

SUMMARY
Peripheral artery disease (PAD) leads to considerable morbidity, yet strategies for therapeutic angiogenesis fall short of being impactful. Inflammatory macrophage subsets play an important role in orchestrating post-developmental angiogenesis, but the underlying mechanisms are unclear. Here, we find that macrophage VEGF-A expression is dependent upon the potent inflammatory cytokine, IL-1β. IL-1β promotes pro-angiogenic VEGF-A165a isoform transcription via activation and promoter binding of STAT3 and NF-κB, as demonstrated by gene-deletion, gain-of-function, inhibition, and chromatin immunoprecipitation assays. Conversely, IL-1β-deletion or inhibition of STAT3 or NF-κB increases anti-angiogenic VEGF-A165b isoform expression, indicating IL-1β signaling may also direct splice variant selection. In an experimental PAD model of acute limb ischemia, macrophage IL-1β expression is required for pro-angiogenic VEGF-A expression and for VEGF-A-induced blood flow recovery via angio- or arteriogenesis. Though further study is needed, macrophage IL-1β-dependent transcription of VEGF-A via STAT3 and NF-κB may have potential to therapeutically promote angiogenesis in the setting of PAD.
Graphical abstract
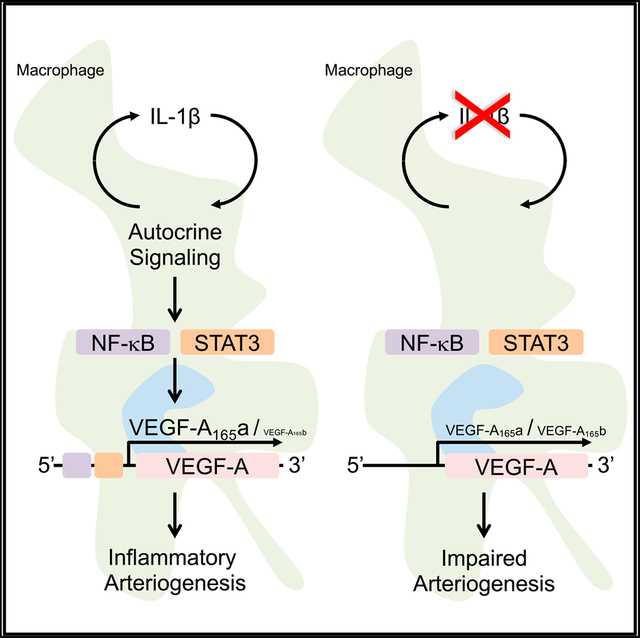
In brief
Mantsounga et al. show inflammatory macrophage IL-1β expression to be required for pro-angiogenic VEGF-A expression and consequent post-developmental angio- or arteriogenesis in an experimental model of peripheral artery disease. Autocrine IL-1β signaling promotes transcription of pro-angiogenic VEGF-A165a isoform expression relative to anti-angiogenic isoform, VEGF-A165b, through activation of STAT3 and NF-κB.
INTRODUCTION
Peripheral artery disease (PAD) caused by atherosclerosis leads to considerable morbidity and mortality due to tissue damage from both acute and chronic occlusive ischemia (Farber and Eberhardt, 2016; Gerhard-Herman et al., 2017; Shishehbor et al., 2016). It is estimated that over 200 million people worldwide have PAD (Fowkes et al., 2013). Treatment is often limited to mechanical revascularization (i.e., surgical bypass or angioplasty), which can be incomplete due to the complexity of lesions (Shishehbor et al., 2016). Therapeutic angiogenesis is an attractive revascularization concept, yet clinical trials have fallen short of impacting outcomes (Iyer and Annex, 2017).
Multiple cell types contribute to post-developmental angiogenesis (Carmeliet and Jain, 2011). Cells of the monocyte or macrophage lineage appear to play an important role in orchestrating angiogenesis after ischemic injury (Guo et al., 2018; Morrison et al., 2014; Takeda et al., 2011). The induction of a pro-inflammatory environment in the earliest stages of tissue injury recruits macrophages polarized toward a classic inflammatory phenotype, referred to as “M1” because of the association with Th1 T cell cytokines (Geissmann et al., 2003). In humans, this inflammatory macrophage population is characterized by the high expression of the chemokine receptor, C-C motif chemokine receptor 2 (CCR2), and the lipopolysaccharide (LPS) co-receptor, CD14, inducible nitric oxide synthase (NOS2), interleukin-1β (IL-1β), and interleukin-6 (IL-6) (Willenborg et al., 2012). At later stages of wound healing, a subset of macrophages transition to an “alternatively activated” phenotype, referred to as “M2” because of the association with a Th2 T cell cytokine environment. Alternatively activated macrophages express high levels of the chemokine receptor, CX3CR1, and the type III Fcγ receptor, CD16, and they contribute to wound healing. Effective vascularization during healing has been largely attributed to the alternatively activated phenotype (Charo, 2007; Gordon and Taylor, 2005; Pollard, 2009).
The pro-angiogenic factor, vascular endothelial growth factor A (VEGF-A), plays a critical role in regulating angiogenesis during both homeostasis and disease (Apte et al., 2019). Early studies of VEGF-A identified potential pro-inflammatory activity, including upregulation of chemokines and adhesion molecules for leukocyte recruitment and trafficking (Reinders et al., 2003). During acute limb ischemia or wound injury, early infiltrating inflammatory macrophages appear to be an important source of VEGF-A (Willenborg et al., 2012). CCR2-deficient mice demonstrate reduced blood vessel formation attributable to an inflammatory macrophage defect, and myeloid deficiency of VEGF-A decreased vascularization in the early, inflammatory phases of wound healing. Engagement of myeloid leukocyte β2 integrins triggers a molecular switch in inflammatory macrophages that results in the stabilization of multiple transcripts, including GM-CSF, VEGF-A, and MMP-9 (Morrison et al., 2014; Ramgolam et al., 2010; Wang et al., 2006; Zhang et al., 2012). The major monocyte chemokine, C-C motif chemokine ligand 2 (CCL2), ligand to CCR2, is a well-established β2 integrin activator that is vital to arteriogenesis in animal models of acute hindlimb ischemia (Carr et al., 1996; Ito et al., 1997; van Royen et al., 2003). In fact, CCL2 stimulation both promotes and couples with β2 integrin adhesion to activate myeloid VEGF-A post transcriptional stability with consequent increases in VEGF-A protein and arteriogenesis (Morrison et al., 2014).
Alternative splicing has also been recognized to regulate VEGF-A activity via production of two separate isoform families with opposing biological activity. The splicing event occurs in terminal exon 8, where a proximal splice site generates a pro-angiogenic VEGF-A165a isoform and a distal splice site in the 3′-untranslated region leads to an anti-angiogenic VEGF-A165b isoform (Woolard et al., 2004). The two isoforms are the same size but differ by six amino acids; exon 8a encodes CDKPRR while exon 8b encodes SLTRKD. VEGF-A165a binds to VEGFR2 leading to autophosphorylation and consequent downstream pro-angiogenic effector signaling, whereas VEGF-A165b results in altered tyrosine phosphorylation patterns that do not promote angiogenesis (Peiris-Pages, 2012). Inflammatory macrophage expression of VEGF-A165b appears to inhibit revascularization of ischemic limbs, in part, through an autocrine VEGFR1-induced inflammatory-like phenotype (Ganta et al., 2019; Kikuchi et al., 2014). Mechanisms that determine splice variant preference are largely unknown. Moreover, the vast majority of studies involving VEGF-A have not used isoform-specific primers or antibodies to quantify respective differences in expression during inflammation and angiogenesis.
Inflammatory cytokines like IL-1β and tumor necrosis factor alpha (TNF-α) induce transcription of VEGF-A in some cell types, including smooth muscle cells, tumor cell lines, and cardiac myocytes (Gille et al., 1997; Li et al., 1995; Ryuto et al., 1996; Tanaka et al., 2000). While early infiltrating inflammatory macrophages appear critical for arteriogenesis in the experimental PAD model (Morrison et al., 2014), the relationship between pro-angiogenic VEGF-A expression and the macrophage phenotypic profile (i.e., inflammatory versus alternatively activated) remains unclear. The goal of this study is to define the inflammatory mechanism(s) that drive pro-angiogenic VEGF-A expression and consequent VEGF-A-dependent arteriogenesis.
RESULTS
IL-1β and VEGF-A expression levels are increased in inflammatory relative to alternatively activated macrophages
To establish the relationship between VEGF-A expression and macrophage phenotypic profile, we polarized primary macrophages, using lipopolysaccharide and interferon gamma (LPS+IFN-γ) or interleukin-4 and interleukin-13 (IL-4+IL-13). Bone marrow-derived macrophages (BMDMs) from C57BL/6J mice adopt an inflammatory phenotype when activated by LPS+IFN-γ, marked by high expression of NOS2, IL-1β, and TNF-α (Figures 1 and S1) (Kim and Nair, 2019). BMDMs take on an alternatively activated state when activated by IL-4+IL-13, marked by high expression of arginase 1 (ARG1), found in inflammatory zone 1 (FIZZ1), and the chitinase-like protein, YM1 (Figure S1) (Kim and Nair, 2019). To assess differential expression patterns between polarized BMDMs, we performed RNA-seq analysis on BMDMs stimulated with LPS+IFN-γ and IL-4+IL-13. IL-1β was found to be the most significant, most differentially downregulated mRNA when moving from an inflammatory (LPS+IFN-γ) to alternatively activated (IL-4+IL-13) phenotype (Figure 1A). Somewhat surprisingly, we found that VEGF-A and transcripts encoding several key VEGF-A promoter-driving transcription factors, including HIF-1α, STAT3, and the NF-κB subunit, v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA), demonstrated a comparable pattern to IL-1β. Quantitative RT-PCR confirmed that expression levels of IL-1β, VEGF-A, HIF-1α, STAT3, and RELA mRNA were significantly increased in inflammatory relative to alternatively activated BMDMs (Figures 1B–1F). In summary, VEGF-A expression correlates closely with IL-1β based on the expression profiles of polarized macrophages.
Figure 1. IL-1β and VEGF-A expression levels are increased in inflammatory relative to alternatively activated macrophages.
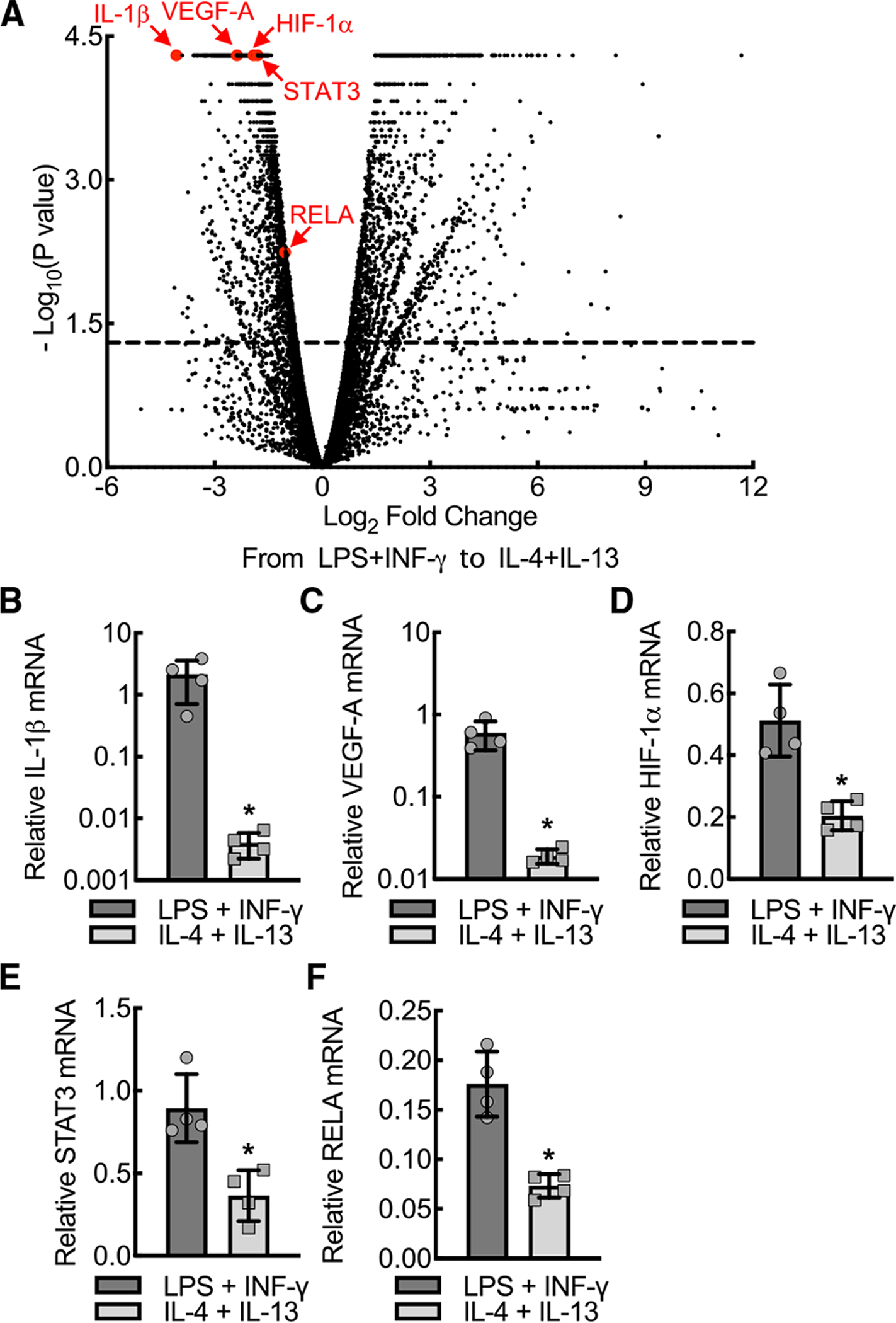
(A) Volcano plot demonstrating RNA-seq transcriptome data displaying expression profiles from mouse bone marrow-derived macrophages (BMDMs) treated with either LPS+IFN-γ or IL-4+IL-13 for 24 h. IL-1β, VEGF-A, HIF-1α, STAT3, and RELA are marked by arrows and red dots. Dashed line, p = 0.05.
(B–F) Quantitative RT-PCR of relative (normalized to HPRT) mRNA expression for IL-1β, VEGF-A, HIF-1α, STAT3, and RELA from the BMDM lysates treated as in (A) (*, p < 0.05 by t test; n = 4 mice total; two males and two females). Data, mean ± SD.
