

Abstract
Purpose:
Pancreatic ductal adenocarcinoma (PDAC) remains a significant health issue. For most patients, there are no options for targeted therapy, and existing treatments are limited by toxicity. The HOPE trial (Harnessing Organoids for Personalized Therapy) was a pilot feasibility trial aiming to prospectively generate patient-derived organoids (PDO) from patients with PDAC and test their drug sensitivity and correlation with clinical outcomes.
Experimental Design:
PDOs were established from a heterogeneous population of patients with PDAC including both basal and classical PDAC subtypes.
Results:
A method for classifying PDOs as sensitive or resistant to chemotherapy regimens was developed to predict the clinical outcome of patients. Drug sensitivity testing on PDOs correlated with clinical responses to treatment in individual patients.
Conclusions:
These data support the investigation of PDOs to guide treatment in prospective interventional trials in PDAC.
Introduction
Despite advances in other tumor types, standard treatment for pancreatic ductal adenocarcinoma (PDAC) remains chemotherapy, which often yields modest responses and excess toxicity. With the exception of the recent Pancreas Cancer Olaparib Ongoing (POLO) trial (1) which led to the approval of olaparib in the maintenance setting for patients with germline pathogenic variants of BRCA genes, treatment selection remains empiric. In the metastatic setting, the choice between first-line regimens is based primarily on patient factors such as age and performance status (2). In most instances, the selection between the standard-of-care regimens, FOLFIRINOX [5-fluorouracil (5-FU), leucovorin, irinotecan, and oxaliplatin], and gemcitabine/nab-paclitaxel is arbitrary. To date, there is no head-to-head comparison of these regimens to identify factors contributing to response to therapy.
In patients with other cancer types, such as lung cancer, precision medicine based on cancer genomics routinely guides clinical care and allows for tailored treatments that increase response rates and reduce toxicity (3). In contrast, genomic-based precision medicine has had limited impact on the survival of patients with PDAC (4–11). This was illustrated in the Know Your Tumor program where out of 1,856 patients with PDAC, overall survival was improved in only the 46 patients with actionable mutations who received matched therapy (12).
A complementary approach for personalized medicine utilizes patient-derived models for phenotypic-based screening of drugs (13). Using avatar models, we have shown a high correlation between the response to drugs in models and clinical response in the patients from whom the models were derived (14–17). However, similar to genomic-based treatment approaches, there are a number of barriers including the feasibility of generating models from patients, cost, time, finding effective agents in a recalcitrant cancer, and administration of candidate drug(s) to the patient. For these reasons, it is also unlikely that phenotypic screening in avatar models will be broadly used in the clinical care of patients with PDAC.
More recently, several groups including ours have developed methods to generate patient-derived organoids (PDO) from primary tumors as PDAC models (18–20). These models recapitulate the histopathologic and molecular architecture of the disease and are broadly used for molecular and genomic studies, drug screening, as well as biomarker discovery (21). In addition, several studies have retrospectively tested the role of PDOs to predict response to conventional chemotherapy and have reported a high positive predictive value (22, 23). Furthermore, we and others have investigated the response correlation between avatars and organoid models derived from the same patients and found a high correlation between these two models (24).
Herein, we report the results of the HOPE trial (Harnessing Organoids for PErsonalized Therapy) in patients with PDAC. This prospective trial assessed the feasibility of generating PDOs from patients with PDAC in real time using scant biopsy samples collected in the course of routine clinical care. We enumerate clinical and analytical parameters that predict the successful generation of PDOs, prospectively correlate the response to conventional anticancer agents between PDOs and the donor subject, and develop a model for predicting disease control.
Patients and Methods
Study design
This was a pilot trial designed to assess the feasibility of generating PDOs suitable for drug testing in patients with pancreatic cancer at our institution including all histologic subtypes, all stages of disease, and various modes of tissue procurement including fine-needle biopsy (FNB) of primary tumors, surgical specimens, core biopsies of metastatic lesions, and fluid including ascites and pleural effusions. In addition, in patients where drug sensitivity in the PDO could be assessed, the study was designed to detect correlation between in vitro drug responses and disease control. Participants were required to give written informed consent before inclusion in the study. The study protocol was approved by the Dana-Farber Cancer Institute Institutional Review Board (Boston, MA), and was conducted in accordance with the principles of the Declaration of Helsinki. Participants were enrolled from March 2018 to January 2020.
Participant selection
Adults (age ≥18 years) with cytologically or histologically confirmed or suspected pancreatic cancer on the basis of clinical, radiologic, and analytic evidence; Eastern Cooperative Oncology Group performance status ≤3; and willingness and ability to comply with scheduled visits, treatment plans, and laboratory tests were enrolled in the study. Patients with a coagulopathy that could not be corrected at the time of biopsy or those who were on anticoagulants that could not be discontinued safely for biopsy as determined by the treating physician were excluded from the study.
Study aims
Primary aims: (i) Success rate growing PDOs suitable for drug testing, and (ii) Time until a PDO drug sensitivity report could be generated; this was revised to time until initial drug testing was performed. Exploratory aims: (i) To correlate clinical, biochemical, and radiographic outcomes in patients with drug sensitivity reports generated on matched PDOs, and (ii) To correlate patients’ PDO drug sensitivity results to genomic/RNA profiling of their tumor and PDO.
Biopsies
Endoscopic ultrasound (EUS)-FNBs were performed using a linear echoendoscope and a 22-gauge FNB needle. Interventional radiology (IR) biopsies were performed under CT or US guidance using an 18-gauge core biopsy device with an 11 or 22 mm throw. Biopsies were performed in almost all cases as part of routine diagnostic procedures with a median of four passes. Cores were preferentially sent to pathology for diagnosis with remaining extra cores used for PDO generation. Obtaining tissue for PDO generation was only allowed on subsequent biopsies if no PDO was grown successfully from an earlier biopsy.
Clinical outcomes
We assessed clinical outcomes in patients in whom PDOs were successfully generated in order to correlate these outcomes retrospectively with PDO drug sensitivity data. Imaging in patients on this trial was performed as standard of care, and was not available at uniform intervals. Therefore, disease control [stable disease + partial response (SD+PR)] from a particular line of therapy was determined by comparing the first scan prior to starting a new line of therapy with the first available scan after starting a new line of therapy. Response was measured by RECIST v1.1 performed by our radiologist, and in one case by RECIST v1.0 per the clinical trial the subject was enrolled on. Patients with progressive disease (PD) at the first imaging assessment after starting a new line of treatment were deemed not to have disease control for that line of therapy.
Analysis of association of clinical parameters to PDO growth
The Fisher exact test was used to model the outcome of PDO growth, lack of expansion, and insufficient cells as a function of all available categorical clinical variables. The Kruskal–Wallis test was used for the long axis dimension of biopsy targets. Analyses were performed using R package (SciCruch registry ID: SCR_001905).
