SMC1A and it has been shown to be the only target of ATM whose phosphorylation directly affects cell survival as lack of other substrates of ATM including p53, NBS1, and BRCA1 has no significant effects on radiosensitivity.28,29 Loss of SMC1A phosphorylation abolishes the S-phase delay induced by radiation and leads to increased radiosensitivity. Moreover, the role of SMC1A in the cellular response to oxidative stress is also considered to be significant.46–48 SMC1A is overexpressed in several malignancies and shown to be involved in the regulation of the EMT phenotype and stem cell pluripotency.19–28 Consistent with this notion, here we demonstrate the role of SMC1A in sensitizing prostate cancer cells toward x-irradiation by manipulating the intracellular redox status, cancer stem-like cells, and EMT phenotype.
In response to a single dose of x-irradiation to two metastatic castration-resistant prostate tumor cell lines (DU145 and PC3), we provide evidence that SMC1A is an important determinant of cell survival. SMC1A suppression is involved in radiosensitizing prostate cancer cells via multiple mechanisms. Lower number of sphere formation in x-irradiated SMC1A shRNA expressing prostate cancer cells showed that SMC1A sensitized the cells by altering the stem-like cell properties of prostate cancer cells. Radiation resistant cells have shown to form significantly more spheres compared with the control cells, indicating that CSCs are closely associated with radioresistance.14
Radiation therapy kills the cells directly by damaging the DNA (double or single-stranded breaks) or indirectly producing free radicals including hydroxyl radical, hydrogen peroxide, and superoxide which cause damage to biomolecules.10–13 Since cohesin has shown to involve in DSB repair through sister-chromatid homologous recombination (HR) and non-homologous end joining (NHEJ)30,46–48 the two major pathways of repairing DSBs, we dissected the effect of SMC1A suppression in altering these repair pathways. Our data showed that suppression of SMC1A sensitized the cells toward x-irradiation by attenuating the NHEJ and HR pathways. SMC1A suppression also manipulate the intracellular redox status as increased cellular level of ROS and lower glutathione levels were detected in SMC1A shRNA expressing DU145 and PC3 cells compared to their corresponding NT shRNA expressing cells.
Consistently, we demonstrate here the impact of SMC1A on proteins that regulate EMT. We demonstrated that downregulation of Vimentin, OCT4, SOX2, ALDH1, and CD44 and upregulation of E-cadherin/N-cadherin ratio were found in radiation sensitive SMC1A shRNA expressing prostate cancer cells indicating that EMT is correlated with radioresistance. SMC1A may also have a value in the staging of prostate cancer and have prognostic value as well since CSCs are known to have mesenchymal traits and mobility which are associated with metastasis.23–25,51,52 In summary, we demonstrate for the first time that suppression of SMC1A radio-sensitized prostate cancer cells by manipulating the intracellular redox status, cancer stem-like population, and EMT phenotype. Our results show that since SMC1A regulate the CSCs and EMT properties and involved in DDR pathways, it potentially not only sensitizes prostate cancer cells toward radiation, but also may delay prostate cancer recurrence after radiation treatment.26–28 Increasing evidence supports the notion that SMC1A-associated activity might be involved in the acquisition of resistance for various cancer therapies.19,26–28
5 |. CONCLUSIONS
SMC1A, a member of cohesin known as nuclear protein has been found to be associated with cell membrane, microtubules, and centrosomes which may provide a basis for its additional role including cellular response to stress, tumor progression, and metastasis. SMC1A downregulation in prostate cancer cells could help in augmenting the radiosensitivity and could be useful in reversing intrinsic or acquired drug-resistance of cancer cells toward radiation and chemotherapy.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication included work performed in the Analytical Cytometry, Pathology, Small Animal Imaging and Biostatistics Cores supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572. We thank Xueli Liu for her assistance with statistical analysis of data; Loren Quintanar and Brian Armstrong (Microscopy Core) for their help with Microscopy studies. This work is supported in part by shared resources grant from the Beckman Research Institute of City of Hope (SY).
