

Abstract
Alpha-pinene (AP), produced by pine trees and other plants, is the main component of turpentine and is used as a fragrance and flavor ingredient. Exposure occurs via the use of personal care and household cleaning products and in the lumber industry. Despite widespread exposure, toxicity data for AP are limited. The objective of this work was to develop and validate a method to quantitate AP in rodent blood and mammary glands, in support of toxicokinetic and toxicology studies of AP. The method uses 100 µL of blood or 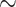 100 mg of mammary gland with analysis by headspace (HS) gas chromatography--mass spectrometry. The samples are diluted with internal standard (2H3-AP, IS) and sealed in HS vials; mammary glands are homogenized within the vial. The vials are equilibrated briefly at 60°C before a HS sample is analyzed. The method was validated in Sprague Dawley rat blood over the range 5–500 ng/mL and mammary gland over the range 100–5,000 ng/g. The method was linear (r ≥ 0.99), accurate (mean relative error (RE) ≤ ±13.4%) and precise (relative standard deviation (RSD) ≤ 7.1%) in both matrices. Recoveries incorporating IS were ≥88.7% at all concentrations in both tissues. Standards as high as 1,500 ng/mL in blood and 20,000 ng/g in mammary gland could be analyzed using lower injection volume or extrapolating the calibration curve beyond the upper limit of quantitation (mean %RE ≤ ±18.7; %RSD ≤ 2.2). Loss of AP occurred during overnight autosampler storage as well as frozen storage in as few as 15 days, but incorporation of IS prior to storage corrected for the loss such that calculated concentrations were within 84.7–117% of day 0 concentrations following frozen storage up to ≥32 days in both matrices. Matrix evaluation was performed in Hsd:Sprague Dawley®SD® rat and B6C3F1 mouse blood and mammary glands (mean %RE ≤ ±9.2; %RSD ≤ 4.3). These data demonstrate that the method is suitable for determination of AP in rodent blood and mammary glands.
100 mg of mammary gland with analysis by headspace (HS) gas chromatography--mass spectrometry. The samples are diluted with internal standard (2H3-AP, IS) and sealed in HS vials; mammary glands are homogenized within the vial. The vials are equilibrated briefly at 60°C before a HS sample is analyzed. The method was validated in Sprague Dawley rat blood over the range 5–500 ng/mL and mammary gland over the range 100–5,000 ng/g. The method was linear (r ≥ 0.99), accurate (mean relative error (RE) ≤ ±13.4%) and precise (relative standard deviation (RSD) ≤ 7.1%) in both matrices. Recoveries incorporating IS were ≥88.7% at all concentrations in both tissues. Standards as high as 1,500 ng/mL in blood and 20,000 ng/g in mammary gland could be analyzed using lower injection volume or extrapolating the calibration curve beyond the upper limit of quantitation (mean %RE ≤ ±18.7; %RSD ≤ 2.2). Loss of AP occurred during overnight autosampler storage as well as frozen storage in as few as 15 days, but incorporation of IS prior to storage corrected for the loss such that calculated concentrations were within 84.7–117% of day 0 concentrations following frozen storage up to ≥32 days in both matrices. Matrix evaluation was performed in Hsd:Sprague Dawley®SD® rat and B6C3F1 mouse blood and mammary glands (mean %RE ≤ ±9.2; %RSD ≤ 4.3). These data demonstrate that the method is suitable for determination of AP in rodent blood and mammary glands.
Introduction
Alpha-pinene (AP) is a bicyclic monoterpene that occurs naturally in coniferous trees, some herbs and other plants. It is the main component of turpentine and is also used as a fragrance ingredient in perfumes, personal care products, household cleaners and air fresheners (1). The general population may be exposed to AP via the use of these commercial products, and occupational exposure can occur during the sawing or processing of fresh wood or in use of turpentine as a solvent (2–4). Industrial exposures as high as 550 mg/m3 have been reported (5).
AP is highly soluble in blood and adipose tissue, which suggests rapid absorption following inhalation, and accumulation in peripheral fat and other tissues (6). It is metabolized primarily by hydroxylation and glucuronidation and is eliminated by the kidneys (3, 4). The limited toxicity data available for AP suggest that it may irritate the skin and mucous membranes (2, 3), and long-term exposure could impair lung function (5) and increase risk of developing respiratory cancer (7, 8). In 2-week and 3-month toxicity studies conducted by the National Toxicology Program (NTP), following administration of AP to rats and mice by inhalation, the major target organs were generally consistent across species and included the liver, the urinary system (kidney in rats and bladder in mice) and the male reproductive system (1). With limited toxicity data and continued widespread exposure to AP, the NTP evaluates the toxicity and toxicokinetic properties of AP (9). Quantitative determination of internal dose after AP exposure is essential in interpreting toxicological outcome. The objective of this work was to develop and validate a method to quantitate AP in rodent blood and mammary gland in support of NTP studies. Limited methods have been published for the determination of AP in plant material (10), pharmaceutical products (11, 12), air and blood (13) by gas chromatography (GC) with detection by flame ionization, mass spectrometry (MS) or tandem mass spectrometry (MS-MS). However, the reported methods required sample preparation that could lead to loss of volatile constituents such as AP during handling, and no methods for analysis of mammary gland were found. Hence, a novel, simple method with minimal sample handling was developed and validated employing headspace (HS) GC--MS, in which the samples remain sealed during preparation and AP is sampled directly from the HS.