Macrophage VEGF-A expression is dependent on IL-1β expression during inflammatory conditions
To develop an animal model of conditional IL-1β-deletion, we generated a mouse strain with a LoxP flanked exon 4 of the IL-1β allele, using a clustered regularly interspaced short palindromic repeat and its associated protein9 (CRISPR/Cas9) strategy (Figure S2) (Farboud et al., 2019). LoxP insertion sites, 157 base pairs upstream of exon 4 and 103 base pairs downstream of exon 4, were selected based on distance from the exon, availability of CRISPR guides to ensure low off-target effect, and conservation profile. Cre-mediated excision of exon 4 resulted in deletion of a coding sequence involving 67 amino acids along with a frameshift, leading to premature termination of translation in exon 5.
To determine whether inflammatory macrophage VEGF-A expression is dependent on IL-1β expression, we crossed these IL-1βfl/fl mice with mice that express the tamoxifen-inducible MerCreMer fusion protein under control of the macrophage-specific promoter, colony stimulating factor 1 receptor (Csf1rmercremer) (Qian et al., 2011). BMDMs from tamoxifen-injected mice demonstrated 75% decreases in both IL-1β mRNA expression and mature IL-1β protein secretion when subjected to strong inflammasome stimulation by the combination of LPS priming and cholesterol crystal exposure (Figures 2A and 2B). In the BMDMs from vehicle control mice, both VEGF-A mRNA and VEGF-A protein secretion demonstrated significant increases during inflammasome stimulation, comparable to the IL-1β expression pattern. Moreover, both VEGF-A mRNA and protein secretion were significantly reduced by about 70% upon macrophage IL-1β-deletion (Figures 2C and 2D).
Figure 2. Macrophage VEGF-A expression is dependent on IL-1β expression during inflammatory conditions.
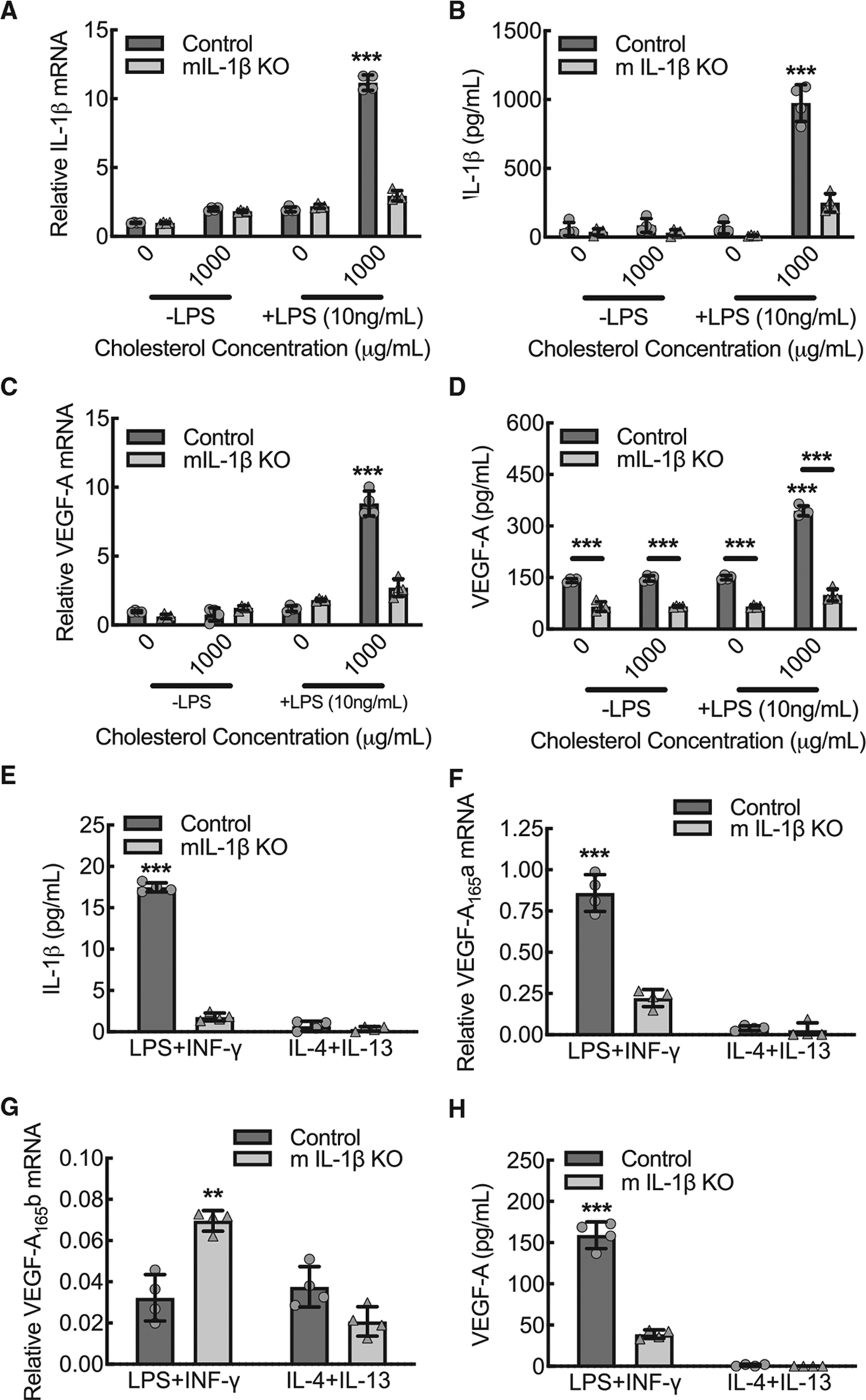
(A) BMDMs from control or myeloid IL-1β-deleted mice (mIL-1β KO) were primed with or without LPS (10 ng/mL) and exposed to cholesterol crystals (0 or 1000 μg/mL) for 24 h to induce inflammasome activation, followed by quantitative RT-PCR for relative IL-1β mRNA expression (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(B) BMDMs treated as in (A), followed by ELISA on cell culture supernatants for secreted, mature IL-1β protein (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(C) BMDMs treated as in (A), followed by quantitative RT-PCR for relative VEGF-A mRNA expression (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(D) BMDMs treated as in (A), followed by ELISA on cell culture supernatants for secreted VEGF-A protein (***, p < 0.0001 compared to all others or between control and mIL-1β KO by ANOVA; n = 4 mice total, two males and two females).
(E) Primary mouse BMDMs from control or mIL-1β KO were treated with either LPS+IFN-γ or IL-4+IL-13 for 24 h, followed by ELISA on culture supernatants for secreted, mature IL-1β protein (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(F) BMDMs treated as in (E) followed by quantitative RT-PCR for relative VEGF-A165a mRNA expression (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(G) BMDMs treated as in (E) followed by quantitative RT-PCR for relative VEGF-A165b mRNA expression (**, p < 0.001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(H) BMDMs treated as in (E) followed by ELISA on culture supernatants for secreted VEGF-A protein (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females). Data, mean ± SD.
Next, we validated the dependence of macrophage VEGF-A expression on IL-1β expression under conditions of LPS+IFN-γ versus IL-4+IL-13 stimulation. With the exception of IL-1β, IL-1β-deleted BMDMs demonstrated comparable expression patterns for the polarization markers, TNF-α, NOS2, ARG1, FIZZ1, YM1, relative to wild-type control (Figure S3). Secretion of mature IL-1β protein was increased approximately 30-fold in wild-type BMDMs stimulated by LPS+IFN-γ relative to those stimulated by IL-4+IL-13, and macrophage IL-1β-deletion led to a 90% reduction in secreted mature IL-1β protein (Figure 2E). Effective vascularization after injury has been largely attributed to the inflammation suppressed, alternatively activated, macrophages (Charo, 2007; Gordon and Taylor, 2005; Pollard, 2009). To quantify VEGF-A isoform expression patterns with LPS+IFN-γ stimulation or IL-1β-deletion, we used primer-specific quantitative RT-PCR for the pro-angiogenic VEGF-A165a and the anti-angiogenic VEGF-A165b. VEGF-A165a mRNA expression was upregulated over 6-fold in BMDMs stimulated by LPS+IFN-γ relative to those stimulated by IL-4+IL-13, and macrophage IL-1β-deletion led to a 75% reduction in VEGF-A165a mRNA expression in LPS+IFN-γ polarized BMDMs (Figure 2F). Anti-angiogenic VEGF-A165b mRNA expression was not affected by BMDM polarization state; however, macrophage IL-1β-deletion led to a 2-fold increase in VEGF-A165b Mrna expression in LPS+IFN-γ-stimulated BMDMs, suggesting that IL-1β promotes selective expression of VEGF-A165a relative to VEGF-A165b (Figure 2G). Comparable to the IL-1β expression pattern, VEGF-A protein secretion was significantly increased in wild-type BMDMs stimulated by LPS+IFN-γ, and macrophage IL-1β-deletion led to a >75% reduction in VEGF-A secretion (Figure 2H). To confirm that IL-1β drives VEGF-A expression through an autocrine signaling loop, we cultured BMDMs from global IL-1R-deleted mice and quantified VEGF-A expression in the context of stimulation with LPS+IFN-γ or IL-4+IL-13. Comparable to the IL-1β-deleted BMDMs, LPS+IFN-γ-stimulated BMDMs from IL-1R-deleted mice expressed decreased VEGF-A protein, decreased pro-angiogenic VEGF-A165a isoform mRNA, and increased anti-angiogenic VEGF-A165b mRNA (Figure S4).
Macrophage IL-1β-dependent upregulation of VEGF-A is transcriptional and associated with NF-κB and STAT3 activity
To determine whether the IL-1β-dependent VEGF-A mRNA expression was transcriptional, we quantified nascent RNA from wild-type control and IL-1β-deleted BMDMs under conditions of LPS+IFN-γ or IL-4+IL-13 stimulation. IL-1β-deleted BMDMs stimulated by LPS+IFN-γ and both wild-type control and IL-1β-deleted BMDMs stimulated with IL-4+IL-13 demonstrated approximately a 75% reduction in nascent VEGF-A mRNA relative to wild-type BMDMs stimulated by LPS+IFN-γ, indicating that differences in VEGF-A mRNA expression were attributable to changes in transcription (Figure 3A).
Figure 3. Macrophage IL-1β drives nascent VEGF-A mRNA transcription, nuclear-localized NF-κB and STAT3 activity but not HIF-1a activity, and gain-of-function mutations for STAT3 or IKK2 increase pro-angiogenic VEGF-A165a isoform expression.
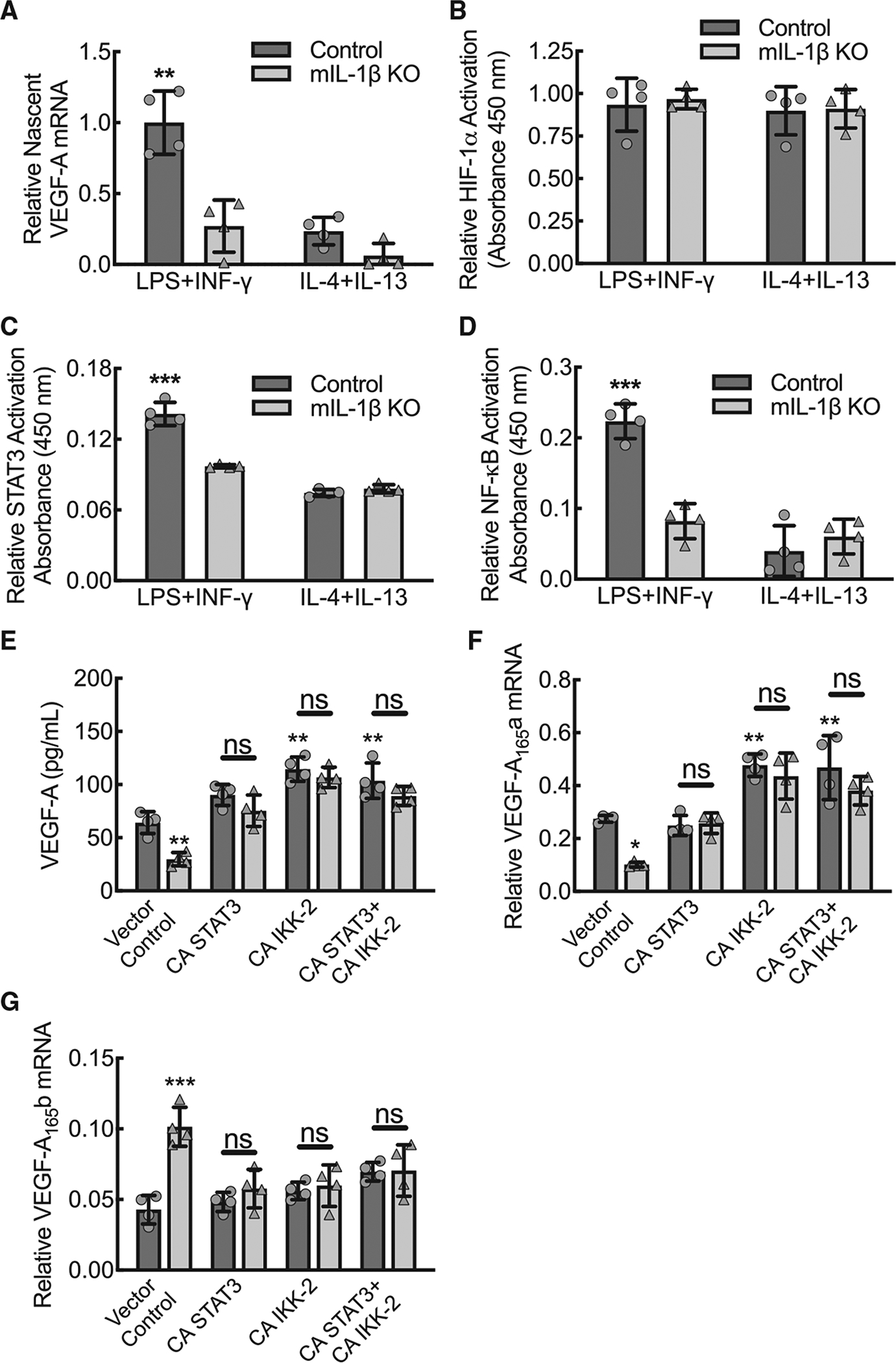
(A) BMDMs from control or myeloid IL-1β-deleted mice (mIL-1β KO) were treated with either LPS+IFN-γ or IL-4+IL-13 for 12 h followed by 20-min 5-ethynyl Uridine pulse, Click-iT Nascent RNA purification, and quantitative RT-PCR for relative nascent VEGF-A mRNA (**, p ≤ 0.0002 compared to all others by ANOVA; n = 4 biological replicates from two mice, one male and one female).
