

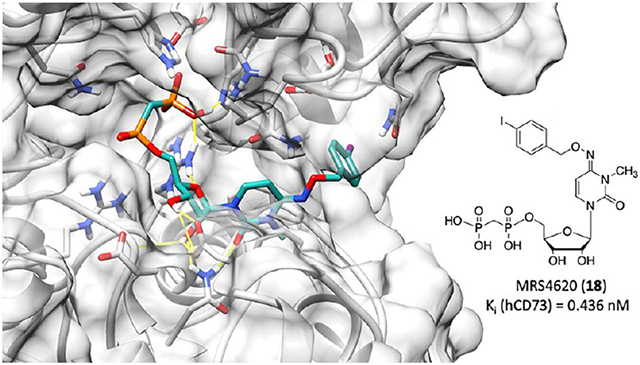
Abstract
We recently reported N4-substituted 3-methylcytidine-5′-α,β-methylenediphosphates as CD73 inhibitors, potentially useful in cancer immunotherapy. We now expand the structure–activity relationship of pyrimidine nucleotides as human CD73 inhibitors. 4-Chloro (MRS4598 16; Ki = 0.673 nM) and 4-iodo (MRS4620 18; Ki = 0.436 nM) substitution of the N4-benzyloxy group decreased Ki by ~20-fold. Primary alkylamine derivatives coupled through a p-amido group with a varying methylene chain length (24 and 25) were functionalized congeners, for subsequent conjugation to carrier or reporter moieties. X-ray structures of hCD73 with two inhibitors indicated a ribose ring conformational adaptation, and the benzyloxyimino group (E configuration) binds to the same region (between the C-terminal and N-terminal domains) as N4-benzyl groups in adenine inhibitors. Molecular dynamics identified stabilizing interactions and predicted conformational diversity. Thus, by N4-benzyloxy substitution, we have greatly enhanced the inhibitory potency and added functionality enabling molecular probes. Their potential as anticancer drugs was confirmed by blocking CD73 activity in tumor tissues in situ.
Graphical Abstract
INTRODUCTION
In cancer immunotherapy, the body’s immune system is mobilized to attack and eliminate a cancerous tumor. In this context, immunosuppresive adenosinergic signaling in the tumor microenvironment attenuates the therapeutic effect.1–3 Cyclic AMP-dependent signaling through the A2A and A2B adenosine receptors (ARs) on immune cells (T cells, dendritic cells, NK cells, macrophages, and neutrophils) prevents their activation. Adenosine also enhances the immunosuppressive effects of myeloid-derived suppressor cells and regulatory T cells (Treg) as well as angiogenesis and tumor cell invasion. Therefore, a means of weakening the extracellular adenosine effects would be beneficial in cancer immunotherapy either as a co-therapy or as a monotherapy. Two approaches are currently being explored to counteract adenosine effects in the tumor microenvironment, e.g., through preventing its production from AMP by ecto-5′-nucleotidase (also known as CD73) or by blocking its receptor interaction with specific antagonists. Various clinical trials in which a small molecule CD73 inhibitor or an anti-CD73 monoclonal antibody is combined with cancer immunotherapy, such as anti-PD-L1 therapy, are planned or in progress.4–11 CD73 mouse knockout also inhibited tumor angiogenesis, which was dependent on multiple AR subtypes.12
CD73 also mediates both adenosine-dependent and -independent effects in various organs, and CD73 blockade may produce beneficial or deleterious effects.13 Rare genetic defects resulting in nonfunctional CD73 in humans cause widespread calcification in arteries and joints.14 Also, pathological downregulation of CD73 is associated with postmenopausal bone loss.15 CD73 inhibitors may eventually also prove to be useful in treating neurodegeneration and other central nervous system disorders, such as Parkinson’s disease, early Alzheimer’s disease and epilepsy, or fibrosis, or increasing the myocardial immune response to infections.16–21 Nevertheless, the presence of CD73 was found to be important in neuronal development.22,23 It should be noted that there are interspecies differences in the effects of a lack of CD73. Knockout of this enzyme in the mouse does not produce the same phenotype of calcification and arteriomegaly as seen in humans lacking CD73.24
The affinity of the prototypical ADP-competitive CD73 inhibitor adenosine-5′-α,β-methylenediphosphonate [AOPCP, 1 (Chart 1)] has been enhanced in recently reported inhibitors.11,25-28 Bhattarai and co-workers included N6-benzyl and 2-chloro substitution of the adenine moiety to increase the inhibitory potency to subnanomolar levels.25-27 Several other classes of nucleotides and nucleotide mimics as CD73 inhibitors have been reported. For example, a pyrazolo[3.4-b]pyridine nucleotide (6a, Quemliclustat) was recently reported by Arcus Biosciences and has entered a Phase 1 clinical trial for advanced pancreatic cancer in 2019 (ClinicalTrials.gov: NCT04104672, accessed November 30, 2021).11,29,30 OP-5244 6b was recently reported by ORIC Pharmaceuticals.9 Compounds 6a and 6b display oral bioavailability. Also, substitution of the phosphonate moiety with malonate groups (7) or introduction of novel chemotypes as CD73 inhibitors has produced significant inhibitory potency.31–33
Chart 1. Structures of CD73 Inhibitors [showing the IC50 or Ki at membrane-bound human (h) CD73, except for 7, which was measured at soluble hCD73]26–29,31–33a.
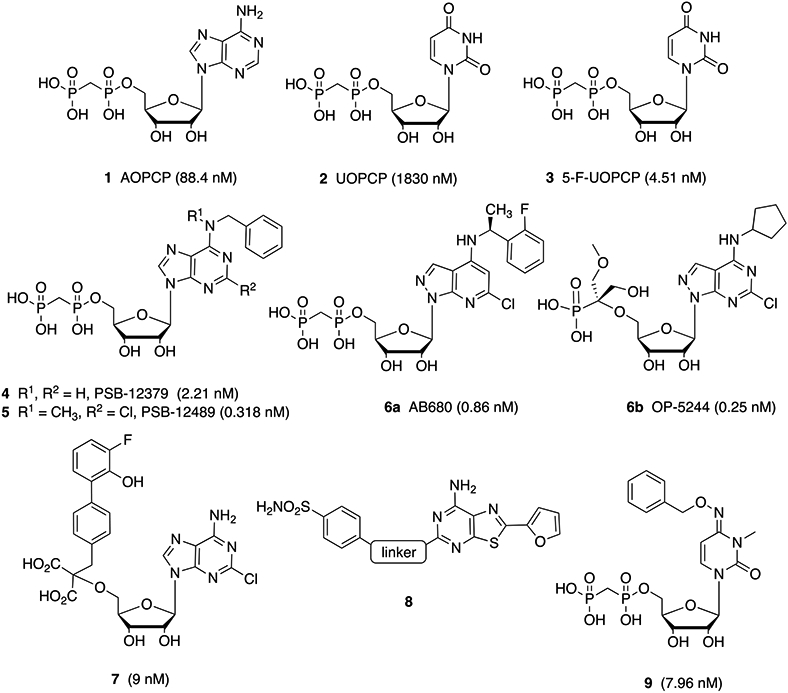
a Compound 8 is a dual-acting A2AAR inverse agonist and CD73 inhibitor. The linker moiety of 8 consists of various alkylamino and cycloalkylamino groups.
We recently extended this approach by replacing the adenine-like nucleobase entirely, in pyrimidine 5′-nucleoside 3-methylcytidine-5′-α,β-methylenediphosphonate derivatives, such as compound 9.28 The cytidine 3-methyl group that is common to these potent inhibitors was shown previousy to completely block affinity at the UDP-selective P2Y6 receptor.34 These N4-substituted 3-methylcytidine-5′-α,β-methylenediphosphonates do not produce adenosine derivatives as nucleoside metabolites, i.e., making AR activation less likely. We now further explore the structure–activity relationship (SAR) of these pyrimidine nucleotides as inhibitors of human (h) CD73.
RESULTS AND DISCUSSION
Chemical Synthesis.
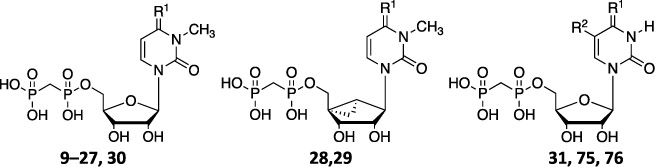
Compounds 1–3, 9, 75, and 76 (Table 1) were prepared as previously reported.26–28 As shown in Scheme 1, synthesis of compounds 10–21 required the preparation of various aryl-substituted benzyloxyamines (32–43). These benzyloxyamines were produced following the previously reported procedure,34,35 starting from the corresponding substituted benzyl bromides. Similar methods were used to prepare phenylethyl and phenylpropyl homologues (structures not shown) of the benzyl derivatives.
Table 1.
Ki Values of Compounds 10–31 and Archival Compounds 9, 75, and 76 in the Inhibition of Soluble hCD73 or as Otherwise Noted (triethylammonium salts were prepared and tested)c
| ||
|---|---|---|
| No. | Structure R1 = (other changes) |
Ki ± SEM (nM, n=3) |
| 9 |
|
10.6 ± 0.4a, 3.67 ± 0.26a,b |
| 10 |
|
1.63 ± 0.28 |
| 11 |
|
1.58 ± 0.06 |
| 12 |
|
1.27 ± 0.18 |
| 13 |
|
0.894 ± 0.175 |
| 14 |
|
3.50 ± 0.11 |
| 15 |
|
1.14 ± 0.06 |
| 16 |
|
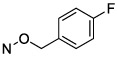
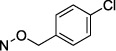
0.673 ± 0.091 |
| 17 |
|
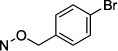
0.441 ± 0.157 |
| 18 |
|
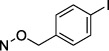
0.436 ± 0.078 |
| 19 |
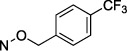
|
1.08 ± 0.34 |
| 20 |
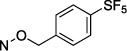
|
0.511 ± 0.041 |
| 21 |
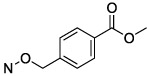
|
0.848 ± 0.229 |
| 22 |
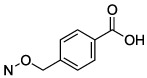
|
14.1 ± 2.9 |
| 23 |
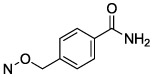
|
1.76 ± 0.43 |
| 24 |
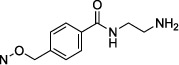
|
0.626 ± 0.076 |
| 25 |
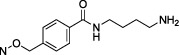
|
1.78 ± 0.33 |
| 26 |
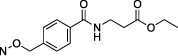
|
2.51 ± 0.51 |
| 27 |
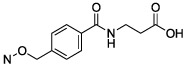
|
2.61 ± 0.94 (n=2) |
| 28 | O | > 50 μM (28% inhibition) |
| 29 |
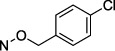
|
59.2 ± 5.58 |
| 30 |
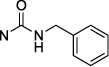
|
71.5 ± 17.6 |
| 31 | O, R2 = F | 2540 ± 710 |
| 75 | O, R2 = CH3 | 338 ± 56a,b |
| 76 |
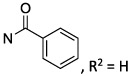
|
4.58 ± 0.55a, 13.9 ± 1.6a,b |
Scheme 1. Substitution of the Pyrimidine Scaffold with Diverse 4-Benzyloxyimino Substituentsa.
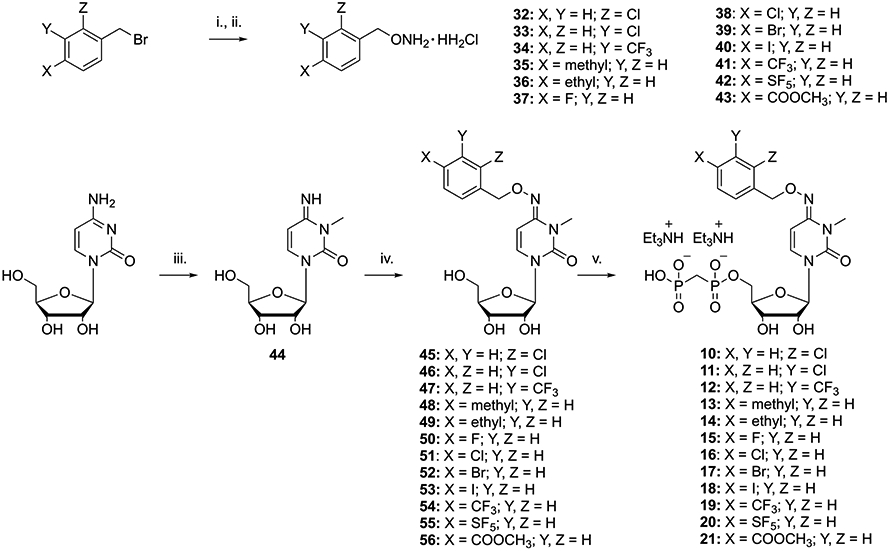
a Reagents and conditions: (i) (Boc)2NOH,33 DBU, DMF, 50 °C, 2 h; (ii) 4 N HCl in dioxane, rt, 12 h; (iii) MeI, DMAc, rt, 4 h, 95%; (iv) the desired benzylhydroxylamine (32–43), pyridine, 80 °C, 12 h, 35–97%; (v) methylenebis(phosphonic dichloride), (CH3O)3PO, 0 °C, 3 h, 5–40%.
3-Methylcytidine (44) was prepared using iodomethane in dimethylacetamide (DMAc) without adding a base. Reaction of 44 with the desired substituted benzyloxyamine (32–43) in pyridine at 80 °C gave the desired intermediates 45–56, respectively, in good yields. 5′-O-Phosphonylation of the latter was performed using methylenebis(phosphonic dichloride) in trimethyl phosphate to afford the desired final compounds 10–21, respectively, in acceptable yields.
The envisaged synthetic route for the preparation of derivatives 22–25 is shown in Scheme 2. Hydrolysis of the methyl ester moiety of compound 56 using sodium hydroxide furnished the p-carboxybenzyl nucleoside intermediate 57, which was 5′-phosphonylated as previously indicated to afford 22.
Scheme 2. Synthetic Strategies for Extension at the 4-Carboxyl Group on a 4-Benzyloxyimino Substituenta.
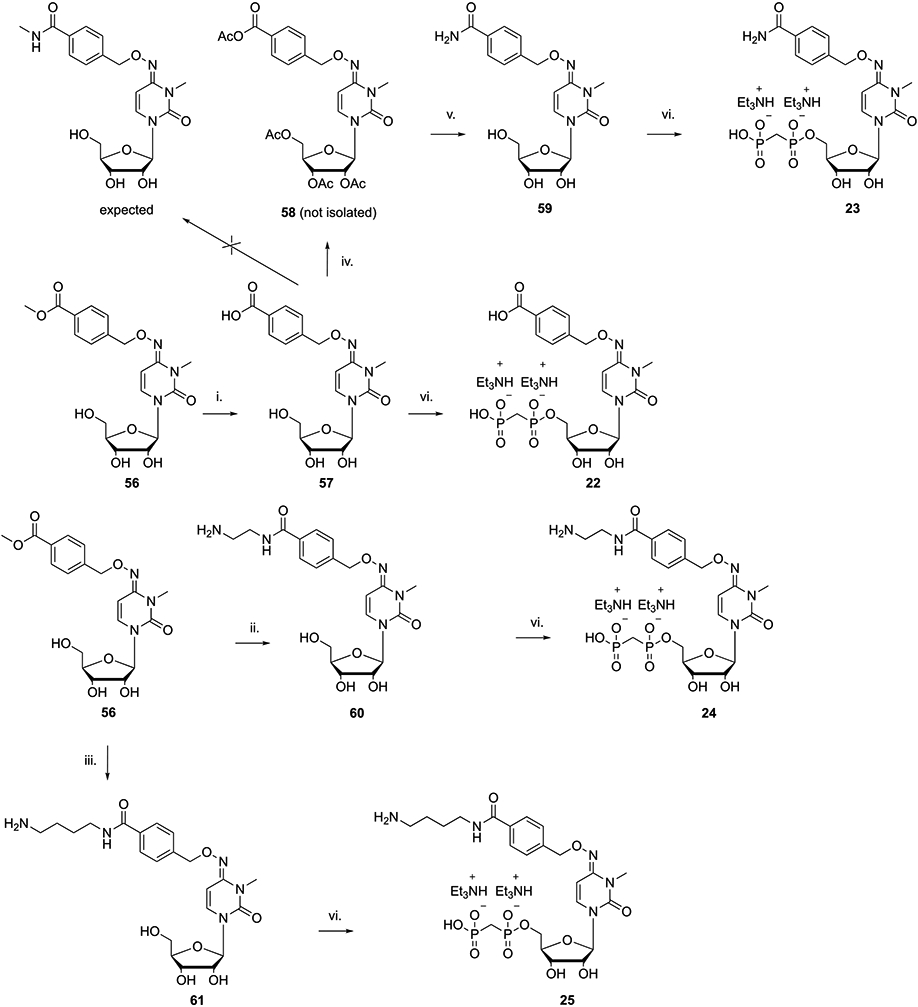
a Reagents and conditions: (i) 1 N NaOH, MeOH, rt, 20 min, 77%; (ii) ethylenediamine, rt, 2 h, 95%; (iii) 1,4-diaminobutane, rt, 2 h, 90%; (iv) MeNH2, HATU, DIPEA, DMF, rt, 12 h; (v) NH3/MeOH, rt, 12 h, 64% for steps iv and v; (vi) methylenebis(phosphonic dichloride), (CH3O)3PO, 0 °C, 3 h, 8–30%.
Starting from compound 57, we planned to prepare its methylamide derivative, using HATU and DIPEA as a coupling agent and base, respectively, to condense the carboxylic acid moiety with methylamine. LC-MS of the isolated product indicated that multiple acetylations had occurred, i.e., 58 (probably because of the presence of acetic acid traces used during the previous workup). 58 was directly reacted with ammonia to remove the acetyl groups and form an amide 59. Nucleoside product 59 was used to prepare final phosphonate derivative 23. The two amine congeners 24 and 25 were synthesized by amination of 56 with ethylenediamine and 1,4-diaminobutane, respectively. The crude nucleoside amides thus obtained were directly subjected to 5′-O-phosphonylation without further purification, but using an increased amount of methylenebis(phosphonic dichloride) to ensure full conversion of the starting compounds to the desired products.
