

Abstract
Purpose:
In acute myeloid leukemia (AML), recurrent DNA methyltransferase 3A (DNMT3A) mutations are associated with chemoresistance and poor prognosis, especially in advanced-age patients. Gene-expression studies in DNMT3A-mutated cells identified signatures implicated in deregulated DNA damage response and replication fork integrity, suggesting sensitivity to replication stress. Here, we tested whether pharmacologically induced replication fork stalling, such as with cytarabine, creates a therapeutic vulnerability in cells with DNMT3A(R882) mutations.
Experimental Design:
Leukemia cell lines, genetic mouse models, and isogenic cells with and without DNMT3A(mut) were used to evaluate sensitivity to nucleoside analogues such as cytarabine in vitro and in vivo, followed by analysis of DNA damage and signaling, replication restart, and cell-cycle progression on treatment and after drug removal. Transcriptome profiling identified pathways deregulated by DNMT3A(mut) expression.
Results:
We found increased sensitivity to pharmacologically induced replication stress in cells expressing DNMT3A(R882)-mutant, with persistent intra–S-phase checkpoint activation, impaired PARP1 recruitment, and elevated DNA damage, which was incompletely resolved after drug removal and carried through mitosis. Pulse-chase double-labeling experiments with EdU and BrdU after cytarabine washout demonstrated a higher rate of fork collapse in DNMT3A(mut)-expressing cells. RNA-seq studies supported deregulated cell-cycle progression and p53 activation, along with splicing, ribosome biogenesis, and metabolism.
Conclusions:
Together, our studies show that DNMT3A mutations underlie a defect in recovery from replication fork arrest with subsequent accumulation of unresolved DNA damage, which may have therapeutic tractability. These results demonstrate that, in addition to its role in epigenetic control, DNMT3A contributes to preserving genome integrity during replication stress.
See related commentary by Viny, p. 573
Translational Relevance.
The success of precision oncology hinges upon identification of predictive biomarkers and mechanistic understanding of underlying molecular pathophysiology. Alterations in the DNMT3A gene, especially missense mutations at arginine 882 (R882), are recurrent in acute myeloid leukemia (AML) and associated with poor prognosis due to relative resistance to anthracycline-based chemotherapy. Although dose-dense regimens can partially overcome resistance, this approach is not suitable for advanced-age and frail patients due to excessive toxicity. We report that DNMT3AR882 expression sensitizes to replication-stalling chemotherapeutics cytarabine and cladribine, through disrupted DNA damage repair, which coincides with impaired PARP1 recruitment to chromatin. This leads to replication fork collapse, cell-cycle delay, and mitotic defects. Our study supports low-intensity cytarabine-based regimens in the management of AML with DNMT3AR882 mutations and warrants further credentialling of PARP inhibitor combinations for AML with wild-type DNMT3A, to maximize clinical benefit and improve outcomes in leukemia patients unfit for standard high-intensity chemotherapy.
Introduction
Acute myeloid leukemia (AML), a genetically heterogeneous malignant clonal disorder of the hematopoietic system, is the most common leukemia in adults (1, 2). Its incidence sharply increases with age, with median age at diagnosis 67–68 years. Due to intensive basic and clinical research, the long-term survival in younger patients (<60) has significantly increased in the last 50 years and is currently approaching 30%–40%. Development of targeted therapies has led to remarkable successes in some genetic subtypes of AML leading to improved survival and quality of life. However, for most AML subtypes targeted approaches are still unavailable, leaving cytotoxic chemotherapy the next best option (3). To date, therapeutic targeting for AML with DNMT3A mutations, which are overwhelmingly detected in patients over age 60, has not shown much promise. Additional stratification and therapeutic strategies based on mechanistically informative mutational profiles may be needed to refine matching patients to optimal treatment regimens.
Recurrent mutations in the DNA methyltransferase 3A (DNMT3A) gene are found in 20%–35% of de novo AMLs and are associated with poor prognosis (4–10), in part attributable to relative resistance to anthracyclines (10–12). Although chemotherapy dose intensification demonstrated some survival benefit (10, 12), outcomes remain unsatisfactory in most patients with DNMT3A alterations due to advanced age, comorbidities, and inability to tolerate treatment (1, 6, 13). Cytarabine (Ara-C) remains a foundation of AML treatment (14), with low-dose Ara-C being a strategy of choice in many advanced-age patients (3, 15, 16). Clinical trials of low-intensity regimens combining cytarabine and cladribine, nucleoside analogue chain terminators that stall DNA replication, with and without venetoclax, are safe and effective (3, 17) and, in a recent study, tended to benefit patients with DNMT3A mutations (18). The most common mutation type, representing up to 70% of all DNMT3A alterations in AML, is a single amino-acid substitution at arginine 882 (R882, most commonly R882H or R882C) in the catalytic domain (8–10, 19). Hence, we and others performed DNA methylation analyses (11, 20–22) and gene-expression profiling (11, 23) in primary AML samples and in animal models carrying DNMT3A mutations. These studies uncovered gene signatures of altered cell-cycle control such as negative enrichment of the CHK1-regulated G2–M checkpoint (24, 25), which may indicate persistent replication stress, in cells expressing mutant DNMT3A (11, 23). Importantly, DNMT3A protein has been detected in direct association with stalled replication forks in cells treated with hydroxyurea (26). Yet, its role in the context of replication fork stalling such as after cytarabine-based chemotherapy remains unknown.
Once cytarabine is converted into its active metabolite ara-CTP, it is incorporated into nascent DNA during replication where it is a poor substrate for chain extension (27). This results in chain termination, replication fork stalling, and DNA damage response (DDR) often leading to p53-dependent apoptosis (28, 29). Single-stranded DNA breaks (SSB) resulting from stalled replication forks activate ATR-dependent CHK1 phosphorylation necessary to preserve replication fork integrity (30). If unresolved, SSBs are converted to double-stranded DNA breaks (DSB) and activate ATM-dependent phosphorylation cascade of CHK2, H2A.X, and p53, which in turn mediate DNA DSB repair and cell-cycle arrest and/or apoptosis (31, 32).
This study investigated the role of mutant DNMT3A (DNMT3Amut) in recovery of stalled replication forks and therapeutic responses to replication stress-inducing drugs. We show that cells expressing DNMT3Amut are more sensitive to cytarabine-induced replication stress, and that this effect is independent of the cell- and tissue-type context. This was accompanied by persistent intra-S checkpoint activation and accumulation of unrepaired DNA damage, which was carried through mitosis, leading to mitotic defects (33). Mechanistic studies of replication fork dynamics in cytarabine-treated cells demonstrated a higher rate of replication fork collapse in cells with DNMT3Amut. These results provide mechanistic insights into the regulation of replication fork recovery after DNA damage, and suggest additional strategies, such as patient stratification or novel pharmacological combinations including with PARP inhibitors or p53 signaling potentiators, to augment therapeutic responses to cytarabine and similar drugs used to treat AML.
Materials and Methods
Cell lines
Human leukemia cell lines K-562, KU-812, SET-2, and KO-52 were grown in RPMI supplemented with 10% FBS and penicillin/streptomycin. U2OS cells were maintained in DMEM supplemented with 4.5 g/L glucose, nonessential amino acids, 10% FBS, and penicillin/streptomycin. Cells were lentivirally transduced to express wild-type or R882C-mutant DNMT3A or with empty vector control (pMIGR1 expressing GFP as a selectable marker, or pPICH expressing dsRed). For robustness, all experiments were performed with both sets of isogenic cell lines, one with pMIGR1 and one with pPICH, as independent biological replicates.