Materials and Methods
Chemicals and reagents
AP (CASRN 80-56-8; Lot No. A-9211; 99.9% purity) was purchased from John D. Walsh Co., Inc (Ringwood, NJ), and 2H3-AP (internal standard, IS) was purchased from AromaLAB GmbH (Planegg Germany). The molecular structures of AP and 2H3-AP are shown in Figure 1. Male Sprague Dawley (SD) and male and female Hsd: Sprague Dawley®SD® (HSD) rat blood, male and female B6C3F1 mouse blood, and mammary glands from female SD rats, HSD rats and B6C3F1 mice were purchased from BioIVT (Westbury, NY). Ethanol (100%) was from Decon Labs (King of Prussia, PA), and veterinarian-grade saline was purchased from commercial sources.
Figure 1.
Structures of alpha-pinene (left) and 2H3-alpha-pinene (right).
Method development and validation
HS GC--MS methodologies were optimized with respect to chromatographic performance, sensitivity, selectivity and dynamic range. Analyte volatility and logistics of sample handling were carefully considered with respect to sample preparation; different spiking volumes and solvent compositions were tested to determine their effect on AP sensitivity, and sample amounts and homogenization processes were optimized for mammary gland.
The final method was fully validated in male SD rat blood and in female SD rat mammary gland by analyzing calibration standards and quality control (QC) samples in replicate over multiple analysis days to demonstrate linearity, selectivity, sensitivity, recovery, and intra- and inter-assay precision and accuracy over the desired concentration ranges. The concentration ranges were 5–500 ng/mL in blood and 100–5,000 ng/g in mammary gland. Selectivity was assessed by analyzing six matrix blanks without IS and six method blanks with IS for background interferences at the alpha-pinene retention time. Accuracy was evaluated as percent relative error (%RE) and precision as relative standard deviation (RSD). The limit of quantitation (LOQ) was the lowest matrix standard that meets accuracy and precision criteria. The limit of detection (LOD) was defined as three times the standard deviation of the lowest concentration. Absolute recovery was calculated as the percentage of the AP peak area in matrix to the AP peak area in solvent; relative recovery was expressed as the percentage of the response ratio (AP:IS) in matrix to that in solvent. Intraday precision and accuracy were evaluated at three concentration levels in blood and two levels in mammary gland, using three independent standards prepared on the same day. Interday precision and accuracy were evaluated at the same concentration levels, using multiple sets of independent standards prepared and analyzed over multiple analysis days. The method was evaluated for secondary matrices, male and female HSD rat and B6C3F1 mouse blood, and female HSD rat and B6C3F1 mouse mammary glands.
Preparation of stock solutions
Standard stock solutions of AP were prepared at 0.2 and 0.5 mg/mL in ethanol. Intermediate solutions (0.004 and 0.01 mg/mL) were prepared as dilutions of the stocks with a diluent containing IS in 50:50 ethanol:saline. This diluent contained 250 ng/mL IS for spiking in blood and 500 ng/mL IS for spiking in mammary gland. Spiking solutions were prepared from the alternating AP intermediate solutions by diluting with the diluent containing IS and were used to prepare the matrix calibration standards and QC samples. The ranges for these spiking solutions were 10–1,000 and 100–5,000 ng/mL for spiking in blood and mammary gland, respectively. All standard solutions were stored sealed in amber glass bottles at ambient temperature when not in use.
Preparation of blood calibration standards
Blood calibration standards and QC samples were prepared by fortifying 100 μL of blank male SD rat blood with 50 μL of the appropriate spiking standard (containing IS) in a 2-mL vial to obtain blood AP concentration of 5, 10, 25, 50, 100, 250 and 500 ng/mL. QC samples were prepared at 10, 100 and 250 ng/mL. For matrix blanks, 50 μL of the IS diluent (250 ng/mL in 50:50 ethanol:saline) was substituted for the spiking solution. Solvent standards were prepared at the same concentrations in the same manner as the matrix standards, except 100 μL of water was used instead of blood. Each vial was sealed with a metal crimp-top cap and analyzed by HS GC--MS.
Secondary matrix evaluation in male and female HSD rat blood and male and female B6C3F1 mouse blood
Six replicate matrix samples containing AP were prepared at 10 ng/mL in each secondary matrix and analyzed using a primary (male SD rat blood) calibration curve. Six matrix blanks (no IS) and six method blanks (with IS) were also prepared in each secondary matrix to evaluate selectivity.
