

Abstract
Friedreich's ataxia is a rare disorder resulting from deficiency of frataxin, a mitochondrial protein implicated in the synthesis of iron-sulfur clusters. Preclinical studies in mice have shown that gene therapy is a promising approach to treat individuals with Friedreich's ataxia. However, a recent report provided evidence that AAVrh10-mediated overexpression of frataxin could lead to cardiotoxicity associated with mitochondrial dysfunction. While evaluating an AAV9-based frataxin gene therapy using a chicken β-actin promoter, we showed that toxic overexpression of frataxin could be reached in mouse liver and heart with doses between 1 × 1013 and 1 × 1014 vg/kg. In a mouse model of cardiac disease, these doses only corrected cardiac dysfunction partially and transiently and led to adverse findings associated with iron-sulfur cluster deficiency in liver. We demonstrated that toxicity required frataxin's primary function by using a frataxin construct bearing the N146K mutation, which impairs binding to the iron-sulfur cluster core complex. At the lowest tested dose, we observed moderate liver toxicity that was accompanied by progressive loss of transgene expression and liver regeneration. Together, our data provide insights into the toxicity of frataxin overexpression that should be considered in the development of a gene therapy approach for Friedreich's ataxia.
Keywords: Friedreich's ataxia, gene therapy, adeno-associated virus, frataxin, iron-sulfur cluster, mitochondria, toxicity, liver regeneration, transgene overexpression, loss of transgene expression
Graphical abstract
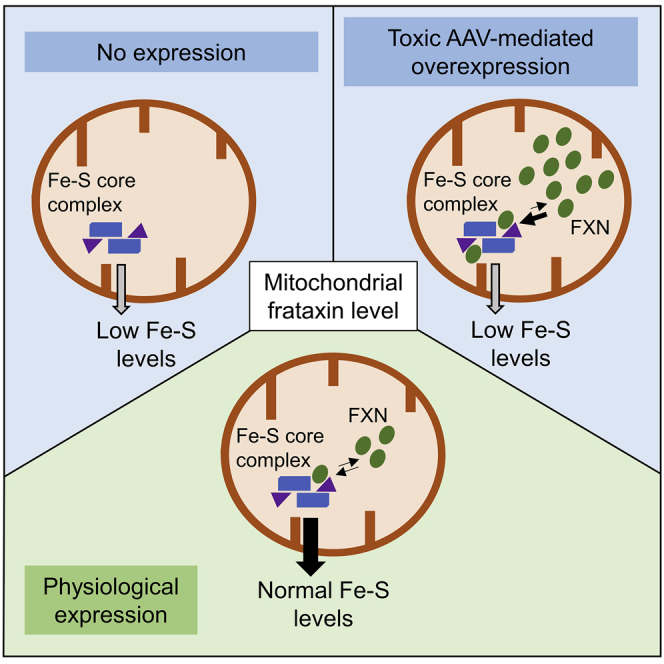
Martelli and colleagues provide evidence of AAV9-mediated toxicity of high frataxin expression in mice. The authors demonstrate that toxicity is associated with frataxin's primary function in iron-sulfur cluster biogenesis and should therefore be carefully considered in the development of a safe and effective gene therapy for Friedreich's ataxia.
Introduction
Friedreich's ataxia (FRDA), the most prevalent autosomal recessive ataxia in Caucasians, is a devastating early-onset neurodegenerative disease characterized by a progressive spinocerebellar and sensory ataxia, hypertrophic cardiomyopathy, and increased incidence of diabetes.1,2 The major FRDA-causing mutation is a GAA-repeat expansion within the first intron of the FXN gene, resulting in decreased transcription of the gene and a reduced amount of the encoded protein, frataxin (FXN).3 FXN is a mitochondrial and ubiquitously expressed protein implicated in the biogenesis of iron-sulfur (Fe-S) clusters, inorganic cofactors with essential roles in various cellular functions such as mitochondrial respiration.4 FXN is involved in the early steps of Fe-S cluster assembly within mitochondria via its binding and regulation of the Fe-S cluster core complex consisting of the cysteine desulfurase NFS1, the scaffold protein ISCU, and accessory proteins ISD11 and the mitochondrial acyl carrier protein (ACP).5, 6, 7, 8, 9 Reduction of FXN level in cells causes impairment of Fe-S cluster biogenesis and reduction of the level of Fe-S cluster client proteins, leading to multiple cellular metabolic dysfunctions and ultimately cell death.10,11
Preclinical evaluations in cardiac and neuronal mouse models of FRDA have shown that AAV-based gene therapy is a promising therapeutic approach for the treatment of the disease.12, 13, 14, 15 The first gene therapy proof of concept was obtained with the intravenous injection of an AAVrh10 construct expressing HA-tagged FXN under the control of a ubiquitous chicken b-actin promoter (CAG) in a cardiac mouse model of FRDA.12 The study showed the approach can correct the severe cardiac phenotype rapidly and efficiently. However, a recent report showed that systemic injection of high doses of the same AAVrh10-FXN construct in mice could be toxic.16 Transgene protein levels higher than 20-fold the endogenous mouse FXN protein level were associated with cardiotoxicity characterized by mitochondrial dysfunction with impaired activities of respiratory complexes I and II.16 In parallel, no detectable toxicity was reported in the liver of mice treated with this AAVrh10 construct despite expression up to 90-fold of the wild-type FXN protein level.16 How and why FXN led to such toxicity in the heart of injected animals remains unclear.
Herein, we report insights into the toxicity associated with high level of FXN expression that was observed while evaluating a AAV9-based gene therapy approach in mice. Specifically, we showed that with doses ranging from 1 × 1013 to 1 × 1014 vg/kg, an AAV9 construct expressing an untagged FXN under the control of a CAG promoter (AAV9-CAG-FXN) leads to high level of FXN expression and to only partial and transient correction of the cardiac function of a cardiac mouse model of FRDA. The lack of efficacy in that dose range was accompanied by weight loss and premature death in the two highest dose groups and marked adverse findings in the liver that were associated with Fe-S cluster deficiency. In vivo and molecular studies using constructs expressing FXNWT or FXNN146K point mutation showed that toxicity is driven by FXN primary function involving binding to the NFS1-ISD11-ISCU-ACP mitochondrial Fe-S cluster core complex. Furthermore, we provided evidence that even with the lowest tested dose, FXN-induced toxicity occurred in the liver but could be mitigated by liver regeneration while leading to loss of FXN transgene expression.
Results
Transient improvement of cardiac function of MCK-Fxn mice after AAV9-CAG-FXN administration
Evaluation of AAV9-CAG-FXN was performed using the conditional cardiac mouse model of FRDA (MCK-Fxn), in which deletion of the endogenous mouse Fxn gene is triggered by a Cre recombinase under the control of the muscle creatine kinase (MCK) promoter.10 This model reproduces key features of the disease including progressive hypertrophic cardiomyopathy, mitochondrial dysfunction, dysregulation of iron metabolism, and reduced Fe-S cluster biosynthesis.10,12 To evaluate the ability of AAV9-CAG-FXN to correct cardiac dysfunction, we followed a similar protocol as the one reported for AAVrh10-FXN evaluation.12 Seven-week-old MCK-Fxn mice were injected intravenously at a dose of 1 × 1013, 3 × 1013, or 1 × 1014 vg/kg. Efficacy was assessed by longitudinal weight recording and by echocardiographic analysis at baseline, 9, 11, 13, and 15 weeks of age (Figure 1A). Saline-injected MCK-Fxn animals were maintained up to 9 weeks of age, and then euthanized before reaching critical endpoints due to severe cardiac dysfunction. For all mice injected with AAV9-CAG-FXN, weight improvement was observed during the first 3 weeks post-injection compared with the saline group (Figure 1A) and was independent of the dose level. At about 3 weeks after injection, a significant drop in weight reaching critical loss of 10%–20% of the initial body weight was observed in animals of the 1 × 1014 vg/kg dose group (Figure 1A). All 12 mice of this group had to be euthanized between 20 days and 26 days post AAV9-CAG-FXN administration. Later, a similar weight decline was seen with the 3 × 1013 vg/kg dose group (Figure 1A). Eight of the 12 mice of this group had to be euthanized between 26 days and 30 days post-injection, one died prematurely, one was euthanized at 51 days, and two could be maintained throughout the study (57 days post-injection). All mice of the 1 × 1013 vg/kg dose group survived until the end of the study, but weight plateaued below wild-type (WT) values between 25 days and 35 days post-injection, and then progressively declined (Figure 1A). The weight curve of the 1 × 1013 vg/kg dose group was mirrored by the echocardiographic measurements showing an improvement followed by a decline of the ejection fraction (Figure 1B and Table S1), the fractional shortening (Figure 1C and Table S1), and the left ventricular end diastolic volume (Figure 1D and Table S1). Although only two mice could be maintained throughout the study, a similar trend was observed with echocardiographic data of the 3 × 1013 vg/kg dose group (Figures 1B–1D and Table S1). Before premature euthanasia, the 1 × 1014 vg/kg dose group showed similar improvement in the ejection fraction, fractional shortening, and left ventricular volume at diastole as the other injected groups at the 9-week recording (Figures 1B–1D and Table S1), suggesting cardiac function was improving at the time mice had to be euthanized.