Molecular modeling analysis based on the X-ray co-crystal structure of 21 with soluble hCD73 (see below) suggested that the introduction of a carboxylic group with a two-carbon spacer from the benzamide moiety may improve the activity through formation of an H-bond with a lysine residue present in the pocket of the enzyme. On the basis of these predictions, we synthesized compounds 26 and 27 starting from 57 and β-alanine ethyl ester hydrochloride, as reported in Scheme 3.
Scheme 3. β-Alanine Derivatives for Introduction of Distal H-Bonding Groups to Reach Polar and Charged Amino Acid Residues Adjacent to the CD73 Binding Sitea.
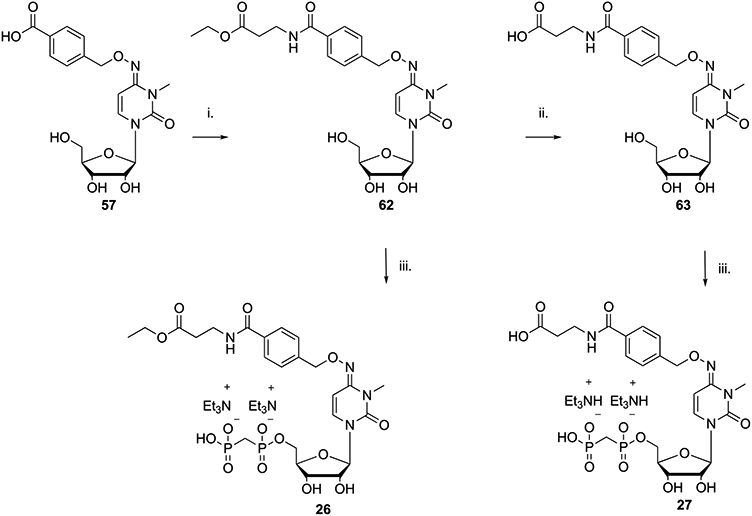
a Reagents and conditions: (i) β-alanine ethyl ester hydrochloride, HATU, DIPEA, DMF, 12 h, rt, ~100%; (ii) 1 N NaOH, MeOH, rt, 12 h, 92%; (iii) methylenebis(phosphonic dichloride), (CH3O)3PO, 0 °C, 3 h, 10%.
A methanocarba ([3.1.0]bicyclohexane) substitution of the ribose ring in nucleosides and nucleotides allows for the pre-establishment of either a north (N) or a south (S) pseudoribose conformation depending on the position of the cyclopropane ring fusion, tailored for the target protein interaction.36 An (S)-methanocarba analogue in this chemical series was already shown to be a weak CD73 inhibitor.28 Furthermore, inspection of the reported CD73 X-ray structures indicates that an (N)-ribose conformation is present.37 Therefore, (N)-methanocarba analogues 28 and 29 were prepared (Scheme 4) by adapting reported procedures.38,39 Sugar protection by acetylation was performed using the standard method with acetic anhydride to afford intermediate 65. Next, the 4-ketone moiety was first activated through reaction with 2,4,6-triisopropylbenzenesulfonyl chloride (TPSCl), followed by nucleophilic displacement by 4-Cl-benzyloxyamine (38) to afford compound 66. N-3 methylation of 66 was performed as described above to afford 67, which was followed by acetyl group removal using a methanolic ammonia solution to afford intermediate 68 in good yield. The synthesis of methanocarba intermediate 67 required addition of K2CO3 as a base, as there was no free 4-amino group on the cytosine moiety that could stabilize the alkylation transition state. The two (N)-methanocarba-5′-O-α,β-methylenediphosphonates 28 and 29 were prepared starting from the corresponding precursors 64 and 68, respectively, using a previously reported method.38
Scheme 4. Substitution of the Flexible Ribose Moiety of 16 with the Conformationally Constrained (N)-Methanocarba Groupa.
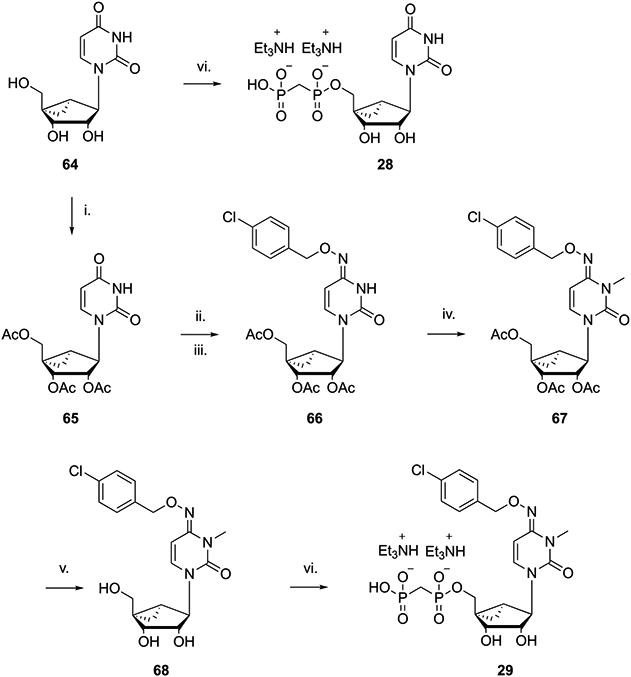
a Reagents and conditions: (i) acetic anhydride, Et3N, DMAP, CH3CN, 2 h, rt, 67%; (ii) TPSCl, Et3N, DMAP, CH3CN, rt, 18 h; (iii) p-Cl-O-benzylhydroxylamine hydrochloride, Et3N, CH3CN, rt, 18 h, 26% for steps ii and iii; (iv) MeI, K2CO3, DMAc, 18 h, rt, 83%; (v) NH3/MeOH, rt, 12 h, 91%; (vi) methylenebis(phosphonic dichloride), (CH3O)3PO, 0 °C, 3 h, 6–16%.
We also envisioned the preparation of a urea analogue (30); its synthesis is highlighted in Scheme 5. Benzylamine was activated through reaction with 1,1′-carbonyldiimidazole (CDI) following the reported procedure40 to afford compound 69. To avoid side reaction products, cytidine was protected as its 2′,3′,5′-tri-O-acetyl derivative (70) through the standard procedure with acetic anhydride. During this reaction, we also observed the formation of the 2′,3′,5′-tri-O-acetyl-N4-acetyl derivative, which was isolated and converted into 70 in a microwave reactor in methanol at 100 °C. N-3 methylation was performed as already described, and reaction of 71 with 69 in the presence of triethylamine furnished intermediate 72. Next, sugar deprotection using a methanolic ammonia solution, followed by phosphonylation, gave the desired product 30. 5-F-UDP (31) was synthesized as reported previously3 and was used for a comparison with its analogue 5-F-UOPCP (3).
Scheme 5. Introduction of a Distal Urea-Type Substituent on the 4-Benzyloxyimino Group of the Pyrimidine Scaffold, to Potentially Enhance H-Bonding in That Region of the CD73 Binding Sitea.
a Reagents and conditions: (i) EtOH, reflux, 12 h, 72%; (ii) acetic anhydride, Et3N, DMAP, CH3CN, 10 min, rt, 95%; (iii) MeI, DMAc, rt, 4 h, 95%; (iv) 69, Et3N, THF, reflux, 12 h, 72%; (v) NH3/MeOH, rt, 12 h, 87%; (vi) methylenebis(phosphonic dichloride), (CH3O)3PO, 0 °C, 3 h, 8.4%.
In Vitro Biological Activity.
We describe here the compounds’ in vitro CD73 inhibitory activity before introducing the extensive structural probing of their protein interactions (below). The inhibitory potency of the various nucleotide analogues was determined with soluble hCD73 by previously reported methods (Table 1).41–43 Many of the analogues were significantly more potent than reference compound N4-benzyloxy-3-methylcytidine-5′-α,β-methylene-diphosphonate 9, as we previously reported.28
Halogen substitution of the benzyl ring of compound 9 produced a significant decrease in the Ki value at soluble hCD73. A chloro substitution at o-10 or m-11 positions produced a 6–7-fold increase in potency, while that of p-chloro analogue 16 was 16-fold enhanced (Figure 1A). Halogen substitution at the para position showed the following order of potency: I and Br > Cl > F. Thus, p-iodo analogue 18 displayed a Ki value of 0.436 nM (Figure 1B). Other neutral para-substituted analogues 13, 14, and 21 exhibited high potencies (0.5–3.5 nM). A p-pentafluorosulfenyl group, which is both highly electron-withdrawing and strongly lipophilic, provided a Ki of 0.511 nM (20). A comparison between methyl ester 21 and free carboxylate 22 indicated that a neutral group is preferred at the benzyl ring para position, although the carboxylate was still suitable as a linking group to form potent primary amino congeners 24 and 25. The corresponding primary amide 23 was 2-fold weaker than methyl ester 21. The Ki values of primary amine congeners 24 and 25 indicated that the two-methylene spacer of 24 (0.626 nM) was preferred. These amine congeners might be useful for conjugation to a variety of carrier or reporter moieties such as fluorophores for enzyme visualization or characterization, solid supports for affinity chromatography, and nanoparticles for altering pharmacokinetic and pharmacodynamic properties.
Figure 1.
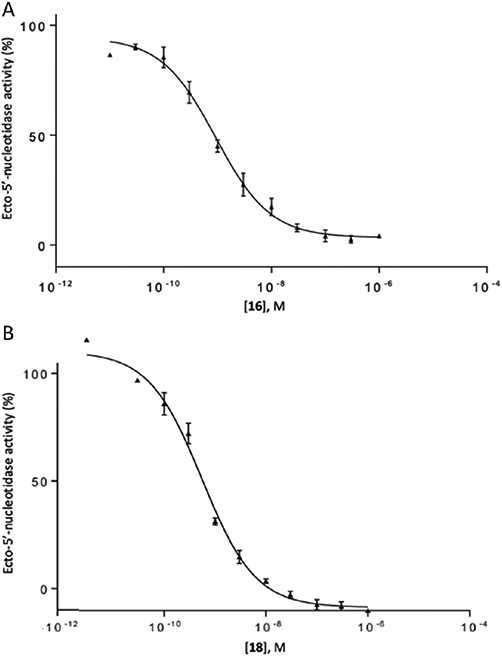
Concentration–response curve for inhibition of soluble hCD73 by 16 (A; Ki = 0.673 ± 0.091 nM) and 18 (B; Ki = 0.436 ± 0.078 nM) (mean of three independent determinations, each in duplicate).
An (N)-methanocarba analogue 29 of p-chloro derivative 16 was an 88-fold weaker inhibitor. Also, simple uridine analogue 28 displayed <50% inhibition between 0.03 nM and 50 μM, compared to the uridine analogue with a Ki value of 1830 ± 530 nM at rat CD73.28 Thus, the enforcement of an N conformation by the [3.1.0]bicyclohexyl ring system is not potency-enhancing compared to ribose in this CD73 inhibitor series, but it does allow moderate inhibition (Ki = 59 nM in 29) when favorable functionalization is present, e.g., 4-(p-chlorobenzyloxyimino). By comparison, the (S)-methanocarba analogue of 28 in the simple uridine series was inactive at 1 μM, but optimized analogues were not prepared.28 The X-ray structures of bound nucleotides in CD73 display an N ribose conformation, consistent with the inhibition by 29.27,37
An N-benzyl urea derivative 30 (71.5 nM) was only a moderately potent CD73 inhibitor compared to 9, indicating the advantage of the 4-oxoimino linkage. One 5′-diphosphate analogue, 5-fluorouracil derivative 31, was found to be a weak inhibitor with a Ki of 2.54 μM.
Later, on the basis of the obtained crystal structures, a molecular modeling investigation suggested that introduction of a β-alanine moiety connected to the carboxylic acid of compound 22 could improve the affinity of our derivatives. Consequently, derivatives 26 and 27, which possess β-alanine-ethyl ester and β-alanine moieties, respectively, were 5-fold more potent than 22.
The off-target activity of selected inhibitors (9, 16, and 18) was determined by the Psychoactive Drug Screening Program (PDSP, The University of North Carolina at Chapel Hill, Chapel Hill, NC).44 The only hit among 46 receptors, channels, and transporters assayed was a weak binding inhibition by p-chloro analogue 16 at the σ1 receptor (Ki = 2.61 ± 0.64 μM).
X-ray Crystallographic Structures.
X-ray crystal structures of hCD7345 (constructs in Figures S1 and S2) complexed with the two potent 5′-α,β-methylenediphosphonate inhibitors 16 (with a 4-Cl-O-benzyl substituent) and 21 (with an O-benzyl 4-methyl ester substituent) were determined in the open and closed states (Table 2 and Figure 2). The two compounds have very similar binding modes (Figure 2B). In both of these N-methyl analogues, the benzyloxyimino group adopts an E configuration, which is most clearly defined in the high-resolution structures of the open crystal forms (Figure S3). In the closed forms, the electron density is well-defined for the binding of 21, indicating high occupancy and no significant conformational flexibility. In the complex with 16 in the closed state, the density of the 4-Cl-O-benzyl substituent is less well-defined. This may be a result of flexibility and/or of the generally poorer quality of this data set with a Wilson B factor of 93.6 Å2 and a resolution of 2.9 Å.
Table 2.
Summary of Key Crystallographic Parameters
| 16 open | 16 closed | 21 open | 21 closed | |
|---|---|---|---|---|
| PDB entry | 7QGA | 7QGM | 7QGL | 7QGO |
| crystallizationbuffer | 10 mM 16, 100 μM ZnCl2, 8% PEG 6000, and 0.1 M MES (pH 6.2) | 10 mM 16, 10 μM ZnCl2, 10% PEG 3350, and 0.1 M BisTris (pH 5.6) | 10 mM 21, 100 μM ZnCl2, 8% PEG 6000, and 0.1 M MES (pH 6.2) | 10 mM 21, 10 μM ZnCl2, 10% PEG 4000, and 0.1 M BisTris (pH 5.7) |
| final buffer | 10 mM 16 and 20% PEG200 | 10 mM 16 and 20% PEG200 | 10 mM 21 and 20% PEG200 | 10 mM 21 and 20% PEG200 |
| dmin (Å) | 1.5 | 2.9 | 1.5 | 2.21 |
| CC1/2a | 0.603 | 0.649 | 0.743 | 0.250 |
| Rwork/Rfree (%) | 0.165/0.184 | 0.261/0.294 | 0.158/0.175 | 0.227/0.269 |
| Bprotein (Å2) | 12.4 | 99.1 | 12.0 | 83.9 |
CC1/2 in the highest-resolution shell. See Table S1 for further details.
Figure 2.

X-ray structures of the complex of 16 and 21 with the closed form of hCD73. (A) Binding mode of 21. (B) Superposition of the binding modes of 16 and 21 based on the Cα atoms of the C-terminal protein domains. (C) Interactions of the N4 substituent of 21 at the cleft between the two domains. (D) Superposition of the crystal structures of 21, 4 (PDB entry 6S7F),27 5 (PDB entry 6S7H),27 and 6 (AB680, PDB entry 6Z9D)11 based on the C-terminal domains. For 6, the N6 substituent and the Phe417 side chain have been modeled in alternative conformations, labeled A and B.
Key features of the interaction of the two inhibitors with CD73 are the polar contacts of the bisphosphonate and ribose groups with both domains (Figure 2A). These interactions closely resemble those of the AOPCP derivatives. The NH2 group of the Asn390 side chain forms hydrogen bonds to the ribose 2′-OH and the carbonyl oxygen of the 3-methylcytidine base (Figure 2B). The carbonyl oxygen thus takes the position of N-3 in the adenine base of AOPCP derivatives. A further important interaction is the hydrophobic stacking interaction of the nucleobase with Phe417 and Phe500. The N4 substituents bind at the cleft between the two domains and interact with both domains, thus stabilizing the closed state (Figure 2C). The cleft has mixed polar and apolar character at this site, but the interactions of the N4 substituents are mostly hydrophobic, which is quite similar to the situation for the N6 substituents in AOPCP derivatives. An exception is the hydrogen bonding interaction of Gln88 with the ester carbonyl group of 21.
A comparison to the complex structures of CD73 with AOPCP derivatives 4–6 shows an overall similar binding mode (Figure 2D). The β-phosphonate groups superimpose well, but shifts of up to ~1 Å are observed for the α-phosphonate, ribose, and base moieties. These differences are caused by a small change in the domain orientation and in addition by local conformational adaptations, in particular, of Phe417 and Gln88. The conformational differences avoid a close contact of Gln88 with the benzyl substituents of 16 and 21 and with Phe417. Indeed, a comparison of pyrimidine inhibitors 16 and 21 bound to the open and closed states shows that the nucleobase, Phe417, and Phe500 are positioned closer to the side of Gln88 in the closed states than in the open states, where Gln88 is not nearby due to the domain orientation (Figure S3C, F). The binding mode observed for pyrimidine-based compounds 16 and 21 matches most closely conformation A of 6a with regard to the positions and binding modes of the nucleobases and the N4 or N6 substituents. The binding modes of 16 and 21 to the open state match that of the unsubstituted CMP substrate of Protein Data Bank (PDB) entry 7PA446 (Figure S3). Small differences are observed in the base position likely resulting from the N4 substitution.
Molecular Modeling.
The crystallographic complex of hCD73 in the closed form bound with compound 16 was subjected to molecular dynamics (MD) simulations (three replicates of 50 ns each) to assess the stability of the binding mode over time. During most of the simulation time, the ligand maintained a geometry compatible with a π–π stacking interaction with Phe417 and Phe500 and the pattern of H-bonds observed in the X-ray state. Asp506 was engaged in H-bonding with the 2′- or 3′-hydroxyl groups during nearly all of the simulation time (99% on average over the three replicates). Asn390 interacted with the ribose 2′-hydroxyl group or with the carbonyl at position 2 of the cytosine for 80% of the simulation time. An H-bond between His118 and the β-phosphonate was found on average in 69% of the trajectory time, and an H-bond between Arg354 and the α-phosphonate occurred during 89% of the trajectory time. Arg395 was engaged in an H-bond with the α-phosphonate or β-phosphonate in 81% of the simulation time on average (Table S2 and Figure 3B). Compound 16, and especially its nucleotide-like scaffold, remained close to its original position during the simulation: the ligand’s heavy atom root-mean-square deviation (RMSD) relative to the X-ray pose showed an average value of 2.12 ± 0.60 Å (±standard deviation) over the three replicates (Table S2), with lower values in the case of replicate 2 (average of 1.70 Å) but reaching maximum values of 3.70 and 3.17 Å in replicates 1 and 3, respectively. Notably, the RMSD reached higher values in the second half of the simulation in replicates 1 and 3, as shown in Figure 3C, and the last frames of these two trajectories showed a deviation of the p-Cl-benzyl substituent compared to replicate 2, which resembled the initial state (Figure 3A). The per-atom root-mean-square fluctuation (RMSF) highlights that the atoms deviating more from the average position belong to the p-Cl-benzyl substituent (Figure 3D). The RMSD of the nucleotide scaffold alone (all heavy atoms except the p-Cl-benzyl substituent) fluctuated with a small deviation around the lower average value of 1.71 ± 0.27 Å (Figure 3E).
