

Abstract
Keratoconus is a common corneal defect with a complex genetic basis. By whole exome sequencing of affected members from 11 multiplex families of European ancestry, we identified 23 rare, heterozygous, potentially pathogenic variants in 8 genes. These include nonsynonymous single amino acid substitutions in HSPG2, EML6 and CENPF in two families each, and in NBEAL2, LRP1B, PIK3CG and MRGPRD in three families each; ITGAX had nonsynonymous single amino acid substitutions in two families and an indel with a base substitution producing a nonsense allele in the third family. Only HSPG2, EML6 and CENPF have been associated with ocular phenotypes previously. With the exception of MRGPRD and ITGAX, we detected the transcript and encoded protein of the remaining genes in the cornea and corneal cell cultures. Cultured stromal cells showed cytoplasmic punctate staining of NBEAL2, staining of the fibrillar cytoskeletal network by EML6, while CENPF localized to the basal body of primary cilia. We inhibited the expression of HSPG2, EML6, NBEAL2 and CENPF in stromal cell cultures and assayed for the expression of COL1A1 as a readout of corneal matrix production. An upregulation in COL1A1 after siRNA inhibition indicated their functional link to stromal cell biology. For ITGAX, encoding a leukocyte integrin, we assayed its level in the sera of 3 affected families compared with 10 unrelated controls to detect an increase in all affecteds. Our study identified genes that regulate the cytoskeleton, protein trafficking and secretion, barrier tissue function and response to injury and inflammation, as being relevant to keratoconus.
Introduction
Keratoconus, a relatively common ocular disease, affects work-age young adults and is a leading cause of cornea transplantation worldwide (1,2). Clinically, keratoconus manifests as a bilateral thinning of the cornea and its conical protrusion, resulting in severe astigmatism, myopia, corneal scarring and loss of vision (3–5). Diagnosis normally occurs in the second–third decade of life, prompted by progressive myopic astigmatism, characteristic clinical features upon ocular examination and changes in corneal topography. Although there are no curative treatments for keratoconus, if recognized at an early age, its progressive corneal weakening can be stabilized with corneal UVA/riboflavin cross-linking (6). Clinical diagnosis in the early stages can be challenging as patients present with progressive myopia and astigmatism, a common optometric presentation in young adults. Therefore, identifying at-risk individuals and early disease presentation by molecular genetic analysis could improve case identification, target precious clinical resources and identify refractive surgery candidates at high risk of post-laser ectasia. Understanding the genetic basis of keratoconus can also help patient stratification, direct the development of new therapies based on disease pathogenesis and lead to personalized keratoconus management.
While a minority of keratoconus cases are syndromic, associated with Leber congenital amaurosis, Down syndrome, Marfan syndrome and other connective tissue anomalies, the most common form of keratoconus is isolated. The incidence (20–230 cases per 100 000 individuals or roughly 1/5000–1/500) and prevalence (270 to ~2000 cases per 100 000 individuals or roughly 0.2–2%) of non-syndromic keratoconus shows population differences (7–11). Environmental factors certainly play a role in these differences since keratoconus is exacerbated by hot dry climates (12), eye rubbing (13–15), allergy (8) and contact lens wear (16). However, a strong genetic component in keratoconus has long been suggested based on the higher concordance of disease in monozygotic over dizygotic twins (17,18,19), a positive family history in 15–18% of cases (20) and a 33% or 167-fold higher than expected risk in first degree relatives as compared with the population (21,22). Indeed, the genetic influence may be significantly larger than its environmental causes. Linkage studies, candidate gene sequencing and genome-wide association studies (GWAS) have identified numerous genes and loci contributing to keratoconus, but very few of these findings are either statistically significant or, if significant, replicated across studies (23–26). Consider, for example, that pathogenic VSX1 variants in keratoconus were later deemed polymorphisms or variants with minor pathogenic consequences (27), and their role remains unclear. Other biologically significant genes that are not yet validated include SOD1, LOX, COL4A3 and COL4A4 (28–31). GWAS on central corneal thickness (CCT), a trait with 95% heritability, used as an endophenotype to increase the success of keratoconus gene discovery, led to five significant associations at FOXO1, FNDC3B, ZFN469, COL5A1 and AKAP13 with keratoconus (22). Among these, variants in the ZFN469 gene, previously identified in brittle cornea syndrome, were also found in keratoconus patients (32); however, another study from Australia found no such enrichment (33). Thus far, no single gene with large effects consistent across studies has been identified.
Keratoconus is not a Mendelian disorder in all cases but many multiplex families show phenotypes consistent with such inheritance. Thus, one genetic model for keratoconus is its etiology from individuals heterozygous for ‘dominant’ susceptibility alleles at many loci, some common, others rare, with their penetrance modified by environmental and lifestyle factors. However, compatibility with monogenic segregation does not imply that multiple genes are not involved. As recently shown for Hirschsprung disease, pathogenesis may not be triggered except with the accumulation of a multiplicity of individually deleterious genetic variants, be they coding or non-coding (34). Thus, keratoconus could arise from the interplay of multiple (oligogenic) genetic variants (35). Generally, the failure of the cornea in keratoconus may be a consequence of multiple dysregulations in the epithelium, stromal keratocytes, assembly of the stromal ECM or a combination of these. Microscopic and biochemical studies suggest changes in the epithelial and stromal layers, loss of stromal cells (36,37), breaks in the Bowman’s layer (38) and collagen fibril anomalies (39,40). Recent proteomic (41–44) and transcriptomic (45–48) studies of the keratoconic cornea underscore the loss of epithelial integrity, impaired cellular response to injury, inflammation and degenerative changes in the stroma as significant in keratoconus. These changes may be causal or consequences of fundamental defects which genomic analysis of keratoconus cases can help resolve.
In this study, we aim to identify specific genes by whole exome sequencing (WES) followed by functional studies; few such WES studies in keratoconus exist and have been performed only in small cohorts of isolated cases (49) or individual families (50,51). Here, we studied 11 Northern Irish families comprising 21 affected members, identifying pathogenic variants in 8 genes, namely, HSPG2, EML6, CENPF, LRP1B, NBEAL2, ITGAX, MRGPRD and PIK3CGi, with multiple variants with features of likely pathogenicity. We present genetic analyses of these 8 genes and their potential functional deficits. Our results suggest that the biological processes of protein trafficking and secretion, barrier tissue function and response to injury and inflammation, fundamentally important to corneal function, are dysregulated in keratoconus. However, we need larger studies of unrelated cases to distinguish between monogenic, oligogenic and polygenic inheritance.
Results
Proband families
Eleven families with a total of 21 cases were recruited for this study (Table 1). Mean age at diagnosis was 19 years, with the minimum and maximum ages being 12 and 46 years, respectively. Clinically, 16 of the 21 cases had stromal thinning, 10 of 21 showed Fleischer ring and 9 of 21 had an irregular retinoscopy reflex. Ten patients reported mild allergies, hay fever or eczema, while 7 of 21 cases had unilateral or bilateral corneal surgery (Table 1). The families were given a BHCMG (BH) ID, with the extension _1 assigned to the proband, _2 to the mother, _3 to the father and _4, etc., to other affected siblings.
Table 1.
Phenotypic features of keratoconus families
| FAM ID | Individual ID | Sex | Age at diagnosis (years) | Corneal surgeryc | AK stagea | Keratometryb | Irregular retinoscopy reflex | Stromal thinning | Fleischer ring | Atopy | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BH 8957 | Proband | M | 17 | - | OU:2 | OD:50.75 OS:49.65 | OU + | OU + | - | - | |
| Maternal aunt | F | 19 | - | OU:2 | OD:51.90 OS:49.30 | OU + | OU + | OU + | - | ||
| BH 8958 | Proband | F | 18 | - | OD:2 OS:1 | OD:49.30 OS:45.50 | OD + | OU + | - | - | |
| Brother | M | 14 | - | OD:2 OS:1 | OD:49.00 OS:45.85 | OU + | OD + | OD + | hay fever | ||
| BH 8959 | Proband | M | 19 | - | OD:3 OS:1 | OD:56.50 OS:45.40 | OU + | OU + | OU + | - | |
| Sister | F | 28 | - | OD:1 OS:2 | OD:46.87 OS:49.60 | OU + | OU + | OU + | eczema | Squint surgery | |
| BH 8960 | Proband | M | 12 | PK OU | NA | NA | NA | NA | NA | eczema | |
| Mother | F | 46 | - | OU:1 | OD:43.50 OS:43.50 | OU + | OU + | OS + | - | Early nuclear sclerotic cataracts | |
| BH 8962 | Proband | M | 16 | - | OU:1 | OD:46.80 OS:47.80 | OU + | OU + | - | - | |
| Maternal cousin | M | 10 | PK OU | NA | NA | NA | NA | NA | asthma | Squint surgery | |
| BH 8964 | Proband | M | 21 | - | OU:1 | OD:47.34 OS:47.50 | OU + | OU + | OD + | asthma | |
| Sister | F | 22 | - | OU:1 | OD:47.75 OS:48.20 | OU + | OD + | OD + | asthma | ||
| BH 8965 | Proband | F | 20 | PK OS | OD:1 | OD:47.75 OS:postop | OD + | OD + | OD + | - | |
| Brother | M | 14 | - | OU:1 | OD:46.40 OS:48.80 | OS + | OS + | - | allergy | ||
| BH 8966 | Proband | F | 13 | - | OD:2 | OD:50.80 OS:52.65 | OU + | OU + | OS + | - | tear duct surgery |
| Maternal cousin | M | 17 | PK OU | NA | NA | NA | NA | NA | - | ||
| BH 8967 | Proband | M | 21 | PK OD DALK OS | OS:3 pre-op | OS:53.00 pre-op | OS + preop | OU + preop | - | eczema | OD hydrops, Dupuytren’s contracture, inguinal hernia |
| Sister | F | 22 | PK OD DALK OS | NA | NA | NA | NA | NA | - | ||
| BH 8969 | Proband | M | 16 | - | OU:1 | OD:47.95 OS:46.57 | OU + | OU + | - | Asthma, eczema, hay fever | |
| Sister | F | 18 | - | OU:2 | OD:49.55 OS:50.90 | OU + | OU + | OS + | - | ||
| BH 8970 | Proband | M | 19 | OU PK | NA | NA | NA | NA | NA | allergy | OD cataract surgery |
aAK: Amsler-Krumeich classification for grading keratoconus.
bMean central K readings measured in diopters (D); OD = right eye, OS = left eye, OU = both eyes.
cCorneal transplantation: PK = penetrating keratoplasty and DALK = deep anterior lamellar keratoplasty; NA = not applicable as post-surgery.