Figure 1.

Transient correction of cardiac dysfunction in MCK-Fxn mice after AAV9-CAG-FXN administration
(A) Upper, Experimental time course of AAV9-CAG-FXN injection and echocardiographic measurements. Lower, Body weight curves obtained from WT untreated (n = 15), MCK-Fxn saline injected (n = 12), MCK-Fxn treated at 1 × 1013 vg/kg (n = 13), 3 × 1013 vg/kg (n = 13), or 1 × 1014 vg/kg (n = 12). (B–D) Echocardiographic measurements of ejection fraction (EF) (B), fractional shortening (FS) (C), and left ventricular volume at diastole (LVVD) (D). Echocardiographic data for each group and time point are provided in Table S1. All data are mean ± SEM.
Hearts of the different groups were collected for analyses. At necropsy, the heart to body weight ratio was calculated to estimate cardiac hypertrophy (Figure 2A). As reported for this model,10 the heart to body weight ratio was significantly higher in saline-injected MCK-Fxn at 9 weeks compared with control animals (Figure 2A). This ratio was significantly lower in AAV9-CAG-FXN-injected mice of the 1 × 1014 vg/kg and 3 × 1013 vg/kg dose groups than in the saline MCK-Fxn group, suggesting an improvement of the cardiac hypertrophy at the time of euthanasia, which is in agreement with the echocardiographic data (Figures 1B–1D). Conversely, the heart to body weight ratio was not distinguishable from saline-injected MCK-Fxn mice in the 1 × 1013 vg/kg dose group at 15 weeks (Figure 2A).
Figure 2.

Time- and dose-dependent correction of MCK-Fxn heart phenotype after AAV9-CAG-FXN treatment
(A) Heart to body weight ratios at the time of necropsy: 57 days post-injection (dpi) for WT untreated (n = 15) and MCK-Fxn treated at 1 × 1013 vg/kg (n = 13), 14 dpi for MCK-Fxn saline group (n = 12), 26–57 dpi for MCK-Fxn treated at 3 × 1013 vg/kg (n = 11), and 20–26 dpi for MCK-Fxn treated at 1 × 1014 vg/kg (n = 12). (B) Vector genome copy (VGC) of AAV9-CAG-FXN in heart samples, as measured by ddPCR at the time of necropsy in WT untreated (n = 15), MCK-Fxn saline (n = 12), and MCK-Fxn treated at 1 × 1013 vg/kg (n = 12), 3 × 1013 vg/kg (n = 10), or 1 × 1014 vg/kg (n = 11). (C) mRNA expression of endogenous mouse Fxn and human FXN transgene in heart samples, as measured by ddPCR in WT untreated (n = 14), MCK-Fxn saline (n = 11), and MCK-Fxn treated at 1 × 1013 vg/kg (n = 13), 3 × 1013 vg/kg (n = 11), or 1 × 1014 vg/kg (n = 11). (D) Detection of Fxn transgene mRNA expression by in situ hybridization (ISH, upper panels) and human FXN protein by immunohistochemistry (IHC, lower panels) in heart sections. Scale bars, 300 μm. (E) Western blot analysis of mitochondrial and Fe-S client proteins from three mouse heart extracts from each group. Vinculin (VINC) was used as a loading control. (F) mRNA expression of cardiac dysfunction and FXN deficiency markers Nppa, Asns, Mthfd2, and Gdf15 as measured by ddPCR in WT untreated (n = 15), MCK-Fxn saline (n = 11), and MCK-Fxn treated at 1 × 1013 vg/kg (n = 13), 3 × 1013 vg/kg (n = 11), or 1 × 1014 vg/kg (n = 12). All data are mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Histopathological assessment was performed on heart sections after hematoxylin and eosin (H&E) staining (Table S2 and Figures S1A–S1C). Saline-injected MCK-Fxn mice showed minimal to moderate cardiomyocyte degeneration/necrosis with minimal to mild fibrosis. Severity of cardiomyocyte degeneration/necrosis and interstitial fibrosis decreased with increasing dose of AAV9-CAG-FXN. However, severity of findings was similar between the MCK-Fxn disease model and mice injected at 1 × 1014 vg/kg and 3 × 1013 vg/kg doses, whereas findings were more severe in heart sections of the 1 × 1013 vg/kg dose group. This could indicate worsening of the heart pathology with time since the lowest dose group was euthanized at 15 weeks and the MCK-Fxn saline group at 9 weeks of age.
Robust transduction and expression of FXN in heart of AAV9-CAG-FXN-injected MCK-Fxn mice
Heart samples were further analyzed by measuring vector genome copy (VGC) and transgene mRNA expression by digital droplet PCR (ddPCR). VGC values and mRNA levels showed a clear dose response (Figures 2B and 2C). At the mRNA level, FXN transgene expression reached ∼50 fold the level of endogenous mouse Fxn measured in heart of untreated WT animals in the 1 × 1013 vg/kg dose group, ∼160 fold in the 3 × 1013 vg/kg dose group, and ∼735 fold in the 1 × 1014 vg/kg dose group (Table 1). Distribution and expression of the FXN transgene was also assessed on heart sections by in situ hybridization (ISH) to specifically measure transgene mRNA and by immunohistochemistry (IHC) to detect human protein expression. Whereas a dose-responsive increase in ISH signal was seen (Figure 2D, upper panels), the human FXN IHC signal was high and consistent across heart sections for all dose groups (Figure 2D, lower panels), suggesting it was saturated due to high expression of FXN.
Table 1.
Summary of VGC and fold human FXN transgene mRNA expression over endogenous mouse Fxn mRNA expression as measured by ddPCR. Data are shown as mean ± SD.
| Mouse | MCK-Fxn | WT | WT | |||||
|---|---|---|---|---|---|---|---|---|
| Transgene | FXNWT | FXNWT | FXNN146K | FXNWT | FXNN146K | |||
| Dose (vg/kg) | 1E13 | 3E13 | 1E14 | 3E13 | 1E13 | |||
| Time at euthanasia | ||||||||
| dpi | 57 | 26–57 | 20-26 | 21 | 21 | 35 | 56 | 56 |
| wpi | 8 | 3.7–8 | 2.8–3.7 | 3 | 3 | 5 | 8 | 8 |
| Vector genome copy number (vg/μg gDNA) | ||||||||
| Heart | 2.4 ± 1.3E+04 | 1.2 ± 1.4E+05 | 4.4 ± 1.8E+05 | 1.1 ± 0.3E+05 | 1.1 ± 0.7E+05 | nd | 1.2 ± 0.6E+04 | 8.8 ± 3.5E+03 |
| Liver | 5.7 ± 2.6E+05 | 6.5 ± 1.4E+06b | 2.0 ± 0.4E+07 | 7.3 ± 1.8E+06 | 7.8 ± 5.0E+06 | 4.0 ± 1.4E+05 | 5.0 ± 2.2E+04 | 1.2 ± 0.3E+06 |
| Fold FXN mRNA expression over mouse Fxn | ||||||||
| Heart | 51 ± 26a | 159 ± 57a | 735 ± 289a | 89 ± 25 | 44 ± 27 | nd | 8 ± 4 | 10 ± 5 |
| Liver | 57 ± 27 | 489 ± 209b | 776 ± 292 | 930 ± 325 | 568 ± 132 | 109 ± 26 | 14 ± 16 | 215 ± 57 |
dpi: days post-injection; wpi: weeks post-injection; nd: not determined.