Cell viability and drug dose–response assays
Cells were plated in 96-well plates at 1 ×104/well (suspension) and 3 × 103/well (adherent) and exposed to cytarabine (Ara-C), fludarabine, or cladribine (Cayman Chemicals), or hydroxyurea (HU; Sigma) in triplicate. Relative cell numbers were quantified at the indicated time points by CellTiter-Glo (Promega) for suspension cultures or by AlamarBlue (Thermo Fisher) for adherent cells. Plates were read with a BMG LABTECH FluoroQuant Optima plate reader. Relative cell viability data normalized to vehicle controls set at 100% were used to calculate IC50 values by fitting a four-parameter (variable slope) dose–response curve. GraphPad Prism version 7 software was used.
Mice
All animal studies were approved by the University of Florida Institutional Animal Care and Use Committee. A knock-in mouse line that conditionally expresses the Dnmt3aR878H allele (equivalent to DNMT3AR882H in humans) from the endogenous locus (Jackson Laboratory stock No. 031514) was described (11). For inducible expression of the mutant form of Dnmt3a and generation of mouse leukemias with Flt3ITD and Npm1c alleles, see Supplementary Methods. For in vivo cytarabine treatment studies, preconditioned congenic CD45.1 recipients were transplanted with CD45.2 donor bone marrow from fully excised Dnmt3a+/R878H:Mx1-Cre+ (Dnmt3amut) and littermate control Dnmt3a+/+:Mx1-Cre+ (Dnmt3awt) mice. Once engrafted, animals were treated with 30 mg/kg cytarabine/day for 5 days IP and vehicle (PBS) (n = 5/group). Bone marrow was analyzed after 48 hours. See also Supplementary Methods.
Colony-forming unit (CFU) assays
Freshly isolated whole bone marrow cells harvested from the Dnmt3a wild-type and mutant mice (described above) were plated in MethoCult M3434 (StemCell), 1 × 104 cells/well in triplicate. Vehicle (PBS) or cytarabine were added directly to the methylcellulose media at indicated concentrations. Colonies were scored 10–14 days after plating and imaged by EVOS FL microscope using a 2× objective.
Immunoblotting
Cells treated with Ara-C were collected after washing in ice-cold PBS and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentrations were determined using BCA protein assay (Pierce). For target detection by Western blotting, 25 μg of total protein was resolved by SDS-PAGE on 4%–12% Bis-Tris gels (Invitrogen), blotted onto PVDF membranes (Millipore) and probed by standard methods. Primary antigen-specific antibodies, listed in Supplementary Table S1, were used at 1:1,000 dilutions and signals were detected after incubation with secondary HRP-linked species-specific antibodies (Santa Cruz Biotech, 1:5,000 dilution) using KwikQuant chemiluminescence reagents and a digital imaging station (Kindle Biosciences). For preparation of chromatin-bound proteins and soluble nuclear extracts, treated cells were trypsinized, washed in ice-cold PBS, and 2 × 106 cells were processed using the Subcellular Protein Fractionation Kit for cultured cells (Pierce) according to the manufacturer's instructions, and analyzed by Western blotting.
Cell-cycle analysis and intracellular flow cytometry
Cell cycle was analyzed based on DNA content (see Supplementary Methods).
Where indicated, cells were incubated with antibodies against phosphorylated histone H3 (pS28) or H2A.X (pS139) conjugated to AlexaFluor647. DNA was counterstained using 1 μg/mL DAPI (Thermo Fisher Scientific) and analyzed by flow cytometry. Primary antibodies are listed in Supplementary Table S2.
Replication restart by dual labeling with EdU and BrdU
Cells in culture were pulsed with EdU (10 μmol/L; C10643, Invitrogen) for 30 minutes. Plates were then washed and treated with 10 μmol/L of Ara-C for 12 hours, washed and released into growth media for the indicated periods of time. BrdU (10 μmol/L; B23151, Invitrogen) was added 30 minutes before harvesting. Trypsinized cells were fixed with 70% ethanol at 4°C overnight, incubated in 2 mol/L HCl solution for 30 minutes at room temperature (RT), and washed in 3% BSA in PBS. EdU was detected using Click-iT EdU AlexaFluor647 picolyl-azide toolkit (C10643, Invitrogen) according to the manufacturer's protocol. Next, cells were incubated with mouse anti-BrdU monoclonal antibody conjugated to Alexa Fluor 488 (1:20, B35130, Invitrogen) in antibody staining solution (1% BSA, 0.2% Tween-20 in PBS) for 1 hour at room temperature. After washing in PBS, cells were incubated with DAPI and analyzed by flow cytometry.
Comet assay
Alkaline comet assays were performed using CometSlides and Comet Assay kit (Trevigen), according to the manufacturer's instructions. See Supplementary Methods.
Immunofluorescence analysis
U2OS cells were seeded 2.5 × 104 cells/well on 12-mm coverslips in 12-well plates, allowed to attach overnight, and treated as indicated. Cells were fixed in 2% paraformaldehyde in PBS for 30 minutes at RT, washed with PBS, permeabilized by 0.5% Triton X-100 in PBS for 10 minutes at RT, and washed with 1 mmol/L glycine to quench unreacted aldehydes. Cells were incubated in blocking buffer (0.1% BSA, 0.05% Tween-20, 0.2% Triton X-100, 10% donkey serum) for 1 hour at RT. Coverslips were incubated with primary antibodies specific to γH2A.X, PARP1, PCNA, or pRPA (see Supplementary Table S3) at manufacturer-recommended dilutions in blocking buffer overnight at 4°C. After washing in PBS, cells were incubated with AlexaFluor488 or AlexaFluor594-conjugated donkey anti-mouse or anti-rabbit secondary antibodies (Invitrogen; 1:1,000) in blocking buffer for 1 hour at RT and washed twice in PBS. Coverslips were mounted using ProLong Gold medium with DAPI (Invitrogen). Slides were examined using a spinning disk confocal (Yokogawa CSU-X1) attached to an inverted microscope (Nikon Eclipse Ti2-E) using a 60× objective (Nikon Plan Apo, 1.4 NA) and a Prime 95B sCMOS camera (Photometrics), with consistent settings maintained across all samples. For quantitative analyses, >40 nuclei were selected at random by thresholding based on the DAPI channel, and background-corrected integrated fluorescence density was calculated using ImageJ software.
BrdU and EdU immunofluorescence analysis
U2OS cells were pulsed with EdU (10 μmol/L; C10340, Invitrogen) for 30 minutes. The cells were then washed and treated with 10 μmol/L of Ara-C for 12 hours and then released in BrdU (10 μmol/L) for 30 minutes at the indicated time points. Cells were fixed and permeabilized as described above. Cells were incubated in 2 mol/L HCl solution for 30 minutes at RT and neutralized in a solution containing 3% BSA in PBS. EdU was detected using the Click-iT EdU AlexaFluor647 imaging kit (C10340, Invitrogen) according to the manufacturer's protocol. Cells were then incubated in blocking buffer (0.1% BSA, 0.05% Tween 20, 0.2% TritonX-100, 10% donkey serum) for 1 hour at RT followed by 1:200 mouse anti-BrdU monoclonal antibody in blocking buffer overnight at 4°C. After washing in PBS, cells were incubated with AlexaFluor488-labeled donkey anti-mouse secondary antibody (Invitrogen; 1: 1,000) in blocking buffer for 1 hour at RT and washed twice in PBS. Coverslips were mounted with ProLong Gold with DAPI (Invitrogen). Slides were examined on Nikon Eclipse Ti2-E spinning disk confocal microscope using a 60× NA 1.4 objective (see above). For quantitative analyses, at least 50 nuclei were selected at random and analyzed using the ImageJ or Nikon Elements software. Colocalization was quantified using Manders' overlap coefficient, which measures the degree of overlap of two separate fluorescence channels relative to the total intensity within each channel. A Manders' coefficient of 0 corresponds to no colocalization, whereas a value of 1 means 100% colocalization.