Funding information
Shared Resources Grant from the Beckman Research Institute, City of Hope; National Cancer Institute of the National Institutes of Health, Grant number: P30CA33572
Abbreviations:
- CSCs
cancer stem-like cells
- DDR
DNA-damage repair
- DSB
double-strand break
- EMT
epithelial to mesenchymal transition
- GSH
glutathione
- mCRPC
metastatic castration resistant prostate cancer
- ROS
reactive oxygen species
- RT
radiation therapy
- SMC1
structural maintenance of chromosome-1
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Wade CA, Kyprianou N. Profiling prostate cancer therapeutic resistance. Int J Mol Sci. 2018;19:pii: E904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein CA. Treatment sequencing in metastatic castration-resistant prostate cancer: a clinical commentary. Clin Genitour Cancer. 2015;13: 407–409. [DOI] [PubMed] [Google Scholar]
- 4.Noonan KL, North S, Bitting RL, et al. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013;24:1802–1807. [DOI] [PubMed] [Google Scholar]
- 5.Suzman DL, Luber B, Schweizer MT, et al. Clinical activity of enzalutamide versus docetaxel in men with castration-resistant prostate cancer progressing after abiraterone. Prostate. 2014;74: 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pieczonka CM, Telonis D, Mouraviev V, et al. Sipuleucel-T for the treatment of patients with metastatic castrate-resistant prostate cancer: considerations for clinical practice. Rev Urol. 2015;17:203–210. [PMC free article] [PubMed] [Google Scholar]
- 7.Sartor O, Coleman R, Nilsson S, et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: results from a phase 3, double-blind, randomised trial. Lancet Oncol. 2014;15:738–746. [DOI] [PubMed] [Google Scholar]
- 8.Srivastava P, Sarma A, Chaturvedi CM. Targeting DNA repair with PNKP inhibition sensitizes radioresistant prostate cancer cells to high LET radiation. PLoS ONE. 2018;13:e0190516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Athar M. Ionizing radiation exposure and basal cell carcinoma pathogenesis. Radiat Res. 2016;185:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Somaiah N, Yarnold J, Lagerqvist A, et al. Homologous recombination mediates cellular resistance and fraction size sensitivity to radiation therapy. Radiother Oncol. 2013;108:155–161. [DOI] [PubMed] [Google Scholar]
- 11.Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol. 2013;3:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pierre S ROS and radiotherapy: more we care. Oncotarget. 2017;8: 35482–35483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. [DOI] [PubMed] [Google Scholar]
- 14.Chang L, Graham PH, Hao J, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cojoc M, Peitzsch C, Kurth I, et al. Aldehyde dehydrogenase is regulated by β-catenin/TCF and promotes radioresistance in prostate cancer progenitor cells. Cancer Res. 2015;75:1482–1494. [DOI] [PubMed] [Google Scholar]
- 16.Kim R-K, Kaushik N, Suh Y. Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget. 2016;7:53430–53442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nambiar DK, Rajamani P, Singh RP. Silibinin attenuates ionizing radiation-induced pro-angiogenic response and EMT in prostate cancer cells. Biochem Biophys Res Comm. 2015;456:262–268. [DOI] [PubMed] [Google Scholar]
- 18.Gordon CJ, Robert K, Lee ME, et al. Antiangiogenic therapy in oncology: current status and future directions. Lancet. 2016;388: 518–529. [DOI] [PubMed] [Google Scholar]
- 19.Yadav S, Sehrawat A, Eroglu Z, et al. Role of SMC1 in overcoming drug resistance in triple negative breast cancer. PLoS ONE. 2013;8:e64338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YF, Jiang R, Li JD, et al. SMC1A knockdown induces growth suppression of human lung adenocarcinoma cells through G1/S cell cycle phase arrest and apoptosis pathways in vitro. Oncol Lett. 2013;5:749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Z, Lin M, Li K, et al. Knocking down SMC1A inhibits growth and leads to G2/M arrest in human glioma cells. Int J Clin Exp Pathol. 2013;6:862–869. [PMC free article] [PubMed] [Google Scholar]
