

Abstract
Streptococcus pneumoniae is a notorious pathogen that affects ∼450 million people worldwide and causes up to four million deaths per annum. Despite availability of antibiotics (i.e., penicillin, doxycycline, or clarithromycin) and conjugate vaccines (e.g., PCVs), it is still challenging to treat because of its drug resistance ability. The rise of antibiotic resistance in S. pneumoniae is a major source of concern across the world. Computational subtractive genomics is one of the most applied techniques in which the whole proteome of the bacterial pathogen is gradually reduced to a limited number of potential therapeutic targets. Whole-genome sequencing has greatly reduced the time required and provides more opportunities for drug target identification. The goal of this work is to evaluate and analyze metabolic pathways in serotype 14 of S. pneumonia to identify potential drug targets. In the present study, 47 potent drug targets were identified against S. pneumonia by employing the computational subtractive genomics approach. Among these, two proteins are prioritized (i.e., 4-oxalocrotonate tautomerase and Sensor histidine kinase uniquely present in S. pneumonia) as novel drug targets and selected for further structure-based studies. The identified proteins may provide a platform for the discovery of a lead drug candidate that may be capable of inhibiting these proteins and, therefore, could be helpful in minimizing the associated risk related to the drug-resistant S. pneumoniae. Finally, these enzymatic proteins could be of prime interest against S. pneumoniae to design rational targeted therapy.
Keywords: Streptococcus pneumoniae, subtractive genomics, serotype 14, metabolic pathways, 4-oxalocrotonate tautomerase and sensor histidine kinase
Graphical Abstract
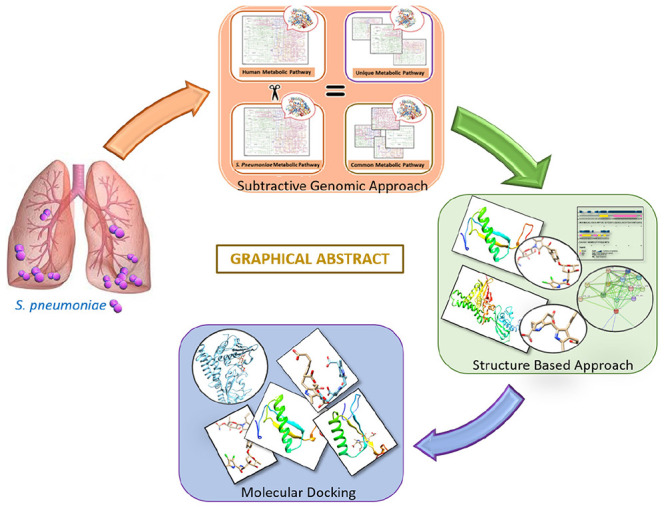
Introduction
Streptococcus pneumoniae is a gram-positive, spherical, alpha-beta hemolytic, and facultatively anaerobic bacterium. It is classified primarily as one of the major pathogenic species resulting in high mortality and morbidity rate. It is responsible for causing meningitis, otitis media, septicemia, bacteremia, and community-acquired pneumonia, otherwise called Invasive Pneumococcal Disease (IPD) (Lo et al., 2019). Nearly ∼600,000 children, ∼200,000 elders, and immuno-compromised individuals die due to pneumococcal diseases caused by streptococci annually (Watt et al., 2009). Surprisingly, in 2019, >600,000 deaths were reported due to pneumonia as well as >385,000 deaths during the COVID-19 pandemic (Elflein, 2021). Antibiotics are used as the most common anti-infection therapy (e.g., beta-lactam antibiotics) for the treatment of pneumonia. Unfortunately, the increasing resistance against common antibiotics as well as the emergence of Multiple Drug Resistant strains (MDRs) worldwide makes the management and treatment of pneumococcal infections highly difficult (Hakenbeck et al., 2012). Surveillance of serotypes and prevalence of drug-resistant strains in the general population is critical to create appropriate prevention and treatment protocol for S. pneumoniae (Ediriweera et al., 2016).
Evidently, the serotype 14 is most frequently responsible for Invasive Pneumococcal Disease (IPD) (Geno et al., 2015) among the 101 defined S. pneumoniae serotypes. The conjugated pneumococcal vaccines are developed against S. pneumoniae infections based on the polysaccharide capsular serotypes (Chiba et al., 2014). Specifically, Polysaccharide Pneumococcal Vaccine (PPV) 23-valent was developed for serotype 14 to cure IPD. Unfortunately, PPSV23 led to poor immunogenicity for pneumococci (Cilloniz et al., 2016). Currently, there is Pneumococcal Polysaccharide Vaccine (PPSV23), 10-valent Pneumococcal Conjugate Vaccine (PCV10), 7-valent Pneumococcal Conjugate Vaccine (PCV7), and 13-valent Pneumococcal Conjugate Vaccine (PCV13) in use. Even after the distribution of multi-valent Pneumococcal Conjugate Vaccine (PCV7), the percentage of serotype 14 infection has increased over time due to the increase in drug resistance (Bouskraoui et al., 2011).
Experimentally, molecular serotyping such as MultiLocus Sequence Typing (MLST) and Pulse-Field Gel Electrophoresis (PFGE) are the gold standard methods used to study the outbreaks and identification of pneumococcal isolates (Enright and Spratt, 1998), but with high associated cost. Thus, accurate determination of the serotypes remained a challenge (Hu et al., 2014). It is therefore imperative that s novel therapeutic drug target is identified against S. pneumoniae. The discovery of a new drug target may lead to better therapeutics (Lodha et al., 2013). Fortunately, the arrival of the post-genomic era and whole-genome sequencing of the pathogens opened up new avenues, such as comparative subtractive genomics, to design new drugs and vaccine candidates. Computational approaches make it possible to identify potential drug targets against such pathogens (Fair and Tor, 2014).
Certain gaps from previous studies against S. pneumoniae, such as metabolic pathways coverage, consideration of hub nodes, and conserved drug targets of bacterial pneumonia (Henriques et al., 2000), are covered in this study. The current study includes comparative and subtractive genomics analysis, subcellular localization, Protein-Protein Interaction (PPI) network analysis, essentiality and druggability of the target proteins, and metabolic pathway analysis. Furthermore, the study proposed that antibacterial lead compounds could be developed against the shortlisted potential drug targets.
Materials and Methods
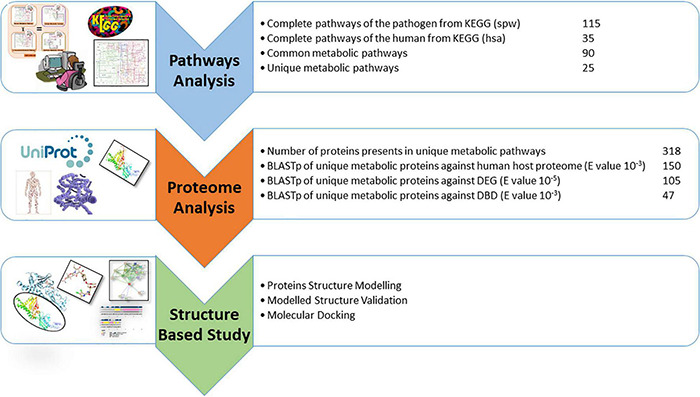
The drug target prioritization and identification against S. pneumoniae was performed by employing a subtractive genomics approach along with the analysis of the metabolic pathways. Various databases and tools, as illustrated in the flow chart (Figure 1), were used for the determination of therapeutic targets against S. pneumoniae.
FIGURE 1.

Flow chart: A general sketch of the current study integrated with the use of various computational approaches and tools to identify potential drug targets candidates.
Data Collection of Proteome and Metabolic Pathways
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000) was used for the metabolic pathways retrieval of the pathogen (S. pneumoniae, i.e., spw01100) and host (Homo sapiens, i.e., hsa01100). The human and S. pneumoniae proteomes were retrieved from the Universal Protein Resource (UniProt) database (Apweiler et al., 2021) with accession numbers UP000005640 and UP000001682 (strain CGSP14), respectively. The Database of Essential Genes (DEG database) (Luo et al., 2021) was used to investigate the essentiality of the drug targets and the DrugBank database (Wishart et al., 2018) was used to assess the druggability of the shortlisted targets.