Analysis of micronucleation and nuclear morphology
Exponentially growing U2OS cells on glass coverslips were pulsed with BrdU (10 μmol/L) for 60 minutes, washed with PBS, treated with 10 μmol/L Ara-C for 24 hours, then released into complete media for the indicated duration of time. Cells were fixed, permeabilized, and extracted in 2 mol/L HCl as discussed above. Coverslips were then incubated in blocking buffer followed by anti-BrdU mouse monoclonal antibody and anti-Lamin B1 rabbit polyclonal antibody (ab16048, Abcam) in blocking buffer overnight at 4°C. After washing in PBS, cells were incubated with AlexaFluor488-labeled or AlexaFluor594-conjugated donkey anti-mouse or anti-rabbit secondary antibodies, washed twice in PBS, and mounted in ProLong Gold mounting medium with DAPI. Images were captured on a Nikon Eclipse Ti2-E spinning disk confocal microscope using a 100× NA 1.49 objective. At least 100 randomly selected BrdU-positive nuclei were counted for the presence of micronuclei, or analyzed for eccentricity using the ImageJ and the Nikon Elements software.
Metaphase spreads
U2OS cells were treated with 10 μmol/L Ara-C for 12 hours, washed in PBS, and released in complete media for 16 hours. The cells were incubated in 50 ng/mL colcemid (KaryoMAX, GIBCO) for 12 hours, harvested and swelled with a hypotonic solution (75 mmol/L KCl) for 15 minutes at 37°C. The cells were then fixed in ice-cold 3:1 mixture of methanol/acetic acid and dropped onto slides. Slides were stained with 5% Giemsa (Sigma), rinsed with distilled water, coverslipped using Permount (Fisher), and scanned on a Keyence BZ-X800 microscope with a 40× NA 0.95 objective.
RNA-seq
RNA integrity and purity were assessed by Qubit and Agilent 2100 Bioanalyzer (Agilent Technologies), and high-quality samples with RIN > 8.0 were sent to Novogene Co. Ltd for library preparation and sequencing on an Illumina NovaSeq 6000 using paired-end 150 chemistry. Sequencing data QC, alignment, quantification, differential expression, and pathway analysis were performed by standard methods as described in Supplementary Methods.
Data availability
Raw and processed RNA-seq data are available at GEO under the accession code GSE153871.
Statistical analysis and rigor
Pairwise comparisons between groups were done using a two-tailed Student t test for normally distributed data, with Welch correction where appropriate, and Mann–Whitney rank-sum test for nonnormally distributed data, after testing for normality and equal variance; P values ≤0.05 were considered significant. Where appropriate, one-way ANOVA with Tukey HSD post-hoc test or two-way ANOVA with post-hoc Šídák correction for multiple hypothesis testing were performed. Data were plotted as mean ± standard deviation (SD) unless indicated otherwise. For categorical data, two-tailed Fisher exact test was applied. GraphPad Prism software version 7 or higher was used. Identity of cell lines was validated by STR profiling; cultures were routinely tested for Mycoplasma contamination. Image scoring was done blindly; investigators were unblinded after data were collected. In vivo studies were repeated twice with comparable results. Unless indicated otherwise, all other experiments were independently performed three or more times for reproducibility.
Results
Cells with DNMT3A R882 mutations are more sensitive to replication-stalling drugs
Our prior studies found that DNMT3A-mutant cells were relatively resistant to anthracyclines and showed no differential sensitivity to DNA cross-linking and DSB-inducing agents (11). To investigate the role of DNMT3A in replication stress sensitivity, we treated a panel of leukemia cell lines with and without DNMT3AR882 mutations with the cytosine analogue cytarabine and purine analogue fludarabine (Fig. 1A and B), purine analogue cladribine, and hydroxyurea (HU), which stalls replication by depleting deoxyribonucleotides (Supplementary Fig. S1A and S1B). Cell lines with DNMT3AR882 SET-2 and KO-52 were more sensitive to replication-stalling drugs than DNMT3AWT cells K-562 and KU-812, reflected by increased apoptosis measured by annexin V and the cell fraction with sub-G1 DNA content (Supplementary Fig. S1C and S1D).
Figure 1.

Expression of mutant DNMT3A confers sensitivity to replication-stalling agents. A and B, Sensitivity to elongation-terminating nucleoside analogues cytarabine (Ara-C, A) and fludarabine (B) and corresponding IC50 values in leukemia cell lines with DNMT3AR882 (red, SET-2 and KO-52) and DNMT3AWT (black, K-562 and KU-812) after 48 hours of treatment, relative to vehicle control, as determined by CellTiter-Glo assay in triplicate. C, Colony formation assay using bone marrow cells derived from Dnmt3aR878H and littermate Dnmt3awt control mice, treated with indicated concentrations of Ara-C and grown in MethoCult M3434, in triplicate. Colonies were scored 10 days after plating and counts normalized to vehicle controls (C). Bone marrow from Dnmt3amut mice formed fewer to no colonies compared with wild-type control when treated with 25 nmol/L cytarabine; cDNA Sanger sequencing traces show complete Cre-mediated excision, resulting in equal expression of WT and R878H alleles. D,Ex vivo cytarabine dose responses and corresponding IC50 of mouse leukemias driven by Flt3ITD:Npm1c, with and without Dnmt3amut, as measured by CellTiter-Glo assay in triplicate. E–G, Sensitivity to cytarabine in vivo. Congenic CD45.1 recipients reconstituted with Dnmt3amut or wild-type control bone marrow cells received five daily doses of 30 mg/kg Ara-C and were analyzed 48 hours later (E). Frequencies of donor-derived (CD45.2+) mature lineage cells (myeloid, B cells, T cells; F) and apoptosis (Annexin V positivity) of the LSK cells (G) in the bone marrow 48 hours after last treatment dose (n = 5–9; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001, two-way ANOVA with Šídák's post hoc multiple comparisons test). H and I, Sensitivity to cytarabine (H) and fludarabine (I) and corresponding IC50 values in U2OS cells ectopically expressing wild-type (blue) and R882C mutant (red) forms of DNMT3A or empty vector control (black), relative to untreated after 48 hours of treatment, by AlamarBlue assay in triplicate. Inset in H shows levels of expression of wild-type and mutant DNMT3A, and endogenous DNMT3A in cells transduced with an empty vector control.
We extended these observations to normal and leukemic murine hematopoiesis. To evaluate the clonogenic potential of hematopoietic stem and progenitor cells in the setting of replication stress, we subjected freshly isolated bone marrow cells from mice with and without conditional Dnmt3aR878H expression (corresponding to human DNMT3AR882H) to CFU assays. Dnmt3aR878H cells plated in the presence of increasing Ara-C concentrations demonstrated a dampened clonogenic potential ex vivo in semisolid media compared with wild-type controls (Fig. 1C). Additionally, in a mouse model of AML driven by Flt3ITD and Npm1c, bone marrow cells from leukemic animals with Dnmt3aR878H were more sensitive to cytarabine than Dnmt3aWT in liquid culture ex vivo (Fig. 1D). In mice reconstituted with Dnmt3aR878H but not WT control bone marrow, administration of cytarabine over 5 days in vivo led to significant depletion of peripheral blood myeloid (CD11b+) cells; a proportional increase in B cells (B220+) likely reflects myeloid cell loss. Consistently, elevated apoptosis of hematopoietic stem and progenitor-enriched LSK cell population was observed (Fig. 1E–G). Together, these data indicate that expression of mutant DNMT3A in hematopoietic cells is associated with increased sensitivity to pharmacologically induced replication stress.