Candidate genes identified through WES analysis
Here, we considered autosomal hypotheses; the finding of only heterozygous variants, as outlined below, implicated dominant effects only. The individual family variant filtering approach in corneal and keratoconus candidate genes identified 1–17 variants in 14 genes (Supplementary Material, Table S2). Among these, we selected EML6, HSPG2 and CENPF with variants in two probands each. A second cohort analysis of all genes with variants in 3 or more probands uncovered an additional five genes, LRP1B, ITGAX, PIK3CG, NBEAL2 and MRGPRD with heterozygous variants. Taken together, we identified 8 candidate genes with 23 rare variants of which 18 had CADD >20 (at 1% false positive rate) and considered potentially damaging (Table 2). All 8 genes have pLI scores from 0 to 0.06. However, these genes, which do tolerate some pathogenic variants, may yet be excellent candidates for keratoconus since this disorder is not a lethal or negatively selected phenotype; in other words, the pLI score is not indicative of its potential phenotypic role when the trait is not lethal and could result from accumulated effects of multiple genes (i.e. a multifactorial disorder).
Table 2.
Filtered sequence variants in 8 putative Keratoconus genes
| Gene symbol | Family ID | rsID (v154) | Variant location | DNA variant | Transcript and exon location | Protein variant | Protein domain | Variant frequency | CADD score | phyloP100way | phastCons100way | Sanger validation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EML6 | BH8970 | rs554072813 | 2:55056533 | c.G766C | NM_001039753:exon6 | p.D256H | WD | 6.32 × 10−6 | 28.5 | 9.42 | 1 | Yes |
| BH8960 | rs374969726 | 2:55122149 | c.C2840T | NM_001039753:exon19 | p.S947L | WD | 4.18 × 10−4 | 26.7 | 7.47 | 1 | No | |
| HSPG2 | BH8967 | rs747403733a | 1:22176673 | c.C7307A | NM_005529:exon57 | p.T2436N | NCAM-like Ig | 1.60 × 10−5 | 21.5 | 1.76 | 0.65 | Yes |
| BH8959 | rs114015043a | 1:22150130 | c.G12982A | NM_005529:exon96 | p.A4328T | LG3 (laminin-like globular domain 3) | 2.31 × 10−3 | 19.16 | 3.02 | 1 | Yes | |
| CENPF | BH8970 | rs140399039 | 1:214795521 | c.T965C | NM_016343:exon7 | p.L322P | coiled coil | 6.05 × 10−4 | 25.5 | 5.02 | 1 | Yes |
| BH8967 | rs145858780 | 1:214819839 | c.G6926A | NM_016343:exon13 | p.R2309H | coiled coil | 9.21 × 10−4 | 1.154 | -2.22 | 0 | Yes | |
| NBEAL2 | BH8959 | rs567205565 | 3:47037281 | c.G1976A | NM_015175:exon14 | p.R659Q | Con A | 5.63 × 10−5 | 28.6 | 7.27 | 1 | Yes |
| BH8960 | rs201015564 | 3:47045708 | c.C6023T | NM_015175:exon37 | p.T2008M | 9.91 × 10−4 | 7.278 | 0.36 | 0 | No | ||
| BH8964 | rs146270553 | 3:47046519 | c.G6352A | NM_015175:exon39 | p.V2118I | BEACH | 7.31 × 10−4 | 23.9 | 5.51 | 1 | Yes | |
| ITGAX | BH8970 | . | 16:31371639 | c.T716C | NM_000887:exon8 | p.L239S | VWF domain | n/a | 22.8 | 3.31 | 0.97 | Yes |
| BH8965 | . | 16:31382761 | c.1948_1954del | NM_000887:exon16 | p.650_652del | truncation | n/a | n/a | 1.51 | 0.32 | Yesb | |
| . | 16:31382761 | c.1956_1957insTT | NM_000887:exon16 | p.S652fs | truncation | n/a | n/a | 1.51 | 0.32 | Yesb | ||
| . | 16:31382761 | c.A1957T | NM_000887:exon16 | p.N653Y | truncation | n/a | n/a | 1.51 | 0.32 | Yesb | ||
| BH8966 | rs142434946 | 16:31391381 | c.G3055A | NM_000887:exon26 | p.V1019M | 4.99 × 10−4 | 16.83 | 1.43 | 0.93 | Yes | ||
| LRP1B | BH8970 | rs149573054a | 2:141299431 | c.C7304T | NM_018557:exon44 | p.T2435I | LDL receptor class B 25 repeat | 4.25 × 10−5 | 25.5 | 9.81 | 1 | Yes |
| BH8967 | rs149644677a | 2:141032021 | c.A13114T | NM_018557:exon85 | p.N4372Y | penultimate EGF like | 5.43 × 10−3 | 23.4 | 2.02 | 1 | Yes | |
| rs199982265a | 2:141250196 | c.A9101G | NM_018557:exon57 | p.N3034S | loss of N-glycosylation | 1.41 × 10−5 | 24.9 | 7.92 | 1 | Yes | ||
| BH8958 | rs759169172a | 2:141208226 | c.G9968A | NM_018557:exon63 | p.R3323H | LDL-receptor class A 21 | 1.37 × 10−4 | 25.9 | 2.84 | 0.99 | Yes | |
| MRGPRD | BH8958 | rs750447253a | 11:68747795 | c.G661A | NM_198923:exon1 | p.V221M | Transmembrane | 2.86 × 10−5 | 12 | -3.5 | 0 | Yes |
| BH8966 | rs143309852a | 11:68747507 | c.A949C | NM_198923:exon1 | p.N317H | Cytoplasmic | 3.83 × 10−3 | 2.586 | 0.14 | 0.01 | Yes | |
| BH8970 | rs143309852a | 11:68747507 | c.A949C | NM_198923:exon1 | p.N317H | Cytoplasmic | 3.83 × 10−3 | 2.586 | 0.14 | 0.01 | Yes | |
| PIK3CG | BH8965 | rs1186998186 | 7:106508317 | c.G311A | NM_002649:exon2 | p.G104E | adaptor binding | 1.21 × 10−5 | 14.47 | 6.29 | 1 | Yes |
| BH8966 | rs1484695035 | 7:106508439 | c.G433C | NM_002649:exon2 | p.E145Q | adaptor binding | 4.07 × 10−6 | 12.4 | 3.24 | 1 | Yes | |
| BH8970 | . | 7:106513176 | c.T2080C | NM_002649:exon4 | p.F694L | PIK helical | 3.98 × 10−6 | 26.3 | 7.96 | 1 | Yes |
aReverse complemented in dbSNP.
bDeletion comprising these 3 variants.
With the exception of HSPG2 (or perlecan), which is a major component of the corneal basement membrane, the remaining genes have unknown roles, if any, in the cornea. We placed the genes in three broad functional categories important to the cornea, based on their own or their closest paralogs’ functions in other tissues: (1) cytoskeletal structure, protein trafficking and secretion by the epithelial and stromal cells (EML6, CENPF and NBEAL2) are centrally important for optimal nutrient distribution and secretion of large proteins in a structurally confined, rigid and avascular cornea; (2) barrier protection (HSPG2) is integral to the cornea as the outermost layer of the eye and (3) response to tissue injury (LRP1B, ITGAX, PIK3CG and MRGPRD) is important to the cornea to restrict immune response and inflammation that can compromise corneal transparency, as discussed below.
Cytoskeletal, protein trafficking and vesicular secretion-related genes
CENPF and EML6 are potentially involved in microtubule association and cytoskeletal functions. EML6 encodes a large protein (1958 aa) of the microtubule-associated protein (MAP) family. We identified the substitutions p.D256H and p.S947L in two Tryp-Asp (WD) dipeptide repeat domains (52) which are highly conserved (Supplementary Material, Fig. S1) and potentially pathogenic, based on their CADD scores (Table 2) The p.D256H variant affects the atypical tandem 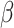 -propellar domain shared by all mammalian EMLs (53), and possibly perturbs microtubule interactions, although no direct evidence supports microtubule binding by EML6. Little is known about EML6 in the eye, except by GWAS association (P = 2.13 × 10−6) with refractive astigmatism, a strong endophenotype of keratoconus, in Asians (54). CENPF encodes a large (3114 amino acid, 350 kDa) centromeric protein of the nuclear matrix and envelope in the G2/S phase and is associated with the kinetochore complex linking chromosomes to microtubules, enabling chromosome movement during cell division (55). The p.L322P change in CENPF affects a highly conserved (Table 2, Supplementary Material, Fig. S1) cytoplasmic localization domain where introduction of a proline can affect protein folding and is potentially damaging. The p.R2309H variant, within a kinetochore-microtubule interaction site, while rare in the general population, is likely mild or has no effect (CADD score = 1.15). Two studies reported extremely rare CENPF nonsense, protein-truncating and missense variants that cause mid to late gestational lethality and milder ciliopathies with rare homozygous recessive or compound heterozygous mutations (56,57). NBEAL2 encodes a cellular scaffold protein (2750 amino acids, 302 kDa), which has not been detected in the cornea before, except it is included in the keratoconus library in NEIbank (neibank.nei.nih.gov). We identified three single amino acid substitutions of which two, p.R659Q and p.V2118I, are deemed pathogenic. The first occurs in a conserved region in a 150 amino acid long concanavalin-A like lectin binding domain that could interact with oligosaccharides (Supplementary Material, Fig. S1). The V21181 substitution affects the highly conserved BEACH domain (Supplementary Material, Fig. S1), found in 8 other related proteins in humans, may regulate vesicular transport and are associated with lysosome-related organelle diseases. Finally, missense, nonsense, and frameshift variants, and small deletions in NBEAL2, including its BEACH domain, cause an extremely rare bleeding disorder known as gray platelet syndrome (GPS) where secretory α granules are defective in microscopically gray-appearing platelets (58–60); none of the GPS variants were detected in keratoconus families.