Clustering of AUCs
Univariate class intervals for AUC were identified using “jenks” in CRAN R-package “classInt” (25). The number of breaks was chosen based on goodness-of-fit measure with accuracy greater than 95% (Supplementary Fig. S4).
DNA and RNA sequencing
Whole-exome sequencing was performed on DNA extracted from PDOs and matched patient blood by Novogene. Whole-exome sequencing libraries were generated using the Agilent SureSelect Human All Exon kit following the manufacturer’s recommendations. In brief, 1 μg of DNA was fragmented using the Covaris hydrodynamic shearing system to generate 180 to 280 bp fragments. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities, and enzymes were removed. After adenylation of 3′ ends of DNA fragments, adapter oligonucleotides were ligated. DNA fragments with ligated adapter molecules on both ends were selectively enriched in a PCR reaction. After PCR reaction, the library was hybridized in liquid phase with biotin-labeled probes, and then magnetic beads with streptomycin were used to capture the exons. Captured libraries were enriched in a PCR reaction to add index tags to prepare for hybridization. Products were purified using the Beckman Coulter–AMPure XP system and quantified using the Agilent high-sensitivity DNA assay on the Agilent Bioanalyzer 2100 System.
Total RNA and DNA were extracted simultaneously using the Zymo-Miniprep Kit and checked for quality of DNA and RNA before starting the library preparation. RNA sequencing (RNA-seq) libraries were constructed using Illumina’s TrueSeq sample prep kit V2 following the manufacturer’s instructions. In brief, purified total RNA was poly-A selected and fragmented, and then cDNA was synthesized followed by end repair, A-tailing, and PCR amplification. Sequencing was done using Illumina HiSeq2500 in Novogene.
Whole-exome sequence analysis
Sequencing reads were aligned using BWA (Version 0.7.8-r455; ref. 26). SAMtools was used for sorting the BAM file, and Picard was used to mark duplicate reads. Single-nucleotide variants (SNV) and insertions and deletions (INDEL) were called using GATK (v3.8). Somatic SNV/INDELs were identified using MuTect (v1.1.4) and Strelka (v1.0.13), and consensus calls were used for the final analysis. Somatic copy-number variation was identified using Control-FREEC (v9.9), and all somatic variants were annotated using ANNOVAR in multiple aspects including protein coding changes, affected genomic regions, and deleteriousness prediction.
RNA-seq analysis
Indexes of the reference genome were built using STAR, and paired-end clean reads were aligned to the reference genome using STAR (v2.5). STAR used the method of Maximal Mappable Prefix which can generate a precise mapping result for junction reads. HTSeq v0.6.1 was used to count the read numbers mapped of each gene. Then the FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. FPKM counts were then normalized using the R package DESeq2 for further analysis.
Clustering of gene expression of PDOs
PDOs were subtyped into previously reported classes based on mRNA expression using R-package “ConsensusClusterPlus.” Consensus clustering was applied on mRNA expression of 46 of 50 tumor-specific transcripts reported in Moffitt and colleagues to define two classes, namely basal-like and classical. Using 467 of 613 transcripts from SAM analysis of Bailey and colleagues, PDOs were classified into four clusters, namely, Squamous, Progenitor, Immunogenic, and ADEX. Clustering was done with Pearson correlation as the internal distance metric.
PDO cultures
PDO cultures were conducted as previously described (19). Human tumor tissues were minced with number 22 blades into 1 to 2 mm fragments and then digested with STEMxyme-1 (250 CLS units per mL) for 30 to 40 minutes. The digestion was stopped by adding an equal volume of 1% BSA in DMEM and then centrifuged at 1,500 RPM for 5 minutes. Pellets were further digested with Accutase for 30 minutes and then collected by centrifugation at 1,500 RPM for 5 minutes. To remove red blood cells, cell pellets were resuspended in 1 mL DMEM and then added on top of 1 mL Ficoll Plus followed by centrifugation (400 × g, 10 minutes). The top layer was collected and mixed with 1 mL 1% BSA in DMEM and then centrifuged at 1,500 RPM for 5 minutes. Supernatant was discarded, and pellets were resuspended in PDO growth medium containing Y-27632, 5% Matrigel, and growth factors such as insulin and FGF2. The suspension was seeded onto 6-well plates precoated with Matrigel. Culture media were replaced every 4 days.
If multiple biopsies were obtained from a single patient, then “best organoid growth” was classified as the most advanced stage a PDO reached from any biopsy as follows: drug testing performed or ready for drug testing, lack of expansion, or insufficient cells. Insufficient cells were defined as one or less tumor cell per 4X field on days 2 and 3 of plating. Lack of expansion was defined as the inability to expand beyond passage two.
Drug screening assays
Established PDO cultures were collected and digested as above. For PDOs that were difficult to dissociate to single cells, TrypLE was used in place of Accutase. Cells were diluted in Matrigel to a density of 25,000 cells/mL, and then 10 μL of the suspension was added into each well of a 96-well plate to a final cell density of 2,500 cells per well. Matrigel was allowed to solidify for 30 minutes, and then 100 μL of organoid growth medium was added. After 4 days of growth, media were replaced with fresh media, and then drugs were added into each well using a Tecan D300e digital dispenser. Organoids were allowed to grow for another 4 days, and then cell growth was measured using Cyto Tox-Glo (Promega).
Morphological and histologic analysis
PDOs were plated at a density of 25,000 cells/mL, and images were taken every day for 12 days. About 200 images were obtained for each cell line. The images were analyzed for growth using OrganoSeg software (27). Raw images were segmented on the software to define the area, perimeter, and eccentricity of cell structures. Data were plotted as a box plot graph using Prism. For preparation of histologic slides, PDOs were grown in chamber slides and at maximum growth fixed in 4% PFA for 2 hours. The PDOs were then incubated with hematoxylin solution for 10 minutes, washed twice with water, and then scraped and sandwiched between two layers of Histogel (Sigma) using a cryomold. The sandwich was then transferred to a tissue cassette and fixed in 10% formalin. Antibodies used for fluorescent immunostaining and their antibody registry IDs were as follows: GATA6 (AB_2798924, Cell Signaling Technology; #5851), p63 (AB_2755007, Agilent; #M7317), KRT5/6 (AB_422594, GeneTex; #GTX17133), KRT17 (AB_869869, Abcam; #ab19067), and KRT19 (AB_307088, Abcam; #ab9221).
Drug sensitivity analysis
PDO cell viability after drug treatment was normalized to the mean number of untreated cells. Response to drug concentrations was analyzed using weighted n-parameter logistic regression, “nplr,” R-package (28), and the AUC was estimated using the Simpson’s rule. Estimated IC50 values are shown in Supplementary Table S4.
Data availability
All sequencing data that support the findings of this study will be deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO); the GEO Series accession number and all other relevant data are available from the corresponding author on request.