Preparation of mammary gland calibration standards
Mammary gland calibration standards and QC samples were prepared by fortifying  100 mg (exact weight recoded) of blank female SD rat mammary gland with 100 μL of the appropriate spiking standard (containing IS) in a 2-mL vial containing 18 2.3-mm stainless steel beads (BioSpec Products, Bartlesville OK). Final AP concentrations in mammary gland were 100, 250, 600, 1,250, 2,500 and 5,000 ng/g. QC samples were prepared at 250 and 2,500 ng/g. For method blanks, 100 μL of the IS diluent (500 ng/mL in 50:50 ethanol:saline) was substituted for the spiking solution. Solvent standards were prepared at the same concentrations in the same manner as the matrix standards, except 100 μL of water was used instead of mammary gland. Each vial was sealed with a metal crimp-top cap and the tissue was homogenized for 30 seconds for two cycles at 1,000 rpm using a Geno/Grinder 2010 (SPEX SamplePrep, Metuchen NJ) prior to analysis by HS GC--MS.
100 mg (exact weight recoded) of blank female SD rat mammary gland with 100 μL of the appropriate spiking standard (containing IS) in a 2-mL vial containing 18 2.3-mm stainless steel beads (BioSpec Products, Bartlesville OK). Final AP concentrations in mammary gland were 100, 250, 600, 1,250, 2,500 and 5,000 ng/g. QC samples were prepared at 250 and 2,500 ng/g. For method blanks, 100 μL of the IS diluent (500 ng/mL in 50:50 ethanol:saline) was substituted for the spiking solution. Solvent standards were prepared at the same concentrations in the same manner as the matrix standards, except 100 μL of water was used instead of mammary gland. Each vial was sealed with a metal crimp-top cap and the tissue was homogenized for 30 seconds for two cycles at 1,000 rpm using a Geno/Grinder 2010 (SPEX SamplePrep, Metuchen NJ) prior to analysis by HS GC--MS.
Secondary matrix evaluation in female HSD rat mammary gland and female B6C3F1 mouse mammary gland
Triplicate matrix samples containing AP were prepared at 250 and 2,500 ng/g in each secondary matrix and analyzed using a primary (female SD rat mammary gland) calibration curve. To evaluate selectivity, three method blanks were prepared in female HSD rat mammary gland. Due to limited sample availability, only one method blank was evaluated for female B6C3F1 mouse mammary gland.
Stability
Stability experiments were performed at two concentrations (10 and 250 ng/mL in male SD rat blood and 250 and 2,500 ng/g in female SD rat mammary gland) prepared in triplicate in the 2-mL sample vials, and the results were evaluated by comparing data to a fresh (day 0) sample. AP stability in SD blood was assessed after storage on the HS GC--MS autosampler overnight (autosampler stability) and at −80°C for up to 32 days (frozen matrix stability). AP stability in frozen SD mammary gland was assessed after storage at −80°C for up to 60 days.
Method extension
Extension of the method to quantitate samples with AP concentrations greater than the validated range was assessed by injecting lower volumes from HS to bring concentrations into the validated range. For blood, triplicate matrix standards were prepared at 1,500 ng/mL and analyzed using a 150-µL injection (instead of 500 µL). For mammary gland, triplicate matrix standards were prepared at 20,000 ng/g and analyzed using 10-, 20- and 50-µL injections (instead of 200-µL). An additional set of triplicate mammary gland standards prepared at 20,000 ng/g were analyzed using the standard 200-µL injection and quantitated by extrapolating beyond the standard 100–5,000 ng/g calibration curve.
HS GC--MS analysis and analyte quantitation
The GC--MS system consisted of a Hewlett Packard (HP) 6890 plus gas chromatograph (Agilent Technologies, Santa Clara, CA) with CTC Analytics CombiPal autosampler (Lake Elmo, MN) and HP 5973 mass selective detector. An Agilent DB-5MS column (30 m × 0.25 mm i.d., 0.25-µm-thick film) was used with helium as the carrier gas at a flow rate of 1.2 mL/min. A 500-µL (blood) or 200-µL (mammary gland) HS sample was injected (splitless) after a sample equilibration time of 10 min at 60°C. The injector and MS transfer line temperatures were 270 and 300°C, respectively. The GC oven was held at 40°C for 5 min, ramped to 75°C at 5°C/min and then to 150°C at 37.5°C/min and held for 1 min. The total run time was 15 min. The EI electron energy was 70 eV and ion source and quadrupole temperatures were both 150°C. Quantitation was achieved in selected ion monitoring (SIM) mode by monitoring ions m/z 136 (M+, AP) and 139 (2H3-AP); m/z 121 and 124 were used as qualifier ions. MSD Chemstation version E.02.02 (Agilent) was used for data acquisition and analysis.