(B) BMDMs from control or mIL-1β KO mice were treated with either LPS+IFN-γ or IL-4+IL-13 for 24 h followed by a modified ELISA for relative HIF-1α activity on nuclear fraction lysates (n = 4 mice total, two males and two females).
(C) BMDMs treated as in (B) followed by a modified ELISA for relative STAT3 activity on nuclear fraction lysates (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(D) BMDMs treated as in (B) followed by a modified ELISA for relative NF-κB activity on nuclear fraction lysates (***, p < 0.0001 compared to all others by ANOVA; n = 4 mice total, two males and two females).
(E) BMDMs from control or mIL-1β KO mice were transfected with plasmid for constitutively active mutations of STAT3 or IKK2 (NF-κB activator) alone or in combination followed by ELISA on culture supernatants for secreted VEGF-A protein (**, p ≤ 0.0075 compared to wild-type control transfected with vector control by ANOVA; n = 4 mice total, two males and two females).
(F) BMDMs treated as in (E) followed by quantitative RT-PCR for relative VEGF-A165a mRNA expression (*, p < 0.05; **, p < 0.005 compared to wild-type control transfected with vector control by ANOVA; n = 4 mice total, two males and two females).
(G) BMDMs treated as in (E) followed by quantitative RT-PCR for relative VEGF-A165b mRNA expression (***, p < 0.0001 compared to wild-type control transfected with vector control by ANOVA; n = 4 mice total, two males and two females). Data, mean ± SD.
To define the IL-1β-dependent molecular mechanisms that determine VEGF-A transcription, we quantified the activity of the three VEGF-A promoter-driving transcription factors, HIF-1α, STAT3, and RELA, identified by RNA-seq analysis (Figure 1A), using nuclear fractionation of BMDM lysates in a modified ELISA assay that involves capture with DNA seed sequences. Nuclear HIF-1α activity was found to be comparable across the spectrum of polarization in both wild-type and IL-1β-deleted BMDMs, indicating that nuclear HIF-1α activity was not dependent on inflammatory phenotype or IL-1β (Figure 3B). Relative to wild-type BMDMs stimulated by LPS+IFN-γ, nuclear STAT3 activity was significantly decreased in wild-type BMDMs stimulated by IL-4+IL-13 and in IL-1β-deleted BMDMs, indicating that nuclear STAT3 activity is increased with inflammation comparable to its expression patterns and that nuclear STAT3 activity is IL-1β-dependent (Figure 3C). Nuclear NF-κB activity was also significantly increased in wild-type BMDMs upon stimulation with LPS+IFN-γ comparable to its expression profile, and nuclear NF-κB activity was found to be IL-1β dependent (Figure 3D). In summary, the data support that nuclear activities of STAT3 and NF-κB increase during inflammatory macrophage polarization in an IL-1β-dependent manner, whereas HIF-1α activity is not impacted by inflammatory state or IL-1β.
NF-κB and STAT3 drive transcription of the pro-angiogenic VEGF-A isoform and bind to the VEGF-A promoter in an IL-1β-dependent manner
To assess the impact of active STAT3 and NF-κB on VEGF-A expression, we transfected LPS+IFN-γ-stimulated BMDMs from wild-type control and IL-1β-deleted mice with plasmids expressing the constitutively active mutations of STAT3 or IKK-2 (NF-κB activator) (Mercurio et al., 1997; Mitchell et al., 2013). Both constitutively active mutants for STAT3 and IKK-2 increased VEGF-A expression in IL-1β-deleted BMDMs comparable to wild-type levels (Figure 3E). By primer-specific quantitative RT-PCR assessment, transfection of IL-1β-deleted BMDMs with the constitutively active mutants led to increased pro-angiogenic VEGF-A165a mRNA expression and suppression of anti-angiogenic VEGF-A165b mRNA expression (Figures 3F and 3G).
Next, we performed chromatin immunoprecipitation (ChIP) assay on BMDMs stimulated by LPS+IFN-γ, using antibodies specific to STAT3 and NF-κB to confirm IL-1β-dependent binding of these transcription factors to the VEGF-A promoter region. Quantitative RT-PCR with primers directed at the regions of the VEGF-A promoter associated with established STAT3 and NF-κB seed sites demonstrated that wild-type BMDMs stimulated by LPS+IFN-γ achieved about 4-fold increased promoter binding using STAT3- and NF-κB-specific antibodies relative to their respective isotype control antibodies (Figures 4A and 4B). Moreover, IL-1β-deleted BMDMs stimulated by LPS+IFN-γ demonstrated no increase over isotype control, supporting that both STAT3 and NF-κB bind to the VEGF-A promoter in an IL-1β-dependent manner.
Figure 4. NF-κB and STAT3 bind to VEGF-A promoter in an IL-1β-dependent manner, and inhibition of either led to decreased expression of pro-angiogenic VEGF-A165a and to increased expression of anti-angiogenic VEGF-A165b.
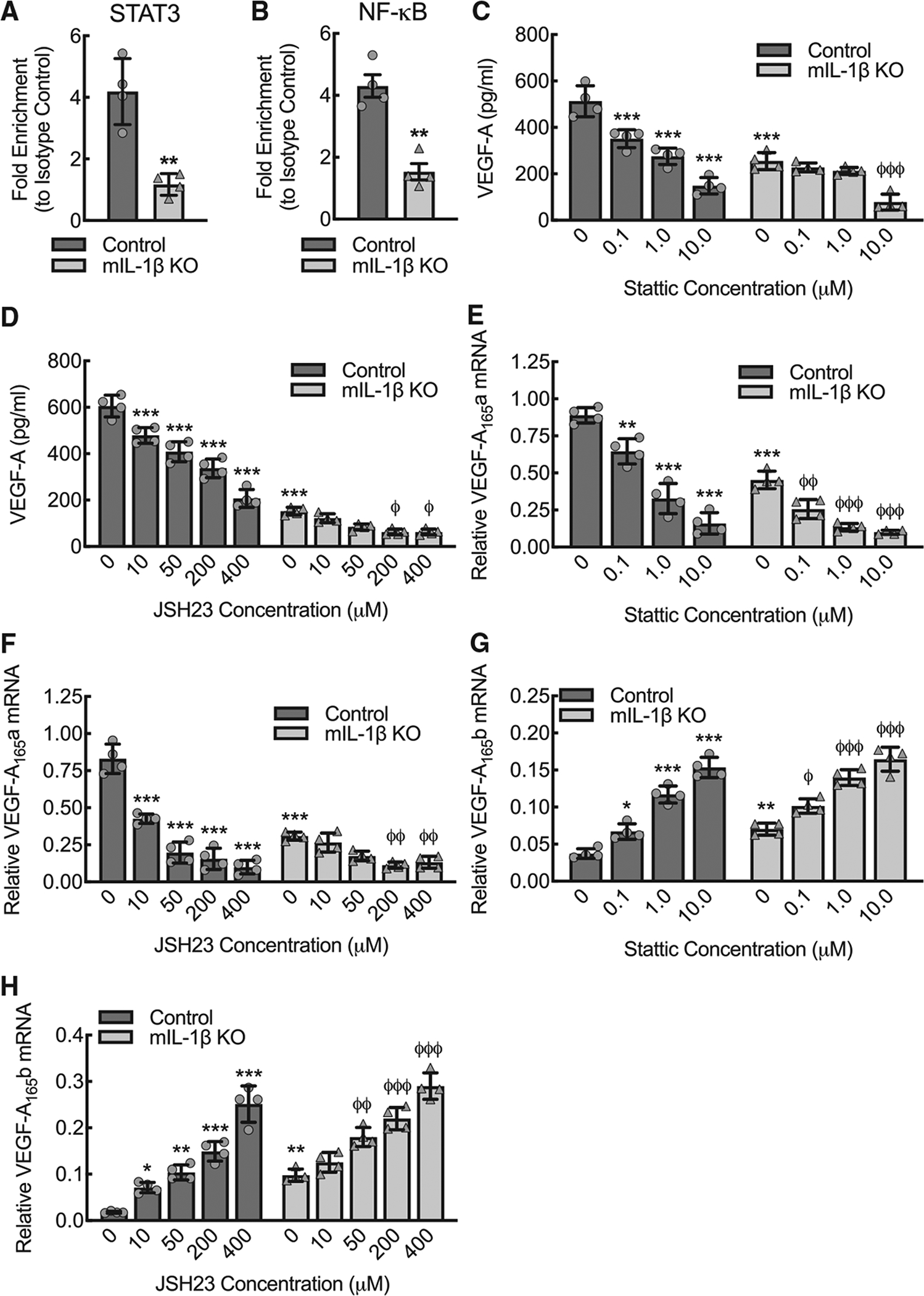
(A) Quantitative RT-PCR of the VEGF-A promoter after chromatin immunoprecipitation (ChIP), using antibody specific to STAT3 relative to isotype control, of lysate from control or myeloid IL-1β-deleted (mIL-1β KO) BMDMs that were treated with LPS+IFN-γ for 24 h (**, p < 0.001 by t test; n = 4 mice total, two males and two females).
(B) Quantitative RT-PCR of the VEGF-A promoter after ChIP, using antibody specific to NF-κB relative to isotype control, of lysate from BMDMs treated as in (A) (**, p = 0.0018 by t test; n = 4 mice total, two males and two females).
(C) BMDMs from control or mIL-1β KO were treated with LPS+IFN-γ for 24 h in the presence or absence of the STAT3 inhibitor, Stattic, at indicated concentrations, followed by ELISA for VEGF-A on culture supernatant (***, p ≤ 0.0001 compared to control at 0 concentration by ANOVA; φφφ, p ≤ 0.0001 compared to mIL-1β KO at 0 concentration by ANOVA; n = 4 mice total, twomales and two females).
(D) BMDMs from control or mIL-1β KO mice were treated with LPS+IFN-γ for 24 h in the presence or absence of the NF-κB inhibitor, JSH23, at indicated concentrations, followed by ELISA for VEGF-A on culture supernatant (***, p ≤ 0.0001 compared to control at 0 concentration by ANOVA; φ, p ≤ 0.01 compared to mIL-1β KO at 0 concentration by ANOVA; n = 4 mice total, two males and two females).
(E) BMDMs treated as in (C) followed by quantitative RT-PCR for relative VEGF-A165a mRNA expression (**, p < 0.001; ***, p ≤ 0.0001 compared to control at 0 concentration by ANOVA; φφ, p ≤ 0.005; φφφ, p ≤ 0.0001; compared tomIL-1β KO at0 concentration by ANOVA; n = 4 mice total, two males and two females).
(F) BMDMs treated as in (D) followed by quantitative RT-PCR for relative VEGF-A165a mRNA expression (***, p < 0.0001 compared to all others by ANOVA; φφ, p ≤ 0.005 compared to mIL-1β KO at 0 concentration by ANOVA; n = 4 mice total, two males and two females).
(G) BMDMs treated as in (C) followed by quantitative RT-PCR for relative VEGF-A165b mRNA expression (*, p < 0.05; **, p < 0.001; ***, p < 0.0001 compared to control at 0 concentration by ANOVA; φ, p < 0.05; φφφ, p ≤ 0.0001; compared to mIL-1β KO at 0 concentration by ANOVA; n = 4 mice total, two males and two females).
(H) BMDMs treated as in (D) followed by quantitative RT-PCR for relative VEGF-A165b mRNA expression (*, p < 0.05; **, p < 0.001; ***, p < 0.0001 compared to control at 0 concentration by ANOVA; φφ, p < 0.001; φφφ, p ≤ 0.0001; compared to mIL-1β KO at 0 concentration by ANOVA; n = 4 mice total, two males and two females). Data, mean ± SD.
To further assess the functional effects of STAT3 and NF-κB binding to the VEGF-A promoter region, we treated LPS+IFN-γ-stimulated BMDMs with Stattic or JSH23, inhibitors of STAT3 and NF-κB, respectively. Inhibitor dose ranges were selected based on the established IC50s (Schust et al., 2006; Shin et al., 2004). Wild-type BMDMs stimulated by LPS+IFN-γ were treated with increasing concentrations of either Stattic or JSH23 and demonstrated dose-dependent reductions in VEGF-A protein secretion (Figures 4C and 4D), indicating that VEGF-A expression requires both STAT3 and NF-κB activity. We then performed primer-specific quantitative RT-PCR to determine the effects of STAT3 and NF-κB inhibition on expression of the pro- and anti-angiogenic VEGF-A isoforms. Both STAT3 and NF-κB inhibition led to a reduction in the pro-angiogenic VEGF-A165a expression and an increase in the anti-angiogenic VEGF-A165b, comparable to the IL-1β-deleted BMDM pattern, indicating that IL-1β-dependent binding of NF-κB and STAT3 to the VEGF-A promoter appears to determine preference for transcribing pro-angiogenic VEGF-A165a (Figures 4E–4H).
VEGF-A-induced blood flow recovery consequent to new angiogenesis and arteriogenesis is dependent on macrophage IL-1β expression in response to acute hindlimb ischemia
We sought to understand whether loss of macrophage IL-1β-dependent pro-angiogenic VEGF-A expression would significantly impair perfusion recovery in a femoral artery ligation model of hindlimb ischemia. Myeloid IL-1β deletion resulted in decreased blood flow recovery consistent with impaired angiogenesis and arteriogenesis (Figures 5A and 5B). Sex was analyzed as a biological variable using ANOVA, and we found no significant sex-specific changes in this preclinical PAD model (Figure S5A). To assess whether myeloid IL-1β deletion impacted hemodynamics, the mean arterial pressure (MAP) was quantified before (day −2) and after (days 3 and 21) ligation of the femoral artery (Figure S5B). MAP was comparable between wild-type control and myeloid IL-1β-deleted mice. Anesthesia induced a modest reduction in MAP that was comparable between control and myeloid IL-1β-deleted mice (Figure S5C). Traditional histology of ischemic muscle tissue using hematoxylin and eosin staining demonstrated comparable inflammatory cell infiltrate between wild-type and myeloid IL-1β-deleted animals at day 3 post femoral artery ligation (Figures 5C and 5D). Immunofluorescence staining of ischemic gastrocnemius muscle tissue on day 3 post ligation demonstrated that the percent of cells associated with the macrophage marker, CD68, was comparable between wild-type and myeloid IL-1β-deleted animals, whereas percent of cells associated with endothelial marker, CD31, was significantly reduced in myeloid IL-1β-deleted animals (Figures 5E–5H). To quantify vessel proliferation consequent to angiogenesis and arteriogenesis, we quantified the CD31+ vessel areas and the SMA+ vessel areas in gastrocnemius muscle at day 21 post femoral artery ligation and calculated the ratio of ischemic to nonischemic contralateral control muscle vessel area in both wild-type control and myeloid IL-1β-deleted mice (Figure S6). The ratio of CD31+ vessel area in ischemic to contralateral control limb revealed a 30% increase in wild-type control mice but a 30% reduction in myeloid IL-1β-deleted mice. The ratio of SMA+ vessel area, as a marker of new arterial growth, in ischemic limb to contralateral control limb revealed a 2.5-fold increase in wild-type control mice but essentially no increase in myeloid IL-1β-deleted mice. In summary, these data indicate that early endothelial cell recruitment to the ischemic muscle is reduced in myeloid IL-1β-deleted mice despite adequate inflammatory cell recruitment, leading to consequent reductions in new blood vessel (capillary and arterial) growth. Taken in sum, the data support that early endothelial cell recruitment to the ischemic muscle is reduced, consistent with other studies of VEGF-A-mediated post-developmental angiogenesis and arteriogenesis (Amano et al., 2004).