-propellar domain shared by all mammalian EMLs (53), and possibly perturbs microtubule interactions, although no direct evidence supports microtubule binding by EML6. Little is known about EML6 in the eye, except by GWAS association (P = 2.13 × 10−6) with refractive astigmatism, a strong endophenotype of keratoconus, in Asians (54). CENPF encodes a large (3114 amino acid, 350 kDa) centromeric protein of the nuclear matrix and envelope in the G2/S phase and is associated with the kinetochore complex linking chromosomes to microtubules, enabling chromosome movement during cell division (55). The p.L322P change in CENPF affects a highly conserved (Table 2, Supplementary Material, Fig. S1) cytoplasmic localization domain where introduction of a proline can affect protein folding and is potentially damaging. The p.R2309H variant, within a kinetochore-microtubule interaction site, while rare in the general population, is likely mild or has no effect (CADD score = 1.15). Two studies reported extremely rare CENPF nonsense, protein-truncating and missense variants that cause mid to late gestational lethality and milder ciliopathies with rare homozygous recessive or compound heterozygous mutations (56,57). NBEAL2 encodes a cellular scaffold protein (2750 amino acids, 302 kDa), which has not been detected in the cornea before, except it is included in the keratoconus library in NEIbank (neibank.nei.nih.gov). We identified three single amino acid substitutions of which two, p.R659Q and p.V2118I, are deemed pathogenic. The first occurs in a conserved region in a 150 amino acid long concanavalin-A like lectin binding domain that could interact with oligosaccharides (Supplementary Material, Fig. S1). The V21181 substitution affects the highly conserved BEACH domain (Supplementary Material, Fig. S1), found in 8 other related proteins in humans, may regulate vesicular transport and are associated with lysosome-related organelle diseases. Finally, missense, nonsense, and frameshift variants, and small deletions in NBEAL2, including its BEACH domain, cause an extremely rare bleeding disorder known as gray platelet syndrome (GPS) where secretory α granules are defective in microscopically gray-appearing platelets (58–60); none of the GPS variants were detected in keratoconus families.
Corneal barrier protection-related gene
We placed HSPG2/perlecan in this category as it is a major component of the corneal basement membrane, and pericellular matrices of stromal cells, detected in the keratoconus library in NEIbank (neibank.nei.nih.gov), helps to maintain a stratified epithelium, and provides a physical extracellular matrix barrier for the eye (61). The p.T2436N change, rare (1.6 × 10−5) in the control population, occurs in the long immunoglobulin repeat-carrying domain IV, believed to form a scaffold platform for protein–protein interactions in the ECM. The second p.A4328T substitution affects the terminal domain V and implicated in cell adhesion, regulation of angiogenesis and autophagy (62). Both variants may be damaging but neither are highly conserved (Table 2). Other HSPG2 variants have been identified in chondrodysplasias such as Schwartz-Jampel syndrome type 1 with autosomal recessive inheritance in which affected individuals can display microcornea (63).
Response to injury and inflammation-related genes
The LRP1B, ITGAX, PIK3CG and MRGPRD genes may function in corneal response to injury and inflammation. We identified 4 missense variants in LRP1B encoding a large 4599 aa long single-pass type I transmembrane LDL receptor family member. Like other LDL receptors, LRP1B regulates cholesterol internalization via clathrin-mediated endocytosis, lysosomal transport, degradation and metabolism. Three of the variants with frequencies ranging from 4.25 × 10−5 to 1.3 × 10−4 are extremely rare in the control population. However, all four encode potentially damaging substitutions, based on their CADD scores, and occur in highly conserved regions within the large extracellular segment of 14 EGF-like domains, 32 LDL receptor class A and 36 LDL receptor class B domains.
ITGAX, encoding a leukocyte-specific integrin subunit (CD11c), carries four different variants in three families: two families with missense variants, p.L239S (BH9870) and p.V1019M (BH8966), while the third family has an indel, and a base substitution at the same site with consequent protein truncation. ITGAX/CD11c, not expressed by resident cells of the cornea, is expressed by monocytes and granulocytes, where it dimerizes with the 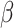 2 integrin subunit to form the C3b complement component receptor 4 on leukocytes (64). Using enzyme-linked immunosorbent assay (ELISA), we detected significantly higher levels of secreted ITGAX/CD11c in the plasma of the four patients from these three families compared with 10 healthy controls. It is not clear why circulating ITGAX/CD11c should increase in patients with an amino acid substitution or an ITGAX truncation (Figure 1). A possible explanation is that these variants either directly or indirectly affect complexation of CD11c with the
2 integrin subunit to form the C3b complement component receptor 4 on leukocytes (64). Using enzyme-linked immunosorbent assay (ELISA), we detected significantly higher levels of secreted ITGAX/CD11c in the plasma of the four patients from these three families compared with 10 healthy controls. It is not clear why circulating ITGAX/CD11c should increase in patients with an amino acid substitution or an ITGAX truncation (Figure 1). A possible explanation is that these variants either directly or indirectly affect complexation of CD11c with the 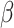 2 subunit, causing increased shedding of ITGAX/CD11c in circulation, a hypothesis that requires further study.
2 subunit, causing increased shedding of ITGAX/CD11c in circulation, a hypothesis that requires further study.
Figure 1 .
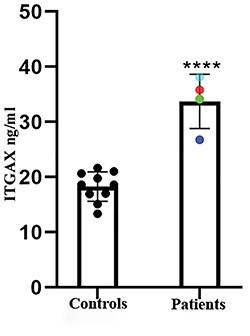
Increased circulating ITGAX/CD11c in keratoconus probands compared with unaffected controls. ELISA measurements of ITGAX in the sera of 10 controls and 4 patients. Each data point represents the mean ± standard error of the mean (SEM) of 3 technical replicates. Data points in color indicate individual patients with the following family IDs:  -BH8965,
-BH8965,  -BH8970,
-BH8970,  -BH8966,
-BH8966,  -BH8965.
-BH8965.
We identified three rare variants in PIK3CG, two of which are predicted to have mild consequences. The third, p.F694L (BH8970) substitution is extremely rare (4 × 10−6) in the control population, conserved in vertebrates (Supplementary Material, Fig. S1) and potentially damaging based on its CADD score and may affect substrate presentation to the kinase complex. PIK3CG is a phosphoinositide 3 kinase that phosphorylates PIP2 to PIP3, interacts with GPCRs and regulates AKT signaling, affecting a range of functions from platelet aggregation, thrombosis, response to tissue injury and immune cell functions. We identified two variants in MRGPRD in three families, none considered to be damaging (Table 2). MRGPRD encodes a GPCR known to be expressed by primary sensory neurons and possibly regulating itch and pain sensations (65,66). Further studies are needed to define the functions of MRGPRD in the eye and its potential connection with corneal health.
Expression of candidate genes in the cornea and corneal tissues
Earlier RNA sequencing studied by us detected all these candidate gene transcripts except MRGPRD, in human donor and keratoconus corneas. Nevertheless, here, we used Taqman qPCR to assess the expression of these 8 genes in a human corneal epithelial cell line hTCEpi, and low passage primary donor stromal fibroblasts for subsequent functional studies (Figure 2). We considered Ct values 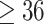 as not expressed, values between 29 and 34 as very low expression and values <29 as high expression (Table 3). HSPG2 is expressed at high levels in the hTCEpi and cultured stromal fibroblasts. CENPF is expressed more strongly in stromal fibroblasts than in hTCEpi epithelial cells, while we detected low levels of EML6 transcripts in both cell types. NBEAL2 is robustly expressed in the epithelial cell layers. PIK3CG and MRGPRD expressions are undetectable in both cell cultures, while ITGAX and LRP1B are barely detectable in epithelial and stromal cultures, respectively.
as not expressed, values between 29 and 34 as very low expression and values <29 as high expression (Table 3). HSPG2 is expressed at high levels in the hTCEpi and cultured stromal fibroblasts. CENPF is expressed more strongly in stromal fibroblasts than in hTCEpi epithelial cells, while we detected low levels of EML6 transcripts in both cell types. NBEAL2 is robustly expressed in the epithelial cell layers. PIK3CG and MRGPRD expressions are undetectable in both cell cultures, while ITGAX and LRP1B are barely detectable in epithelial and stromal cultures, respectively.
Figure 2 .
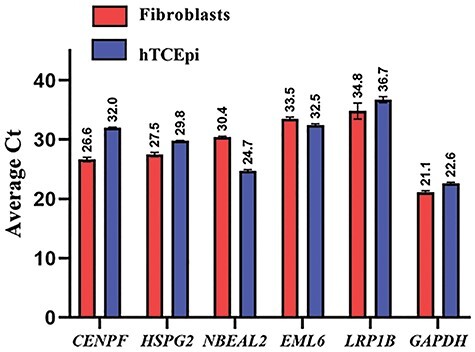
Expression of candidate genes in the human corneal epithelial cell line hTCEpi and primary donor corneal cell cultures (Supplementary Material, Table S1). Cycle threshold (Ct) values from Taqman qPCR assays are shown for selected candidate genes and GAPDH as a housekeeping gene as mean  SEM of 3 technical replicates.
SEM of 3 technical replicates.
Table 3.
Expression and localization of the 8 putative keratoconus genes in the cornea
| Functional relevance to the cornea and keratoconus | Gene symbol | Localization of protein | Gene expression (Average Ct) | |||
|---|---|---|---|---|---|---|
| human cornea | HTcEp1 cultures | Stromal fibroblast | HTcEp1 cultures | Stromal fibroblast | ||
| Cytoskeletal protein trafficking; secretory vesicular | CENPF | All epithelial layers & stroma cytoplasmic and nuclear | Nuclear and cytoplasmic | Cytoplasmic and nuclear; basal body of primary cilia | 32 ± 0.213 | 26.6 ± 0.745 |
| EML6 | All epithelial layers & stroma | nd* | Cytoskeletal network | 32.5 ± 0.382 | 33.5 ± 0.616 | |
| NBEAL2 | All epithelial layers & stromal keratocyte cell bodies | Nuclear and cytoplasmic | Cytoplasmic punctate | 24.7 ± 0.416 | 30.4 ± 0.320 | |
| Barrier protection | HSPG2 | Not tested | Not tested | Not tested | 29.8 ± 0.056 | 27.5 ± 0.706 |
| Injury and Inflammation response | PIK3CG | All epithelial layer cytoplasmic with stronger basal cell staining | Cytoplasmic | nd | nd | nd |
| LRP1B | All epithelial layer cytoplasmic | Nuclear | Nuclear | 36.7 ± 0.738 | 34.8 ± 2.35 | |
| ITGAX | nd | nd | nd | 36.6 ± 0.429 | 35.3 ± 1.47 | |
| MRGPRD | nd | nd | nd | nd | nd | |
* nd: not detected.