Results
Patient characteristics
The study enrolled a total of 76 patients whose baseline characteristics are listed in Supplementary Table S1. The study design and consort diagram are depicted in Fig. 1A and B. The consort diagram is per patient and shows that 67 patients had tissue delivered to our lab and 13 patients underwent more than one biopsy (Supplementary Table S2, sheet 1). Chemotherapy regimens prior to and following PDO generation are described in Supplementary Table S3. Most patients received standard-of-care regimens such as FOLFIRINOX or gemcitabine/nab-paclitaxel, but treatments approved in other cancer types such as pembrolizumab, trametinib, and lapatinib are also represented.
Figure 1.
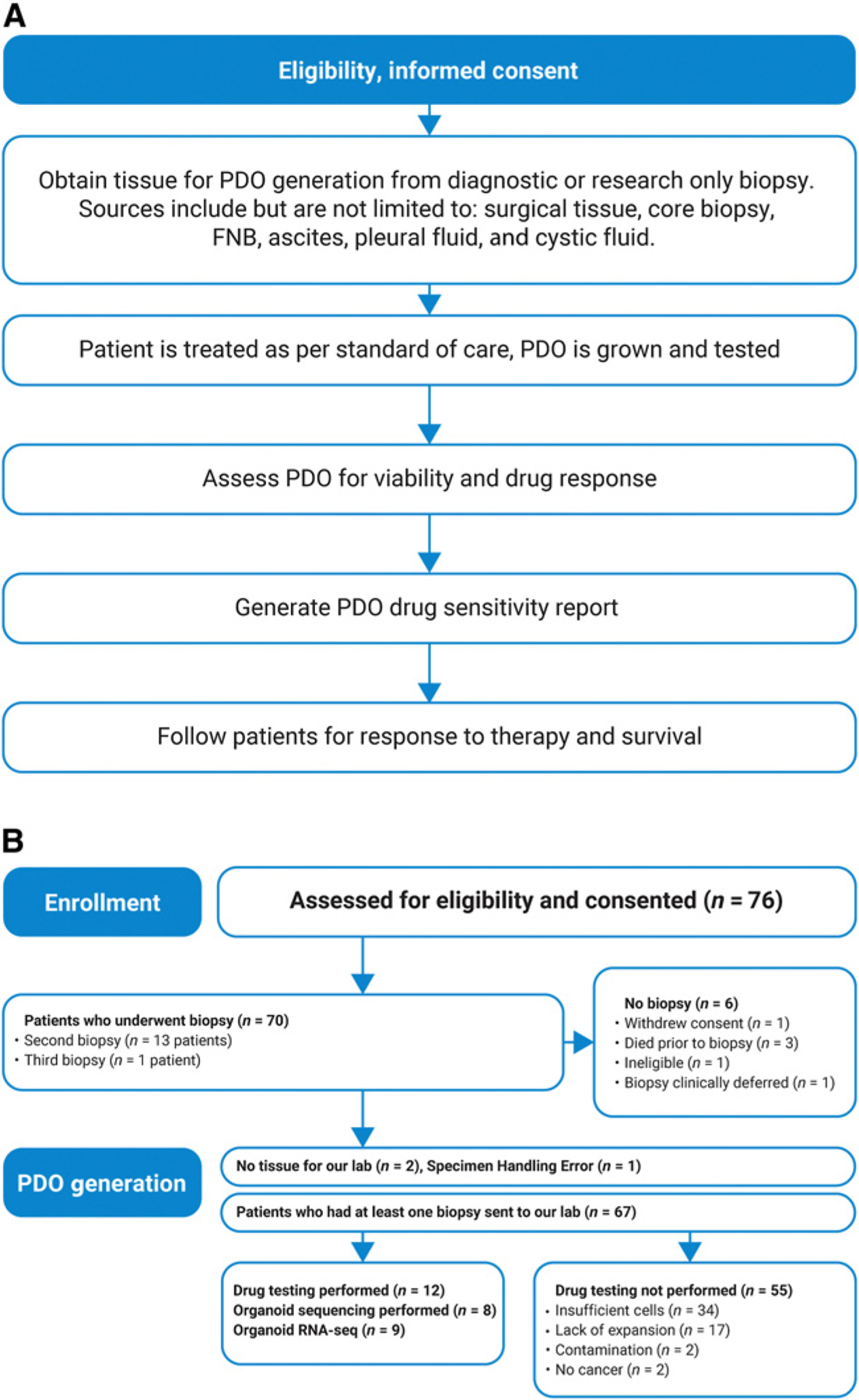
Design of the HOPE trial. A, Study schema. B, Consort diagram. This per-patient analysis includes all patients consented for the study regardless of confirmed diagnosis or adequate availability of tissue. Follow-up was performed on all patients when possible to assess clinical attributes that may contribute to successful PDO generation, and 43 patients were still living at last follow-up.
PDO generation and characterization
Out of 76 patients enrolled, 46 had a confirmed pathologic diagnosis of pancreatic adenocarcinoma from a biopsy performed on study. A total of 75 specimens were used for generation of PDOs. These were expanded to a minimum of two passages in 31 of 75 specimens (41%). The remaining 59% of specimens did not initiate a culture because either they contained insufficient cells as defined by one or fewer tumor cells per 4X field on days 2 and 3 of plating, or cultures were terminated due to bacterial contamination (Fig. 1B). Eighteen of 75 (24%) specimens did not expand beyond passage two in culture and therefore did not yield sufficient cells for drug sensitivity analysis, and these were categorized as “lack of expansion.” The remaining 13 specimens (17%) were expanded for drug sensitivity testing. In total, we established 13 PDOs suitable for drug testing from 12 of 76 (16%) individuals enrolled in the study. Only in one case were two PDOs grown from the same subject; these were from synchronous biopsies of the primary tumor and a liver metastasis. In this case, drug sensitivity testing was performed on the PDO derived from the metastatic biopsy. Among the 12 patients in whom drug testing was achieved, the time until initial testing ranged from 35 to 265 days with a median of 96 days (Supplementary Table S2).
To better understand the factors influencing the success of organoid growth and to improve patient selection for future clinical trials, we analyzed radiologic and histologic variables that could affect these outcomes (Supplementary Table S2, sheet 2). We describe three possible outcome classes of PDO generation: “drug testing performed,” “lack of expansion,” and “insufficient cells.” The categorical variables analyzed using the Fisher exact test included: biopsy modality (surgery, IR-guided core biopsies, EUS-guided FNB, paracentesis/pleurocentesis), tumor location (primary pancreatic tumor or metastasis), tumor grade, stage at diagnosis, and tumor cellularity. Additional variables that could not be analyzed with statistical significance due to low sample size include descriptors of matched pathology specimens such as degree of apoptosis, necrosis, and differentiation. Tumor size was analyzed using the Kruskal–Wallis test. Two variables, biopsy modality (P = 0.047) and tumor cellularity (P = 0.067), were marginally linked to the success of organoid growth. It is important to note that biopsy modality and tumor location were interdependent; only 3 of 27 EUS-guided FNBs were of metastases, and the remainder were taken from primary pancreatic tumors; conversely, all 37 IR-guided core biopsies were from metastases. When tumor cellularity was >20%, we were able to expand to a minimum of 2 passages in 14 (50%) of 28 cases, compared with only 9 (29%) of 31 specimens with ≤20% cellularity, highlighting the importance of tumor cellularity in the successful generation of PDOs.