Figure 5. VEGF-A–induced blood flow recovery consequent to new angiogenesis and arteriogenesis is dependent on macrophage IL-1β expression in response to acute hindlimb ischemia.
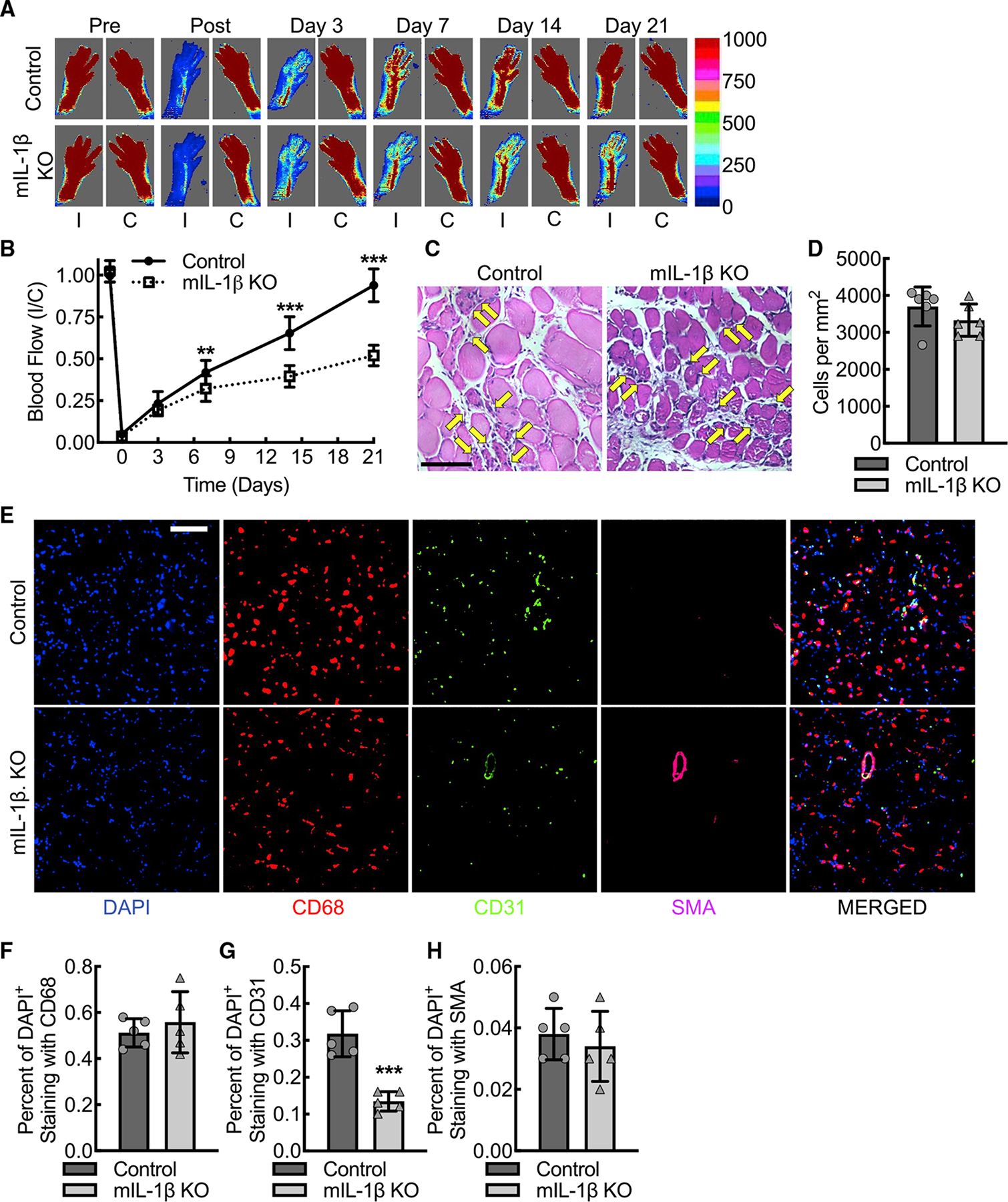
(A and B) Laser Doppler images of flow in the ischemic (I) and contralateral control (C) hindlimbs of control or myeloid IL-1β-deleted mice (mIL-1β KO) at indicated time points before and after femoral artery ligation along with quantitative analysis (B) (**, p = 0.002; ***, p < 0.0001 between control and mIL-1β KO for each time point by ANOVA; n = 12 mice total, six males and six females).
(C and D) Standard histology stained for hematoxylin and eosin of day 3 ischemic gastrocnemius muscle tissue from control or mIL-1β KO mice along with quantification (D) of number of infiltrating inflammatory cells, indicated by yellow arrows (n = 6 mice total, three males and three females). Bar, 100 microns.
(E–H) Immunofluorescence micrographs of ischemic muscle tissue at day 3 post femoral artery ligation from control or mIL-1β KO mice along with quantitation of DAPI+CD68+ (F), DAPI+CD31+ (G), and DAPI+SMA+ (H) cells (***, p = 0.0003 by t test; n = 6 mice, three males and three females). Bar, 100 microns. Data, mean ± SD.
To confirm that myeloid cells have an impact on the total levels of IL-1β and VEGF-A in ischemic muscle tissue, we isolated and homogenized ischemic gastrocnemius muscle tissue from wild-type and myeloid IL-1β-deleted mice. On day 3 post femoral artery ligation, there was decreased mature IL-1β protein in the ischemic muscle tissue from myeloid IL-1β-deleted mice, supporting myeloid cells as a major early source of IL-1β after acute limb ischemia (Figures 6A and 6B). Consistent with our prior work on macrophage VEGF-A-dependent arteriogenesis in the setting of acute limb ischemia (Morrison et al., 2014), this phenotype of reduced blood flow recovery in the myeloid IL-1β-deleted mice was also associated with early reductions in ischemic muscle tissue VEGF-A expression on day 3 post femoral artery ligation (Figures 6A and 6C). Comparable to findings from myeloid IL-1β-deleted BMDMs, mRNA isolated from ischemic gastrocnemius muscle on day 3 post ligation demonstrated a reduction in the pro-angiogenic VEGF-A165a isoform and an increase in the anti-angiogenic VEGF-A165b isoform, supporting IL-1β′s role in determining pro-angiogenic splice variant transcription in vivo (Figures 6D and 6E).
Figure 6. During acute hindlimb ischemia, muscle tissue expression of pro-angiogenic VEGF-A is dependent on macrophage IL-1β despite intact activation of hypoxia-driven transcriptional mechanisms via HIF-1a.
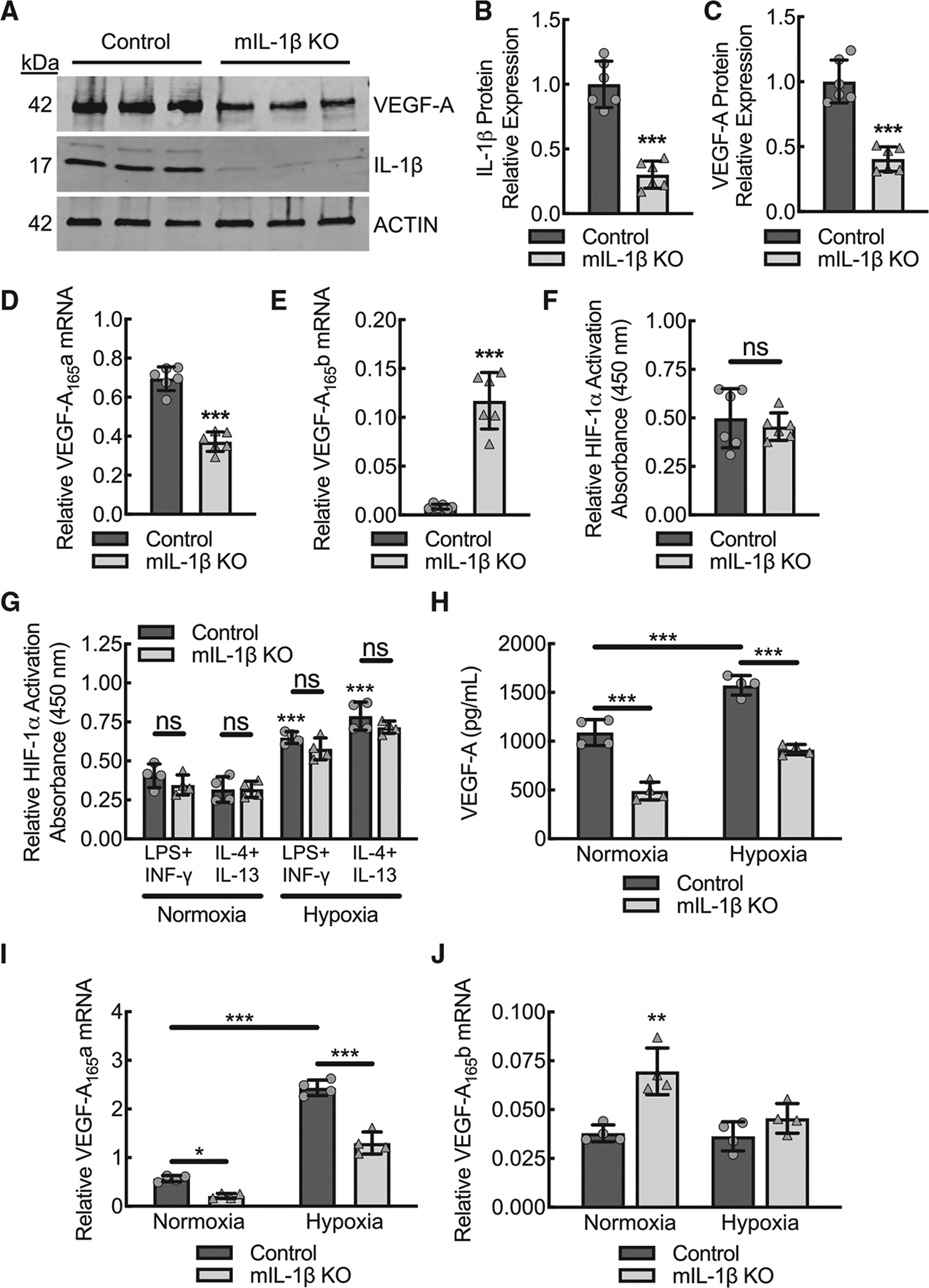
(A–C) Representative VEGF-A and IL-1β immune-blots from ischemic muscle tissue at day 3 post femoral artery ligation from control or myeloid IL-1β-deleted mice (mIL-1β KO) with each lane containing muscle lysate from a separate animal along with quantification (B, C) of β-actin-normalized protein (***, p < 0.0001 by t test; n = 6 mice, three males and three females).
(D) Quantitative RT-PCR for relative VEGF-A165a mRNA expression from ischemic muscle of animals treated as in (A) (***, p < 0.0001 by t test; n = 6 mice total, three males and three females).
(E) Quantitative RT-PCR for relative VEGF-A165b mRNA expression from ischemic muscle of animals treated as in (A) (***, p < 0.0001; n = 6 mice total, three males and three females).
(F) Modified ELISA for relative HIF-1α activity on nuclear fraction lysates from day 3 ischemic muscle tissue of animals treated as in (A) (n = 6 mice total, three males and three females).
(G) BMDMs from control or mIL-1β KO mice were treated with either LPS+IFN-γ or IL-4+IL-13 in the setting of normoxia or hypoxia (1% O2) followed by modified ELISA for relative HIF-1α activity on nuclear fraction lysates (***, p ≤ 0.0001 compared to normoxia control treated with LPS+IFN-γ by ANOVA; n = 4 mice total, two males and two females).
(H) BMDMs from control or mIL-1β KO mice were treated with LPS+IFN-γ in the setting of normoxia or hypoxia (1% O2) followed by ELISA on culture supernatants for secreted VEGF-A protein (***, p ≤ 0.0001 by ANOVA; n = 4 mice total, two males and two females).
(I) BMDMs treated as in (H) followed by quantitative RT-PCR for relative VEGF-A165a mRNA expression (*, p < 0.05; ***, p < 0.0001 by ANOVA; n = 4 mice total, two males and two females).
(J) BMDMs treated as in (H) followed by quantitative RT-PCR for relative VEGF-A165b mRNA expression (**, p < 0.009 compared with all others by ANOVA n = 4 mice total, two males and two females). Data, mean ± SD.
HIF-1α is a powerful transcriptional regulator for detecting and adapting to cellular oxygen levels during ischemia and a major upstream mediator of VEGF-A expression and signaling (Choudhry and Harris, 2018). To assess whether impaired muscle expression of VEGF-A in myeloid IL-1β-deleted mice reflected impaired HIF-1α activity, we quantified relative HIF-1α activity on nuclear fractions isolated from gastrocnemius muscle tissue lysates at day 3 post femoral artery ligation. Relative to wild-type mice, IL-1β-deleted mice had comparable nuclear HIF-1α activity in ischemic muscle tissue on day 3 post ligation (Figure 6F). To assess induction of HIF-1α activity by hypoxia in BMDMs, wild-type control and IL-1β-deleted BMDMs were stimulated by LPS+IFN-γ or IL-4+IL-13 in the setting of hypoxia (1% O2) and demonstrated comparable increases in HIF-1α activity relative to normoxia (Figure 6G). We assessed whether VEGF-A expression was increased in IL-1β-deleted BMDMs by stimulation with hypoxia (Figures 6H–6J). Both wild-type and IL-1β-deleted BMDMs stimulated by LPS+IFN-γ in the setting of hypoxia demonstrated increased pro-angiogenic VEGF-A expression; however, in the IL-1β-deleted BMDMs, the hypoxia-driven component to VEGF-A expression was insufficient to fully overcome the impairment in VEGF-A expression conferred by the loss of IL-1β.