Fold change was determined using the mean value of mouse Fxn expression as measured in WT untreated group.
Only animals euthanized between 26 and 30 dpi were used due to loss of transgene expression in liver of animals euthanized at later time points.
Altogether, our data indicate that doses between 1 × 1013 and 1 × 1014 vg/kg of our AAV9-CAG-FXN construct lead to broad and high expression of FXN in heart but could at best provide partial and transient correction of the cardiac dysfunction in the MCK-Fxn model.
AAV9-CAG-FXN injection leads to transient correction of Fe-S cluster deficit in heart of MCK-Fxn mice
Heart samples were further analyzed by western blot to detect levels of Fe-S cluster-dependent enzymes and activities as an indirect way to assess the integrity of Fe-S cluster biogenesis and the ability of human FXN to correct the molecular phenotype.11,12 Puccio and collaborators identified perturbations in several Fe-S cluster-dependent proteins in cardiac tissue isolated from MCK-Fxn mice.10,12 In particular, the level of succinate dehydrogenase subunit B (SDHB), a Fe-S cluster-containing subunit of mitochondrial respiratory complex II, was reduced. Furthermore, decreased activity of the Fe-S cluster-containing lipoic acid synthase resulted in low lipoic acid (LA) modification of pyruvate dehydrogenase (PDHE2) and a-ketoglutarate dehydrogenase (KGDHE2) E2 subunits. As expected, these deficiencies were observed in saline-injected MCK-Fxn mice (Figure 2E). In contrast, both SDHB and LA signals were improved in the 1 × 1014 vg/kg dose group (Figure 2E), in agreement with the improved cardiac function that was measured by echocardiography just prior to the premature euthanasia (Figures 2B–2D). For the 3 × 1013 vg/kg dose group, results were mixed with some improvement of the LA signal but no obvious increase in SDHB compared with the saline MCK-Fxn group (Figure 2E). In the 1 × 1013 vg/kg dose group, both LA and SDHB signals were indistinguishable from the saline-injected group (Figure 2E), despite the strong and distributed FXN transgene expression (Figures 2E and 2D).
We then measured the mRNA expression of markers associated with FXN deficiency in the MCK-Fxn model: the natriuretic peptide A gene (Nppa) and Gdf15, two markers of cardiac dysfunction, as well as Asns and Mthfd2, two genes known to be upregulated in FXN deficiency.11,12,17 As previously reported, all genes were significantly upregulated in the saline-injected MCK-Fxn group at 9 weeks of age (Figure 2F). In the 3 × 1013 vg/kg and 1 × 1014 vg/kg dose groups, Nppa, Asns, and Mthfd2 were significantly reduced compared with the saline group (Figure 2F), whereas Gdf15 was significantly lower in the 3 × 1013 vg/kg dose group only (Figure 2F). This might reflect the fact that at the time of euthanasia, most mice of both 3 × 1013 and 1 × 1014 vg/kg dose groups showed improvement of the cardiac function as seen by echocardiography (Figures 1B–1D and Table S1). Conversely, no significant change in expression was observed in the lowest dose group compared to the saline control group, reflecting the cardiac dysfunction measured by echocardiography for this group at the end of the study (Figures 1B–1D and Table S1).
Together, our analyses of the in vivo and molecular heart phenotype showed that all the tested doses of AAV9-CAG-FXN had the ability to correct the cardiac dysfunction in the first weeks after injection. For the lowest 1 × 1013 vg/kg dose group, cardiac correction could not be maintained during the study. This observation of a transient effect might be explained by a progressive increase of FXN expression after AAV injection, going from efficacious levels to toxic levels. Because most mice dosed at 3 × 1013 and 1 × 1014 vg/kg were euthanized when the heart function was improving, we investigated the possibility that other factors than the cardiac phenotype contributed to weight loss and reduced survival in these groups.
Toxic overexpression of FXN leads to Fe-S cluster deficit in liver of MCK-Fxn mice
Because AAV9 has a high tropism for liver, we performed analyses on the livers of MCK-Fxn mice. High transduction of liver in AAV9-CAG-FXN-injected MCK-Fxn mice was confirmed by the measurement of VGC and transgene mRNA expression (Figures 3A and 3B). In both cases, a dose-responsive increase was observed, and the fold human FXN transgene mRNA expression over endogenous mouse Fxn mRNA level was ∼57 for the 1 × 1013 vg/kg dose group, ∼490 for the 3 × 1013 vg/kg dose group, and ∼775 for the 1 × 1014 vg/kg dose group (Table 1). A dose-responsive increase was also seen by ISH (Figure 3C, left panels), and high FXN protein expression was detected by IHC (Figure 3C, right panels). Of note, depending on when animals were euthanized, different levels of FXN IHC staining were observed in liver samples of the 3 × 1013 vg/kg dose group (Figure S2A). The significance of this observation will be discussed in a section below.
Figure 3.

Mice requiring early euthanasia display liver pathology and Fe-S cluster deficiency
(A) Vector genome copy (VGC) of AAV9-CAG-FXN in liver samples, as measured by ddPCR in WT untreated (n = 15), MCK-Fxn saline (n = 12), and MCK-Fxn treated at 1 × 1013 vg/kg (n = 13), 3 × 1013 vg/kg (n = 10), or 1 × 1014 vg/kg (n = 12). (B) mRNA expression of endogenous mouse Fxn and human FXN transgene in liver extracts, as measured by ddPCR in WT untreated (n = 15), MCK-Fxn saline (n = 12), and MCK-Fxn treated at 1 × 1013 vg/kg (n = 13), 3 × 1013 vg/kg (n = 9), or 1 × 1014 vg/kg (n = 12). (C) Detection of Fxn transgene mRNA expression by in situ hybridization (ISH, upper panels) and human FXN protein by immunohistochemistry (IHC, lower panels) in liver sections. Scale bars, 200 μm (ISH), 300 μm (IHC). (D) Measurements of liver-specific glutamate dehydrogenase (GLDH) from sera taken at time of necropsy: 57 days post-injection (dpi) for WT untreated (n = 15) and MCK-Fxn treated at 1 × 1013 vg/kg (n = 12), 14 dpi for MCK-Fxn saline group (n = 10), 26–57 dpi for MCK-Fxn treated at 3 × 1013 vg/kg (n = 11) and 20–26 dpi for MCK-Fxn treated at 1 × 1014 vg/kg (n = 13). All data are mean ± SEM. ∗∗∗∗p < 0.0001. (E) Western blot analysis of mitochondrial and Fe-S client proteins from three mouse liver extracts from each group. Vinculin (VINC) was used as a loading control.
Histopathological assessment of liver tissues indicated the presence of a significantly higher number of microscopic findings in the injected groups when compared with WT or saline-injected MCK-Fxn mice (Table S2 and Figures S1D–S1F). Minimal and mild single cell hepatocyte degeneration/necrosis was consistently observed in all AAV9-CAG-FXN injected groups. All three dose groups also showed minimal to mild hepatocellular hypertrophy and vacuolation that was variable in incidence and distribution. The highest incidence of hepatocyte hypertrophy was seen in the highest dose group and was centrilobular. The highest severity changes were observed in the 1 × 1014 vg/kg dose group with moderate centrilobular hepatocellular vacuolation (one mouse of 12), and in the 1 × 1013 vg/kg dose group with a marked midzonal hepatocellular necrosis (two mice of 13) (Table S2 and Figures S1D–S1F). Clinical pathology showed higher levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and glutamate dehydrogenase (GLDH) markers in sera of all injected groups (Figures 3D and S2B). However, whereas AST and ALT were significantly higher in all groups, GLDH values were only significantly increased in the 3 × 1013 vg/kg dose group when compared with saline-injected MCK-Fxn mice (Figure 3D).
We then assessed levels of Fe-S cluster client proteins and activities by western blot. Strikingly, the levels of LA and SDHB in liver were strongly impacted by AAV9-CAG-FXN transduction in the 1 × 1014 vg/kg and 3 × 1013 vg/kg dose groups when compared with WT non-injected, saline-injected MCK-Fxn animals or even the 1 × 1013 vg/kg dose group (Figure 3E). Mitochondrial PDHE2 and prohibitin (PHB) levels were not affected (Figure 3E), suggesting that FXN high expression in liver might trigger a specific deficit of Fe-S cluster client proteins.