Subtractive Genomics Approach
In the current study, a “Subtractive Genomic Approach,” which is one of the most commonly applied methodologies, was applied. Subtractive Genomics is a powerful approach that applies the sequence subtraction between the host, the pathogen proteomes, and metabolic pathways. It provides necessary information on a set of proteins essential for the microorganism that do not exist in the respective host. Subtractive Genomics plays a role of great importance in potential drug target identification that is unique and essential to the pathogen survival without altering the host’s (human) systematic metabolic pathways (Fenoll et al., 2009).
Host and Pathogen Metabolic Pathway Analysis
As mentioned above, the KEGG database (Kanehisa and Goto, 2000) was used for the genome-wide pathways analysis of S. pneumoniae and human. A standalone comparison was performed between the host and pathogen to identify unique and commonly found metabolic pathways. Unique pathways are classified as those pathways that are present only in the pathogen, while common pathways are those pathways that are present in both organisms (i.e., pathogen and host) (Linares et al., 2010). In the present study, the common metabolic pathways proteins were discarded while the protein sequences of unique metabolic pathways were retrieved from the UniProt for further downstream study.
Non-homologous Protein Identification
Accordingly, the protein sequences retrieved from the unique metabolic pathways were subjected to BLASTp (with E-value 10-3) against Homo sapiens. The BLASTp resulted in “Hits” (Homologous sequences between host and pathogen) and “No-Hits” (Non-homologous sequences). For further analysis, the non-homologous sequences that showed no similarity with the human host were selected.
Essential Non-homologous Genes Identification
Proteins that play a major role in cellular metabolism are said to be essential for any organism’s survival (Deng et al., 2011). Thus, to shortlist proteins essential to pathogen’s survival, BLASTp of non-homolog S. pneumoniae proteins was performed against DEG with cut-off parameters of E value 10–5. Consequently, the obtained Hits protein sequences (homologous proteins) were used for further analysis while “No-Hits” (non-homologous proteins) protein sequences were discarded.
Druggability of Essential Protein
Furthermore, the essential non-homologous proteins were assessed through BLASTp with an E value 10–3 against the DrugBank database to determine the drug–target-like ability of shortlisted proteins to finally identify novel drug targets against S. pneumoniae. Similarly, the identified Hits protein sequences were retrieved and analyzed for the prioritization of a potent drug target while obtained “No-Hits” proteins sequences were discarded.
Sub-Cellular Location Prediction
The subcellular location of final shortlisted drug-like protein targets was determined by using PSORTb v.3.0. tool (Yu et al., 2010). The PSORTb predicts results based on the sub-cellular localization, i.e., cytoplasmic membrane, cell wall, cytoplasmic, extracellular, and unknown.
Protein Structure Prediction, Validation, and Conservancy Analysis
The structures from the proteins shortlisted from the above approach were searched for in PDB. The suitable template for the protein structure modeling was found through BLASTp against the PDB. It resulted in the shortlisting of various proteins with different query coverage and percent identity that can be further filtered to select a suitable template for protein structure modeling. Eventually, the 3D structures of shortlisted drug targets were then modeled by using the Homology modeler tool. Based on DOPE score, the best optimized structure from five modeled structures was selected. In order to perform the docking experiment against shortlisted proteins, PSIPRED (Buchan and Jones, 2019), ProSA-web (Wiederstein and Sippl, 2007), and PROCHECK (Laskowski et al., 2006) were applied for the model structure evaluation based on secondary structure analysis, error in 3D model identification, and tertiary structure stereochemistry analysis, respectively.
The shortlisted proteins were further analyzed to investigate the conservancy of these proteins in the S. pneumoniae strains. Therefore, BLASTp of shortlisted proteins was performed and resultant matched sequences were retrieved. These sequences were further aligned through Clustal Omega tool to study conservation among these proteins from other serotypes of S. pneumoniae.
Active Site Prediction
As the structure was modeled, there is a need to find the active site where a ligand could bind to alter its function. The DogSite Scorer1 was used to find the possible binding sites of the modeled protein. The DogSite Scorer identifies active site pockets based on the physico-chemical properties of the protein residues. These active sites can be selected to dock ligand against respective proteins.
Network Topology Studies
Furthermore, to identify whether these proteins can be a hub protein and validate their functional interactions, the PPIs network of pathogen proteins as potential drug targets were generated through the STRING database (Szklarczyk et al., 2021). The STRING is a database of experimentally known and predicted PPIs, including direct (physical) and indirect (functional) network. Node degrees and clustering coefficient are used to classify these PPIs as hub proteins.
Ligand Prediction
As the protein structure was not available, the ligand information was also missing. The ProBis server (Konc et al., 2015) was used to find the probable ligands and also used for the assessment of the fundamental interaction between the ligand and drug targets. The ProBis predicts the best possible ligand for proteins through molecular simulation based on a theoretical approach. On the basis of the proposed ligand, one may design new drugs in further drug discovery stages.
Molecular Docking Studies
In molecular docking, the most effective ligand shows the minimal score of docking for its target proteins. The proteins with the modeled structure were used as target proteins while identified ligands were docked. The ligand was docked following the standard docking parameters of 250 times Lamarckian GA settings resulting in 27,000 generations (Tanchuk et al., 2016). The MOE software was used to investigate the docked ligand-protein interactions, and to visualize the hydrogen bond and hydrophobic interactions of the ligand with docked protein within the range of 5 Å.
Results
Identification of Metabolic Pathways in Streptococcus pneumoniae
Currently, the KEGG database has 115 metabolic pathways for S. pneumoniae serotype 14 and 375 for human, as shown in Table 1. Unique and common metabolic pathways among pathogen and host (human) were identified through standalone BLASTp. Consequently, 25 unique pathways are identified for the pathogen that are crucial for the persistence of S. pneumoniae (Table 2) with 90 pathways commonly present in both organisms (Table 3).
TABLE 1.
Steps involved in the current study: Subtractive filtering of proteins and metabolic pathways against S. pneumoniae.
| S. no | Steps involved in the current study | Streptococcus pneumoniae |
| 1 | Complete pathways of the pathogen from KEGG (spw) | 115 |
| 2 | Complete pathways of the human from KEGG (hsa) | 375 |
| 3 | Common metabolic pathways | 90 |
| 4 | Unique metabolic pathways | 25 |
| 5 | Number of proteins presents in unique metabolic pathways | 318 |
| 6 | Removal of redundant KEGG IDs | 207 |
| 7 | BLASTp of unique metabolic proteins against human host proteome (E value 10–3) | 150 |
| 8 | BLASTp of unique metabolic proteins against DEG (E value 10–5) | 105 |
| 9 | BLASTp of unique metabolic proteins against DBD (E value 10–3) | 47 |
TABLE 2.
Unique metabolic pathways: List of all unique metabolic pathways and uniquely present proteins in these metabolic pathways present in S. pneumoniae.
| S. no | Metabolic pathways | Pathway Ids | Proteins in the pathway |
| 1 | Aminobenzoate degradation | spw00627 | 2 |
| 2 | Antimicrobial resistance genes | spw01504 | 17 |
| 3 | Bacterial secretion system | spw03070 | 13 |
| 4 | Benzoate degradation | spw00362 | 2 |
| 5 | C5-Branched dibasic acid metabolism | spw00660 | 5 |
| 6 | Carbapenem biosynthesis | spw00332 | 2 |
| 7 | Cationic antimicrobial peptide (CAMP) resistance | spw01503 | 6 |
| 8 | Chloroalkane and chloroalkene degradation | spw00625 | 2 |
| 9 | Cyanoamino acid metabolism | spw00460 | 4 |
| 10 | D-Alanine metabolism | spw00473 | 4 |
| 11 | Lysine biosynthesis | spw00300 | 12 |
| 12 | Methane metabolism | spw00680 | 9 |
| 13 | Monobactam biosynthesis | spw00261 | 4 |
| 14 | Naphthalene degradation | spw00626 | 2 |
| 15 | Peptidoglycan biosynthesis | spw00550 | 25 |
| 16 | Peptidoglycan biosynthesis and degradation protein | spw01011 | 24 |
| 17 | Phosphotransferase system | spw02060 | 45 |
| 18 | Photosynthesis proteins | spw00194 | 8 |
| 19 | Quorum sensing | spw02024 | 52 |
| 20 | Streptomycin biosynthesis | spw00521 | 3 |
| 21 | Two-component system | spw02022 | 37 |
| 22 | Two-component system | spw02020 | 16 |
| 23 | Vancomycin resistance | spw01502 | 6 |
| 24 | Xylene degradation | spw00622 | 2 |
| 25 | beta-Lactam resistance | spw01501 | 16 |
TABLE 3.