Figure 3.

(A) Superposition of the last frames of three 50 ns trajectories of the compound 16-hCD73 complex in the closed form. The trajectories have been aligned by superposing the Cα atoms on the initial state. Proteins are represented by a gray ribbon, interacting residues as gray lines, and ligands of replicates 1–3 as orange, green, and blue sticks, respectively. (B) Percentage of H-bonds between compound 16 and hCD73 residues during the three replicates. Only residues interacting on average >10% of the simulations are shown. (C) RMSDs of compound 16 heavy atoms over time in the three replicates. (D) RMSF of compound 16 heavy atoms in the three replicates. The atom numbers of atoms with higher RMSFs are reported in the two-dimensional depiction of the p-Cl-benzyl moiety. (E) RMSDs of the heavy atoms of the nucleotide scaffold (all heavy atoms except the p-Cl-benzyl) in the three replicates.
To investigate the conformational exploration of the p-Cl-benzyl substituent of 16 within the surrounding protein environment, we clustered the trajectories on the basis of the RMSD of this moiety (Figure 4A). In particular, the trajectories were combined, then aligned superposing the ligand nucleotide-like scaffold to its initial conformation, and sampled every 200 ps. The RMSD of the p-Cl-benzyl substituent heavy atoms was used as a parameter to cluster the trajectories using Wordom’s QT-like method.47 The most populated clusters (including at least 5% of total frames) were considered, resulting in three examples. Cluster 1 presented the ligand in a conformation comparable to the X-ray structure (Figure 4C), and it collected the initial frames of replicates 1 and 3 and approximately the entire replicate 2 (cluster population equal to 47% of the whole combined simulations). In this conformation, the p-Cl-benzyl substituent was surrounded (distance of ≤4 Å) by Gln88, Asp121, Asn186, Pro416, and Phe417 in at least 40% of the cluster frames (Figure 4B). Cluster 2 (cluster population of ~24%) collected an ensemble of intermediate conformations from replicate 1 and those explored in the second half of replicate 3: the ligand p-Cl-benzyl substituent pointed toward the N-terminal domain of hCD73 (Figure 4E) and was in contact with Asp121 and Asn186 in almost all of the cluster frames (Figure 4D). Cluster 3 was represented during only the second half of replicate 1 (cluster population of ~19%), and in this conformation, the p-Cl-benzyl substituent faced a cleft between the N-terminal and C-terminal domains opposite the cleft occupied by the the p-Cl-benzyl substituent in the X-ray structure (Figure 4G). The substituent was mainly (>40% of the cluster frames) in contact with Asn186, Pro187, Phe500, and Asn503 (Figure 4F). It is interesting to note that in the described clusters, the p-Cl-benzyl substituent was often in the proximity of an Asn or Gln residue, which could potentially stabilize the moiety through polar interactions. Moreover, the flexibility of the p-Cl-benzyl moiety was not surprising, considering that two alternative positions comparable to those identified in clusters 1 and 3 have already been shown in the X-ray structure of inhibitor 6a bound to hCD73 (PDB entry 6Z9D11), whose 1-(2-fluorophenyl)ethyl substituent is reported in two alternate states (Figure S7). Also, in a comparison of the binding modes of different AOPCP derivatives, the N6 substituent was positioned at different sites of the cleft between the two domains and often had poorer density and higher B-factors indicating flexibility.26,27,44 The ligand conformation represented in cluster 2 seemed to be an intermediate state between these two opposite conformations, and the transition from cluster 1 to cluster 3 conformations going through cluster 2 can be seen in Video S1 (trajectory of replicate 1). The three conformations appear to mainly originate from the three rotational preferences around the N–O–C–C torsion angle (Figure S8).
Figure 4.

MD simulation of compound 16 bound to the closed form of hCD73. (A) Conformational distance (RMSD) matrix, reporting the frame number on the x- and y-axes, and RMSDs of p-Cl-benzyl heavy atoms on a colorimetric scale going from dark to bright colors [low to high RMSD values (Å)]. The frames of three 50 ns replicates have been merged for the sake of the analysis and sampled every 200 ps (750 frames total) after superposition of the nucleotide-like scaffold of compound 16 to the starting conformation. The frames of each replicate are identified by bars parallel to the x- and y-axes (replicates 1–3 in orange, green, and blue, respectively). White numbers on the matrix highlight the most represented clusters (population of >5%): cluster 1 (349 frames, ~47%), cluster 2 (183 frames, ~24%), and cluster 3 (145 frames, ~19%). (B) Residues in contact (distance of ≤4 Å) with compound 16 in cluster 1. The persistence of each contact is indicated by the percentage of frames of the cluster where that contact is present. (C) Center of cluster 1 (replicate 1, frame 1, 0.02 ns). The ligand is depicted as cyan sticks, and the protein is colored gray. Residues in contact with the ligand in >40% frames of the cluster are shown. (D) Residues in contact (distance of ≤4 Å) with compound 16 in cluster 2. (E) Center of cluster 2 (replicate 3, frame 1611, 32.22 ns). The ligand is depicted as cyan sticks, and the protein is colored gray. Residues in contact with the ligand in >40% frames of the cluster are shown. (F) Residues in contact (distance of ≤4 Å) with compound 16 in cluster 3. (G) Center of cluster 3 (replicate 1, frame 991, 19.82 ns). The ligand is depicted as cyan sticks, and the protein is colored gray. Residues in contact with the ligand in >40% frames of the cluster are shown.
Following the results of the compound 16 simulations, we applied the same simulation and analysis protocol to compound 18, the most active of the series. In this case, the complex between the enzyme and the compound was obtained by building the ligand in place of compound 16, i.e., substituting the Cl atom with I. The same pattern of H-bonds observed for compound 16 was also maintained in this case, with persisting H-bonds with His118, Arg354, Asn390, Arg395, and Asp506 (Table S2 and Figure 5B). Compound 18 showed an increased geometrical stability, with an average heavy atom RMSD of 1.62 ± 0.41 Å over the three replicates. In this case, the average RMSD of the whole ligand was closer to the RMSD of the nucleotide scaffold (RMSDave = 1.57 ± 0.28 Å): the p-I-benzyl moiety deviated less from its initial conformation, just in the last frames of replicates 2 and 3 (Figure 5A, C-E). The cluster analysis identified a main cluster collecting ~90% of the frames, with the ligand oriented as in the starting pose (Figure S5B, C). In a second cluster (population of ~7%), the p-I-benzyl moiety pointed toward the N-terminal domain of the enzyme, surrounded principally by Asp121, Asn186, and Pro187 (Figure S5D, E), similar to the previous observation of cluster 2 of compound 16. Interestingly, the presence of Gln or Asn surrounding the p-I-benzyl substituent was confirmed in both clusters, with these residues potentially able to interact with and stabilize the I substituent of the highly potent compound 18.
Figure 5.

(A) Superposition of the last frames of three 50 ns trajectories of the compound 18-hCD73 complex in the closed form. The trajectories have been aligned by superposing the Cα atoms to the initial state. Proteins are represented as a gray ribbon, interacting residues as gray lines, and ligands of replicates 1–3 as orange, green, and blue sticks, respectively. (B) Percentage of H-bonds between compound 18 and hCD73 residues during the three replicates. Only residues interacting with on average >10% of the simulations are shown. (C) RMSDs of compound 18 heavy atoms over time in the three replicates. (D) RMSFs of compound 18 heavy atoms in the three replicates. The atom numbers of atoms with higher RMSFs are reported in the two-dimensional depiction of the p-I-benzyl moiety. (E) RMSDs of the heavy atoms of the nucleotide scaffold (all heavy atoms except the p-I-benzyl) in the three replicates.
Analogously, we analyzed the X-ray structure of the complex between hCD73 and compound 21, and we observed the presence of two main clusters (Figure S6A): cluster 1 (population of ~49%), collecting the first part of replicates 1 and 3 and almost all of replicate 2, where the p-methoxycarbonyl-benzyl maintained the original position (Figure S6B, C), and cluster 2 (population of ~40%). Cluster 2 appeared in the second part of replicates 1 and 3 and showed a conformation of the substituent pointing toward the enzyme’s N-terminal domain (Figure S6D, E), which is similar to the intermediate state described in clusters 2 of compounds 16 and 18. The evolution of replicate 2 in its final frames (Figure 6A and Figure S6A) can be described by a further small cluster (population of ~3%), where the p-methoxycarbonyl-benzyl was rotated ~180° around the benzyloxy bond to interact with Asn503 (Figure S6F, G). Regardless of the flexibility and conformational variability of the p-methoxycarbonyl-benzyl substituent, the nucleotide-like scaffold was stable, as assessed by an average RMSD of 1.20 ± 0.28 Å (Table S2 and Figure 6C-E), which correlated with H-bonds to His118, Arg354, Asn390, Arg395, and Asp506 being maintained during most of the simulation time (Figure 6B). The methyl ester of compound 21 is an H-bond acceptor, and in fact, it engaged in additional transient H-bonds during the simulations, i.e., with Gln88 and Asn186 (average persistence over the three replicates of >10%).
Figure 6.

(A) Superposition of the last frames of three 50 ns trajectories of the compound 21-hCD73 complex in the closed form. The trajectories have been aligned by superposing the Cα atoms with the initial state. Proteins are represented as a gray ribbon, interacting residues as gray lines, and ligands of replicates 1–3 as orange, green, and blue sticks, respectively. (B) Percentage of H-bonds between compound 21 and hCD73 residues during the three replicates. Only residues interacting with on average >10% of the simulations are shown. (C) RMSDs of compound 21 heavy atoms over time in the three replicates. (D) RMSFs of compound 21 heavy atoms in the three replicates. The atom numbers of atoms with higher RMSFs are reported in the two-dimensional depiction of the p-methoxycarbonyl-benzyl moiety. (E) RMSDs of the heavy atoms of the nucleotide scaffold (all heavy atoms except the p-methoxycarbonyl-benzyl) in the three replicates.
In addition to analyzing the benzylic substituent behavior and the persistence of interactions typical of the bound state, we also exploited the collected MD trajectories to monitor the puckering of the ribose ring in a dynamic environment. In fact, the X-ray structures of the complexes between hCD73 and compound 16 and 21 showed that the ligands assumed an N conformation in the bound state, as shown by their phase angle of pseudorotation (P) and degree of deformation from the plane (νmax) reported in the pseudorotational cycle in Figure 7B). This agrees with the general trend of nucleotide-like compounds in the hCD73 active site already deposited in the PDB (Figure 7D). The N puckering was maintained by all of the compounds during the MD simulations, where the ring only transiently assumed the S conformation (Figure S9A, C, E). This suggested application of a chemical constraint of the ribose ring to maintain the enzyme-preferred N conformation. Therefore, the ribose furan ring was substituted with a bicyclo[3.1.0]hexane (N)-methanocarba moiety, driven by the previous successful experience provided by this bicyclic structure to freeze the puckering of A3 adenosine receptor agonists in their receptor-preferred N conformation,36,48 thereby increasing the affinity. However, compound 29, the (N)-methanocarba analogue of compound 16, did not show an expected increase in binding affinity (Ki ~ 59 nM) but instead was ~90-fold less potent than ribose analogue 16 (Ki ~ 0.67 nM). This could be explained by the protein environment hosting the O4′ position of the ligand: the X-ray structure of the compound 16-hCD73 complex showed an H-bond between this atom and Arg395, which was not directly present in the case of compound 21 and did not persist during MD simulations. However, an examination of residues in contact with atom O4′ in the MD trajectories (Figure 7F) showed that Arg395 was in the proximity of the atom in nearly all of the simulations (all compounds, all replicates), suggesting that the local polar binding site environment was more favorable with O4′ present than for the (N)-methanocarba bicyclic ring.
Figure 7.

(A, C, and E) Puckering of the ribose ring of compounds 16, 18, and 21, respectively, during MD simulations (combination of three 50 ns replicates). The phase angle of pseudorotation (P) and degree of deformation of the plane (νmax) are reported on the angular and radial coordinates, respectively. (B) Puckering of the ribose ring of compounds 16 (dark green) and 21 (light green), in the X-ray structures. (D) Puckering of all of the nucleoside/nucleotide-like ligands bound to hCD73 deposited so far in the PDB. (F) Number of contacts (percentage of frames) between the O4′ atom of compounds 16, 18, and 21 and hCD73 residues during the three MD replicates.
In addition to these modeling studies, the availability of the X-ray structures reported here was the starting point of prospective molecular docking studies, aimed to suggest the synthesis of new compounds.26,27,44 Docking of compound 24 (Ki ~ 0.63 nM), among the most active in the series, confirmed a binding mode comparable to the crystallographic pose of compounds 16 and 21 but placed the primary charged amino group in the proximity of Lys97 (Figure 8A). This suggested a substitution of the primary amine with a carboxylate moiety, giving compound 27, to exploit an ionic interaction with the protein Lys, as shown by the docking pose of the compound (Figure 8B). However, this did not result in an increase in the binding affinity (Ki ~ 2.61 nM), and this could be explained by the solvent exposure of this possible ionic interaction.
Figure 8.

(A) Docking pose of compound 24 at the closed form hCD73 active site. (B) Docking pose of compound 27 at the hCD73 active site. In both cases, the ligand is shown as cyan sticks, with the protein colored gray. Protein residues within 4 Å of the ligand are shown as sticks. Hydrogen bonds are shown as yellow dashed lines, ionic interactions as magenta dashed lines, and π–π interactions as cyan dashed lines.
In Situ ecto-5′-Nucleotidase Activity Assay.
The effect of potent CD73 inhibitors on ecto-5′-nucleotidase activity in human tissues was evaluated in situ by using surgically removed human head and neck squamous cell carcinoma (HNSCC) and palatine tonsils28 as appropriate enzyme sources. Staining of the HNSCC cryosections with hematoxylin and eosin (H&E) enabled the visualization of the main histological structures (Figure 9A). The tissue-specific distribution of AMPase activity and the expression level of CD73 in the tumor microenvironment (TME) were determined by using lead nitrate-based enzyme histochemistry (Figure 9B) and immunofluorescence staining (Figure 9C), respectively. Histological analysis of the HNSCC sample identified two different areas of differentiation, with significant AMPase activity and CD73 immunoreactivity being detected on nonkeratinized poorly differentiated squamous carcinoma cells (Figure 9, top insets), but not on well-differentiated squamous carcinoma cells and neoplastic cells (Figure 9, bottom insets). High CD73 activity was also detected on other benign components of the TME, including the basal epithelial layer, blood vessels, tumor-infiltrating leukocytes, and muscle cells. We have shown earlier that CD73 is also expressed in human tonsils, with high AMPase activity detected in the germinal centers, in connective tissues, and, to a lesser extent, in the interfollicular area (see also Figure 10B).28,49
Figure 9.

Distribution of AMPase activity and the expression level of CD73 in the TME. A tumor was obtained from the patient with squamous cell carcinoma of mobile tongue. (A) Staining of tissue cryosections with H&E enabled the visualization of the malignant and benign cells in the TME. (B) Tissue-specific distribution of AMPase activity determined by incubating the slides with 100 μM AMP in the presence of Pb(NO3)2 followed by microscopic detection of the nucleotide-derived inorganic phosphate (Pi) as a brown precipitate. (C) For immunofluorescence staining, tumor sections were stained with the anti-CD73 antibody, together with Alexa Fluor-488-conjugated pan-cytokeratin mAb serving as a marker of well-differentiated epithelial tumor cells. Single channels are shown in grayscale, and the right-hand panels display merged images with nuclei counterstained with DAPI. Abbreviations: BEL, basal epithelial layer; BV, blood vessels; MC(l), muscular cells (longitudinally oriented); MC(t), muscular cells (transverse orientation); TC(p), tumor cells (poorly differentiated); TC(w), tumor cells (well differentiated); TIL, tumor-infiltrating leukocytes. Scale bars are 1 mm (A–C) and 300 μm (top and bottom insets).
Figure 10.

Effect of CD73 inhibitors on AMPase activity in human tumors and tonsils. The effect of CD73 inhibitors on AMPase activity was determined in situ by incubating human (A) HNSCC and (B) tonsillar cryosections with 100 μM AMP and 1.5 mM Pb(NO3)2 in the absence (Control) and presence of the indicated concentrations of tested compounds. AMPase activity was determined in the selected regions, corresponding to pan-cytokeratin−/CD73+ poorly differentiated tumor cells (A) and CD73+ tonsillar germinal centers (B). (C) Mean pixel intensities of AMP-specific brown staining were quantified in the selected regions using QuPath version 0.2.3 and expressed as arbitrary units (AU) (mean ± standard error of the mean). Compared with the control, *P < 0.05 and **P < 0.01, determined by one-way analysis of variance with Dunnett’s multiple-comparison test. Scale bars are 1 mm (A, top image; B, inset), 4 mm (B, top image), and 300 μm (A, insets).