Together, these data indicate that liver dysfunction could be the primary cause of body weight loss and premature death observed in the 3 × 1013 and 1 × 1014 vg/kg dose groups.
FXN overexpression-mediated toxicity correlates with FXN ability to bind the Fe-S cluster mitochondrial core complex
Belbellaa et al. showed that high overexpression of frataxin in mouse heart lowered the activity of SDH and NADH-ubiquinone oxidoreductase, two enzymes that require Fe-S clusters, whereas cytochrome c oxidase, a heme-dependent enzyme, was unaffected.16 In addition to the liver phenotype in AAV9-CAG-FXN-treated MCK-Fxn mice described above, this suggests that overexpression of FXN could inhibit Fe-S cluster biosynthesis, and paradoxically leads to a phenotype similar to FXN depletion. To further explore this possibility, we performed additional experiments in WT mice and evaluated the levels of multiple Fe-S cluster-containing proteins. In addition, we generated a construct expressing FXN(N146K) point mutation. This mutation is found in some individuals with FRDA and has been shown to maintain FXN mitochondrial localization and folding but to impair binding to the Fe-S cluster core complex.6,8,18, 19, 20 Furthermore, because the protein sequences of human and mouse FXN are different, in particular in the mitochondrial targeting sequence, the comparison between FXNWT and FXNN146K was also a way to assess whether the toxicity could be independent of the FXN function and could for instance result from an altered mitochondrial import or of peptides generated during human FXN maturation.
We first explored whether the deleterious effect of high FXN expression occurs in WT mice and showed that following intravenous injection of AAV9-CAG-FXN at 3 × 1013 vg/kg in 7-week-old WT animals, weight loss at around 3 weeks post-injection could be reproduced (Figure 4A). In contrast, injection of 7-week-old WT mice with AAV-CAG-FXN(N146K) at 3 × 1013 vg/kg had no impact on the weight curve of injected animals (Figure 4A). Tissues of the different groups were harvested 3 weeks post-injection to perform comparative analyses.
Figure 4.

Toxicity associated with FXN overexpression requires FXN binding to the Fe-S cluster core complex
(A) Body weight curves obtained from WT untreated (n = 4), AAV9-CAG-FXN-injected (n = 8), and AAV9-CAG-FXN(N146K)-injected (n = 8) mice dosed at 3 × 1013 vg/kg. Necropsy was performed 3 weeks post-injection. (B) Vector genome copy (VGC) in liver samples, as measured by ddPCR in WT treated with AAV9-CAG-FXN (n = 8) or AAV9-CAG-FXN(N146K) (n = 8) at 3 × 1013 vg/kg, 3 weeks post-injection. (C) mRNA expression of endogenous mouse Fxn and human FXN transgene in liver samples, as measured by ddPCR in WT treated with AAV9-CAG-FXN (n = 8) or AAV9-CAG-FXN(N146K) (n = 8) at 3 × 1013 vg/kg, 3 weeks post-injection. (D) Western blot analysis of mitochondrial and Fe-S client proteins from three mouse liver extracts, 3 weeks post-injection. Vinculin (VINC) was used as a loading control.
Similar VGCs were measured between animals injected with FXNWT and FXNN146K constructs in both liver and heart (Figures 4B and S3A). At the mRNA level, although AAV9-CAG-FXN(N146K) injected mice displayed slightly lower FXN expression compared with the FXNWT construct (Figures 3B and S3B), both constructs showed mean mRNA expression levels in liver higher than the mean FXN expression measured in livers of MCK-Fxn injected with AAV9-CAG-FXN at the same dose. Indeed, the mean fold transgene mRNA expression over endogenous mouse Fxn in liver was of ∼930 fold and ∼568 fold for FXNWT and FXNN146K constructs, respectively (Table 1). Conversely, transgene mRNA expression in heart was higher in MCK-Fxn animals than in WT mice, although VGC values were similar between groups (Table 1). This might suggest that the pathological state of MCK-Fxn mice could influence cardiac expression of the FXN transgene. FXN expression in both liver and heart was further confirmed by IHC showing strong and homogeneous signal in liver and an evenly distributed but patchy staining in heart (Figures S3C and S4A). Histopathological analysis of H&E sections revealed that AAV9-CAG-FXN expression was associated with an increased incidence and/or severity of centrilobular to midzonal hepatocellular hypertrophy (minimal to mild), karyocytomegaly of hepatocytes (minimal), hepatocellular lipid-like vacuolation (minimal to mild), single cell necrosis of hepatocytes (multifocal, minimal), and mononuclear/mixed cell infiltrate (multifocal, minimal) (Table S3, and Figure S5). Conversely, liver from mice injected with AAV9-CAG-FXN(N146K) was not distinguishable from non-injected controls (Table S3). No test article-related findings were seen in heart sections of these animals. Levels of GLDH were slightly but significantly elevated in AAV9-CAG-FXN-injected animals, whereas AST and ALT markers were not distinguishable from WT or AAV9-CAG-FXN(N146K)-injected mice (Figure S4B).
Western blot analysis showed that levels of LA on PDHE2 and KGDHE2 as well as of SDHB were reduced in livers of WT mice injected with AAV9-CAG-FXN (Figure 4D). The protein levels of mitochondrial aconitase (ACO2) and respiratory complex III Rieske subunit, two other mitochondrial Fe-S cluster clients, were also impaired (Figure 4D). Conversely, PDH2, PHB, and glycine cleavage system H (GCSH), which are all non-Fe-S cluster-containing mitochondrial proteins, were not affected by the AAV-mediated FXN expression (Figure 4D). Strikingly, despite similar FXN protein levels detected by western blot between FXNWT and FXNN146K, the introduction of the N146K point mutation could prevent all Fe-S cluster-related dysregulations observed with the FXNWT construct since levels of LA, SHDB, ACO2, and Rieske were maintained (Figure 4D). In heart, neither FXNWT nor FXNN146K had an impact on Fe-S cluster client protein levels at this dose and time point (Figure S3D).
Together, our data demonstrate that toxicity associated with FXN overexpression is not mediated by differences between human and mouse FXN but rather by the binding of FXN to the mitochondrial Fe-S cluster core complex that leads to Fe-S cluster deficit the same way FXN depletion does.
FXN overexpression-mediated toxicity can trigger liver regeneration
As mentioned above, MCK-Fxn mice injected with AAV9-CAG-FXN at 3 × 1013 vg/kg led to heterogeneous phenotypes with some animals euthanized earlier than others. Further microscopic evaluation of the liver sections stained for FXN showed that whereas animals from early time points (from 26 to 30 days post-injection) displayed homogeneous FXN staining, the three animals euthanized later in the study (at 51 and 57 days post-injection) had a patchy FXN staining (Figure S2A). This observation correlated with lower VGC and lower GLDH serum levels in mice euthanized at the end of the study (Figures S2C and S2D). The decrease in FXN staining observed in liver was not observed in heart of the same animals (Figure S2E), thus indicating that variation of staining in liver was not a result of variability in AAV injection. These data suggest that a concomitant loss of transgene expression and an improvement of liver function might have occurred in surviving mice. On this basis, we hypothesized that hepatocytes expressing high levels of FXN could be lost due to FXN toxicity while being replaced by transgene non-expressing or low-expressing cells via liver regeneration.