Rescue of blood flow recovery in myeloid IL-1β-deleted mice using wild-type BMDMs confirms the important contribution of IL-1β derived from macrophages relative to other leukocytes
The Csf1r promoter-driven Cre recombinase system may affect neutrophils and some subsets of T cells, potentially confounding the macrophage specificity of in vivo findings (McCubbrey et al., 2017). To address this concern, we performed a clodronate liposome macrophage depletion followed by intravenous injection of cultured BMDMs to demonstrate that macrophages are sufficient to rescue the phenotype of the myeloid IL-1β-deleted animals in this model of experimental PAD (Jetten et al., 2013; Moreno, 2018; Pilny et al., 2019). To track the BMDMs in vivo, we crossed wild-type and myeloid IL-1β-deleted mouse lines to the Ai9 (RCL-tdT) Cre recombinase reporter strain (Madisen et al., 2010), leading to robust cultured BMDM expression of tdTomato, in addition to F4/80 and CD68, after tamoxifen-induced recombination (Figures S7A–S7D). IL-1β-deleted mice underwent femoral artery ligation in the setting of clodronate liposome macrophage depletion followed by transplant of wild-type Ai9 BMDMs and demonstrated >80% hindlimb blood flow recovery within 21 days, comparable to the established wild-type phenotype (Figures 7A and 7B). IL-1β-deleted animals that received autologous IL-1β-deleted Ai9 BMDMs demonstrated <50% perfusion recovery, consistent with the previously established myeloid IL-1β-deleted phenotype. To confirm that the intravenously injected BMDMs were recruited to the ischemic muscle tissue, we carried out immunofluorescence microscopy of ischemic gastrocnemius muscle tissue on day 3 post femoral artery ligation. The percent of cells associated with tdTomato expression was comparable among IL-1β-deleted animals that received donor BMDMs from either wild-type control or IL-1β-deleted Ai9 mice, whereas the percent of cells associated with CD31 positivity was significantly reduced in mice receiving myeloid IL-1β-deleted Ai9 BMDMs (Figures 7C–7E). To confirm the effectiveness of our clodronate liposome macrophage depletion, we carried out additional immunofluorescence co-staining for CD68 and found the percent of CD68 positive cells that also co-stained with tdTomato to be >98% (Figures S7E and S7F). This supports that an effective clodronate liposome depletion results in <2% residual CD68-positive cells, which likely represent the combination of residual macrophages with other small leukocyte subpopulations expressing CD68. In summary, primary macrophage rescue appears sufficient to overcome the myeloid IL-1β-deleted animal phenotype, supporting that the phenotype in the experimental acute limb ischemia model is largely driven by macrophages and macrophage-specific IL-1β expression.
Figure 7. Clodronate liposome treatment followed by BMDM transplant in the context of acute hindlimb ischemia confirms the important contribution of IL-1β from macrophages relative to other leukocytes.
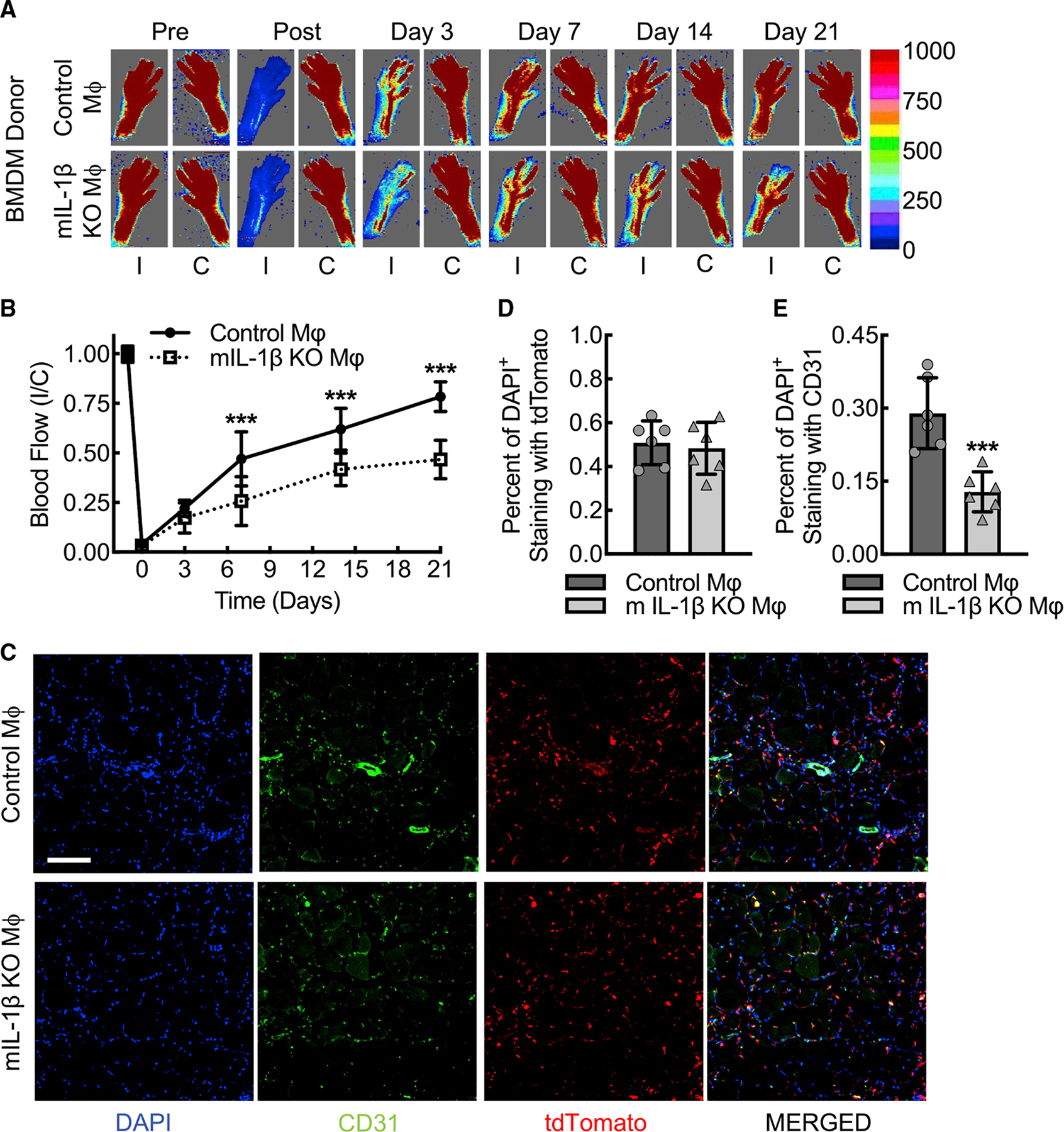
(A and B) Laser Doppler images of flow in the ischemic (I) and contralateral control (C) hindlimbs from myeloid IL-1β-deleted mice (mIL-1β KO) that underwent clodronate liposome macrophage depletion followed by transplant of tdTomato-labeled BMDMs from either wild-type (Control Mφ) or IL-1β-deleted (mIL-1β KO Mφ) Ai9 mice along with quantitative analysis (B) (***, p ≤ 0.0003 compared between control Mφ and mIL-1β KO Mφ for each time point by ANOVA; n = 6 mice total, three males and three females).
(C–E) Immunofluorescence micrographs of ischemic muscle tissue at day 3 post femoral artery ligation mice treated as in (A) along with quantitation of DAPI+tdTomato+ (D) and DAPI+CD31+ (E) cells (***, p = 0.0008 by t test; n = 6 mice, three males and three females). Bar, 100 microns. Data, mean ± SD.
DISCUSSION
These results identify a mechanistic relationship between IL-1β and VEGF-A signaling that is critical for early inflammatory macrophages to set the stage for angiogenesis and arteriogenesis, leading to more complete limb perfusion recovery. In the setting of acute ischemic injury or other perturbations of tissue homeostasis, the early innate inflammatory macrophage response cytokine, IL-1β, appears to participate in an autocrine feedback loop to upregulate VEGF-A expression required for angiogenic responses. By establishing IL-1β signaling as a critical initiator of an angiogenesis program, these mechanistic studies challenge the current paradigm that only inflammation suppressed, or alternatively activated, macrophages can carry out angiogenesis and wound healing.
Though other studies of exogenous IL-1β- and TNF-α-induced transcription of VEGF-A have been demonstrated in cultured cell systems, including those of smooth muscle cells, tumor cell lines, and cardiac myocytes (Gille et al., 1997; Li et al., 1995; Ryuto et al., 1996; Tanaka et al., 2000), the mechanisms of exogenous IL-1β transcriptional regulation were either undefined or differed from what we found here with macrophage systems (i.e., mitogen-activated protein kinase signaling, AP-2, AP-1, or SP-1 versus STAT3 and NF-κB). Prior studies assessing the role of global IL-1β knock-out mice have shown that angiogenesis is reduced in both hindlimb and tumor angiogenesis models (Amano et al., 2004; Voronov et al., 2003); however, those prior studies postulated an IL-1β-dependent HIF-1α activity responsible for VEGF-A expression. Using a conditional myeloid IL-1β-deletion model, we found macrophage HIF-1α activity in the nucleus was not IL-1β dependent and that the HIF-1α-driven component to VEGF-A expression was insufficient to fully overcome the impairment in VEGF-A expression conferred by the loss of the inflammatory cytokine, IL-1β. This highlights the significant and previously underappreciated role that early inflammatory signaling contributes to VEGF-A expression in setting the stage for angiogenesis during acute ischemia.
We demonstrated that autocrine IL-1β signaling in inflammatory macrophage subsets results in selective pro-angiogenic VEGF-A isoform expression and consequently VEGF-A-dependent arteriogenesis via transcription factors STAT3 and NF-κB. This coordinated IL-1β-dependent transcriptional regulation involving STAT3 and NF-κB in macrophage pro-angiogenic VEGF-A expression is validated in vivo in an experimental model of PAD. Though VEGF-A is known to have transient systemic effects of reducing blood pressure (Yang et al., 1996), we found that the higher levels of VEGF-A in the muscle tissue during hindlimb ischemia in wild-type mice or in myeloid IL-1β-deleted mice rescued with wild-type BMDMs did not confer changes in MAP, comparable to other similar studies in vivo (Clayton et al., 2008). Much of the increased VEGF-A signaling likely remains local and paracrine, so the increased VEGF-A does not appear to have a measurable, sustained systemic effect in this model system.
Gain-of-function and small molecule inhibitor studies confirmed that both transcription factors appear necessary to promote the levels of IL-1β-dependent inflammatory macrophage VEGF-A expression required for activation of arteriogenesis. ChIP assays confirmed autocrine IL-1β-dependent binding of STAT3 and NF-κB to the promoter region of VEGF-A. STAT3-dependent VEGF-A expression has been described in human tumor cell lines and aberrant expression of constitutively active STAT3 mutant in cells can upregulate VEGF-A expression, indicating that STAT3 may play an important role in crosstalk between oncogenic and angiogenic events (Niu et al., 2002). Here we find that STAT3 together with NF-κB stands at the convergence of inflammatory and angiogenic events. Given the recent findings of reduced incidence of lung cancer in patients with atherosclerosis by IL-1β inhibition in the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (Ridker et al., 2017), it is interesting to speculate whether suppression of a macrophage IL-1β-STAT3/NF-κB-VEGF-A axis in tumor angiogenesis may be part of the preventive therapeutic mechanism. Additional studies are clearly needed in this area of IL-1β-dependent angiogenesis in the context of both cancer biology and atherosclerotic vascular disease.
Recent studies have demonstrated clinical PAD to be associated with the anti-angiogenic VEGF-A165b isoform, and VEGF-A165b appears upregulated in conditions with impaired limb revascularization (Kikuchi et al., 2014). The mechanism appears to involve VEGF-A165b inhibition of VEGFR1 signaling, inducing an inflammatory phenotype that impairs ischemic muscle vascularization. However, there is reduced expression of VEGF-A165b in patients with critical limb ischemia, relative to those with intermittent claudication or normal peripheral blood flow, suggesting our understanding of mechanisms governing splice variant signaling in vivo is somewhat incomplete (Ganta et al., 2019; Kikuchi et al., 2014; Ngo et al., 2014). Here we found the M1-promoting conditions of LPS+IFN-γ upregulated pro-angiogenic VEGF-A165a in an IL-1β-dependent manner. Interestingly and somewhat surprisingly, IL-1β-gene deletion led to reduced VEGF-A165a mRNA expression and conversely increased relative VEGF-A165b mRNA expression, suggesting that IL-1β-dependent STAT3 and NF-κB transcriptional activity may also, in part, mediate the VEGF-A exon 8 splice site and consequent isoform preference. Moreover, these data raise the question whether there may be ineffective IL-1β signaling or an uncoupling of IL-1β from transcription of the pro-angiogenic VEGF-A isoform in pathologic conditions involving upregulation of VEGF-A165b or impaired angiogenesis. Further research in this area is required to better define these counter regulatory mechanistic relationships.
Limitations of the study
While we demonstrated pro-angiogenic VEGF-A expression and post-developmental arteriogenesis in the PAD model are dependent on macrophage IL-1β, we did not directly confirm that IL-1β acts through VEGF-A to promote revascularization. IL-1β-mediated recruitment of endothelial cell precursors to the ischemic limb was previously demonstrated to be VEGF-A-dependent (Amano et al., 2004). Future studies rescuing macrophage STAT3- and NF-κB-induced expression of pro-angiogenic VEGF-A in the presence or absence of blocking anti-VEGF-A antibody may help determine whether VEGF-A is sufficient to promote arteriogenesis in response to hindlimb ischemia in the absence of macrophage IL-1β signaling. Further, translational relevance of our pathway will require confirmation in human inflammatory macrophage subsets that the IL-1β-STAT3/NF-κB-VEGF-A axis is conserved.
In summary, macrophage-derived IL-1β serves as an important early inflammatory activator of angiogenesis through transcriptional regulation of pro-angiogenic VEGF-A by both STAT3 and NF-κB. Moreover, we have demonstrated that manipulation of this molecular pathway in macrophages is possible and that macrophages can be used to rescue impaired angiogenesis and arteriogenesis in a model of acute limb ischemia. Though further study into the translational potential of this mechanism is required, we are enthusiastic about the potential for this molecular pathway to be therapeutically targeted for the enhancement of wound healing during acute ischemic injury in patients who have underlying PAD.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alan R. Morrison (alan_morrison@brown.edu).