Calibration curves were generated by plotting the peak area ratios of AP to IS as a function of analyte concentration. The regression model for AP was a linear weighted least squares algorithm with a weighting factor of 1/concentration squared (1/x2). The concentration of AP was calculated using the response ratio and the regression equation. Blood data are expressed as nanograms per milliliter, while mammary gland data are expressed as nanogram per gram.
Results and Discussion
Method development
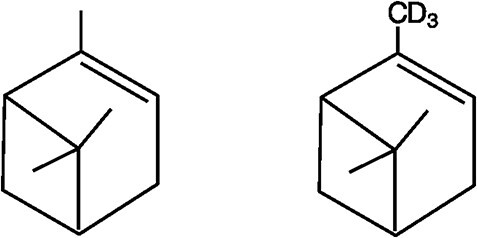
HS GC--MS parameters were carefully evaluated to optimize sensitivity for AP in ethanol. The EI spectra for AP and 2H3-AP are shown in Figure 2. Regardless of source temperature and voltage, fragment ion m/z 93 (C7H9+) was always the most abundant ion for AP. Because this ion is not unique to AP, the less abundant molecular ion (m/z 136) was chosen for SIM, and the source temperature and injection volume were optimized to improve sensitivity. An effort was also made using a triple quadrupole HS GC--MS-MS (Agilent G7000B) with multiple reaction monitoring (MRM) to improve sensitivity and selectivity, but it was difficult to find unique MRM transitions without interference, and sensitivity was not any better than with SIM using the single quadrupole GC--MS.
Figure 2.
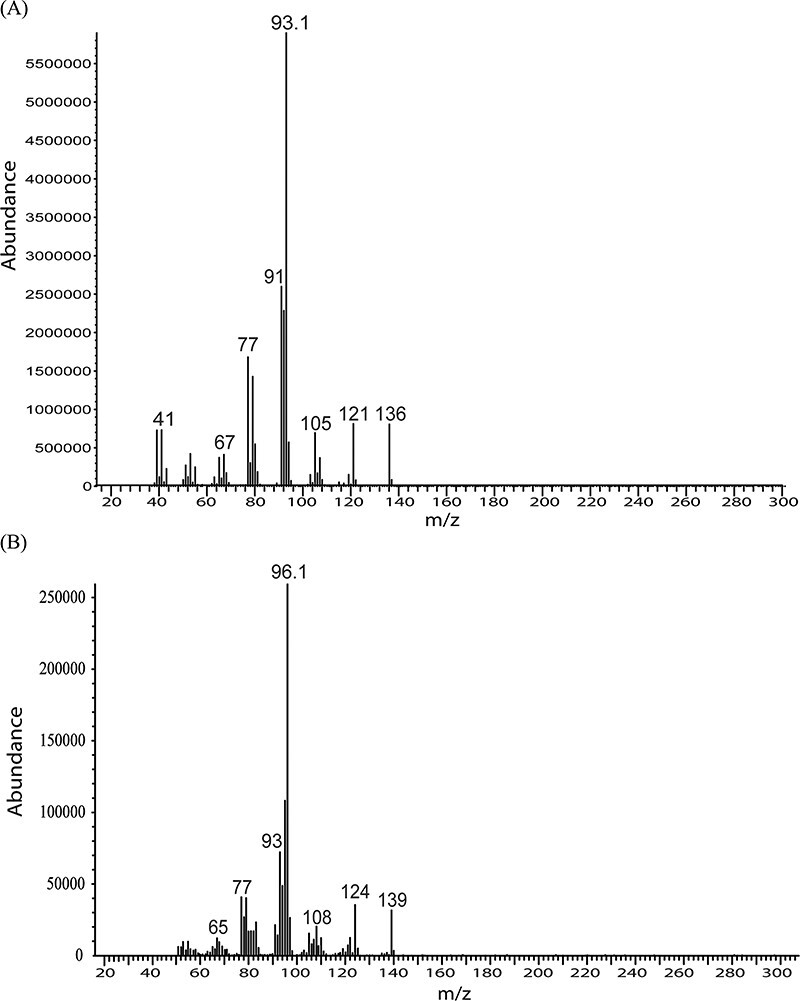
EI spectra for alpha pinene (top) and 2H3-alpha pinene (bottom).
HS volumes, spiking volumes and solvent compositions were also tested to determine their effect on AP sensitivity in matrix and in saline. It was found that the response for AP and 2H3-AP increased dramatically with increasing amounts of ethanol. For blood, fortification of a 100-μL aliquot with 50 µL AP in 50:50 ethanol:saline was found to give optimum AP response, and AP was found to be stable in this diluent. For mammary gland, the homogenization process was optimized using bead beating within the sealed HS vial such that the vial would not have to be opened prior to sampling. Different sizes and amounts of beads, ratios of liquid to matrix and speed and time of homogenization were tested. Ultimately, it was found that a 100-mg sample of mammary gland, fortified with 100 µL AP in 50:50 ethanol:saline, could be consistently homogenized using 18 2.3-mm stainless steel beads with 2 × 30-second cycles at 1,000 rpm.