To investigate this hypothesis, we performed additional studies by injecting 7-week-old WT mice with AAV9-CAG-FXN at 1 × 1013 vg/kg, a dose that did not have an impact on survival and allowed long-term evaluation of transgene liver expression. In a first 8-week study, a group injected with AAV9-CAG-FXN(N146K) was also included for comparison. Although no significant difference in weight between the groups was observed at the end of the study (Figure 5A), a transient decrease in weight was seen in the AAV9-CAG-FXN cohort around 3–4 weeks post-injection (Figure 5A). Both heart and liver tissues were harvested for analyses. In heart samples, no significant difference between FXNWT and FXNN146K constructs was measured for VGC, FXN mRNA, or protein expression, indicating similar doses were injected for both constructs (Figures S6A–S6C). In liver, much lower VGC, FXN mRNA, and protein expressions were detected in the AAV-CAG-FXN-injected animals than in the AAV-CAG-FXN(N146K) group (Figures 5B–5E). Accordingly, a patchy FXN IHC staining was obtained in the FXNWT group, whereas it was more homogeneous in animals expressing the FXNN146K construct (Figures 5D and S7A). Assessment of liver H&E sections indicated that FXNWT expression was associated with an increased incidence and/or severity of single cell necrosis of hepatocytes, mononuclear and mixed cell infiltrates, increased mitosis, and karyo/cytomegaly of hepatocytes when compared with AAV9-CAG-FXN(N146K) administration, which was similar to non-injected controls (Table S4). Additionally, no test article-related microscopic findings were observed in the heart of injected mice.
Figure 5.

Recovery from moderate FXN-mediated liver toxicity correlates with loss of transgene expression and liver regeneration
(A) Body weight curves obtained from WT untreated (n = 4), AAV9-CAG-FXN-injected (n = 8), and AAV9-CAG-FXN(N146K)-injected (n = 8) mice dosed at 1 × 1013 vg/kg. Necropsy was performed 8 weeks post-injection. All data are mean SEM. (B) Vector genome copy (VGC) in liver samples, as measured by ddPCR in WT untreated (n = 4), treated with AAV9-CAG-FXN at 1 × 1013 vg/kg, 5 weeks (n = 10) or 8 weeks (n = 8) post-injection (wpi), or with AAV9-CAG-FXN(N146K) at 1 × 1013 vg/kg, 8 wpi (n = 8). (C) mRNA expression of endogenous mouse Fxn and human FXN transgene in liver samples, as measured by ddPCR in WT untreated (n = 4), treated with AAV9-CAG-FXN at 1 × 1013 vg/kg, 5 weeks (n = 10) or 8 weeks (n = 8) post-injection (wpi), or with AAV9-CAG-FXN(N146K) at 1 × 1013 vg/kg, 8 wpi (n = 8). (D) Detection of human FXN protein by immunohistochemistry in liver section from WT mice dosed with AAV9-CAG-FXN or AAV9-CAG-FXN(N146K). Necropsy was performed 5 or 8 weeks post-injection (wpi). Scale bars, 300 μm. (E) Western blot analysis of mitochondrial and Fe-S client proteins from mouse liver extracts. Necropsy was performed 5 or 8 weeks post-injection (wpi). Vinculin (VINC) was used as a loading control. (F) Quantification of percentage of phospho-histone H3 (pHH3)-positive cells in liver sections of WT untreated (n = 7), dosed with AAV9-CAG-FXN at 1 × 1013 vg/kg, 5 weeks (n = 9) or 8 weeks (n = 7) post-injection (wpi), or dosed with AAV9-CAG-FXN(N146K) at 1 × 1013 vg/kg, 8 wpi (n = 7). (G) Representative image of pHH3 IHC (upper panel) and FXN IHC (lower panel) on consecutive liver sections of WT mouse dosed with AAV9-CAG-FXN at 1 × 1013 vg/kg. Arrows indicate corresponding pHH3-positive nuclei. Scale bars, 200 μm.
Another cohort of 7-week-old WT mice was injected with AAV9-CAG-FXN at 1 × 1013 vg/kg and euthanized 5 weeks post-injection, a time at which mice were starting to recover weight after a transient loss (Figure 5A). Analysis of VGC and FXN mRNA levels in this new cohort showed intermediate values between the ones for FXNWT and FXNN146K constructs at 8 weeks post-injection (Figures 5B and 5C). This supports the idea that FXNWT liver expression was lost over time in AAV9-CAG-FXN-injected animals. At 5 weeks post-injection, FXN staining by IHC was variable between animals with mice displaying more patchy staining than others, but overall, it was more homogeneous than the signal observed at 8 weeks post-injection (Figures 5D and S7B). A progressive decrease of transgene expression in liver was further confirmed by western blot showing a barely detectable FXN signal at 8 weeks post-injection when compared with the 5-week post-injection time point or the FXNN146K construct (Figure 5E). Interestingly, no deficit in LA could be detected at either 5 weeks or 8 weeks post-injection in liver extracts, and only a slight decrease of SDHB signal could be seen in some animals dosed with AAV9-CAG-FXN at 5 weeks post-injection (Figure 5E). Histopathological analysis after H&E staining revealed minimal to moderate single cell necrosis of hepatocytes, minimal increased karyocytomegaly/binucleation, mild to moderate degeneration/vacuolation of hepatocytes, and minimal to mild increased mitoses in AAV-injected animals (Table S5 and Figure S8). Markers were measured in serum samples of mice at 3, 5, and 8 weeks after injection. GLDH was highly increased in the AAV9-CAG-FXN cohort at the 5-week time point when compared with non-injected controls, and slightly increased at 8 weeks post-injection (Figure S9A). AST and ALT were not significantly different from control mice at any of the time points (Figures S9B and S9C). Furthermore, no difference in total protein, albumin (ALB), or alkaline phosphatase (ALP) levels was seen in animals at 5 weeks after dosing (Figure S9D).
Based on the increased mitoses observed in the histopathological analysis, liver cell proliferation in AAV9-CAG-FXN injected animals was further investigated by IHC of phospho-histone H3 (pHH3), a marker of cell division. The number of pHH3-positive cells was increased in AAV9-CAG-FXN-injected mice at 5 weeks post-injection, in particular in mildly to moderately affected livers as seen in the histopathological assessment (Figures 5F and S9E). Evaluation on consecutive liver sections indicated that occasional pHH3-positive cells appeared to be larger cells with lower FXN IHC signal intensity and larger round nuclei (Figure 5G), thus further suggesting that cell division occurred in favor of transgene low- or non-expressing cells.
Together, our data demonstrate that FXN-expressing cells were lost with time in animals injected with the FXNWT construct because of liver injury associated with FXN toxicity. Consequently, cell proliferation was induced to regenerate the liver and restore function. Our results also confirmed the ability of the N146K mutation to mitigate most of the effects observed in liver, pointing again to the intrinsic role of FXN function in the toxic mechanism.
Discussion
Our work provides insights into the toxicity associated with AAV-mediated supraphysiological expression of FXN in vivo. We observed toxicity in heart of MCK-Fxn mice treated with our lowest AAV9 tested dose, with a cardiac function that first improved and then deteriorated 4 weeks after injection. This suggests a transition from efficacious transgene expression levels to toxic levels in heart. Surprisingly, toxicity was not observed in WT mice with a similar dose and time frame. However, FXN transgene mRNA levels reached in heart of MCK-Fxn mice were higher than the ones measured in WT mice with similar doses (see Table 1). Whereas the difference in expression could correlate with VGC differences at 1 × 1013 vg/kg, similar VGCs were obtained at 3 × 1013 vg/kg, suggesting a potential impact of the heart pathological state on transgene expression. Effect of cardiac disease on AAV9-mediated transgene expression was recently reported in a mouse model of heart ischemia.21 Therefore, we could speculate that transgene expression is affected by the phenotype of MCK-Fxn mice, leading to expression above the toxicity threshold in the cardiac model that was not achieved in the WT background with the same dose. These data point to the need to consider the pathological state of target tissues in the evaluation of toxicity associated with FXN gene therapy. Another potential reason for the difference in toxicity-related outcome is that heart in MCK-Fxn mice relies on transduced cells to correct and maintain function, whereas in WT animals, the non-transduced wild-type cardiomyocytes could still contribute to the cardiac activity. Hence, toxicity in AAV9-transduced cells could have acute consequences on the phenotype of MCK-Fxn mice and subclinical consequences in WT animals. However, no histopathological microscopic findings could be seen in heart of dosed WT animals, suggesting no toxicity had occurred with the tested AAV9 doses. Taking into consideration the fold expression levels shown in Table 1, it appears that transgene mRNA expression 8-fold over mouse Fxn expression would be safe for the heart. This value is in agreement with the 9-fold FXN protein level threshold reported by Belbellaa et al. with AAVrh10.16 Furthermore, the 50-fold transgene mRNA expression over mouse Fxn measured in MCK-Fxn mice is also in line with the report suggesting that cardiotoxicity could be reached over a 20-fold FXN protein expression.16
In addition to the cardiotoxicity, we report marked toxic effects in liver of MCK-Fxn and WT mice dosed with AAV9-CAG-FXN. This toxicity was either acute, leading to weight loss and early mortality, or moderate with a transient liver dysfunction and a progressive loss of transgene expression. The time component of the moderate liver toxicity is a challenge for the detection as well as for the definition of a tolerable expression level. Data obtained with the FXNN146K construct could however provide some indication on the threshold between moderate and acute toxicity. Assuming the FXN N146K point mutation prevents any loss of expression with time, the 215-fold FXN mRNA expression over mouse Fxn measured in our 8-week study using 1 × 1013 vg/kg would be a condition leading to moderate toxicity. Around and above 500-fold transgene expression, our data show the liver toxicity would be acute. It is interesting that AAVrh10 that has a good tropism for liver did not show similar acute toxicity in reported experiments.16 However, the protein transgene expression obtained with AAVrh10 reached up to 90-fold over endogenous levels, indicating that a threshold for acute toxicity might have not been attained with the tested doses. It is possible that a moderate toxicity occurred in the AAVrh10-dosed mice but might have been missed because evaluation was performed 8 weeks after AAV administration, when liver damages might have resolved after induced regeneration.16 Additional longitudinal experiments would be required to further evaluate this type of toxicity with other capsids. For this purpose, our data show that GLDH is a better serum marker than AST, ALT, ALB, or AP to detect both the acute and moderate FXN-related liver toxicities.