To determine if differential sensitivity to replication-stalling drugs was impacted by the tissue context, we generated immortalized mouse embryonic fibroblasts (MEF) from Dnmt3aR878H and wild-type control littermates. Similarly to cells of hematopoietic origin, Dnmt3amut MEFs were more sensitive to cytarabine and fludarabine (Supplementary Fig. S1E and S1F), suggesting that this phenotype is generalizable independently of the tissue origin.
Cells expressing mutant DNMT3A accumulate unresolved DNA damage after cytarabine treatment
To investigate the molecular mechanism of sensitivity to replication-stalling drugs, we lentivirally transduced wild-type and R882C-mutant forms of DNMT3A into U2OS osteosarcoma cells, a well-established model for DNA damage and repair studies, and confirmed differential cell killing by cytarabine and fludarabine (Fig. 1H and I; Supplementary Fig. S1G and S1H). At the same time, steady-state U2OS cell growth was unaffected (Supplementary Fig. S1I). Of note, ectopic expression of DNMT3Amut but not DNMT3AWT in the context of an endogenous DNMT3A mutation such as in OCI/AML-3 cells further sensitized them to cytarabine (Supplementary Fig. S1J).
We next investigated the DDR to replication fork stalling induced by a low dose of cytarabine (approximately IC20) in cells with DNMT3AR882C. Cells overexpressing mutant DNMT3A showed CHK1 phosphorylation persisting over 24 hours of continuous drug exposure and a concomitant increase in p53 phosphorylation, whereas in DNMT3AWT-expressing cells there was a rapid decline in pCHK1 and a less pronounced p53 activation (Fig. 2A; Supplementary Fig. S2A). The intensified signaling in DNMT3Amut-expressing cells reflected accumulation of DNA damage as measured by an increased fraction of mobile DNA per nucleus in an alkaline comet assay (Fig. 2B and C) and confirmed by immunofluorescent detection of phosphorylated histone H2A.X (γH2A.X) (Supplementary Fig. S2B and S2C), whereas overexpression of DNMT3AWT appeared to be protective in both assays. Consistently, SET-2 and KO-52 cell lines with DNMT3AR882 demonstrated accumulation of γH2A.X and cleaved PARP indicative of apoptosis along with persisting low-level pCHK1, whereas in DNMT3AWT K-562 and KU-812 cells CHK1 activation was rapidly resolved (Supplementary Fig. S2D). These results indicate that cells with mutant DNMT3A accumulate DNA damage after cytarabine.
Figure 2.

Cells expressing mutant DNMT3A accumulate DNA damage due to incomplete DNA repair upon cytarabine treatment. A, Immunoblot analysis of the DNA damage signaling at indicated timepoints after cytarabine treatment in U2OS cells with wild-type and mutant DNMT3A or empty vector control. B and C, Detection of damaged DNA by alkaline Comet assay at 24 hours (B) or at indicated timepoints of continuous cytarabine exposure (C) in U2OS cells lentivirally expressing wild-type (blue) and mutant (red) DNMT3A or empty vector control (black). D, Incomplete DNA repair in cells expressing mutant DNMT3A, 12 and 24 hours after cytarabine removal following 12-hour treatment. At least 40 comets per condition were scored using OpenComet plugin for ImageJ software (NIH) by calculating percentage of DNA in the comet tail on the basis of comet head and tail integral intensity (*, P < 0.05; **, P < 0.01; ****, P < 0.0001; Mann–Whitney rank-sum test; graphs represent mean ± SEM). E, Analysis of DDR in U2OS cells lentivirally expressing wild-type and mutant DNMT3A or empty vector control at 12 hours of cytarabine exposure and 36 and 48 hours after drug washout (UT, untreated; R, at release). F, Recruitment of PARP1 to chromatin-bound fraction from soluble nuclear extract after 12 hours of treatment with 10 μmol/L cytarabine in cells overexpressing wild-type or mutant forms of DNMT3A. G, Analysis of PARP1 foci formation (green) and DAPI (blue) by immunofluorescent staining in U2OS cells overexpressing wild-type and mutant DNMT3A or empty vector control 24 hours after cytarabine washout. Representative immunofluorescence microscopy images and quantification of PARP1 signal intensity in at least 50 nuclei per condition (****, P < 0.0001, Mann–Whitney rank-sum test). All experiments were independently replicated at least twice. H, PARP inhibitor olaparib increases sensitivity to cytarabine in U2OS cells expressing wild-type (blue) but not mutant (red) DNMT3A. Cell viability after 48 hours of treatment relative to vehicle control was measured by AlamarBlue assay in triplicate, in a single experiment (**, P < 0.01; ***, P < 0.001, unpaired Student t test).
Deficiency in DNA repair is a common explanation for accumulation of DNA damage in drug-treated cells. Accordingly, DNA damage resolution after cytarabine washout was markedly delayed in DNMT3AR882C-overexpressing cells and coincided with sustained p53 activation (Fig. 2D and E). Of note, per-cell levels of a DNA damage marker γH2A.X were also significantly higher (Supplementary Fig. S2E and S2F) consistent with a possible defect in DNA damage repair. Mechanistically, this coincided with impaired recruitment of PARP1, a key component in directing DNA repair (34), to chromatin and PARP1 foci formation in DNMT3Amut-expressing cells treated by low-dose cytarabine insufficient to induce apoptosis, which was evident at 12 hours of drug exposure and persisted 24 hours after washout (Fig. 2F and G). Overexpression of DNMT3AWT promoted PARP1 recruitment and foci formation, suggesting a likely role in the resolution of cytarabine-induced DNA damage through PARP1-mediated DNA repair mechanisms. Consistently, PARP inhibitor olaparib sensitized DNMT3AWT-overexpressing cells to low-dose cytarabine yet had no effect on DNMT3Amut-overexpressing cells (Fig. 2H). Similarly, primary samples from DNMT3AWT AMLs were sensitive to olaparib ex vivo, whereas DNMT3AR882 cells remained unaffected (Supplementary Fig. S2G).
Cells expressing mutant DNMT3A are prone to replication stress and fork collapse
We hypothesized that increased sensitivity to DNA damage during S phase and accumulation of unresolved DNA breaks was associated with replication stress. Indeed, this was found in cells expressing mutant DNMT3A as evidenced by high levels of phospho-RPA, which persisted even after drug washout (Fig. 3A and B), and coincided with γH2A.X accumulation (Supplementary Fig. S3A and S3B), whereas DNMT3AWT appeared to alleviate drug-induced replication stress. Consistently, DNMT3Amut-expressing cells treated with cytarabine were characterized by a higher number and a larger area of PCNA foci (Fig. 3C and D; ref. 35). Of note, because Ara-C inhibits elongation, BrdU incorporation, an otherwise ideal means to visualize replication foci, could not be used in these studies.
Figure 3.

Cells expressing mutant DNMT3A experience prolonged replication stress after cytarabine treatment. A and B, Replication stress and DNA damage after cytarabine treatment in U2OS cells transduced to express wild-type and mutant DNMT3A or empty vector control. Representative immunofluorescence microscopy images after 12-hour exposure (A, pRPA pSer33, red, and DSB marker γH2A.X, green) and pRPA signal quantification at steady state, 12 hours of treatment, and 12 hours after drug washout in at least 50 cells per condition (B). C and D, Analysis of replication foci by PCNA immunofluorescence staining (green) and DAPI (blue) after 12 hours of cytarabine treatment (C) and PCNA signal quantification in untreated, treated, and 12 hours after drug removal in at least 50 cells per condition (D) in U2OS cells with wild-type and mutant DNMT3A. E–G, Replication restart analysis by EdU and BrdU double-labeling pulse-chase experiments. U2OS cells overexpressing wild-type and mutant DNMT3A or empty vector control were pulsed with EdU (red), treated with cytarabine for 12 hours, and washed and pulsed with BrdU (green) at indicated timepoints (E). Representative immunofluorescence microscopy images of double-labeled cells (F, 16 hours after drug removal) and quantification of BrdU and EdU signal colocalization (Manders coefficient) in at least 25 nuclei per condition at indicated time points after drug removal (G). (***, P < 0.001; ****, P < 0.0001, Mann–Whitney rank-sum test). Each experiment was independently replicated at least three times. H, Recovery of stalled replication forks was measured by DNA fiber assay in U2OS cells expressing wild-type or mutant DNMT3A or empty vector control. Cells were pulsed with CldU (red), treated with Ara-C for 12 hours, allowed to recover for 16 hours after drug washout, and pulsed with IdU (green) prior to harvest. Proportion of recovered forks (double-labeled red/green tracks) was calculated in triplicate, in a single experiment; at least 300 fibers were scored per condition in each replicate (*, P < 0.05; Student t test with Welch correction). Representative examples of recovered forks are shown.
