

Abstract
Severe immune-related adverse events (irAEs) occur in up to 60% of patients with melanoma treated with immune checkpoint inhibitors (ICIs). However, it is unknown whether a common baseline immunological state precedes irAE development. Here we applied mass cytometry by time of flight, single-cell RNA sequencing, single-cell V(D)J sequencing, bulk RNA sequencing and bulk T cell receptor (TCR) sequencing to study peripheral blood samples from patients with melanoma treated with anti-PD-1 monotherapy or anti-PD-1 and anti-CTLA-4 combination ICIs. By analyzing 93 pre- and early on-ICI blood samples and 3 patient cohorts (n = 27, 26 and 18), we found that 2 pretreatment factors in circulation—activated CD4 memory T cell abundance and TCR diversity—are associated with severe irAE development regardless of organ system involvement. We also explored on-treatment changes in TCR clonality among patients receiving combination therapy and linked our findings to the severity and timing of irAE onset. These results demonstrate circulating T cell characteristics associated with ICI-induced toxicity, with implications for improved diagnostics and clinical management.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Although ICIs have revolutionized cancer treatment, approximately 10–60% of ICI-treated patients with melanoma currently develop severe immune-related toxicities, with the rate of toxicity closely linked to the specific therapy administered1–9. Also known as irAEs, ICI-induced toxicities impact a range of organ systems, including the lungs, liver, heart, skin, pituitary gland and gastrointestinal tract4, and can be associated with substantial morbidity requiring urgent medical intervention. Such morbidities can lead to the suspension of anticancer treatment, and in the most severe cases, death1,4,10–14. The biological drivers of irAEs are poorly characterized and there is no method in standard clinical practice to identify which patients are at highest risk for developing them1,4.
Accordingly, several groups have investigated potential biomarkers of ICI-induced toxicity based on blood or tumor analysis15–29. However, these studies have generally been focused on early on-treatment prediction or single organ systems15–23,27–29, with only modest performance for predicting irAEs in the pretreatment setting independent of the affected organ system24,25. Recently, a candidate pneumonitis-only irAE biomarker using tumor immunohistochemistry was reported; however, this biomarker was indirectly identified from The Cancer Genome Atlas, which lacks toxicity annotations, and was evaluated in a case-control setting without the inclusion of low-grade irAEs23. Another group identified a single-nucleotide polymorphism within the gene encoding microRNA-146a that was associated with severe irAE development30. Still, other groups have identified ICI response biomarkers without examining irAEs31–37.
Given the considerable heterogeneity of ICI-induced irAEs, including variation in their timing, severity and location, determining the factors that cause them has remained challenging. Pre-existing autoantibodies15, autoreactive tissue-resident T cells29 and T cells with specificity for viral antigens stemming from chronic viral infection14,27 have all been implicated in irAEs. Changes in the gut microbiome leading to increased colonic interleukin-1ß expression were also recently reported in ICI-induced colitis26. Given these observations, several groups have investigated parallels between irAEs and autoimmune disease29. Indeed, case reports have shown that ICIs can cause frank autoimmunity38–40, suggesting that irAEs could represent subclinical autoimmunity in a subset of patients. However, whether a common immunological state precedes distinct manifestations of ICI-induced toxicity is unknown.
In this study, we set out to systematically evaluate immunological features in the peripheral blood associated with ICI-induced toxicity in patients with metastatic melanoma. Across distinct single-cell and bulk profiling modalities, we identified common T cell features linked to the development of severe irAEs within three months of treatment initiation. These features were independent of key clinical variables, including durable clinical response and treatment with anti-PD-1 monotherapy or anti-PD-1 and anti-CTLA-4 combination therapy. Leveraging these findings, we developed predictive models of irAE development and explored their utility for pretreatment and early on-treatment identification of ICI-induced toxicity.
Results
Clinical cohort characteristics.
To study candidate risk factors associated with severe (grade 3+) irAE development, we identified 78 patients with metastatic melanoma, 71 of whom were evaluable after exclusion criteria were applied (Fig. 1 and Supplementary Table 1). Among these patients, 33 were treated with anti-PD-1 monotherapy, 38 were treated with anti-PD-1 plus anti-CTLA-4 combination therapy and 90% had no previous ICI history (Supplementary Table 1). All patients were monitored closely during and after ICI treatment for irAE development (median follow-up time of 14.9 months; median time to grade 3+ irAE of 1.5 months). Most patients experienced one or more irAEs, ranging from mild (grade 1) to life-threatening (grade 4) and affecting diverse organ systems, which were classified by board-certified clinicians according to standardized criteria (CTCAE v.5.0; Methods). We stratified the 71 patients into 3 nonoverlapping cohorts, a single-cell discovery cohort and a larger bulk cohort divided into training and validation sets (Fig. 1 and Supplementary Table 1).
Fig. 1 |. Study schema.
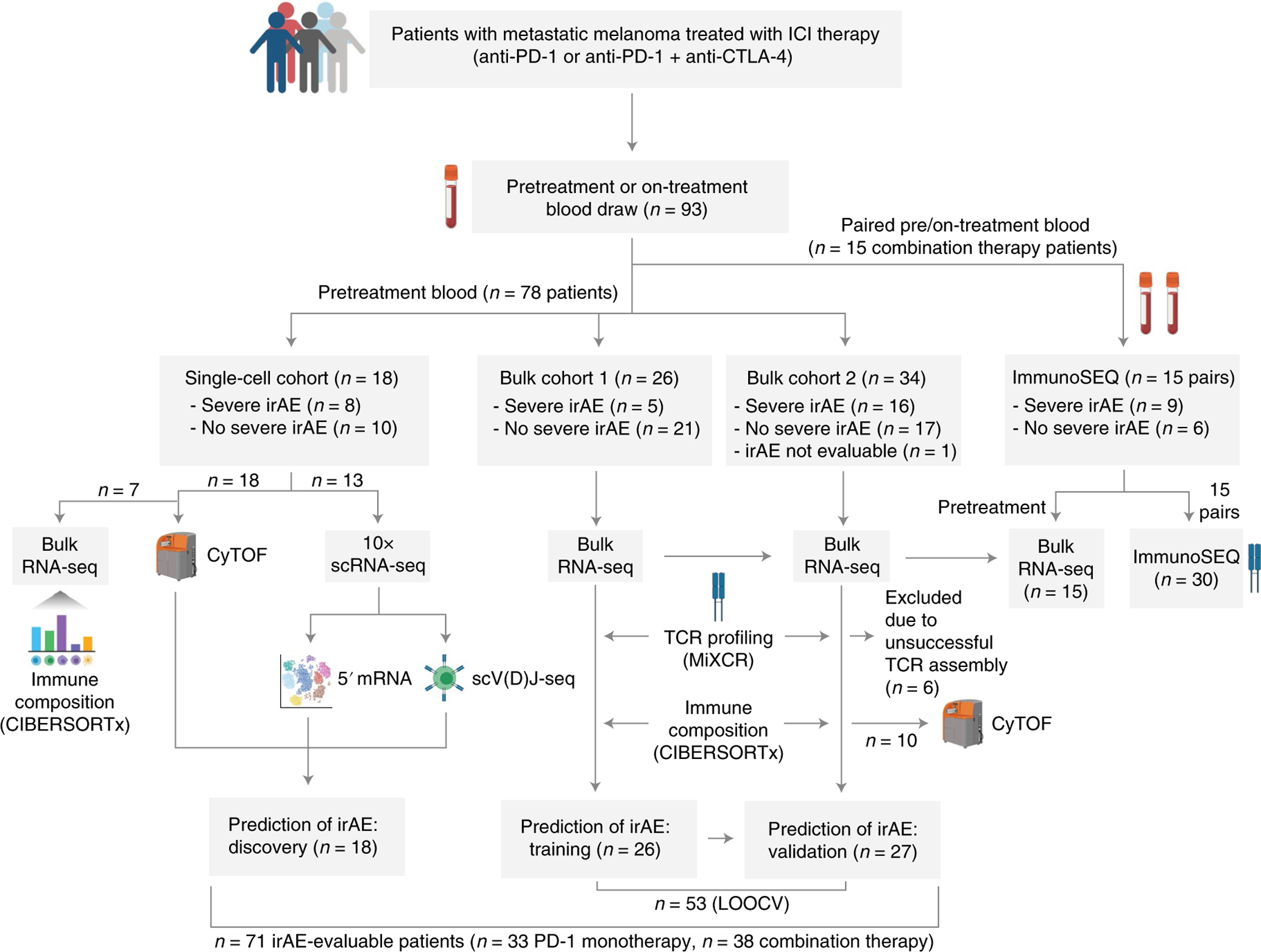
Overview of patients included in this study, summary of their irAE status, exclusion criteria and downstream analyses that were performed. Among 78 total eligible patients, 71 were evaluable for irAE analysis after exclusion criteria were applied. Further details are provided in the Methods and Supplementary Tables 1–3, 7–9 and 18. Created using icons from BioRender.com.
Determinants of severe irAEs from pretreatment blood.
We started by performing high-dimensional single-cell profiling of pretreatment peripheral blood samples from 18 patients (single-cell discovery cohort, Figs. 1 and 2a; Supplementary Tables 2 and 3) of which 8 patients experienced severe irAEs after treatment initiation. By applying mass cytometry by time of flight (CyTOF) to profile 35 leukocyte markers in each sample (Supplementary Table 4), we analyzed 20 distinct subpopulations from nearly 800,000 evaluable cells, encompassing 7 major mononuclear lineages (B cells, plasmablasts, CD4 and CD8 T cells, natural killer (NK) cells, natural killer T (NKT) cells, monocytes) (Fig. 2b,c, Supplementary Figs. 1 and 2a, Supplementary Table 5 and Methods). Next, we interrogated each subpopulation with respect to severe irAE outcomes (Fig. 2c). Of all subpopulations, only CD4 effector memory T (TEM) cells were significant after multiple hypothesis correction, with higher levels in pretreatment blood associated with severe irAE development (P = 0.0002; Q = 0.004; Fig. 2c,d, Supplementary Figs. 1b and 2b,c and Supplementary Table 3).
Fig. 2 |. Analysis of pretreatment peripheral blood for cellular determinants of severe irAEs using mass cytometry.
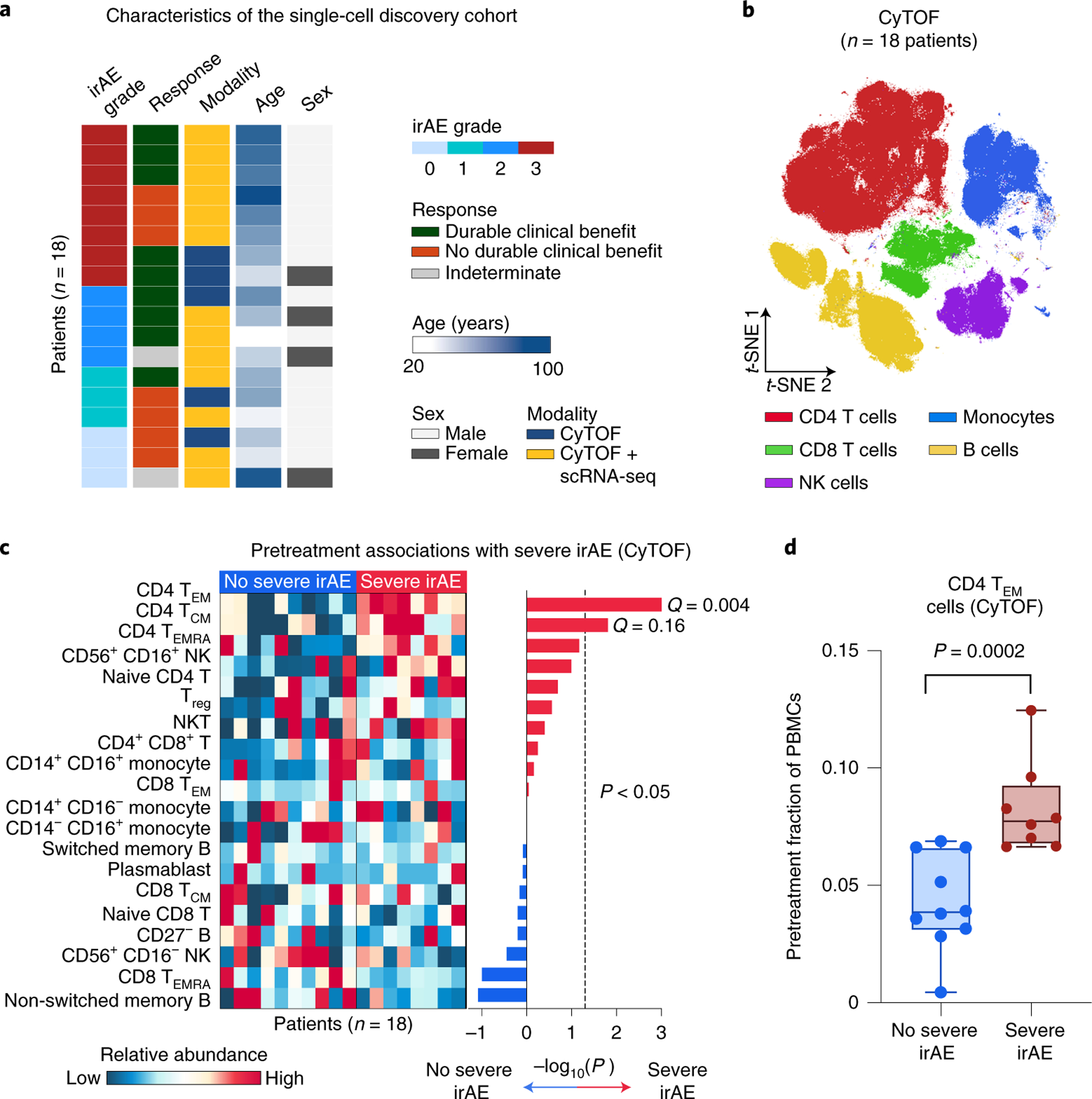
a, Characteristics of the single-cell discovery cohort (Fig. 1), including the highest irAE grade experienced and durable clinical response status after the start of immunotherapy (related to Supplementary Tables 2 and 3). b, viSNE projection of peripheral blood cells analyzed by CyTOF. t-SNE, t-distributed stochastic neighbor embedding. c, Left: Heatmap showing the relative abundance of 20 cell states identified by CyTOF in 18 patients (Supplementary Table 5), grouped by future irAE status. Right: Association of cell state abundance with severe irAE development. Statistical significance was determined by a two-sided, unpaired Wilcoxon rank-sum test and expressed as directional −log10 P values. For associations with no severe irAE, −log10 P values were multiplied by −1. Q values were determined by the Benjamini–Hochberg method. d, Frequencies of CD4 TEM cells (CyTOF) in the pretreatment peripheral blood of patients stratified by future irAE status (no severe irAE, n = 10 patients; severe irAE, n = 8 patients). The box center lines, box bounds and whiskers denote the medians, first and third quartiles and minimum and maximum values, respectively. Statistical significance was determined by a two-sided, unpaired Wilcoxon rank-sum test.
To corroborate this finding, we examined the same peripheral blood samples from 13 patients using 5′ droplet-based 10x Chromium single-cell RNA sequencing (scRNA-seq) paired with single-cell V(D)J sequencing (scV(D)J-seq) of TCR and B cell receptor (BCR) clonotypes. After quality control (Extended Data Fig. 1a and Methods), the 5′ assay yielded 24,807 cells and 7 major lineages classified on the basis of canonical marker gene expression (Fig. 3a). Employing unsupervised clustering, we identified 32 distinct transcriptional states across the 7 cell types (Fig. 3a and Methods). We then calculated the association between cell state abundance and the development of severe irAEs. Remarkably, across these 32 cell states, we found that CD4 T cell state 5, which lacks expression of CCR7 and SELL (CD62L) and is consistent with CD4 TEM cells, was most strongly associated with severe irAE development (nominal P = 0.05, two-sided, unpaired Wilcoxon rank-sum test; Fig. 3b). This state was also most correlated with CD4 TEM levels measured by CyTOF (Fig. 3b). When considering the joint probability of this result via permutation testing, we calculated an empirical P value of 0.003 (Methods). Further analysis revealed that CD4 T cell state 3, which is closely related to state 5 by unsupervised hierarchical clustering (Extended Data Fig. 1b), also showed an expression profile consistent with CD4 TEM (Fig. 3c and Extended Data Fig. 1c). When combined with state 5, the resulting cluster (CD4 T 5 + 3) was more significantly associated with severe irAE development and CD4 TEM levels enumerated by CyTOF (Fig. 3b and Supplementary Table 3). In fact, across all 82 possible pairwise combinations of cell states within each major cell type, CD4 T 5 + 3 achieved both the highest Spearman correlation against CD4 TEM levels enumerated by CyTOF and the strongest association with severe irAE development (Extended Data Fig. 1d,e and Methods).
Fig. 3 |. Analysis of pretreatment peripheral blood for cellular determinants of severe irAEs using single-cell RNA and V(D)J sequencing.