Growth of PDAC PDOs in basal and classical subtypes
To further understand whether our platform is compatible with the expansion of different subtypes of PDAC, we performed IHC on four biopsies and six PDOs and RNA-seq on nine PDOs (summarized in Fig. 2A). Previous studies have used IHC to classify PDAC as classical (GATA6 positive; ref. 29) or basal types (positive for KRT5/6, p63, or KRT17; refs. 30, 31). By IHC, the PDO and biopsy from patient 3 were positive for GATA6 and negative for KRT5/6, KRT17, and p63, consistent with the classical subtype, whereas matched biopsies and PDOs from patients 6 and 10 expressed basal subtype markers (Fig. 2C; Supplementary Fig. S1). Patients 19 and 38 stained positive for all markers suggesting heterogeneous intratumoral cellular differentiation (Fig. 2D; Supplementary Fig. S1). We further classified PDOs based on previously published PDAC subtype classifiers (6, 32, 33) using mRNA expression profiles. Based on 46 (out of 50) tumor-specific transcripts identified by Moffitt and colleagues, PDOs were classified into two groups, “basal-like” and “classical,” by applying consensus clustering with Pearson correlation as the internal distance metric (34, 35). In addition, using DECODER, an integrated framework, we estimated the weights of different cellular components based on gene weights derived from The Cancer Genome Atlas pancreatic adenocarcinoma database for each patient. Figure 2A shows the proportion of estimated basal and classical components and suggests intrinsic heterogeneity within epithelial cells. A comparison of subtype classifications between Moffitt, DECODER, and IHC shows a very high level of concordance between all three approaches. Taken together, IHC and RNA-seq data demonstrate the feasibility using our methodology of growing and expanding PDOs that represent the molecular subtypes of the original tumor tissues for both the classical and basal subtypes of PDAC.
Figure 2.
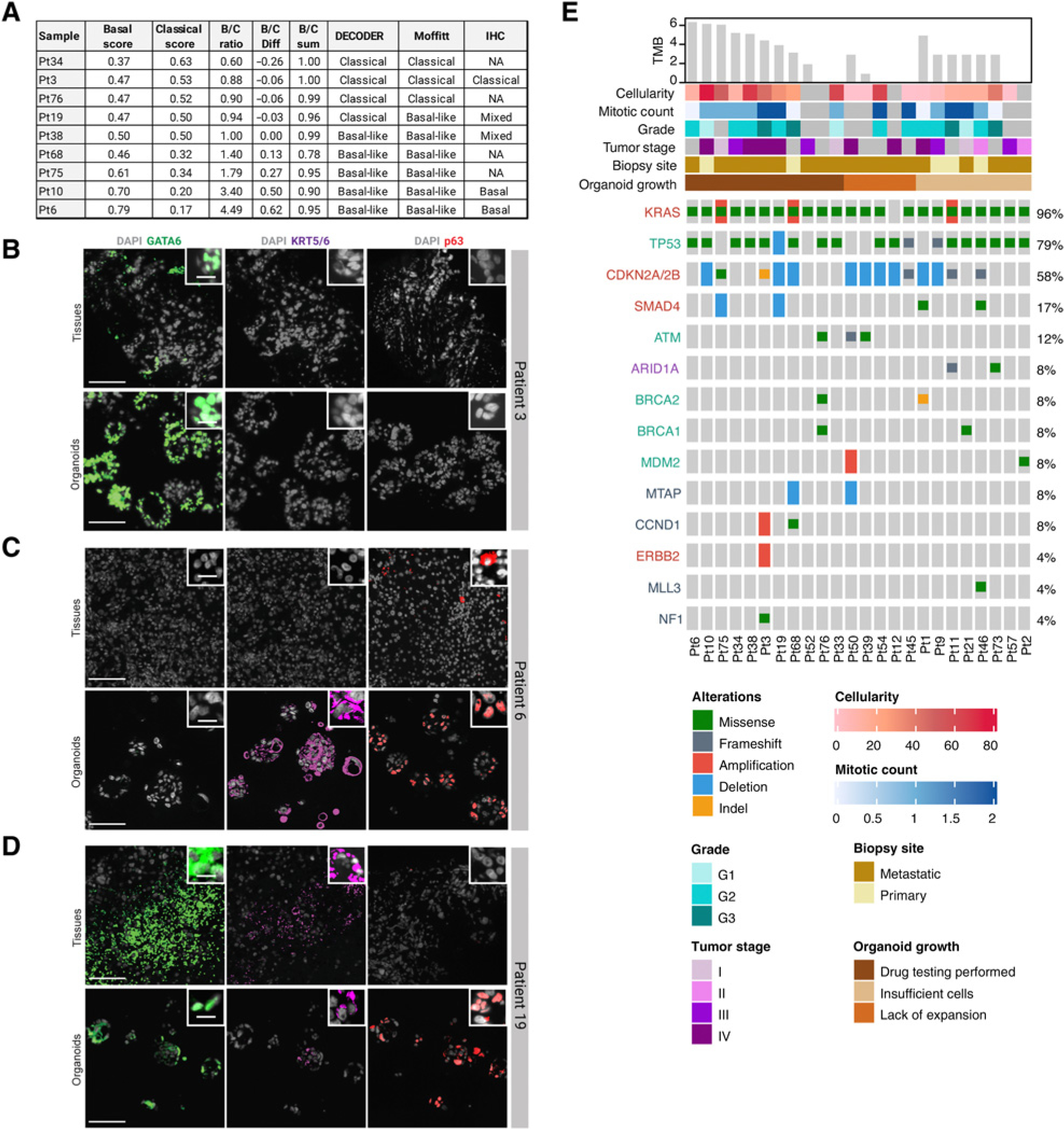
Identification of clinically relevant DNA alterations and molecular subtypes of PDAC PDOs. A, A comparison of molecular subtyping using two algorithms on RNA-seq data from PDOs: Moffitt and DECODER, and IHC on patient tissue and PDOs. B–D, Nuclear staining of GATA6 (classical subtype marker in green) and KRT5/6 and p63 (basal subtype marker in red) expressed in biopsies and PDOs for patients 3, 6, and 19. E, Oncoplot of clinically relevant DNA alterations identified on biopsies using targeted sequencing annotated with relevant clinical parameters.