Method validation
Small background peaks were consistently detected in the matrix and method blanks, but the responses were low and did not interfere with method performance. Representative chromatograms are presented in Figures 3 and 4 for blood and mammary, respectively. It should be noted that peak shapes were somewhat broad due to the large HS injections, but the peaks were consistent, easily integrated, and did not negatively impact the performance of the method. Method validation data are summarized in Table I for both blood and mammary in SD rat. The LOQ for AP was 5.00 ng/mL for blood and 100 ng/g for mammary gland; the LODs, estimated as 3X the standard deviation of LOQ, were 0.499 ng/mL in blood and 17.7 ng/g in mammary gland.
Figure 3.
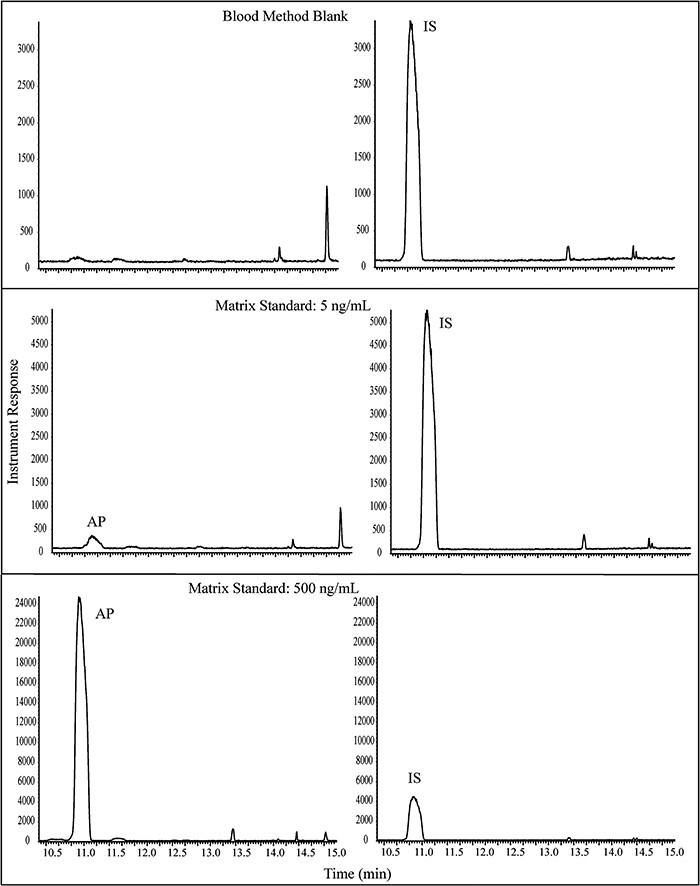
Representative GC--MS chromatograms for AP: blank rat blood with IS, 5 ng/mL AP in blood, and 500 ng/mL AP in blood. Panes on the left are SIM for m/z 136 (AP); panes on the right are SIM for m/z 139 (IS).
Figure 4.
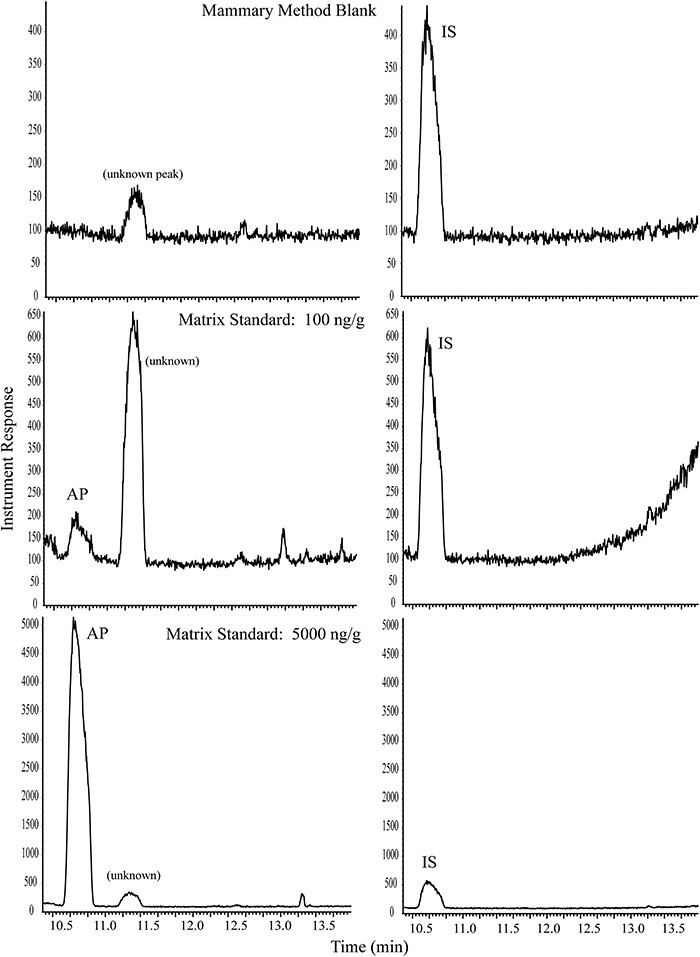
Representative GC--MS chromatograms for AP: blank rat mammary gland with IS, 100 ng/g AP in mammary gland, and 5,000 ng/g AP in mammary gland. Panes on the left are SIM for m/z 136 (AP); panes on the right are SIM for m/z 139 (IS).