Common metabolic pathways: List of all common metabolic pathways, commonly shared by both organisms (S. pneumoniae and Human).
| S. no | Metabolic pathways | Pathway ids | Protein in pathways |
| 1 | ABC transporters | pw02010 | 97 |
| 2 | Alanine, aspartate, and glutamate metabolism | spw00250 | 17 |
| 3 | Amino acid related enzymes | spw01007 | 27 |
| 4 | Amino sugar and nucleotide sugar metabolism | spw00520 | 35 |
| 5 | Aminoacyl-tRNA biosynthesis | spw00970 | 84 |
| 6 | Arachidonic acid metabolism | spw00590 | 1 |
| 7 | Arginine and proline metabolism | spw00330 | 7 |
| 8 | Arginine biosynthesis | spw00220 | 7 |
| 9 | Ascorbate and aldarate metabolism | spw00053 | 11 |
| 10 | Bacterial toxins | spw02042 | 4 |
| 11 | Base excision repair | spw03410 | 9 |
| 12 | Biotin metabolism | spw00780 | 5 |
| 13 | Butanoate metabolism | spw00650 | 7 |
| 14 | CD molecules | spw04090 | 1 |
| 15 | Chaperones and folding catalysts | spw03110 | 18 |
| 16 | Chloroalkane and chloroalkene degradation | spw00625 | 2 |
| 17 | Chromosome and associated proteins | spw03036 | 29 |
| 18 | Citrate cycle (TCA cycle) | spw00020 | 4 |
| 19 | Cysteine and methionine metabolism | spw00270 | 21 |
| 20 | Cytoskeleton proteins | spw04812 | 3 |
| 21 | D-Glutamine and D-glutamate metabolism | spw00471 | 3 |
| 22 | DNA repair and recombination proteins | spw03400 | 67 |
| 23 | DNA replication | spw03030 | 15 |
| 24 | DNA replication proteins | spw03032 | 25 |
| 25 | Enzymes | spw01000 | 1 |
| 26 | Exosome | spw04147 | 26 |
| 27 | Fatty acid biosynthesis | spw00061 | 15 |
| 28 | Folate biosynthesis | spw00790 | 9 |
| 29 | Fructose and mannose metabolism | spw00051 | 20 |
| 30 | Galactose metabolism | spw00052 | 27 |
| 31 | Glutathione metabolism | spw00480 | 8 |
| 32 | Glycerolipid metabolism | spw00052 | 10 |
| 33 | Glycerophospholipid metabolism | spw00564 | 8 |
| 34 | Glycine, serine, and threonine metabolism | spw00260 | 16 |
| 35 | Glycolysis/Gluconeogenesis | spw00010 | 26 |
| 36 | Glycosyltransferases | spw01003 | 6 |
| 37 | Glyoxylate and dicarboxylate metabolism | spw00630 | 7 |
| 38 | Homologous recombination | spw03440 | 21 |
| 39 | Inositol phosphate metabolism | spw00562 | 4 |
| 40 | Ion channels | spw04040 | 1 |
| 41 | Lipid biosynthesis proteins | spw01004 | 12 |
| 42 | Lysine degradation | spw00310 | 2 |
| 43 | Membrane trafficking | spw04131 | 5 |
| 44 | Messenger RNA biogenesis | spw03019 | 14 |
| 45 | Mismatch repair | spw03430 | 18 |
| 46 | Mitochondrial biogenesis | spw03029 | 27 |
| 47 | Nicotinate and nicotinamide metabolism | spw00760 | 8 |
| 48 | Nitrogen metabolism | spw00910 | 4 |
| 49 | Non-coding RNAs | spw03100 | 74 |
| 50 | Nucleotide excision repair | spw03420 | 8 |
| 51 | One carbon pool by folate | spw00670 | 10 |
| 52 | Other glycan degradation | spw00511 | 10 |
| 53 | Oxidative phosphorylation | spw00190 | 18 |
| 54 | Pantothenate and CoA biosynthesis | spw00770 | 11 |
| 55 | Pentose and glucuronate interconversions | spw00040 | 9 |
| 56 | Pentose phosphate pathway | spw00030 | 19 |
| 57 | Peptidases and inhibitors | spw01002 | 38 |
| 58 | Phenylalanine, tyrosine, and tryptophan biosynthesis | spw00400 | 18 |
| 59 | Porphyrin and chlorophyll metabolism | spw00860 | 1 |
| 60 | Prenyltransferases | spw01006 | 3 |
| 61 | Prokaryotic defense system | spw02048 | 25 |
| 62 | Propanoate metabolism | spw00640 | 12 |
| 63 | Protein export | spw03060 | 14 |
| 64 | Protein kinases | spw01001 | 10 |
| 65 | Protein phosphatases and associated proteins | spw01009 | 3 |
| 66 | Purine metabolism | spw00230 | 36 |
| 67 | Pyrimidine metabolism | spw00240 | 30 |
| 68 | Pyruvate metabolism | spw00620 | 22 |
| 69 | RNA degradation | spw03018 | 10 |
| 70 | RNA polymerase | spw03020 | 6 |
| 71 | Riboflavin metabolism | spw00740 | 6 |
| 72 | Ribosome | spw03011 | 63 |
| 73 | Ribosome biogenesis | spw03009 | 52 |
| 74 | Secretion system | spw02044 | 13 |
| 75 | Selenocompound metabolism | spw00450 | 7 |
| 76 | Sphingolipid metabolism | spw00600 | 6 |
| 77 | Starch and sucrose metabolism | spw00500 | 40 |
| 78 | Sulfur metabolism | spw00920 | 5 |
| 79 | Sulfur relay system | spw04122 | 5 |
| 80 | Taurine and hypotaurine metabolism | spw00430 | 4 |
| 81 | Terpenoid backbone biosynthesis | spw00900 | 8 |
| 82 | Thiamine metabolism | spw00730 | 13 |
| 83 | Transcription factors | spw03000 | 41 |
| 84 | Transcription machinery | spw03021 | 11 |
| 85 | Transfer RNA biogenesis | spw03016 | 52 |
| 86 | Translation factors | spw03012 | 14 |
| 87 | Transporters | spw02000 | 264 |
| 88 | Valine, leucine, and isoleucine biosynthesis | spw00290 | 11 |
| 89 | Valine, leucine, and isoleucine degradation | spw00280 | 3 |
| 90 | Vitamin B6 metabolism | spw00750 | 4 |
Metabolic Pathway Analysis
Furthermore, BLASTp of proteins that are uniquely present in the shortlisted unique metabolic pathways was performed against the human proteome to encounter further cross-reactivity of drug-like compounds with the host protein. Metabolic pathway analysis through KEGG showed 318 proteins present in the unique metabolic pathways in which the shortlisted proteins are playing significant roles, as shown in Table 2.
Non-homologous Proteins Identification
As described above, a total of 318 proteins were retrieved from the unique metabolic pathways. These 318 proteins were subjected to BLASTp to determine the non-homologous protein sequences against the host proteome. The BLASTp revealed 150 proteins classified as non-homologous proteins that are present only in the pathogen. These proteins are further analyzed in subsequent steps.
Identification of Essential Proteins
The essentiality of all non-homologous proteins was determined by using BLASTp from the DEG database with the E value of 10–5. About 105 non-homolog proteins were classified as essential proteins required for the survival of S. pneumoniae and could be proposed as the potential drug targets (Supplementary Table 1). These non-homologous essential proteins can be safely recommended as possible therapeutic targets for pathogens. Theoretically, bacteria may survive but expected to be less virulent if such proteins are targeted, or numerous important processes could be inhibited, resulting in pathogenicity being eradicated. The DEG database is being updated periodically, however, it is limited to the studies of S. pneumoniae survival in rich growth medium only. Despite the DEG’s limitation it is a well cited database and provided reliable results for known organisms. The essentiality of S. pneumoniae proteins can be further evaluated through experimental studies for the survival of pathogen in environmental or other biological conditions such as saliva (van Opijnen et al., 2009; Liu et al., 2017, 2021).