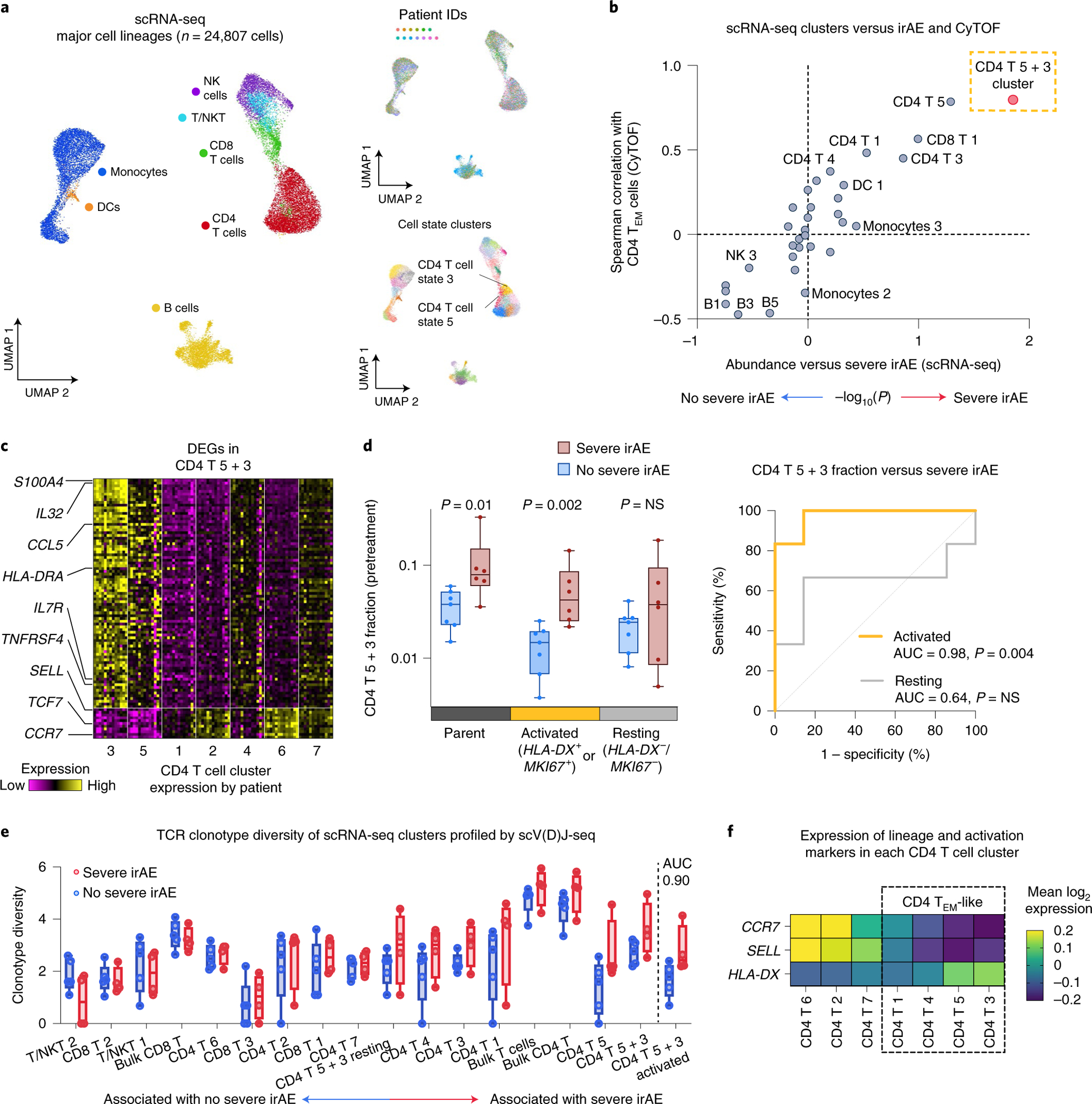
a, UMAP of peripheral blood cells profiled by scRNA-seq from 13 patients coanalyzed by CyTOF (Fig. 2a and Supplementary Tables 1–3), colored by cell type, patient and state (n = 32). T/NKT, NK-like T cells. b, Cell state abundances (scRNA-seq) versus future irAE status and CD4 TEM cell frequencies (CyTOF). The former was quantified by a two-sided, unpaired Wilcoxon rank-sum test and expressed as −log10 P values. For associations with no severe irAE, −log10 P values were multiplied by −1. CD4 T cell states 5 and 3 are indicated together as CD4 T 5 + 3. c, Heatmap of DEGs (Padj < 0.05) between CD4 T cell states 5 and 3 and other CD4 T cell states. Within each state, the columns represent the mean expression from individual patients converted to z-scores. d, Left: Frequencies of candidate activated and resting subsets of CD4 T 5 + 3 cell states in 13 patients stratified by no severe (n = 7) and severe (n = 6) irAE status. Activation markers with counts per million (CPM) > 0 were considered expressed. Significance was determined by a two-sided, unpaired Wilcoxon rank-sum test. Right: Receiver operating characteristic curve plot showing the performance of the CD4 T 5 + 3 subsets (from the left panel) for predicting severe irAE development. NS, not significant. e, Pretreatment TCR clonotype diversity within each T cell state, total T cells, CD8 T cells, CD4 T cells and activated versus resting CD4 T 5 + 3 cells (defined as in d), grouped by future irAE status. TCR diversity was calculated for all patients with at least 100 TCR clones (n = 9; Methods). States are ordered by the AUC between TCR diversity and severe irAE status. f, Mean expression of key lineage and activation genes in CD4 T cell states. States within the box are consistent with TEM and TEM-like phenotypes. The box center lines, box bounds and whiskers indicate the medians, first and third quartiles and minimum and maximum values, respectively.
Differential gene expression analysis against other CD4 T cell states revealed that CD4 T 5 and 3 are enriched for markers of activated effector cells including HLA-DRA, MKI67, TNFRSF4 (OX40), CCL5 and IL32 and depleted in markers of TCM cells (SELL/CD62L) and naive T cells (CCR7, TCF7)41–43 (Fig. 3c and Extended Data Fig. 1c). Using Seurat Azimuth for reference-guided cell labeling44, we confirmed that CD4 TEM cells are most associated with severe irAE and most similar to the CD4 T 5 + 3 population identified by de novo analysis (Extended Data Fig. 2 and Methods). Moreover, when the CD4 T 5 + 3 population was subdivided into activated and resting subsets based on the expression of canonical activation markers (HLA-DX, MKI67), the activated subset showed the strongest association with severe irAE development (P = 0.002, two-sided, unpaired Wilcoxon rank-sum test; Fig. 3d, Extended Data Fig. 3a and Supplementary Table 6). We verified this finding using reference-guided annotation with Azimuth and with CyTOF (Extended Data Fig. 3a,b and Supplementary Table 6), suggesting that activated CD4 TEM cells preferentially underlie severe ICI toxicity.
Given this observation, we wondered whether pretreatment TCR diversity in activated CD4 TEM cells might also correlate with severe ICI toxicity. Indeed, single-cell TCR clonotype diversity (Shannon entropy45,46) of activated CD4 T 5 + 3 cells was elevated in patients who experienced severe irAEs (area under the receiver operating characteristic curve (AUC) = 0.90, P = 0.05; Fig. 3e and Methods). This suggests that TCR richness, defined as the number of unique clonotypes within a sample and a key component of diversity metrics including Shannon entropy45,46, eclipses the loss of diversity resulting from clonal expansion when activated CD4 TEM cells are quantified relative to total peripheral blood mononuclear cells (PBMCs) (Extended Data Fig. 4a,b). In other words, among total PBMCs, the TCR richness of activated CD4 TEM cells underlies an overall increase in pretreatment TCR diversity in patients destined to develop severe irAE. Notably, definitions of clonotype diversity that incorporate richness have substantial precedent in previous literature, including studies of circulating and tumor-infiltrating T cells35,37,47,48, providing a strong foundation for their application in this work.
While this association between TCR diversity and severe irAE development was diminished or absent in other T cell subpopulations, when combining all evaluable T cells, we observed a striking trend between bulk TCR diversity in pretreatment samples and severe irAE development (AUC = 0.80; Fig. 3e). Moreover, this association was primarily attributable to CD4 T cells with an effector memory profile (low CCR7 and SELL) (Fig. 3f and Extended Data Fig. 4c–f). In contrast, differences in peripheral blood BCR diversity linked to severe irAE development were less pronounced (Extended Data Fig. 4g). Collectively, these findings suggest that a more diverse TCR repertoire at baseline in CD4 TEM cells, broadly reflected in bulk peripheral blood, is associated with the development of severe ICI toxicity.
Extended analysis of T cell features associated with irAEs.
Having identified candidate pretreatment determinants of severe irAE development, we next set out to verify our findings in a larger independent group of patients. Based on sample size estimates (Methods), we applied bulk RNA sequencing (bulk RNA-seq) to pretreatment peripheral blood samples from 53 additional patients with metastatic melanoma spanning two cohorts (n = 26 and 27) treated with single-agent (anti-PD-1, n = 29) or combination-agent (anti-PD-1 and anti-CTLA-4, n = 24) checkpoint blockade (Fig. 1 and Supplementary Tables 1 and 7). To assess circulating immunological features in bulk transcriptomic profiles, we applied CIBERSORTx49, a machine learning approach for the enumeration of cell subsets from bulk tissue expression profiles49–52, and MiXCR, a computational approach for V(D)J clonotype assembly and quantitation from bulk RNA-seq data53 (Supplementary Tables 8 and 9). By direct comparison to cytometry assays, we confirmed the accuracy of CIBERSORTx for deconvolution of major blood lineages, including the specificity of an activated CD4 memory T cell (TM) signature for activated CD4 TEM cells using peripheral blood from 17 patients with melanoma (CyTOF) (Extended Data Fig. 5, Supplementary Fig. 3 and Methods).
Remarkably, of 13 PBMC subsets evaluable by CIBERSORTx, only activated CD4 TM cell levels were associated with severe irAE development (Fig. 4a; P < 0.05 in each cohort, one-sided, unpaired Wilcoxon rank-sum test; Supplementary Table 9 and Methods). Moreover, higher TCR clonotype diversity in bulk peripheral blood also predicted severe irAE development, analogous to our findings in the single-cell discovery cohort (Fig. 4b; P = 0.0004, two-sided, unpaired Wilcoxon rank-sum test; Supplementary Table 9). Baseline TCR diversity further correlated with irAE severity across ICI treatment types, whether assessed by Shannon entropy or the Gini–Simpson index45 (Extended Data Fig. 6, Supplementary Table 9 and Methods), again reflecting its association with severe irAE development.
Fig. 4 |. Integrative modeling for early irAE detection from bulk peripheral blood.
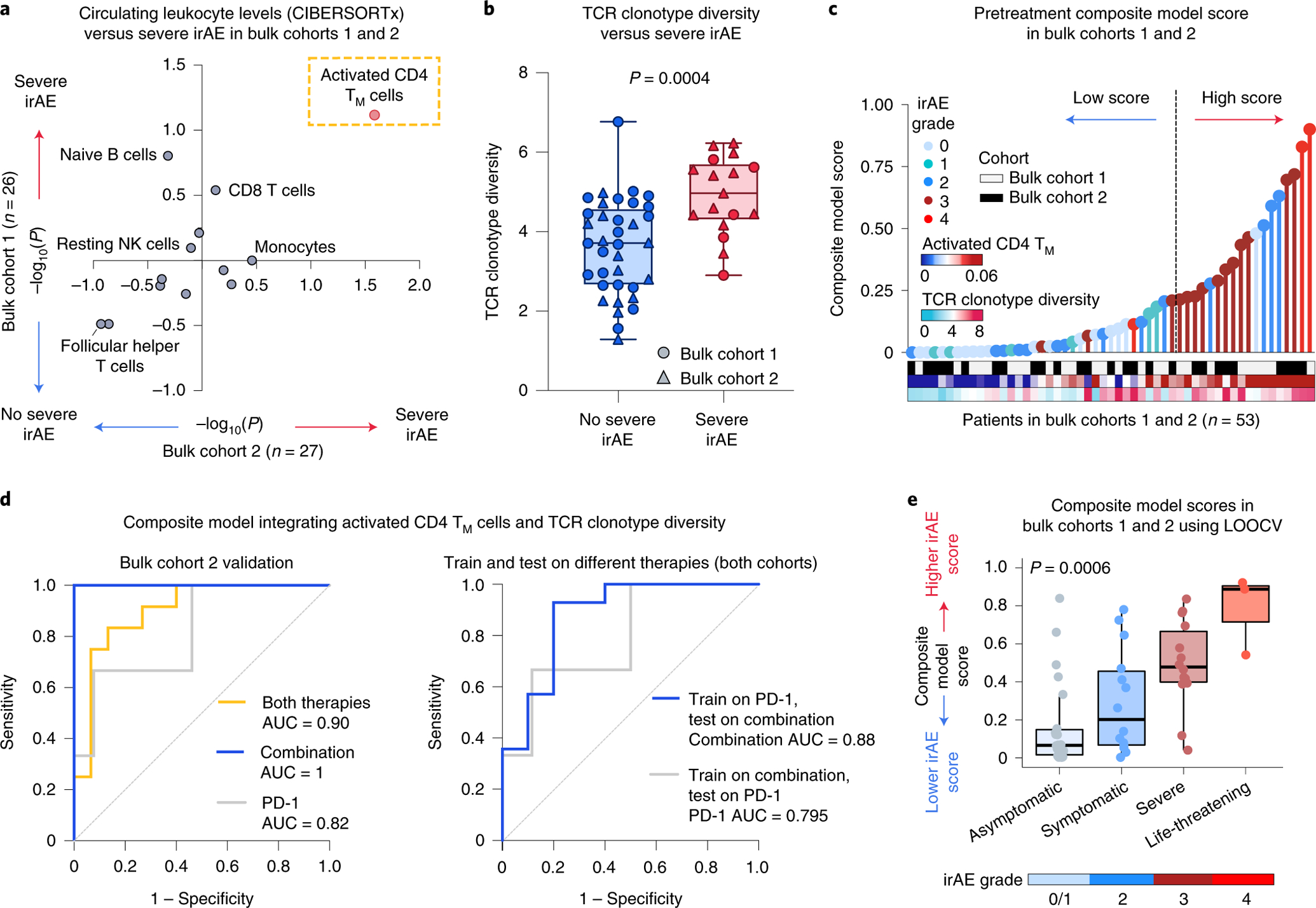
a, Association between pretreatment peripheral blood leukocyte composition (CIBERSORTx) and severe irAE development in bulk cohort 1 (n = 26 patients) and bulk cohort 2 (n = 27 patients) (Fig. 1 and Supplementary Tables 7 and 9). Significance was determined by a two-sided, unpaired Wilcoxon rank-sum test and expressed as −log10 P values. For associations with no severe irAE, −log10 P values were multiplied by −1. b, TCR clonotype diversity (Shannon entropy) in both bulk cohorts (n = 53 patients), stratified by future irAE status (no severe irAE, n = 36; severe irAE, n = 17). The box center lines, box bounds and whiskers denote the medians, first and third quartiles and minimum and maximum values, respectively. Significance was determined by a two-sided, unpaired Wilcoxon rank-sum test. c, Development of a composite model for the prediction of severe irAEs, integrating activated CD4 TM cell abundance and TCR clonotype diversity from pretreatment peripheral blood transcriptomes (Methods), with model scores trained on bulk cohort 1 and shown across both cohorts (Supplementary Table 9). The cut-point for high/low scores was optimized using Youden’s J statistic on bulk cohort 1 (Methods). d, Left: ROC plot showing composite model performance in bulk cohort 2 (held-out validation), whether applied to all patients (both therapies, n = 27), combination therapy patients (n = 11) or PD-1 monotherapy patients (n = 16). Right: ROC plot showing composite model performance in bulk cohorts 1 and 2, whether trained on PD-1 patients (n = 29) and tested on combination therapy patients (n = 24) or vice versa. The AUC is shown for each ROC curve. e, Composite model scores for all bulk cohort patients (n = 53) after model training for severe irAE development with LOOCV (Extended Data Fig. 7a and Supplementary Table 9), grouped by the highest irAE grade per patient. The box center lines, box bounds and whiskers indicate the medians, first and third quartiles and minimum and maximum values within 1.5× the interquartile range of the box limits, respectively. Statistical significance was determined by a Kruskal–Wallis test.
Given these results, we wondered whether a composite model integrating both features—activated CD4 TM cell abundance and bulk TCR diversity—might outperform either feature alone (Fig. 4c and Methods). Indeed, using a logistic regression framework to train a bivariable model on bulk cohort 1, the resulting composite model yielded an AUC of 0.85 in bulk cohort 1 (P = 0.02) and an AUC of 0.90 (P = 0.0004) in the held-out bulk cohort 2 (Fig. 4d and Supplementary Table 10), outperforming either feature alone (Supplementary Table 11). Furthermore, when evaluating all 53 patients by leave-one-out cross-validation (LOOCV), the composite model exhibited strong classification performance, underscoring its robustness (AUC = 0.82, P = 0.0002; Extended Data Fig. 7a,b). It also outperformed pathways identified by gene set enrichment analysis (GSEA) and previously published candidate biomarkers15,23,24 assessed in bulk transcriptomic data (Extended Data Fig. 7c and Supplementary Table 12). Additionally, higher pretreatment composite model scores were not significantly associated with durable clinical benefit, emphasizing specificity for irAE biology (Supplementary Table 13).
We next asked if the composite model could predict severe irAEs independent of key patient parameters. Indeed, regardless of ICI therapy type, ICI response status, age, sex, melanoma subtype or affected organ system, both in held-out patients (bulk cohort 2) and across both bulk cohorts evaluated by LOOCV, the model remained predictive (Fig. 4d, Extended Data Fig. 7d and Supplementary Tables 10 and 14). Moreover, in patients treated with combination ICIs, the standard of care for high-performing patients with metastatic melanoma54, the composite model yielded an AUC of 1.0 in bulk cohort 2 (P = 0.04) and an AUC of 0.86 by LOOCV for predicting severe irAE development across all evaluable patients (P = 0.01) (Fig. 4d, left and Extended Data Fig. 7a). While PD-1 monotherapy was associated with a lower rate of irAE development3,6–9,55, the model showed generalizability to severe irAE prediction in PD-1-treated patients (Fig. 4d, left and Extended Data Fig. 7a). Moreover, when trained on patients treated with PD-1 monotherapy alone, the model generalized to combination ICIs (AUC = 0.88 for grade 3+irAEs), with the same also being true in reverse (AUC = 0.795 for PD-1) (Fig. 4d, right).
We next explored model performance as a function of irAE grade and organ system involvement. Although trained on a single categorical outcome (severe versus no severe irAEs), pretreatment composite model scores increased as a function of irAE severity (Fig. 4e and Extended Data Fig. 7e,f), the number of symptomatic (grade 2+) irAEs experienced per patient (Extended Data Fig. 7g and Supplementary Table 9) and the number of affected organ systems per patient (Extended Data Fig. 7h–j and Supplementary Table 15). The model was also effective when trained to distinguish grade 2 from grade 0/1 irAEs (Extended Data Fig. 7e,f), albeit with reduced performance for patients treated with PD-1 monotherapy (Supplementary Table 16).
To test whether the pretreatment composite model could predict time-to-severe irAE, we next assigned patients to high versus low groups by defining an optimal cut-point in bulk cohort 1 (Methods). In held-out bulk cohort 2, patients in the high group experienced severe irAEs within a median of 1.74 months after treatment initiation, whereas the vast majority of patients in the low group never experienced a severe irAE (P < 0.0001, hazard ratio (HR) = 11.6; Extended Data Fig. 8a). Similar results were seen for each therapy type separately, whether assessed in bulk cohort 2 (P < 0.025, HR = 8.3 and 14.8 for combination and PD-1, respectively; Extended Data Fig. 8b,c) or across cohorts by LOOCV (P = 0.0028 and HR = 12.2 for combination therapy, Fig. 5a; P = 0.03 and HR = 9.0 for PD-1 therapy). The model also predicted time-to-severe irAE in multivariable models independently of therapy type, age, sex and other key parameters (Supplementary Table 17).
Fig. 5 |. Correlates of severe irAE onset in patients treated with combined CTLA-4 and PD-1 blockade.