Subsequent competitive analysis of CD73-mediated AMPase activities was performed by incubating human tissue cryosections with AMP in the presence of Pb(NO3)2 and different concentrations of CD73 inhibitors. The lead orthophosphate precipitated during the course of ecto-5′-nucleotidase activity was visualized as a brown deposit. Figure 10 depicts representative images of tumor (panel A) and tonsillar (panel B) AMP-specific staining determined in the absence (control) and presence of the indicated compounds. Given the uneven distribution of CD73 in the tissues, we selected the same matched areas of poorly differentiated squamous carcinoma cells (Figure 10A) and tonsillar germinal centers (B) in the control and treated samples and further compared their mean pixel intensities using QuPath version 0.2.3 (Table S3). Treatment of tissue cryosections with the increasing concentrations of compounds 18 and 20 (50–250 nM), but not with equimolar concentrations of classical CD73 inhibitor AOPCP (compound 1), decreased the catalytic activities of both tumor and tonsillar CD73 in a concentration-dependent manner (Figure 10C).
CONCLUSIONS
We have explored the SAR of nucleotide inhibitors of CD73 in the pyrimidine series, building on our recent publication and focusing mainly on cytidine functionalization at the N4 position.28 We have substantially improved the potency compared to those of our previous inhibitors and demonstrated efficacy in actual cancer tissue. Although this study is restricted to various substituted N4-benzyloxy groups, future work will evaluate the N4 position more broadly. This work also contains extensive structural characterization, including four new X-ray structures with our inhibitors bound. Molecular dynamics identified stabilizing interactions and predicted conformational diversity. The most potent analogue 18 contained 4-iodo substitution of the N4-benzyloxy group with a Ki value of 0.436 nM, which represents a 24-fold enhancement over 9. Many of the substituted benzyl derivatives presented here are 10–20-fold more potent than the previously most potent inhibitors in this series. Inclusion of a ribose substitution that enforces an N conformation, similar to that present in crystallographic CD73 structures,33 greatly decreased the inhibitory potency. Thus, the rigid methanocarba ring system is not a means of enhancing the pyrimidine nucleotide potency at CD73. Furthermore, we have introduced additional functionality that can be useful in bivalent molecular probes.50,51 The utility of novel compounds as potential anticancer drugs was further ascertained by their ability to efficiently block CD73 activity in the human HNSCC tissue in situ. The ability to potently inhibit CD73 using pyrimidine-based nucleotide analogues provides new opportunities for translational studies for cancer treatment.
EXPERIMENTAL PROCEDURES
Chemical Synthesis.
Reagents and Instrumentation.
All reagents were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO). Anhydrous solvents were obtained directly from commercial sources. All reactions were carried out under a nitrogen atmosphere using anhydrous solvents. Room temperature (rt) refers to 25 ± 2 °C. NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts are given in parts per million (δ), calibrated to the residual solvent signals or TMS and frequency calibrated internally (Xi) for 31P NMR. High-resolution mass measurements were performed on a Micromass/Waters LCT Premier Electrospray Time of Flight (TOF) mass spectrometer coupled with a Waters HPLC system, unless noted. Ion-exchange chromatography (IE-LPLC) was performed using 10 mm × 100 mm Pharmacia column packed with GE Bioscience SOURCE 15Q resins, which was connected to an Agilent 1100 HPLC system. The IE-LPLC mobile phase condition is as follows: 100% H2O from 0 to 5 min, 0% to 100% linear gradient of 1 M TEAB/H2O from 5 to 40 min, eluent flow rate of 2 mL/min. RP-HPLC was performed using a Phenomenex Luna 5 μm C18(2)100A, AXIA, 21.2 mm × 250 mm column. Purity was determined using an Agilent Eclipse XDB-C18, 5 μm, 4.6 mm × 250 mm column and a linear gradient from 0% to 100% acetonitrile/5 mM tetrabutylammonium dihydrogen phosphate (TBAP) as the mobile phase at a flow rate of 1 mL/min. All of the derivatives tested for the biological activity showed >95% purity by HPLC analysis (detection at 254 nm).
General Procedure for the Synthesis of Nucleotides (10–30).
To a solution of methylenebis(phosphonic dichloride) (3 equiv) in trimethylphosphate (3 mL) at 0 °C was added a solution of the desired compound (1 equiv) in trimethylphosphate (2 mL) at 0 °C. The so obtained mixture was stirred at 0 °C for 3 h, and then a sample was withdrawn for LC-MS to check the disappearance of the nucleoside. Then, the reaction was quenched with 7 mL of a cold 1 M aqueous triethylammonium hydrogen carbonate buffer solution (pH 8.4–8.6), and the mixture stirred at rt for 1 h. The solvent was removed by lyophilization, and the residue dissolved in water and purified first by ion-exchange chromatography and then by RP-HPLC to afford the pure product.
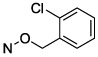
N4-(2-Chlorobenzyloxy)-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (10).
The product was obtained as a white solid after lyophilization (9.8 mg, 20%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.45–7.55 (m, 2H), 7.29–7.40 (m, 3H), 6.44 (d, J = 8.3 Hz, 1H), 5.93 (d, J = 4.5 Hz, 1H), 5.12 (s, 2H), 4.35 (s, 2H), 4.21 (s, 1H), 4.10 (s, 2H), 3.11–3.22 (m, 15H), 2.18 (t, J = 19.7 Hz, 2H), 1.26 (t, J = 7.1 Hz, 18H); 31P NMR (D2O) δ 18.20, 14.21; HRMS calcd for C18H23ClN3O11P2 (M – H)– 554.0496, found 554.0490.
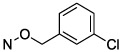
N4-(3-Chlorobenzyloxy)-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (11).
The product was obtained as a white solid after lyophilization (3.7 mg, 2.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.49 (s, 1H), 7.32–7.42 (m, 3H), 6.45 (d, J = 8.4 Hz, 1H), 5.93 (d, J = 4.9 Hz, 1H), 5.03 (s, 2H), 4.30–4.39 (m, 2H), 4.21 (s, 1H), 4.12 (s, 2H), 3.11–3.29 (m, 9H), 2.18 (t, J = 19.7 Hz, 2H), 1.26 (t, J = 6.7 Hz, 9H); 31P NMR (D2O) δ 20.50, 12.50; HRMS calcd for C18H23ClN3O11P2 (M – H)− 554.0496, found 554.0503.
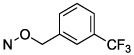
N4-[(3-Trifluoromethyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphate Triethylammonium Salt (12).
The product was obtained as a white solid after lyophilization (4.2 mg, 9.5%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.64 (s, 1H), 7.56 (d, J = 7.6 Hz, 2H), 7.45 (t, J = 7.6 Hz, 1H), 7.22 (d, J = 8.3 Hz, 1H), 6.32 (d, J = 8.3 Hz, 1H), 5.80 (d, J = 4.7 Hz, 1H), 4.95 (s, 2H), 4.20 (d, J = 4.8 Hz, 2H), 4.08 (s, 1H), 3.97 (s, 2H), 3.00–3.11 (m, 15H), 2.02 (t, J = 19.7 Hz, 2H), 1.13 (t, J = 7.1 Hz, 18H); 31P NMR (D2O) δ 29.00, 24.00; 19F NMR (D2O) δ −62.40; HRMS calcd for C19H23F3N3O11P2 (M – H)− 588.0760, found 588.0756.
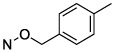
N4-[(3-Methyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (13).
The product was obtained as a white solid after lyophilization (21.1 mg, 40%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 10/90 to 25/75 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.21–7.25 (m, 3H), 7.16 (d, 2H, J = 7.8 Hz), 6.32 (d, 1H, J = 8.4 Hz), 5.84 (d, 1H, J = 5.0 Hz), 4.87 (s, 2H), 4.22–4.24 (m, 2H), 4.11 (s, 1H), 4.01–4.02 (m, 2H), 3.06–3.12 (m, 15H), 2.23 (s, 3H), 2.07 (t, 2H, J = 19.8 Hz), 1.17 (t, 18H, J = 7.3 Hz); 31P NMR (D2O) δ 18.23, 14.66; HRMS calcd for C19H26N3O11P2 (M – H)− 534.1043, found 534.1053.
N4-[(3-Ethyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (14).
The product was obtained as a white solid after lyophilization (24.6 mg, 37%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 10/90 to 25/75 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.29 (d, 2H, J = 7.9 Hz), 7.18–7.23 (m, 3H), 6.32 (d, 1H, J = 8.3 Hz), 5.84 (d, 1H, J = 5.0 Hz), 4.87 (s, 2H), 4.20–4.24 (m, 2H), 4.10 (s, 1H), 4.00–4.02 (m, 2H), 3.06–3.11 (m, 15H), 2.54 (dd, 2H, J = 7.6, 15.2 Hz), 2.07 (t, 2H, J = 19.8 Hz), 1.17 (t, 18H, J = 7.3 Hz), 1.08 (t, 3H, J = 7.6 Hz); 31P NMR (D2O) δ 18.21, 14.67; HRMS calcd for C20H28N3O11P2 (M – H)− 548.1199, found 548.1209.
N4-(4-Fluorobenzyloxy)-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (15).
The product was obtained as a white solid after lyophilization (12.9 mg, 27.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.36 (t, J = 6.8 Hz, 2H), 7.22 (d, J = 7.7 Hz, 1H), 7.02 (t, J = 8.7 Hz, 2H), 6.30 (d, J = 8.1 Hz, 1H), 5.81 (s, 1H), 4.84 (s, 2H), 4.16–4.27 (m, 2H), 3.91–4.09 (m, 3H), 3.00–3.12 (m, 15H), 2.00 (t, J = 19.7 Hz, 2H), 1.13 (t, J = 7.2 Hz, 18H); 31P NMR (D2O) δ 21.00, 13.00; 19F NMR (D2O) δ −75.50; HRMS calcd for C18H23FN3O11P2 (M – H)− 538.0792, found 538.0789.
N4-(4-Chlorobenzyloxy)-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (16).
The product was obtained as a white solid after lyophilization (35.4 mg, 33.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.23 (s, 4H), 7.19 (d, J = 8.4 Hz, 1H), 6.23 (d, J = 8.3 Hz, 1H), 5.79 (d, J = 5.0 Hz, 1H), 4.82 (s, 2H), 4.18–4.22 (m, 2H), 4.08 (d, J = 1.8 Hz, 1H), 4.12 (s, 2H), 3.98–4.00 (m, 2H), 3.08 (dd, J = 7.3, 14.6 Hz, 12H), 2.99 (s, 3H), 2.06 (t, J = 19.7 Hz, 2H), 1.12 (t, J = 7.3 Hz, 18H); 31P NMR (D2O) δ 22.00, 15.00; HRMS calcd for C18H23ClN3O11P2 (M – H)− 554.0496, found 554.0501.
N4-(4-Bromobenzyloxy)-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (17).
The product was obtained as a white solid after lyophilization (3.5 mg, 20.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.50 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.16 Hz, 3H), 6.36 (d, J = 8.2 Hz, 1H), 5.82 (d, J = 4.5 Hz, 1H), 4.91 (s, 2H), 4.31 (s, 1H), 4.22 (s, 1H), 4.00–4.12 (m, 3H), 3.10 (d, J = 7.2 Hz, 21H), 1.95 (t, J = 19.1 Hz, 2H), 1.20 (s, 27H); 31P NMR (D2O) δ 21.00, 12.00; HRMS calcd for C18H23BrN3O11P2 (M – H)− 597.9991, found 597.9988.
N4-(4-Iodobenzyloxy)-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (18).
The product was obtained as a white solid after lyophilization (8.2 mg, 22.1%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.69 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.2 Hz, 1H), 7.12 (d, J = 8.0 Hz, 2H), 6.32 (d, J = 8.2 Hz, 1H), 5.82 (d, J = 5.0 Hz, 1H), 4.88 (s, 2H), 4.25 (t, J = 5.5 Hz, 2H), 4.12 (s, 1H), 4.01 (s, 2H), 3.10 (t, J = 6.7 Hz, 6H), 3.03 (s, 3H), 2.05 (t, J = 19.5 Hz, 2H), 1.18 (t, J = 6.6 Hz, 9H); 31P NMR (D2O) δ 23.00, 12.00; HRMS calcd for C18H23IN3O11P2 (M – H)− 645.9852, found 645.9861.
N4-[4-(Trifluoromethyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (19).
The product was obtained as a white solid after lyophilization (1.4 mg, 10.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.58 (d, J = 6.3 Hz, 2H), 7.43 (d, J = 6.3 Hz, 2H), 7.22 (d, J = 6.5 Hz, 1H), 6.32 (d, J = 6.6 Hz, 1H), 5.79 (d, J = 3.9 Hz, 1H), 4.94 (s, 2H), 4.23 (s, 1H), 4.17 (d, J = 4.1 Hz, 1H), 4.00–4.05 (m, 2H), 3.92 (s, 1H), 3.02 (dd, J = 5.8, 11.6 Hz, 6H), 2.98 (s, 3H), 1.91 (t, J = 15.2 Hz, 2H), 1.10 (t, J = 5.8 Hz, 9H); 31P NMR (D2O) δ 22.00, 12.50; 19F NMR (D2O) δ −62.00; HRMS calcd for C19H23F3N3O11P2 (M – H)− 588.0760, found 588.0755.
N4-[4-(Pentafluorosulfanyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (20).
The product was obtained as a white solid after lyophilization (8.0 mg, 23.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.75 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.3 Hz, 1H), 6.37 (d, J = 8.3 Hz, 1H), 5.82 (d, J = 5.0 Hz, 1H), 4.99 (s, 2H), 4.22 (dd, J = 4.4, 11.8 Hz, 2H), 4.11 (s, 1H), 4.00 (s, 2H), 3.09 (dd, J = 7.3, 14.6 Hz, 6H), 3.02 (s, 3H), 2.05 (t, J = 19.7 Hz, 2H), 1.14 (t, J = 14.6 Hz, 9H); 31P NMR (D2O) δ 22.00, 12.00; 19F NMR (D2O) δ −75.55; HRMS calcd for C18H23F5N3O11P2S (M – H)− 646.0449, found 646.0452.
N4-[4-(Methoxycarbonyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (21).
The product was obtained as a white solid after lyophilization (38.7 mg, 32.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 30/70 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.80 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 8.3 Hz, 1H), 6.30 (d, J = 8.3 Hz, 1H), 5.77 (d, J = 5.0 Hz, 1H), 4.91 (s, 2H), 4.21 (t, J = 5.2 Hz, 2H), 4.07 (s, 1H), 3.98 (s, 2H), 3.74 (s, 3H), 3.09 (dd, J = 7.1, 14.4 Hz, 12H), 2.92 (s, 3H), 2.04 (t, J = 19.7 Hz, 2H), 1.13 (t, J = 7.2 Hz, 18H); 31P NMR (D2O) δ 17.50, 14.00; HRMS calcd for C20H26N3O13P2 (M – H)− 578.0941, found 578.0943.
N4-[4-(Carbonyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (22).
The product was obtained as a white solid after lyophilization (11.9 mg, 29.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 5/95 to 15/85 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.75 (d, J = 7.8 Hz, 2H), 7.39 (d, J = 7.7 Hz, 2H), 7.22 (d, J = 8.2 Hz, 1H), 6.37 (d, J = 8.2 Hz, 1H), 5.83 (s, 1H), 4.96 (s, 2H), 4.23 (s, 2H), 4.09 (s, 1H), 4.00 (s, 2H), 3.74 (s, 3H), 3.06 (dd, J = 7.2, 14.3 Hz, 21H), 2.05 (t, J = 19.6 Hz, 2H), 1.14 (t, J = 7.2 Hz, 27H); 31P NMR (D2O) δ 21.00, 15.40; HRMS calcd for C19H24N3O13P2 (M – H)− 564.0784, found 564.0781.
N4-[4-(Carbamoyl)benzyloxy]-3-methylcytidine-5′-α,β-methylenediphosphate Triethylammonium Salt (23).
The product was obtained as a white solid after lyophilization (1.6 mg, 9.6%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 5/95 to 15/85 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.71 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 8.3 Hz, 1H), 6.37 (d, J = 8.4 Hz, 1H), 5.83 (d, J = 5.3 Hz, 1H), 4.98 (s, 2H), 4.21–4.26 (m, 2H), 4.09 (s, 1H), 4.01 (s, 2H), 3.08 (d, J = 7.0 Hz, 6H), 3.05 (s, 3H), 2.02 (t, J = 19.9 Hz, 2H), 1.16 (t, J = 6.9 Hz, 9H); 31P NMR (D2O) δ 19.90, 13.50; HRMS calcd for C19H25N4O12P2 (M – H)− found 563.0950.
N4-{4-[(2-Aminoethyl)carbamoyl]benzyloxy}-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (24).
The product was obtained as a white solid after lyophilization (4.1 mg, 10.5%, two-step yield; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 5/95 to 15/85 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.69 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.4 Hz, 1H), 6.28 (d, J = 8.3 Hz, 1H), 5.80 (d, J = 4.8 Hz, 1H), 4.96 (s, 2H), 4.22 (d, J = 4.7 Hz, 1H), 4.18 (t, J = 4.9 Hz, 1H), 4.01 (s, 2H), 3.60 (t, J = 5.7 Hz, 2H), 3.16–3.04 (m, 17H), 2.00 (t, J = 18.7 Hz, 2H), 1.17 (t, J = 7.3 Hz, 18H); 31P NMR (D2O) δ 20.00, 13.00; HRMS calcd for C21H30N5O12P2 (M – H)− 606.1366, found 606.1362.
N4-{4-[(2-Aminobutyl)carbamoyl]benzyloxy}-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (25).
The product was obtained as a white solid after lyophilization (1.8 mg, 8.0%, two-step yield; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 5/95 to 15/85 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.66 (d, J = 8.3 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.3 Hz, 1H), 6.36 (d, J = 8.3 Hz, 1H), 5.85 (d, J = 5.2 Hz, 1H), 4.99 (s, 2H), 4.20–4.28 (m, 2H), 4.12 (s, 1H), 4.03 (d, J = 5.0 Hz, 2H), 3.35 (t, J = 6.1 Hz, 2H), 3.07–3.15 (m, 4H), 2.95 (t, J = 7.1 Hz, 2H), 2.04 (t, J = 19.2 Hz, 2H), 1.63 (d, J = 3.04 Hz, 4H), 1.19 (t, J = 7.2 Hz, 5H); 31P NMR (D2O) δ 19.92, 13.29; HRMS calcd for C23H34N5O12P2 (M – H)− 634.1679, found 634.1685.
N4-{4-[(3-Ethoxy-3-oxopropyl)carbamoyl]benzyloxy}-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (26).