Table I.
Method Validation Data for AP in Blood and Mammary Gland
| Validation parameter | Blood | Mammary |
|---|---|---|
| Matrix concentration range (ng/mL or ng/g)a | 5.00–500 | 100–5,000 |
| LODb (ng/mL or ng/g) | 0.499 | 17.7 |
| LOQc (ng/mL or ng/g) | 5.00 | 100 |
| Correlation coefficient (r) | ≥0.99 | ≥0.99 |
| Absolute recoveryd (%) | 67.1–79.0 | 1.26–3.70 |
| Relative recoverye (%) | 96.7–101 | 88.7–102 |
| Precision and accuracyf | ||
| Intraday precision (%RSD)g | ≤0.78 | ≤2.0 |
| Intraday accuracy (Mean %RE)h | ≤±13.4 | ≤±6.3 |
| Interday precision (%RSD) | ≤7.1 | ≤5.7 |
| Interday accuracy (Mean %RE) | ≤±9.6 | ≤±0.9 |
| Method extension (up to 1,500 ng/mL or 20,000 ng/g)i | ||
| Precision (%RSD) | 0.32 | 16.0 |
| Accuracy (mean %RE) | 18.7 | ≤±6.5 |
| Secondary matrix evaluation (HSD rat and B6C3F1 mouse blood and mammary)j | ||
| Male HSD precision (%RSD) | 2.4 | N/A |
| Male HSD accuracy (mean %RE) | 0.2 | N/A |
| Female HSD precision (%RSD) | 4.3 | ≤2.1 |
| Female HSD accuracy (mean %RE) | −1.2 | ≤±3.8 |
| Male B6C3F1 precision (%RSD) | 2.4 | N/A |
| Male B6C3F1 accuracy (mean %RE) | −3.9 | N/A |
| Female B6C3F1 precision (%RSD) | 1.7 | ≤2.0 |
| Female B6C3F1 accuracy (mean %RE) | −9.2 | ≤±3.2 |
Full validation was performed in SD rat blood and mammary gland, with matrix evaluation in HSD rat and B6C3F1 mouse blood and mammary.
Blood data are expressed as ng/mL; mammary data are expressed as ng/g.
LOD = limit of detection.
LOQ = lower limit of quantitation; lowest standard at which RE ≤ ±20% and RSD ≤ 20% for n = 6.
Absolute recovery expressed as percentage of AP peak area in matrix to AP peak area in solvent.
Relative recovery expressed as percentage of response ratio (AP:IS) in matrix to that in solvent.
Precision and accuracy determined for QCs at three levels in blood: 10, 100 and 250 ng/mL, and two levels in mammary gland: 250 and 2,500 ng/g. n = 3 for intraday; n = 9 for interday.
%RSD = percent relative deviation.
%RE = percent relative error.
Precision and accuracy determined for triplicate QCs at 1,500 ng/mL (blood) and 20,000 ng/g (mammary gland) analyzed using smaller injection volumes (150 µL for blood and 10–50 µL for mammary).
Precision and accuracy for secondary matrices determined for six replicate QCs at 10 ng/mL for blood and triplicate QCs at 250 and 2,500 ng/g for mammary gland.
N/A = not applicable (only female tissue assessed).
The calibration curves in blood and mammary gland were linear with correlation coefficients r > 0.99. Precision, estimated as RSD, and accuracy, estimated as RE, were determined for QC samples at three levels in blood (10, 100 and 250 ng/mL) and two levels in mammary gland (250 and 2,500 ng/g). Intra-day (n = 3) and inter-day (n = 9, across 3 days) precisions were less than 7.1% in each matrix, and accuracies were within ±13.4%. Recovery was determined at all calibration levels in each matrix both with and without IS. The mean absolute recovery (without IS) was 74% for blood, but only 2% for mammary gland, likely due to high lipophilicity of AP. However, sensitivity was adequate, and mean recoveries incorporating the IS were 99.1% in blood and 93.1% in mammary gland. For blood, a set of matrix standards was analyzed at the beginning of an analytical batch, and another at the end, to assess instrument drift. The relative difference between the determined concentrations for the second set compared to the calibration set was within ±8.0%, indicating no instrument drift.