Finally, we demonstrated a direct correlation between the primary function of FXN and the AAV-mediated toxicity observed in vivo. High expression of FXN recapitulated features observed in FXN deficiency, with a specific deficit of Fe-S cluster-dependent activities. It is possible that high amount of FXN might impair the dynamic of Fe-S cluster biosynthesis by affecting the equilibrium between different complexes involving NFS1 and ISCU. For instance, high concentration of FXN might favor the formation of the FXN-bound Fe-S core complex and might constrain ISCU and NFS1 in specific conformations, hence preventing further maturation of the nascent Fe-S cluster and/or its transfer to acceptor proteins. It is very likely that this toxic mechanism would happen in any cell type and species since the Fe-S cluster assembly is a ubiquitous process relying on the same evolutionary-conserved machinery and components. What might change between tissues or cell types is the amount of FXN that can be tolerated, based on mitochondrial metabolism or Fe-S cluster demand. Investigating the toxicity in other cells such as neurons of the dorsal root ganglion (DRG), the dentate nucleus, or the motor cortex, primarily affected neuronal cell types in FRDA, would be important to understand whether different thresholds can be identified. In the context of DRG transduction, the intrinsic toxicity of FXN overexpression reported herein should be considered in addition to the AAV-induced pathology that was recently reported.22
In summary, our work provides further evidence as well as mechanistic insights into the toxicity associated with FXN overexpression. We show that toxicity is intrinsic to FXN function and that multiple organs can be affected, with different levels of overexpression leading to either moderate or acute liver dysfunction. These findings call for careful design and preclinical evaluation of gene therapy approaches for FRDA, to ensure a safe and efficacious expression of the FXN transgene in the targeted tissues.
Materials and methods
Animal procedures
Fxnflox/null:MCK-cre (MCK-Fxn) mice (strain #029720) and WT C57Bl/6 mice (strain #000664) were obtained from The Jackson Laboratory (Farmington, CT, USA) at 5 weeks of age. Animal housing facility was controlled for temperature and humidity, with a 12-h light/dark cycle and free access to water and a standard rodent chow. Seven-week-old males (MCK-Fxn or C57Bl/6) were anesthetized with isoflurane prior to a single intravenous injection into the orbital vein. For the MCK-Fxn efficacy study, doses of 1 × 1013, 3 × 1013, or 1 × 1014 vg/kg of AAV9-CAG-FXNwt were injected. WT C57BL/6 mice were injected at 7 weeks of age with 1 × 1013 or 3 × 1013 vg/kg of AAV9-CAG-FXNwt or AAV9-CAG-FXNN146K preparations. Animals were observed twice a week and body weight was recorded weekly. At the end of each study, mice were anesthetized with isoflurane, perfused with PBS, and tissues of interest were harvested. For gDNA, RNA, and protein analyses, tissues were snap frozen in liquid nitrogen. For histochemical analysis, a piece of each tissue was collected in a cassette and fixed in 10% formalin (#426-797, Thermo Fisher Scientific, Waltham, MA) for 48 h. Blood was collected from awake animals via submandibular route using a 5-mm lancet (#GR5MM Goldenrod, Braintree Scientific, Braintree, MA) or at the end of each study via cardiac puncture before PBS perfusion.
For echocardiography, mice were induced to anesthetic state using isoflurane (3%) in an induction chamber, and then anesthetic status was maintained at ∼2% isoflurane during animal preparation and image acquisition. While anesthetized, an animal was transferred onto a water-circulating heated blanket to remove hair around the left lateral and ventral thoracic area using a chemical hair removal agent. After hair removal, each animal was transferred onto a heated (∼34–35°C) platform for echocardiography. Transthoracic parasternal long axis B-mode and parasternal short axis M-mode images of the heart were acquired using 18–38 MHz transducer (VisualSonics MS400) in Vevo 2100 ultrasound machine to assess left ventricular (LV) structure and function. Anesthesia level was adjusted to maintain heart rate between 450 and 550 bpm during image acquisition. After image acquisition, the animal was transferred to a warm cage for recovery. Post image acquisition, images were analyzed using Vevo lab analysis software, and the following parameters were collected from image analysis: LV ejection fraction, fractional shortening, LV end diastolic and systolic volume, LV end diastolic and systolic area, LV posterior and anterior wall thickness, LV corrected mass, and heart rate.
All animal-involved procedures were reviewed and approved by Pfizer's Institutional Animal Care and Use Committee (IACUC) and conducted in an Assessment and Accreditation of Laboratory Animal Care (AAALAC) International accredited facility.
Constructs and AAV production
Codon-optimized WT and N146K human FXN cDNAs coding for full-length mitochondrial frataxin (Uniprot: Q16595-1) were synthesized (Genewiz, South Plainfield, NJ, USA) and cloned into an AAV plasmid between a chicken beta-actin (CAG) promoter consisting of the cytomegalovirus (CMV) enhancer, the CAG promoter sequence and a beta-globin intron, and an SV40 polyA sequence. AAV9 vectors were produced by triple transfection method in HEK293 cells.23 Titers of AAV preparations were obtained by quantitative real-time PCR using Taqman probes targeting the human FXN transgene sequence. Titers were of 2.14 × 1013 vg/mL (MCK-Fxn study) and 3.41 × 1012 vg/mL (WT C57Bl/6 studies) for AAV9-CAG-FXNWT and of 4.07 × 1012 vg/mL for AAV9-CAG-FXNN146K. Quality control of AAV preparations was performed by SDS-PAGE and silver staining, analysis of DNA content on agarose gel, and evaluation of endotoxin contamination with Endosafe PTS test (<0.5 EU/m).
Histology
All mouse tissues were fixed in 10% neutral buffered formalin and processed to paraffin blocks using standard histologic procedures. Tissue sections were cut on a rotary microtome at 5 μm and mounted on SuperFrost Plus slides (#12-550-15, ThermoFisher, Waltham, MA) for H&E, IHC, and ISH.