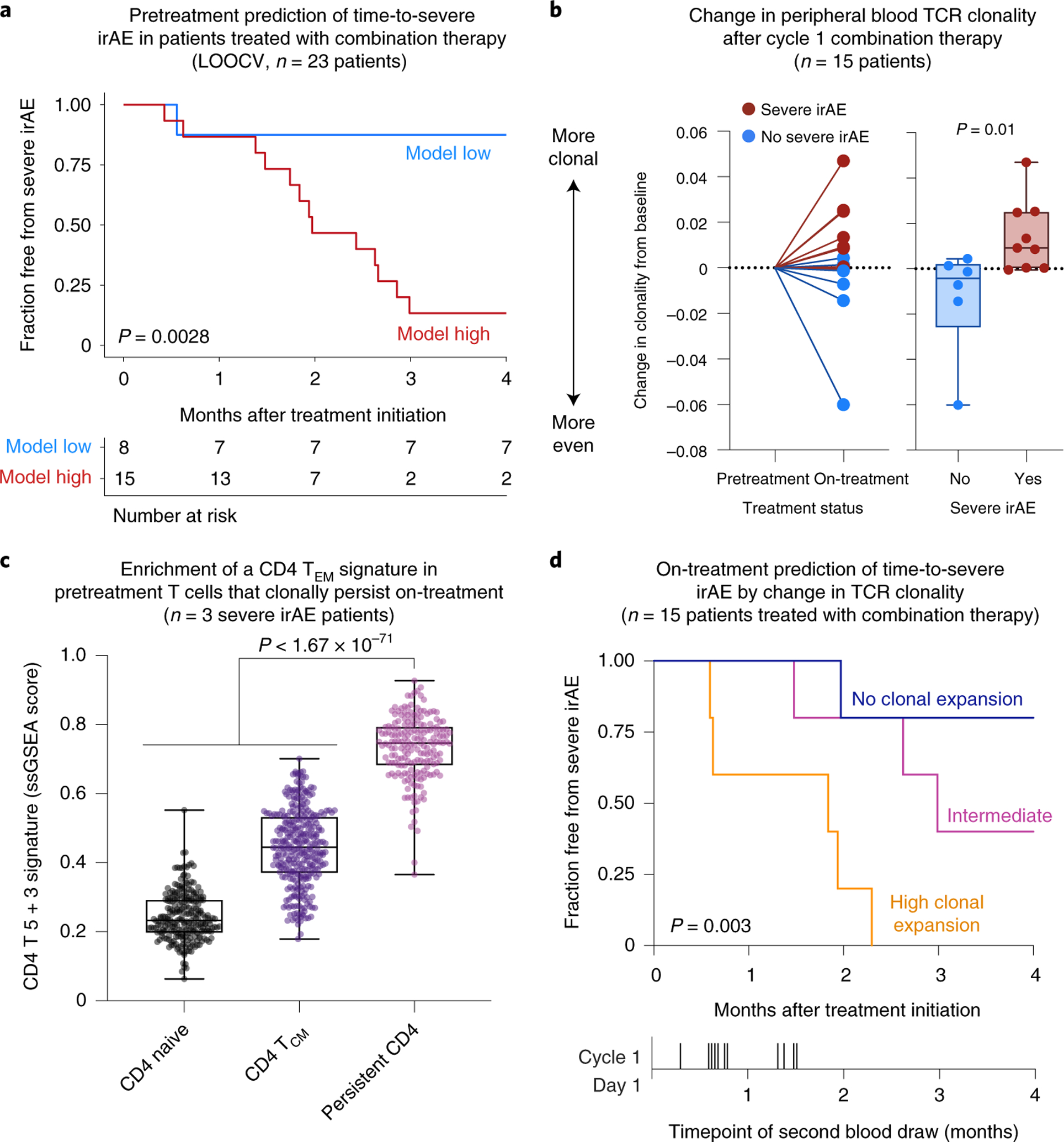
a, Pretreatment prediction of time-to-severe irAE onset in patients treated with combination therapy. The cut-point was optimized using composite model scores trained with LOOCV (Methods). Only patients from bulk cohorts 1 and 2 who did not experience early progression were analyzed (n = 23; Methods). b, TCR clonal dynamics in relation to severe irAE development in patients treated with combination therapy (Supplementary Table 18). Left: Change in TCR clonality from baseline after initiation of combination therapy as measured by 1 – Pielou’s evenness, with future irAE status indicated by color. Right: Same as the left but showing change in clonality according to future irAE status. Significance was determined by a two-sided, unpaired Wilcoxon rank-sum test. c, Enrichment of a CD4 T 5 + 3 gene signature in CD4 T cells from pretreatment PBMC samples obtained from 3 patients analyzed in b, all of whom developed severe irAEs and showed TCR clonal expansion after ICI initiation (Extended Data Fig. 9d). The box center lines, box bounds and whiskers indicate the medians, first and third quartiles and minimum and maximum values, respectively. The points denote cells profiled by scRNA-seq and annotated either by Azimuth (CD4 naive, n = 245 cells; CD4 TCM, n = 320 cells) or by their clonal persistence from baseline to early on-treatment time points (persistent CD4, n = 190 cells). The most persistent CD4 clonotypes in this analysis showed evidence of clonal expansion (Extended Data Fig. 9f,g). Significance was determined relative to persistent cells by a two-sided, unpaired Wilcoxon rank-sum test. ssGSEA, single-sample GSEA. d, Differences in freedom from severe irAE stratified by the degree of TCR clonal expansion after initiating combination therapy, as measured by the change in 1 – Pielou’s evenness. Patients were grouped into the following tertiles as detailed in Methods: no clonal expansion (n = 5), intermediate (n = 5) and high clonal expansion (n = 5). Statistical significance in a,d was assessed by a two-sided log-rank test.
Peripheral TCR clonal expansion linked to severe irAEs.
Previous case reports of patients with melanoma experiencing deadly ICI-mediated toxicity have shown evidence of clonally expanded self- or virus-reactive T cells in the affected tissue, linking self- and pathogen-recognizing T cell clones to lethal toxicity10,14. Accordingly, we hypothesized that pretreatment TCR clonotypes in peripheral blood might show a greater propensity to expand in patients destined to develop severe irAE after ICI treatment initiation. To examine this, we applied immunoSEQ to profile bulk TCR-ß repertoires in paired pretreatment and early on-treatment PBMC samples collected from 15 patients with metastatic melanoma treated with combination therapy (Methods). Using a TCR clonality index that is robust to variation in the number of clones captured (Pielou’s evenness45), we confirmed significant concordance between MiXCR (bulk RNA-seq) and immunoSEQ (DNA) in pretreatment samples from these 15 patients, underscoring the integrity of our composite model in bulk cohorts 1 and 2 (Extended Data Fig. 9a). We then assessed TCR clonal expansion (that is, clonal dominance) after treatment initiation, as measured by an increase in 1 – Pielou’s evenness. In support of our hypothesis, we observed both significantly increased TCR clonal expansion and persistence of baseline clones in patients who developed severe irAE compared to those who did not (Fig. 5b, Extended Data Fig. 9b,c and Supplementary Table 18). In severe irAE patients for whom we performed scRNA-seq and single-cell TCR sequencing (scTCR-seq) (n = 3), we also observed preferential expansion of the activated CD4 TEM compartment among clones detected in both blood draws (Extended Data Fig. 9d–g and Methods). Moreover, persistent CD4 T cell clones were highly enriched for the CD4 T 5 + 3 population identified by scRNA-seq analysis (Fig. 5c and Extended Data Fig. 9e).
We additionally explored whether the degree of TCR clonal expansion early on-treatment correlated with the timing of severe irAE development. Indeed, whether assessed in tertiles by log-rank test or by rank via Cox proportional hazards regression, patients with a greater magnitude of TCR clonal expansion developed severe irAE sooner (P = 0.003, log-rank test; Fig. 5d and Supplementary Table 19). These results were significant independently of the time between blood draws and when restricting the analysis to on-treatment blood draws obtained within one month of cycle 1 ICI (Extended Data Fig. 9h and Supplementary Table 19).
Circulating leukocytes in autoimmune disease.
Lastly, we asked whether the baseline peripheral blood profile of patients at risk for severe irAE development parallels clinical autoimmunity. To this end, we applied CIBERSORTx to examine 15 leukocyte subsets in bulk peripheral blood transcriptomes spanning 6 studies56–61 and 587 patients with either systemic lupus erythematosus (SLE) or inflammatory bowel disease (IBD) relative to 191 healthy controls (Supplementary Table 20). Using a meta-analytical framework to integrate P values across studies and pathologies (Extended Data Fig. 10 and Methods), we found that circulating activated CD4 TM cells were most significantly associated with autoimmune disorders relative to healthy individuals (Fig. 6). These data suggest that severe irAEs might represent a subclinical or latent autoimmune state that is clinically unmasked on ICI administration, in line with recent case reports38–40 and multi-institutional data showing that patients with autoimmunity treated with immune checkpoint blockade have a propensity to experience flares in their autoimmune symptoms62–64.
Fig. 6 |. Large-scale assessment of circulating leukocytes in autoimmune diseases.
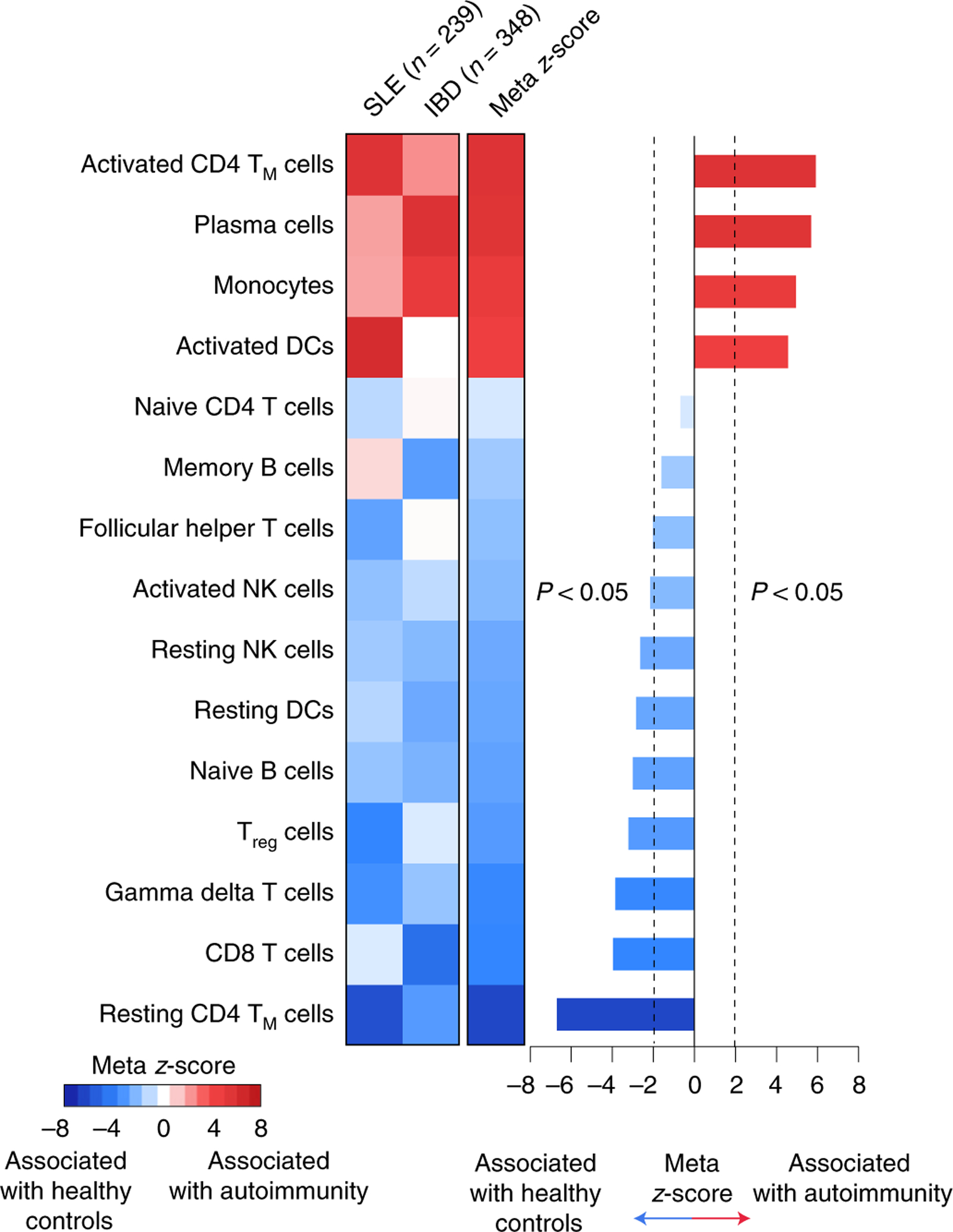
Enrichment of circulating leukocyte levels in two autoimmune disorders relative to healthy controls. Leukocyte composition was determined by CIBERSORTx. Significance was determined by a two-sided, unpaired Wilcoxon rank-sum test and integrative meta z-score. Details of the analytical workflow and underlying datasets are provided in Extended Data Fig. 10 and Supplementary Table 20, respectively.
Discussion
In this study, we identified two baseline features—activated CD4 TM cell abundance and a more clonally diverse TCR repertoire in the peripheral blood—as promising determinants of ICI-induced irAEs in patients with metastatic melanoma. Although previous studies have linked (1) activated T cells and clonally expanded TCRs in postmortem tissue to fatal irAEs (myocarditis, encephalitis)10,14 and (2) effector CD4 T cells to organ-specific irAEs (destructive thyroiditis, hepatitis)27,28, this work extends the scope of these findings to pretreatment T cell characteristics of irAE development in diverse organ systems. Integration of these features into a composite model predicted greater risk for severe irAEs and demonstrated sufficient granularity to distinguish different irAE grades and burdens.
We also identified a striking correlation between early T cell clonal expansion and the timing of severe irAE onset in patients treated with combination therapy. Future studies are needed to further characterize this finding and elucidate the relative contributions of CD4 and CD8 T cells to irAE-associated clonal dynamics.
Consistent with the possibility of a common immunological mechanism underlying both irAE development and autoimmunity, we additionally observed elevated levels of activated CD4 TM cells in patients with SLE or IBD. While it is reasonable to predict that patients with previous autoimmunity would be enriched for higher activated CD4 T cell levels and higher rates of severe irAE from ICI, none of the patients in our cohort had documented pre-existing autoimmunity. Moreover, such patients may develop compensatory immune regulatory mechanisms before starting ICI that change their baseline irAE risk. Nevertheless, it is important to study this connection in greater detail in future studies and determine whether circulating activated CD4 TM cells exhibit an increased propensity for recognizing self-antigens in patients at risk for severe ICI toxicity. Indeed, the risk of flare is greater in patients with autoimmune disease treated with combination immunotherapy, particularly those with gastrointestinal or rheumatological conditions63. More reliably identifying these at-risk patients during ICI decision-making could improve their outcomes.
This study has several limitations. First, it employed a retrospective design using banked clinical samples. Second, patients received either anti-PD-1 monotherapy or anti-PD-1 plus anti-CTLA-4 combination therapy, which are associated with different risk profiles for severe irAE development. Third, while most irAEs occur within the first three months of ICI treatment initiation, a subset can occur later65. Whether our findings generalize to late-onset irAEs will need to be investigated since the median time-to-severe irAE development in our cohorts was 6.4 weeks (consistent with clinical trial data65), with no irAEs occurring beyond 3 months (Supplementary Tables 3 and 9). Fourth, the timing of on-treatment peripheral blood collection during immunotherapy with respect to treatment initiation was not homogeneous. Finally, it is yet unclear whether our findings will generalize to ICI-related irAE risk in other cancer types.
Future studies should address these limitations, along with greater application of single-cell profiling both before and early during immunotherapy. In addition, it will be important to confirm our findings in larger multi-institutional cohorts and assess whether the circulating immunological determinants of ICI-induced toxicity vary based on the organs most likely to be involved. If prospectively validated, these findings could facilitate treatment adaptation to improve the risk profile of immune checkpoint blockade, with implications for the prediction and potential prevention of ICI-mediated toxicities.
Methods
Study design and participants.
The samples analyzed in this study were collected with informed consent for research use and were approved by the Yale University School of Medicine and Washington University School of Medicine institutional review boards, in accordance with the Declaration of Helsinki (2013) as part of observational registry studies focusing on melanoma. Eligible patients were aged >18 years with metastatic melanoma treated with ICI treatment consisting of either anti-PD-1 blockade (nivolumab or pembrolizumab) or combination immune checkpoint blockade (anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab); Fig. 1). Ninety percent of patients were naive to any previous immune checkpoint blockade at the time of pretreatment blood collection (Supplementary Table 1).All patients underwent routine clinical assessment for irAEs and response by board-certified medical oncologists. Surveillance occurred before each cycle of ICI treatment (approximately every 3 weeks), and in several cases, more frequently (for example, by inpatient medical staff in patients admitted to the hospital for severe irAEs). It also continued, when applicable, after completion of the treatment course. All irAEs were classified according to the United States Health and Human Services Common Terminology Criteria for Adverse Events (CTCAE) v.5.0, with grade ≥2 and ≥3 considered symptomatic and severe, respectively. Within and across patient cohorts, irAEs spanned diverse organ systems including the gastrointestinal tract, skin, liver, pituitary, thyroid, adrenal, musculoskeletal, ocular, pancreatic and cardiac systems (Extended Data Fig. 7i and Supplementary Tables 1 and 15). Three patients experienced a systemic inflammatory syndrome related to ICI administration (YUGIM, YUHERN and YUTORY; Supplementary Table 1). All severe irAEs occurred within three months of ICI initiation, a landmark period during which no patients in this cohort died. Response was scored as durable clinical benefit, no durable benefit or not evaluable as defined previously66,67. We identified three cohorts of patients who met the aforementioned eligibility criteria and had pretreatment PBMC samples collected just before the first cycle of anti-PD-1 or combination ICI administration (median 0 d; range 0–2 months). PBMCs from each cohort (pretreatment for all patients and pre/on-treatment pairs for 15 patients) were analyzed as depicted in Fig. 1 and Supplementary Table 1.
Blood collection and processing.
Peripheral blood specimens were collected in K2EDTA Vacutainer tubes (Becton Dickinson) and processed within 1 h of phlebotomy. PBMC extraction was by either an ammonium chloride or Lymphoprep (STEMCELL Technologies) protocol. The Lymphoprep protocol was applied according to the manufacturer’s instructions. With the ammonium chloride protocol, 4–8 ml of blood was mixed with 20 ml of cold ammonium chloride lysing buffer (0.1 M of ammonium chloride, 0.01 M of Tris-HCl) and incubated for 5 min at room temperature. Cells were then centrifuged at 300g for 5 min and washed with 5 ml of cold PBS. PBMC samples were cryopreserved in 10% dimethyl sulfoxide/90% FBS. Cryovials were placed in Nalgene Mr. Frosty containers (Thermo Fisher Scientific) for 24 h, then stored in liquid nitrogen until cellular and RNA processing for expression analysis.
Mass cytometry.
Metal-conjugated antibodies were either purchased preconjugated from Fluidigm or purchased purified from BioLegend, Thermo Fisher Scientific or Cell Signaling Technology and subsequently conjugated to metals using Maxpar Antibody Labeling Kits (Fluidigm) according to the manufacturer’s instructions. All CyTOF antibodies are provided in Supplementary Table 4.
PBMCs from each of 28 patients were prepared for CyTOF as indicated in Supplementary Table 1. Cryopreserved cell suspensions were first thawed by holding cryovials in a 37 °C water bath for 1–2 min without submerging the cap. Subsequently, 1–3 × 106 PBMCs in single-cell suspension were incubated with Human TruStain FcX (BioLegend) at room temperature for 10 min to block nonspecific antibody binding, followed by incubation with metal-conjugated antibodies against cell surface molecules (Supplementary Table 4) for 20 min on ice. Cells were also incubated with Cell-ID Cisplatin (Fluidigm) according to the manufacturer’s instructions to identify viable cells. After treatment with intracellular fixation and permeabilization buffers (Thermo Fisher Scientific), cells were incubated with metal-conjugated antibodies against intracellular proteins (Supplementary Table 4). Cells were then washed and stained with Cell-ID Intercalator-Ir (Fluidigm) diluted in PBS containing 1.6% paraformaldehyde (Electron Microscopy Sciences) and stored at 4 °C until acquisition. After a wash step, sample acquisition was then performed using the Helios System (Fluidigm) at an event rate of <400 s−1.