PDOs display phenotypic and genetic homology with donor tumors
To determine whether PDOs recapitulate the biology and histopathologic architecture of donor tumors, we analyzed PDO sections by hematoxylin and eosin (H&E) staining (Fig. 3A). H&E images of PDOs demonstrated a fairly homogeneous appearance, composed of rounded spheres of pancreatobiliary-type malignant cells with hyper-chromatic nuclei and abundant amphophilic cytoplasm, with occasional mucin droplets. For all cases, conserved features from tissue to PDOs include nuclear pleomorphism, occasional mitotic figures, and a moderate amount of cytoplasm. In addition to histopathologic correspondence, molecular correspondence was observed between PDOs and matched donor tumors. Comparison of DNA alterations in seven PDOs and their matched tumors (Fig. 3B) revealed an overall concordance of genomic alterations including KRAS mutations, with the exception of two mutations that were absent in corresponding PDOs: a frameshift mutation in DNMT3A (D391fs*16) in patient 6 and a missense mutation in CHEK1 (R160H) in patient 10. The absence of the alteration in CHEK1 in patient 10 may be because tissue sequencing was performed on a biopsy of the primary pancreatic tumor, whereas the PDO was derived from a liver metastasis.
Figure 3.
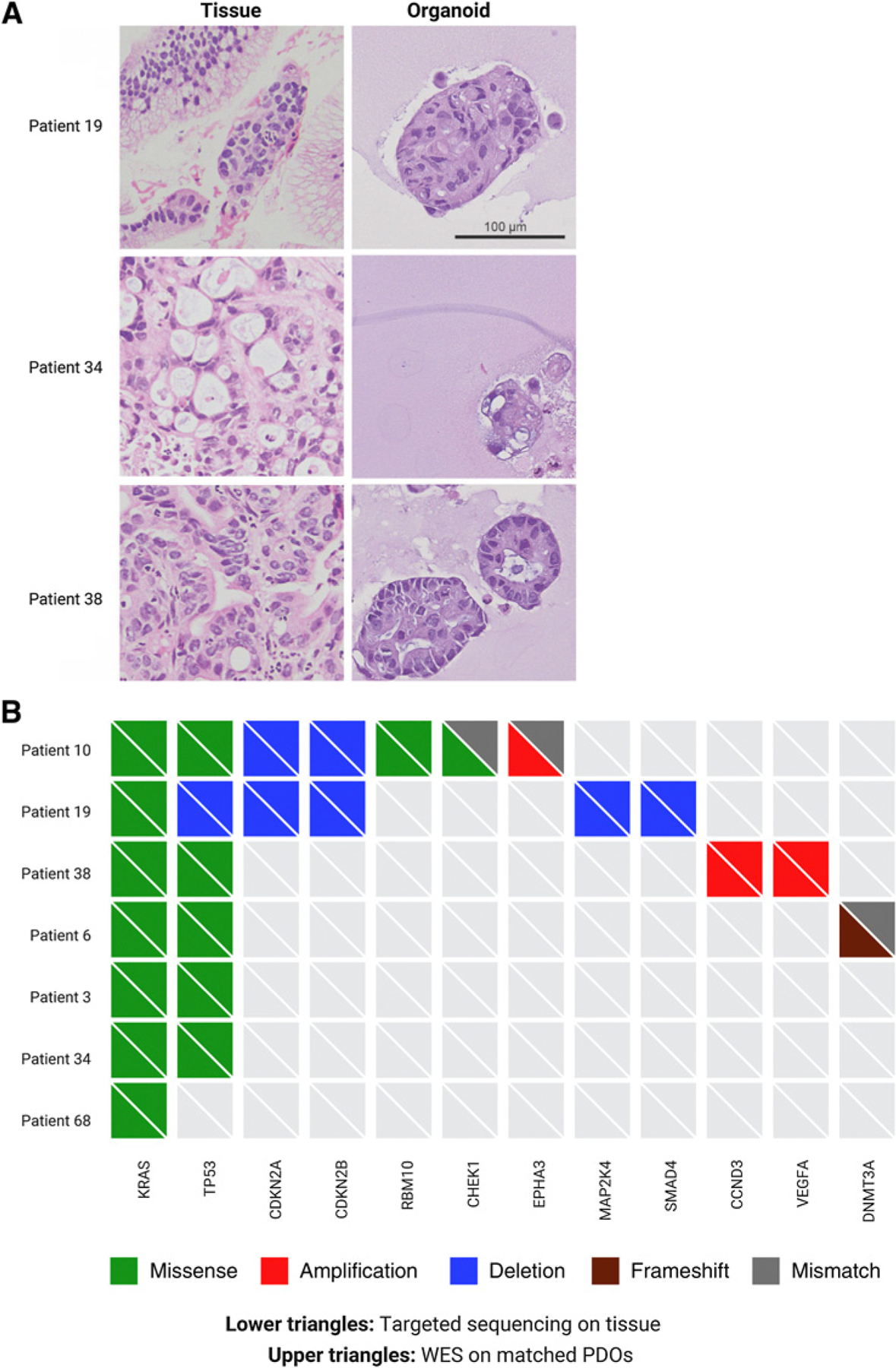
Comparison of tumor histology and DNA alterations between tumor tissue and matched PDOs. A, H&E images of tumor tissue from patients 19, 34, and 38, with matched PDO H&E images. B, Comparison of DNA alterations identified in patient tissue using the FoundationOne CDx-targeted sequencing panel (bottom triangles) with DNA alterations identified in matched PDOs using whole-exome sequencing (top triangles).
DNA sequencing of PDOs reveals clinically relevant mutations
We profiled 24 tumor biopsies and 9 PDOs, remaining PDOs were not sequenced, and DNA alterations could not be compared. Recurrent alterations, shown in Fig. 2E, were observed in known pancreatic cancer genes including KRAS, CDKN2A/2B, SMAD4, PREX1, GNAS, MYC, GATA6, ERBB2; genes in DNA damage repair pathways including TP53, BRCA1, BRCA2, ATM, and PALB2; and genes in SWI/SWF chromatin remodeling pathways including SMARCA2 and ARID1A. The four common events, alterations in KRAS (96%), TP53 (79%), loss of CDKN2A/2B (58%), and loss of SMAD4 (17%), were observed in a majority of tumors where sequencing was performed. Tumor mutational burden varied from 1 to 6 mutations/Mb. In patients who had targeted sequencing of tumor tissue performed or whole-exome sequencing on their matched PDOs, 29% were found to have somatic point mutations/INDELs in one of the DNA damage repair genes, including BRCA1/2 and ATM, which may predict sensitivity to PARP inhibitors.
Furthermore, we investigated the mutational signatures of nine PDOs using nonnegative matrix factorization based on a six-class mutational spectrum in their trinucleotide context (Supplementary Fig. S2). There are two notable COSMIC signatures in our data (Supplementary Fig. S3). The primary signature “A” of C > T transition at CpG sites is associated with age at diagnosis followed by the signature “B” that is predictive of defective homologous recombination–based repair arising due to somatic mutations in BRCA1/2 and short INDELs.