We next attempted to localize the proteins encoded by EML6, CENPF, NBEAL2, PIK3CG, HSPG2 (although, HSPG2/perlecan is a known entity of the cornea) and LRP1B in the human cornea and cultured corneal cells. We excluded ITGAX and MRGPRD as by qPCR these are not expressed in the cornea. Donor cornea sections show strong immunofluorescence staining of CENPF in the epithelium, in particular the basal epithelial layer and a subset of the stromal keratocytes in the central cornea (Figure 3A). The staining is primarily nuclear, consistent with CENPF being a centromeric protein. CENPF is known to localize to the basal body of primary cilium and primary cilia have been visualized in the mouse cornea (67). However, as there are no reports of CENPF in primary cilia of corneal cells, we visualized primary cilia in serum-starved, quiescent donor corneal stromal fibroblasts by immunostaining for polymerized acetylated tubulin and detected CENPF in the basal body by immunostaining (Figure 3B). EML6 shows extracellular staining of all layers of the epithelium and lamellar structures in the stroma (Figure 4A). Stromal fibroblast cultures show staining of fibrillar EML6 structures that form a cytoskeletal nest around the nucleus, with little colocalization with microtubules, except where the EML6 fibrils seemed to traverse microtubules (Figure 4B, video in supplemental materials). We detected strong NBEAL2 staining in all layers of the corneal epithelium and some stromal keratocytes (Figure 5A). Cultured stromal fibroblasts show punctate cytoplasmic staining of NBEAL2 with some colocalization with tubulin in the cell periphery (Figure 5Bvideo in supplemental materials). We immuno-localized HSPG2 in frozen sections of donor corneas and show robust staining of the basement membrane and pericellular matrices of stromal cells (Supplementary Material, Fig. S3). PIK3CG and LRP1B showed strong cytoplasmic immunolocalization in all layers of the corneal epithelium (Supplementary Material, Fig. S3).
Figure 3 .
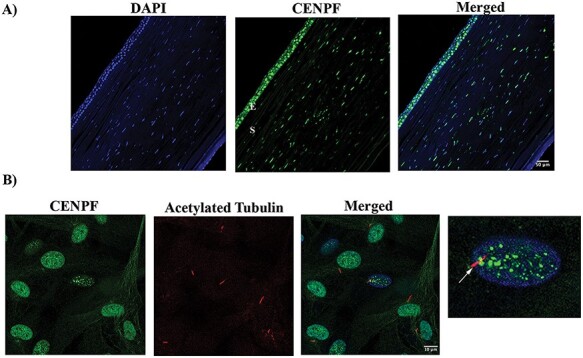
CENPF localizes to the epithelial and stromal layers and to primary cilia in cultured stromal Keratocytes. (A) CENPF (green) strong staining in the epithelial and stromal cell layers in donor (DN) corneas. A representative of three different DN corneas is shown. (E—Epithelial layer, S—Stromal layer). (B) CENPF (green) and acetylated tubulin (red) staining in 10-day serum-starved DN corneal stromal fibroblasts. CENPF is located at the basal body (arrow) of the cilium. A representative of three independent localization experiments is shown.
Figure 4 .
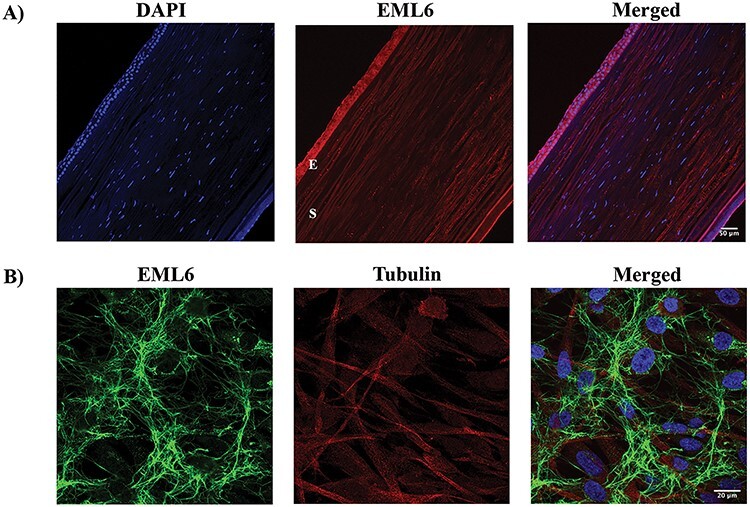
EML6 localizes to the epithelial and stromal layers of the cornea. (A) EML6 (red) is abundantly expressed in all layers of the DN cornea. A representative of three different DN corneas is shown (E—Epithelial layer, S—Stromal layer). (B) EML6 (green) and alpha tubulin (red) staining in corneal stromal fibroblasts, EML6 forms a cytoskeletal network around the nucleus. A representative image from three different cell cultures is shown.
Figure 5 .
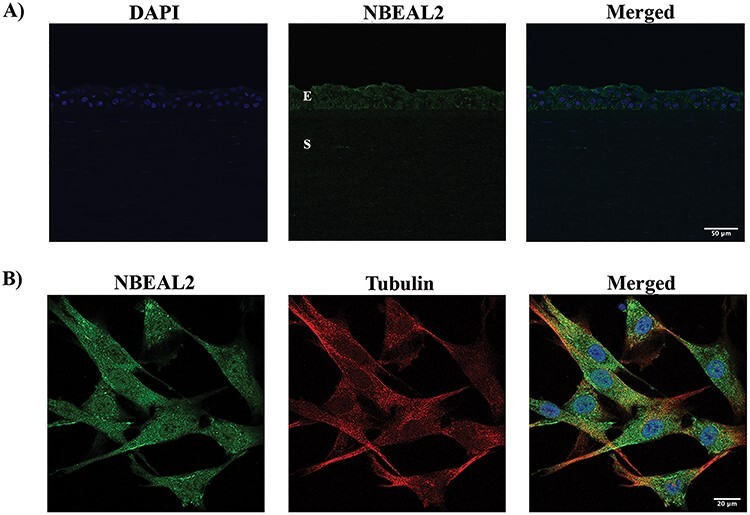
NBEAL2 primarily localizes to the epithelial layers; NBEAL2 is also detectable in cultured donor stromal fibroblasts. (A) NBEAL2 staining (green) in a representative DN cornea is shown. NBEAL2 localizes to all epithelial layers of the cornea (E—Epithelial layer, S—Stromal layer). (B) NBEAL2 (green) and alpha tubulin (red) stained in DN fibroblasts show some co-localization in the cell periphery. A representative image from three different cell cultures is shown.
We used siRNA to knockdown the expression of HSPG2, CENPF, EML6 and NBEAL2 in two dimensional (D) cultures of primary donor stromal cell fibroblasts, as by qPCR these genes were detectably expressed by stromal cells. We normalized the expression data to that of electroporation and off-target control RNA treatment as a control, because this treatment had a small effect on gene expression when compared with untreated cells. We examined the expression of COL1A1 as a readout of stromal cell functions, as producing and maintaining an ECM rich stroma is a major function of these cells. We detected a small, but significant increase in COL1A1 gene expression after inhibition of CENPF and HSPG2 (Figure 6A). However, siRNA inhibition of EML6 and NBEAL2 had no significant effect on COL1A1 expression (Figure 6B). Overall, these findings suggest a functional link between CENPF and HSPG2 and stromal cell biology. It is worth noting that upregulated collagen expression is not necessarily a phenotype of healthy stromal cells. Perturbation of cellular homeostasis can also cause an induction of an ECM-remodeling response to increase collagen gene expression. Finally, lack of significant change in COL1A1 expression after knockdown of EML6 or NBEAL2 does not mean that these two genes have no relevance in the cornea. Additional phenotypic measurements may be needed to gauge the functional contributions of EML6 and NBEAL2 in the cornea.
Figure 6 .
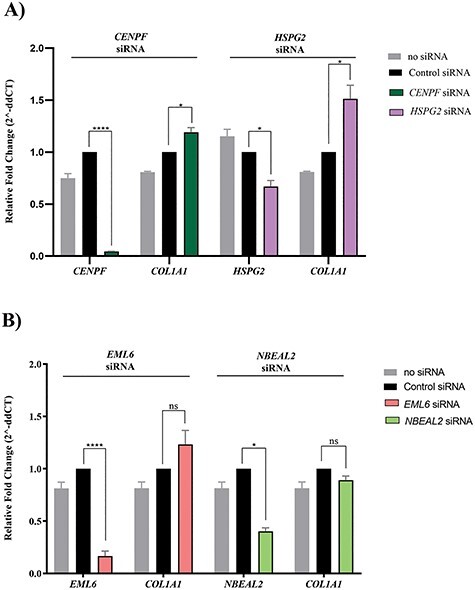
COL1A1 expression in cultured donor stromal fibroblasts after siRNA knockdown of (A) CENPF and HSPG2, and (B) EML6 and NBEAL2. Cells were electroporated with 100 nM SMARTpool siRNAs (Dharmacon) for each targeted gene individually, and COL1A1 expression, measured as a functional readout of stromal cells. Inhibition of CENPF (95%), HSPG2 (44%), EML6 (84%) and NBEAL2 (60%). Only knockdown of CENPF and HSPG2 resulted in 20–51% increase in COL1A1 expression, respectively. Gene expression was measured by Taqman qPCR and relative expression normalized to 18S RNA determined using the 2^-DDCt (cycle threshold) method. Error bars represent SEs of the mean for 3 replicates and all pairwise comparisons are with transfections using control siRNAs. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns—not significant).