Blood samples prepared at 1,500 ng/mL and injected using a 150-µL injection (instead of 500 µL) gave an average back-calculated value of 1,780 ng/mL (18.7% RE, 0.32% RSD), demonstrating the ability to accurately quantitate samples containing higher concentrations of AP than the validated calibration range. Likewise, mammary gland samples prepared at a concentration higher than the validated range (20,000 ng/g) could be accurately quantitated by injecting a smaller volume (as low as 10 µL) or by extrapolating beyond the validated range; average back-calculated values were within ±6.5% RE, ≤16.0% RSD.
Matrix evaluation in HSD rat and B6C3F1 mouse blood and mammary gland
Successful matrix evaluations were achieved for AP in male and female HSD rat blood, male and female B6C3F1 mouse blood, female HSD rat mammary gland and female B6C3F1 mouse mammary gland. Blood samples were prepared at 10 ng/mL (n = 6 on a single day) and quantitated against the primary (male SD rat blood) calibration curve; RE and RSD were ≤±9.2% and ≤4.3% RSD, respectively. Mammary gland samples were prepared at 250 and 2,500 ng/g (n = 3 on a single day) and quantitated against the primary (female SD rat mammary gland) calibration curve; RE and RSD were ≤±3.8% and ≤2.1%, respectively. No significant or interfering peaks were detected in any of the blood or mammary gland control blanks, demonstrating that the method was selective in these secondary matrices.
Stability
Studies were performed to evaluate both post-preparative (autosampler) and frozen storage stability of AP in biological matrix. Stability samples were prepared in triplicate at two concentrations (10 and 250 ng/mL in blood and 250 and 2,500 ng/g in mammary gland) in the 2-mL HS vials (containing beads in the case of mammary samples), diluted with IS and sealed. The mammary samples were homogenized as described previously. Autosampler stability samples were equilibrated for 10 min at 60°C but then left on the HS GC--MS autosampler overnight before analysis the next day. Frozen matrix stability samples were stored at −80°C until analysis; on analysis day they were thawed, equilibrated for 10 min at 60°C and analyzed by HS GC--MS. The determined concentrations of the stability samples were compared to those of freshly prepared samples (day 0), and data are presented in Table II. Loss of AP occurred during overnight autosampler storage as well as frozen (−80°C) storage in as few as 15 days, but incorporation of the IS prior to storage corrected for the loss such that calculated concentrations were within 98.9–99.4% of day 0 concentrations following overnight autosampler storage in blood, and 84.7–117% of day 0 concentrations following frozen storage up to 32 days in blood and 60 days in mammary gland. It is therefore critical that the labeled IS be added to the samples upon collection to correct for the loss due to volatilization.
Table II.
Matrix Stability Data for AP in Blood and Mammary Gland
| Mean % of day 0 (% RSD) | ||
|---|---|---|
| Stability endpointa | Blood | Mammary |
| Autosampler (overnight) | 98.9–99.4 (%RSD ≤ 0.93%) | N/A |
| Frozen matrix (−80°C, at least 32 d)b | 105–117 (%RSD ≤ 5.4%) | 84.7–110 (%RSD ≤ 7.0%) |
Autosampler and frozen matrix stability determined for triplicate QCs at two levels: 10 and 250 ng/mL (blood) and 250 and 2,500 ng/g (mammary gland).
Frozen matrix stability was assessed after storage for 32 days for blood, and 60 days for mammary gland.
Conclusion
A method was developed and validated to quantitate AP in rat and mouse blood and mammary gland using a simple HS GC--MS method. The quantitation ranges are 5–500 ng/mL for blood and 100–5,000 ng/g for mammary gland. In addition, samples above the upper limits of quantitation can be analyzed using a lower injection volume or by extrapolating beyond the curve. Loss of AP occurred during overnight autosampler storage as well as frozen (−80°C) storage in as few as 15 days, but incorporation of the IS prior to storage corrected for the loss. It is therefore critical that the labeled IS be added to the samples upon collection to correct for the loss due to volatilization. The method is suitable for determination of AP in rodent blood and mammary glands in support of toxicokinetic and toxicology studies.
Acknowledgment
The authors are grateful to Drs Esra Mutlu and Pei-Li Yao for their review of this manuscript.
Contributor Information
Melanie A Rehder Silinski, Discovery Sciences Unit, RTI International, P.O. Box 12194, Research Triangle Park, Durham, NC 27709, USA.
Joseph Licause, Discovery Sciences Unit, RTI International, P.O. Box 12194, Research Triangle Park, Durham, NC 27709, USA.
Teruyo Uenoyama, Discovery Sciences Unit, RTI International, P.O. Box 12194, Research Triangle Park, Durham, NC 27709, USA.
James C Blake, Discovery Sciences Unit, RTI International, P.O. Box 12194, Research Triangle Park, Durham, NC 27709, USA.
Reshan A Fernando, Discovery Sciences Unit, RTI International, P.O. Box 12194, Research Triangle Park, Durham, NC 27709, USA.