Materials availability
IL-1βfl/fl mouse lines generated in this study will be made available upon request to the lead contact through use of a material transfer agreement (MTA) with Ocean State Research Institute, Inc. The MTA will be necessary to guide 1) general terms on how the mice will be used; 2) liabilities; 3) husbandry and shipping costs; 4) prevention of inappropriate distribution; 5) appropriate acknowledgement of the source; and 5) insurances that all planned animal subjects use is compliant with NIH and VA regulations.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table.
Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NF-κB p65, monoclonal | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| STAT3, monoclonal | Cell Signaling Technology | Cat# 9139; RRID: AB_331757 |
| VEGF-A, polyclonal | Sigma | Cat# SAB2502119 |
| Cleaved IL-1 β, mouse-specific monoclonal | Cell Signaling Technology | Cat# 52718; RRID: AB_2799421 |
| IL-1 β (A6), mouse-specific monoclonal | Cell Signaling Technology | Cat# 12242; RRID: AB_2715503 |
| Anti-β-Actin Antibody (C4) | Santa Cruz | Cat# sc-47778; RRID: AB_2714189 |
| APC anti-mouse CD68 Antibody | BioLegend | Cat# 137008; RRID: AB_10575300 |
| Anti-Actin, α-Smooth Muscle - Cy3™ antibody, Mouse monoclon | MilliporeSigma | Cat# C6198; RRID: AB_476856 |
| CD31 (PECAM-1) Monoclonal Antibody (390), FITC, eBioscience™ | Thermo Fisher | Cat# 11-0311-85; RRID: AB_465013 |
| Rabbit anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher | Cat# A21088; RRID: AB_10373119 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher | Cat# A10043; RRID: AB_2534018 |
| IRDye® 800CW Donkey anti-Mouse IgG Secondary Antibody | Thermo Fisher | Cat# 926–32212; RRID: AB_621847 |
| Biological samples | ||
| Bone marrow-derived macrophages | Davies and Gordon (2005); Healy et al. (2020) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| JSH 23 (NF-κB inhibitor) | Abcam | Cat# ab144824 |
| Stattic (STAT3 inhibitor) | Abcam | Cat# ab120952 |
| Lipopolysaccharides from E. coli 0111:B4 | Sigma Aldrich | Cat# L2630 |
| Recombinant Mouse IFN-γ (carrier-free) | BioLegend | Cat# 575304 |
| Recombinant Mouse IL-13 (carrier-free) | BioLegend | Cat# 575904 |
| Recombinant Mouse IL-4 | BioLegend | Cat# 574304 |
| Cholesterol | MilliporeSigma | Cat# C3045 |
| ProLong™ Gold Antifade Mountant with DAPI | Thermo Fisher | Cat# P36931 |
| Critical commercial assays | ||
| Mouse VEGF DuoSet ELISA | R&D Systems | Cat# DY493; RRID: AB_2884000 |
| ELISA MAX™ Deluxe Set Mouse IL-1 β | BioLegend | Cat# 432604 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| HIF-1 alpha Transcription Factor Assay Kit | Abcam | Cat# ab133104 |
| STAT3 Transcription Factor Assay Kit (Colorimetric) | Abcam | Cat# ab207229 |
| NFkB p65 Transcription Factor Assay Kit | Abcam | Cat# ab133112 |
| Chromatin Immunoprecipitation (ChIP) Assay Kit | Millipore | Cat# 17–295 |
| Standard Macrophage Depletion Kit (Clodrosome® + Encapsome®) | Fisher Scientific | Cat# NC1361099 |
| Mouse Macrophage Nucleofector™ Kit | Lonza | Cat# VPA-1009 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| Click-iT Nascent RNA Capture Kit | Invitrogen/Life Technologies | Cat# C10365 |
| Deposited data | ||
| Raw and analyzed RNA-Seq data | This manuscript | GEO: GSE191152 |
| Experimental models: Organisms/strains | ||
| Mus musculus: C57BL/6J | The Jackson Laboratory | Cat# 000664; RRID: IMSR_JAX:000664 |
| Mus musculus: Csf1tmlercmmer, FVB-Tg(Csf1r-cre/Esr1*)1Jwp/J | The Jackson Laboratory | Cat# 019098; RRID: IMSR_JAX:019098 |
| Mus musculus: IL-1R−/−, B6.129S7-Il1r1tm1Imx/J | The Jackson Laboratory | Cat# 003245; RRID: IMSR_JAX:003245 |
| Mus musculus: Ai9 (RCL-tdT), B6.Cg-Gt(ROSA) 26Sottm9(CAG-tdTomato)Hze/j | The Jackson Laboratory | Cat# 007909; RRID: IMSR_JAX:007909 |
| Mus musculus: IL-1βfl/fl | The Morrison Laboratory and The Brown University Mouse Transgenic and Gene Targeting Facility | This manuscript. |
| Oligonucleotides | ||
| HPRT, 5’-GACCGGTCCCGTCATGCCGA-3’ (sense) and 5’-TGGCCTCCCATCTCCTCCATGACA-3’ (antisense) | Integrated DNA Technologies | N/A |
| GAPDH, 5’-GTGTGAACGGATTTGGCCG-3’ (sense) and 5’-GTGATGGGCTTCCCGTTGAT-3’ (antisense) | Integrated DNA Technologies | N/A |
| IL-1 β, 5’-AAAGATGAAGGGCTGCTTCC-3’ (sense) and 5’-GTCCACGGGAAAGACACAGG-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A, 5’-ACTGGACCCTGGCTTTACTGC-3’ (sense) and 5’-TGATCCGCATGATCTGCATGGTG-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A165a, 5’-CAGAAAATCACTGTGAGCCTTGTT-3’ (sense) and 5’-CTTGGCTTGTCACATCTGCAA-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A165b, 5’-CAGAAAATCACTGTGAGCCTTGTT-3’ (sense) and 5’-CTTTCCGGTGAGAGGTCTGC-3’ (antisense) | Integrated DNA Technologies | N/A |
| TNF-α, 5’-CACGTCGTAGCAAACCACCAA-3’ (sense) and 5’-AGCAAATCGGCTGACGGTGT-3’ (antisense) | Integrated DNA Technologies | N/A |
| NOS2, 5’-CGGCAAACATGACTTCAGGC-3’ (sense) and 5’-GCACATCAAAGCGGCCATAG-3’ (antisense) | Integrated DNA Technologies | N/A |
| ARG1, 5’-ACATTCGCTTGCGAGACGTA-3’ (sense) and 5’-ATCACCTTGCCAATCCCCAG-3’ (antisense) | Integrated DNA Technologies | N/A |
| FIZZ1, 5’-CCTGCTGGGATGACTGCTAC-3’ (sense) and 5’-CAGTGGTCCAGTCAACGAGT-3’ (antisense) | Integrated DNA Technologies | N/A |
| YM1, 5’-GCTTTTGACGAAGAATCTGTGGAC-3’ (sense) and 5’ TCAGGGATCTTGTACCCAGAC 3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A promoter STAT3 ChIP, 5’-TCCACAGGTCGTCTCACTCC-3’ (sense) and 5’-GCAGTCATCAGGAGCTAGGG-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A promoter NF-κB ChIP, 5’-TACAGCCTCTGTTATGCCACG-3’ (sense) and 5’-TGGGTTTCCTCACCTGATCC-3’ (antisense) | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pcDNA3.1-(empty)-TAG | Addgene | Cat# 138209; RRID: Addgene_138209 |
| Stat3-C Flag pRc/CMV | Addgene | Cat# 8722; RRID: Addgene_8722 |
| IKK-2 S177E S181E | Addgene | Cat# 11105; RRID: Addgene_11105 |
| Software and algorithms | ||
| Prism 9 Version 9.3.1 (350), December 7, 2021 | GraphPad | https://www.graphpad.com/support/faq/prism-931-release-notes/ |
| Image J Version: 2.0.0-rc-69/1.52p Build:269a0ad53f Date 2018-12-04T11:30:09+000 | Schneider et al. (2012) | https://imagej.net/software/fiji/downloads |
| moorLDI Version 6.1 | Moor Instruments | https://www.moor.co.uk/en-us/products/control/moorldi2-research-software/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6J (Cat# 000664), Csf1rmercremer (Cat# 019098) (Qian et al., 2011), IL-1R−/− (Cat# 003245)(Glaccum et al., 1997) and Ai9 (RCL-tdT) (Cat# 007909)(Madisen et al., 2010), male and female mice, aged 10–12 weeks, were obtained from The Jackson Laboratory. The IL-1βfl/fl mice were generated by CRISPR/Cas9 technology by Brown University’s Mouse Transgenic and Gene Targeting Facility and were then crossed with Csf1rmercremer mice to generate a myeloid-specific, tamoxifen-inducible IL-1β-deleted mice. For cell tracking, wild-type control and myeloid IL-1β-deleted mice were crossed to the Ai9 (RCL-tdT) reporter strain. For BMDM isolations, animals 10–12 weeks of age were used. For hind limb ischemia, mice of 12–14 weeks of age were used. Male and female animals were used in equal numbers, and sex was analyzed as a biological variable, by 2way ANOVA with sex as a variable. Blood pressure was measured in indicated conscious and anesthetized mice using the tail-cuff method with an automated monitor (CODA-3.0, Kent Scientific Corporation, CT, USA). Measurements were taken at 5 second intervals, and the final blood pressure represented an average of 6 consecutive values at indicated time points before and after femoral artery ligation to induce hind limb ischemia. All experiments were performed in accordance with the Providence VA Medical Center guidelines, and the Institutional Animal Care and Use Committee approved all housing protocols and experimental animal procedures.
Bone marrow derived macrophage (BMDM) isolation and polarization
BMDMs were obtained by in vitro differentiation of primary femur and tibia bone marrow cells using an established protocol(Davies and Gordon, 2005; Healy et al., 2020). In brief, femurs and tibias from the indicated mouse lines mice were dissected, cleaned, dis-infected in 70% ethanol, and washed with fully supplemented RPMI 1640 medium. Red blood cells were removed by ammonium-chloride potassium lysis buffer and subsequent centrifugation. Remaining BM cells were then cultured at a density of 3.5 × 106 cells/10 cm Petri dish in fully supplemented RPMI with 30% (vol/vol) L929 cell-conditioned medium. Cells were matured to phenotypic macrophages over 6–7 days as confirmed by >98% expression of F4/80 and CD68. Non-adherent cells were washed away with PBS while adherent cells were recovered by gentle pipetting in PBS with 1 mM EDTA. Polarization of BMDMs was carried out by incubation with LPS (100 ng/mL; Sigma-Aldrich L2630) and IFN-γ (50 ng/mL; BioLegend 575304) or with IL-4 (10 ng/mL; BioLegend 574304) and IL-13 (10 ng/mL; BioLegend cat.575904) up to 24 hours. In some experiments, BMDMs were treated with the STAT3 inhibitor, Stattic (Abcam, ab120952), or the NF-κB inhibitor, JSH23 (Abcam, ab144824).
Hindlimb ischemia model
Femoral artery ligation surgeries for hindlimb ischemia experiments were performed at the Providence VA Medical Center animal facility. Briefly, mice were anesthetized by inhaled isoflurane (2%) in an induction chamber and then transferred to the surgery platform, where they were kept under anesthesia by a continuous inhalation through a snugly fitted nose cone. The femoral artery was then ligated at two positions, spaced 5 mm apart: one just below the inguinal ligament and the second distal to the superficial epigastric artery. All branches between the two ligatures were ligated and the femoral artery segment was completely excised. Analgesic (Buprenorphine 0.05–0.1 mg/kg, Par Pharmaceuticals) was administered intraperitoneally at the time of surgery, four hours post-surgery, and then twice a day for the next 72 hours. In some animals, early inflammatory assessment was performed on Day 3 post ligation, where gastrocnemius ischemic muscle tissue was dissected, harvested, and segmented at the midpoint of the muscle for subsequent analysis. The mid-gastrocnemius was homogenized for subsequent TRIzol-based fractionation and isolation of DNA, RNA, and protein(Chomczynski, 1993). RNA was treated with a DNase kit (Life Technologies) to remove genomic contaminant prior to reverse transcription. Immediately adjacent segments to the mid-gastrocnemius were fixed for both cryomolding and paraffin embedding, followed by immunofluorescence or traditional hematoxylin and eosin histology, respectively. In some mice, mid-gastrocnemius tissue sections from both ischemic muscle and contralateral nonischemic control muscle were harvested on Day 21 post femoral artery ligation for additional immunofluorescence histology.
Laser Doppler blood flow imaging
Mice were anesthetized by inhaled isoflurane (2%) as described above and placed on a heating platform where body temperature was kept at 37±0.5°C to minimize the influence of body temperature on blood flow imaging. Blood flow images of the hind paws were acquired using a Moor Laser Doppler Imager (MoorLDI2, Moor Instruments). The data were analyzed with moorLDI image processing software (moorLDI V6.1) and reported as the ratio of blood flow in the ischemic to the non-ischemic, contralateral control hind paws. Blood flow was quantified before surgery, immediately after surgery, and at postoperative Days 3, 7, 14, and 21.
METHOD DETAILS
Cholesterol crystal inflammasome assay
Cholesterol crystals were generated as previously described (Duewell et al., 2010). Cholesterol (Sigma-Aldrich C3045) was dissolved in 1-propanol at a concentration of 2 mg/ml. Crystals were precipitated overnight at room temperature by dilution to 40% 1-propanol with water. Crystals were pelleted by centrifugation and dried at 70°C. Crystal pellets were then re-suspended in 0.1% FBS in PBS at a concentration of 50 mg/ml. Average crystal size of 1–2 μm was confirmed by microscopy. BMDMs (+/− priming with low dose (10 ng/ml) LPS for 2 hours) were then either exposed to 1000 μg/ml cholesterol crystals or vehicle control for 24 hours. Culture supernatants were collected, and IL-1β and VEGF-A expression were quantified by ELISA.
Mouse IL-1β and mouse VEGF-A ELISA kits
Conditioned media from either polarized BMDMs or inflammasome-stimulated (cholesterol crystal assay) were used to evaluate IL-1β (BioLegend, 432604) and VEGF-A (R&D Systems, DY493) protein concentrations, according to the manufacturer’s instructions. Each animal measurement reflects the average of experimental triplicates.