Next, we examined resumption of DNA synthesis using pulse-chase double-labeling experiments with EdU (to identify replicating cells) and, following cytarabine wash-out, BrdU (marking replication restart). Cells were able to resume replication regardless of the DNMT3A genotype, with similar overall kinetics (Supplementary Fig. S3C and S3D) despite persisting DNA damage at replication forks. Confocal imaging revealed that DNMT3A mutant cells were more likely to suffer fork collapse seen as lack of colocalization between EdU (replication foci precytarabine) and BrdU (replication restart; Fig. 3E–G). Yet, the resolution of confocal microscopy is insufficient to track single replication forks. DNA combining assays to visualize individual forks using CldU (ongoing replication preexposure) and IdU (restart after drug washout) found significantly fewer recovered forks in DNMT3Amut-overexpressing cells after cytarabine treatment, seen as double-labeled DNA fibers compared with DNMT3AWT or empty vector controls (Fig. 3H). Similarly, DNMT3Amut cells showed impaired fork recovery after HU treatment, a well-established replication-stalling agent (Supplementary Fig. S3E). These results indicate a replication fork restart defect in the presence of mutant DNMT3A that is not restricted to cytarabine.
Cells expressing mutant DNMT3A progress through the cell cycle despite persisting DNA damage, leading to mitotic catastrophe
The ability of cytarabine-treated U2OS cells expressing wild-type or mutant DNMT3A to resume cycle progression was determined. The analysis was facilitated by partial cell synchronization at the G1–S phase boundary by Ara-C treatment. By 18 hours after drug washout, cells completed replication and began entering mitosis marked by histone H3 phosphorylation (Fig. 4A; Supplementary Fig. S4A and S4B). Notably, cells with DNMT3A mutation continued to advance through the cell cycle, albeit with a lag (Supplementary Fig. S4C), despite unresolved DNA damage. As a result, there was an increased frequency of chromosome breaks in metaphase preparations of mutant DNMT3A-expressing cells (Fig. 4B and C) along with hallmarks of mitotic catastrophe such as micronucleation and abnormal nuclear morphology (Fig. 4D–G).
Figure 4.

Mutant DNMT3A-expressing cells carrying DNA breaks after cytarabine treatment progress through mitosis. A, Cell-cycle profiles (by DNA content) in U2OS cells with wild-type (blue) and mutant (red) DNMT3A or empty vector control (black) after cytarabine treatment and after washout. Fixed and permeabilized cells were stained with DAPI and analyzed by flow cytometry. Results are representative of three independent experiments, each performed in duplicate. B and C, Persistent DNA breaks in mitosis after cytarabine treatment in U2OS cells lentivirally overexpressing wild-type and mutant DNMT3A or empty vector control. Representative images of metaphase spreads (B, Wright–Geimsa stain) and frequency of metaphases with chromosomal abnormalities from five independent experiments each scoring at least 200 metaphases per genotype (C) 16 hours after drug washout (*, P < 0.05; ****, P < 0.0001, unpaired t test; graphs represent mean ± SEM). D–G, Mitotic catastrophe as a consequence of abnormal mitoses in cells with persistent DNA damage after cytarabine wash-out. Cells were pulsed with BrdU for 1 hour, exposed to cytarabine for 24 hours, and released from drug treatment; 48 hours later, cells were fixed and stained for BrdU (D, E) and Lamin-B1 (F, G). Representative microphotographs of nuclear morphology (D, DAPI was used to visualize all nuclei in blue) and percentage of BrdU+ cells with micronuclei (E, **, P < 0.01, unpaired t test; graphs represent mean ± SEM from three independent replicate experiments each scoring at least 40 nuclei). Representative microphotographs of nuclear lamina abnormalities visualized by Lamin-B1 staining (red) in BrdU+ cells (green; F) and fraction of BrdU+ cells with abnormally shaped nuclei (G). At least 100 nuclei per genotype were scored in each of two independent experiments (*, P < 0.05; ***, P < 0.001, pairwise two-tailed Fisher exact tests, where each normal nucleus was categorized as 0 and each abnormal as 1).
Collectively, our studies indicate that cells with mutant DNMT3A exhibit a defect in repair and recovery of stalled replication forks, resulting in accumulation of DNA breaks carried through mitosis. The accentuated cytarabine sensitivity is further highlighted by our observation that doubling Ara-C concentration prevented mutant DNMT3A-expressing cells from completing replication seen as G2 peak degradation in cell-cycle analysis, together with a new sub-G1 population indicative of cell death, in contrast to DNMT3A wild-type cells that maintained a normal cell-cycle distribution (Supplementary Fig. S4D).
Gene-expression profiling after cytarabine treatment identifies signatures of deregulated cell cycle and DNA replication in cells expressing mutant DNMT3A
To gain a broader view of cytarabine response, we performed RNA-sequencing to elucidate gene-expression signatures deregulated by mutant DNMT3A (Supplementary Table S4). Unsupervised hierarchical clustering of the 2,000 most variable genes demonstrated predominant gene activation induced by cytarabine treatment and robust separation from untreated controls, with consistency between biological triplicates (Supplementary Fig. S5A). Pairwise comparisons between cytarabine-treated and untreated control cells found widespread gene activation that was largely shared between genotypes yet featured a significant proportion of uniquely regulated genes in DNMT3Amut-expressing samples, confirmed by qPCR (Fig. 5A; Supplementary Fig. S5B and S5C; Supplementary Table S5).
Figure 5.

Gene-expression profiling identifies pathways deregulated in mutant DNMT3A-expressing cells treated with Ara-C. A, Venn diagram showing differentially expressed genes (FC ≥ 1.5, FDR < 0.05) between vehicle and Ara-C–treated (10 μmol/L, 24 hours) cells that are common or unique to each of the experimental groups: U2OS cells expressing wild-type (blue) or mutant (red) DNMT3A, or empty vector control (black); data from three independent biological replicates. B, HALLMARK gene sets (MSigDB) significantly enriched among commonly upregulated or downregulated genes in all three groups (FDR < 0.1). C, Significantly enriched KEGG pathways and HALLMARK gene sets among genes uniquely downregulated in DNMT3Amut cells after cytarabine treatment (FDR<0.1). D, STRING functional protein network analysis corresponding to genes uniquely downregulated in the DNMT3Amut cells 24 hours after treatment with Ara-C, and their UniProt annotations. E, Enrichment of GO terms among genes differentially expressed (P < 0.05) in cytarabine-treated DNMT3Amut cells compared with treated empty vector controls.