To reduce technical variation between samples, we used Ce beads in each sample and normalized the files together using Bead Normalizer v0.3 (https://github.com/nolanlab/bead-normalization/wiki/Installing-the-Normalizer). To further minimize technical variability, we limited our sample processing and acquisition batches to four, used the same reagent lots across all samples and made no major adjustments to Helios calibration. We also noted that Astrolabe does not compare numerical intensities between samples; rather it analyzes each sample separately, with the assumption that a given subset is the same whether the underlying marker intensities are shifted or not. Thus, the platform has been reported to be resistant to batch effects68.
Mass cytometry data analysis.
CyTOF data were initially analyzed with Cytobank v8.0 and v8.1 (Beckman Coulter) using the FlowSOM algorithm for hierarchical cluster optimization and the viSNE algorithm (5,000 iterations, perplexity = 100) for visualization of high-dimensional data69,70. Subsequent cell subpopulation identification and data visualization were performed using the Astrolabe Cytometry Platform v3.6 and v4.0 (Astrolabe), which leverages the Ek’Balam algorithm71, a knowledge-based hierarchical annotation strategy coupled with unsupervised clustering, for automated labeling of cell subpopulations. In total, 20 cell subpopulations spanning major mononuclear lineages in peripheral blood were identified and quantified (Supplementary Table 5). For each patient sample, cell subpopulation levels were normalized to sum to 1, with unclassifiable cells based on protein marker expression excluded from the analysis. To corroborate Astrolabe, we used Cytobank to perform blinded manual gating of major cell populations including CD4 TEM cells (Supplementary Figs. 1 and 2a,b). The total abundance of CD4 TEM cells, whether calculated as a fraction of total PBMCs or circulating T cells, but not as a fraction of CD4 T cells, was significantly associated with severe irAE development (Supplementary Fig. 2c).
Flow cytometry.
PBMCs collected from five healthy donors were analyzed by flow cytometry (Extended Data Fig. 5e, Supplementary Fig. 3 and Supplementary Table 1). Briefly, 2–5 million PBMC cells were treated with TruStain FcX Fc Receptor Blocking Solution (BioLegend) for 10 min at room temperature to block Fc receptors and then stained with fluorophore-tagged surface antibodies for 30 min at room temperature. The following antibodies were used to stain the cells: FITC-conjugated anti-human CD45 (clone 2D1; BioLegend); AF700-conjugated anti-human CD3 (clone OKT3; BioLegend); APC-conjugated anti-human CD4 (clone OKT4; BioLegend); PE/Cy7-conjugated anti-human CD8 (clone SK1; BioLegend); APC-Cy7-conjugated anti-human CD19 (clone HIB19; BioLegend); PerCp/Cy5.5-conjugated anti-human CD14 (clone HCD14; BioLegend); and BV605-conjugated anti-human CD56 (clone 5.1H11; BioLegend). Cells were then washed twice with ice cold PBS-based buffer (1× PBS, 2% FBS, 1 mM of EDTA) and stained with 4′,6-diamidino-2-phenylindole (DAPI) (BioLegend) to evaluate cell viability. Antibody capture beads (BD Biosciences) were used to compensate each fluorophore in the experiment. Stained cells were analyzed by flow cytometry with operator assistance using a MoFlo Legacy instrument (Beckman Coulter) at the Siteman Flow Cytometry Core at the Washington University School of Medicine. After exclusion of DAPI-positive cells and putative doublets based on forward and side scatter analysis, major lymphocyte populations including B cells, CD4 T cells, CD8 T cells and NK cells were enumerated as a percentage of total lymphocytes using FlowJo v.10 (FlowJo LLC).
scRNA-seq and scV(D)J-seq library preparation and sequencing.
Single-cell suspensions from PBMC samples were obtained as described in above and prepared to a concentration of 700–1,200 viable cells μl−1 using a hemacytometer (Thermo Fisher Scientific) or Coulter Counter (Beckman Coulter Life Sciences) for cell counting, according to the manufacturers’ instructions. Single-cell suspensions subsequently underwent library preparation for scRNA-seq with paired scV(D) J-seq for TCR and BCR clonotypes using the 5′ transcriptome kit (10x Genomics) according to the manufacturer’s instructions. Complementary DNA libraries were sequenced on a NovaSeq instrument (Illumina) with 2 × 92 base pair (bp) paired-end reads targeting a mean of 20,000 reads per cell.
scRNA-seq analysis (discovery cohort).
Raw scRNA-seq reads were barcode-deduplicated and aligned to the hg38 reference genome using Cell Ranger v.3.1.0, yielding sparse digital count matrices, which were analyzed to identify cell types and cellular states using Seurat v.3.1.5 or v.3.2.1 (ref.72). Outlier cells were identified and removed based on the following criteria: (1) >25% mitochondrial content or (2) cells with less than 100 or greater than 1,500–3,000 expressed genes, depending on sample-level distributions. After normalization (NormalizeData) and variable feature identification (FindVariableFeatures with n = 2,000 features), we applied FindIntegrationAnchors (dims = 1:30) to identify anchors and IntegrateData (with default parameters) to perform batch correction. Once integrated, we applied principal component analysis (PCA) and uniform manifold approximation and projection (UMAP) using the 2,000 most variable genes and the top 30 principal components. FindClusters was applied to identify cell types and cellular states with a resolution parameter set to 3, yielding 37 clusters.
All identified clusters were assigned to major cell lineages based on the expression of canonical marker genes: CD3D/CD3Ehi = T cells; CD8A/CD8Bhi and NKG7/GNLYlo = CD8 T cells; non-CD8 T cells with high IL7R expression and low NKG7/GNLY = CD4 T cells; NKG7/GNLYhi and CD3D/CD3Elo = NK cells; CD14 or FCGR3Ahi = monocytes; FCER1Ahi = dendritic cells (DCs); MS4A1hi = B cells; HBBhi = red blood cells; PPBPhi = platelets. Cells with high expression of CD3D/E and GNLY/NKG7 that were not annotated as CD8/CD4 T cells were included in a T or NKT cell group, denoted T/NKT. Clusters annotated as red blood cells or platelets were omitted from further analysis. To assess the effective doublet rate, we cross-referenced cellular barcodes with single-cell BCR (scBCR) and TCR (scTCR) clonotypes. By determining (1) the percentage of non-T cells anomalously mapped to TCR clonotypes (denoted m) and (2) the frequency (that is, recovery rate) of annotated T cells with a matching scTCR clonotype (denoted f), we calculated an effective double rate (m/f) of 2.2%. The effective doublet rate calculated for scBCR clonotypes mapping to non-B cells was the same (also 2.2%). Since the effective doublet rate was reasonably low, we eliminated all single cells with aberrant expression of TCR or BCR clonotypic sequences. We then repeated PCA, UMAP and FindClusters as described above, yielding 32 clusters. Two red blood cell clusters, marked by very high HBB expression, remained and were removed from the analysis, followed by one final round of PCA, UMAP and FindClusters, yielding a final set of 32 clusters (that is, states) and the low-dimensional embedding shown in Fig. 3a and Extended Data Fig. 1a.
All 32 states were assessed for their association with severe irAE development (x axis of Fig. 3b) and CD4 TEM abundance as measured by CyTOF (y axis of Fig. 3b). Among them, CD4 T cluster 5 was most strongly correlated with both variables (Fig. 3b). To determine the statistical significance of this result, we calculated the joint probability of (1) being ranked first by each measure and (2) achieving a P value and Spearman correlation coefficient at least as strong as CD4 T cluster 5. To calculate this probability empirically, we implemented a permutation scheme, where cell fractions associated with each scRNA-seq cluster were independently shuffled across all patient samples, then evaluated for (1) and (2) above. By repeating this process 10,000 times, we calculated an empirical P value of 0.003 for CD4 T cluster 5. We also performed a pairwise combinatorial analysis, restricting pairs of cell states to the same major cell type to maintain biological coherence (B cells, CD4 T cells, CD8 T cells, NK cells, monocytes) and compared each of 82 possible cell cluster combinations to CD4 TEM levels enumerated by CyTOF and severe irAE development (Extended Data Fig. 1d,e). CD4 T cell clusters 5 and 3 emerged as the top-ranking pair. Using the abovementioned statistical approach, we calculated an empirical P value of 0.002 for this result. To identify the differentially expressed genes (DEGs) in Fig. 3c, we applied Seurat FindMarkers with default parameters to the CD4 T 5 + 3 population versus other CD4 T cell states.
To evaluate the relative utility of unsupervised clustering for delineating cellular determinants of irAE development, we leveraged a reference-guided annotation framework44 within Seurat v.4.0.1 (Azimuth) to project our scRNA-seq dataset onto a PBMC atlas of 161,764 cells spanning 6 major lineages and 27 finer-grained subsets defined with scRNA-seq and codetection of over 220 protein markers44. First, we preprocessed the query dataset following the quality control steps described above, yielding 24,807 cells. We then normalized the query dataset by SCTransform, applied FindTransferAnchors to the query and reference datasets using a precomputed supervised PCA transformation with 50 dimensions, then applied MapQuery to map the cell type labels and UMAP structure from the reference to the query dataset.
Among the 27 cell states identified by Azimuth (Extended Data Fig. 2a), CD4 TEM was most strongly associated with severe irAE development and most correlated with CD4 TEM cells enumerated by CyTOF (Extended Data Fig. 2c). Among two other CD4 TEM-like subsets identified by Azimuth (CD4 CTL, CD4 proliferating), CD4 proliferating showed the highest expression of HLA-DX and lowest expression of SELL (Extended Data Fig. 2d), which is consistent with an activated CD4 TEM phenotype. Additionally, when examining Azimuth-imputed protein expression from antibody-derived tag data, only CD4 TEM and CD4 proliferating states showed hallmarks of TEM cells (CD45ROhiCD45RAloCD27lo; Extended Data Fig. 2e). Indeed, a population combining CD4 TEM and CD4 proliferating was most associated with severe irAE development (Extended Data Fig. 2c). Hypergeometric testing was applied to assess overlap in cellular barcodes between the combined CD4 TEM + CD4 proliferating population (Azimuth) and states defined by de novo clustering. CD4 T 5 + 3 emerged as the top hit (Benjamini–Hochberg-adjusted P = 2.5 × 10−7). Despite strong overlap between unsupervised and supervised approaches, CD4 T 5 + 3 was more associated with severe irAE development and CyTOF than populations labeled by reference-guided annotation (Extended Data Fig. 2f).
Bulk RNA-seq library preparation, sequencing and quantification.
Cryopreserved cell suspensions were thawed as described above. RNA was subsequently extracted using the RNeasy PowerLyzer Tissue & Cells Kit (QIAGEN) and quality was assessed with a 2100 Bioanalyzer System (Agilent Technologies). All samples were sufficiently high quality for TruSeq RNA Exome analysis (DV200 > 30%) and were prepared using the TruSeq RNA Exome Kit (Illumina) according to the manufacturer’s instructions. After hybrid capture, cDNA libraries were pooled and sequenced on a HiSeq 2500 instrument (Illumina) using 2 × 150 bp paired-end reads with a target of 20–25 million reads per sample. Raw reads were quantified with Salmon v.0.12.0 using the GENCODE v.29 reference transcriptome; the following command line arguments were used with otherwise default parameters: --seqBias--gcBias--posBias--validateMappings--rangeFactorizationBins 4. Read counts were normalized to gene-level transcripts per million (TPM) using tximport v.1.10.1 (ref.73). Only samples with a mapping rate ≥60% and successful TCR assembly (see the V(D)J receptor profiling and clonotype analysis below) were included for further analysis, with the exception of 3 samples with mapping rates >40% (but < 60%) and successful TCR assembly, which were included. In total, 53 sequenced samples (88%) in bulk cohorts 1 and 2 satisfied these criteria (Fig. 1 and Supplementary Table 8).
Bulk RNA-seq deconvolution.
To determine leukocyte composition in bulk RNA-seq profiles of PBMCs, we applied CIBERSORTx v.1.0.41 (https://cibersortx.stanford.edu)49 with the LM22 signature matrix49,51 to the TPM matrix of each cohort (Fig. 1 and Supplementary Table 1). CIBERSORTx was separately applied with B-mode batch correction and no quantile normalization to each sequencing batch. LM22, which consists of highly optimized reference profiles for distinguishing 22 functionally defined human hematopoietic subsets51, has been widely validated against flow cytometry for accurate enumeration of leukocyte subsets in whole blood and PBMCs, whether profiled by RNA-seq or microarray49,51,66. CIBERSORTx and the performance of the LM22-activated CD4 TM cell profile14,49,74 were further corroborated in this work through gene expression analysis (CCR5, SELL, TCF7 and CD27; Extended Data Fig. 5a) and comparison between CIBERSORTx, mass cytometry, flow cytometry and scRNA-seq using PBMC samples from patients with melanoma (Extended Data Fig. 5b–f and Supplementary Table 1). All LM22 subsets except the granulocyte and macrophage subsets were evaluated in this work (n = 15; Fig. 4a), with their relative fractions renormalized to sum to 1 for each sample. While a total of 15 subsets were evaluated, 2 were sparsely detected by CIBERSORTx (regulatory T (Treg) cells, gamma delta T cells) and could not be assessed by Wilcoxon rank-sum test in Fig. 4a.
V(D)J receptor profiling and clonotype analysis.
For the single-cell discovery cohort, raw scV(D)J-seq reads were mapped with Cell Ranger v.3.1.0 to reference refdata-cellranger-vdj-GRCh38-altsensembl-4.0.0 and the resulting clonotype assemblies were downloaded from the Loupe V(D)J browser v.3.0.0 (10x Genomics). Given that activated TM cells arise from clonal expansion75,76, the former are expected to have lower TCR diversity than their naive counterparts77, provided that (1) cells from both populations are equally sampled (that is, their counts are equivalent) or (2) variation in total T cell counts is normalized out (Extended Data Fig. 4a). However, by disregarding variation in total T cell frequency, such sampling ignores richness—the number of unique species (clonotypes) within a population and a key factor underlying immune repertoire diversity. As such, we primarily used Shannon entropy45,46 to characterize immune repertoire diversity in this work, an information theoretic metric that combines evenness and richness in a single measure (Extended Data Fig. 4a).
For each evaluable patient sample in the single-cell discovery cohort (Figs. 1 and 2a and Supplementary Tables 2 and 3), the TCR clonotype repertoire was randomly sampled (without replacement) to equalize the number of evaluable PBMC cells across patients while addressing technical variation in TCR recovery. To maximize the pool of TCR clones available for sampling, patients with <100 TCR clones were excluded (n = 4; YUTAUR, YUTORY, YUHERN and YUTHEA). We then calculated Shannon entropy (R package vegan v.2.5–6 (ref.78)) relative to total PBMCs for each T cell subset and averaged the resulting values across 100 iterations of this procedure for the remaining 9 patients (Fig. 3e and Extended Data Fig. 4b,d–f). Shannon entropy was analyzed as described above for scBCR clonotypes across IGK, IGL and IGH chains in the same nine patients (Extended Data Fig. 4g).
For bulk cohorts 1 and 2, after adapter sequence trimming using Skewer v.0.2.2 (ref.79), TCR clonotypes were assembled and quantitated with MiXCR v.3.0.125 using the following command: mixcr align -p rna-seq -s hsa -O allowPartialAlignments=true data_R1.fastq.gz data_R2.fastq.gz alignments.vdjca (Supplementary Table 8). For each patient sample, TCR clonotype diversity was measured in aggregate for TCR-α and TCR-β chains using Shannon entropy (R package vegan v.2.5–6 (ref.78)) and compared between patients based on irAE severity (Fig. 4b,c, Extended Data Fig. 6a,c and Supplementary Table 9). We additionally applied the Gini–Simpson index45, which was calculated using the R package immunarch v.0.6.5 (https://doi.org/10.5281/zenodo.3367200), to evaluate bulk TCR diversity according to irAE severity (Extended Data Fig. 6b,d and Supplementary Table 9). Of note, TCR richness is a key component for calculating both Shannon entropy and the Gini–Simpson index.
Analysis of T cell clonal dynamics from bulk PBMCs.
Bulk TCR-ß chain profiling was performed on paired pretreatment and early on-treatment PBMCs from 15 patients treated with combination ICIs (Supplementary Tables 1 and 18). No patients had on-treatment peripheral blood collected after the onset of severe irAE. Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (QIAGEN) and submitted for survey-resolution immunoSEQ (Adaptive Biotechnologies). Data from productive TCR-ß chain rearrangements were exported using the immunoSEQ Analyzer online tool and evaluated for TCR-ß repertoire richness and diversity using Pielou’s evenness45, with increased 1 – evenness associated with increased clonality. The Pielou’s evenness results from immunoSEQ profiling were compared with bulk RNA-seq (MiXCR), which revealed concordance (Extended Data Fig. 9a). We also verified that all pretreatment and on-treatment samples were properly paired by cross-comparison of TCR-ß CDR3 sequences. Clonal expansion was inferred by analyzing the difference in clonality, defined as 1 – Pielou’s evenness in each sample, between paired on- and pretreatment time points (Fig. 5b and Extended Data Fig. 9b). More specifically, to calculate the change in clonality from baseline, pretreatment clonality was subtracted from on-treatment clonality in a paired fashion, thereby normalizing all pretreatment samples to zero (Fig. 5b, left). The data from Fig. 5b were also analyzed without normalizing on-treatment samples to paired pretreatment samples in Extended Data Fig. 9b.
To assess freedom from severe irAE, the degree of clonal expansion, denoted δ, was evenly divided into tertiles using the R package dplyr v.1.0.7 (Fig. 5d and Extended Data Fig. 9h). This yielded the following groups: no clonal expansion, δ < 0, n = 5; intermediate, 0 < δ < 0.009, n = 5; and high clonal expansion, δ > 0.009, n = 5. We applied these thresholds to the full immunoSEQ cohort (n = 15; Fig. 5d) and to patients with blood samples obtained on ICI treatment day 1 and <1 month later (n = 7; Extended Data Fig. 9h). Additionally, when represented in rank space, the degree of clonal expansion was significantly associated with time-to-severe irAE development in Cox regression models and was independent of the time between blood draws, the number of productive TCR clones detected and the age and sex of each patient (Supplementary Table 19).
Analysis of persistent T cell clones.
Paired pretreatment peripheral blood scRNA-seq and scTCR-seq were performed for three patients (Fig. 5b) who experienced severe irAEs with variable levels of clonal expansion: YUALOE, YUNANCY and YUHONEY (Fig. 5c, Extended Data Fig. 9d–g and Supplementary Table 18). Of note, samples from these three patients were not previously profiled by scRNA-seq or scV(D)J-seq in the single-cell discovery cohort. Sequencing libraries were generated and processed for quality control identically to those described in the single-cell discovery cohort. Mapping was performed with Cell Ranger v.5.0.1.