Frataxin IHC was performed on Leica Bond RX (Leica Biosystems, Buffalo Grove, IL) using an Alexa Fluor 488-conjugated mouse monoclonal antibody directed against human frataxin (#ab156033, Abcam, Cambridge, MA). Tissue sections were baked at 60°C for 30 min, deparaffinized, and pretreated with Epitope Retrieval 2 (#AR9640, Leica Biosystems) for 20 min at 100°C. Slides were incubated in peroxide block using the Bond DAB Polymer Refine detection kit (#DS9800, Leica Biosystems) for 10 min followed by primary antibody incubation at 1 μg/mL for 30 min, and incubation with a rabbit anti-Alexa Fluor 488 secondary antibody (#A11094, Thermo Fisher Scientific) at 0.5 μg/mL for 10 min. Frataxin was detected using the anti-rabbit HRP polymer in the Bond DAB Polymer Refine detection kit for 10 min and visualized using DAB chromogen for 10 min. An Alexa Fluor 488-conjugated mouse IgG2a Ƙ negative control antibody was also run in parallel at 1 μg/mL (#IC003G, R&D Systems, Minneapolis, MN).
pHH3 IHC was performed on Leica Bond III. Sections were baked at 60°C for 30 min, deparaffinized, and pretreated with Epitope Retrieval 2 for 20 min at 100°C. Background Buster (#NB306, Innovex Biosciences, Richmond, CA) was applied for 10 min followed by a rabbit polyclonal antibody to Phospho-Histone H3 (#9701, Cell Signaling Technology, Danvers, MA) for 15 min at 0.13 μg/mL. Slides were incubated with the Refine kit peroxide block for 10 min prior to application of the Refine Kit anti-rabbit HRP polymer for 8 min. The signal was visualized through incubation with the Refine Kit DAB chromogen for 10 min. A rabbit negative isotype control was run at the same concentration (#02-6102, ThermoFisher, Waltham, MA). All slides were counterstained with hematoxylin, dehydrated through graded ethanol and xylene, and permanently mounted with coverslips.
Frataxin ISH was performed using either the RNAScope 2.5 LSx Assay – Brown reagent kit (#322700 or #322100, Advanced Cell Diagnostics, Newark, CA) according to the manufacturer's guidelines on a Leica Bond RX (Leica Biosystems). The human frataxin probe (#814408) was designed by Advanced Cell Diagnostics. Positive control assays were performed using a mouse PPIB probe (#313918, Advanced Cell Diagnostics), and negative control assays were performed using an Escherichia coli DapB probe (#312038, Advanced Cell Diagnostics).
DNA, RNA, and protein analyses
Tissues were pulverized using a mortar and pestle in the presence of liquid nitrogen. A portion of pulverized tissue was transferred into an RNAse-free 2-mL tube using a disposable smartSpatula (#Z561762 Sigma, St. Louis, MO). One stainless-steel bead (#1026563, Qiagen, Venlo, Netherlands) and 1 mL of Trizol (#5596026, Invitrogen, Carlsbad, CA) were added to each tube. Tissue was homogenized using Qiagen TissueLyser II (Frequency 25 1/s x 2 times 2.5 min). After homogenization, samples were transferred to a phasemaker tube (#A33248, Invitrogen, Carlsbad, CA). Two hundred microliters of chloroform (#BP1145, Thermo Fisher Scientific) were added to the tube and vortexed for 15 s, then incubated for 5 min at room temperature. Samples were then centrifuged at 4°C x 14,000 g for 5 min. RNA extraction was completed using PureLink RNA mini kit (#12183025, Invitrogen) according to manufacturer's instructions. First-strand cDNA was prepared from extracted RNA using iScript Reverse Transcription Supermix (#1708840, Bio-Rad, Hercules, CA). Cycling conditions for cDNA generation were 25°C for 1 min x 1, 42°C for 30 min x 1, and 85°C for 5 min x 1.
For genomic DNA extraction, pulverized tissue was resuspended in TEN Lysis buffer (0.1 M NaCl, 0.01 M Tris, pH 8.0, 0.001 M EDTA, pH 8.0, 1% SDS) and vortexed at full speed for 15 s. Seven and half microliters of Proteinase K (#25530-049, Invitrogen) were added, and samples were incubated in a Thermomixer for 3 h at 56°C x 1,000 rpm. Samples were then cooled at room temperature for 10 min. Two microliters of RNAse A (#EN0531, Thermo Fisher Scientific) were then added and incubated for 5 min at room temperature. One volume of phenol:chloroform:isoamyl alcohol (25:24:1 ratio) (#P2609, Sigma) was added and samples were vortexed for 20 s at full speed. Samples were centrifuged at 15,0000 x g for 5 min at room temperature. The upper aqueous phase was transferred to a new tube and gDNA was precipitated with 0.5x volume of 7.5 M NH4OAc (#A2000, Teknova, Hollister, CA) and 3 volumes of 100% ethanol. Samples were kept at −20°C overnight and then centrifuged at 16,000 x g for 30 min at 4°C to pellet DNA. Pellets were washed once with cold 70% ethanol and resuspended in nuclease-free water. Genomic DNA was diluted to 10 ng/μL prior to analysis. All reactions were prepared inside an Airclean 600 PCR workstation.
ddPCR reactions were set up using dUTP-free ddPCR Supermix for Probes (#186-3024, Bio-Rad, Hercules, CA) according to manufacturer's instructions, with droplets prepared using the Auto DG droplet generator. Amplification of target cDNA or genomic DNA was performed using the following cycling conditions: 95°C for 10 min x 1, 94°C for 30 s x 40, 60°C for 1 min x 40, 98°C for 10 min x 1. Droplets were then read using a QX200 Droplet Reader and data were analyzed using Quantasoft software (Bio-Rad) following manufacturer's recommendations for quality control and analysis. The following primer/probe sets were used: primer/probe set targeting the codon-optimized cDNA of WT and N146K human FXN was designed and synthesized (Applied Biosystems, Sparta, NJ); mouse Fxn (FAM20X, Applied Biosystems, #dMmuCPE5107916); Asns (FAM 20X, #Mm00803785_m1, Applied Biosysems, Foster City, CA); Nppa (FAM 20X, #Mm01255747_g1, Applied Biosystems); Mthfd2 (FAM 20X, #Mm00485276_m1, Applied Biosystems), and Gdf15 (FAM 20X, #Mm00442228_m1, Applied Biosystems); Gapdh (HEX 20X, #dMmuCPE5195283, Bio-Rad); Hprt (HEX 20X, #dMmuCPE5095493, Bio-Rad); and Tbp (VIC 20X, #Mm00446973_m1, Applied Biosystems); mouse Tfrc Copy Number Reference Assay (VIC-Tamara 20X) (#4458366, Applied Biosystems).
For protein extraction, pulverized tissue was resuspended in 1X NP40 extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl and 0.2% NP-40) containing 1x Protease Inhibitor Cocktail (#04693159001, Roche, Basel, Switzerland). Samples were sonicated briefly for 8–15 rounds at 25% amplitude, 0.2 s on/0.2 s off, and then incubated on ice for 20 min. After incubation, samples were centrifuged at 10,000 x g for 10 min at 4°C. One volume of 2.5X protein sample buffer (PSB 5X: 100 mL contains 2.5g Tris base, 50 mL glycerol, 10g SDS and is adjusted at pH 6.8 with HCl 37%) was added directly to the tube containing the NP40 buffer and pellet. Samples were resuspended by pipetting up and down, and sonication was repeated as necessary to shear DNA and resuspend membrane proteins. Determination of protein concentration was performed using Pierce 660 nm Protein Assay Reagent (#22660, Thermo Scientific, Waltham, MA) on NP40 extracts and adjusted according to the final volumes.
Samples were run on 4%–12% NuPage Bis-Tris gels in MES Buffer (#NP0002, Thermo Fisher Scientific) using 2.5–5 μg of protein per lane. Samples were transferred onto nitrocellulose membranes using the Trans-Blot Turbo Transfer system (Bio-Rad) and blocked in TBS-T (0.05%) + 5% non-fat milk (#9999S, Cell Signaling Technology, Danvers, MA). Primary antibodies used were Anti-FXN (#ab110328, Abcam), Anti-Lipoic Acid (#437695, Calbiochem, San Diego, CA), Anti-Vinculin (#SAB4200080, Sigma), Anti-SDHB (#ab14714, Abcam), Anti-PDHE2 (#ab172617, Abcam), Anti-Prohibitin (PHB) (#ab28172, Abcam), Anti-ACO2 (#ab129069, Abcam), and Anti-GCSH (#ab192613, Abcam). Secondary antibodies used were Goat Anti-Mouse HRP (#G21040, Invitrogen) and Goat Anti-Rabbit HRP (#65-6120, Invitrogen).