Pathway analysis using Enrichr to search against HALLMARK gene set collection (MSigDB) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Supplementary Table S6) identified p53 signaling as most significantly overrepresented among commonly upregulated genes in all groups (Fig. 5B), consistent with genotoxic stress, yet was most pronounced in the DNMT3Amut-overexpressing group evidenced by robust induction of its direct transcriptional target CDKN1A (Supplementary Fig. S5C). Consistently, pharmacologically induced p53 stabilization, an emerging therapeutic paradigm in AML (36), further sensitized DNMT3Amut cells to cytarabine. This was seen in OCI/AML-3 leukemia cells, in DNMT3Amut-overexpressing U2OS cells, and in primary DNMT3Amut AML samples from a recently completed BeatAML clinical trial (ref. 37; Supplementary Fig. S5D–S5F). At the same time, genes associated with the MYC pathway, G2–M checkpoint, and E2F targets were repressed in all groups, indicative of ongoing replication stress and proliferation delay, reinforcing our biochemical findings (Figs. 2A and E and 4A). Conversely, genes uniquely upregulated in DNMT3Amut-expressing cells after cytarabine treatment did not show similarly robust gene set enrichment apart from high expression of genes implicated in stemness and hematopoietic development such as CD34 (38), PDGFRA (39), HOXB6 (40), and IKZF2 (ref. 41; Supplementary Fig. S5B and S5G). Analysis of genes with decreased expression after cytarabine uniquely in the mutant DNMT3A-expressing cells highlighted further repression of proliferation-related MYC targets and identified disruption of vital leukemia and stem cell regulatory and metabolic pathways such as mitochondrial oxidative phosphorylation (OXPHOS) and fatty acid metabolism, RNA splicing, and protein synthesis and degradation (Fig. 5C; refs. 42–45). Furthermore, functional networks formed by proteins encoded by these exclusively DNMT3Amut-downregulated genes were characterized by prominent clusters annotated as proteasome, mitochondria, spliceosome, and ribosome biogenesis (Fig. 5D), suggesting distinctive shifts in cell physiology and molecular liabilities.
Pairwise comparison between cytarabine-treated DNMT3A-mutant and empty vector control cells found deregulation of cell cycle-related pathways, emphasizing G1–S phase transition and DNA replication as well as cell adhesion (Fig. 5E; ref. 46). Gene set enrichment analysis of gene ontology (GO) data sets additionally detected negative enrichment of signatures related to chromatin packaging and myeloid cell differentiation, in line with previous reports and a prominent role of DNMT3A in myeloid biology (Supplementary Fig. S5H; refs. 4, 9, 11, 20, 47).
Discussion
Mutations in DNMT3A are among the most common genetic alterations in de novo AML and are associated with adverse outcomes (5–8, 10, 12, 13, 19). In particular, the R882 mutation conveys an unfavorable prognosis in patients ≥60 years of age (48). This is at least in part due to relative resistance to anthracyclines stemming from another defect in DDR (10–12) and inability to tolerate dose-dense regimens, straining an already limited list of therapeutic options available for this patient group (3, 16). Here we show that cells with DNMT3A mutations may be sensitive to replication stress–related DNA damage induced by nucleoside analogues such as cytarabine, in agreement with a recent clinical trial (18). Low-dose cytarabine-, cladribine-based, or other similar regimens are well tolerated and hence suitable for older patients and those with poor performance status.
A growing body of evidence recognizes emergence of “collateral sensitivities,” or increased susceptibility to a second therapy as a fitness trade-off in cancer cells following the development of resistance to initial treatment or other perturbations (49, 50). This raises a possibility of rationally constructing synthetic lethality-based treatment approaches exploiting such antagonistic pleiotropic mechanisms (51, 52). The concept that the same genetic defect may be protective in the context of a specific type of DNA damage yet make cells vulnerable to a different type of genotoxic insult has been described (53). Similarly, our studies demonstrate that although the presence of DNMT3A(R882) decreases the sensitivity to DNA torsional stress induced by anthracyclines and possibly other DNA intercalators (11), it renders the cells more susceptible to the deleterious effects of replication fork stalling, summarized in a working model in Fig. 6.
Figure 6.
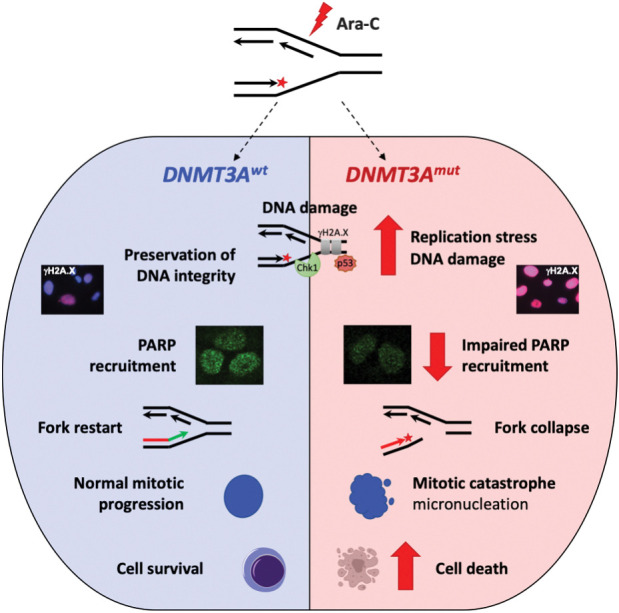
Cells expressing DNMT3A with R882 mutations are more sensitive to DNA damage at replication forks. In cells with mutant DNMT3A, treatment with replication-stalling drugs such as nucleoside analogue cytarabine leads to prolonged replication stress and accumulation of DNA damage, which coincides with attenuated chromatin recruitment of PARP and propensity to replication fork collapse. DNA damage persists through mitosis, resulting in mitotic defects, such as formation of micronuclei or mitotic catastrophe.
Accentuated replication stress in cells expressing mutant DNMT3A is further supported by previous findings of negatively enriched E2F target and CHEK1-regulated G2–M checkpoint gene sets reported in independent AML cohorts as well as in mice (11, 23), which reflect G1–S cell-cycle phase transition rate (24, 25) and G2–M checkpoint adaptation (54). Accordingly, after low-dose cytarabine exposure (approximately IC20), mutant DNMT3A-expressing cells demonstrated delayed cell-cycle progression that culminated in entering mitosis with damaged DNA. Surviving cells demonstrated increased formation of micronuclei, structures containing shattered chromosomes, indicative of mitotic catastrophe (33, 55). Furthermore, incremental increase in cytarabine concentration resulted in disproportionate apoptotic response, highlighting heightened replication stress in DNMT3A-mutant cells as a potential therapeutic vulnerability that can be unveiled by replication-stalling nucleoside analogues.
Such distinctive sensitivity to cytarabine arises following a defect in recovery from replication fork arrest that leads to accumulation of persistent, unrepaired DNA damage observed in cells with mutant DNMT3A. Although previous studies have attributed specific DNA damage repair pathways as defining therapeutic responses to replication stress–inducing nucleoside analogues (28, 56–59), the exact mechanisms that underlie impaired resolution of cytarabine-induced DNA lesions in cells with mutant DNMT3A remain uncertain. Interestingly, other than a slight upregulation of genes annotated to be repressed during UV response indicative of a possible disruption of nucleotide excision repair (Supplementary Fig. S5C; ref. 34) our gene-expression studies did not detect signatures of impaired DNA damage repair. Prior in-depth studies reported only modest changes in DNA methylation (11, 21, 22, 60) that predominantly affected lineage-specific regulatory elements (23, 47, 61–63) yet did not affect DNA damage and repair pathways. Whether a methylation-independent mechanism of mutant DNMT3A action might be at play remains to be determined. In support of this, wild-type DNMT3A protein was found to directly associate with stalled replication forks (26) and likely enriched in the PCNA interactome (64). Consistently, our results support a protective role played by wild-type DNMT3A against Ara-C-induced DNA damage and replication stress. There are many examples of noncatalytic accessory functions of epigenetic enzymes regulating both histone (65, 66) and DNA modifications (67–69). Future studies interrogating the replication fork-associated proteome (70) in cells with and without mutant DNMT3A, in addition to differential recruitment of DNA repair proteins to chromatin and proximity-labeling proteomics approaches (71), will be necessary to clarify its role in preserving DNA integrity. This emerging knowledge will be instructive to developing further therapeutic combination strategies such as with PARP inhibitors.