Determination of PDO drug sensitivity and clinical correlation
PDO sensitivities to gemcitabine, 5-FU, oxaliplatin, SN-38 (the active metabolite of irinotecan), paclitaxel, and other drugs were tested in 12 PDOs, based on which 49 independent area under the dose–response curves were calculated and normalized. Not all drugs were tested in each PDO, rather the panel was customized according to what drugs the subject had received prior to PDO generation and was likely to receive immediately following PDO generation. Independent AUC assessments were calculated for each drug tested on each PDO line and then compared to develop a personalized rank for each patient. This is illustrated in Fig. 4A and B, where comparisons of dose–response curves and AUCs for multiple drugs tested in patient 3 are shown, suggesting the highest sensitivity to irinotecan, followed by trametinib. The trametinib sensitivity was consistent with tumor and PDO genetic analysis that showed both KRAS mutation and amplification of ERBB2, a potent activator of RAS/MAPK signaling. Subject 3, from whom this PDO was derived, had stable disease on FOLFIRINOX (which contains irinotecan) and a PR on trametinib/lapatinib giving an early indication that AUC values estimated from PDOs are likely to provide insight into clinical responses to treatment.
Figure 4.
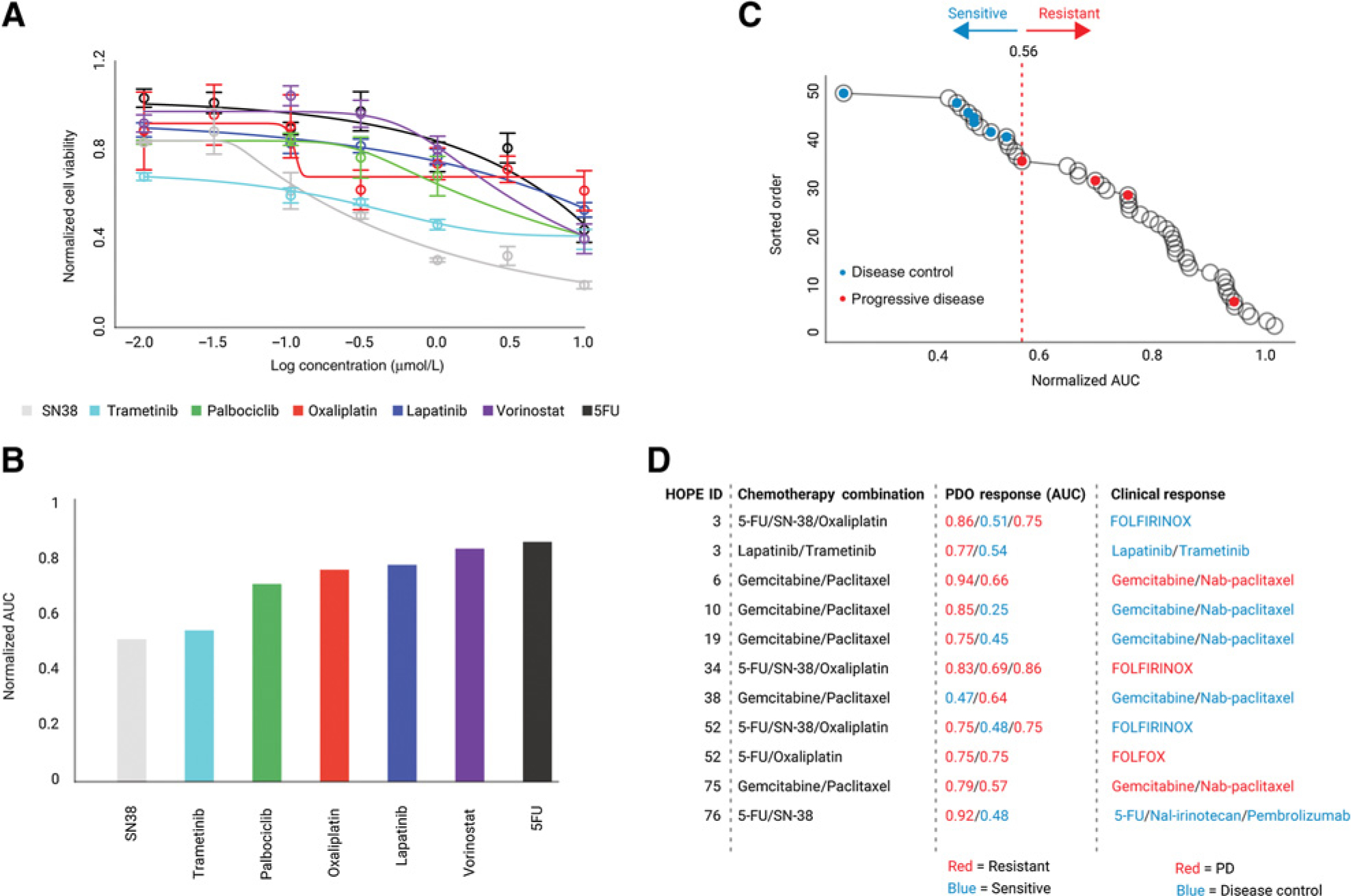
Correlation of PDO drug dose response to clinical response. A, Drug dose–response curves for drug testing on PDOs from patient 3. Drugs were selected based on treatments the patient received and was likely to receive, with a dose range of three orders of magnitude from 0.01 to 10 μmol/L. B, AUC calculated for drug responses in A. Dose–response curves were fitted using weighted n-parameter logistic regression, and AUC was estimated using Simpson rule. C, Correlation of AUC and disease control. Ranked AUC values are shown as black circles. Classification of AUC values using the Jenks natural breaks algorithm is shown by dotted lines. Clinical classification is superimposed on AUC circles, with blue representing disease control and red representing PD. D, For each patient with known clinical response to treatment: (a) drug combination received, (b) AUC estimated for the component drug responses in PDOs, and (c) clinical response classified as disease control (SD or PR) versus PD.
PDOs from different patients demonstrated a spectrum of sensitivities to drugs tested, with AUCs ranging from 0.25 (highly sensitive) to 1.0 (resistant). To understand the relationship between clinical response and PDO sensitivity results, all AUC values were ordered and then classified into groups using the Jenks Natural Breaks algorithm (36) which identifies boundaries by minimizing intragroup variance and maximizing intergroup variance (Fig. 4C). On the basis of goodness-of-fit analysis, the AUC values were separated into four classes, the minimum number needed to observe optimal classification of the data (Supplementary Fig. S4). We then annotated PDO and clinical responses in Fig. 4C as follows: Each black circle represents the AUC value per drug per PDO; for clinical responses, if disease control (PR + SD by RECIST v1.1) was achieved, the corresponding PDO treatment with the lowest AUC in the combination was colored blue, whereas if there was PD, it was colored red. Instances of clinical correlation (Supplementary Table S3) included regimens that patients received immediately prior to and following PDO generation with the exception of subject 3 who received two regimens following PDO generation with clinical correlation available and annotated for both. When AUC values were annotated with patient responses, the Jenks break of 0.56 (segregation break) segregated PDO/AUC values to match clinical responses classified as disease control and PD. The segregation of disease control and PD suggests that PDOs can be defined as “sensitive” to a treatment regimen if any one of the drugs in the regimen yielded an AUC < 0.56 and resistant only if all of the drugs in the regimen yielded an AUC ≥ 0.56. This is summarized in Fig. 4D where treatment regimens are listed alongside PDO AUC data and clinical responses. These findings highlight the promise of utilizing PDO drug sensitivity data to increase response rates and minimize toxicity by avoiding inefficacious drugs.