The product was obtained as a white solid after lyophilization (4.2 mg, 10.4%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.60 (d, J = 8.2 Hz, 2H), 7.41 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 8.5 Hz, 1H), 6.35 (d, J = 8.4 Hz, 1H), 5.81 (d, J = 5.2 Hz, 1H), 4.95 (s, 2H), 4.18–4.24 (m, 2H), 3.99–4.08 (m, 4H), 3.54 (t, J = 6.4 Hz, 2H), 3.02–3.09 (m, 15 H), 2.07 (t, J = 6.4 Hz, 2H), 2.01 (t, J = 19.6 Hz, 2H), 1.14 (t, J = 7.3 Hz, 18H), 1.06 (t, J = 7.1 Hz, 3H); 31P NMR (D2O) δ 19.15, 13.79; HRMS calcd for C24H35N4O14P2 (M – H)– 665.1625, found 665.1625.
N4-{4-[(2-Carboxyethyl)carbamoyl]benzyloxy}-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (27).
The product was obtained as a white solid after lyophilization (3.3 mg, 9.1%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 10/90 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.61 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.5 Hz, 1H), 6.35 (d, J = 8.4 Hz, 1H), 5.78 (d, J = 5.2 Hz, 1H), 4.94 (s, 2H), 4.26 (t, J = 4.6 Hz, 1H), 4.19 (t, J = 5.1 Hz, 1H), 3.96–4.05 (m, 3H), 3.45 (t, J = 7.0 Hz, 2H), 3.01–3.05 (m, 21H), 2.36 (t, J = 7.1 Hz, 2H), 1.92 (t, J = 19.5 Hz, 2H), 1.13 (t, J = 6.6 Hz, 27H); 31P NMR (D2O) δ 21.66, 11.49; HRMS calcd for C22H29N4O14P2 (M – H)– 635.1155, found 635.1162.
{[({(2R,3S,4R,5S)-4-[2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl]-2,3-dihydroxy-1λ5-bicyclo[3.1.0]hexan-1-yl}methoxy)(hydroxy)phosphoryl]methyl}phosphonic Acid Triethylammonium Salt (28).
The product was obtained as a white solid after lyophilization (1.6 mg, 6.1%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 0/100 to 10/90 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.91 (d, J = 7.8 Hz, 1H), 5.82 (d, J = 7.8 Hz, 1H), 4.28 (dd, J = 5.1, 11.0 Hz, 1H), 3.86 (d, J = 6.6 Hz, 1H), 3.70 (d, J = 11.0 Hz, 1H), 3.60 (dd, J = 5.2, 10.5 Hz, 1H), 2.55–3.30 (sbr, 6H), 2.04 (t, J = 19.8 Hz, 2H), 1.48 (d, J = 7.8 Hz, 1H), 1.14–1.25 (m, 11H), 0.81 (d, J = 7.3 Hz, 1H); 31P NMR (D2O) δ 21.00, 13.03; HRMS calcd for C12H18N2O10P2 (M – H)– 411.0358, found 411.0355.
[({[(2R,3S,4R,5S)-4-(4-{[(4-Chlorobenzyl)oxy]imino}-3-methyl-2-oxo-3,4-dihydropyrimidin-1(2H)-yl)-2,3-dihydroxy-1λ5-bicyclo[3.1.0]hexan-1-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid Triethylammonium Salt (29).
The product was obtained as a white solid after lyophilization (6.9 mg, 16.0%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/65 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.32 (s, 4H), 7.26 (d, J = 8.3 Hz, 1H), 6.28 (d, J = 8.2 Hz, 1H), 4.87 (s, 2H), 4.53–4.58 (m, 2H), 4.23 (dd, J = 5.4, 10.9 Hz, 1H), 3.79 (d, J = 6.8 Hz, 1H), 3.64 (dd, J = 4.9, 10.9 Hz, 1H), 3.05 (s, 21H), 1.95 (t, J = 19.6 Hz, 2H), 1.38 (t, J = 4.4 Hz, 2H), 1.15 (s, 27H), 0.75 (t, J = 6.6 Hz, 1H); 31P NMR (D2O) δ 22.00, 12.00; HRMS calcd for C20H26ClN3O10P2 (M – H)– 564.0704, found 564.0702.
4-[(Benzylcarbamoyl)imino]-3-methylcytidine-5′-α,β-methylenediphosphonate Triethylammonium Salt (30).
The product was obtained as a white solid after lyophilization (7.6 mg, 8.4%; RP-HPLC acetonitrile/10 mM triethylammonium acetate, 15/85 to 35/75 in 40 min at a rate of 5 mL/min): 1H NMR (D2O, 400 Hz) δ 7.64 (d, J = 8.2 Hz, 1H), 7.25–7.7.46 (m, 5H), 6.13 (d, J = 8.2 Hz, 1H), 5.97 (d, J = 3.1 Hz, 1H), 4.42 (s, 2H), 4.35 (s, 2H), 4.24 (s, 1H), 4.14 (s, 2H), 3.31 (s, 3H), 3.19 (dd, J = 7.3, 14.6 Hz, 12H), 2.16 (t, J = 19.7 Hz, 2H), 1.26 (t, J = 7.0 Hz, 18H); 31P NMR (D2O) δ 20.00, 13.00; HRMS calcd for C19H25N4O11P2 (M – H)– 547.0995, found 547.0995.
General Procedure for the Synthesis of Substituted O-Benzylhydroxylamine Hydrochloride (32–43).
Compounds 32–43 were synthesized following the reported procedure33 starting from the corresponding benzyl bromide.
O-2-Chlorobenzylhydroxylamine Hydrochloride (32).
The product was obtained as a white solid (120 mg) in 63% yield: 1H NMR (MeOD, 400 Hz) δ 7.35–7.55 (m, 4H), 5.20 (s, 2H).
O-3-Chlorobenzylhydroxylamine Hydrochloride (33).
The product was obtained as a white solid (105 mg) in 74% yield: 1H NMR (MeOD, 400 Hz) δ 7.36–7.47 (m, 4H), 5.02 (s, 2H).
O-3-(Trifluoromethyl)benzylhydroxylamine Hydrochloride (34).
The product was obtained as a white solid (130 mg) in 68% yield: 1H NMR (MeOD, 400 Hz) δ 7.61–7.78 (m, 4H), 5.12 (s, 2H).
O-3-Methylbenzylhydroxylamine Hydrochloride (35).
The product was obtained as a white solid (272 mg) in 97% yield: 1H NMR (MeOD, 400 Hz) δ 7.33 (d, 2H, J = 7.9 Hz), 7.26 (d, 2H, J = 7.9 Hz), 4.98 (s, 2H), 2.37 (s, 3H).
O-3-Ethylbenzylhydroxylamine Hydrochloride (36).
The product was obtained as a white solid (198 mg) in 84% yield: 1H NMR (MeOD, 400 Hz) δ 7.37 (d, 2H, J = 7.8 Hz), 7.29 (d, 2H, J = 8.0 Hz), 4.99 (s, 2H), 2.68 (dd, 2H, J = 7.5, 15.1 Hz), 1.24 (t, 3H, J = 7.6 Hz).
O-4-Fluorobenzylhydroxylamine Hydrochloride (37).
The product was obtained as a white solid (150 mg) in 75% yield: 1H NMR (MeOD, 400 Hz) δ 7.48 (dd, J = 5.4, 8.4 Hz, 2H), 7.16 (t, J = 8.7 Hz, 2H), 5.03 (s, 2H).
O-4-Chlorobenzylhydroxylamine Hydrochloride (38).
The product was obtained as a white solid (100 mg) in 70% yield: 1H NMR (MeOD, 400 Hz) δ 7.45–7.59 (m, 4H), 5.04 (s, 2H).
O-4-Bromobenzylhydroxylamine Hydrochloride (39).
The product was obtained as a white solid (120 mg) in 74% yield: 1H NMR (MeOD, 400 Hz) δ 7.60 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 5.02 (s, 2H).
O-4-Iodobenzylhydroxylamine Hydrochloride (40).
The product was obtained as a white solid (124 mg) in 64% yield: 1H NMR (MeOD, 400 Hz) δ 7.82 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 4.98 (s, 2H).
O-4-(Trifluoromethyl)benzylhydroxylamine Hydrochloride (41).
The product was obtained as a white solid (145 mg) in 76% yield: 1H NMR (MeOD, 400 Hz) δ 7.95 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 5.12 (s, 2H).
O-4-(Pentafluorosulfanyl)benzylhydroxylamine Hydrochloride (42).
The product was obtained as a white solid (114 mg) in 59% yield: 1H NMR (MeOD, 400 Hz) δ 7.90 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.3 Hz, 2H), 5.10 (s, 2H).
O-4-(Methylbenzoate)benzylhydroxylamine Hydrochloride (43).
The product was obtained as a white solid (123 mg) in 64% yield: 1H NMR (MeOD, 400 Hz) δ 8.09 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.1 Hz, 2H), 5.11 (s, 2H), 3.92 (s, 3H).
3-Methylcytidine (44).
To a solution of cytidine (1.0 g, 4.1 mmol) in N,N-dimethylacetamide (DMAc, 25 mL) was added methyl iodide (0.77 mL, 12.3 mmol), and the mixture was stirred at rt for 4 h. The solvent was removed under reduced pressure, and the resulting crude mixture resuspended in dichloromethane with the formation of a white precipitate that was filtered, affording 1.0 g of the pure compound (95% yield): 1H NMR (MeOD, 400 Hz) δ 8.5 (d, J = 7.8 Hz, 1H), 6.15 (d, J = 7.9 Hz, 1H), 5.87 (d, J = 2.6 Hz, 1H), 4.14–4.21 (m, 2H), 4.08 (t, J = 3.1 Hz, 1H), 3.95 (dd, J = 2.12, 12.4 Hz, 1H), 3.79 (dd, J = 2.12, 12.4 Hz, 1H), 3.49 (s, 3H); HRMS calcd for C10H17N3O5 (M + H)+ 258.1087, found 258.1090.
General Procedure for the Synthesis of Compounds 45–56.
A solution of compound 44 (N3-methylcytidine, 0.8 mmol) and the desired O-benzylhydroxylamine (32–43, 1.6 mmol) in dry pyridine was stirred at 80 °C for 12 h. Then the reaction mixture was concentrated under reduced pressure and purified with a silica gel column.
N4-(2-Chlorobenzyloxy)-3-methylcytidine (45).
Compound 45 was synthesized following the procedure described above starting from O-(2-chlorobenzyl)hydroxylamine hydrochloride. The crude was purified by silica gel column (8/92 methanol/dichloromethane) to afford 96 mg of a white solid (97% yield): 1H NMR (MeOD, 400 Hz) δ 7.45–7.50 (m, 1H), 7.40–7.43 (m, 1H), 7.25–7.35 (m, 3H), 6.30 (d, J = 8.3 Hz, 1H), 5.89 (d, J = 4.8 Hz, 1H), 5.11 (s, 2H), 4.10–4.17 (m, 2H), 3.95 (t, J = 3.3 Hz, 1H), 3.80 (dd, J = 2.7, 12.1 Hz, 1H), 3.70 (dd, J = 2.7, 12.1 Hz, 1H), 3.18 (s, 3H); HRMS calcd for C17H21ClN3O6 (M + H)+ 398.1119, found 398.1121.
N4-(3-Chlorobenzyloxy)-3-methylcytidine (46).
Compound 46 was synthesized following the procedure described above starting from O-(3-chlorobenzyl)hydroxylamine hydrochloride. The crude was purified by silica gel column (5/95 methanol/dichloromethane) to afford 66 mg of a white solid (74% yield): 1H NMR (MeOD, 400 Hz) δ 7.38 (s, 1H), 7.25–7.35 (m, 4H), 6.26 (d, J = 8.3 Hz, 1H), 5.89 (d, J = 4.8 Hz, 1H), 5.00 (s, 2H), 4.10–4.16 (m, 2H), 3.95 (t, J = 3.3 Hz, 1H), 3.80 (dd, J = 2.7, 12.1 Hz, 1H), 3.70 (dd, J = 2.7, 12.1 Hz, 1H), 3.18 (s, 3H); HRMS calcd for C17H21ClN3O6 (M + H)+ 398.1178, found 398.1121.
N4-(3-Trifluoromethylbenzyloxy)-3-methylcytidine (47).
Compound 47 was synthesized following the procedure described above starting from O-(3-trifluoromethylbenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 29 mg of a white solid (59% yield): 1H NMR (MeOD, 400 Hz) δ 7.50–7.65 (m, 5H), 7.31 (d, J = 8.4 Hz, 1H), 6.23 (d, J = 8.3 Hz, 1H), 5.05 (s, 2H), 4.10–4.16 (m, 2H), 3.95 (t, J = 3.2 Hz, 1H), 3.80 (dd, J = 2.7, 12.1 Hz, 1H), 3.68 (dd, J = 2.7, 12.1 Hz, 1H), 3.15 (s, 3H); HRMS calcd for C18H21F3N3O6 (M + H)+ 431.1298, found 431.1304.
N4-(3-Methylbenzyloxy)-3-methylcytidine (48).
Compound 48 was synthesized following the procedure described above starting from O-(3-methylbenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 181 mg of a white solid (62% yield): 1H NMR (MeOD, 400 Hz) δ 7.23–7.29 (m, 3H), 7.14 (d, 2H, J = 7.8 Hz), 6.22 (d, 1H, J = 8.3 Hz), 5.87 (d, 1H, J = 4.7 Hz), 4.93 (s, 2H), 4.09–4.14 (m, 2H), 3.94 (dd, 1H, J = 3.2, 6.4 Hz), 3.79 (dd, 1H, J = 2.6, 12.1 Hz), 3.69 (dd, 1H, J = 3.3, 12.1 Hz), 3.19 (s, 3H), 2.33 (s, 3H); HRMS calcd for C18H24N3O6 (M + H)+ 378.1665, found 378.1659.
N4-(3-Ethylbenzyloxy)-3-methylcytidine (49).
Compound 49 was synthesized following the procedure described above starting from O-(3-ethylbenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 150 mg of a white solid (72% yield): 1H NMR (MeOD, 400 Hz) δ 7.27 (d, 3H, J = 8.4 Hz), 7.17 (d, 2H, J = 7.9 Hz), 6.23 (d, 1H, J = 8.3 Hz), 5.87 (d, 1H, J = 4.7 Hz), 4.94 (s, 2H), 4.09–4.14 (m, 2H), 3.94 (dd, 1H, J = 3.2, 6.4 Hz), 3.79 (dd, 1H, J = 2.7, 12.1 Hz), 3.69 (dd, 1H, J = 3.3, 12.2 Hz), 3.20 (s, 3H), 2.63 (dd, 2H, J = 7.6, 15.2 Hz), 1.22 (t, 3H, J = 7.6 Hz); HRMS calcd for C19H26N3O6 (M + H)+ 392.1822, found 392.1827.
N4-(4-Fluorobenzyloxy)-3-methylcytidine (50).
Compound 50 was synthesized following the procedure described above starting from O-(4-fluorobenzyl)hydroxylamine hydrochloride. The crude was purified by silica gel column (5/95 methanol/dichloromethane) to afford 56 mg of a white solid (35% yield): 1H NMR (MeOD, 400 Hz) δ 7.32–7.41 (m, 2H), 7.29 (d, J = 8.4 Hz, 1H), 7.00–7.10 (m, 2H), 6.21 (d, J = 8.2 Hz, 1H), 5.85 (d, J = 4.6 Hz), 4.95 (s, 2H), 4.05–4.15 (m, 2H), 3.95 (t, J = 3.2 Hz, 1H), 3.75 (dd, J = 2.7, 12.1 Hz, 1H), 3.68 (dd, J = 2.7, 12.1 Hz, 1H), 3.16 (s, 3H); HRMS calcd for C17H21FN3O6 (M + H)+ 382.1409, found 382.1414.
N4-(4-Chlorobenzyloxy)-3-methylcytidine (51).
Compound 51 was synthesized following the procedure described above starting from O-(4-chlorobenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 96 mg of a white solid (97% yield): 1H NMR (MeOD, 400 Hz) δ 7.33–7.36 (m, 4H), 7.30 (d, J = 8.4 Hz, 1H), 6.21 (d, J = 8.2 Hz, 1H), 5.88 (d, J = 4.6 Hz, 1H), 4.98 (s, 2H), 4.08–4.15 (m, 2H), 3.95 (t, J = 3.2 Hz, 1H), 3.78 (dd, J = 2.7, 12.1 Hz, 1H), 3.69 (dd, J = 2.7, 12.1 Hz, 1H), 3.19 (s, 3H); HRMS calcd for C17H21ClN3O6 (M + H)+ 397.1041, found 397.1050.
N4-(4-Bromobenzyloxy)-3-methylcytidine (52).
Compound 52 was synthesized following the procedure described above starting from O-(4-bromobenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (6/94 methanol/dichloromethane) to afford 40 mg of a white solid (39% yield): 1H NMR (MeOD, 400 Hz) δ 7.49 (d, J = 8.3 Hz, 2H), 7.26–7.32 (m, 3H), 6.22 (d, J = 8.2 Hz, 1H), 5.88 (d, J = 4.6 Hz, 1H), 4.98 (s, 2H), 4.08–4.15 (m, 2H), (t, J = 3.2 Hz, 1H), 3.80 (dd, J = 2.7, 12.1 Hz, 1H), 3.70 (dd, J = 2.7, 12.1 Hz, 1H), 3.17 (s, 3H); HRMS calcd for C17H21BrN3O6 (M + H)+ 442.0614, found 442.0614.
N4-(4-Iodobenzyloxy)-3-methylcytidine (53).
Compound 53 was synthesized following the procedure described above starting from O-(4-iodobenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 108 mg of a white solid (92% yield): 1H NMR (MeOD, 400 Hz) δ 7.30 (d, J = 8.3 Hz, 2H), 7.26–7.32 (m, 3H), 6.22 (d, J = 8.2 Hz, 1H), 5.87 (d, J = 4.6 Hz, 1H), 4.98 (s, 2H), 4.08–4.15 (m, 2H), 3.94 (t, J = 3.2 Hz, 1H), 3.78 (dd, J = 2.7, 12.1 Hz, 1H), 3.69 (dd, J = 2.7, 12.1 Hz, 1H), 3.17 (s, 3H); HRMS calcd for C17H21IN3O6 (M + H)+ 490.0471, found 490.0475.