In addition to DNA replication, cell-cycle regulation, and proliferation-related gene sets, which are likely secondary to DNA damage after cytarabine, our gene-expression studies uncovered signatures of deregulated cell adhesion, proteostasis, as well as repressed OXPHOS and fatty acid metabolism, and RNA splicing, all of which are critical for hematopoietic stem cell function and are commonly deregulated during aging and in myeloid malignancies. Notably, deregulation of ribosome biogenesis and protein degradation may reflect loss of stemness (72, 73), whereas increased reliance on OXPHOS and fatty acid metabolism is characteristic of chemoresistant AML (43, 46, 74) and together with altered splicing engenders potential therapeutic vulnerabilities (44, 75, 76). It will be interesting to investigate if these processes contribute to the leukemic progression in individuals with clonal hematopoiesis with DNMT3A mutations (77–82) to serve as biomarkers for high risk of AML development.
In summary, our studies demonstrate that cells expressing mutant DNMT3A have a defect in recovery from replication fork arrest and subsequent accumulation of unresolved DNA damage, which may have therapeutic tractability. These results demonstrate, in addition to its role in epigenetic control, DNMT3A contributes to preserving genome integrity during DNA replication and suggest that cytarabine-induced replication fork stalling may further synergize with other agents aimed at DNA damage and repair, replication, p53 pathway potentiation, or cell survival, as well as splicing and oxidative phosphorylation (44, 45, 75, 76, 83). Future studies will determine if such combinatorial treatment approaches can be developed to usher in the era of precision oncology for AML patients with mutant DNMT3A.
Authors' Disclosures
J. Li reports grants from Leukemia and Lymphoma Society during the conduct of the study. P.B. Staber reports grants and personal fees from F. Hoffmann-La Roche AG and personal fees from Amgen, Takeda, AbbVie, Janssen Cilag, Incyte, BMS, MSD, AstraZeneca, and Beigen outside the submitted work. J.D. Licht reports grants from Epizyme outside the submitted work. J.E. Bird reports grants from NIH during the conduct of the study. O.A. Guryanova reports grants from NCI/NIH, Thomas H. Maren Junior Investigator Fund, University of Florida College of Medicine, National Institute for Diabetes and Digestive and Kidney Diseases (NIDDK)/NIH, Harry T Mangurian Foundation, and Ocala Royal Dames for Cancer Research during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge generous support by NIH awards CA178191 and DK121831 (O.A. Guryanova), CA188561 (R. Opavsky), and CA252400 (B.K. Law). O.A. Guryanova was supported in part by the Ocala Royal Dames for Cancer Research, the Harry T. Mangurian Foundation, and the Thomas H. Maren Junior Investigator Fund. B.K. Law was supported in part by the Florida Breast Cancer Foundation and the Ocala Royal Dames for Cancer Research. S. Bhaduri-McIntosh was supported by the Children's Miracle Network. J. Li and D. Dupéré-Richer are Leukemia and Lymphoma Society (LLS) Special Fellows; J.D. Licht is supported in part by the LLS Specialized Center of Research (SCOR). Immunophenotyping was performed at the Flow Cytometry and Imaging Core, UF Interdisciplinary Center for Biotechnology Research (ICBR) (RRID: SCR_019119). The authors thank Lidia Kulemina, PhD (UF Health Cancer Center) for critical reading of the manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
K. Venugopal: Conceptualization, data curation, formal analysis, investigation, methodology, writing–original draft. Y. Feng: Formal analysis, investigation, and methodology. P. Nowialis: Formal analysis. H. Xu: Formal analysis and investigation. D.E. Shabashvili: Investigation. C.M. Berntsen: Investigation. P. Kaur: Investigation. K.I. Krajcik: Investigation. C. Taragjini: Investigation. Z. Zaroogian: Investigation. H.L. Casellas Román: Investigation. L.M. Posada: Investigation. C. Gunaratne: Investigation. J. Li: Methodology. D. Dupéré-Richer: Methodology. R.L. Bennett: Methodology. S. Pondugula: Formal analysis. A. Riva: Methodology. C.R. Cogle: Conceptualization. R. Opavsky: Conceptualization. B.K. Law: Conceptualization, writing–review and editing. S. Bhaduri-McIntosh: Resources, supervision, and methodology. S. Kubicek: Resources, data curation, formal analysis, and investigation. P.B. Staber: Resources, data curation, investigation, and methodology. J.D. Licht: Conceptualization, methodology, writing–review and editing. J.E. Bird: Conceptualization, methodology, writing–review and editing. O.A. Guryanova: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52. [DOI] [PubMed] [Google Scholar]
- 2. Estey EH. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am J Hematol 2018;93:1267–91. [DOI] [PubMed] [Google Scholar]
- 3. Tamamyan G, Kadia T, Ravandi F, Borthakur G, Cortes J, Jabbour E, et al. Frontline treatment of acute myeloid leukemia in adults. Crit Rev Oncol Hematol 2017;110:20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brunetti L, Gundry MC, Goodell MA. DNMT3A in leukemia. Cold Spring Harb Perspect Med 2017;7:a030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 2011;43:309–15. [DOI] [PubMed] [Google Scholar]
- 9. Venugopal K, Feng Y, Shabashvili D, Guryanova OA. Alterations to DNMT3A in hematologic malignancies. Cancer Res 2021;81:254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med 2016;22:1488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sehgal AR, Gimotty PA, Zhao J, Hsu JM, Daber R, Morrissette JD, et al. DNMT3A mutational status affects the results of dose-escalated induction therapy in acute myelogenous leukemia. Clin Cancer Res 2015;21:1614–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balasubramanian SK, Aly M, Nagata Y, Bat T, Przychodzen BP, Hirsch CM, et al. Distinct clinical and biological implications of various DNMT3A mutations in myeloid neoplasms. Leukemia 2018;32:550–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lowenberg B, Pabst T, Vellenga E, van Putten W, Schouten HC, Graux C, et al. Cytarabine dose for acute myeloid leukemia. N Engl J Med 2011;364:1027–36. [DOI] [PubMed] [Google Scholar]
- 16. Sekeres MA, Guyatt G, Abel G, Alibhai S, Altman JK, Buckstein R, et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv 2020;4:3528–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kadia TM, Borthakur G, Pemmaraju N, Daver N, DiNardo CD, Sasaki K, et al. Phase II study of venetoclax added to cladribine + low dose AraC (LDAC) alternating with 5-azacytidine demonstrates high rates of minimal residual disease (MRD) negative complete remissions (CR) and excellent tolerability in older patients with newly diagnosed acute myeloid leukemia (AML). Blood 2020;136:17–9. [Google Scholar]
- 18. Kadia TM, Cortes J, Ravandi F, Jabbour E, Konopleva M, Benton CB, et al. Cladribine and low-dose cytarabine alternating with decitabine as front-line therapy for elderly patients with acute myeloid leukaemia: a phase 2 single-arm trial. Lancet Haematol 2018;5:e411–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaidzik VI, Schlenk RF, Paschka P, Stolzle A, Spath D, Kuendgen A, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML study group (AMLSG). Blood 2013;121:4769–77. [DOI] [PubMed] [Google Scholar]
- 20. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2011;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014;25:442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeong M, Park HJ, Celik H, Ostrander EL, Reyes JM, Guzman A, et al. Loss of dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep 2018;23:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glass JL, Hassane D, Wouters BJ, Kunimoto H, Avellino R, Garrett-Bakelman FE, et al. Epigenetic identity in AML depends on disruption of nonpromoter regulatory elements and is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov 2017;7:868–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 1997;277:1495–7. [DOI] [PubMed] [Google Scholar]
- 25. Branigan TB, Kozono D, Schade AE, Deraska P, Rivas HG, Sambel L, et al. MMB-FOXM1-driven premature mitosis is required for CHK1 inhibitor sensitivity. Cell Rep 2021;34:108808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, et al. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 2015;59:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamba JK. Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009;10:1657–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ewald B, Sampath D, Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 2008;27:6522–37. [DOI] [PubMed] [Google Scholar]