Discussion
Exome sequence analysis of genomic DNA from 21 members of 11 multiplex non-syndromic keratoconus families identified 23 pathogenic variants in 8 genes (CENPF, EML6, NBEAL2, HSPG2, LRP1B, ITGAX, PIK3CG and MRGPRD). Given the small sample size, a usual statistical approach would not be effective (powered). Therefore, we used an algorithm to identify genes with multiple variants in likely candidates and prioritized those associated with corneal abnormalities by cross-checking OMIM diseases, GWAS, mouse models, ClinVar or the Human Gene Mutation Database. In addition, we selected genes with variants in three or more families. The final 8 genes we consider are likely to have functional consequences in keratoconus based on the predicted change in the encoded protein, sequence conservation across 100 vertebrates and their rare frequencies in the adult unselected population. However, stringent functional studies on each variant would require cell lines either derived from the patients in question or control cells with the variants introduced by gene-editing. In the latter, multiple variants must be introduced in the same cells to gauge their combined deleterious effects. Future studies will pursue some of these in-depth functional analyses. Here, we investigated the functional importance of a subset of the 8 genes by siRNA knockdown of their expression in cultured stromal cells and assessed their effects on COL1A1 expression, as a representative ECM gene. Thus, knockdown of HSPG2 and CENPF led to increased expression of COL1A1, suggesting perturbation of corneal ECM homeostasis and their relevance to stromal cell functions. Although additional studies are required to more fully understand their role in keratoconus.
All of the identified pathogenic variants, except the S947L substitution in EML6, were confirmed by Sanger sequencing. Furthermore, 8 variants in five genes, EML6 (p.D256H and p.S947L), CENPF (p.L322P), NBEAL2 (p.R659Q and p.V2118I), LRP1B (p.T2435I and p.N3034S) and PIK3CG (p.F694L) in four of the families, appear to be particularly consequential (Supplementary Material, Fig. S1). Additionally, ITGAX, which we demonstrate harbors variants with a detectable plasma phenotype, is not detectable in the cornea and requires additional studies to elucidate its role in the corneal pathology in keratoconus. It certainly raises the possibility that pathogenic variants, affecting resident or infiltrating immune cells, can contribute to keratoconus.
The cornea has multiple protective mechanisms to remove abnormal cytotoxic proteins, DNA and lipids produced by oxidative damage-induced reactive oxygen and nitrogen species. Keratoconus has long been postulated to be the result of disruptions in these protective mechanisms with downstream extracellular matrix remodeling, collagen structural anomalies, scarring and vison loss (68). SOD1 (super oxide dismutase), LOX, COL5A1, COL4A3 and COL4A4 are some of the previously identified candidate genes that are consistent with this idea. From our data here, LRP1B may have a significant role in lysosomal degradation of oxidative stress-associated lipid byproducts and their removal from the cornea. All four variants in our three families are predicted to be damaging and causal in keratoconus. NBEAL2, also associates with lysosome related organelles (69), interacts with the guanine nucleotide exchange factor DOCK7 and the ER export factor SEC16A (70) to regulate actin reorganization and vesicular transport. Disruptions in lysosome-related organelle functions are connected to lysosomal storage diseases and other ocular abnormalities (71), and we hypothesize that keratoconus may have shared pathogenesis with milder variants.
UV light and environmental stress-induced cell death and autophagy is highly relevant to the cornea. Our earlier proteomic studies of keratoconus had suggested alterations in cell survival and AKT signaling in keratoconus (72,73). This is further emphasized by our finding of significant PIK3CG variants in keratoconus families, as PIK3CG encodes a subunit of the phosphoinositide kinase that phosphorylates PIP2 to PIP3 and is integral to AKT signaling, WNT signaling: it has been previously implicated in keratoconus (74) and cell survival. Consistently, FOXO1, identified as a CCT and keratoconus susceptibility gene by a GWAS meta-analysis (22), is a major regulator of cellular response to stress and nutrient deprivation and is itself regulated by AKT (75). In addition, our recent keratoconus transcriptomic analyses identified dysregulated expression of NRF2-mediated antioxidant genes (48), further supporting the connection between impaired resolution of oxidative stress and keratoconus.
Our findings of EML6 and CENPF variants reveal a link between keratoconus and cytoskeletal functions. Microtubules have an important, but poorly understood role in protein traffic and export, and energy and nutrient dissemination (76), factors of relevance to the avascular, nutrient-restricted cornea. Moreover, the stratified epithelium and the mesenchymal keratocytes show tissue-level anterior to posterior polarity where morphogen gradients, cell–cell and cell-matrix communications are regulated by cytoskeletal proteins. Related to the sea urchin EMAP, the mammalian EML family of six MAPs are relatively understudied. EML1–4 are known to associate with and regulate microtubule dynamics, but EML5 or 6 have not been demonstrated to associate with microtubules (53). Our immunofluorescence staining shows discrete points of overlap between the EML6 cytoskeletal network and microtubules in cultured keratocytes. We detected cytoplasmic and nuclear staining of CENPF in the epithelium and stroma of the cornea, and typical staining of the basal body of primary cilium in cultured stromal keratocytes. In quiescent cells, the primary cilium is a microtubule containing antenna-like organelle near the plasma membrane, and a specialized area of hedgehog, TGFβ, Wnt-frizzled and other ligand-receptor interactions (77,78). These signal networks and the primary cilium play important roles in the development and patterning of the cornea (67). Rare CENPF variants have been reported to cause ciliopathies (56,57,79), which raises the possibility that the CENPF variants we detected in keratoconus perturb primary cilia functions in the cornea and that keratoconus falls within the ciliopathy spectrum. Most intriguingly, this implies that multiple genes that regulate planar cell polarity and tissue-level polarity (80,81) in the developing and adult cornea may be important in keratoconus.
Our genetic and functional data suggest that although single variants can have large impact in some families, keratoconus largely occurs through the effects of multiple variants that disrupt major physiological processes in the cornea. We speculate that the heterozygous non-synonymous variants we detected in the Northern Irish families are likely to impact major biological processes in the cornea through gain of function effects, except for the protein-truncation indel in ITGAX. Notably, each affected family member carries more than one genetic variant, and even in families where disease appears to be Mendelian inheritance compatible, causation is likely oligogenic. Multiple pathogenic variants residing in an individual within a high susceptibility genome, such as individuals with low CCT from common polygenic contributions, can result in keratoconus but not when it resides in a low susceptibility genome. This modulation of penetrance by genetic variation could equally occur through environmental factors affecting the same processes. In the future, it will be useful to perform whole genome genotyping to assess polygenic background risk in all keratoconus patients to evaluate the genetic contributions of individual rare pathogenic and sub-pathogenic variants in candidate genes.
Taken together, our findings of pathogenic variants in EML6 and CENPF link microtubule and primary cilia-related functions to keratoconus. On the other hand, variants in PIK3CG are consistent with our previous studies that suggest AKT cell signaling to be an important cell survival regulatory network in keratoconus. Finally, pathogenic variants in the leukocyte integrin ITGAX suggest a role for a peripheral blood-derived regulator in keratoconus and perhaps a long sought-after potential serum biomarker for some forms of keratoconus.
Materials and Methods
Familial keratoconus recruitment and diagnosis
This study was approved by local Research Ethics Committees and conducted in accordance with the Declaration of Helsinki; all participants provided written informed consent. We used familial keratoconus patients identified at the Department of Ophthalmology, Royal Victoria Hospital, Belfast, UK (Belfast Health and Social Care Trust, UK). Families with at least 2 affected members with non-syndromic keratoconus and available DNA were recruited to this study (Table 1). Disease diagnosis was based on clinical examination and corneal topography (32,82). Specifically, we used characteristic slit-lamp biomicroscopy findings (corneal thinning, Vogts’ striae or a Fleischer ring) and dilated retinoscopy signs (scissoring red reflex and the oil droplet sign). Corneal topography, using either the Tomey KC screening (Topographic Modeling System, software version 2.4.2 J, Tomey Corp, Nagoya, Japan), the Orbscan II (Bausch & Lomb Surgical, Orbtek Inc., Salt Lake City, Utah, USA) or Pentacam (Oculus Optikgeräte GmbH, Wetzlar, Germany) systems confirmed diagnosis. Corneal transplantation (penetrating or deep anterior lamellar keratoplasty) for keratoconus was sufficient to confirm diagnosis.
DNA extraction, WES and variant detection
DNA was extracted from leukocytes from whole blood samples using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI) following the manufacturer’s recommendations. DNA samples were sent to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) for WES. Each individual sequenced was processed using the Agilent SureSelect XT kit to capture ~52 Mb CCDS exonic and flanking introni regions (83). Paired end 100 bp reads with the Illumina HiSeq2500 platform were performed. Each read was aligned to the GRCh37 human genome reference with the Burrows-Wheeler Alignment version 0.5.10-tpx (84). Local realignment around indels and base call quality score recalibration was performed using the Genome Analysis Toolkit (GATK) version 2.3-9-ge5ebf34, with HaplotypeCaller/CombineGVCF/GenotypeGVCF workflows (85). Variants were then filtered using the Variant Quality Score Recalibration method (86). Single nucleotide variants (SNVs) were annotated by the MQRankSum, HaplotypeScore, QD, FS, MQ, ReadPosRankSum adaptive error model (6 max Gaussians allowed, worst 3% used for training the negative model). HapMap3.3 (The International HapMap 3 Consortium, 2010) and Omni2.5 were used as training data with HapMap3.3 used as the true positive set. SNVs were filtered to obtain all variants up to the 99th percentile of positive control sites (1% false negative rate). For indels, the annotations of QD, FS, Haplotype Score and ReadPosRankSum were used in the adaptive error model (4 max Gaussians allowed, worst 12% used for training the negative model, indels that had annotations more than 10 standard deviations from the mean were excluded from the Gaussian mixture model). A set of curated indels obtained from the GATK resource bundle (Mills_and_1000G_gold_standard.indels.b37.vcf) were used as training and truth sites. Indels were filtered to obtain all variants up to the 95th percentile of true positive sites (5% false negative rate).
Variant filtering
Using the PhenoDB Variant Analysis Tool (87), we prioritized rare variants, which defined as those with a minor allele frequency (MAF) <1%. Variant allele frequencies were obtained from the Exome Variant Server (release ESP6500SI-V2), 1000 Genomes Project (88), ExAC/gnomAD (89) and in our in-house BHCMG samples (87). These included functional (missense, nonsense, canonical splice site variants and coding indels) heterozygous and homozygous variants in each proband.