To analyze persistent clones—which we defined as productive TCR-ß CDR3 nucleotide sequences shared between paired pretreatment and on-treatment blood samples—we interrogated the immunoSEQ data for shared clonotypes with at least 2 templates in 1 blood draw (pretreatment or on-treatment) and at least 1 template in the other blood draw (60% of all shared clones, on average). This allowed us to preferentially focus on persistent clones that either expanded or contracted. The resulting sequences were cross-referenced with the TCR-ß CDR3 nucleotide sequences from the pretreatment scTCR-seq libraries, which were further cross-referenced with scRNA-seq data and filtered for cells annotated as T cells by Azimuth (applied as described above) (Extended Data Fig. 9d). In total, 1,504 single-cell transcriptomes with paired immunoSEQ clonotype data were identified. Significant Spearman correlations between pretreatment single-cell and immunoSEQ TCR clonotype frequencies were observed for each patient (ρ > 0.59; P < 0.0002), underscoring the integrity of our data. Importantly, to maximize stringency and avoid classification artifacts, when annotating CD4 and CD8 T cell-derived clonotypes, we only considered TCR clonotypes with uniform expression of positive lineage markers (CD4 > 0 and CD8A/B = 0 for CD4 T cells; CD8A or CD8B > 0 and CD4 = 0 for CD8 T cells). In all, 69% of all cross-referenced clonotypes could be unambiguously labeled by this approach (Extended Data Fig. 9e). For the plot shown in Fig. 5c, we calculated the mean log2 fold change between CD4 T 5 and 3 versus the remaining CD4 T cell clusters in the single-cell discovery cohort (Extended Data Fig. 1b) and then selected the top 20 genes for subsequent analysis. Enrichment of this gene set was determined using single-sample GSEA(R package escape v.1.0.1 (ref.80)), which we applied to T cells labeled by Azimuth or labeled as described above for persistent CD4/CD8 T cells. For the analysis shown in Extended Data Fig. 9f,g, productive frequencies of persistent T cell clones measured by immunoSEQ were grouped into CD4 and CD8 T cells, with differences in productive frequencies displayed on a per-clonotype basis (Extended Data Fig. 9g) or in aggregate (Extended Data Fig. 9f) and compared to bulk clonal expansion from baseline (Fig. 5b).
Integrative models to predict irAE development.
Activated CD4 TM cell abundance and bulk TCR clonotype diversity were individually associated with severe irAE development (Fig. 4a,b and Supplementary Table 11). Accordingly, we explored integrative modeling as a means of improving performance. While several techniques were assessed, including nonlinear modeling with random forests, logistic regression (glm in R) achieved comparable performance and was selected owing to the relative simplicity and robustness of a generalized linear model. Before training, we tested each feature in bulk cohorts 1 and 2 for outliers using the ROUT test81 with a false discovery rate = 10%. Of 88 data points (2 features × 53 samples), 3 outliers were detected, all from activated CD4 TM cells in bulk cohort 1. Regardless of the training cohort, all detected outliers were invariably from among these three samples. Therefore, for each integrative model, we determined the maximum fraction maxF of activated CD4 TM cell levels from among all non-outlier samples in the training cohort. We then used maxF as a ceiling for all samples.
The composite model was trained to predict severe irAE (grade 3+) development in several ways, as summarized in Supplementary Table 10. These include: training on bulk cohort 1 and testing on held-out bulk cohort 2 (Fig. 4d, left); training on one therapy type and testing on another (Fig. 4d, right); and training across bulk cohorts using LOOCV. For all models assessed by LOOCV, the analysis was repeated n times, where n is the total number of patients. In each iteration, the model was trained on each patient except the ith patient and evaluated on the held-out ith patient. To mitigate overfitting when dividing patients into high and low groups by LOOCV, we applied Youden’s J statistic to determine the threshold that optimized sensitivity and specificity in each training cohort, then allocated the held-out ith patient on the basis of this threshold.
Composite model scores were assessed by receiver operating characteristic (ROC) analysis. Models trained to discriminate severe from non-severe irAEs were used to predict future development of severe irAE (Fig. 4d, Extended Data Fig. 7a and Supplementary Table 11), irAE grade (Fig. 4c,e and Extended Data Fig. 7b,e), the number of irAE-impacted organ systems (Extended Data Fig. 7h–j and Supplementary Table 15) and the time-to-severe irAE development (Fig. 5a, Extended Data Fig. 8 and Supplementary Table 17). They were also assessed in different patient subgroups (Fig. 4d, Extended Data Fig. 7d and Supplementary Table 14) and compared to pathways and previously published biomarkers evaluated in bulk RNA-seq (Extended Data Fig. 7c). Composite models were additionally validated at different irAE grade thresholds (Extended Data Fig. 7f and Supplementary Table 16) and tested separately by therapy type to predict irAE development (Figs. 4d and 5a, Extended Data Figs. 7a,d–f and 8b,c and Supplementary Table 16).
Assessment of circulating leukocyte composition in autoimmune disorders.
Peripheral blood gene expression datasets profiled by bulk RNA-seq or microarrays and spanning 239 patients with SLE, 348 patients with IBD and 191 paired healthy controls, were downloaded from the Gene Expression Omnibus56–61 (GEO) (Supplementary Table 20). RNA-seq data from Hung et al.58 were downloaded as a preprocessed expression matrix and TPM-normalized before analysis. Affymetrix microarray datasets (n = 5) were downloaded as CEL files, MAS5-normalized (affy v.3.12 (ref.82) in R), mapped to Entrez gene identifiers using a custom chip definition file specific to each platform (http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/) and converted to HUGO gene symbols. One dataset (Burczynski et al.56) did not have raw CEL files available; instead we obtained preprocessed expression data from GEO. In cases for which multiple probe sets mapped to the same gene symbol, we selected the probe set with highest mean log2 expression across samples for further analysis. In the Palmer et al.60 dataset, some samples identified as controls were from subjects with Escherichia coli infection, celiac disease or progression to Crohn’s disease; these were excluded from the analysis. For replicate samples in the Carpintero et al.57 dataset, the most recent sample was selected. For the Peters et al.61 dataset, only pretreatment blood samples from patients with Crohn’s disease (week 0) were further analyzed. CIBERSORTx49 was applied with LM22 (refs.49,51) to the Hung et al.58 bulk RNA-seq dataset as described above, while microarray datasets were either run with (1) quantile normalization and B-mode batch correction (non-HG-U133 platforms) or (2) quantile normalization and no batch correction (HG-U133 platforms). Leukocyte subsets were limited to mononuclear subsets found in peripheral blood (granulocytes and macrophages were omitted) and were renormalized to sum to one for each sample.
Within each dataset, a two-sided, unpaired Wilcoxon rank-sum test was applied to evaluate the levels of each leukocyte subset in peripheral blood between individuals with the disease and healthy controls from the same study (Extended Data Fig. 10 and Supplementary Table 20). The resulting P values were converted into two-sided z-scores while taking the directionality of the association into account. Within a given disease phenotype (SLE or IBD), z-scores were combined across datasets using Lipták’s method83 weighted by sample size (Extended Data Fig. 10). Lastly, SLE- and IBD-specific meta z-scores were combined via the Stouffer’s method84 (Extended Data Fig. 10), yielding a pan SLE/IBD meta z-score for each leukocyte subset (Fig. 6).
Candidate toxicity biomarkers from previous literature and pathway analysis.
We benchmarked the composite model against previously published irAE biomarkers15,23,24 and enriched pathways for severe irAE prediction (Extended Data Fig. 7c). Each candidate biomarker was assessed separately in bulk cohorts 1 and 2 by determining the AUC by ROC analysis. The following pretreatment irAE biomarkers, which were measured by protein expression in previous literature, were assessed by RNA surrogates in the peripheral blood in this study: ADPGK and LCP1 (ref.23), which we evaluated individually and with bivariable linear regression (also done by Jing et al.23); CD74 and GNAL15 expression; and the CYTOX score24, which we evaluated as the geometric mean expression of genes encoding the same 11 cytokines (CSF3, CSF2, CX3CL1, FGF2, IFNA2, IL12A, IL1A, IL1B, IL1RA,IL2, IL13). Separately, we applied preranked GSEA v.4.1.0 via GSEAPreranked v.7.1.0 (ref.85) to identify the most irAE-enriched pathways in bulk cohorts 1 and 2 from the Molecular Signatures Database v7.4 hallmark pathway collection (Supplementary Table 12). As input, we defined transcriptome-wide gene lists for bulk cohorts 1 and 2 that were rank-ordered by log2 fold change between patients who developed severe irAE and those who did not. Gene sets with q < 0.25 were considered statistically significant (Supplementary Table 12). The two most-enriched gene sets in patients with severe irAEs versus patients with no severe irAEs (MYC_TARGETS_V1; OXIDATIVE_ PHOSPHORYLATION) were compared to the composite model in bulk cohorts 1 and 2 (Extended Data Fig. 7c).
Statistics.
All statistical tests were two-sided unless stated otherwise. The Wilcoxon rank-sum test was used to assess statistical differences between two groups. When assessing >2 groups simultaneously, the nonparametric Kruskal–Wallis test was used. The Benjamini–Hochberg method was applied for multiple hypothesis testing unless stated otherwise. We implemented a permutation scheme to assess scRNA-seq cluster correlation with severe irAE development and CyTOF CD4 TEM abundance as described above. A Fisher’s exact test was applied to assess statistical differences between two categorical variables. ROC analysis was performed to assess classification accuracy, which was quantified by AUC. Statistical significance of the AUC was determined by a two-sided z-test. Youden’s J statistic was used to identify the optimal cut-point after ROC analysis. Linear concordance was determined by Pearson (r) or Spearman (ρ) correlation and a two-sided t-test was used to assess whether the result was significantly nonzero. Kaplan–Meier and Cox regression analyses were used to assess covariates with respect to time-to-severe irAE. Significance levels and HRs for Kaplan–Meier analyses were determined using a two-sided log-rank test. The composite models and related analyses in Fig. 5a, Extended Data Fig. 8 and Supplementary Table 17 include patients from bulk cohorts 1 and 2 (Fig. 1 and Supplementary Tables 7 and 9) with the exception of two patients (YUDIME and YUMEDIC) who did not develop severe irAEs but experienced early disease progression leading to therapy switch before three months had elapsed. These two patients were included in other analyses, since they each received 63 d (2.1 months) of immune checkpoint blockade, a time period within which 76% of all severe irAEs occurred in our patient population.
For Cox regressions, the results were analyzed based on the Wald statistic (z-score) and significance was assessed by the Wald test. The proportional hazards assumption was confirmed for each covariate included in a Cox regression before analysis by evaluating the Schoenfeld residuals. Lipták’s83 and Stouffer’s methods84 were used for integrative statistical analyses, as appropriate. Sample size calculations for bulk cohorts 1 and 2 were performed using pwr v.1.3–0 in R86. In the single-cell discovery cohort, the association between CD4 TEM cell abundance (CyTOF) and severe irAE development had an effect size of 1.99 (Fig. 2c,d). Bulk cohorts 1 and 2 were designed to satisfy this effect size requirement at α = 0.05 and 1- β = 0.8 while emphasizing specificity in bulk cohort 1 (number of patients without severe irAEs > number of patients with severe irAEs) and balance in bulk cohort 2 (number of patients without severe irAEs ≈ number of patients with severe irAEs). All statistical analyses were performed using R v.3.5.1+ or Prism 8+ (GraphPad Software).
Extended Data
Extended Data Fig. 1 |. Quality control and extended characterization of cell states identified by unsupervised clustering of scRNA-seq data.
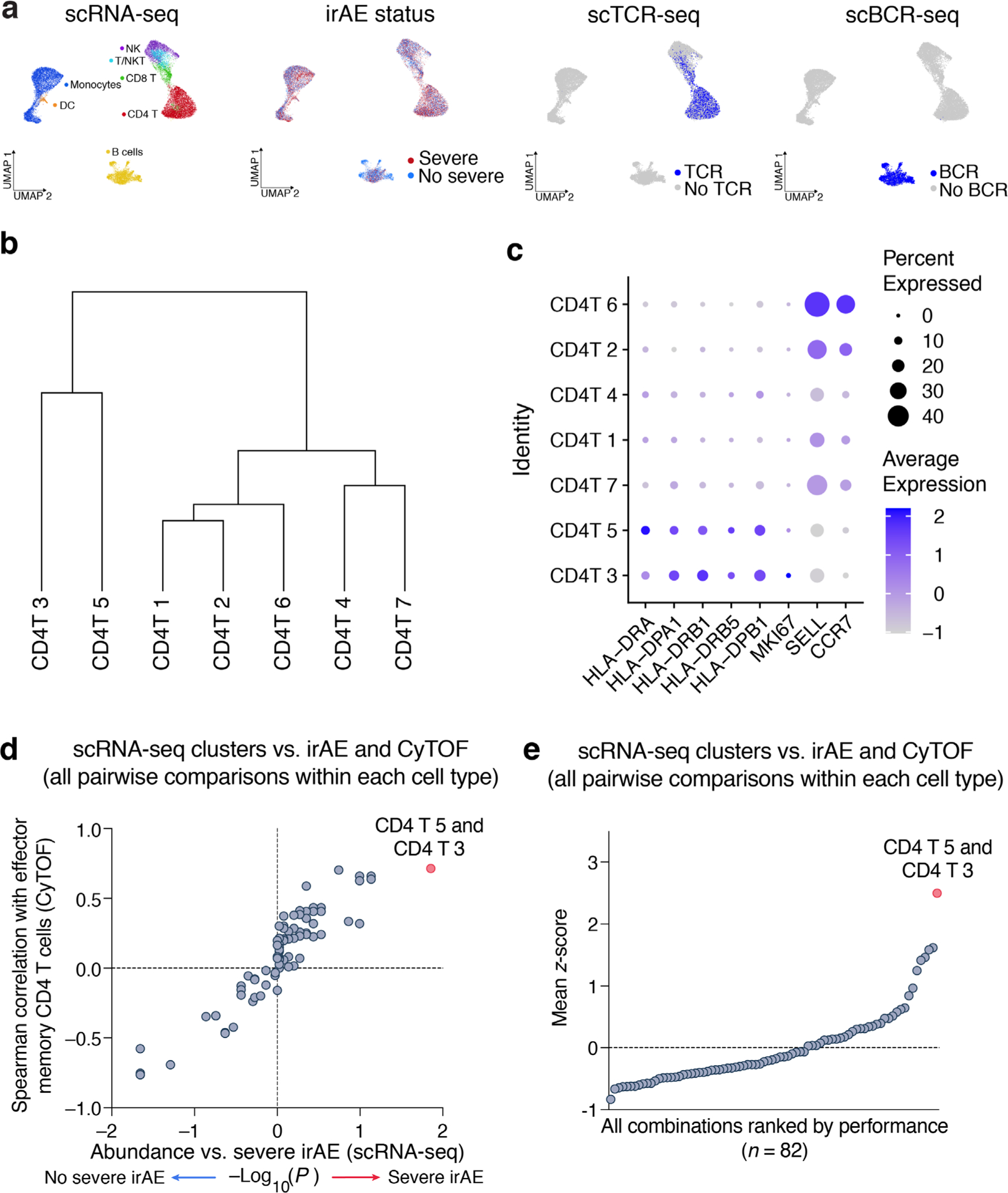
a, UMAP representation of pretreatment peripheral blood leukocytes profiled by droplet-based scRNA-seq (10x Genomics) from 13 patients with metastatic melanoma, colored by major cell lineages, severe irAE status, TCR expression by scV(D)J-seq, and BCR expression by scV(D)J-seq (related to Fig. 3a). b, Unsupervised hierarchical clustering (average linkage) of the mean log2 transcriptome per CD4 T cell cluster identified from scRNA-seq data. c, Dot plot showing the average expression of key activation (HLA-DX, MKI67) and lineage markers (SELL, CCR7) in CD4 T cell clusters. d, Same as Fig. 3b but showing all pairwise combinations of scRNA-seq clusters within each of the major cell types analyzed (B cells, CD4 T cells, CD8 T cells, NK cells, monocytes). Across 82 possible pairwise combinations, CD4 T 5 + 3 achieved the highest Spearman correlation against CD4 TEM levels enumerated by CyTOF and the strongest association with severe irAE development. Cells annotated as ‘T/NKT’ were collapsed into CD8 T cells. e, Same as panel d but showing all pairwise combinations ranked by the mean of each feature following unit variance normalization (mean of 0 and standard deviation of 1). In this analysis, the −log10 P-value for the association with severe irAE (two-sided, unpaired Wilcoxon rank sum test) was normalized to unit variance without considering the direction of the association.
Extended Data Fig. 2 |. Analysis of scRNA-seq states identified by reference-guided annotation.