- 29. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014;14:359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berti M, Cortez D, Lopes M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 2020;21:633–51. [DOI] [PubMed] [Google Scholar]
- 31. Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene 2003;22:9063–74. [DOI] [PubMed] [Google Scholar]
- 32. Forment JV, O'Connor MJ. Targeting the replication stress response in cancer. Pharmacol Ther 2018;188:155–67. [DOI] [PubMed] [Google Scholar]
- 33. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004;23:2825–37. [DOI] [PubMed] [Google Scholar]
- 34. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010;40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell 2007;129:665–79. [DOI] [PubMed] [Google Scholar]
- 36. Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med 2018;10:eaao3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satterthwaite AB, Burn TC, Le Beau MM, Tenen DG. Structure of the gene encoding CD34, a human hematopoietic stem cell antigen. Genomics 1992;12:788–94. [DOI] [PubMed] [Google Scholar]
- 39. Demoulin JB, Montano-Almendras CP. Platelet-derived growth factors and their receptors in normal and malignant hematopoiesis. Am J Blood Res 2012;2:44–56. [PMC free article] [PubMed] [Google Scholar]
- 40. Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2013;27:1000–8. [DOI] [PubMed] [Google Scholar]
- 41. Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, et al. IKZF2 drives leukemia stem cell self-renewal and inhibits myeloid differentiation. Cell Stem Cell 2019;24:153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014;509:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov 2017;7:716–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, et al. RNA splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell 2016;19:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science 2020;368:eaaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raffel S, Klimmeck D, Falcone M, Demir A, Pouya A, Zeisberger P, et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020;136:1507–19. [DOI] [PubMed] [Google Scholar]
- 47. Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, Shank K, et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016;30:1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol 2012;30:742–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao B, Sedlak JC, Srinivas R, Creixell P, Pritchard JR, Tidor B, et al. Exploiting temporal collateral sensitivity in tumor clonal evolution. Cell 2016;165:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell 2018;173:1413–25 e14. [DOI] [PubMed] [Google Scholar]
- 51. Lin KH, Rutter JC, Xie A, Pardieu B, Winn ET, Bello RD, et al. Using antagonistic pleiotropy to design a chemotherapy-induced evolutionary trap to target drug resistance in cancer. Nat Genet 2020;52:408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Acar A, Nichol D, Fernandez-Mateos J, Cresswell GD, Barozzi I, Hong SP, et al. Exploiting evolutionary steering to induce collateral drug sensitivity in cancer. Nat Commun 2020;11:1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eccles LJ, Bell AC, Powell SN. Inhibition of non-homologous end joining in Fanconi anemia cells results in rescue of survival after interstrand crosslinks but sensitization to replication associated double-strand breaks. DNA Repair 2018;64:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kalsbeek D, Golsteyn RM. G2/M-phase checkpoint adaptation and micronuclei formation as mechanisms that contribute to genomic instability in human cells. Int J Mol Sci 2017;18:2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Imreh G, Norberg HV, Imreh S, Zhivotovsky B. Chromosomal breaks during mitotic catastrophe trigger gammaH2AX-ATM-p53-mediated apoptosis. J Cell Sci 2016;129:1950. [DOI] [PubMed] [Google Scholar]
- 56. Lai TH, Mitchell S, Wu PJ, Orwick S, Liu C, Ravikrishnan J, et al. HSP90 inhibition depletes DNA repair proteins to sensitize acute myelogenous leukemia to nucleoside analog chemotherapeutics. Leuk Lymphoma 2019;60:2308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strom SS, Estey E, Outschoorn UM, Garcia-Manero G. Acute myeloid leukemia outcome: role of nucleotide excision repair polymorphisms in intermediate risk patients. Leuk Lymphoma 2010;51:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fordham SE, Matheson EC, Scott K, Irving JA, Allan JM. DNA mismatch repair status affects cellular response to Ara-C and other anti-leukemic nucleoside analogs. Leukemia 2011;25:1046–9. [DOI] [PubMed] [Google Scholar]
- 59. Boeckemeier L, Kraehenbuehl R, Keszthelyi A, Gasasira MU, Vernon EG, Beardmore R, et al. Mre11 exonuclease activity removes the chain-terminating nucleoside analog gemcitabine from the nascent strand during DNA replication. Sci Adv 2020;6:eaaz4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, et al. CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 2017;168:801–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Izzo F, Lee SC, Poran A, Chaligne R, Gaiti F, Gross B, et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet 2020;52:378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tovy A, Reyes JM, Gundry MC, Brunetti L, Lee-Six H, Petljak M, et al. Tissue-biased expansion of DNMT3A-mutant clones in a mosaic individual is associated with conserved epigenetic erosion. Cell Stem Cell 2020;27:326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lim JY, Duttke SH, Baker TS, Lee J, Gambino KJ, Venturini NJ, et al. DNMT3A haploinsufficiency causes dichotomous DNA methylation defects at enhancers in mature human immune cells. J Exp Med 2021;218:e20202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Srivastava M, Chen Z, Zhang H, Tang M, Wang C, Jung SY, et al. Replisome dynamics and their functional relevance upon DNA damage through the PCNA interactome. Cell Rep 2018;25:3869–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, et al. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell 2017;66:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH, et al. A UTX-MLL4-p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol Cell 2017;67:308–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol 2008;28:752–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nowialis P, Lopusna K, Opavska J, Haney SL, Abraham A, Sheng P, et al. Catalytically inactive Dnmt3b rescues mouse embryonic development by accessory and repressive functions. Nat Commun 2019;10:4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, et al. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell 2016;30:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sirbu BM, Couch FB, Cortez D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 2012;7:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bosch JA, Chen CL, Perrimon N. Proximity-dependent labeling methods for proteomic profiling in living cells: an update. Wiley Interdiscip Rev Dev Biol 2021;10:e392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hidalgo San JL, Sunshine MJ, Dillingham CH, Chua BA, Kruta M, Hong Y, et al. Modest declines in proteome quality impair hematopoietic stem cell self-renewal. Cell Rep 2020;30:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chavez JS, Rabe JL, Loeffler D, Higa KC, Hernandez G, Mills TS, et al. PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J Exp Med 2021;218: e20201169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tcheng M, Roma A, Ahmed N, Smith RW, Jayanth P, Minden MD, et al. Very long chain fatty acid metabolism is required in acute myeloid leukemia. Blood 2021;137:3518–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036–46. [DOI] [PubMed] [Google Scholar]
- 76. Escobar-Hoyos L, Knorr K, Abdel-Wahab O. Aberrant RNA splicing in cancer. Annu Rev Cancer Biol 2019;3:167–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018;559:400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 2014;111:2548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018;24:1015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017;547:104–8. [DOI] [PubMed] [Google Scholar]
- 82. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Leon TE, Rapoz-D'Silva T, Bertoli C, Rahman S, Magnussen M, Philip B, et al. EZH2-deficient T-cell acute lymphoblastic leukemia is sensitized to CHK1 inhibition through enhanced replication stress. Cancer Discov 2020;10:998–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed RNA-seq data are available at GEO under the accession code GSE153871.