Analysis strategy
Individual family variant filtering
For each proband, we included heterozygous or homozygous variants that were rare (MAF < 1%) and in coding exons (missense, nonsense, stoploss or indels) or canonical splicing. Next, we classified the variants per mode of inheritance, as compound heterozygote (1–18 variants/proband), autosomal recessive homozygote (0–5 variants/proband) or autosomal dominant (37–246 variants/proband): numbers in parenthesis are the numbers of variants observed across the genes tested. From among these, we selected the ones in genes associated with corneal abnormalities by cross-checking OMIM diseases, GWAS databases, mouse models, ClinVar or the Human Gene Mutation Database (HGMD) and present in 2 or more probands. We also searched for variants in genes association with cornea-related traits in GWAS and in those where the encoded proteins have been reported as significantly changed in the keratoconus corneal proteome (24,25,43,90).
Cohort analysis
We searched for all genes with variants irrespective of their known association with cornea phenotypes but required their presence in three or more probands.
Extracting and culturing corneal stromal cells
Cadaverous donor corneas, unsuitable for transplantation (Lions Eye Institute for Transplant and Research, Tampa, FL), were extracted with 2 mg/ml of collagenase type-I (Invitrogen; Carlsbad, CA) as described (72,73), further digested with 0.25% Trypsin–EDTA (Invitrogen) and plated in DMEM: F12 media containing 5% FBS and 1% antibiotic/antimycotic solution. To evoke the keratocyte phenotype, fibroblasts were switched to low-glucose serum-free (LGSF) DMEM supplemented with 1% insulin, transferrin, selenium (Sigma-Aldrich Corp., St. Louis, MO, USA) and 1 mM phosphoascorbic acid (73). All cultures were used within 4–5 passages.
Immunofluorescence staining of cell cultures
Cells were seeded into 8-well chamber slides at a density of 30 000 cells per well, allowed to adhere overnight and fixed in 4% paraformaldehyde in PBS for 10 minutes on ice, washed with Tris buffered saline (TBS) and 0.01% Tween. Cells were permeabilized with 100% Ethanol, blocked in 5% bovine serum albumin in TBS for 1 hour at room temperature and incubated overnight with primary antibody (Supplementary Material, Table S1) in blocking buffer. The slides were washed three times with TBS and Tween and further incubated with 5 μg/ml Alexa Fluor secondary antibody (Supplementary Material, Table S1) diluted in TBS. The nuclei were counterstained with DAPI and images acquired with a Zeiss LSM 700 microscope. For co-staining of CENPF and acetylated tubulin (polymerized), cells were serum starved for 10 days using LGSF DMEM supplemented with 1% insulin, transferrin, selenium (I3146; Sigma-Aldrich Corp., St. Louis, MO, USA), 1 mM phosphoascorbic acid and 1% antibiotic/antimycotic solution.
Immunohistology of tissues
Sections of paraffin-embedded human donor corneas were blocked with 10% animal serum in TBS, followed by overnight incubation with primary antibody (Supplementary Material, Table S1) at 4°C. The slides were washed three times with TBS, further incubated with a secondary antibody (Supplementary Material, Table S1) for 2 hours, nuclei counterstained with DAPI and images acquired with a Zeiss LSM 700 microscope.
siRNA inhibition and gene expression by Taqman qPCR
100  M SMARTpool siRNAs (Dharmacon, Lafayette, CO) for CENPF, EML6, HSPG2 and NBEAL2 (Supplementary Material, Table S1) were electroporated into donor stromal fibroblast cultures in duplicate at a density of 105 cells/ml using a Neon Transfection System (Thermo Fisher Scientific, Waltham, MA). A protocol for maximum transfection efficiency was used (Pulse voltage 1300 V, pulse width 20 ms, number of pulses 2). Forty-eight hours after electroporation total RNA was isolated in TRIzol (Life Technologies, Carlsbad, CA), treated with DNAse I (New England BioLabs, Ipswich, MA) and 500
M SMARTpool siRNAs (Dharmacon, Lafayette, CO) for CENPF, EML6, HSPG2 and NBEAL2 (Supplementary Material, Table S1) were electroporated into donor stromal fibroblast cultures in duplicate at a density of 105 cells/ml using a Neon Transfection System (Thermo Fisher Scientific, Waltham, MA). A protocol for maximum transfection efficiency was used (Pulse voltage 1300 V, pulse width 20 ms, number of pulses 2). Forty-eight hours after electroporation total RNA was isolated in TRIzol (Life Technologies, Carlsbad, CA), treated with DNAse I (New England BioLabs, Ipswich, MA) and 500  g used to synthesize cDNA using the iScript™ cDNA synthesis kit (Biorad, Hercules, CA). Each cDNA (20 ng) was subjected to qPCR for selected genes (Supplementary Material, Table S1), and 18S RNA or GAPDH as housekeeping genes, in duplicate or triplicate wells, using TaqMan assays (Applied Biosystems, Foster City, CA) on a One Step Plus instrument (Applied Biosystems, Foster City, CA). The 2-ΔΔCt (threshold cycle) method was used to calculate relative expression in fold change.
g used to synthesize cDNA using the iScript™ cDNA synthesis kit (Biorad, Hercules, CA). Each cDNA (20 ng) was subjected to qPCR for selected genes (Supplementary Material, Table S1), and 18S RNA or GAPDH as housekeeping genes, in duplicate or triplicate wells, using TaqMan assays (Applied Biosystems, Foster City, CA) on a One Step Plus instrument (Applied Biosystems, Foster City, CA). The 2-ΔΔCt (threshold cycle) method was used to calculate relative expression in fold change.
Enzyme-linked immunosorbent assay
ITGAX levels in sera were measured using a Human ITGAX ELISA kit (Biobool, E020155). In brief, ITGAX antibody precoated NUNC Maxisorp plates were incubated with the serum samples, washed and incubated with the detection antibody, washed, incubated with Streptavidin-HRP and developed with TMB. Then, the reaction was stopped and absorbance measured at 450 nm using a microplate reader (VERSAmax, San Jose, CA).
Supplementary Material
Acknowledgements
Clinical data collection and DNA from Northern Ireland Research and Development Office RRG (grant number 4.46) and Fight for Sight (grant number 1787) to C.E.W. Sequencing and other support from Baylor-Hopkins Center for Mendelian Genomics to N.S. and the National Institute of Health/National Eye Institute support (grant number EY026104) to S.C.
Conflict of Interest statement. No conflicts of interest for any of the authors are recognized.
Contributor Information
Vishal Shinde, Department of Ophthalmology, NYU Grossman School of Medicine, New York, NY 10016, USA.
Nara Sobreira, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University, Baltimore, MD 21287, USA.
Elizabeth S Wohler, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University, Baltimore, MD 21287, USA.
George Maiti, Department of Ophthalmology, NYU Grossman School of Medicine, New York, NY 10016, USA.
Nan Hu, Department of Ophthalmology, NYU Grossman School of Medicine, New York, NY 10016, USA.
Giuliana Silvestri, Department of Ophthalmology, Belfast Health and Social Care Trust, Belfast BT12 6BA UK.
Sonia George, Department of Ophthalmology, Belfast Health and Social Care Trust, Belfast BT12 6BA UK.
Jonathan Jackson, Department of Ophthalmology, Belfast Health and Social Care Trust, Belfast BT12 6BA UK.
Aravinda Chakravarti, Center for Human Genetics and Genomics, NYU Grossman School of Medicine, New York, NY 10016, USA.
Colin E Willoughby, Department of Ophthalmology, Belfast Health and Social Care Trust, Belfast BT12 6BA UK; Genomic Medicine, Biomedical Sciences Research Institute, Ulster University, Coleraine BT52 1SA, UK.
Shukti Chakravarti, Department of Ophthalmology, NYU Grossman School of Medicine, New York, NY 10016, USA; Department of Pathology, NYU Grossman School of Medicine, New York, NY 10016, USA.