Druggability of Therapeutic Targets
Eventually, the non-homologous essential 105 proteins were BLASTp-ed against the DrugBank database and any sequence similarities with the drug target proteins in the DrugBank were found. Only proteins with significant sequence similarity to FDA-approved therapeutic targets were chosen, while the remaining were eliminated from the database. As a result, only 47 proteins were identified as being essential, non-homologous, and drug target-like against S. pneumoniae. These 47 proteins showed significant similarities with the FDA approved drug targets found in DrugBank and, therefore, subsequently followed up in the next step. On the other hand, the excluded 58 proteins at this stage have not shown any significant similarity to the drug targets found in DrugBank. Although those 58 proteins were excluded they still may be studied as potential drug targets by the scientific community owing to their essentiality and non-homologous nature. The list of these 47 drug target-like proteins are provided in Supplementary Table 2.
Subcellular Localization Prediction
Protein localization is important to understand throughout the drug development process because it influences the design of novel drugs and vaccines. Cell membrane proteins, for example, are frequently employed as vaccine targets, while cytoplasmic proteins are frequently used as therapeutic targets. Among these 47 essential proteins, 30 were found to be cytoplasmic proteins, 11 were cytoplasmic membrane proteins, three were cell wall proteins, and one was identified as an extracellular protein, as shown in Table 4. Figure 2 showed the location distribution of all essential proteins in S. pneumoniae. The step-wise filtering of the proteins during the current study is shown in Table 1.
TABLE 4.
Subcellular localization: Distribution of essential non-homologous proteins in a different area of cell.
| S. no | PSORTb results | No. of proteins |
| 1 | Cell wall | 3 |
| 2 | Cytoplasmic | 30 |
| 3 | Cytoplasmic Membrane | 11 |
| 4 | Extracellular | 1 |
| 5 | Unknown | 2 |
FIGURE 2.

Subcellular localization: PSORTb results showing the subcellular distribution of essential proteins occur in S. pneumoniae.
Novel Drug Targets Prediction and Significance of Selected Proteins
The literature is full of examples of cytoplasmic proteins as proven therapeutic targets because of easy reach by the drugs (Anis Ahamed et al., 2021). Additionally, ∼70% FDA approved drugs are reported as enzymatic proteins because of their significant role in multiple pathways. Finally, among 47 potential drug targets, two proteins were shortlisted as essential, non-homologous, druggable targets against S. pneumoniae i.e., 4-oxalocrotonate tautomerase (B2IPH4) and sensor histidine kinase CiaH (B2INS3). Based on their cytoplasmic subcellular localization, length > 100 amino acids, their enzymatic nature, and involvement in essential metabolic pathways, these identified proteins were subjected to further structure-based studies. Figure 3 showed the comprehensive outcome of the current study.
FIGURE 3.
Current study summary for target protein identification: Stepwise analysis of subtractive genomic approach for drug targets identification in S. pneumoniae.
4-Oxalocrotonate Tautomerase
4-oxalocrotonate tautomerase enzyme (EC 5.3.2.-4-OT) belongs to a family of isomerases that readily convert hydroxymuconate to the αβ-unsaturated ketone (van Diemen et al., 2017). It is involved in benzonate degradation and xylene degradation pathways uniquely found in S. pneumoniae.
Through this enzyme, bacteria utilize aromatic compounds and amino acids to essential hydrocarbons which are the sole source of carbon and energy that is further used in the citric acid cycle.
Sensor Histidine Kinase CiaH
Sensor histidine kinase CiaH (EC:2.7.13.3) is an ATP binding signal transduction protein involved in the Two-component system of S. pneumoniae (Singh et al., 2011). These sensor histidine kinases sense the changes in the environment (in stress or presence of drug) surrounding the pathogen and provide such signals that alter the inside mechanism of bacterial cells, preparing it to utilize these changes. It has been reported that the changes in these sensor kinases showed resistance to many antibacterial drugs such as cefotaxime (Möglich, 2019). The protein helps in the catalysis of reactions such as,
Environmental variables influence the synthesis of virulence proteins, and two-component regulatory mechanisms are involved in detecting these influences. Tatsuno et al. (2014) reported the effected regulation of Streptococcus and significant growth reduction in knockout CiaH strains. Additionally, CiaH is highly conserved in S. pneumoniae, and deletion of the gene encoding its cognate response regulator (ciaRpn) made pneumococcal strains more susceptible to oxidative stress (Kaiser et al., 2020). Furthermore, it has been widely studied for its mutations resulting in the beta-lactamase resistance (Schweizer et al., 2017; Peters et al., 2021). It is used as a drug target to inhibit ESKAPE pathogens (Velikova et al., 2016), Staphylococcus aureus (Xie et al., 2020), and Corynebacterium diphtheriae (Khalid et al., 2018). However, CiaH protein has never been studied as drug target against S. pneumoniae and thus in the current study it is proposed as a potential drug target.
Conservancy Analysis of Shortlisted Proteins
Furthermore, the conservancy analysis was performed for 4-oxalocrotonate tautomerase and sensor histidine kinase CiaH proteins. The BLASTp of 4-oxalocrotonate tautomerase resulted in the local alignment of XylH with P67530 S. pneumoniae (serotype 4), P67531 (strain ATCC BAA-255/R6), A5MAV1 (strain SP14-BS69), and J0V2K5 (from strain 2070335). The alignments of these identified proteins were further analyzed through multiple sequence alignment using Clustal Omega. The percent matric analysis showed that these proteins are 100–98% conserved among them (Supplementary Figure 1). Additionally, the BLASTp of sensor histidine kinase CiaH showed similarities with P0A4I6 (strain ATCC BAA-255/R6), P0A4I5 (serotype 4 strain ATCC BAA-334/TIGR4), A0A0H2ZQ10 (serotype 2 strain D39/NCTC 7466), J1DIP7 (strain 2070335), A5M9S1 (SP14-BS69), and A5M9S2 (strain SP14-BS69), respectively. These proteins showed conservancy of 100–93% when analyzed through MSA (Supplementary Figure 2).
Comparative Structure Prediction and Evaluation
The 3D structures of shortlisted proteins were not yet available in the PDB. Therefore, its homology modeling was performed by taking the FASTA sequence of the protein from the UniProt database with the accession numbers B2IPH4 and B2INS3 as mentioned in the database. Structural and functional studies of 4-oxalocrotonate tautomerase and sensor histidine kinase CiaH, were further evaluated by performing BLASTp against the PDB database to find a possible template. For 4-oxalocrotonate tautomerase, the template PDB ID: 6FPS was identified with 41% sequence similarity (Figure 4A). Likewise, for sensor histidine kinase CiaH, template PDB ID: 4I5S with a 40% sequence similarity was selected (Figure 4B). Using these identified template proteins, the 3D structure of 4-oxalocrotonate tautomerase and sensor histidine kinase CiaH was modeled. Structures with high DOPE score (i.e., modeled structure 4) were further evaluated for ligand screening.
FIGURE 4.

Modeled structure of proteins (drug targets): Structure modeled through Homology modeler for 4-oxalocrotonate tautomerase (A) and sensor histidine kinases (B) using the respective template.
Validation of the Modeled Structure
Different tools were used to verify the modeled protein structure. In the following, the structure verification procedure is discussed.
Confirmation of Proteins Through PSIPRED
The PSIPRED resulted in the prediction of a higher number of alpha helices than beta sheets formation as shown in Supplementary Figure 3 for both XylH and CiaH. The PSIPRED results verified the protein on the basis of their sequence for the alpha helices and beta sheets formation as modeled through modeler tool.
PROCHECK Validation of Proteins
The PROCHECK was used to generate a Ramachandran plot for the modeled protein structures. For XylH, the Ramachandran plot showed about 93.8% residues in the favorable region and four residues in additionally allowed regions (6.2%). While in the case of CiaH, validation showed about 84.4% residues in the favorable region, with seven residues in the disallowed region and 51 and 7 residues in the additionally allowed and generously allowed regions responsible for about 12.2 and 1.7%, respectively, as shown in Supplementary Figure 4.
Structure Validation Through ProSA
Additionally, the cross validation of modeled structure through ProSA tool predicted a Z-score value of –2.66 for XylH and –4.62 for CiaH, indicating the model falls within the range of NMR/X-ray crystallography derived structures (Supplementary Figure 5).