- 22.Jianwei W, Shaojun Y, Liming C, et al. Role of SMC1A overexpression as a predictor of poor prognosis in late stage colorectal cancer. BMC Cancer. 2015;15:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhodes JM, McEwan M, Horsfield JA. Gene regulation by cohesin in cancer: is the ring an unexpected party to proliferation? Mol Cancer Res. 2011;9:1587–1607. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Scannell DR, Eisen MB, et al. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell. 2011;146: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Jiao W, Sun L, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30–35. [DOI] [PubMed] [Google Scholar]
- 26.Pan XW, Gan SS, Ye JQ, et al. SMC1A promotes growth and migration of prostate cancer in vitro and in vivo. Int J Oncol. 2016;49: 1963–1972. [DOI] [PubMed] [Google Scholar]
- 27.Zhou P, Xiao N, Wang J, et al. SMC1A recruits tumor associated-fibroblasts (TAFs) and promotes colorectal cancer metastasis. Cancer Lett. 2016;385:39–45. [DOI] [PubMed] [Google Scholar]
- 28.Kitagawa R, Bakkenist CJ, McKinnon PJ, et al. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yazdi PT, Wang Y, Zhao S, Patel N, et al. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivey RG, Moore HD, Voytovich UJ, et al. Blood-based detection of radiation exposure in humans based on novel phospho-Smc1 ELISA. Radiat Res. 2011;175:266–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yadav S, Berz D, Somlo G, et al. Abstract 2160: targeting SMC1 in combination therapy for triple negative breast cancer. AACR Cancer Res. 2016;76. [Google Scholar]
- 32.Singhal SS, Wickramarachchi D, Yadav S, et al. Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol Cancer Ther. 2011;1:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gangkak G, Bhattar R, Mittal A. Immunohistochemical analysis of estrogen receptors in prostate and clinical correlation in men with benign prostatic hyperplasia. Investig Clin Urol. 2017;58:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia LM, Leblanc J, Wilkins D, et al. Fitting the linear-quadratic model to detailed data sets for different dose ranges. Phys Med Biol. 2006;51:2813–2823. [DOI] [PubMed] [Google Scholar]
- 35.Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-Cytometry. In: Armstrong D, editor. Advanced Protocols in Oxidative Stress II. Methods in Mol Biol (Methods and Protocols), vol. 594. Totowa, NJ: Humana Press; 2010. http://cran.r-project.org/ [DOI] [PubMed] [Google Scholar]
- 36.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armenia J, Wankowicz S, Liu D, Gao J. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50:645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson D, Van Allen EM, Wu YM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;16:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grasso CS, Wu YM, Robinson DR. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar A, Coleman I, Morrissey C. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22: 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beltran H, Prandi D, Mosquera JM. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22: 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279:53272–53281. [DOI] [PubMed] [Google Scholar]
- 45.Bauerschmidt C, Woodcock M, Stevens DL, et al. Cohesin phosphorylation and mobility of SMC1 at ionizing radiation-induced DNA double-strand breaks in human cells. Exp Cell Res. 2011;317: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schär P, Fäsi M, Jessberger R. SMC1 coordinates DNA double-strand break repair pathways. Nucleic Acids Res. 2004;32:3921–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tew KD, Townsend DM. Glutathione-S-Transferases as determinants of cell survival and death. Antioxid Redox Signal. 2012;17: 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothkamm K, Horn S. Gamma-H2AX as protein biomarker for radiation exposure. Ann Ist Super Sanita. 2009;45:265–271. [PubMed] [Google Scholar]
- 50.Oorschot B, Hovingh SE, Rodermond H, Güçlü A, Losekoot N. Decay of γ-H2AX foci correlates with potentially lethal damage repair in prostate cancer cells. Oncol Rep. 2013;29:2175–2180. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen LV, Vanner R, Dirks P, et al. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.