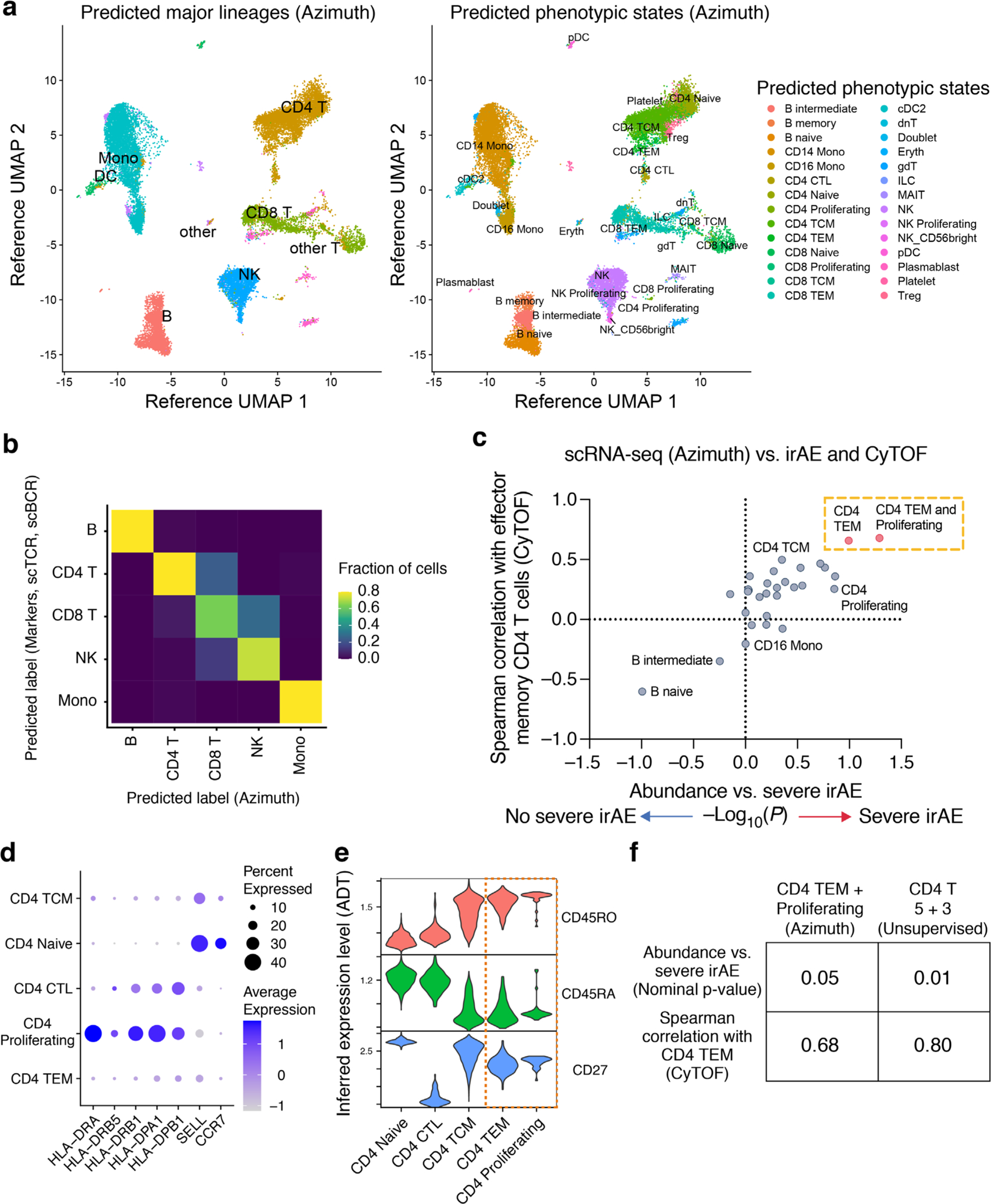
a, UMAP projections of scRNA-seq data generated in this work, embedded and labeled by Azimuth using a reference PBMC atlas of 162k cells profiled by scRNA-seq and 228 antibodies (Methods). b, Confusion matrix showing the agreement between phenotypic labels determined by marker genes and unsupervised clustering (rows; related to Fig. 3a and Extended Data Fig. 1a) versus reference-guided annotation with Azimuth (columns). In total, 85% of single cells assigned to a major lineage group by Azimuth (B cells, CD4 T, CD8 T, NK cells, monocytes) were assigned to the same identity by canonical marker gene assessment. Given the absence of NKT cells in the reference atlas used for Azimuth, the T/NKT cluster defined by unsupervised analysis was relabeled as CD8 T cells. c, Same analysis as in Fig. 3b but shown for all 27 phenotypic states identified by Azimuth. Among these states, CD4 TEM was most associated with severe irAE and CyTOF-enumerated CD4 TEM. A population combining CD4 TEM and CD4 Proliferating states was also strongly associated with severe irAE. The latter showed the highest expression of HLA-DX and lowest expression of SELL (panel d), consistent with an activated CD4 TEM phenotype. d, Dot plot depicting key activation and lineage markers among CD4 T cell states annotated by Azimuth. e, Violin plots showing protein expression levels imputed by Azimuth using antibody-derived tag (ADT) data, supporting the combination of CD4 TEM and CD4 Proliferating states in panels c and f. f, Performance of top-ranking cell subsets identified by Azimuth and unsupervised clustering for prediction of severe irAEs. The combined CD4 T 5 + 3 clusters (Fig. 3b) were more associated with severe irAE and CyTOF than the top-ranking reference-guided population (panel c). Statistical significance was calculated using a two-sided, unpaired Wilcoxon rank sum test. Data in all panels shown are from the 13 samples profiled by scRNA-seq in Fig. 3.
Extended Data Fig. 3 |. Analysis of activated, resting, and parental T cell subsets in relation to severe irAE development.
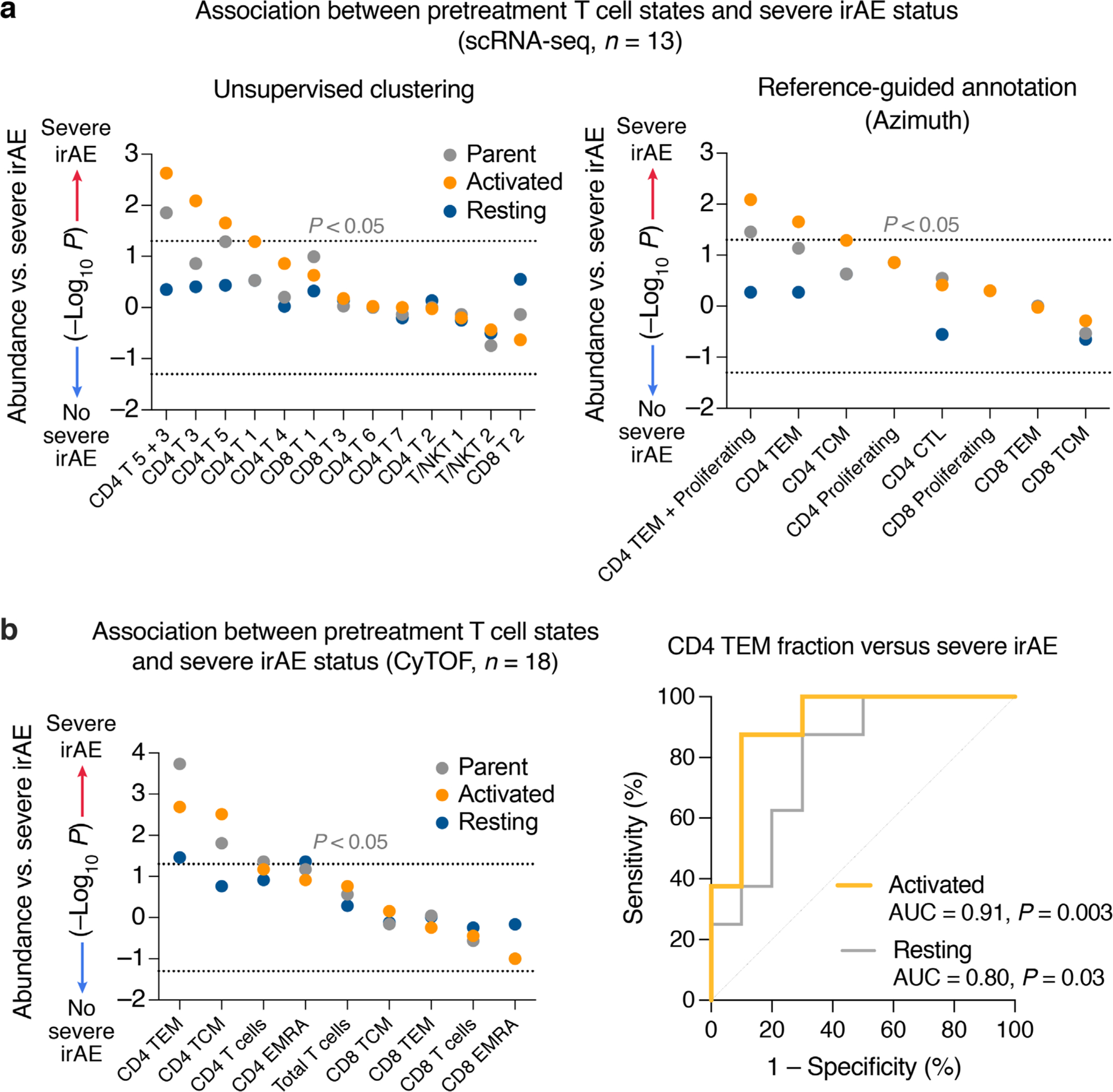
a, Association between severe irAE development and pretreatment levels of T cell states identified by unsupervised clustering (left) and memory-like T cell states identified by Azimuth (right) in 13 PBMC samples profiled by scRNA-seq (Figs. 1 and 3a). Activated cells were defined as those expressing HLA-DX or MKI67 (CPM > 0); resting cells were defined by the absence of HLA-DX and MKI67 expression (CPM = 0). b, Left: Association between severe irAE development and pretreatment levels of memory T cell subsets, total CD4 and CD8 T cells, and total T cells quantified by CyTOF, for all 18 patients analyzed in the single-cell discovery cohort (Figs. 1 and 2a). Activated phenotypes were defined as CD38+ or HLA-DR+ or Ki67+. Resting phenotypes were defined as CD38−HLA-DR−Ki67−. Right: ROC plot showing the performance of activated and resting CD4 TEM subsets (left panel) for predicting severe irAE development. Cell fractions were assessed relative to total PBMC content. Statistical significance in a, b was determined by a two-sided, unpaired Wilcoxon rank sum test and nominal −log10 P-values are displayed. −log10 P-values were further multiplied by −1 for associations with no severe irAE. See also Supplementary Table 6.
Extended Data Fig. 4 |. Extended characterization of immune repertoire diversity from single-cell V(D)J sequencing data.
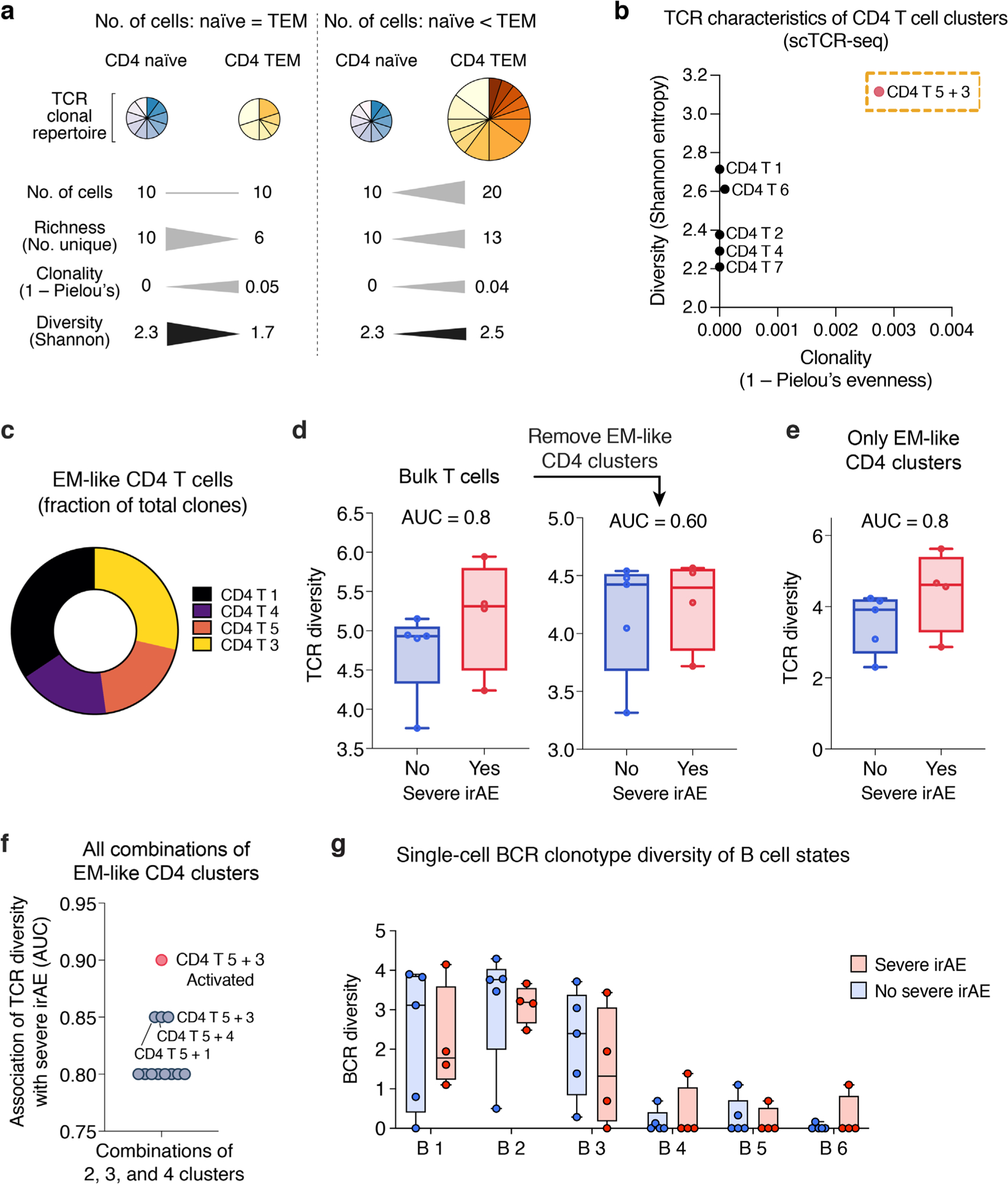
a, Key TCR diversity measures and the impact of cell abundance, TCR richness, and distinct clonal repertoires on such measures. Hypothetical CD4 naïve and TEM cell subsets are shown as examples. Triangles depicting differences in magnitude are not drawn to scale. b, Mean Shannon entropy versus mean clonality (1 ‒ Pielou’s evenness) for each CD4 T cell state identified by unsupervised clustering of scRNA-seq data. CD4 T 5 + 3 (Fig. 3b,c), a TEM state enriched for activated cells, shows elevated clonality relative to other CD4 states, as expected for this phenotype77, while also showing higher diversity (Shannon entropy), indicating elevated richness. c, Distribution of EM-like CD4 T cell states (from Fig. 3f) with available scTCR clonotype data. d, Association between severe irAE development and TCR diversity (Shannon entropy) in pseudo-bulk T cells from pretreatment blood, shown for all T cell states identified by scRNA-seq (left) and after the removal of the EM-like states indicated in panel c (no severe irAE, n = 5 patients; severe irAE, n = 4 patients). e, Same as d but shown for EM-like states alone. f, Area under the curve (AUC) for the association between pretreatment peripheral TCR diversity (Shannon entropy) and severe irAE development, shown for all combinations of the constituent cell states in e, including the combined CD4 T 5 + 3 cluster after restricting to activated cells (CPM > 0 for HLA-DX or MKI67). Of note, no other combination of activated EM-like states achieved an AUC > 0.85 in this analysis. g, BCR clonotype diversity (Shannon entropy), shown for each B cell state identified by unsupervised clustering (Fig. 3a). In b, d–f, only patients with at least 100 TCR clones were analyzed (n = 9; Methods). The same patients were analyzed in g for consistency. In panels d, e, and g, center lines, bounds of the box, and whiskers indicate medians, 1st and 3rd quartiles, and minimum and maximum values, respectively.
Extended Data Fig. 5 |. Validation of CiBERSORTx by single-cell analysis.
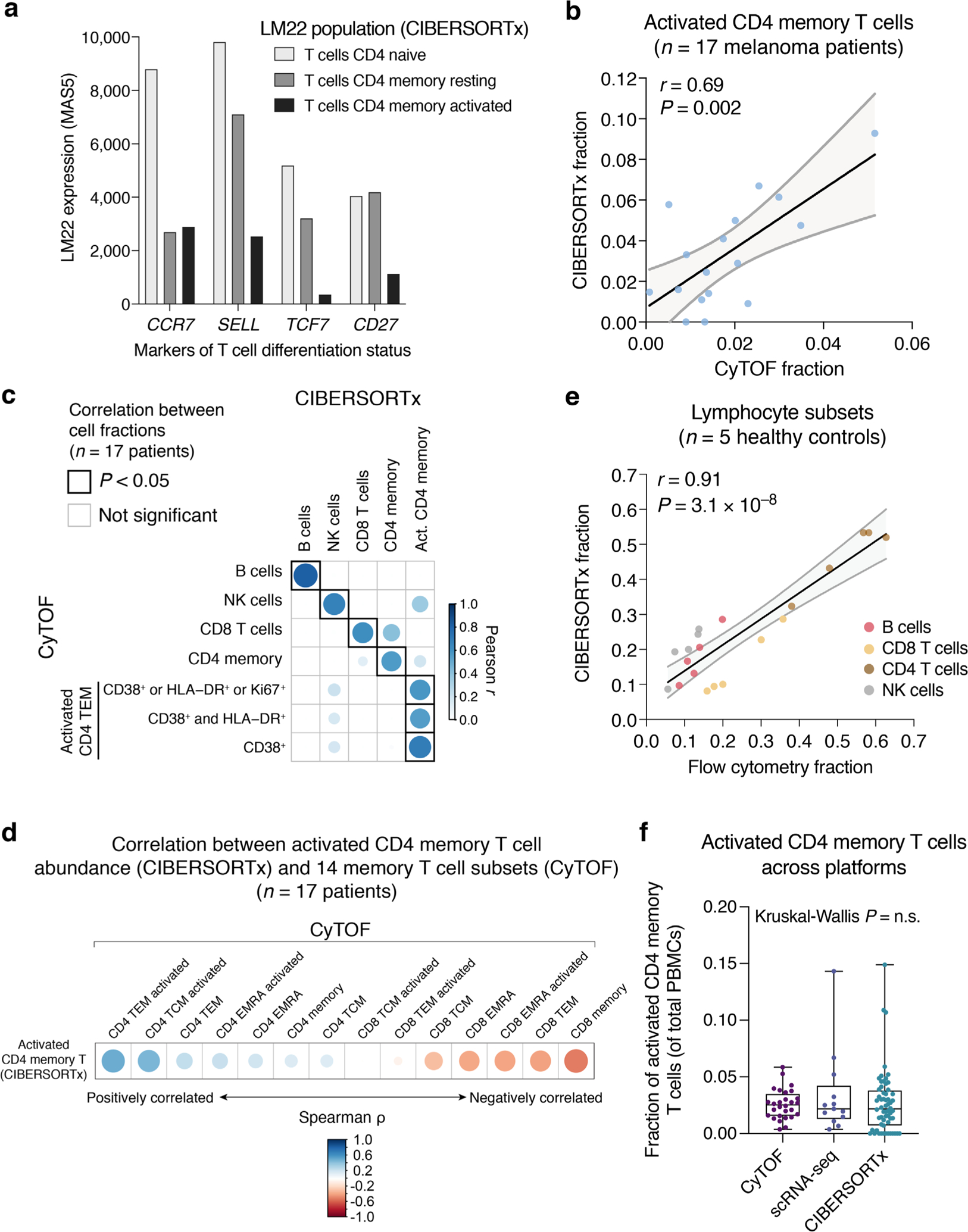
a, Expression of developmentally-regulated marker genes in major CD4 T cell subsets from the LM22 signature matrix (MAS5 normalized), showing that the LM22 reference signature for activated CD4 memory T cells has a TEM profile. b, CIBERSORTx versus mass cytometry for enumeration of activated CD4 memory T cells in the pretreatment peripheral blood of 17 metastatic melanoma patients (Supplementary Table 1). A linear regression line with 95% confidence band is shown. Concordance and significance were determined by Pearson r and a two-sided t test, respectively. While activated CD4 memory T cells quantitated by CyTOF were defined by CD38 expression in this plot, other activated CD4 TEM subsets were also significantly correlated with CIBERSORTx (panel c). c, Cross correlation plot of lymphocyte subset frequencies determined by CyTOF and CIBERSORTx. Act., Activated. d, Correlation between activated CD4 memory T cell levels inferred by CIBERSORTx and 14 memory T cell states profiled by CyTOF, including CD38+ activated subsets manually gated within each population, in PBMCs from 17 metastatic melanoma patients (Supplementary Table 1). e, Scatter plot depicting the global correlation of lymphocyte subsets enumerated by CIBERSORTx and flow cytometry in peripheral blood samples from five healthy subjects profiled by bulk RNA-seq (Supplementary Table 1). A linear regression line with 95% confidence band is shown. Concordance and significance were determined by Pearson r and a two-sided t test, respectively. As monocytes were variably underestimated by cytometry compared to complete blood counts, all results in b–e are expressed as a function of total lymphocytes. f, Distribution of activated CD4 memory T cell levels quantitated by CyTOF (CD38+, HLA-DR+ or Ki67+ CD4 TEM cells, n = 28 patients), scRNA-seq (HLA-DX+ or MKI67+ cells within CD4 T clusters 5 and 3, n = 13 patients), and CIBERSORTx (n = 60 patients) across all irAE-evaluable samples profiled by each modality in this work (Supplementary Table 1). Box center lines, bounds of the box, and whiskers indicate medians, 1st and 3rd quartiles, and minimum and maximum values, respectively. Statistical significance was determined by a Kruskal-Wallis test. n.s., not significant (P > 0.05).