Network Topology for Protein-Protein Interactions
Many functional and physical interactions between various types of proteins develop, and these interactions are critical for many biological processes involving cellular machinery. Filtering and analyzing functional genomic data for annotating functional, structural, and evolutionary information of proteins may be performed using this information. Investigating the predicted PPIs might open up new avenues for experimental research and computer-assisted drug discovery in the future (Schwartz et al., 2009). The PPI and functional annotation of selected proteins (XylH and CiaH) were determined using the STRING server. The STRING results showed different nodes and edges of each protein and showed that the prioritized target proteins may act as hub protein inter-acting with more than ten proteins. Therefore, targeting such proteins may affect the activity of all interactor proteins.
The interaction of XylH protein with other proteins in S. pneumoniae was identified using the STRING database, which was submitted with the protein sequence of XylH. It resulted in 16 PPI networks for 4-oxalocrotonate tautomerase (Figure 5A) represented as (XylH in red node) with neighbor proteins as SPD_0131 (Conserved hypothetical protein), SPD_0182 (Conserved hypothetical protein), SPD_0072 (Catechol 2,3-dioxygenase), SPD_1834 (Acetaldehyde dehydrogenase), TrmE (Trna modification gtpase trme), glyA (Glycine hydroxymethyltransferase), PolA (In addition to polymerase activity), prfA (Peptide chain release factor 1), tdk (Thymidine kinase), SPD_0905 (Acetyltransferase), SPD_0908 (L-threonylcarbamoyladenylate synthase), SPD_0911 (Uncharacterized protein), PvaA (Pneumococcal vaccine antigen A), and HemK (Release factor glutamine methyltransferase). The XylH protein has a total number of edges of 62, expected number of edges of 23, number of nodes of 16, and an average nodes degree of 7.75, according to the results. The enrichment p-value for Protein-Protein Interactions is 7.79e-12, with an average local clustering coefficient of 0.908. These proteins are involved in a variety of critical functions. As a result, targeting the XylH protein may result in the loss of function of the other associated proteins. As a result, we can suggest this protein as a therapeutic target.
FIGURE 5.

Protein-protein interactions: Schematic PPI network generated through the STRING database for XylH (A) and CiaH (B).
Similarly, the CiaH protein sequence was uploaded to the STRING database. The STRING resulted in 28 PPI networks for sensor histidine kinase CiaH, verifying that the selected proteins are hub proteins (Figure 5B). The CiaH was represented by a red node having interactions with CiaR (DNA-binding response regulator ciaR), SPD_2068 (DNA-binding response regulator), SPD_0574 (DNA-binding response regulator), ArgF (Ornithine carbamoyltransferase), SPD_1908 (Response regulator), SPD_0081 (Response regulator), VncS (Histidine kinase), VncR (DNA-binding response regulator VncR), SPD_1446 (DNA-binding response regulator), SPD_2020 (DNA-binding response regulator protein), SPD_0344 (DNA-binding response regulator), SPD _1085 (response regulator saer), SPD_0574 (DNA-binding response regulator), ComD (sensor histidine kinase comd), ComC1 (Competence stimulating peptide precursor 1), PtsH (phosphocsrrier protein), SPD_0698 (Uncharacterized protein), SPD_0695 (Oxidoreductase), SPD_0699 (Uncharacterized protein), SPD_0694 (Uncharacterized protein), SPD_0697 (Acetyltransferase), PepN (Aminopeptidase), mapZ (Conserved hypothetical protein), SPD_1429 (Uncharacterized protein), CppA (C3-degrading proteinase), SPD_0585 (Uncharacterized protein), SPD_1522 (Replication initiation and membrane attachment protein), and FtsL (Cell division protein ftsl). It has a total of 28 number of nodes, 90 edges, 6.43 average node degree, average clustering coefficient as 0.824, expected number of edges as 38, and PPI enrichment p-value of 6.97e-13.
Active Site Prediction
Accordingly, as the protein structures were modeled, there is a need to find an interactive interface for the binding of the ligand. For that purpose, the DogSite Scorer tool was used to identify such binding sites. It predicted only one binding pocket for XylH protein with a drug score of 0.82 as shown in Supplementary Figure 6A. Table 5 showed the residues present in the selected binding pocket. As for CiaH, it predicted 16 binding pockets. Among these pockets, the binding site with a high drug score 0.81 was selected, as shown in Supplementary Figure 6B. Table 5 showed the residues present in the selected binding pocket.
TABLE 5.
Active site: Residues present in the Active site of XylH and CiaH protein.
| XylH protein active site residues |
CiaH protein active site residues |
|||||
| S. no | Chain | Position | Residue | Chain | Position | Residue |
| 1 | A | 1 | Met | A | 211 | Leu |
| 2 | A | 2 | Val | A | 212 | Glu |
| 3 | A | 3 | Lys | A | 218 | Gln |
| 4 | A | 4 | Trp | A | 219 | Ser |
| 5 | A | 5 | Lys | A | 223 | Asn |
| 6 | A | 6 | Lys | A | 226 | His |
| 7 | A | 7 | Ser | A | 227 | Glu |
| 8 | A | 8 | Lys | A | 228 | Leu |
| 9 | A | 9 | Leu | A | 229 | Arg |
| 10 | A | 10 | Val | A | 230 | Thr |
| 11 | A | 11 | Glu | A | 231 | Pro |
| 12 | A | 12 | Glu | A | 233 | Ala |
| 13 | A | 13 | Ala | A | 234 | Val |
| 14 | A | 14 | Ile | A | 235 | Leu |
| 15 | A | 15 | Met | A | 236 | Gln |
| 16 | A | 16 | Pro | A | 237 | Asn |
| 17 | A | 17 | Phe | A | 238 | Arg |
| 18 | A | 28 | Leu | A | 256 | Ser |
| 19 | A | 31 | Lys | A | 259 | Ser |
| 20 | A | 32 | Lys | A | 260 | Ser |
| 21 | A | 35 | Ala | A | 262 | Glu |
| 22 | A | 36 | Lys | A | 263 | Glu |
| 23 | A | 39 | Thr | A | 266 | Asn |
| 24 | A | 48 | Ala | A | 267 | Met |
| 25 | A | 49 | Pro | A | 269 | Phe |
| 26 | A | 50 | Gln | A | 270 | Leu |
| 27 | A | 51 | Ser | A | 273 | Ser |
| 28 | A | 52 | Ala | A | 274 | Leu |
| 29 | A | 53 | Val | A | 280 | Arg |
| 30 | A | 54 | His | A | 281 | Asp |
| 31 | A | 56 | Val | A | 282 | Asp |
| 32 | A | 284 | Ile | |||
| 33 | A | 289 | Ala | |||
| 34 | A | 290 | Glu | |||
| 35 | A | 294 | Ser | |||
| 36 | A | 295 | Phe | |||
| 37 | A | 298 | Thr | |||
| 38 | A | 301 | Thr | |||
| 39 | A | 332 | Lys | |||
| 40 | A | 351 | Glu | |||
| 41 | A | 385 | Arg | |||
| 42 | A | 402 | Leu | |||
| 43 | A | 407 | Ala | |||
| 44 | A | 410 | Ile | |||
| 45 | A | 413 | Ala | |||
Protein-Ligand Interactions Study Through Docking
The protein-ligand interactions were analyzed through AutoDock Vina.
Ligand Identification
The identification of protein binding site and their corresponding ligands have an important role in drug target identification and drug research. The protein binding sites are structurally and functionally important regions on the protein surface on which different type of drugs interact to perform a desired action (Konc and Janežič, 2017). The ProBis server was used for the ligand identification. For 4-oxalocrotonate tautomerase, DPM commonly named as DIPYRROMETHANE COFACTOR with IUPAC name: 3-[5-[[3-(2-carboxyethyl)-4-(carboxymethyl)-5-methyl-1H-pyrrol-2-yl]methyl]-4-(carboxymethyl)-1H-pyrrol-3-yl] propanoic acid was used. The ligand was identified from a template protein PDB ID: 3EQ1 (from Human) and for sensor histidine kinase CiaH, XAM commonly named as Amycolamicin antibiotic, IUPAC name (1R,4aS,5S,6S,8aR)-5-{[(5S)-1-(3-O-acetyl-4-O-carbamoyl-6-deoxy-2-O-methyl-alpha -L-talopyranosyl)-4-hydroxy-2-oxo-5-(propan-2-yl)-2,5-dihydro-1H-pyrrol-3-yl]carbonyl}-6-methyl-4-methylidene-1,2, 3,4,4a,5,6,8a-octahydronaphthalen-1-yl-2,6-dideoxy-3-C-[(1S)- 1-{[(3,4-dichloro-5-methyl-1H-pyrrol-2-yl)carbonyl]amino} ethyl]-beta-D-ribo-hexopyranosideligand was identified from a template PDB ID: 4URL (from Escherichia coli BL21) (Supplementary Figure 7).