Discussion
The HOPE trial prospectively tested the feasibility of generating PDO models from patients with PDAC. Our analysis included all patients enrolled in the trial regardless of whether they eventually had a confirmed diagnosis or positive biopsy. The goal was to determine the potential of this approach to personalize the treatment of patients with PDAC in real time under “real world” conditions. Our decision to attempt PDO generation for all patients enrolled in the study led to the identification of factors related to the type of specimen and cellular content that can be used as inclusion criteria in future studies to achieve a better success rate. Critical morphologic and genomic characteristics of individual patients’ tumors were conserved in PDOs, and both basal and classical subtypes of PDAC could be propagated as PDOs. Importantly, the response of PDOs to conventional chemotherapy as well as targeted agents correlated with disease control. Despite concerns regarding our success rate generating PDOs from limited biopsy tissue, our findings are important as proof of principle that it is possible to generate PDO drug-sensitivity profiles and personalize treatment options within 12 to 16 weeks from the time of biopsy. This is a clinically relevant timeframe as restaging scans are often performed 2 to 3 months after an initial treatment is begun and a decision to continue treatment or move to second-line therapy is made at that time. It is also important to note that our success rate of 16% performing drug testing is likely an underestimate, as out of the 76 patients enrolled, only 46 (60%) had a confirmed diagnosis of adenocarcinoma from a study biopsy. For several patients, no biopsy was performed, no tissue was available for our lab, or histology was not pancreatic adenocarcinoma.
When our lab received sufficient tumor cells, our success rate generating PDOs suitable for drug testing was 13 of 31 samples (42%). This can be compared to a study that included only patients with a confirmed diagnosis and adequate cellularity, where 66% of patients had PDOs that could be passaged 5 times (37). Other similar studies focused primarily on surgical specimens (38) or a variety of specimens from multiple centers but with very few pretreated patients and unspecified inclusion criteria (22). In distinction to other studies, we did not require research-only biopsies with a minimum number of cores or onsite cytopathology, a majority of our specimens were not from surgery, none were from rapid autopsy, and we did not require a minimum number of cells in our lab to attempt PDO generation. This approach to tissue procurement more closely reflects clinical practice. In addition, our primary endpoint was the success rate generating PDAC PDOs suitable for drug testing, not simply establishing PDOs. Finally, we used media without Wnt family proteins which were chosen on the basis of our previous work (19) to prevent overpopulation of normal pancreatic epithelial cells at the expense of tumor cells and to maintain fidelity to the tumor cell phenotype of donor tissue (38). These factors likely explain much of the difference in the various success rates reported. Future efforts will be required to evaluate how the contribution of complex aspects of the tumor microenvironment affects drug sensitivity. In addition, although we have focused our efforts in this study on the scientific aspects of utilizing PDOs to tailor therapy, additional work needs to be done on related health economics and regulatory issues to ultimately approve future predictive assays. It is our hope that with technical advances in PDO culture systems, such assays may someday become clinically validated, affordable, and widely available.
Because our study was conducted at a single institution and complete clinical information was available in the majority of cases, we were able to analyze clinical and pathologic factors that correlated with the success of obtaining sufficient tissue and generating PDOs suitable for drug testing. As expected, and reported in Supplementary Table S2, the overall success rate generating PDOs was highly dependent on whether or not there were sufficient tumor cells for PDO initiation, and hence the cellularity of tumors. The success rate of obtaining sufficient cells in specimens with cellularity > 20% was nearly 2-fold higher than in specimens with cellularity ≤ 20%. Surgical specimens and IR-guided core biopsies of the liver provided sufficient cells for PDO generation at a higher rate than other specimens, which is expected given the larger amount of tissue obtained. Success in generating PDOs suitable for drug testing from metastatic sites was approximately 2-fold higher as compared with primary pancreatic tissue (10/44 = 23% vs. 3/31 = 10%).
In a comprehensive analysis of PDO drug sensitivity profiles on 12 PDOs, we observed that there is not only a diversity of responses to a drug across PDOs, but also a diversity of responses of individual agents within a therapeutic regimen in a given PDO, and a high degree of correlation between clinical and PDO responses. The correlation we report is consistent with recent data collected retrospectively in other cancer types (23, 39). In distinction to these previous studies, we have proposed a novel method to classify AUCs as assessed by PDO dose–response profiles to predict clinical disease control, allowing this information to be translated to the clinic. We chose AUC as an index for clinical response prediction because AUC integrates responses from a range of concentrations rather than a single point and does not require a high drug concentration exposure for complete cell killing, thereby reducing the amount of material required for testing and expanding potential clinical utility. We chose disease control (PR+SD) rather than PR alone, since this is a more clinically relevant metric for a difficult-to-treat disease where some patients receive clinical benefit from chemotherapy regimens that can prolong survival and control symptoms without a radiologic response. We acknowledge the small number of comparisons, and this method will need to be validated in a larger prospective study.
One limitation of our study is that PDO models were tested primarily against individual drugs because there is currently no validated method to determine appropriate drug concentrations for multiagent in vitro testing designed to detect synergy. However, justification for testing of individual agents is provided by a previous study (40) that suggests that most clinical responses to combination therapy can be explained by the action of a single agent in the combination and are not the result of synergy, with the possible relevant exception of 5-FU/oxaliplatin. In this study, to allow for clinical correlation, single agents chosen for drug testing were prioritized based on drugs that individual patients received or were likely to receive in the future. Hence, sensitivity to olaparib was not evaluated in PDOs from patients carrying mutations in DNA damage response (DDR) pathways who had not received a PARP inhibitor and were not likely to receive one. In addition, our previous study using organoids derived from patient-derived xenograft tumors (41) did not show enhanced olaparib sensitivity as a single agent in PDOs harboring DDR mutations. It is likely that an improved experimental design will be needed to evaluate the correlation of PDO and clinical responses to olaparib, an interesting subject for future studies.