Veronica G Robinson, Division of the National Toxicology Program, NIEHS, P.O. Box 12233, Research Triangle Park, Durham, NC 27709, USA.
Suramya Waidyanatha, Division of the National Toxicology Program, NIEHS, P.O. Box 12233, Research Triangle Park, Durham, NC 27709, USA.
Funding
This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, Intramural Research project ZIA ES103316-04, and performed for the NTP, National Institute of Environmental Health Sciences, National Institutes of Health, U.S. Department of Health and Human Services, under contract HHSN273201400022C (RTI International, RTP, NC).
Conflict of Interests
The authors declare that they have no conflict of interest.
References
- 1. National Toxicology Program . (2016) NTP Technical Report on the toxicity studies of α-pinene (CASRN 80-56-8) administered by inhalation to F344/N rats and B6C3F1/N mice. NTP TOX 81, National Toxicology Program, Public Health Service, U.S. Department of Health and Human Service. ISSN: 2378-8992, May 2016. https://ntp.niehs.nih.gov/ntp/htdocs/st_rpts/tox081_508.pdf?utm_source=direct&utm_medium=prod&utm_campaign=ntpgolinks&utm_term=tox081 accessed Apr 16, 2020).
- 2. Filipsson A.F. (1996) Short term inhalation exposure to turpentine: toxicokinetics and acute effects in men. Occupational and Environmental Medicine, 53, 100–105. doi: 10.1136/oem.53.2.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Falk A.A., Hagberg M.T., Lof A.E., Wigaeus-Hjelm E.M., Zhiping W. (1990) Uptake, distribution and elimination of α-pinene in man after exposure by inhalation. Scandinavian Journal of Work, Environment & Health, 16, 372–378. doi: 10.5271/sjweh.1771 [DOI] [PubMed] [Google Scholar]
- 4. Eriksson K., Levin J. (1990) Identification of cis- and trans-verbenol in human urine after occupational exposure to terpenes. International Archives of Occupational and Environmental Health, 62, 379–383. doi: 10.1007/BF00381368 [DOI] [PubMed] [Google Scholar]
- 5. Hedenstierna G., Alexandersson R., Wimander K., Rosén G. (1983) Exposure to terpenes: effects on pulmonary function. International Archives of Occupational and Environmental Health, 51, 191–198. doi: 10.1007/BF00377751 [DOI] [PubMed] [Google Scholar]
- 6. Falk A., Gullstrand E., Löf A., Wigaeus-Hjelm E. (1990) Liquid/air partition coefficients of four terpenes. British Journal of Industrial Medicine, 47, 62–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kauppinen T.P., Partanen T.J., Nurminen M.M., Nickels J.I., Hernberg S.G., Hakulinen T.R., et al. (1986) Respiratory cancers and chemical exposures in the wood industry: a nested case-control study. British Journal of Industrial Medicine, 43, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schrader W., Döring S., Joppek W. (2004) Mass spectrometric studies of DNA adducts from a reaction with terpenoids. Angewandte Chemie International Edition, 43, 6657–6660. doi: 10.1002/anie.200461022 [DOI] [PubMed] [Google Scholar]
- 9. National Toxicology Program . (2020) Testing status of alpha-pinene M030014. National Toxicology Program, U.S. Department of Health and Human Service. https://ntp.niehs.nih.gov/whatwestudy/testpgm/status/ts-m030014.html?utm_source=direct&utm_medium=prod&utm_campaign=ntpgolinks&utm_term=ts-m030014 (accessed Aug 24, 2020).
- 10. Nezhadali A., Zarrabi Shirvan B. (2010) Separation, identification and determination of volatile compounds of Ziziphora persica Bunge using HS-SPME/GC-MS. International Journal of Environmental Science and Developoment, 1, 115–118. doi: 10.7763/IJESD.2010.V1.23 [DOI] [Google Scholar]
- 11. Eldeen E.M.S., Etman M.A., Aboul-Enein H.Y. (2012) Validation of a rapid gas chromatographic method for determination of seven volatile compounds in a urinary tract antiseptic soft gelatin capsules. Gazi University Journal of Science, 25, 623–629. [Google Scholar]
- 12. Esfahanizadeh M., Ayatollahi S.A., Goodarzi A., Bayat M., Ata A., Kobarfard F. (2018) Development and validation of a GC/MS method for simultaneous determination of 7 monoterpens in two commercial pharmaceutical dosage forms. Iranian Journal of Pharmaceutical Research, 17, 24–32. [PMC free article] [PubMed] [Google Scholar]
- 13. Sumitomo K., Akutsu H., Fukuyama S., Minoshima A., Kukita S., Yamamura Y., et al. (2015) Conifer-derived monoterpenes and forest walking. Mass Spectrometry, 4, 1–4. doi: 10.5702/massspectrometry.A0042 [DOI] [PMC free article] [PubMed] [Google Scholar]