Molecular Docking With AutoDock
Molecular docking analysis was performed with the identified ligand from ProBis server and modeled structure of CiaH and XylH protein through the AutoDock 4.2 tool. The XAM compound resulted in the binding score of –5.09 kcal/mol while DPM showed more potency toward XylH i.e., –7.59 kcal/mol. Furthermore, the post-docking analysis was performed to study the interaction of protein compounds complex. The analysis showed that XAM mediates five hydrogen bonds with Glu377, Lys424, and Arg393 whereas DPM mediates one hydrogen bond through its side chain oxygen with Val55 residue, as shown in Figure 6. The detailed interaction analysis of XAM and DPM is highlighted in Table 6.
FIGURE 6.
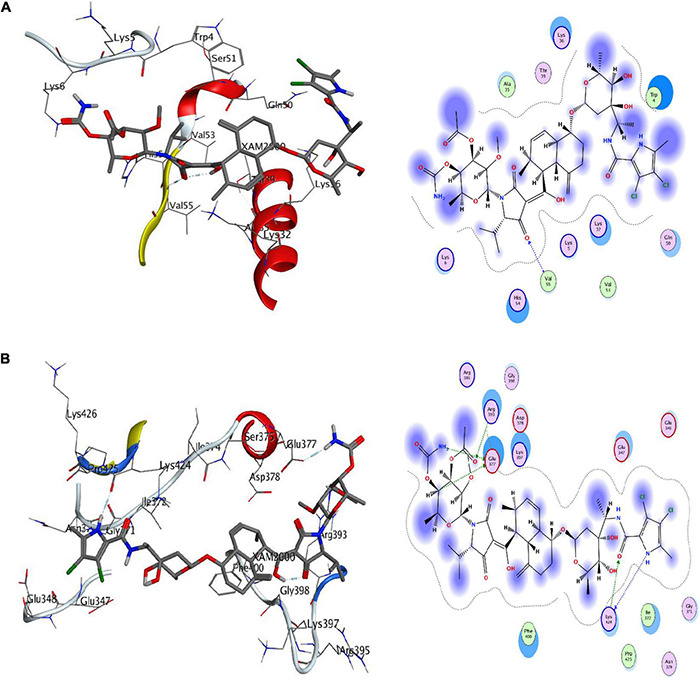
Docking of ligands with their respective drug targets. (A) 3D and 2D interaction of DPM with Ciah protein, and (B) XAM ligand for XylH protein.
TABLE 6.
Post-docking interactional analysis of XAM and DPM with XylH and CiaH protein.
| Compounds | Ligand | Receptor | Interaction | Distance | E (kcal/mol) | Binding affinity (kcal/mol) |
| XAM | C4 1 | OE2 GLU377 | H-donor | 3.21 | –0.7 | –7.59 |
| NAV 20 | O LYS424 | H-donor | 2.98 | –4.5 | ||
| NCA 115 | OE2 GLU377 | H-donor | 3.09 | –5.0 | ||
| OAQ 24 | CB LYS424 | H-acceptor | 3.54 | –0.7 | ||
| OBY 120 | NH2 ARG393 | H-acceptor | 3.45 | –0.6 | ||
| DPM | OBI 88 | N VAL55 | H-acceptor | 3.11 | –2.6 | –5.09 |
Discussion
Streptococcus pneumoniae is the most common cause of pneumonia in children and the number of antimicrobial-resistant cases of S. pneumoniae has increased globally. The situation tends to the worst due to limited medication for acute and chronic illnesses (Yang et al., 2019). Recently, computational methods gained more attention for the development of numerous alternative approaches for treating the resistant pathogens (Boeckmann et al., 2003). The subtractive genomics has been widely applied on various pathogens for the prediction and identification of therapeutic targets. Despite the methodological advancements, the high throughput experimental results are yet not available for the majority of pathogens. As a result, the efforts to find essential drug targets now mostly rely on bioinformatics based predictions (Uddin and Rafi, 2017). In silico based subtractive genomic analysis has been widely applied for strain-specific drug target identifications, particularly for the resistant pathogens (Uddin et al., 2019).
In the current study, a subtractive genomics based metabolic pathways analysis was applied for the prediction of drug targets against the clinically relevant S. pneumoniae serotype 14. In the present study, metabolic pathway analysis of S. pneumoniae against human host was performed that resulted in the prediction of 25 metabolic pathways uniquely present in S. pneumoniae. The proteins involved in these pathways were retrieved and further studied for homologous proteins identification and also to identify proteins that are only found in pathogens. The computational subtractive genomics is a powerful tool to prioritize those proteins that are essential and play a role in the pathogenicity. However, the unique pathways found exclusively in bacteria may still share the common proteins that are found in the bacterial pathogens as well as in human host. Similarly, there are common pathways between the pathogen and human host that may also have unique proteins and those unique proteins may also act as drug targets. That common proteins may still be looked for and studied to find the drug targets since the mere similarity among proteins is not the only criterion to exclude them from the search of drug targets. There are many examples of successful drugs which target common proteins of the pathogens that are shared by the human host. One such drug target is a bacterial enzyme known as Pantothenate synthetase that has a homolog present in human host, however, with low similarity (∼40%) and it is safely considered as a drug target for numerous other bacterial pathogens including M. tuberculosis (Suresh et al., 2020; Rahman et al., 2021). It is the matter of focus of the current study that we considered the unique and non-homologous proteins only from the bacterial pathogen to proceed in our work pipeline. This could be considered as a limitation of the current applied protocol. However, one may explore the unique pathways for common proteins and also the common pathways for the unique proteins depending upon the similarity thresholds.
Furthermore, the filtration for essential proteins/genes were performed by the DEG database. The essential genes from the DEG database are identified by in vitro rich medium for the growth of the bacteria, therefore, it may result in identifying those proteins which may not be essential during the in vivo (i.e., in the host) infection phase. This is the major limitation of working with the DEG. Even with this limitation the DEG has been well studied in literature examples and produced the reliable results that led to the identification of novel in vivo drug targets (Umland et al., 2012).
In the next step, the drug target-like proteins were searched with the help of DrugBank database. Consequently, a total of 47 essential druggable and unique proteins were prioritized as potential drug targets against S. pneumoniae serotype 14. Finally, only two enzymes out of the 47 shortlisted proteins were selected for further ligand discovery as potential drug candidates against S. pneumonia. The selected two proteins are 4-oxalocrotonate tautomerase and sensor histidine kinases. These two proteins were selected because of their role in benzoate degradation and two-component system as both are critical for the growth and survival of the bacteria (Gupta et al., 2020). Though the focus of the current study is on the above mentioned two proteins, the other proteins can also be equally considered for further study to characterize them as potential drug targets against which lead compounds can be developed as potent drugs.
The 4-oxalocrotonate tautomerase enzyme is involved in benzonate and xylene degradation pathways which is unique to S. pneumonia. The benzonate and xylene degradation pathways degrade the aromatic compounds and amino acids into essential hydrocarbons to fulfill its carbon and energy requirements (Medellin et al., 2020; Baas et al., 2021). On the other hand, the sensor histidine kinase is involved in the Two-Component System (TCS) of S. pneumonia and regulates certain physiological conditions. The TCS is a basic signaling mechanism that allows bacteria to detect environmental signals and develop an appropriate stress response by expressing genes that allow for environmental adaptability. The function of these proteins elaborate their importance and hence can be considered as potential drug targets (Rosales-Hurtado et al., 2020). The shortlisted proteins were modeled through modeler and validated through PROCHECK, PSIPRED, and ProSA. Furthermore, the inhibitors, i.e., XAM (against CiaH) and DPM (for XylH), were predicted against through probis server and were screened using AutoDock tool. Subsequently, the interaction analysis was performed through MOE tool. These compounds showed favorable potency against each respective protein with the estimated binding affinities of –5.09 kcal/mol for XAM against CiaH and –7.59 kcal/mol of DPM against XylH. The results we achieved in the current study are presented here and are open for the experimental validation of compounds against respective drug targets in future by the scientific community.