N4-(4-Trifluorobenzyloxy)-3-methylcytidine (54).
Compound 54 was synthesized following the procedure described above starting from O-(4-trifluoromethylbenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 111 mg of a white solid (83% yield): 1H NMR (MeOD, 400 Hz) δ 7.64 (d, J = 8.0 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.3 Hz, 1H), 6.27 (d, J = 8.2 Hz, 1H), 5.88 (d, J = 4.6 Hz, 1H), 5.07 (s, 2H), 4.08–4.14 (m, 2H), 3.95 (t, J = 3.2 Hz, 1H), 3.79 (dd, J = 2.7, 12.1 Hz, 1H), 3.70 (dd, J = 2.7, 12.1 Hz, 1H), 3.16 (s, 3H); HRMS calcd for C18H21F3N3O6 (M + H)+ 432.1375, found 432.1382.
N4-[4-(Pentafluorosulfanyl)benzyloxy]-3-methylcytidine (55).
Compound 55 was synthesized following the procedure described above starting from O-(4-pentafluorosulfanylbenzyl)hydroxylamine hydrochloride. The crude was purified by a silica gel column (2/98 methanol/dichloromethane) to afford 68 mg of a white solid (69% yield): 1H NMR (MeOD, 400 Hz) δ 7.78 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.3 Hz, 1H), 6.26 (d, J = 8.3 Hz, 1H), 5.86 (d, J = 4.6 Hz, 1H), 5.07 (s, 2H), 4.11–4.16 (m, 2H), 3.95 (t, J = 3.2 Hz, 1H), 3.79 (dd, J = 2.7, 12.1 Hz, 1H), 3.71 (dd, J = 2.7, 12.1 Hz, 1H), 3.16 (s, 3H); HRMS calcd for C17H21F5N3O6S (M + H)+ 490.1071, found 490.1071.
Methyl 4-{[({-1-[(2R,3R,4S,5R)-3,4-Dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl]-3-methyl-2-oxo-2,3-dihydropyrimidin-4(1H)-ylidene}amino)oxy]methyl}benzoate (56).
Compound 56 was synthesized following the procedure described above starting from methyl 4-[(aminooxy)methyl]benzoate hydrochloride. The crude was purified by a silica gel column (5/95 methanol/dichloromethane) to afford 26 mg of a white solid (48% yield): 1H NMR (MeOD, 400 Hz) δ 7.79 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.31 (d, J = 8.3 Hz, 1H), 6.26 (d, J = 8.3 Hz, 1H), 5.86 (d, J = 4.7 Hz, 1H), 5.05 (s, 2H), 4.10–4.16 (m, 2H), 3.94 (t, J = 3.2 Hz, 1H), 3.89 (s, 3H), 3.79 (dd, J = 2.7, 12.1 Hz, 1H), 3.69 (dd, J = 2.7, 12.1 Hz, 1H), 3.16 (s, 3H); HRMS calcd for C19H24N3O8 (M + H)+ 422.1558, found 422.1547.
Synthesis of Compound 57 through Hydrolysis of the Methyl Ester Moiety.
To a solution of compound 56 (84 mg, 0.2 mmol) in MeOH (2 mL) was added a 1 N solution of NaOH (2 mL), and the reaction mixture was stirred at rt for 40 min. Glacial acetic acid was added until neutrality. Then, the mixture was diluted with water and extracted with ethyl acetate (3 × 15 mL). The combined organic layer was dried over sodium sulfate, filtered, and evaporated to afford 62 mg (77%) of the pure compound as a white solid: 1H NMR (MeOD, 400 Hz) δ 8.05 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.3 Hz, 1H), 6.37 (d, J = 8.3 Hz, 1H), 5.99 (d, J = 4.7 Hz, 1H), 5.14 (s, 2H), 4.20–4.27 (m, 2H), 4.05 (d, J = 3.2 Hz, 1H), 3.87–3.93 (m, 1H), 3.78–3.84 (m, 1H), 3.28 (s, 3H); HRMS calcd for C18H22N3O8 (M + H)+ 408.1407, found 408.1408.
Synthesis of Compound 59 through Hydrolysis of the Acetylated Intermediate Formed by Serendipity.
To a solution of compound 57 (30 mg, 0.07 mmol, with traces of acetic acid), HATU (42 mg, 0.11 mmol), and DIPEA (19 μL, 0.11 mmol) in anhydrous DMF was added methylamine hydrochloride (6.5 mg, 0.09 mmol), and the reaction mixture was stirred at rt for 12 h. Next, the solvent was removed, and the crude purified by a silica gel column (5% MeOH/DCM) to afford an acetylated intermediate as a white solid (58). The latter was treated with methanolic ammonia for 2 h at rt to deblock the acetylated group and eventually recover the starting material. TLC analysis showed formation of a new compound that was obtained pure directly after removal of the solvent. NMR and LC-MS confirmed the formation of compound 59 (19 mg, 64%, two-step yield): 1H NMR (MeOD, 400 Hz) δ 7.85 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 8.3 Hz, 1H), 6.27 (d, J = 8.2 Hz, 1H), 5.88 (d, J = 4.6 Hz, 1H), 5.06 (s, 2H), 4.10–4.16 (m, 2H), 3.95 (d, J = 3.1 Hz, 1H), 3.77–3.82 (m, 1H), 3.67–3.73 (m, 1H), 3.18 (s, 3H); HRMS calcd for C18H22N4O7 (M + H)+ 407.1567, found 407.1567.
General Procedure for the Synthesis of Derivatives 60 and 61 by Amination of Methyl Ester 56.
A solution of compound 56 (20 mg, 0.047 mmol; 50 mg, 0.119 mmol) in the required amine (ethylene diamine for compound 60 or 1,4-diaminobutine for compound 61, 2 mL) was stirred at rt for 2 h. LC-MS analysis showed full conversion to the amide product. The solvent was removed under reduced pressure, and the so obtained crude mixture directly used for the next step.
Synthesis of the β-Alanine-ethyl Ester Derivative 62.
A mixture of compound 57 (50 mg, 0.2 mmol), β-alanine-ethyl ester hydrochloride (18 mg, 0.2 mmol), HATU (68 mg, 0.2 mmol), and DIPEA (32 μL, 0.2 mmol) in DMF (4 mL) was stirred at rt for 12 h. Next, the solvent was removed, and the crude product purified by a silica gel column (2% methanol in dichloromethane) to afford the pure compound as a white powder (60 mg, quantitative): 1H NMR (MeOD, 400 Hz) δ 7.77 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.3 Hz, 1H), 6.25 (d, J = 8.3 Hz, 1H), 5.88 (d, J = 4.7 Hz, 1H), 5.04 (s, 2H), 4.10–4.17 (m, 4H), 3.96 (d, J = 3.2 Hz, 1H), 3.81–3.82 (m, 1H), 3.78–3.79 (m, 1H), 3.62–3.69 (m, 2H), 3.15 (s, 3H), 2.65 (t, J = 7.4 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H); HRMS calcd for C23H31N4O9 (M + H)+ 507.2091, found 507.2093.
Synthesis of the β-Alanine Derivatives 63.
Compound 62 (40 mg, 0.1 mmol) was dissolved in a mixture of methanol (1 mL) and a 1 N NaOH water solution (1 mL), and the mixture was stirred at rt for 12 h. Next, phosphate buffer (pH 7.4) was added to neutralize the mixture. The solvent was removed, and the crude product dissolved in MeOH with formation of a precipitate that was removed by filtration. The collected filtrate was dried under reduced pressure to afford the desired compound as a white solid (38 mg, 92%): 1H NMR (MeOD, 400 Hz) δ 7.78 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.3 Hz, 1H), 6.26 (d, J = 8.3 Hz, 1H), 5.88 (d, J = 4.7 Hz, 1H), 5.04 (s, 2H), 4.13 (s, 2H), 3.96 (d, J = 3.2 Hz, 1H), 3.78–3.81 (m, 1H), 3.72–3.73 (m, 1H), 3.61–3.64 (m, 2H), 3.16 (s, 3H), 2.62 (t, J = 6.7 Hz, 2H): HRMS calcd for C21H27N4O9 (M + H)+ 479.1778, found 479.1777.
(2R,3S,4R,5S)-1-(Acetoxymethyl)-4-[2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl]-1λ5-bicyclo[3.1.0]hexane-2,3-diyl Diacetate (65).
Compound 64 was prepared following the reported procedure.3,4 A mixture of compound 64 (100 mg, 0.4 mmol), acetic anhydride (148 μL, 1.6 mmol), triethylamine (219 μL, 1.6 mmol), and DMAP (24 mg, 0.2 mmol) in acetonitrile (3 mL) was stirred at rt for 2 h. The solvent was removed under reduced pressure, and the residue purified by a silica gel column (50% ethyl acetate in hexane) to afford 100.0 mg of the pure compound as a white solid (67%): 1H NMR (CDCl3, 400 Hz) δ 9.95 (s, 1H), 7.36 (d, J = 7.9 Hz, 1H), 5.67 (dd, J = 7.7, 12.9 Hz, 2H), 5.15 (d, J = 7.2 Hz, 1H), 4.58 (s, 1H), 4.34 (d, J = 12.0 Hz, 1H), 3.96 (d, J = 12.0 Hz, 1H), 2.04 (s, 3H), 1.99 (s, 6H), 1.47 (dd, J = 3.8, 8.4 Hz, 1H), 1.21 (dd, J = 5.3, 9.6 Hz, 2H), 0.95 (t, J = 7.0 Hz, 1H); HRMS calcd for C17H22N2O8 (M + H)+ 382.1381, found 382.1384.
(2R,3S,4R,5S)-1-(Acetoxymethyl)-4-(4-{[(4-chlorobenzyl)oxy]-imino}-2-oxo-3,4-dihydropyrimidin-1(2H)-yl)-1λ5-bicyclo[3.1.0]-hexane-2,3-diyl Diacetate (66).
Compound 66 was synthesized following the reported procedure33 starting from compound 65 and O-4-chlorobenzylhydroxylamine hydrochloride (38). Purification was performed by a silica gel column (50% ethyl acetate in hexane) to afford 36 mg of the pure product as a white powder (26% yield for two steps): 1H NMR (CDCl3, 400 Hz) δ 8.13 (s, 1H), 7.30 (dd, J = 8.3, 19.6 Hz, 4H), 6.77 (d, J = 8.1 Hz, 1H), 5.65 (d, J = 6.3 Hz, 1H), 5.56 (dd, J = 2.0, 8.1 Hz, 1H), 5.14 (d, J = 7.1 Hz, 1H), 4.97 (s, 2H), 4.62 (s, 1H), 4.42 (d, J = 12.0 Hz, 1H), 3.90 (d, J = 12.0 Hz, 1H), 2.07 (s, 3H), 2.04 (s, 6H), 1.42 (dd, J = 4.0, 8.7 Hz, 1H), 1.22–1.29 (m, 2H), 0.94 (t, J = 6.8 Hz, 1H); HRMS calcd for C24H28ClN3O8 (M + H)+ 520.1487, found 520.1481.
1-(Acetoxymethyl)-4-(4-{[(4-chlorobenzyl)oxy]imino}-3-methyl-2-oxo-3,4-dihydropyrimidin-1(2H)-yl)-1λ5-bicyclo[3.1.0]hexane-2,3-diyl Diacetate (67).
To a mixture of compound 66 (36.0 mg, 0.07 mmol) and K2CO3 (15 mg, 0.10 mmol) in N,N-dimethylacetamide (DMAc, 2.0 mL) was added methyl iodide (13 μL, 0.20 mmol), and the mixture was stirred at rt for 18 h. The solvent was removed under reduced pressure, and the resulting crude mixture purified by a silica gel column (50% ethyl acetate in hexane) to afford 31 mg of the pure product (83%): 1H NMR (CDCl3, 400 Hz) δ 7.30 (s, 4H), 6.80 (d, J = 8.2 Hz, 1H), 6.16 (d, J = 8.2 Hz, 1H), 5.69 (d, J = 7.2 Hz, 1H), 5.14 (d, J = 7.2 Hz, 1H), 4.96 (s, 2H), 4.58 (s, 1H), 4.41 (d, J = 12.0 Hz, 1H), 3.92 (d, J = 12.0 Hz, 1H), 3.19 (s, 3H), 2.10 (s, 3H), 2.04 (s, 6H), 1.42 (dd, J = 4.0, 8.7 Hz, 1H), 1.25 (t, J = 4.9 Hz, 2H), 0.94 (t, J = 7.3 Hz, 1H); HRMS calcd for C25H30ClN3O8 (M + H)+ 534.1643, found 534.1647.
4-{[(4-Chlorobenzyl)oxy]imino}-1-[(1S,2R,3S,4R)-3,4-dihydroxy-(hydroxymethyl)-5λ5-bicyclo[3.1.0]hexan-2-yl]-3-methyl-3,4-di-hydropyrimidin-2(1H)-one (68).
A solution of compound 67 (30.7 mg, 0.06 mmol) in a NH3 methanol solution (2.0 mL) was stirred at rt for 18 h. The solvent was removed, and the crude purified by a silica gel column (5% methanol in dichloromethane) to afford 21 mg of the pure product (91%): 1H NMR (MeOD, 400 Hz) δ 7.38 (d, J = 8.2 Hz, 1H), 7.32 (d, J = 1.0 Hz, 4H), 6.17 (d, J = 8.2 Hz, 1H), 4.95 (s, 2H), 4.65 (s, 1H), 4.55 (d, J = 6.7 Hz, 1H), 4.16 (d, J = 11.7 Hz, 1H), 3.75 (d, J = 6.7 Hz, 1H), 3.23 (d, J = 11.7 Hz, 1H), 3.17 (s, 3H), 26–1.33 (m, 2H), 0.64 (t, J = 3.3 Hz, 1H); HRMS calcd for C19H24ClN3O5 (M + H)+ 408.1326, found 408.1330.
Synthesis of Compound 70 by Acylation of Cytidine.
A mixture of cytidine (200 mg, 0.8 mmol), acetic anhydride (310 μL, 3.3 mmol), triethylamine (458 μL, 3.3 mmol), and DMAP (50 mg, 0.4 mmol) in acetonitrile (4 mL) was stirred at rt for 10 min with formation of both tri-O-acetyl-cytidine and tetra-O,N4-acetyl-cytidine. The solvent was removed under reduced pressure, and the two derivatives were separated by a silica gel column (10% methanol in dichlomethane). Next, the tetra-O,N4-acetyl-cytidine was dissolved in MeOH (3 mL) and heated at 105 °C for 3 h in a microwave reactor with full conversion of the latter to tri-O-acetyl-cytidine (purified by the same method used previously).5 The total yield for two steps was 95% (289 mg).
tri-O-Acetyl-3-methylcytidine (71).
Compound 71 was synthesized as reported above for the 3-methylcytidine, starting from compound 70: 1H NMR (CDCl3, 400 Hz) δ 7.4 (s, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.00 (d, J = 8.0 Hz, 1H), 5.95 (d, J = 4.7 Hz, 1H), 5.27–5.36 (m, 2H), 4.29–4.33 (m, 3H), 3.44 (s, 3H), 2.12 (s, 3H), 2.08 (t, J = 4.9 Hz, 6H); HRMS calcd for C16H23N3O8 (M + H)+ 384.1407, found 384.1410.
tri-O-Acetyl-4-[(benzylcarbamoyl)imino]-3-methylcytidine (72).
N-Benzyl-1H-imidazole-1-carboxamide (69) was synthesized as reported previously.38 A solution of compound 71 (73 mg, 0.2 mmol), compound 69 (77 mg, 0.4 mmol), and triethylamine (27 μL, 0.2 mmol) in THF (3 mL) was stirred at reflux for 12 h. The solvent was removed under reduced pressure, and the crude mixture purified by a silica gel column (2% methanol in dichloromethane) to afford the pure product as a white solid (72 mg, 72%): 1H NMR (CDCl3, 400 Hz) δ 7.26–7.33 (m, 5H), 7.13 (d, J = 8.2 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 6.00 (d, J = 4.4 Hz, 1H), 5.27–5.36 (m, 2H), 4.45 (d, J = 5.9 Hz, 2H), 4.32 (s, 3H), 3.33 (s, 3H), 2.12 (s, 3H), 2.09 (s, 3H), 2.08 (s, 3H); HRMS calcd for C24H29N4O9 (M + H)+ 517.1935, found 517.1940.
4-[(Benzylcarbamoyl)imino]-3-methylcytidine (73).
A suspension of compound 72 (72 mg, 0.1 mmol) in a methanolic ammonia solution (2 mL) was stirred at rt for 12 h. The solvent was removed, and the crude mixture purified by a silica gel column (7% methanol in dichloromethane) to afford the pure product as a white solid (47 mg, 87%): 1H NMR (MeOD, 400 Hz) δ 7.72 (d, J = 8.2 Hz, 1H),7.22–7.33 (m, 5H), 6.18 (d, J = 8.2 Hz, 1H), 5.90 (d, J = 3.7 Hz, 1H), 4.38 (s, 2H), 4.12 (dd, J = 4.4, 8.2 Hz, 2H), 3.98 (d, J = 3.0 Hz, 1H), 3.83 (dd, J = 2.8, 12.2 Hz, 1H), 3.71 (dd, J = 2.8, 12.2 Hz, 1H), 3.35 (s, 3H); HRMS calcd for C18H23N4O6 (M + H)+ 391.1618, found 391.1616.
Tissue Collection.
A human tumor was obtained from a 44-year-old male patient with squamous cell carcinoma of mobile tongue (oral cavity, T3N2cM0, grade II) undergoing surgery at the Department of Otorhinolaryngology-Head and Neck Surgery of Turku University Hospital. The collection of the tissues is part of an ongoing sample collection performed under the license ETMK 166/2015 with written informed consent from the patients. Palatine tonsils were obtained from an adult patient with chronic tonsillitis undergoing routine tonsillectomy at Turku University Hospital (permission TO6/033/18 from the Hospital Research Council). The excised tumor and tonsil samples were embedded in the cryo-mold with Tissue-Tek O.C.T. compound (Sakura Finetek Europe B.V., Alphen aan den Rijn, The Netherlands), cut at 10 μm onto superfrost glass slides using a cryostat, and stored at −80 °C.