RNA-sequencing
We extracted total RNA from polarized BMDMs with the RNeasy kit (QIAGEN; cat.74106). The RNA was quantified, and the quality verified, using Nanodrop One (Thermo Fisher). RNA-seq was performed on 100 ng total RNA using 76 bp single-end sequencing on an Illumina HiSeq 2500 according to Illumina protocols at the Yale Center for Genomic Analysis. Reads were aligned with TopHat v.2.1.1 to the reference UCSC mouse genome and transcript annotation (GRCm38/mm10), and then TopHat alignments were then processed by Cufflinks v2.2.1 to identify differentially expressed genes(Trapnell et al., 2012).
Quantitative RT-PCR
Total RNA from ischemic muscle tissue (100–150 mg) was extracted using the Trizol kit (Fisher 15596026). Total RNA from BMDMs (1.5million cells/mL) was extracted from RLT Lysis buffer containing 2-βME (QIAGEN; RNeasy Kit cat.74106) according to the manufacturer’s protocol. RNA concentration was measured by Nanodrop One (Thermo Fisher). First-strand complementary DNA was synthesized with iScript cDNA synthesis Kit (Bio-Rad; 1708891). Equal amounts of RNA (200 ng/μl) were used as templates in each reaction. Quantitative RT-PCR was performed with SSoAdvanved UnivSYBR Supermix (Bio-Rad, 1725274) on the StepOne Plus PCR machine (AB Applied Biosystems) using primers. All measurements were made in triplicate and then averaged, using HPRT as the housekeeping gene for BMDMs and GAPDH as the housekeeping gene for muscle tissue lysates. Primers: HPRT, 5’-GACCGGTCCCGTCATGCCGA-3’ (sense) and 5’-TGGCCTCCCATCTCCTCCATGACA-3’ (antisense); GAPDH, 5’-GTGTGAACG GATTTGGCCG-3’ (sense) and 5’-GTGATGGGCTTCCCGTTGAT-3’ (antisense); IL-1β, 5’-AAAGATGAAGGGCTGCTTCC-3’ (sense) and 5’-GTCCACGGGAAAGACACAGG-3’ (antisense); VEGF-A, 5’-ACTGGACCCTGGCTTTACTGC-3’ (sense) and 5’-TGATCCG CATGATCTGCATGGTG-3’ (antisense); VEGF-A165a, 5’-CAGAAAATCACTGTGAGCCTTGTT-3’ (sense) and 5’-CTTGGCTTGTCACATCTGCAA-3’ (antisense); VEGF-A165b, 5’-CAGAAAATCACTGTGAGCCTTGTT-3’ (sense) and 5’-CTTTCCGGTGAGAGGTCTGC-3’ (antisense); TNF-α, 5’-CACGTCGTAGCAAACCACCAA-3’ (sense) and 5’-AGCAAATCGGCTGACGGTGT-3’ (antisense); NOS2, 5’-CGGCAAACATGACTTCAGGC-3’ (sense) and 5’-GCACATCAAAGCGGCCATAG-3’ (antisense); ARG1, 5’-ACATTCGCTTGCGAGAC GTA-3’ (sense) and 5’-ATCACCTTGCCAATCCCCAG-3’ (antisense); FIZZ1, 5’-CCTGCTGGGATGACTGCTAC-3’ (sense) and 5’-CAG TGGTCCAGTCAACGAGT-3’ (antisense); YM1, 5’-GCTTTTGACGAAGAATCTGTGGAC-3’ (sense) and 5’ TCAGGGATCTTGTACC CAGAC 3’ (antisense); VEGF-A promoter STAT3 ChIP, 5’-TCCACAGGTCGTCTCACTCC-3’ (sense) and 5’-GCAGTCATCAGGAGC TAGGG-3’ (antisense); VEGF-A promoter NF-κB ChIP, 5’-TACAGCCTCTGTTATGCCACG-3’ (sense) and 5’-TGGGTTTCCTCACCTGATCC-3’ (antisense).
Nuclear extraction and transcription factor assays
Nuclear and cytoplasmic fractionation of BMDM or muscle tissue lysates was carried out according to manufacturer instructions (Abcam, ab113474). Briefly, cells were collected with cell scraper and pelleted by centrifugation. Pellets were re-suspended in the pre-extraction buffer. After a centrifugation, supernatant containing cytoplasmic fraction was frozen down and stored. The pellet was again centrifuged and re-suspended in nuclear extraction buffer to generate the nuclear fraction. The nuclear fraction was used for HIF-1α, STAT3, and NF-κB p65 transcription assays according to the manufacturer’s instructions (Abcam; ab133104, ab207229, and ab133112, respectively).
Immunoblotting
Proteins from ischemic muscle tissue (100–150 mg) were extracted using Trizol kit (Fisher 15596026). Proteins from BMDMs (1.5million/mL) were collected by using Laemmli sample buffer (Bio-Rad, cat.1610737) containing 2-βME; then boiled at 95°C and aliquoted. Aliquots from tissue lysates or cell lysates were separated by Tris/Glycine/SDS–PAGE (Bio-Rad 1610732) and blotted into PVDF membranes. Membranes were blocked in fluorescent blocking buffer (Rockland Immunochemicals, cat.MB070) and probed at 4°C overnight with antibodies against VEGF-A (1:1000; Sigma SAB2502119); cleaved mature IL-1β (1:500; Cell Signaling Technology 52718); β-Actin (1:1000, sc-47778). After multiple washes, membranes were incubated with fluorescent secondary antibodies (Thermo Fisher A21088, 1:5000; Thermo Fisher A10043, 1:5000; and LI-COR 926–32212, 1:5000). Protein bands were quantified by densitometry, using the LI-COR Imager (Odyssey CLx) and normalization to β-Actin.
Nascent RNA capture assay
BMDMs were subject to polarizing stimuli for 12 hours followed by Nascent RNA Capture using Click-iT chemistry, according to the manufacturer’s instructions (Life technologies, C10365).
Transfections with gain-of-function mutants
BMDMs were transfected with empty vector control (pcDNA3.1-(empty)-TAG, Addgene 138209), constitutively active STAT3 (Stat3-C Flag pRc/CMV, Addgene 8722), or constitutively active IKK-2 (IKK-2 S177E S181E, Addgene 11105) using a mouse macrophage transfection system, according to the manufacturer’s instructions (Lonza, VPA-1009; Mouse Macrophage Nucleofector Kit). Briefly, 1 × 106 cells were combined with 3 μg plasmid DNA individually, or 2.5 μg each together combined in a total volume of 100μL Nucleofector solution and transfected with the Nucleofector 2b Device (Lonza, AAB-1001) per macrophage-specified setting Y-001. Cells were then plated on a sterile 12-well plate and incubated for 12 hours at 37°C in 1 mL of fully supplemented RPMI 1640 and then polarized with LPS and IFN-γ for an additional 12 hours at 37°C. Conditioned media and BMDM lysate RNA were then harvested for ELISA and quantitative RT-PCR, respectively.
Chromatin immunoprecipitation (ChIP)
STAT3 or NF-κB ChIP assays were carried out according to the manufacturer’s protocols (Millipore, 17–295). In brief, LPS+IFN-γ polarized BMDM lysates from 1.5 million cells, underwent histone cross-linking to DNA using formaldehyde (37%), and then the cells were scraped and collected in a PBS solution containing proteases inhibitors. After wash and centrifugation, the cell pellet was lysed in SDS buffer, sonicated, and spun down again. The pellet was then re-suspended in ChIP buffer with Protein A agarose beads pre-coated with salmon sperm DNA and then incubated overnight with antibodies for STAT3 (1:100; Cell Signaling Technology 9139) and its isotype control (1:50; Thermo Fisher; 02–6200) or NF-kB p65 (1:100; Cell Signaling Technology 8242) and its isotype control (1:50, R&D Systems AB-105-C). Samples were then washed and underwent reversal of histone-DNA crosslinking by NaCl (5 M) for 4 hours at 65°C. DNA was recovered with phenol/chloroform extraction and ethanol precipitation. Site-specific primers encompassing either the STAT3 or NF-κB known seed sites in the VEGF-A promoter region were used for quantitative RT-PCR. Results were expressed as relative mRNA expression based on the delta CT of STAT3- or NF-κB-specific antibodies to their respective isotype control antibodies.
Clodronate liposome macrophage depletion
Control and clodronate liposomes (Fisher Scientific, Standard Macrophage Depletion Kit; Fisher Scientific NC1361099) were intraperitoneally injected into the mice at a concentration of 250 μl/25 g two days prior to hindlimb ischemia to deplete macrophages. A second injection of 50 μl was carried out directly into ischemic muscle tissue immediately following femoral artery ligation surgery. For macrophage transplant experiments, 500,000 BMDMs suspended in phosphate buffered saline were intravenously (tail vein) injected 6 hours after femoral artery ligation.
Histology and immunofluorescence microscopy
Paraffin-embedded muscle tissues were sectioned at 10-μm thick, stained with hematoxylin and eosin (H&E), and imaged on a Nikon Eclipse 80i, Nikon Plan Apochromat 20x objective lens (numerical aperture 0.75). For immunofluorescence microscopy, muscle tissue was sectioned at 10-μm-thickness by cryostat. Sections were incubated with CD31-FITC (1:100; Thermo Fisher, 11-0311-85), anti-mouse CD68-APC (1:100; BioLegend, 137008), or anti-mouse α-Smooth Muscle Actin-Cy3 (1:100; MilliporeSigma, C6198), and then mounted with 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Thermo Fisher, P36931). Using a Nikon Eclipse 80i inverted microscope with Nikon Plan Apochromat 10x objective lens (numerical aperture 0.3), 6 fluorescence images were acquired in the regions of interest as determined by cellular infiltration using the DAPI staining. Quantification of cell types was expressed as a ratio of each DAPI+ cellular marker (CD68, CD31, SMA) to the total DAPI stained cells in the field. Each individual data point represented one animal and reflected the average of 3–4 images captured from adjacent tissue sections per slide. All subsequent analyses were performed by Image J software(Schneider et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical data were analyzed with the use of Prism 9 (GraphPad). Results are presented as mean (SD) for continuous variables with normal distribution or equal variance and as median (interquartile range) for continuous variables without normal distribution. The t-test was used to compare normally distributed continuous variables between 2 independent groups. Differences between multiple groups was assessed by ANOVA followed by Tukey’s post hoc multiple comparisons test. The Mann-Whitney U test will be used for continuous variables not normally distributed.
Supplementary Material
Highlights.
Macrophage VEGF-A expression depends on autocrine IL-1β expression and signaling
IL-1β signaling activates both STAT3 and NF-κB to promote VEGF-A transcription
STAT3 and NF-κB upregulate the pro-relative to the anti-angiogenic VEGF-A isoform
In a PAD model, macrophage IL-1β is required for VEGF-A-driven angio/arteriogenesis
ACKNOWLEDGMENTS
Research reported in this publication was supported by Research Project Grants NIH NHLBI R01HL139795 (A.R.M.), R01HL148727 (G.C.), R01HL46716 (F.S.), R01HL128831 (F.S.), R01HL142818 (H.C.), R01HL130230 (Q.L.), IDeA NIH NIGMS P20GM103652 (A.R.M.), F31HL147466 (J.M.B.), R25HL088992 (J.N. and C.P.), and by NIH NIGMS 8P30 GM103410 (The Mouse Transgenic and Gene Targeting Facility at the Brown University). This work was also supported by VA VHA BLR&D 7IK2BX002527 (A.R.M.), VA VHA CSR&D 1I01CX002231 (A.R.M), and VA VHA CSR&D 1I01CX001892 (G.C.). The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the US government. This work was also supported by an AHA Transformational Project (H.C.) and by the Meyerh-off Program at University of Maryland Baltimore County (C.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110309.