The above study predicted the essential proteins that may most likely act as potential drug targets due to their involvement in essential pathways of S. pneumoniae (Lo et al., 2019). Additionally, the metabolic pathways associated with the cytoplasmic proteins may be used to formulate the drug targets, whereas the membrane-associated proteins may be used to formulate peptide vaccines (Masomian et al., 2020). All of the remaining non-homologous essential proteins, on the other hand, might be good therapeutic targets. The vaccines and therapies that target the activities of these proteins may eventually lead to the destruction and eradication of pathogen from the respective hosts. The current study covered all important and potent pharmacological targets in S. pneumoniae that will certainly aid future researchers in developing effective treatment or vaccine candidates. Therefore, various other computational approaches along with this approach in collaboration with experimental researchers could be used in the future to produce potential therapeutic strategy not only against S. pneumoniae but for other pathogens too.
Conclusion
The analysis of the genome and proteome of many pathogens has aided the prediction of drug targets. In the current study, a subtractive genomic-based metabolic pathway analysis approach was applied to predict non-homologous essential druggable proteins against S. pneumoniae participating in the unique metabolic pathway. However, all the non-homologous essential proteins may also act as promising drug targets. Targeting these protein’s functions through novel drug candidates may lead to the destruction and the eradication of pathogen from the respective host. The analysis and results of the study covered all essential, potent drug targets in S. pneumoniae and thus it may facilitate future researchers to develop effective drug compounds and vaccines against strain-specific S. pneumoniae serotype 14.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
RU, AK, and AA-H conceived and designed the study. KK and KJ performed data collection and analysis and contributed to drafting the manuscript. RU provided technical and material support and supervised the study. All authors approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge the Pakistan Science Foundation for providing financial support under research support grant # PSF/Res/S-ICCBS/Med (431). We would also like to thank the University of Nizwa for the generous support of this project and Oman Research Council (TRC) through the funded project (BFP/RGP/HSS/19/198). We thank technical staff for assistance.
Footnotes
Funding
This project was supported by grant from The Oman Research Council (TRC) through the funded project (BFP/RGP/CBS/19/220) and Pakistan Science Foundation under research support grant # PSF/Res/S-ICCBS/Med (431).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.796363/full#supplementary-material
{kind=link}
References
- Anis Ahamed N., Panneerselvam A., Arif I. A., Syed Abuthakir M. H., Jeyam M., Ambikapathy V., et al. (2021). Identification of potential drug targets in human pathogen Bacillus cereus and insight for finding inhibitor through subtractive proteome and molecular docking studies. J. Infect. Public Health 14 160–168. 10.1016/j.jiph.2020.12.005 [DOI] [PubMed] [Google Scholar]
- Apweiler R., Bairoch A., Wu C. H., Barker W. C., Boeckmann B., Ferro S., et al. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49 D480–D489. 10.1093/nar/gkaa1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas B.-J., Medellin B. P., Levieux J. A., Erwin K., Lancaster E. B., Johnson W. H., Jr., et al. (2021). Kinetic and structural analysis of two linkers in the tautomerase superfamily: analysis and implications. Biochemistry 60 1776–1786. 10.1021/acs.biochem.1c00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann B., Bairoch A., Apweiler R., Blatter M.-C., Estreicher A., Gasteiger E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31 365–370. 10.1093/nar/gkg095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskraoui M., Soraa N., Zahlane K., Arsalane L., Doit C., Mariani P., et al. (2011). Étude du portage rhinopharyngé de Streptococcus pneumoniae et de sa sensibilité aux antibiotiques chez les enfants en bonne santé âgés de moins de 2 ans dans la région de Marrakech (Maroc). Arch. Pediatr. 18 1265–1270. 10.1016/j.arcped.2011.08.028 [DOI] [PubMed] [Google Scholar]
- Buchan D. W., Jones D. T. (2019). The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 47 W402–W407. 10.1093/nar/gkz297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba N., Morozumi M., Shouji M., Wajima T., Iwata S., Ubukata K., et al. (2014). Changes in capsule and drug resistance of pneumococci after introduction of PCV7, Japan, 2010–2013. Emerg. Infect. Dis. 20 1132–1139. 10.3201/eid2007.131485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilloniz C., Martin-Loeches I., Garcia-Vidal C., San Jose A., Torres A. (2016). Microbial etiology of pneumonia: epidemiology, diagnosis and resistance patterns. Int. J. Mol. Sci. 17:2120. 10.3390/ijms17122120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Deng L., Su S., Zhang M., Lin X., Wei L., et al. (2011). Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucleic Acids Res. 39 795–807. 10.1093/nar/gkq784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ediriweera D. S., Kasturiratne A., Pathmeswaran A., Gunawardena N. K., Wijayawickrama B. A., Jayamanne S. F., et al. (2016). Mapping the risk of snakebite in Sri Lanka-a national survey with geospatial analysis. PLoS Negl. Trop. Dis. 10:e0004813. 10.1371/journal.pntd.0004813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elflein J. (2021). COVID-19, Pneumonia, and Influenza Deaths Reported in the U.S. November 5 2021. Available online at: https://www.mckinsey.com/industries/healthcare-systems-and-services/our-insights/when-will-the-covid-19-pandemic-end (accessed December 19, 2021). [Google Scholar]
- Enright M. C., Spratt B. G. (1998). A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144 3049–3060. 10.1099/00221287-144-11-3049 [DOI] [PubMed] [Google Scholar]
- Fair R., Tor Y. (2014). Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 6 25–64. 10.4137/PMC.S14459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenoll A., Granizo J., Aguilar L., Giménez M., Aragoneses-Fenoll L., Hanquet G., et al. (2009). Temporal trends of invasive Streptococcus pneumoniae serotypes and antimicrobial resistance patterns in Spain from 1979 to 2007. J. Clin. Microbiol. 47 1012–1020. 10.1128/JCM.01454-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geno K. A., Gilbert G. L., Song J. Y., Skovsted I. C., Klugman K. P., Jones C., et al. (2015). Pneumococcal capsules and their types: past, present, and future. Clin. Microbiol. Rev. 28 871–899. 10.1128/cmr.00024-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. R., Gupta E., Ohri A., Shrivastava S. K., Kachhwaha S., Sharma V., et al. (2020). Comparative proteome analysis of Mycobacterium tuberculosis Strains-H37Ra, H37Rv, CCDC5180, and CAS/NITR204: a step forward to identify novel drug targets. Lett. Drug Des. Discov. 17 1422–1431. [Google Scholar]
- Hakenbeck R., Brückner R., Denapaite D., Maurer P. (2012). Molecular mechanisms of β-lactam resistance in Streptococcus pneumoniae. Future Microbiol. 7 395–410. 10.2217/fmb.12.2 [DOI] [PubMed] [Google Scholar]
- Henriques B., Kalin M., Örtqvist Å., Liljequist B. O., Almela M., Marrie T. J., et al. (2000). Molecular epidemiology of Streptococcus pneumoniae causing invasive disease in 5 countries. J. Infect. Dis. 182 833–839. 10.1086/315761 [DOI] [PubMed] [Google Scholar]
- Hu S., Shi Q., Song S., Du L., He J., Chen C.-I., et al. (2014). Estimating the cost-effectiveness of the 7-valent pneumococcal conjugate vaccine in Shanghai, China. Value Health Reg. Issues 3 197–204. 10.1016/j.vhri.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Kaiser S., Hoppstädter L. M., Bilici K., Heieck K., Brückner R. (2020). Control of acetyl phosphate-dependent phosphorylation of the response regulator CiaR by acetate kinase in Streptococcus pneumoniae. Microbiology 166 411–421. 10.1099/mic.0.000894 [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid Z., Ahmad S., Raza S., Azam S. S. (2018). Subtractive proteomics revealed plausible drug candidates in the proteome of multi-drug resistant Corynebacterium diphtheriae. Meta Gene 17 34–42. [Google Scholar]
- Konc J., Janežič D. (2017). ProBiS tools (algorithm, database, and web servers) for predicting and modeling of biologically interesting proteins. Prog. Biophys. Mol. Biol. 128 24–32. 10.1016/j.pbiomolbio.2017.02.005 [DOI] [PubMed] [Google Scholar]