Immunofluorescence Staining.
Tumor cryosections were processed for the immunofluorescence analysis of CD73 expression, as described elsewhere.48 Briefly, the slides were incubated overnight at 4 °C in a Shandon Sequenza Staining System (Thermo Scientific) with the rabbit anti-human CD73 antibody (h5NT-1L, http://ectonucleotidases-ab.com/) diluted at 1/400 in 200 μL of PBS containing 2% bovine serum albumin (BSA) and 0.1% (v/v) Triton X-100 (blocking buffer). The samples were washed and subsequently incubated for 2 h at rt with the Alexa Fluor-546-conjugated goat anti-rabbit antibody (ThermoFisher Life Technologies), diluted 1/800 in blocking buffer. The Alexa Fluor 488-conjugated pan-cytokeratin monoclonal antibody (catalogue no. MA5-18156, ThermoFisher) was added during the incubation with the secondary antibody for labeling the well-differentiated epithelial tumor cells. The slides were mounted with a ProLong Gold Antifade reagent with DAPI (ThermoFisher) and imaged using a Pannoramic Midi FL Slide Scanner (3DHistech Ltd., Budapest, Hungary) with a 20× objective.
In Situ ecto-5′-Nucleotidase Activity Assay.
For localization of AMPase activities in human tumors and tonsils, the lead nitrate-based enzyme histochemistry was employed.28,49 In brief, tissue cryosections were preincubated for 60 min at rt in 40 mmol/L Trizma-maleate buffer (TMB, pH 7.3) supplemented with 250 mmol/L sucrose, the alkaline phosphatase inhibitor levamisole (2 mmol/L), and different concentrations of CD73 inhibitors. The enzymatic reaction was then performed for 45 min at 37 °C in a final volume of 20 mL of TMB containing 250 mmol/L sucrose, 2 mmol/L levamisole, 1.5 mmol/L Pb(NO3)2, 1 mmol/L CaCl2 100 μmol/L AMP, and tested CD73 inhibitors at the same concentrations as during the preincubation step. The lead orthophosphate precipitated over the course of nucleotidase activity was visualized as a brown deposit by incubating sections in 0.5% (NH4)2S for 10 s, followed by three washes in TMB for 5 min each. Slides were mounted with Aquatex medium (Merck). Tissue sections were also stained with H&E. Whole slide imaging was performed using a Pannoramic-250 Flash slide scanner (3DHistech Ltd.) with a 20× objective. AMPase activity was determined by measuring AMP-specific brown staining intensities from the images using QuPath version 0.2.3.52 Shortly, a project including all images was created. Tissue areas were detected using the threshold classifier. The classifier was run with full resolution (240 nm/pixel) for the whole project. The DAB channel and Gaussian prefiltering were selected. The threshold was set to 1.15 without smoothing, and areas above the threshold were classified as “positive”. Representative areas were manually selected, and the average DAB intensity level was used for AMPase intensity quantification. The script for intensity analysis is shown in Table S3.
X-ray Crystallographic Studies.
For the crystallographic experiments, two constructs of human CD73 were used. These constructs were generated by gene synthesis of the natural signal peptide and the coding region of human CD73 with several variations as indicated in Figures S1 and S2. The synthesized gene included the residues up to the linkers highlighted in green. The remaining C-terminal residues were coded by the expression vectors, pHLsec for the 8.01His construct and pHLsec_StrepII for the 8.1Strep construct, using the EcoRI and KpnI restriction sites.53 For small-scale expression tests, HEK293S GntI− cells were grown in an adherent culture in 24-well TPP plates at 310 K in a humidified atmosphere containing 5% CO2. The cell viability was supported by Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (Gibco), 1% Glutmax (Gibco), and 1% non-essential amino acids (Gibco). The cells were transfected with the expression plasmid after growth to 90% confluency using polyethylenimine (Sigma-Aldrich) as a transfection reagent in a 1.5-fold excess (w/w) with respect to plasmid DNA. For preparative expression, cells from one confluent T175 bottle (Greiner) were seeded into roller bottles (Greiner) with a surface of 2125 cm2 per bottle for 5 days before transfection. The roller bottles were incubated at 310 K with a rotation speed of 0.9 rpm and filled with 250 mL of medium per bottle.
Both constructs were purified using similar procedures as follows. The culture supernatant (1–2 L) was collected 5 days after transfection and separated from detached cells by centrifugation and filtration using a 0.22 μm bottle-top filter (TPP). To prepare the supernatant for purification by immobilized metal affinity chromatography (construct 8.01His), the buffer was exchanged via an ultrafiltration unit (10 kDa molecular weight cutoff, GE Healthcare) using 5 L of buffer A [50 mM Tris and 400 mM NaCl (pH 8.0)]. The resulting protein solution (volume of ~100 mL) was adjusted to 20 mM imidazole by adding elution buffer B [50 mM Tris, 400 mM NaCl, and 500 mM imidazole (pH 8.0)]. After this, the protein solution was loaded onto a HisTrap HP column (1 mL volume, GE Healthcare), washed with 30 mL of 50 mM imidazole in buffer A, and eluted with a linear gradient from buffer A to buffer B over 5 mL. For purification of the 8.1Strep construct, the supernatant of the roller bottle expression was also concentrated to a volume of ~100 mL by ultrafiltration. Then 0.6 mL of a DMEM/biotin blocking solution was added, and the solution was centrifuged at 20000g or filtered through a 0.45 μm filter. The protein was applied in a buffer of 50 mM Tris (pH 8.0) and 150 mM NaCl to a 5 mL StrepTrap HP column (GE Healthcare) and eluted with the same buffer containing 2.5 mM desthiobiotin. After affinity chromatography, fractions containing CD73 were pooled and concentrated to 2 mL using Amicon Ultra centrifugal filter units (10 kDa molecular weight cutoff, Merck Millipore) and further purified on a Superdex 200 16/60 gel-filtration column (120 mL volume, GE Healthcare) using a buffer consisting of 50 mM Tris (pH 8.0) and 100 mM NaCl. Pure protein fractions were pooled, concentrated to 5–10 mg mL−1, and stored at −80 °C in aliquots for further use.
For crystallization, vapor diffusion trials were set up at 19 °C. One microliter of the protein solution containing the respective inhibitor (1 mM) and ZnCl2 (100 μM) was mixed with an equal amount of a reservoir buffer solution to give the concentrations listed in Table S1. Crystals were transferred in a stepwise manner to a cryo buffer composed as reported in Table 2 and Table S1 and finally flash-frozen in liquid nitrogen.
X-ray data collection was carried out at 100 K as specified in Table S1. Data were collected at a Rigaku Synergy R home source and at the BESSY beamlines BL14.1 and BL14.2. The diffraction data were indexed, integrated, and scaled with XDS.54 Coordinates of human CD73 in the closed conformation (PDB entry 4H2I) or in the open conformation (PDB entry 4H2G) were taken as starting models for rigid body refinement. In addition, the pipedream software was used for automatic refinement and ligand placement.55 The models were further improved by iterative cycles of TLS refinement using BUSTER-TNT55 and manual rebuilding with COOT.56 Restraint dictionaries for inhibitors were generated using the GRADE web server (http://grade.globalphasing.org). Relevant crystallographic parameters of the structures are listed in Table S1. Figures were prepared using PYMOL (http://pymol.org).
Molecular Modeling.
Protein Preparation.
The X-ray structures of the complexes between hCD73 and compounds 16 and 21 were prepared using the Protein Preparation Wizard tool of the Schrödinger suite (Maestro 2020-1).57,58 Missing residues (residues 376–382 and 374–382) were built using Prime (knowledge-based method),59,60 used also to revert the Asn residues at positions 53, 311, 333, and 403 into wild-type Asp. The following protomeric and tautomeric states were predicted for His residues: hydrogen at Nδ for His38, His118, His220, His304, His383, and His440; hydrogen at Nε for His33, His103, His243, His437, His456, and His518; and hydrogens at Nδ and Nε for His375. The protein was capped at the N-terminal position with an acetyl group (ACE) and at the C-terminal position with an N-methyl amide (NME). The two catalytic zinc ions and the calcium ion present in the X-ray structures were maintained during the simulations. The ligands were monoprotonated at the β-phosphonate.
Docking.
Compounds 24 and 27 were drawn using the Schrodinger suite (Maestro 2020-1)57 starting from compound 21 and then minimized using the OPLS3 force field and implicit solvent (water).61 The compounds were docked to the hCD73 structure prepared starting from the X-ray complex with compound 21. Glide XP62 was used for docking, employing a grid centered on compound 21, with an inner box of 10 Å and an outer box of 30 Å. Maximum 20 poses were generated for each compound, and a postdocking minimization of the ligand was included in the docking protocol. The poses with the highest score were selected.
Molecular Dynamics Simulations.
The systems for molecular dynamics (MD) simulations were prepared using AmberTools20.63 Antechamber64 was used to assign AM1-BCC65 charges and atom types to the ligands. Few modifications were manually introduced to the atom types: cd, cd, and cc were assigned to carbon atoms at positions 4, 5 and 6 of the pyrimidine ring, respectively, and op, oq, and or atom types were adopted for deprotonated and protonated phosphonate oxygens and the bridging 5′-oxygens.66
The complexes were inserted in a cubic water box (TIP3P67 explicit solvent) with a distance of at least 30 Å between any atom and the edge of the box, and the systems were neutralized with NaCl (concentration of 0.15 M), by using tleap (AmberTools20). The Amber ff14SB68 and GAFF69 force fields were used for the simulations, and the MD simulations were carried out with Acemd3.70
After 500 steps of minimization, each system was subjected to an equilibration stage of 4 ns in the NPT ensemble, with atomic restraints on protein and ligand atoms in the first 3 ns of the simulation. Specifically, restraints of 1 kcal mol−1 Å−2 were applied and gradually decreased (in the case of protein Cα atoms, the restraints were kept fixed to 1 kcal mol−1 Å−2 in the first nanosecond and subsequently gradually decreased from 1 to 3 ns). Starting from each equilibrated system, three 50 ns simulations were collected in the NVT ensemble. A Langevin thermostat was used to keep the temperature at 310 K during the simulations (damping constants of 1 and 0.1 ps−1 for equilibration and production, respectively), and a Monte Carlo barostat was employed to keep the pressure at 1 atm during the equilibration phase. In all of the simulations, a 9 Å cutoff was used for nonbonded interactions, with a switching distance of 7.5 Å for van der Waals interactions, and the particle-mesh Ewald (PME)71 summation was enabled for long-range electrostatic interactions. A time step of 2 fs was adopted, and simulations were saved with a stride of 20 ps (or of 50 ps, in the case of equilibration simulations).
The simulations were carried out on the NIH HPC Biowulf cluster (http://hpc.nih.gov, accessed May/June 15, 2021) using Tesla P100 GPUs.
Trajectory Analysis.
Before the RMSD of the ligand (or ligand nucleotide-like scaffold) heavy atoms was measured, each trajectory was aligned superposing the protein Cα atoms on the starting conformation. Calculation of the RMSFs of the ligand heavy atoms was performed using the average trajectory structure as reference. RMSD and RMSF analysis was conducted with Wordom.47
For the cluster analysis, the three replicates of each system were sampled every 200 ps, aligned superposing the ligand nucleotide-like scaffold heavy atoms, and the RMSD of the para-substituted benzyl moiety was used to build the distance matrix. Wordom’s QT-like method with a 2 Å cutoff was employed for clustering analysis.
Contacts and H-bonds were computed with an in-house script written in Tcl, employing VMD 1.9.3.72 Contacts were calculated as the presence of any residue atom within 4 Å of any ligand atom. H-Bonds were computed using the H-bonds plugin, applying a donor–acceptor distance of 3.5 Å and a donor─H─acceptor angle of 30°.
Puckering analysis was accomplished with an in-house python script, with minor modification of what was reported previously.37,48
Averages and standard deviations were computed through a python script employing Pandas functions.73 Plots were generated using matplotlib74 and gnuplot.75
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. John Lloyd [National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)] for mass spectral determinations, Prof. Jean Sevigny for the antibody against human CD73, Prof. Dr. Henrik Ditzel for MDA-MB-231, and Prof. Dr. Michael Hölzel for MaMel.65-CD73ko cells. The authors acknowledge support from the NIDDK Intramural Research Program (ZIADK031116). The authors thank Bryan L. Roth, National Institute of Mental Health’s Psychoactive Drug Screening Program (The University of North Carolina at Chapel Hill, Contract HHSN-271-2008-00025-C), for screening data. The authors also thank the Biocenter Finland and Cell Imaging and Cytometry Core of Turku Bioscience Centre for imaging instrumentation. Financial support of A.J. by the Deutsche Forschungsgemeinschaft (German Research Foundation, JU 2966/2-1) and the Cells-in-Motion (CiM) cluster of excellence (Münster, Germany) is gratefully acknowledged. We thank the MX Laboratory at the Helmholtz Zentrum Berlin (BESSY II) for beam time as well as for travel support. We also acknowledge the EMBL beamlines of the DESY synchrotron in Hamburg for beamtime.
ABBREVIATIONS USED
- AR
adenosine receptor
- ACN
acetonitrile
- AOPCP
α,β-methylene-ADP, adenosine-5′-O-[(phosphonomethyl)-phosphonic acid], {[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxo-lan-2-yl]methoxyhydroxyphosphoryl}methylphosphonic acid
- BisTris
2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)-propane-1,3-diol
- CD73
cluster of differentiation 73
- CE
capillary electrophoresis
- CMP
cytidine 5′-monophosphate
- DAB
3,3′-diaminobenzidine
- DAPI
4,6-diamidino-2-phenylindole
- DIPEA
diisopropylethylamine
- DMAc
N,N-dimethylacetamide
- DMEM
Dulbecco’s modified Eagle’s medium
- EC
enzyme commission
- eN
ecto-5′-nucleotidase
- eNPP
ecto-nucleoside pyrophosphatases/phosphodiesterase
- eNTPDase
ecto-nucleoside triphosphate diphosphohydrolase
- FBS
fetal bovine serum
- GPI
glycophosphatidylinositol
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]-pyridinium 3-oxide hexafluorophosphate
- H&E
hematoxylin and eosin
- HNSCC
head and neck squamous cell carcinoma
- MES
2-(N-morpholino)ethanesulfonic acid
- MOE
Molecular Operating Environment
- NK cells
natural killer cells
- PD-L1
programmed death ligand 1
- PDSP
Psychoactive Drug Screening Program
- POM
polyoxometalate
- PSB
Pharmaceutical Sciences Bonn
- RMSF
root-mean-square fluctuation
- SF9
Spodoptera frugiperda 9
- TEA
triethylamine
- TEAC
triethylammonium hydrogen carbonate
- TMB
Trizma-maleate buffer
- TME
tumor microenvironment
- TPSCl
2,4,6-triisopropylbenzenesulfonyl chloride
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01852.
1H NMR spectra of all final compounds, details of the X-ray crystallographic studies, molecular modeling procedures and figures, and key to video and PDB files (PDF)
Video S1 (AVI)
Molecular strings file (CSV)
Protein Data Bank file with atomic coordinates of the model of 18 bound to hCD73 (PDB)
The authors declare no competing financial interest.
Contributor Information
Mirko Scortichini, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Riham Mohammed Idris, PharmaCenter Bonn, Pharmaceutical Institute, Pharmaceutical Chemistry I, University of Bonn, D-53121 Bonn, Germany.
Susanne Moschütz, Center for Biotechnology and Biomedicine, Leipzig University, 04103 Leipzig, Germany.
Antje Keim, Center for Biotechnology and Biomedicine, Leipzig University, 04103 Leipzig, Germany.
Veronica Salmaso, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Clemens Dobelmann, European Institute for Molecular Imaging (EIMI), University of Münster, D-48149 Münster, Germany.
Paola Oliva, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Karolina Losenkova, Medicity Research Laboratory, University of Turku, 20520 Turku, Finland.
Heikki Irjala, Department of Otorhinolaryngology-Head and Neck Surgery, Turku University Hospital and Turku University, 20520 Turku, Finland.
Samuli Vaittinen, Department of Pathology, Turku University Hospital and Turku University, 20520 Turku, Finland.
Jouko Sandholm, Turku Bioscience Centre, University of Turku and Åbo Akademi University, 20520 Turku, Finland.
Gennady G. Yegutkin, Medicity Research Laboratory, University of Turku, 20520 Turku, Finland.
Norbert Sträter, Center for Biotechnology and Biomedicine, Leipzig University, 04103 Leipzig, Germany.
Anna Junker, European Institute for Molecular Imaging (EIMI), University of Münster, D-48149 Münster, Germany.
Christa E. Müller, PharmaCenter Bonn, Pharmaceutical Institute, Pharmaceutical Chemistry I, University of Bonn, D-53121 Bonn, Germany.