Extended Data Fig. 6 |. Extended analysis of TCR diversity from pretreatment peripheral blood expression profiles.
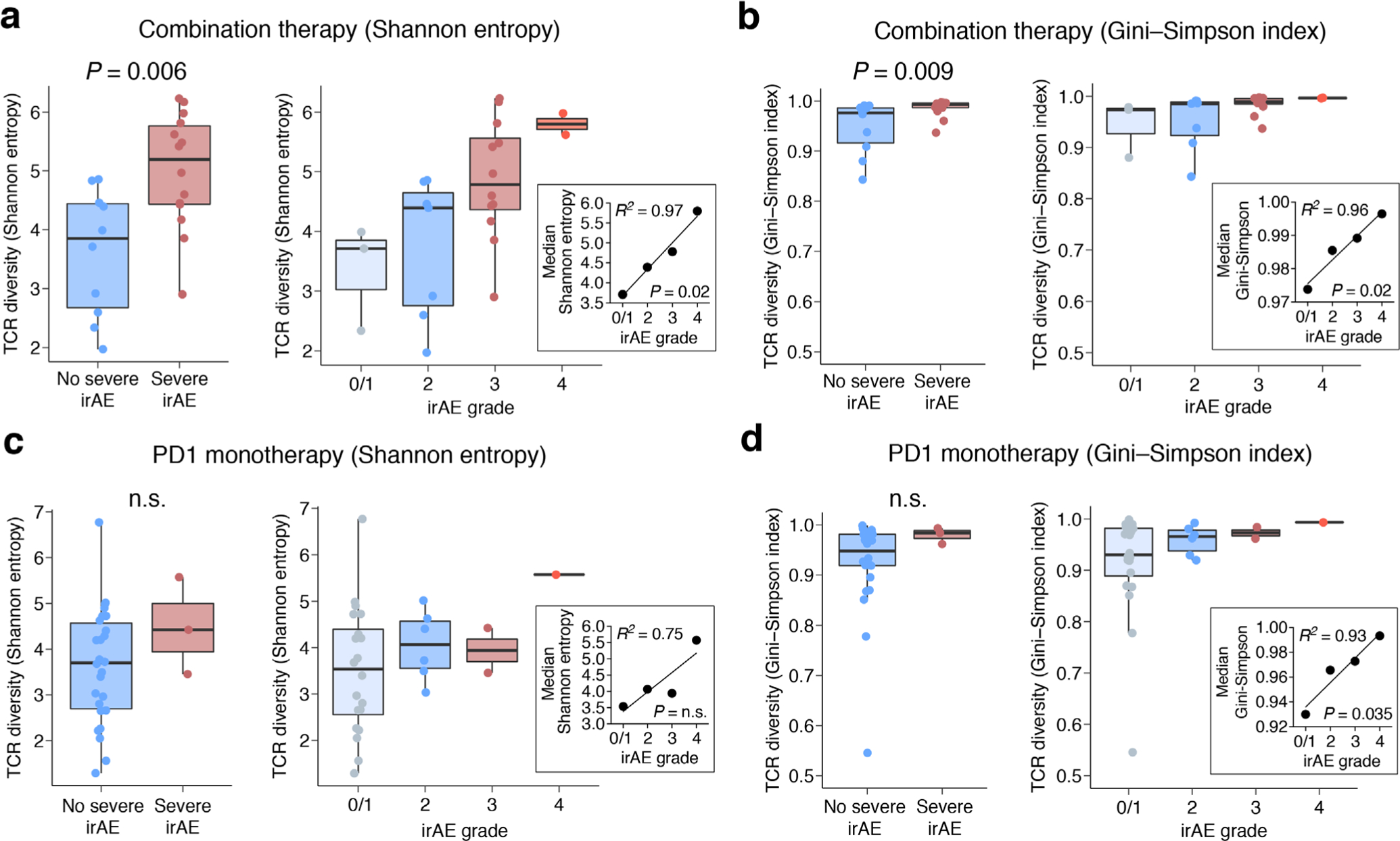
a–d, Association between baseline bulk TCR diversity and the highest irAE grade observed for each patient in bulk cohorts 1 and 2 (Supplementary Tables 7 and 9), shown for two diversity measures (a and c, Shannon entropy; b and d, Gini-Simpson index) and stratified by therapy type. In a and b, patients treated with combination therapy are stratified by future irAE status: no severe irAE (n = 10) versus severe irAE (n = 14 patients) (left) and irAE grade (right): 0/1 (n = 3), 2 (n = 7), 3 (n = 12), and 4 (n = 2). In c and d, patients treated with PD1 monotherapy are stratified by future irAE status: no severe irAE (n = 26) versus severe irAE (n = 3 patients) (left) and irAE grade (right): 0/1 (n = 19), 2 (n = 7), 3 (n = 2), and 4 (n = 1). Two-group comparisons were assessed by a two-sided, unpaired Wilcoxon rank sum test. n.s., not significant (P > 0.05). Linear regression was applied to evaluate the median value of each measure grouped by irAE grade (insets). The significance of linear concordance was determined by a two-sided t test. Grades 0 and 1 reflect no toxicity and asymptomatic toxicity, respectively, and were combined. In all panels, the box center lines, bounds of the box, and whiskers denote medians, 1st and 3rd quartiles, and minimum and maximum values within 1.5 × IQR (interquartile range) of the box limits, respectively.
Extended Data Fig. 7 |. Composite model performance across patients, key patient subgroups, the number of symptomatic irAEs per patient, and organ system involvement.
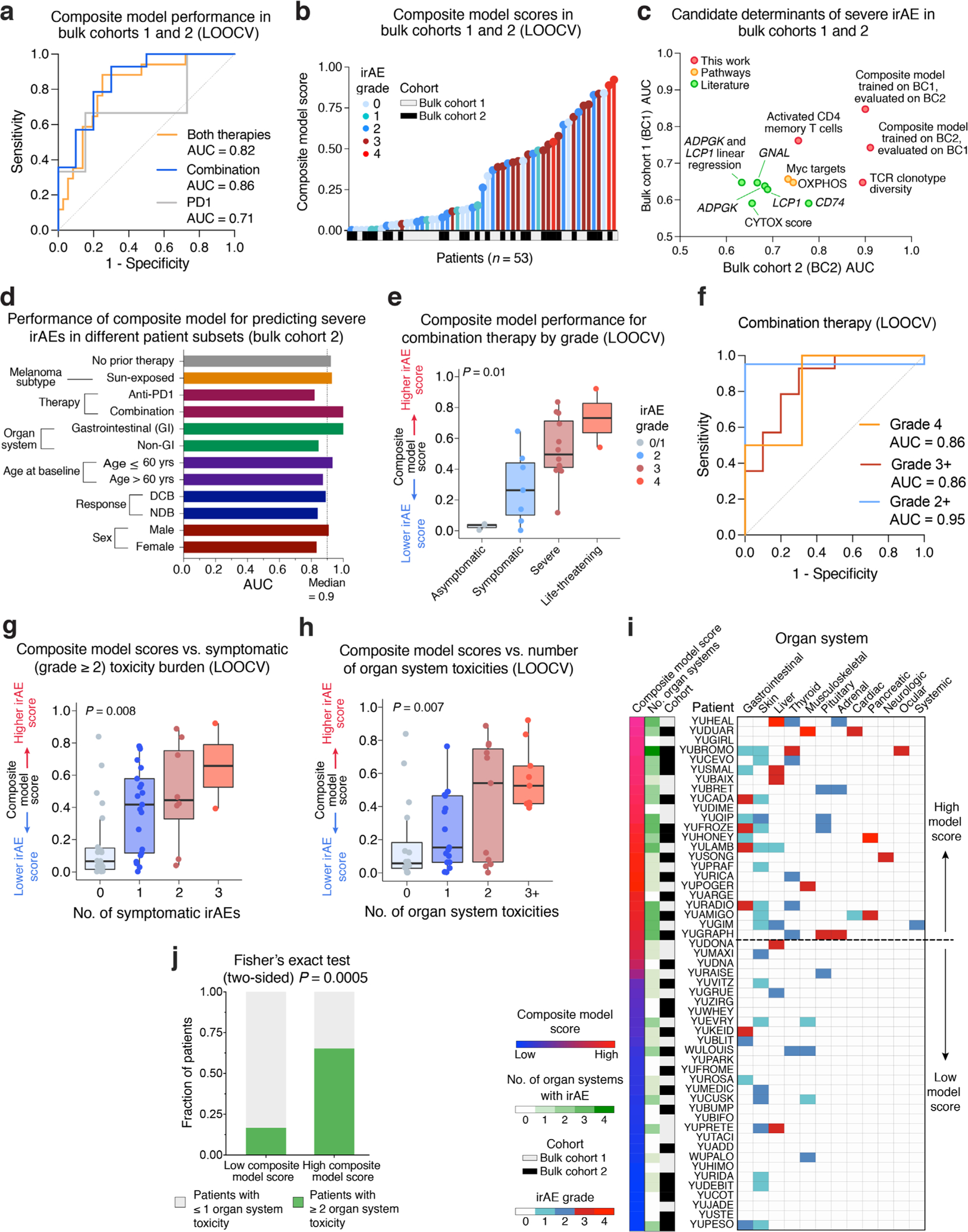
a, Same as Fig. 4d, but applied to both bulk cohorts (n = 53 patients) using leave-one-out cross-validation (LOOCV) (Methods). b, Same as Fig. 4c, but shown for model scores determined by LOOCV. c, Performance of the composite model versus other candidate pretreatment factors for predicting severe irAE development (Methods). The composite model was trained in bulk cohort 1 (BC1) and validated in bulk cohort 2 (BC2) or vice versa, as indicated. d, Performance of the composite model trained on bulk cohort 1 for predicting severe irAEs in different patient subgroups from bulk cohort 2. DCB, durable clinical benefit; NDB, no durable clinical benefit; GI, gastrointestinal. e, Composite model scores determined by LOOCV for all bulk cohort patients treated with combination therapy (n = 24), stratified by future irAE grade: 0/1 (n = 3), 2 (n = 7), 3 (n = 12), and 4 (n = 2). f, Model performance for predicting grade 2 +, 3 +, or 4 irAE development in combination therapy patients using the scores in e. g,h, Composite model scores determined by LOOCV in both bulk cohorts (n = 53 patients) versus the number of symptomatic irAEs (grade 2 +) per patient (g) and the number of organ system toxicities per patient (h). i, Distribution of irAEs across patients and organ systems (Supplementary Table 15). Patients from bulk cohorts 1 and 2 are organized by decreasing composite model scores determined via LOOCV (Methods). The line distinguishing high/low scores was optimized using LOOCV (Methods). j, Fraction of patients in both bulk cohorts that developed irAEs in at least 2 organ systems versus those that did not, stratified by the threshold in panel i (Methods). Significance was determined by a two-sided Fisher’s exact test. In e, g, and h, center lines, bounds of the box, and whiskers indicate medians, 1st and 3rd quartiles, and minimum and maximum values within 1.5 × IQR (interquartile range) of the box limits, respectively. Statistical significance in e, g, and h was determined by a Kruskal-Wallis test.
Extended Data Fig. 8 |. Composite model performance for predicting time to severe irAE in validation bulk cohort 2.
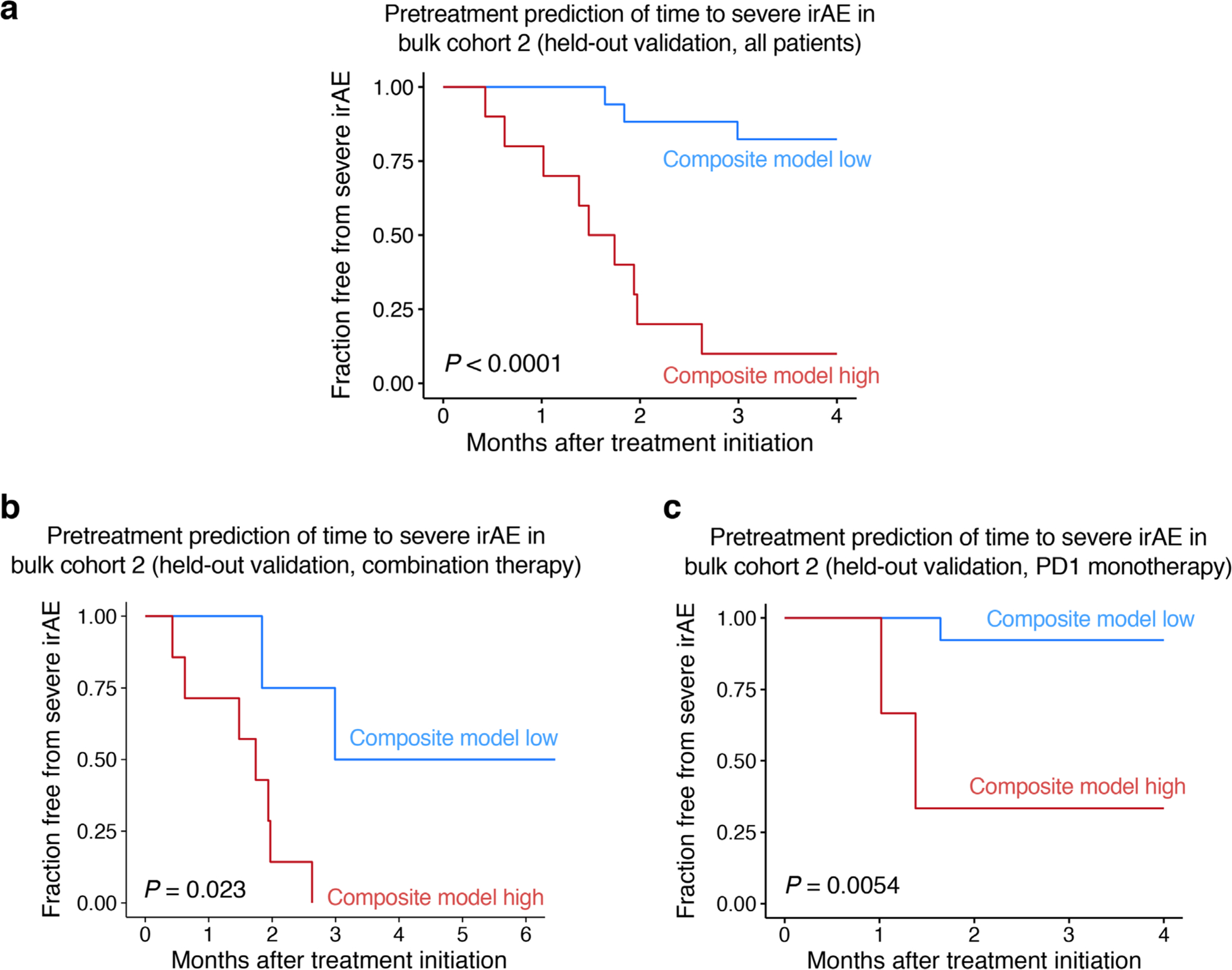
a–c, Kaplan-Meier analysis for freedom from severe irAE in bulk cohort 2 for patients treated with combination or PD1 immune checkpoint blockade (a), combination therapy (b), or PD1 monotherapy (c), stratified by the composite model score (Methods). Statistical significance was calculated by a two-sided log-rank test. In all panels, training was performed in bulk cohort 1 and the cut-point predicting severe irAE was optimized for bulk cohort 1 using Youden’s J statistic (Supplementary Table 10; Methods). Notably, the analyses in a–c were landmarked between treatment initiation and three months following treatment initiation, with all severe irAEs occurring within this period. The Kaplan-Meier plots are shown out to four months given the extended follow-up of patients that did not develop any severe irAE (Supplementary Table 9).
Extended Data Fig. 9 |. Peripheral blood TCR-β profiling with immunoSEQ®.
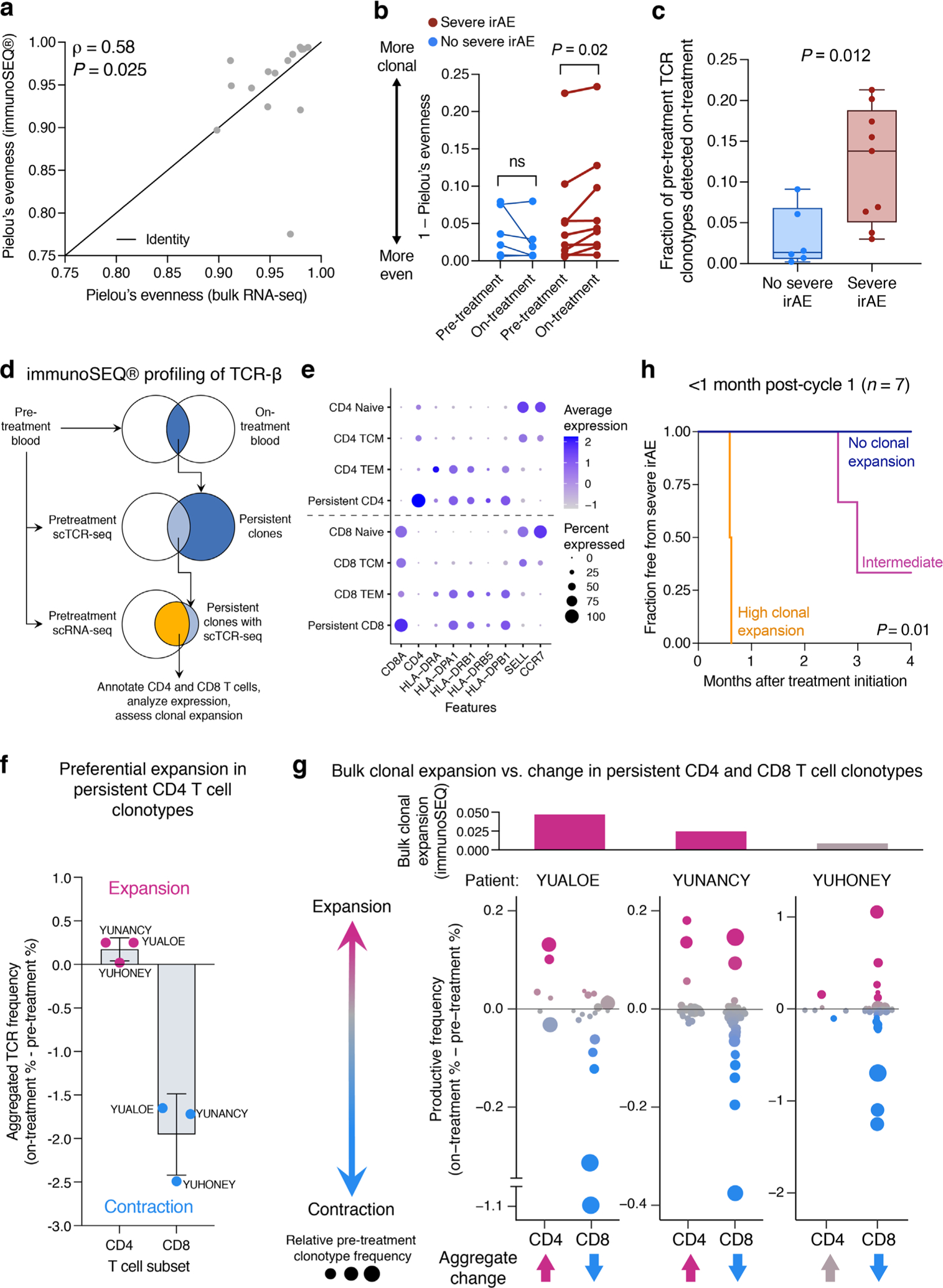
a, Evenness (Pielou’s index) of TCR repertoires assembled by MiXCR (bulk RNA-seq) and immunoSEQ® (genomic DNA) from paired pretreatment PBMC samples (n = 15 combination therapy patients) (Supplementary Tables 1 and 18). Concordance and significance were determined by Spearman ρ and a two-sided t test, respectively. b, Similar to Fig. 5b but showing clonality for each pre- and on-treatment PBMC sample (Supplementary Table 18). Statistical significance was determined by a two-sided, paired Wilcoxon rank sum test. ns, not significant (P > 0.05). c, Fraction of pretreatment peripheral blood TCR clonotypes detected on-treatment in 15 combination therapy patients (Supplementary Table 18), stratified by no severe (n = 6) and severe (n = 9) irAE status. Clonotypes with matching productive CDR3 β-chain nucleotide sequences were considered identical. Center lines, bounds of the box, and whiskers indicate medians, 1st and 3rd quartiles, and minimum and maximum values, respectively. Significance was determined by a two-sided, unpaired Wilcoxon rank sum test. d–g, Clonal dynamics in circulating T cells following combination therapy initiation. d, Persistent T cell clones identified by immunoSEQ® were cross-referenced with scTCR-seq and scRNA-seq data of pretreatment PBMCs from the same three patients (YUALOE, YUNANCY, YUHONEY), all of whom received combination therapy and developed severe ICI-induced toxicity (Supplementary Table 18; Methods). e, Log2 expression of key lineage and activation markers across major T cell states annotated by Azimuth along with persistent clones classified into CD4 and CD8 T cells (Methods). f, Aggregate change from baseline in the productive frequencies of persistent clonotypes, stratified by lineage (n = 2 cell types) and patient (n = 3). The sum of the difference in productive frequencies (on-treatment % – pretreatment %) was calculated from immunoSEQ® data. Bars denote mean + /− SD. g, Top: Change in bulk TCR clonality from baseline (Fig. 5b). Bottom: Same as f but showing the underlying clonotypes, where circle size is proportional to pretreatment clone frequency (immunoSEQ®). h, Same as Fig. 5d but restricted to blood draws taken cycle 1 day 1 of combination therapy and <1 month later (n = 7 patients; Supplementary Table 18).