- Konc J., Miller B. T., Štular T., Lešnik S., Woodcock H. L., Brooks B. R., et al. (2015). ProBiS-CHARMMing: web Interface for Prediction and Optimization of Ligands in Protein Binding Sites. J. Chem. Inf. Model. 55 2308–2314. 10.1021/acs.jcim.5b00534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R., Macarthur M., Thornton J. (2006). PROCHECK: validation of protein structure coordinates. J. Biol. Macromol. 10.1107/97809553602060000882 [Epub ahead of print]. [DOI] [Google Scholar]
- Linares J., Ardanuy C., Pallares R., Fenoll A. (2010). Changes in antimicrobial resistance, serotypes and genotypes in Streptococcus pneumoniae over a 30-year period. Clin. Microbiol. Infect. 16 402–410. 10.1111/j.1469-0691.2010.03182.x [DOI] [PubMed] [Google Scholar]
- Liu X., Gallay C., Kjos M., Domenech A., Slager J., Van Kessel S. P., et al. (2017). High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 13:931. 10.15252/msb.20167449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Kimmey J. M., Matarazzo L., De Bakker V., Van Maele L., Sirard J. C., et al. (2021). Exploration of Bacterial Bottlenecks and Streptococcus pneumoniae Pathogenesis by CRISPRi-Seq. Cell Host Microbe 29 107–120.e6. 10.1016/j.chom.2020.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S. W., Gladstone R. A., Van Tonder A. J., Lees J. A., Du Plessis M., Benisty R., et al. (2019). Pneumococcal lineages associated with serotype replacement and antibiotic resistance in childhood invasive pneumococcal disease in the post-PCV13 era: an international whole-genome sequencing study. Lancet 19 759–769. 10.1016/S1473-3099(19)30297-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodha R., Kabra S. K., Pandey R. M. (2013). Antibiotics for community-acquired pneumonia in children. Cochrane Database Syst. Rev. 2013:CD004874. [DOI] [PubMed] [Google Scholar]
- Luo H., Lin Y., Liu T., Lai F.-L., Zhang C.-T., Gao F., et al. (2021). DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res. 49 D677–D686. 10.1093/nar/gkaa917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masomian M., Ahmad Z., Ti Gew L., Poh C. L. (2020). Development of next generation Streptococcus pneumoniae vaccines conferring broad protection. Vaccines 8:132. 10.3390/vaccines8010132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medellin B. P., Lancaster E. B., Brown S. D., Rakhade S., Babbitt P. C., Whitman C. P., et al. (2020). Structural Basis for the Asymmetry of a 4-Oxalocrotonate Tautomerase Trimer. Biochemistry 59 1592–1603. 10.1021/acs.biochem.0c00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möglich A. (2019). Signal transduction in photoreceptor histidine kinases. Protein Sci. 28 1923–1946. 10.1002/pro.3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K., Schweizer I., Hakenbeck R., Denapaite D. (2021). New Insights into Beta-Lactam Resistance of Streptococcus pneumoniae: serine Protease HtrA Degrades Altered Penicillin-Binding Protein 2x. Microorganisms 9:1685. 10.3390/microorganisms9081685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman N., Shah M., Muhammad I., Khan H., Imran M. (2021). Genome-wide core proteome analysis of Brucella melitensis strains for potential drug target prediction. Mini Rev. Med. Chem. 21 2778–2787. 10.2174/1389557520666200707133347 [DOI] [PubMed] [Google Scholar]
- Rosales-Hurtado M., Meffre P., Szurmant H., Benfodda Z. (2020). Synthesis of histidine kinase inhibitors and their biological properties. Med. Res. Rev. 40 1440–1495. 10.1002/med.21651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz A. S., Yu J., Gardenour K. R., Finley R. L., Jr., Ideker T. (2009). Cost-effective strategies for completing the interactome. Nat. Methods 6 55–61. 10.1038/nmeth.1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer I., Blättner S., Maurer P., Peters K., Vollmer D., Vollmer W., et al. (2017). New aspects of the interplay between penicillin binding proteins, murM, and the two-component system CiaRH of penicillin-resistant Streptococcus pneumoniae serotype 19A isolates from Hungary. Antimicrob. Agents Chemother. 61:e00414-17. 10.1128/AAC.00414-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. P., Mishra M., Chandra A., Dhawan S. (2011). Graphene oxide/ferrofluid/cement composites for electromagnetic interference shielding application. Nanotechnology 22:465701. 10.1088/0957-4484/22/46/465701 [DOI] [PubMed] [Google Scholar]
- Suresh A., Srinivasarao S., Khetmalis Y. M., Nizalapur S., Sankaranarayanan M., Sekhar K. V. G. C. (2020). Inhibitors of pantothenate synthetase of Mycobacterium tuberculosis–a medicinal chemist perspective. RSC Adv. 10 37098–37115. 10.1039/d0ra07398a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A. L., Nastou K. C., Lyon D., Kirsch R., Pyysalo S., et al. (2021). The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49 D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanchuk V. Y., Tanin V. O., Vovk A. I., Poda G. (2016). A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina. Chem. Biol. Drug Des. 87 618–625. 10.1111/cbdd.12697 [DOI] [PubMed] [Google Scholar]
- Tatsuno I., Isaka M., Okada R., Zhang Y., Hasegawa T. (2014). Relevance of the two-component sensor protein CiaH to acid and oxidative stress responses in Streptococcus pyogenes. BMC Res. Notes 7:189. 10.1186/1756-0500-7-189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin R., Rafi S. (2017). Structural and functional characterization of a unique hypothetical protein (WP_003901628. 1) of Mycobacterium tuberculosis: a computational approach. Med. Chem. Res. 26 1029–1041. 10.1007/s00044-017-1822-0 [DOI] [Google Scholar]
- Uddin R., Siddiqui Q. N., Sufian M., Azam S. S., Wadood A. (2019). Proteome-wide subtractive approach to prioritize a hypothetical protein of XDR-Mycobacterium tuberculosis as potential drug target. Genes Genomics 41 1281–1292. 10.1007/s13258-019-00857-z [DOI] [PubMed] [Google Scholar]
- Umland T. C., Schultz L. W., Macdonald U., Beanan J. M., Olson R., Russo T. A. (2012). In vivo-validated essential genes identified in Acinetobacter baumannii by using human ascites overlap poorly with essential genes detected on laboratory media. mBio 3:e00113-12. 10.1128/mBio.00113-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Diemen P. M., Leneghan D. B., Brian I. J., Miura K., Long C. A., Milicic A., et al. (2017). The S. aureus 4-oxalocrotonate tautomerase SAR1376 enhances immune responses when fused to several antigens. Sci. Rep. 7:1745. 10.1038/s41598-017-01421-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T., Bodi K. L., Camilli A. (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6 767–772. 10.1038/nmeth.1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikova N., Fulle S., Manso A. S., Mechkarska M., Finn P., Conlon J. M., et al. (2016). Putative histidine kinase inhibitors with antibacterial effect against multi-drug resistant clinical isolates identified by in vitro and in silico screens. Sci. Rep. 6:26085. 10.1038/srep26085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt J. P., Wolfson L. J., O’brien K. L., Henkle E., Deloria-Knoll M., Mccall N., et al. (2009). Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. Lancet 374 903–911. 10.1016/S0140-6736(09)61203-4 [DOI] [PubMed] [Google Scholar]
- Wiederstein M., Sippl M. J. (2007). ProSA-web: intercation web service for the recognition of errors in three-dimensional structure of proteins. Nucleic Acids Res. 35 W407–W410. 10.1093/nar/gkm290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S., Feunang Y. D., Guo A. C., Lo E. J., Marcu A., Grant J. R., et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46 D1074–D1082. 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q., Wiedmann M. M., Zhao A., Pagan I. R., Novick R. P., Suga H., et al. (2020). Discovery of quorum quenchers targeting the membrane-embedded sensor domain of the Staphylococcus aureus receptor histidine kinase, AgrC. Chem. Commun. 56 11223–11226. 10.1039/d0cc04873a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.-I., Chang T.-H., Lu C.-Y., Chen J.-M., Lee P.-I., Huang L.-M., et al. (2019). Mycoplasma pneumoniae in pediatric patients: Do macrolide-resistance and/or delayed treatment matter? J. Microbiol. Immunol. Infect. 52 329–335. 10.1016/j.jmii.2018.09.009 [DOI] [PubMed] [Google Scholar]
- Yu N. Y., Wagner J. R., Laird M. R., Melli G., Rey S., Lo R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26 1608–1615. 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.