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
REFERENCES
- (1).Vigano S; Alatzoglou D; Irving M; Ménétrier-Caux C; Caux C; Romero P; Coukos G Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol 2019, 10, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Boison D; Yegutkin GG Adenosine metabolism: Emerging concepts for cancer therapy. Cancer Cell 2019, 36, 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Allard B; Allard D; Buisseret L; Stagg J The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol 2020, 17, 611–629. [DOI] [PubMed] [Google Scholar]
- (4).Chen L; Diao L; Yang Y; Yi Y; Rodriguez BL; Li Y; Villalobos PA; Cascone T; Liu X; Tan L; Lorenzi PL; Huang A; Zhao Q; Peng D; Fradette JJ; Peng DH; Ungewiss C; Roybal J; Tong P; Oba J; Skoulidis F; Peng W; Carter BW; Gay CM; Fan Y; Class CA; Zhu J; Rodriguez-Canales J; Kawakami M; Byers LA; Woodman SE; Papadimitrakopoulou VA; Dmitrovsky E; Wang J; Ullrich SE; Wistuba II; Heymach JV; Qin X-F; Gibbons DL CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discovery 2018, 8, 1156–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hay CM; Sult E; Huang Q; Mulgrew K; Fuhrmann SR; McGlinchey KA; Hammond SA; Rothstein R; Rios-Doria J; Poon E; Holoweckyj N; Durham NM; Leow CC; Diedrich G; Damschroder M; Herbst R; Hollingsworth RE; Sachsenmeier KF Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jadidi-Niaragh F Potential of CD73 as a target for cancer immunotherapy. Immunotherapy 2019, 11 (16), 1353–1355. [DOI] [PubMed] [Google Scholar]
- (7).Luke JJ; Powderly JD; Merchan JR; Barve MA; Hotson AN; Mobasher M; Kwei L; Luciano G; Buggy JJ; Piccione E; Miller RA Immunobiology, preliminary safety, and efficacy of CPI-006, an anti-CD73 antibody with immune modulating activity, in a phase 1 trial in advanced cancers. J. Clin. Oncol 2019, 37, 2505. [Google Scholar]
- (8).Harvey JB; Phan LH; Villarreal OE; Bowser JL CD73’s potential as an immunotherapy target in gastrointestinal cancers. Front. Immunol 2020, 11, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Du X; Moore J; Blank BR; Eksterowicz J; Sutimantanapi D; Yuen N; Metzger T; Chan B; Huang T; Chen X; Chen Y; Duong F; Kong W; Chang JH; Sun J; Zavorotinskaya T; Ye Q; Junttila MR; Ndubaku C; Friedman LS; Fantin VR; Sun D Orally bioavailable small-molecule CD73 inhibitor (OP-5244) reverses immunosuppression through blockade of adenosine production. J. Med. Chem 2020, 63 (18), 10433–10459. [DOI] [PubMed] [Google Scholar]; (b) Bowman CE; da Silva RG; Pham A; Young SW An exceptionally potent inhibitor of human CD73. Biochemistry 2019, 58, 3331–3334. [DOI] [PubMed] [Google Scholar]; (c) Chen L; Li J; Sjogren EB; Billedeau RJ Ectonucleotidase inhibitors and methods of use thereof. U.S. Patent US10,472,364 B2, 2019.
- (10).Jeffrey JL; Lawson KV; Powers JP Targeting metabolism of extracellular nucleotides via inhibition of ecto-nucleotidases CD73 and CD39. J. Med. Chem 2020, 63, 13444–13465. [DOI] [PubMed] [Google Scholar]
- (11).Lawson KV; Kalisiak J; Lindsey EA; Newcomb ET; Leleti MR; Debien L; Rosen BR; Miles DH; Sharif EU; Jeffrey JL; Tan JBL; Chen A; Zhao S; Xu G; Fu L; Jin L; Park TW; Berry W; Moschütz S; Scaletti E; Sträter N; Walker NP; Young SW; Walters MJ; Schindler U; Powers JP Discovery of AB680: A potent and selective inhibitor of CD73. J. Med. Chem 2020, 63, 11448–11468. [DOI] [PubMed] [Google Scholar]
- (12).Koszałka P; Gołuńska M; Urban A; Stasiłojć G; Stanisławowski M; Majewski M; Składanowski AC; Bigda J Specific activation of A3, A2A and A1 adenosine receptors in CD73-Knockout mice affects B16F10 melanoma growth, neovascularization, angiogenesis and macrophage infiltration. PLoS One 2016, 11 (3), e0151420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Minor M; Alcedo KP; Battaglia RA; Snider NT Cell type- and tissue-specific functions of ecto-5′-nucleotidase (CD73). Am. J. Physiol. Cell. Physiol 2019, 317, C1079–C1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hilaire C; Ziegler SG; Markello TC; Brusco A; Groden C; Gill F; Carlson-Donohoe H; Lederman RJ; Chen MY; Yang D; Siegenthaler MP; Arduino C; Mancini C; Freudenthal B; Stanescu HC; Zdebik AA; Chaganti RK; Nussbaum RL; Kleta R; Gahl WA; Boehm M NT5E mutations and arterial calcifications. N. Engl. J. Med 2011, 364, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shih Y-RV; Liu M; Kwon SK; Iida M; Gong Y; Sangaj N; Varghese S Dysregulation of ectonucleotidase-mediated extracellular adenosine during postmenopausal bone loss. Science Advances 2019, 5 (8), eaax1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nedeljkovic N Complex regulation of ecto-5′-nucleotidase/CD73 and A2AR-mediated adenosine signaling at neurovascular unit: a link between acute and chronic neuroinflammation. Pharmacol. Res 2019, 144, 99–115. [DOI] [PubMed] [Google Scholar]
- (17).Carmo M; Gonçalves FQ; Canas PM; Oses J-P; Fernandes FD; Duarte FV; Palmeira CM; Tomé AR; Agostinho P; Andrade GM; Cunha RA Enhanced ATP release and CD73-mediated adenosine formation sustain adenosine A2A receptor over-activation in a rat model of Parkinson’s disease. Br. J. Pharmacol 2019, 176 (18), 3666–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Barros-Barbosa AR; Ferreirinha F; Oliveira A; Mendes M; Lobo MG; Santos A; Rangel R; Pelletier J; Sévigny J; Cordeiro JM; Correia-de-Sá P Adenosine A2A receptor and ecto-5′-nucleotidase/CD73 are upregulated in hippocampal astrocytes of human patients with mesial temporal lobe epilepsy (MTLE). Purinergic Signal. 2016, 12, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Meng F; Guo Z; Hu Y; Mai W; Zhang Z; Zhang B; Ge Q; Lou H; Guo F; Chen J; Duan S; Gao Z CD73-derived adenosine controls inflammation and neurodegeneration by modulating dopamine signalling. Brain. 2019, 142 (3), 700–718. [DOI] [PubMed] [Google Scholar]
- (20).Ponce NE; Sanmarco LM; Eberhardt N; García MC; Rivarola HW; Cano RC; Aoki MP CD73 inhibition shifts cardiac macrophage polarization toward a microbicidal phenotype and ameliorates the outcome of experimental Chagas cardiomyopathy. J. Immunol 2016, 197, 814–823. [DOI] [PubMed] [Google Scholar]
- (21).De Leve S; Wirsdörfer F; Jendrossek V The CD73/Ado System—A new player in RT induced adverse late effects. Cancers 2019, 11, 1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Heilbronn A; Maienschein V; Carstensen K; Gann W; Zimmermann H Crucial role of ecto-5′-nucleotidase in differentiation and survival of developing neural cells. NeuroReport 1995, 7, 257–261. [PubMed] [Google Scholar]
- (23).Badimon A; Strasburger HJ; Ayata P; Chen X; Nair A; Ikegami A; Hwang P; Chan AT; Graves SM; Uweru JO; Ledderose C; Kutlu MG; Wheeler MA; Kahan A; Ishikawa M; Wang YC; Loh YE; Jiang JX; Surmeier DJ; Robson SC; Junger WG; Sebra R; Calipari ES; Kenny PJ; Eyo UB; Colonna M; Quintana FJ; Wake H; Gradinaru V; Schaefer A Negative feedback control of neuronal activity by microglia. Nature 2020, 586 (7829), 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Joolharzadeh P; Hilaire C CD73 (Cluster of Differentiation 73) and the differences between mice and humans. Arterioscler., Thromb., Vasc. Biol 2019, 39 (3), 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bhattarai S; Pippel J; Scaletti E; Idris R; Freundlieb M; Rolshoven G; Renn C; Lee SY; Abdelrahman A; Zimmermann H; El-Tayeb A; Müller CE; Sträter N 2-Substituted α,β-methylene-ADP derivatives: Potent competitive ecto-5′-nucleotidase (CD73) inhibitors with variable binding modes. J. Med. Chem 2020, 63 (6), 2941–2957. [DOI] [PubMed] [Google Scholar]
- (26).Bhattarai S; Freundlieb M; Pippel J; Meyer A; Abdelrahman A; Fiene A; Lee S-Y; Zimmermann H; Yegutkin GG; Sträter N; El-Tayeb A; Müller CE α,β-Methylene-ADP (AOPCP) derivatives and analogues: development of potent and selective ecto-5′-nucleotidase (CD73) inhibitors. J. Med. Chem 2015, 58, 6248–6263. [DOI] [PubMed] [Google Scholar]
- (27).Bhattarai S; Pippel J; Meyer A; Freundlieb M; Schmies C; Abdelrahman A; Fiene A; Lee SY; Zimmermann H; El-Tayeb A; Yegutkin GG; Sträter N; Müller CE X-Ray co-crystal structure guides the way to subnanomolar competitive ecto-5′-nucleotidase (CD73) inhibitors for cancer immunotherapy. Adv. Therap 2019, 2 (0), 1900075. [Google Scholar]
- (28).Junker A; Renn C; Dobelmann C; Namasivayam V; Jain S; Losenkova K; Irjala H; Duca S; Balasubramanian R; Chakraborty S; Börgel F; Zimmermann H; Yegutkin GG; Müller CE; Jacobson KA Structure-activity relationship of purine and pyrimidine nucleotides as ecto-5′-nucleotidase (CD73) inhibitors. J. Med. Chem 2019, 62, 3677–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schindler U; Becker A; Lawson K; Jin L; Jeffrey J; Kalisiak j.; Yin F; Zhang K; Chen A; Swinarski D; Walters MJ; Young S; Powers JP; Tan J AB680, a potent and selective CD73 small molecule inhibitor, reverses the AMP/adenosine-mediated impairment of immune effector cell activation by immune checkpoint inhibitors. Abstract P04.03. Eur. J. Cancer 2018, 92, S14. [Google Scholar]
- (30).Ghoteimi R; Nguyen VT; Rahimova R; Grosjean F; Cros-Perrial E; Uttaro JP; Mathé C; Chaloin L; Jordheim LP; Peyrottes S Synthesis of substituted 5′-aminoadenosine derivatives and evaluation of their inhibitory potential toward CD73. ChemMedChem. 2019, 14 (15), 1431–1443. [DOI] [PubMed] [Google Scholar]
- (31).Nocentini A; Capasso C; Supuran CT Small-molecule CD73 inhibitors for the immunotherapy of cancer: a patent and literature review (2017–present). Exp. Opin. Therap. Patents 2021, 31 (10), 867–876. [DOI] [PubMed] [Google Scholar]
- (32).Varano F; Catarzi D; Vincenzi F; Pasquini S; Pelletier J; Lopes Rangel Fietto J; Espindola Gelsleichter N; Sarlandie M; Guilbaud A; Sévigny J; Varani K; Colotta V Structural investigation on thiazolo[5,4-d]pyrimidines to obtain dual-acting blockers of CD73 and adenosine A2A receptor as potential antitumor agents. Bioorg. Med. Chem. Lett 2020, 30 (9), 127067. [DOI] [PubMed] [Google Scholar]
- (33).Beatty JW; Lindsey EA; Thomas-Tran R; Debien L; Mandal D; Jeffrey JL; Tran AT; Fournier J; Jacob SD; Yan X; Drew SL; Ginn E; Chen A; Pham AT; Zhao S; Jin L; Young SW; Walker NP; Leleti MR; Moschütz S; Sträter N; Powers JP; Lawson KV Discovery of potent and selective non-nucleotide small molecule inhibitors of CD73. J. Med. Chem 2020, 63 (8), 3935–3955. [DOI] [PubMed] [Google Scholar]
- (34).Oliva P; Scortichini M; Dobelmann C; Jain S; Gopinatth V; Toti KS; Phung NB; Junker A; Jacobson KA Structure activity relationship of pyrimidine nucleotides containing a 5′-α,β-methylene diphosphonate as P2Y6 receptor agonists. Bioorg. Med. Chem. Lett 2021, 45, 128137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jayasekara PS; Jacobson KA Rapid Synthesis of alkoxyamine hydrochloride derivatives from alkyl bromide and N,N’-di-tert-butoxycarbonylhydroxylamine [(Boc)2NOH]. Synth. Commun 2014, 44, 2344–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jacobson KA; Salmaso V; Suresh RR; Tosh DK Expanding the repertoire of methanocarba nucleosides from purine receptors to diverse targets. RSC Med. Chem 2021, 12, 1808–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Salmaso V; Jacobson KA Survey of ribose ring pucker of signaling nucleosides and nucleotides. Nucleos. Nucleotid. Nucl. Acids 2020, 39, 322–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Toti KS; Jain S; Ciancetta A; Balasubramanian R; Chakraborty S; Surujdin R; Shi ZD; Jacobson KA Pyrimidine nucleotides containing a (S)-methanocarba ring as P2Y6 receptor agonists. Med. Chem. Commun 2017, 8 (10), 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kim HS; Ravi RG; Marquez VE; Maddileti S; Wihlborg AK; Erlinge D; Malmsjö M; Boyer JL; Harden TK; Jacobson KA Methanocarba modification of uracil and adenine nucleotides: high potency of northern ring conformation at P2Y1, P2Y2, P2Y4, and P2Y11 but not P2Y6 receptors. J. Med. Chem 2002, 45 (1), 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rawling T; McDonagh AM; Tattam B; Murray M Synthesis of unsymmetrical biaryl ureas from N-carbamoylimidazoles: kinetics and application. Tetrahedron 2012, 68, 6065–6070. [Google Scholar]
- (41).Freundlieb M; Zimmermann H; Müller CE A new, sensitive ecto-5′-nucleotidase assay for compound screening. Anal. Biochem 2014, 446, 53–58. [DOI] [PubMed] [Google Scholar]
- (42).Servos J; Reiländer H; Zimmermann H Catalytically active soluble ecto-5′-nucleotidase purified after heterologous expression as a tool for drug screening. Drug Dev. Res 1998, 45, 269–276. [Google Scholar]
- (43).Cheng Y-C; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (44).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FR; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Knapp K; Zebisch M; Pippel J; El-Tayeb A; Müller CE; Sträter N Crystal structure of the human ecto-5′-nucleotidase (CD73): Insights into the regulation of purinergic signaling. Structure 2012, 20, 2161–2173. [DOI] [PubMed] [Google Scholar]
- (46).Scaletti E; Huschmann FU; Müller U; Weiss MS; Sträter N Substrate binding modes of purine and pyrimidine nucleotides to human ecto-5′-nucleotidase (CD73) and inhibition by their bisphosphonic acid derivatives. Purinergic Signalling 2021, 17, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Seeber M; Felline A; Raimondi F; Muff S; Friedman R; Rao F; Caflisch A; Fanelli F Wordom: a user-friendly program for the analysis of molecular structures, trajectories, and free energy surfaces. J. Comput. Chem 2011, 32, 1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tosh DK; Salmaso V; Rao H; Campbell R; Bitant A; Gao Z-G; Auchampach JA; Jacobson KA Direct comparison of (N)-methanocarba and ribose-containing 2-arylalkynyladenosine derivatives as A3 receptor agonists. ACS Med. Chem. Lett 2020, 11, 1935–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Losenkova K; Paul M; Irjala H; Jalkanen S; Yegutkin GG Histochemical approach for simultaneous detection of ectonucleotidase and alkaline phosphatase activities in tissues. Methods Mol. Biol 2020, 2041, 107–116. [DOI] [PubMed] [Google Scholar]
- (50).Jacobson KA Functionalized congener approach to the design of ligands for G protein–coupled receptors (GPCRs). Bioconjugate Chem. 2009, 20, 1816–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schmies C; Rolshoven G; Idris RM; Losenkova K; Renn C; Schäkel L; Al-Hroub H; Wang Y; Garofano F; Schmidt-Wolf IGH; Zimmermann H; Yegutkin GG; Müller CE Fluorescent Probes for Ecto-5′-nucleotidase (CD73). ACS Med. Chem. Lett 2020, 11 (11), 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bankhead P; Loughrey MB; Fernández JA; Dombrowski Y; McArt DG; Dunne PD; McQuaid S; Gray RT; Murray LJ; Coleman HG; James JA; Salto-Tellez M; Hamilton PW QuPath: Open source software for digital pathology image analysis. Sci. Rep 2017, 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Aricescu AR; Lu W; Jones EY Acta Crystallogr. D Biol. Crystallogr 2006, 62 (10), 1243–1250. [DOI] [PubMed] [Google Scholar]
- (54).Kabsch W XDS. Acta Crystallogr. 2010, D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Blanc E; Roversi P; Vonrhein C; Flensburg C; Lea SM; Bricogne G Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. 2004, D60, 2210–2221. [DOI] [PubMed] [Google Scholar]
- (56).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Maestro, release 2020-1; Schrödinger, LLC: New York, 2019. [Google Scholar]
- (58).Madhavi Sastry G; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (59).Jacobson MP; Pincus DL; Rapp CS; Day TJF; Honig B; Shaw DE; Friesner RA A hierarchical approach to all-atom protein loop prediction. Proteins: Struct., Funct., Genet 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- (60).Jacobson MP; Friesner RA; Xiang Z; Honig B On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol 2002, 320 (3), 597–608. [DOI] [PubMed] [Google Scholar]
- (61).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (62).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra Precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49 (21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- (63).Case DA; Aktulga HM; Belfon K; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TE III; Cruzeiro VWD; Darden TA; Duke RE; Giambasu G; Gilson MK; Gohlke H; Goetz AW; Harris R; Izadi S; Izmailov SA; Jin C; Kasavajhala K; Kaymak MC; King E; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Machado M; Man V; Manathunga M; Merz KM; Miao Y; Mikhailovskii O; Monard G; Nguyen H; O’Hearn KA; Onufriev A; Pan F; Pantano S; Qi R; Rahnamoun A; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Skrynnikov NR; Smith J; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xue Y; York DM; Zhao S; Kollman PA Amber 2021; University of California, San Francisco: San Francisco, 2021. [Google Scholar]
- (64).Wang J; Wang W; Kollman PA; Case DA Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- (65).Jakalian A; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- (66).Steinbrecher T; Latzer J; Case DA Revised AMBER parameters for bioorganic phosphates. J. Chem. Theory Comput 2012, 8, 4405–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79 (2), 926. [Google Scholar]
- (68).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (70).Harvey MJ; Giupponi G; Fabritiis GD ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput 2009, 5, 1632–1639. [DOI] [PubMed] [Google Scholar]
- (71).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (72).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (73).McKinney W Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference; SciPy, 2010; pp 56–61. [Google Scholar]
- (74).Hunter JD Matplotlib: A 2D graphics environment. Comput. Sci. Eng 2007, 9, 90–95. [Google Scholar]
- (75).Williams T; Kelley C Gnuplot 5.0.[Sl]; http://www.gnuplot.info.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.