Extended Data Fig. 10 |. Schema of large-scale assessment of peripheral blood leukocytes in autoimmune disorders versus healthy controls.
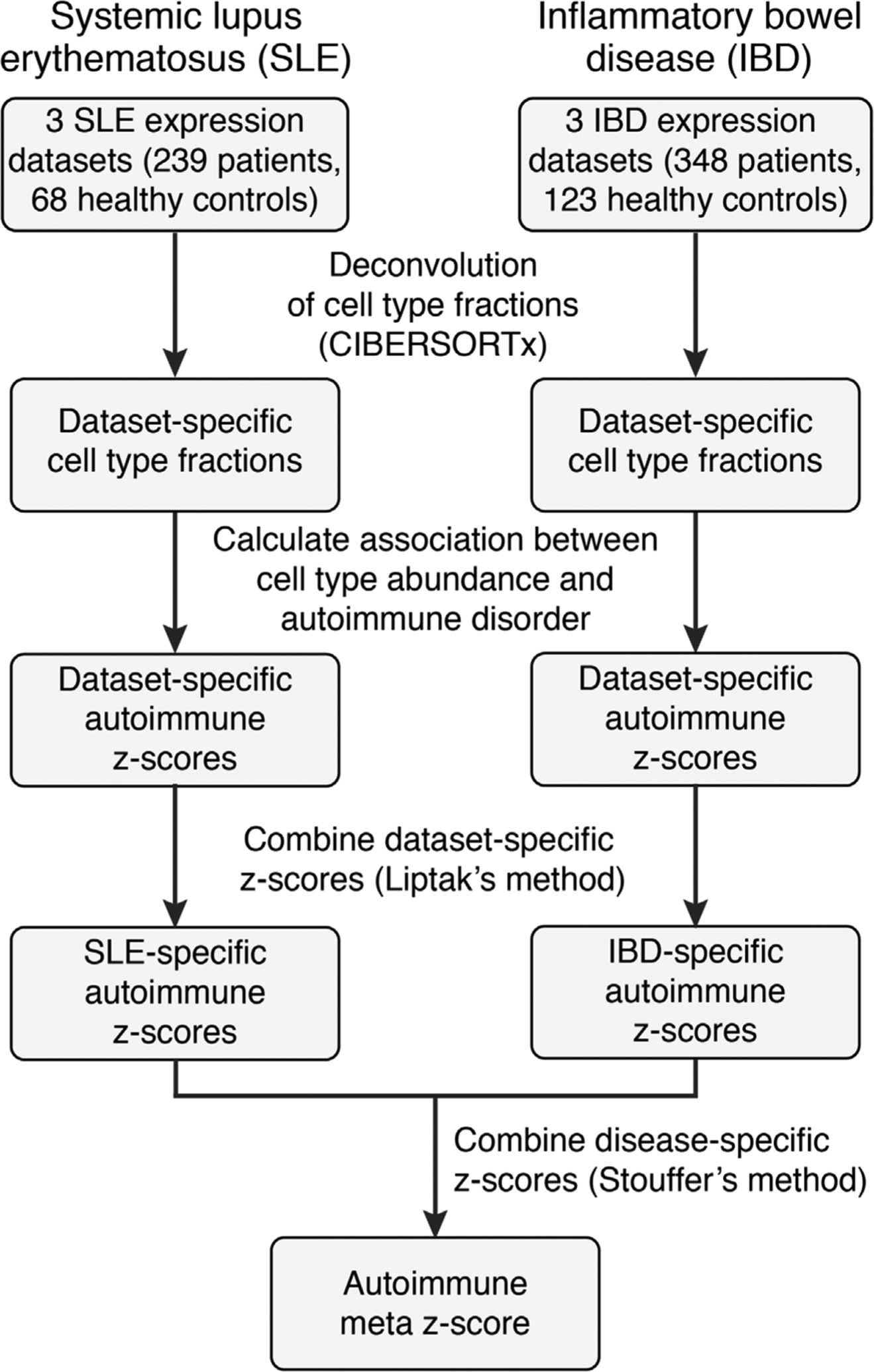
Schema describing the workflow and statistical meta-analysis for evaluating the enrichment of individual circulating leukocyte subsets in autoimmune disorders relative to healthy controls (Fig. 6; Methods). In brief, CIBERSORTx was applied to enumerate 15 leukocyte subsets in bulk RNA-seq or microarray profiles of peripheral blood samples from patients with either systemic lupus erythematosus57–59 (SLE; n = 239) or inflammatory bowel disease56,60,61 (IBD; n = 348) compared to healthy controls (Supplementary Table 20). For each dataset and cell subset, a two-sided, unpaired Wilcoxon rank sum test was applied to assess the difference in relative abundance between healthy and disease phenotypes. Results were subsequently combined across studies by meta-z statistics (Methods).
Supplementary Material
Acknowledgements
We thank the patients and families involved in this study. We thank A. Nassar and L. Chen for technical assistance with the CyTOF experiments, including provision of resources, staining and processing of samples. We thank G. Ansstas and C. Kaufman for clinical samples. We also thank T. Ley, S. Devarakonda and M. Tal for providing critical feedback on the manuscript. This work was supported by grants from the National Cancer Institute (no. K08CA238711 to A.A.C., no. K08CA237727 to D.Y.C., no. R01CA238471 to K.D., no. R21CA218950 to R.H. and no. R00CA187192 to A.M.N.), National Heart, Lung, and Blood Institute (no. T35HL007649 to A.N.), National Institute of Arthritis and Musculoskeletal and Skin Diseases (no. R01AR077926 to K.D.), a fellowship from the Natural Sciences and Engineering Research Council of Canada (A.X.L.), the Cancer Research Foundation Young Investigator Award (A.A.C.), V Foundation for Cancer Research V Scholar Award (A.A.C.), Washington University Alvin J. Siteman Cancer Research Fund (A.A.C.), Yale Cancer Center Meyers Award (M.S. and R.H.), a 10x Genomics Pilot Program Award (R.H.), the Melanoma Research Alliance (no. 137453 and no. 828544 to R.H.), the Virginia and D.K. Ludwig Fund for Cancer Research (A.M.N.), Stinehart-Reed Foundation (A.M.N.), Stanford Bio-X Interdisciplinary Initiatives Seed Grants Program (IIP) (A.M.N.) and Donald E. and Delia B. Baxter Foundation (A.M.N.).
Competing interests
A.A.C. has patent filings related to cancer biomarkers and digital cytometry and has served as an advisor/consultant to Roche, AstraZeneca, Daiichi Sankyo, Tempus, Geneoscopy, NuProbe, Fenix Group International and Guidepoint. A.A.C. has stock options in Geneoscopy, research support from Roche and ownership interests in Droplet Biosciences. M.S. has consulted for Idera Pharmaceuticals, Regeneron Pharmaceuticals, Apexigen, Alligator Bioscience, Verastem Oncology, Agenus, Rubius Therapeutics, Bristol Myers Squibb, Genentech-Roche, Boston Pharmaceuticals, Servier Laboratories, Adaptimmune Therapeutics, Immunocore, Dragonfly Therapeutics, Pierre Fabre Pharmaceuticals, Molecular Partners, Boehringer Ingelheim, Innate Pharma, Nektar Therapeutics, Pieris Pharmaceuticals, Numab Therapeutics, Abbvie, Zelluna Immunotherapy, Seattle Genetics/Seagen, Genocea Biosciences, GI Innovation, Chugai-Roche, BioNTech, Eli Lilly, Modulate Therapeutics, Array Biopharma, AstraZeneca and Genmab. M.S. has stock options in EvolveImmune, NextCure, Repertoire Immune Medicines, Adaptive Biotechnologies, Actym Therapeutics and Amphivena Therapeutics and has stock ownership in GlaxoSmithKline and Johnson & Johnson. A.M.N. has patent filings related to expression deconvolution, digital cytometry and cancer biomarkers and has served as an advisor/consultant to Roche, Merck and CiberMed. The other authors declare no conflicts of interest.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-021-01623-z.
Code availability
Custom scripts for the training and validation of composite models, evaluating freedom from severe toxicity and generating related figures are publicly available from https://doi.org/10.25936/f3np-k536.
Extended data is available for this paper at https://doi.org/10.1038/s41591-021-01623-z.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-021-01623-z.
Data availability
Bulk and single-cell expression data generated in this work have been deposited in GEO under accession no. GSE186144. All requests for raw data will be promptly reviewed by the corresponding authors to determine whether the request is subject to any confidentiality obligations. Any data that can be shared will be released via a material transfer agreement. The published expression datasets analyzed in this work (Supplementary Table 20) are available from GEO with accession nos. GSE50772, GSE61635, GSE72509, GSE126124, GSE3365 and GSE100833. Processed CyTOF, MiXCR and immunoSEQ data are publicly available from https://doi.org/10.25936/f3np-k536. Additional data supporting the findings in this work are available in the main text, figures, extended data and supplementary files.
References
- 1.Kumar V et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front. Pharmacol 8, 49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larkin J et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi FS et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 19, 1480–1492 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Postow MA, Sidlow R & Hellmann MD Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med 378, 158–168 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Wolchok JD et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 377, 1345–1356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamid O et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol 30, 582–588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robert C et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 20, 1239–1251 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Robert C et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Schachter J et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390, 1853–1862 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Johnson DB et al. Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med 375, 1749–1755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang DY et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol 4, 1721–1728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haanen JBAG et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol 28, iv119–iv142 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Das S & Johnson DB Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 7, 306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson DB et al. A case report of clonal EBV-like memory CD4+ T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat. Med 25, 1243–1250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahir SA et al. Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc. Natl Acad. Sci. USA 116, 22246–22251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahabi V et al. Gene expression profiling of whole blood in ipilimumab-treated patients for identification of potential biomarkers of immune-related gastrointestinal adverse events. J. Transl. Med 11, 75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarhini AA et al. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J. Immunother. Cancer 3, 39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujimura T et al. Serum levels of soluble CD163 and CXCL5 may be predictive markers for immune-related adverse events in patients with advanced melanoma treated with nivolumab: a pilot study. Oncotarget 9, 15542–15551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das R et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J. Clin. Invest 128, 715–720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subudhi SK et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc. Natl Acad. Sci. USA 113, 11919–11924 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaput N et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol 30, 2012 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Dubin K et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun 7, 10391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jing Y et al. Multi-omics prediction of immune-related adverse events during checkpoint immunotherapy. Nat. Commun 11, 4946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim SY et al. Circulating cytokines predict immune-related toxicity in melanoma patients receiving anti-PD-1-based immunotherapy. Clin. Cancer Res 25, 1557–1563 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Pavan A et al. Peripheral blood markers identify risk of immune-related toxicity in advanced non-small cell lung cancer treated with immune-checkpoint inhibitors. Oncologist 24, 1128–1136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews MC et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med 27, 1432–1441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchinson JA et al. Virus-specific memory T cell responses unmasked by immune checkpoint blockade cause hepatitis. Nat. Commun 12, 1439 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yasuda Y et al. CD4+ T cells are essential for the development of destructive thyroiditis induced by anti-PD-1 antibody in thyroglobulin-immunized mice. Sci. Transl. Med 13, eabb7495 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Morad G, Helmink BA, Sharma P & Wargo JA Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184, 5309–5337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marschner D et al. MicroRNA-146a regulates immune-related adverse events caused by immune checkpoint inhibitors. JCI Insight 5, e132334 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auslander N et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med 24, 1545–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieg C et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med 24, 144–153 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Liu D et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med 25, 1916–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacquelot N et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat. Commun 8, 592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riaz N et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang AC et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fairfax BP et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med 26, 193–199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bieber AK, Yin L & Lo Sicco K Pruritus and tense bullae after discontinuation of pembrolizumab in a patient with renal cell carcinoma. JAMA 324, 1453–1454 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Lopez AT & Geskin L A case of nivolumab-induced bullous pemphigoid: review of dermatologic toxicity associated with programmed cell death protein-1/programmed death ligand-1 inhibitors and recommendations for diagnosis and management. Oncologist 23, 1119–1126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singer S, Nelson CA, Lian CG, Dewan AK & LeBoeuf NR Nonbullous pemphigoid secondary to PD-1 inhibition. JAAD Case Rep 5, 898–903 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croft M, So T, Duan W & Soroosh P The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev 229, 173–191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell JJ et al. CCR7 expression and memory T cell diversity in humans. J. Immunol 166, 877–884 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Shifrut E et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiffelle J et al. T-cell repertoire analysis and metrics of diversity and clonality. Curr. Opin. Biotechnol 65, 284–295 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Rényi A On the foundations of information theory. Rev. Int. Stat. Inst 33, 1–14 (1965). [Google Scholar]
- 47.Robert L et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res 20, 2424–2432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sims JS et al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc. Natl Acad. Sci. USA 113, E3529–E3537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newman AM et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med 21, 938–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newman AM et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolotin DA et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Eggermont AMM, Crittenden M & Wargo J Combination immunotherapy development in melanoma. Am. Soc. Clin. Oncol. Educ. Book 38, 197–207 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Boutros C et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol 13, 473–486 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Burczynski ME et al. Molecular classification of Crohn’s disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J. Mol. Diagn 8, 51–61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpintero MF et al. Diagnosis and risk stratification in patients with anti-RNP autoimmunity. Lupus 24, 1057–1066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hung T et al. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 350, 455–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kennedy WP et al. Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci. Med 2, e000080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmer NP et al. Concordance between gene expression in peripheral whole blood and colonic tissue in children with inflammatory bowel disease. PLoS ONE 14, e0222952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters LA et al. A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat. Genet 49, 1437–1449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Menzies AM et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol 28, 368–376 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Brown LJ et al. Combination anti-PD1 and ipilimumab therapy in patients with advanced melanoma and pre-existing autoimmune disorders. J. Immunother. Cancer 9, e002121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson DB et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol 2, 234–240 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Tang S-Q et al. The pattern of time to onset and resolution of immune-related adverse events caused by immune checkpoint inhibitors in cancer: a pooled analysis of 23 clinical trials and 8,436 patients. Cancer Res. Treat 53, 339–354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nabet BY et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell 183, 363–376.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steele NG et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat. Cancer 1, 1097–1112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amir ED et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol 31, 545–552 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Van Gassen S et al. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Amir ED et al. Development of a comprehensive antibody staining database using a standardized analytics pipeline. Front. Immunol 10, 1315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soneson C, Love MI & Robinson MD Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen GM et al. Integrative bulk and single-cell profiling of pre-manufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov 11, 2186–2199 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaech SM, Wherry EJ & Ahmed R Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol 2, 251–262 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Sprent J & Surh CD T cell memory. Annu. Rev. Immunol 20, 551–579 (2002). [DOI] [PubMed] [Google Scholar]
- 77.van den Broek T, Borghans JAM & van Wijk F The full spectrum of human naive T cells. Nat. Rev. Immunol 18, 363–373 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Dixon P VEGAN, a package of R functions for community ecology. J. Veg. Sci 14, 927–930 (2003). [Google Scholar]
- 79.Jiang H, Lei R, Ding S-W & Zhu S Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Borcherding N et al. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun. Biol 4, 122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Motulsky HJ & Brown RE Detecting outliers when fitting data with nonlinear regression—a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gautier L, Cope L, Bolstad BM & Irizarry RA affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 83.Lipták T On the combination of independent tests. Magyar Tud. Akad. Mat. Kutató Int. Közl 3, 171–197 (1958). [Google Scholar]
- 84.Stouffer SA, Suchman EA, Devinney LC, Star SA, & Williams RM Jr. in Studies in Social Psychology in World War II (Princeton Univ. Press, 82–154 1949). [Google Scholar]
- 85.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cohen J Statistical Power Analysis for the Behavioral Sciences (L. Erlbaum Associates, 1988). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk and single-cell expression data generated in this work have been deposited in GEO under accession no. GSE186144. All requests for raw data will be promptly reviewed by the corresponding authors to determine whether the request is subject to any confidentiality obligations. Any data that can be shared will be released via a material transfer agreement. The published expression datasets analyzed in this work (Supplementary Table 20) are available from GEO with accession nos. GSE50772, GSE61635, GSE72509, GSE126124, GSE3365 and GSE100833. Processed CyTOF, MiXCR and immunoSEQ data are publicly available from https://doi.org/10.25936/f3np-k536. Additional data supporting the findings in this work are available in the main text, figures, extended data and supplementary files.