DECLARATION OF INTERESTS
The authors have no disclosures to declare.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Amano K, Okigaki M, Adachi Y, Fujiyama S, Mori Y, Kosaki A, Iwasaka T, and Matsubara H (2004). Mechanism for IL-1 beta-mediated neovascularization unmasked by IL-1 beta knock-out mice. J. Mol. Cell. Cardiol 36, 469–480. [DOI] [PubMed] [Google Scholar]
- Apte RS, Chen DS, and Ferrara N (2019). VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, and Jain RK (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MW, Alon R, and Springer TA (1996). The C-C chemokine MCP-1 differentially modulates the avidity of beta 1 and beta 2 integrins on T lymphocytes. Immunity 4, 179–187. [DOI] [PubMed] [Google Scholar]
- Charo IF (2007). Macrophage polarization and insulin resistance: PPAR-gamma in control. Cell Metab 6, 96–98. [DOI] [PubMed] [Google Scholar]
- Chomczynski P (1993). A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques 15, 532–534, 536–537. [PubMed] [Google Scholar]
- Choudhry H, and Harris AL (2018). Advances in hypoxia-inducible factor biology. Cell Metab 27, 281–298. [DOI] [PubMed] [Google Scholar]
- Clayton JA, Chalothorn D, and Faber JE (2008). Vascular endothelial growth factor-A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ. Res 103, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JQ, and Gordon S (2005). Isolation and culture of murine macrophages. Methods Mol. Biol 290, 91–103. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber A, and Eberhardt RT (2016). The current state of critical limb ischemia: a systematic review. JAMA Surg 151, 1070–1077. [DOI] [PubMed] [Google Scholar]
- Farboud B, Severson AF, and Meyer BJ (2019). Strategies for efficient genome editing using CRISPR-Cas9. Genetics 211, 431–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDer-mott MM, Norman PE, Sampson UK, Williams LJ, Mensah GA, et al. (2013). Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet 382, 1329–1340. [DOI] [PubMed] [Google Scholar]
- Ganta VC, Choi M, Farber CR, and Annex BH (2019). Antiangiogenic VEGF165b regulates macrophage polarization via S100A8/S100A9 in peripheral artery disease. Circulation 139, 226–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, and Littman DR (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Gerhard-Herman MD, Gornik HL, Barrett C, Barshes NR, Corriere MA, Drachman DE, Fleisher LA, Fowkes FG, Hamburg NM, Kinlay S, et al. (2017). 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: a report of the American College of Cardiology/American heart association task force on clinical practice guidelines. Circulation 135, e726–e779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille J, Swerlick RA, and Caughman SW (1997). Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO J 16, 750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, and Morrissey PJ (1997). Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol 159, 3364–3371. [PubMed] [Google Scholar]
- Gordon S, and Taylor PR (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol 5, 953–964. [DOI] [PubMed] [Google Scholar]
- Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V, Ot-suka F, Gannon RL, Braumann RE, Dickinson MH, et al. (2018). CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Invest 128, 1106–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy A, Berus JM, Christensen JL, Lee C, Mantsounga C, Dong W, Watts JP Jr., Assali M, Ceneri N, Nilson R, et al. (2020). Statins disrupt macrophage Rac1 regulation leading to increased atherosclerotic plaque calcification. Arterioscl. Thromb. Vasc. Biol 40, ATVBAHA119313832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito WD, Arras M, Winkler B, Scholz D, Schaper J, and Schaper W (1997). Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ. Res 80, 829–837. [DOI] [PubMed] [Google Scholar]
- Iyer SR, and Annex BH (2017). Therapeutic angiogenesis for peripheral artery disease: lessons learned in translational science. JACC Basic Transl. Sci 2, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten N, Donners MM, Wagenaar A, Cleutjens JP, van Rooijen N, de Winther MP, and Post MJ (2013). Local delivery of polarized macrophages improves reperfusion recovery in a mouse hind limb ischemia model. PLoS One 8, e68811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi R, Nakamura K, MacLauchlan S, Ngo DT, Shimizu I, Fuster JJ, Katanasaka Y, Yoshida S, Qiu Y, Yamaguchi TP, et al. (2014). An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med 20, 1464–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, and Nair MG (2019). Macrophages in wound healing: activation and plasticity. Immunol. Cell Biol 97, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Perrella MA, Tsai JC, Yet SF, Hsieh CM, Yoshizumi M, Patterson C, Endege WO, Zhou F, and Lee ME (1995). Induction of vascular endothelial growth factor gene expression by interleukin-1 beta in rat aortic smooth muscle cells. J. Biol. Chem 270, 308–312. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubbrey AL, Allison KC, Lee-Sherick AB, Jakubzick CV, and Janssen WJ (2017). Promoter specificity and efficacy in conditional and inducible transgenic targeting of lung macrophages. Front. Immunol 8, 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, et al. (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278, 860–866. [DOI] [PubMed] [Google Scholar]
- Mitchell M, Backus K, and Hynes MM (2013). Connecticut Resident Deaths: Leading Causes of Death by Sex, Race, and Hispanic Ethnicity, 2006–2010 (State of Connecticut Department of Public Health; ). [Google Scholar]
- Moreno SG (2018). Depleting macrophages in vivo with clodronate-liposomes. Methods Mol. Biol 1784, 259–262. [DOI] [PubMed] [Google Scholar]
- Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Ceneri N, Zhang J, Zhuang ZW, Sinusas AJ, Pardi R, et al. (2014). Chemokine-coupled beta2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med 211, 1957–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo DT, Farb MG, Kikuchi R, Karki S, Tiwari S, Bigornia SJ, Bates DO, LaValley MP, Hamburg NM, Vita JA, et al. (2014). Antiangiogenic actions of vascular endothelial growth factor-A165b, an inhibitory isoform of vascular endothelial growth factor-A, in human obesity. Circulation 130, 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, et al. (2002). Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21, 2000–2008. [DOI] [PubMed] [Google Scholar]
- Peiris-Pages M (2012). The role of VEGF 165b in pathophysiology. Cell Adh. Migr 6, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilny E, Smolarczyk R, Jarosz-Biej M, Hadyk A, Skorupa A, Ciszek M, Krakowczyk L, Kulach N, Gillner D, Sokol M, et al. (2019). Human ADSC xenograft through IL-6 secretion activates M2 macrophages responsible for the repair of damaged muscle tissue. Stem Cell Res. Ther 10, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JW (2009). Trophic macrophages in development and disease. Nat. Rev. Immunol 9, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, and Pollard JW (2011). CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramgolam VS, DeGregorio SD, Rao GK, Collinge M, Subaran SS, Markovic-Plese S, Pardi R, and Bender JR (2010). T cell LFA-1 engagement induces HuR-dependent cytokine mRNA stabilization through a Vav-1, Rac1/2, p38MAPK and MKK3 signaling cascade. PLoS one 5, e14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders ME, Sho M, Izawa A, Wang P, Mukhopadhyay D, Koss KE, Geehan CS, Luster AD, Sayegh MH, and Briscoe DM (2003). Proinflammatory functions of vascular endothelial growth factor in alloimmunity. J. Clin. Invest 112, 1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, and Group CT (2017). Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842. [DOI] [PubMed] [Google Scholar]
- Ryuto M, Ono M, Izumi H, Yoshida S, Weich HA, Kohno K, and Kuwano M (1996). Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP-1. J. Biol. Chem 271, 28220–28228. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, and Berg T (2006). Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR, and Kim Y (2004). Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett 571, 50–54. [DOI] [PubMed] [Google Scholar]
- Shishehbor MH, White CJ, Gray BH, Menard MT, Lookstein R, Rosenfield K, and Jaff MR (2016). Critical limb ischemia: an expert statement. J. Am. Coll. Cardiol 68, 2002–2015. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, et al. (2011). Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kanai H, Sekiguchi K, Aihara Y, Yokoyama T, Arai M, Kanda T, Nagai R, and Kurabayashi M (2000). Induction of VEGF gene transcription by IL-1 beta is mediated through stress-activated MAP kinases and Sp1 sites in cardiac myocytes. J. Mol. Cell. Cardiol 32, 1955–1967. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Royen N, Hoefer I, Bottinger M, Hua J, Grundmann S, Voskuil M, Bode C, Schaper W, Buschmann I, and Piek JJ (2003). Local monocyte chemoattractant protein-1 therapy increases collateral artery formation in apolipoprotein E-deficient mice but induces systemic monocytic CD11b expression, neointimal formation, and plaque progression. Circ. Res 92, 218–225. [DOI] [PubMed] [Google Scholar]
- Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, and Apte RN (2003). IL-1 is required for tumor invasive-ness and angiogenesis. Proc. Natl. Acad. Sci. U S A 100, 2645–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JG, Collinge M, Ramgolam V, Ayalon O, Fan XC, Pardi R, and Bender JR (2006). LFA-1-dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J. Immunol 176, 2105–2113. [DOI] [PubMed] [Google Scholar]
- Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, Brachvogel B, Hammerschmidt M, Nagy A, Ferrara N, et al. (2012). CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 120, 613–625. [DOI] [PubMed] [Google Scholar]
- Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui TG, Sugiono M, Waine E, Perrin R, et al. (2004). VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res 64, 7822–7835. [DOI] [PubMed] [Google Scholar]
- Yang R, Thomas GR, Bunting S, Ko A, Ferrara N, Keyt B, Ross J, and Jin H (1996). Effects of vascular endothelial growth factor on hemodynamics and cardiac performance. J. Cardiovasc. Pharmacol 27, 838–844. [DOI] [PubMed] [Google Scholar]
- Zhang J, Modi Y, Yarovinsky T, Yu J, Collinge M, Kyriakides T, Zhu Y, Sessa WC, Pardi R, and Bender JR (2012). Macrophage beta2 integrin-mediated, HuR-dependent stabilization of angiogenic factor-encoding mRNAs in inflammatory angiogenesis. Am. J. Pathol 180, 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table.
Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NF-κB p65, monoclonal | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| STAT3, monoclonal | Cell Signaling Technology | Cat# 9139; RRID: AB_331757 |
| VEGF-A, polyclonal | Sigma | Cat# SAB2502119 |
| Cleaved IL-1 β, mouse-specific monoclonal | Cell Signaling Technology | Cat# 52718; RRID: AB_2799421 |
| IL-1 β (A6), mouse-specific monoclonal | Cell Signaling Technology | Cat# 12242; RRID: AB_2715503 |
| Anti-β-Actin Antibody (C4) | Santa Cruz | Cat# sc-47778; RRID: AB_2714189 |
| APC anti-mouse CD68 Antibody | BioLegend | Cat# 137008; RRID: AB_10575300 |
| Anti-Actin, α-Smooth Muscle - Cy3™ antibody, Mouse monoclon | MilliporeSigma | Cat# C6198; RRID: AB_476856 |
| CD31 (PECAM-1) Monoclonal Antibody (390), FITC, eBioscience™ | Thermo Fisher | Cat# 11-0311-85; RRID: AB_465013 |
| Rabbit anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher | Cat# A21088; RRID: AB_10373119 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher | Cat# A10043; RRID: AB_2534018 |
| IRDye® 800CW Donkey anti-Mouse IgG Secondary Antibody | Thermo Fisher | Cat# 926–32212; RRID: AB_621847 |
| Biological samples | ||
| Bone marrow-derived macrophages | Davies and Gordon (2005); Healy et al. (2020) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| JSH 23 (NF-κB inhibitor) | Abcam | Cat# ab144824 |
| Stattic (STAT3 inhibitor) | Abcam | Cat# ab120952 |
| Lipopolysaccharides from E. coli 0111:B4 | Sigma Aldrich | Cat# L2630 |
| Recombinant Mouse IFN-γ (carrier-free) | BioLegend | Cat# 575304 |
| Recombinant Mouse IL-13 (carrier-free) | BioLegend | Cat# 575904 |
| Recombinant Mouse IL-4 | BioLegend | Cat# 574304 |
| Cholesterol | MilliporeSigma | Cat# C3045 |
| ProLong™ Gold Antifade Mountant with DAPI | Thermo Fisher | Cat# P36931 |
| Critical commercial assays | ||
| Mouse VEGF DuoSet ELISA | R&D Systems | Cat# DY493; RRID: AB_2884000 |
| ELISA MAX™ Deluxe Set Mouse IL-1 β | BioLegend | Cat# 432604 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| HIF-1 alpha Transcription Factor Assay Kit | Abcam | Cat# ab133104 |
| STAT3 Transcription Factor Assay Kit (Colorimetric) | Abcam | Cat# ab207229 |
| NFkB p65 Transcription Factor Assay Kit | Abcam | Cat# ab133112 |
| Chromatin Immunoprecipitation (ChIP) Assay Kit | Millipore | Cat# 17–295 |
| Standard Macrophage Depletion Kit (Clodrosome® + Encapsome®) | Fisher Scientific | Cat# NC1361099 |
| Mouse Macrophage Nucleofector™ Kit | Lonza | Cat# VPA-1009 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| Click-iT Nascent RNA Capture Kit | Invitrogen/Life Technologies | Cat# C10365 |
| Deposited data | ||
| Raw and analyzed RNA-Seq data | This manuscript | GEO: GSE191152 |
| Experimental models: Organisms/strains | ||
| Mus musculus: C57BL/6J | The Jackson Laboratory | Cat# 000664; RRID: IMSR_JAX:000664 |
| Mus musculus: Csf1tmlercmmer, FVB-Tg(Csf1r-cre/Esr1*)1Jwp/J | The Jackson Laboratory | Cat# 019098; RRID: IMSR_JAX:019098 |
| Mus musculus: IL-1R−/−, B6.129S7-Il1r1tm1Imx/J | The Jackson Laboratory | Cat# 003245; RRID: IMSR_JAX:003245 |
| Mus musculus: Ai9 (RCL-tdT), B6.Cg-Gt(ROSA) 26Sottm9(CAG-tdTomato)Hze/j | The Jackson Laboratory | Cat# 007909; RRID: IMSR_JAX:007909 |
| Mus musculus: IL-1βfl/fl | The Morrison Laboratory and The Brown University Mouse Transgenic and Gene Targeting Facility | This manuscript. |
| Oligonucleotides | ||
| HPRT, 5’-GACCGGTCCCGTCATGCCGA-3’ (sense) and 5’-TGGCCTCCCATCTCCTCCATGACA-3’ (antisense) | Integrated DNA Technologies | N/A |
| GAPDH, 5’-GTGTGAACGGATTTGGCCG-3’ (sense) and 5’-GTGATGGGCTTCCCGTTGAT-3’ (antisense) | Integrated DNA Technologies | N/A |
| IL-1 β, 5’-AAAGATGAAGGGCTGCTTCC-3’ (sense) and 5’-GTCCACGGGAAAGACACAGG-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A, 5’-ACTGGACCCTGGCTTTACTGC-3’ (sense) and 5’-TGATCCGCATGATCTGCATGGTG-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A165a, 5’-CAGAAAATCACTGTGAGCCTTGTT-3’ (sense) and 5’-CTTGGCTTGTCACATCTGCAA-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A165b, 5’-CAGAAAATCACTGTGAGCCTTGTT-3’ (sense) and 5’-CTTTCCGGTGAGAGGTCTGC-3’ (antisense) | Integrated DNA Technologies | N/A |
| TNF-α, 5’-CACGTCGTAGCAAACCACCAA-3’ (sense) and 5’-AGCAAATCGGCTGACGGTGT-3’ (antisense) | Integrated DNA Technologies | N/A |
| NOS2, 5’-CGGCAAACATGACTTCAGGC-3’ (sense) and 5’-GCACATCAAAGCGGCCATAG-3’ (antisense) | Integrated DNA Technologies | N/A |
| ARG1, 5’-ACATTCGCTTGCGAGACGTA-3’ (sense) and 5’-ATCACCTTGCCAATCCCCAG-3’ (antisense) | Integrated DNA Technologies | N/A |
| FIZZ1, 5’-CCTGCTGGGATGACTGCTAC-3’ (sense) and 5’-CAGTGGTCCAGTCAACGAGT-3’ (antisense) | Integrated DNA Technologies | N/A |
| YM1, 5’-GCTTTTGACGAAGAATCTGTGGAC-3’ (sense) and 5’ TCAGGGATCTTGTACCCAGAC 3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A promoter STAT3 ChIP, 5’-TCCACAGGTCGTCTCACTCC-3’ (sense) and 5’-GCAGTCATCAGGAGCTAGGG-3’ (antisense) | Integrated DNA Technologies | N/A |
| VEGF-A promoter NF-κB ChIP, 5’-TACAGCCTCTGTTATGCCACG-3’ (sense) and 5’-TGGGTTTCCTCACCTGATCC-3’ (antisense) | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pcDNA3.1-(empty)-TAG | Addgene | Cat# 138209; RRID: Addgene_138209 |
| Stat3-C Flag pRc/CMV | Addgene | Cat# 8722; RRID: Addgene_8722 |
| IKK-2 S177E S181E | Addgene | Cat# 11105; RRID: Addgene_11105 |
| Software and algorithms | ||
| Prism 9 Version 9.3.1 (350), December 7, 2021 | GraphPad | https://www.graphpad.com/support/faq/prism-931-release-notes/ |
| Image J Version: 2.0.0-rc-69/1.52p Build:269a0ad53f Date 2018-12-04T11:30:09+000 | Schneider et al. (2012) | https://imagej.net/software/fiji/downloads |
| moorLDI Version 6.1 | Moor Instruments | https://www.moor.co.uk/en-us/products/control/moorldi2-research-software/ |