Serum analysis
Serum was obtained by collecting blood in 0.8-mL MiniCollect Serum Clot Activator Tubes (#450472 Greiner Bio-One, Austria) and allowing to clot for 30 min at room temperature. Samples were centrifuged for 10 min at 10,000 rpm at 4°C. Supernatant was transferred to a new Eppendorf tube and stored at −80°C until analysis. Serum samples were analyzed in the ADVIA 1800 Chemistry System. ALT was measured using Siemens Advia Chemistry reagent (#10318168, Munich, Germany). AST was measured using Siemens Advia Chemistry reagent (#10341132), and GLDH was measured using Randox (#GL441/GL442, Crumlin, UK). GLDH was measured using an optimized standard method according to the recommendations of the Deutsche Gesellschaft für Klinische Chemie (DGKC). Alkaline phosphatase was measured using Siemens Advia Chemistry reagent (#10916067) and the bichromatic (410/478 nm) rate technique. Albumin was measured using Siemens Advia Chemistry reagent (#10211832); serum albumin quantitively binds to BCG to form albumin-BCG complex and was measured at 596/694 nm. Total protein was measured using Siemens Advia Chemistry reagent (#10311878) reading the reaction at 545 nm.
Statistical analysis
Data were compiled and graphed using GraphPad Prism software (version 9.0.0, GraphPad Software, San Diego, CA). Statistical analyses were performed in GraphPad Prism using one-way ANOVA and Dunnett's multiple comparison test.
Acknowledgments
The authors would like to acknowledge Pfizer Biomedicine Design team for the generation of AAV vectors, Madhu Sirivelu and James Stejskal for clinical pathology interpretation and assays, and Joan Kristel Aguilar for performing phospho-histone H3 staining.
Author contributions
C.H., D.H-S., B.T.A., S.S., L.O.W., M.P., and A.M designed and supervised experiments, C.H., T.L.P., A.K., R.G., R.C.K., K.A.B, X.C., T.M., B.T.A., I.P., T.F., L.M, T-W.C, K.L., C.S., and A.M. conducted experiments or collected data, C.H., T.L.P., A.K., R.G., R.C.K., K.A.B., X.C., D.H-S., T.M., B.T.A., I.P., L.O.W., and A.M. analyzed and interpreted data, C.H., T.L.P., and A.M. drafted the manuscript, C.H., T.L.P., R.C.K., K.A.B., D.H-S., L.O.W., C.B., M.P., and A.M. performed critical revision of the manuscript.
Declaration of interests
All authors are or were employees of Pfizer Inc. at the time of their contribution to this manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.02.002.
Supplemental information
References
- 1.Harding A.E. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- 2.Pandolfo M. Friedreich ataxia: the clinical picture. J. Neurol. 2009;256:3–8. doi: 10.1007/s00415-009-1002-3. [DOI] [PubMed] [Google Scholar]
- 3.Campuzano V., Montermini L., Molto M.D., Pianese L., Cossee M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 4.Lill R., Freibert S.A. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 2020;89:471–499. doi: 10.1146/annurev-biochem-013118-111540. [DOI] [PubMed] [Google Scholar]
- 5.Tsai C.L., Barondeau D.P. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- 6.Schmucker S., Martelli A., Colin F., Page A., Wattenhofer-Donze M., Reutenauer L., Puccio H. Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One. 2011;6:e16199. doi: 10.1371/journal.pone.0016199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colin F., Martelli A., Clemancey M., Latour J.M., Gambarelli S., Zeppieri L., Birck C., Page A., Puccio H., Ollagnier de Choudens S. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 2013;135:733–740. doi: 10.1021/ja308736e. [DOI] [PubMed] [Google Scholar]
- 8.Fox N.G., Yu X., Feng X., Bailey H.J., Martelli A., Nabhan J.F., Strain-Damerell C., Bulawa C., Yue W.W., Han S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019;10:2210–2217. doi: 10.1038/s41467-019-09989-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gervason S., Larkem D., Mansour A.B., Botzanowski T., Muller C.S., Pecqueur L., Le Pavec G., Delaunay-Moisan A., Brun O., Agramunt J., et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019;10:3566–3577. doi: 10.1038/s41467-019-11470-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puccio H., Simon D., Cossee M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 11.Martelli A., Schmucker S., Reutenauer L., Mathieu J.R.R., Peyssonnaux C., Karim Z., Puy H., Galy B., Hentze M.W., Puccio H. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell. Metab. 2015;21:311–322. doi: 10.1016/j.cmet.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Perdomini M., Belbellaa B., Monassier L., Reutenauer L., Messaddeq N., Cartier N., Crystal R.G., Aubourg P., Puccio H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich's ataxia. Nat. Med. 2014;20:542–547. doi: 10.1038/nm.3510. [DOI] [PubMed] [Google Scholar]
- 13.Gerard C., Xiao X., Filali M., Coulombe Z., Arsenault M., Couet J., Li J., Drolet M.C., Chapdelaine P., Chikh A., Tremblay J.P. An AAV9 coding for frataxin clearly improved the symptoms and prolonged the life of Friedreich ataxia mouse models. Mol. Ther. Methods Clin. Dev. 2014;1:14044–14054. doi: 10.1038/mtm.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piguet F., de Montigny C., Vaucamps N., Reutenauer L., Eisenmann A., Puccio H. Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of Friedreich ataxia. Mol. Ther. 2018;26:1940–1952. doi: 10.1016/j.ymthe.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salami C.O., Jackson K., Jose C., Alyass L., Cisse G.I., De B.P., Stiles K.M., Chiuchiolo M.J., Sondhi D., Crystal R.G., Kaminsky S.M. Stress-induced mouse model of the cardiac manifestations of Friedreich's ataxia corrected by AAV-mediated gene therapy. Hum. Gene Ther. 2020;31:819–827. doi: 10.1089/hum.2019.363. [DOI] [PubMed] [Google Scholar]
- 16.Belbellaa B., Reutenauer L., Messaddeq N., Monassier L., Puccio H. High levels of frataxin overexpression lead to mitochondrial and cardiac toxicity in mouse models. Mol. Ther. Methods Clin. Dev. 2020;19:120–138. doi: 10.1016/j.omtm.2020.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belbellaa B., Reutenauer L., Monassier L., Puccio H. Correction of half the cardiomyocytes fully rescue Friedreich Ataxia mitochondrial cardiomyopathy through cell-autonomous mechanisms. Hum. Mol. Genet. 2019;28:1274–1285. doi: 10.1093/hmg/ddy427. [DOI] [PubMed] [Google Scholar]
- 18.Gellera C., Castellotti B., Mariotti C., Mineri R., Seveso V., Didonato S., Taroni F. Frataxin gene point mutations in Italian Friedreich ataxia patients. Neurogenetics. 2007;8:289–299. doi: 10.1007/s10048-007-0101-5. [DOI] [PubMed] [Google Scholar]
- 19.Bridwell-Rabb J., Winn A.M., Barondeau D.P. Structure-function analysis of Friedreich's ataxia mutants reveals determinants of frataxin binding and activation of the Fe-S assembly complex. Biochemistry. 2011;50:7265–7274. doi: 10.1021/bi200895k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bellanda M., Maso L., Doni D., Bortolus M., De Rosa E., Lunardi F., Alfonsi A., Noguera M.E., Herrera M.G., Santos J., et al. Exploring iron-binding to human frataxin and to selected Friedreich ataxia mutants by means of NMR and EPR spectroscopies. BBA - Proteins Proteomics. 2019;1867:140254–140265. doi: 10.1016/j.bbapap.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Olloqui P., Rodriguez-Madoz J.R., Di Scala M., Abizanda G., Vales A., Olague C., Iglesias-Garcia O., Larequi E., Aguado-Alvaro L.P., Ruiz-Villalba A., et al. Effect of heart ischemia and administration route on biodistribution and transduction efficiency of AAV9 vectors. J. Tissue Eng. Regen. Med. 2020;14:123–134. doi: 10.1002/term.2974. [DOI] [PubMed] [Google Scholar]
- 22.Hordeaux J., Buza E.L., Dyer C., Goode T., Mitchell T.W., Richman L., Denton N., Hinderer C., Katz N., Schmid R., et al. Adeno-associated virus-induced dorsal root ganglion pathology. Hum. Gene Ther. 2020;31:808–818. doi: 10.1089/hum.2020.167. [DOI] [PubMed] [Google Scholar]
- 23.Grieger J.C., Soltys S.M., Samulski R.J. Production of recombinant Adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol. Ther. 2016;24:287–297. doi: 10.1038/mt.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.