References
- 1. Cassidy, D., Beltz, J., Jhanji, V. and Loughnan, M.S. (2013) Recent advances in corneal transplantation for keratoconus. Clin. Exp. Optom., 96, 165–172. [DOI] [PubMed] [Google Scholar]
- 2. Jhanji, V., Sharma, N. and Vajpayee, R.B. (2011) Management of keratoconus: current scenario. Br. J. Ophthalmol., 95, 1044–1050. [DOI] [PubMed] [Google Scholar]
- 3. Ihalainen, A. (1986) Clinical and epidemiological features of keratoconus genetic and external factors in the pathogenesis of the disease. Acta Ophthalmol. Suppl., 178, 1–64. [PubMed] [Google Scholar]
- 4. Rabinowitz, Y.S. (1998) Keratoconus. Surv. Ophthalmol., 42, 297–319. [DOI] [PubMed] [Google Scholar]
- 5. Soiberman, U., Foster, J.W., Jun, A.S. and Chakravarti, S. (2017) Pathophysiology of keratoconus: what do we know today. Open Ophthalmol. J., 11, 252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wollensak, G., Spoerl, E. and Seiler, T. (2003) Riboflavin/ ultraviolet-a-induced collagen crosslinking for the treatment of keratoconus. Am J. Ophthalmol., 135, 620–627. [DOI] [PubMed] [Google Scholar]
- 7. Godefrooij, D.A., deWit, G.A., Uiterwaal, C.S., Imhof, S.M. and Wisse, R.P. (2017) Age-specific incidence and prevalence of keratoconus: a nationwide registration study. Am J. Ophthalmol., 175, 169–172. [DOI] [PubMed] [Google Scholar]
- 8. Hashemi, H., Heydarian, S., Hooshmand, E., Saatchi, M., Yekta, A., Aghamirsalim, M., Valadkhan, M., Mortazavi, M., Hashemi, A. and Khabazkhoob, M. (2020) The prevalence and risk factors for keratoconus: a systematic review and meta-analysis. Cornea, 39:263–270. [DOI] [PubMed] [Google Scholar]
- 9. Nielsen, K., Hjortdal, J., Aagaard Nohr, E. and Ehlers, N. (2007) Incidence and prevalence of keratoconus in Denmark. Acta Ophthalmol. Scand., 85, 890–892. [DOI] [PubMed] [Google Scholar]
- 10. Assiri, A.A., Yousuf, B.I., Quantock, A.J. and Murphy, P.J. (2005) Incidence and severity of keratoconus in Asir province, Saudi Arabia. Br. J. Ophthalmol., 89, 1403–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jonas, J.B., Nangia, V., Matin, A., Kulkarni, M. and Bhojwani, K. (2009) Prevalence and associations of keratoconus in rural Maharashtra in Central India: the Central India eye and medical study. Am J. Ophthalmol., 148, 760–765. [DOI] [PubMed] [Google Scholar]
- 12. Cingu, A.K., Cinar, Y., Turkcu, F.M., Sahin, A., Ari, S., Yuksel, H., Sahin, M. and Caca, I. (2013) Effects of vernal and allergic conjunctivitis on severity of keratoconus. Int. J. Ophthalmol., 6, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bawazeer, A.M., Hodge, W.G. and Lorimer, B. (2000) Atopy and keratoconus: a multivariate analysis. Br. J. Ophthalmol., 84, 834–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krachmer, J.H. (2004) Eye rubbing can cause keratoconus. Cornea, 23, 539–540. [DOI] [PubMed] [Google Scholar]
- 15. Balasubramanian, S.A., Pye, D.C. and Willcox, M.D. (2013) Effects of eye rubbing on the levels of protease, protease activity and cytokines in tears: relevance in keratoconus. Clin. Exp. Optom., 96, 214–218. [DOI] [PubMed] [Google Scholar]
- 16. Macsai, M.S., Varley, G.A. and Krachmer, J.H. (1990) Development of keratoconus after contact lens wear. Patient characteristics. Arch. Ophthalmol., 108, 534–538. [DOI] [PubMed] [Google Scholar]
- 17. Parker, J., Ko, W.W., Pavlopoulos, G., Wolfe, P.J., Rabinowitz, Y.S. and Feldman, S.T. (1996) Videokeratography of keratoconus in monozygotic twins. J. Refract. Surg., 12, 180–183. [DOI] [PubMed] [Google Scholar]
- 18. Weed, K.H., MacEwen, C.J. and McGhee, C.N. (2006) The variable expression of keratoconus within monozygotic twins: Dundee University Scottish Keratoconus Study (DUSKS). Cont. Lens Anterior Eye, 29, 123–126. [DOI] [PubMed] [Google Scholar]
- 19. Tuft, S.J., Hassan, H., George, S., Frazer, D.G., Willoughby, C.E. and Liskova, P. (2012) Keratoconus in 18 pairs of twins. Acta Ophthalmol., 90, e482–e486. [DOI] [PubMed] [Google Scholar]
- 20. Szczotka-Flynn, L., Slaughter, M., McMahon, T., Barr, J., Edrington, T., Fink, B., Lass, J., Belin, M. and Iyengar, S.K. (2008) Disease severity and family history in keratoconus. Br. J. Ophthalmol., 92, 1108–1111. [DOI] [PubMed] [Google Scholar]
- 21. Wang, Y., Rabinowitz, Y.S., Rotter, J.I. and Yang, H. (2000) Genetic epidemiological study of keratoconus: evidence for major gene determination. Am. J. Med. Genet., 93, 403–409. [PubMed] [Google Scholar]
- 22. Lu, Y., Vitart, V., Burdon, K.P., Khor, C.C., Bykhovskaya, Y., Mirshahi, A., Hewitt, A.W., Koehn, D., Hysi, P.G., Ramdas, W.D.et al. (2013) Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nat. Genet., 45, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nielsen, K., Hjortdal, J., Pihlmann, M. and Corydon, T.J. (2012) Update on the keratoconus genetics. Acta Ophthalmol., 91:106–113. [DOI] [PubMed] [Google Scholar]
- 24. Chang, H.Y. and Chodosh, J. (2013) The genetics of keratoconus. Semin. Ophthalmol., 28, 275–280. [DOI] [PubMed] [Google Scholar]
- 25. Bykhovskaya, Y., Margines, B. and Rabinowitz, Y.S. (2016) Genetics in keratoconus: where are we? Eye Vis (Lond)., 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mas Tur, V., MacGregor, C., Jayaswal, R., O'Brart, D. and Maycock, N. (2017) A review of keratoconus: diagnosis, pathophysiology, and genetics. Surv. Ophthalmol., 62, 770–783. [DOI] [PubMed] [Google Scholar]
- 27. Dash, D.P., George, S., O'Prey, D., Burns, D., Nabili, S., Donnelly, U., Hughes, A.E., Silvestri, G., Jackson, J., Frazer, D.et al. (2010) Mutational screening of VSX1 in keratoconus patients from the European population. Eye (Lond.), 24, 1085–1092. [DOI] [PubMed] [Google Scholar]
- 28. Stabuc-Silih, M., Strazisar, M., Hawlina, M. and Glavac, D. (2010) Absence of pathogenic mutations in VSX1 and SOD1 genes in patients with keratoconus. Cornea, 29, 172–176. [DOI] [PubMed] [Google Scholar]
- 29. Saee-Rad, S., Hashemi, H., Miraftab, M., Noori-Daloii, M.R., Chaleshtori, M.H., Raoofian, R., Jafari, F., Greene, W., Fakhraie, G., Rezvan, F.et al. (2011) Mutation analysis of VSX1 and SOD1 in Iranian patients with keratoconus. Mol. Vis., 17, 3128–3136. [PMC free article] [PubMed] [Google Scholar]
- 30. Bykhovskaya, Y., Li, X., Epifantseva, I., Haritunians, T., Siscovick, D., Aldave, A., Szczotka-Flynn, L., Iyengar, S.K., Taylor, K.D., Rotter, J.I.et al. (2012) Variation in the lysyl oxidase (LOX) gene is associated with keratoconus in family-based and case-control studies. Invest. Ophthalmol. Vis. Sci., 53, 4152–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kokolakis, N.S., Gazouli, M., Chatziralli, I.P., Koutsandrea, C., Gatzioufas, Z., Peponis, V.G., Droutsas, K.D., Kalogeropoulos, C., Anagnou, N., Miltsakakis, D.et al. (2014) Polymorphism analysis of COL4A3 and COL4A4 genes in Greek patients with keratoconus. Ophthalmic Genet., 35, 226–228. [DOI] [PubMed] [Google Scholar]
- 32. Lechner, J., Porter, L.F., Rice, A., Vitart, V., Armstrong, D.J., Schorderet, D.F., Munier, F.L., Wright, A.F., Inglehearn, C.F., Black, G.C.et al. (2014) Enrichment of pathogenic alleles in the brittle cornea gene, ZNF469, in keratoconus. Hum. Mol. Genet., 23, 5527–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lucas, S.E.M., Zhou, T., Blackburn, N.B., Mills, R.A., Ellis, J., Leo, P., Souzeau, E., Ridge, B., Charlesworth, J.C., Brown, M.A.et al. (2017) Rare, potentially pathogenic variants in ZNF469 are not enriched in keratoconus in a large Australian cohort of European descent. Invest. Ophthalmol. Vis. Sci., 58, 6248–6256. [DOI] [PubMed] [Google Scholar]
- 34. Tilghman, J.M., Ling, A.Y., Turner, T.N., Sosa, M.X., Krumm, N., Chatterjee, S., Kapoor, A., Coe, B.P., Nguyen, K.H., Gupta, N.et al. (2019) Molecular genetic anatomy and risk profile of Hirschsprung's disease. N. Engl. J. Med., 380, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kriszt, A., Losonczy, G., Berta, A., Vereb, G. and Takacs, L. (2014) Segregation analysis suggests that keratoconus is a complex non-mendelian disease. Acta Ophthalmol., 92, e562–e568. [DOI] [PubMed] [Google Scholar]
- 36. Efron, N. and Hollingsworth, J.G. (2008) New perspectives on keratoconus as revealed by corneal confocal microscopy. Clin. Exp. Optom., 91, 34–55. [DOI] [PubMed] [Google Scholar]
- 37. Erie, J.C., Patel, S.V., McLaren, J.W., Nau, C.B., Hodge, D.O. and Bourne, W.M. (2002) Keratocyte density in keratoconus. A confocal microscopy study(a). Am J. Ophthalmol., 134, 689–695. [DOI] [PubMed] [Google Scholar]
- 38. Shetty, R., Vunnava, K.P., Dhamodaran, K., Matalia, H., Murali, S., Jayadev, C., Murugeswari, P., Ghosh, A. and Das, D. (2019) Characterization of corneal epithelial cells in keratoconus. Transl. Vis. Sci. Technol., 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hayes, S., Boote, C., Tuft, S.J., Quantock, A.J. and Meek, K.M. (2007) A study of corneal thickness, shape and collagen organisation in keratoconus using videokeratography and X-ray scattering techniques. Exp. Eye Res., 84, 423–434. [DOI] [PubMed] [Google Scholar]
- 40. Mikula, E., Winkler, M., Juhasz, T., Brown, D.J., Shoa, G., Tran, S., Kenney, M.C. and Jester, J.V. (2018) Axial mechanical and structural characterization of keratoconus corneas. Exp. Eye Res., 175, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nielsen, K., Vorum, H., Fagerholm, P., Birkenkamp-Demtroder, K., Honore, B., Ehlers, N. and Orntoft, T.F. (2006) Proteome profiling of corneal epithelium and identification of marker proteins for keratoconus, a pilot study. Exp. Eye Res., 82, 201–209. [DOI] [PubMed] [Google Scholar]
- 42. Joseph, R., Srivastava, O.P. and Pfister, R.R. (2011) Differential epithelial and stromal protein profiles in keratoconus and normal human corneas. Exp. Eye Res., 92, 282–298. [DOI] [PubMed] [Google Scholar]