In summary, we have prospectively generated PDOs from unselected patients with PDAC in a clinically meaningful timeframe while enumerating clinical, histologic, and radiologic predictors of success. We have shown a high degree of correlation between clinical outcomes in patients with PDAC and individual PDO drug sensitivity data, and outlined criteria based on the AUC of individual drugs in PDOs to rationally select therapy for patients with PDAC using a novel method. The data we report give a strong indication that drug sensitivities in PDO models have the capability to predict clinical responses; however, additional work is needed to test this method in a larger patient population. Based on what we have learned, we anticipate selecting patients in the future for PDO generation who have liver metastases amenable to core biopsies with a minimum number of cores available.
Supplementary Material
Translational Relevance.
We report a high degree of correlation between pancreatic ductal adenocarcinoma patient-derived organoid (PDO) drug sensitivity and clinical responses. This finding supports the utility of PDOs to tailor therapy for individual patients to improve clinical outcomes.
Acknowledgments
The authors are grateful to the patients who participated in this study and their families. They also acknowledge the input and suggestions of the entire Muthuswamy Hidalgo laboratory, the support of the division of Hematology and Oncology at BIDMC including the GI oncology group, our clinical and research nurses, members of the BIDMC CCTO (Cancer Clinical Trials Office), and James Grady for assistance with graphic design and layout.
Funding for this study was generously provided by Judy and Kim Davis. M. Hidalgo is supported by ERC-2014-ADG-670582. L. Huang is supported by the Hirshberg Foundation for Pancreatic Cancer Research. This work was also conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL 1TR002541) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the National Institutes of Health.
Authors’ Disclosures
J.E. Grossman reports other support from Agenus outside the submitted work. L. Huang reports grants from Hirshberg Foundation for Pancreatic Cancer Research during the conduct of the study; in addition, L. Huang has a patent for US20170267977A1 pending, licensed, and with royalties paid from StemCell Technologies. B. Bockorny reports grants from NanoView Biosciences and other support from Erytech Pharma outside the submitted work. A.J. Bullock reports grants from Agenus, Novocure, Geistlich Pharma, MedImmune, and Ipsen, as well as personal fees from Exelixis and Eisai outside the submitted work. M.L.B. Peters reports grants from NIH/NCI, personal fees from Agios and Exelixis, and other support from Ambry Genetics, BeiGene, Berg, Halozyme, AstraZeneca, and Exelixis outside the submitted work. R.B. Davis reports grants from Harvard Catalyst/NIH during the conduct of the study. D. Pleskow reports other support from Boston Scientific, Olympus, Fuji, and National Pancreas Foundation outside the submitted work. T.M. Berzin reports personal fees from Medtronic and Boston Scientific outside the submitted work. S.K. Muthuswamy reports personal fees from KAHR Bio outside the submitted work; in addition, S.K. Muthuswamy has a patent for PCT/CA2015/050723 pending and licensed to Stem Cell Technologies. M. Hidalgo reports grants from BIDMC during the conduct of the study. M. Hidalgo also reports personal fees and other support from BMS; other support from Nelum and Champions Oncology; personal fees from InxMed, Genchen, Khar, Oncomatrix, and MiNKi outside the submitted work. No disclosures were reported by the other authors.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 2019;381:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tempero MA. NCCN guidelines updates: pancreatic cancer. J Natl Compr Canc Netw 2019;17:603–5. [DOI] [PubMed] [Google Scholar]
- 3.Ettinger DS, Aisner DL, Wood DE, Akerley W, Bauman J, Chang JY, et al. NCCN guidelines insights: non-small cell lung cancer, version 5.2018. J Natl Compr Canc Netw 2018;16:807–21. [DOI] [PubMed] [Google Scholar]
- 4.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biankin AV, Maitra A. Subtyping pancreatic cancer. Cancer Cell 2015;28:411–3. [DOI] [PubMed] [Google Scholar]
- 6.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 7.Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin Cancer Res 2017;23:6094–100. [DOI] [PubMed] [Google Scholar]
- 8.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov 2018;8:1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular profiling of patients with pancreatic cancer: Initial results from the know your tumor initiative. Clin Cancer Res 2018;24:5018–27. [DOI] [PubMed] [Google Scholar]
- 10.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2019;16:207–20. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network. Electronic address, a. a. d. h. e. & cancer genome atlas research, N. integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020;21:508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov 2014;4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, et al. Apilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther 2011;10:1311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther 2011;10:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morelli MP, Calvo E, Ordonez E, Wick MJ, Viqueira BR, Lopez-Casas PP, et al. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J Clin Oncol 2012;30:e45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garralda E, Paz K, Lopez-Casas PP, Jones S, Katz A, Kann LM, et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin Cancer Res 2014;20:2476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med 2015;21:1364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov 2017;7:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science 2019;364:952–5. [DOI] [PubMed] [Google Scholar]
- 22.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018;359:920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendoo DMA, Denroche RE, Zhang A, Radulovich N, Jang GH, Lemire M, et al. Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput Biol 2019;15:e1006596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bivand R classInt: Choose Univariate Class Intervals. R urlpackage version 0.4–2. 2019. [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borten MA, Bajikar SS, Sasaki N, Clevers H, Janes KA. Automated brightfield morphometry of 3D organoid populations by OrganoSeg. Sci Rep 2018;8:5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Commo F, Bot MB. R package nplr n-parameter logistic regressions. [cited 2016]. Available from: https://cran.r-project.org/web/packages/nplr/vignettes/nplr.pdf.
- 29.O’Kane GM, Grunwald BT, Jang GH, Masoomian M, Picardo S, Grant RC, et al. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin Cancer Res 2020;26:4901–10. [DOI] [PubMed] [Google Scholar]
- 30.Roa-Pena L, Leiton CV, Babu S, Pan CH, Vanner EA, Akalin A, et al. Keratin 17 identifies the most lethal molecular subtype of pancreatic cancer. Sci Rep 2019;9:11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayashi A, Fan J, Chen R, Ho Y-j, Makohon-Moore AP, Lecomte N, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nat Cancer 2020;1:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng XL, Moffitt RA, Torphy RJ, Volmar KE, Yeh JJ. De novo compartment deconvolution and weight estimation of tumor samples using DECODER. Nat Commun 2019;10:4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn 2003;52:91–118. [Google Scholar]
- 35.Wilkerson MD, Hayes DN. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenks GF. Optimal classification for cloropleth maps. Volume Occasional Paper No. 2. Department of Geography: University of Kansas; 1977. [Google Scholar]
- 37.Tiriac H, Bucobo JC, Tzimas D, Grewel S, Lacomb JF, Rowehl LM, et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest Endosc 2018;87:1474–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Driehuis E, van Hoeck A, Moore K, Kolders S, Francies HE, Gulersonmez MC, et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc Natl Acad Sci U S A 2019;116:26580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, et al. Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 2020;26:17–26. [DOI] [PubMed] [Google Scholar]
- 40.Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017;171:1678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang L, Bockorny B, Paul I, Akshinthala D, Frappart PO, Gandarilla O, et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020;5:e135544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data that support the findings of this study will be deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO); the GEO Series accession number and all other relevant data are available from the corresponding author on request.