- 43. Chaerkady, R., Shao, H., Scott, S.G., Pandey, A., Jun, A.S. and Chakravarti, S. (2013) The keratoconus corneal proteome: loss of epithelial integrity and stromal degeneration. J. Proteome, 87, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shinde, V., Hu, N., Renuse, S., Mahale, A., Pandey, A., Eberhart, C., Stone, D., Al-Swailem, S.A., Maktabi, A. and Chakravarti, S. (2019) Mapping keratoconus molecular substrates by multiplexed high-resolution proteomics of Unpooled corneas. OMICS, 23:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabza, M., Karolak, J.A., Rydzanicz, M., Szczesniak, M.W., Nowak, D.M., Ginter-Matuszewska, B., Polakowski, P., Ploski, R., Szaflik, J.P. and Gajecka, M. (2017) Collagen synthesis disruption and downregulation of core elements of TGF-beta, hippo, and Wnt pathways in keratoconus corneas. Eur. J. Hum. Genet., 25, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szczesniak, M.W., Kabza, M., Karolak, J.A., Rydzanicz, M., Nowak, D.M., Ginter-Matuszewska, B., Polakowski, P., Ploski, R., Szaflik, J.P. and Gajecka, M. (2017) KTCNlncDB-a first platform to investigate lncRNAs expressed in human keratoconus and non-keratoconus corneas. Database (Oxford). 2017:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. You, J., Corley, S.M., Wen, L., Hodge, C., Hollhumer, R., Madigan, M.C., Wilkins, M.R. and Sutton, G. (2018) RNA-Seq analysis and comparison of corneal epithelium in keratoconus and myopia patients. Sci. Rep., 8, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shinde, V., Hu, N., Mahale, A., Maiti, G., Daoud, Y., Eberhart, C.G., Maktabi, A., Jun, A.S., Al-Swailem, S.A. and Chakravarti, S. (2020) RNA sequencing of corneas from two keratoconus patient groups identifies potential biomarkers and decreased NRF2-antioxidant responses. Sci. Rep., 10, 9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu, L., Yang, K., Fan, Q., Gu, Y., Zhang, B., Pang, C. and Ren, S. (2020) Exome sequencing identification of susceptibility genes in Chinese patients with keratoconus. Ophthalmic Genet. 41:518–525. [DOI] [PubMed] [Google Scholar]
- 50. Khaled, M.L., Bykhovskaya, Y., Gu, C., Liu, A., Drewry, M.D., Chen, Z., Mysona, B.A., Parker, E., McNabb, R.P., Yu, H.et al. (2019) PPIP5K2 and PCSK1 are candidate genetic contributors to familial keratoconus. Sci. Rep., 9, 19406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin, Q., Zheng, L., Shen, Z. and Jie, L. (2019) A novel splice-site variation in COL5A1 causes keratoconus in an Indian family. J. Ophthalmol., 2019, 2851380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith, T.F., Gaitatzes, C., Saxena, K. and Neer, E.J. (1999) The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci., 24, 181–185. [DOI] [PubMed] [Google Scholar]
- 53. Fry, A.M., O'Regan, L., Montgomery, J., Adib, R. and Bayliss, R. (2016) EML proteins in microtubule regulation and human disease. Biochem. Soc. Trans., 44, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 54. Li, Q., Wojciechowski, R., Simpson, C.L., Hysi, P.G., Verhoeven, V.J., Ikram, M.K., Hohn, R., Vitart, V., Hewitt, A.W., Oexle, K.et al. (2015) Genome-wide association study for refractive astigmatism reveals genetic co-determination with spherical equivalent refractive error: the CREAM consortium. Hum. Genet., 134, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Monda, J.K. and Cheeseman, I.M. (2018) The kinetochore-microtubule interface at a glance. J. Cell Sci., 131, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Filges, I., Bruder, E., Brandal, K., Meier, S., Undlien, D.E., Waage, T.R., Hoesli, I., Schubach, M., deBeer, T., Sheng, Y.et al. (2016) Stromme syndrome is a ciliary disorder caused by mutations in CENPF. Hum. Mutat., 37, 711. [DOI] [PubMed] [Google Scholar]
- 57. Waters, A.M., Asfahani, R., Carroll, P., Bicknell, L., Lescai, F., Bright, A., Chanudet, E., Brooks, A., Christou-Savina, S., Osman, G.et al. (2015) The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J. Med. Genet., 52, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Albers, C.A., Cvejic, A., Favier, R., Bouwmans, E.E., Alessi, M.C., Bertone, P., Jordan, G., Kettleborough, R.N., Kiddle, G., Kostadima, M.et al. (2011) Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat. Genet., 43, 735–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gunay-Aygun, M., Falik-Zaccai, T.C., Vilboux, T., Zivony-Elboum, Y., Gumruk, F., Cetin, M., Khayat, M., Boerkoel, C.F., Kfir, N., Huang, Y.et al. (2011) NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet alpha-granules. Nat. Genet., 43, 732–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kahr, W.H., Hinckley, J., Li, L., Schwertz, H., Christensen, H., Rowley, J.W., Pluthero, F.G., Urban, D., Fabbro, S., Nixon, B.et al. (2011) Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat. Genet., 43, 738–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Noonan, D., Fulle, A., Valante, P., Cai, S., Horigan, E., Sasaki, S., Yamada, Y. and hassell, J. (1991) The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin a chain, LDL-receptorm and N-CAM. J. Biol. Chem., 266, 22939–22947. [PubMed] [Google Scholar]
- 62. Cailhier, J.F., Sirois, I., Laplante, P., Lepage, S., Raymond, M.A., Brassard, N., Prat, A., Iozzo, R.V., Pshezhetsky, A.V. and Hebert, M.J. (2008) Caspase-3 activation triggers extracellular cathepsin L release and endorepellin proteolysis. J. Biol. Chem., 283, 27220–27229. [DOI] [PubMed] [Google Scholar]
- 63. Nicole, S., Davoine, C.S., Topaloglu, H., Cattolico, L., Barral, D., Beighton, P., Hamida, C.B., Hammouda, H., Cruaud, C., White, P.S.et al. (2000) Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat. Genet., 26, 480–483. [DOI] [PubMed] [Google Scholar]
- 64. Bilsland, C.A., Diamond, M.S. and Springer, T.A. (1994) The leukocyte integrin p150,95 (CD11c/CD18) as a receptor for iC3b. Activation by a heterologous beta subunit and localization of a ligand recognition site to the I domain. J. Immunol., 152, 4582–4589. [PubMed] [Google Scholar]
- 65. Zylka, M.J., Dong, X., Southwell, A.L. and Anderson, D.J. (2003) Atypical expansion in mice of the sensory neuron-specific Mrg G protein-coupled receptor family. Proc. Natl. Acad. Sci. U. S. A., 100, 10043–10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Olson, W., Abdus-Saboor, I., Cui, L., Burdge, J., Raabe, T., Ma, M. and Luo, W. (2017) Sparse genetic tracing reveals regionally specific functional organization of mammalian nociceptors. elife, 6, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Portal, C., Rompolas, P., Lwigale, P. and Iomini, C. (2019) Primary cilia deficiency in neural crest cells models anterior segment dysgenesis in mouse. elife, 8, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kenney, M.C. and Brown, D.J. (2003) The cascade hypothesis of keratoconus. Cont. Lens Anterior Eye, 26, 139–146. [DOI] [PubMed] [Google Scholar]
- 69. Cullinane, A.R., Schaffer, A.A. and Huizing, M. (2013) The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic, 14, 749–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mayer, L., Jasztal, M., Pardo, M., Aguera de Haro, S., Collins, J., Bariana, T.K., Smethurst, P.A., Grassi, L., Petersen, R., Nurden, P.et al. (2018) Nbeal2 interacts with Dock7, Sec16a, and Vac14. Blood, 131, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Klintworth, G.K. (2009) Corneal dystrophies. Orphanet J. Rare Dis., 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Foster, J., Wu, W.H., Scott, S.G., Bassi, M., Mohan, D., Daoud, Y., Stark, W.J., Jun, A.S. and Chakravarti, S. (2014) Transforming growth factor beta and insulin signal changes in stromal fibroblasts of individual keratoconus patients. PLoS One, 9, e106556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Foster, J.W., Shinde, V., Soiberman, U.S., Sathe, G., Liu, S., Wan, J., Qian, J., Dauoud, Y., Pandey, A., Jun, A.S.et al. (2018) Integrated stress response and decreased ECM in cultured stromal cells from keratoconus corneas. Invest. Ophthalmol. Vis. Sci., 59, 2977–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Karolak, J.A., Gambin, T., Rydzanicz, M., Polakowski, P., Ploski, R., Szaflik, J.P. and Gajecka, M. (2020) Accumulation of sequence variants in genes of Wnt signaling and focal adhesion pathways in human corneas further explains their involvement in keratoconus. PeerJ., 8, e8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Manning, B.D. and Toker, A. (2017) AKT/PKB Signaling: navigating the network. Cell, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Burute, M. and Kapitein, L.C. (2019) Cellular logistics: unraveling the interplay between microtubule organization and intracellular transport. Annu. Rev. Cell Dev. Biol., 35, 29–54. [DOI] [PubMed] [Google Scholar]
- 77. Bangs, F. and Anderson, K.V. (2017) Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol., 9, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang, L. and Dynlacht, B.D. (2018) The regulation of cilium assembly and disassembly in development and disease. Development, 145, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ozkinay, F., Atik, T., Isik, E., Gormez, Z., Sagiroglu, M., Sahin, O.A., Corduk, N. and Onay, H. (2017) A further family of Stromme syndrome carrying CENPF mutation. Am. J. Med. Genet. A, 173, 1668–1672. [DOI] [PubMed] [Google Scholar]
- 80. Butler, M.T. and Wallingford, J.B. (2017) Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol., 18, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lawrence, P.A. and Casal, J. (2018) Planar cell polarity: two genetic systems use one mechanism to read gradients. Development, 145, 1–10. [DOI] [PubMed] [Google Scholar]
- 82. Lechner, J., Bae, H.A., Guduric-Fuchs, J., Rice, A., Govindarajan, G., Siddiqui, S., Abi Farraj, L., Yip, S.P., Yap, M., Das, M.et al. (2013) Mutational analysis of MIR184 in sporadic keratoconus and myopia. Invest. Ophthalmol. Vis. Sci., 54, 5266–5272. [DOI] [PubMed] [Google Scholar]
- 83. Lek, M., Karczewski, K.J., Minikel, E.V., Samocha, K.E., Banks, E., Fennell, T., O'Donnell-Luria, A.H., Ware, J.S., Hill, A.J., Cummings, B.B.et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M.et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. DePristo, M.A., Banks, E., Poplin, R., Garimella, K.V., Maguire, J.R., Hartl, C., Philippakis, A.A., delAngel, G., Rivas, M.A., Hanna, M.et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sobreira, N., Schiettecatte, F., Boehm, C., Valle, D. and Hamosh, A. (2015) New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum. Mutat., 36, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Genomes Project, C., Abecasis, G.R., Auton, A., Brooks, L.D., DePristo, M.A., Durbin, R.M., Handsaker, R.E., Kang, H.M., Marth, G.T. and McVean, G.A. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. MacArthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., Junkins, H., McMahon, A., Milano, A., Morales, J.et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.