

Abstract
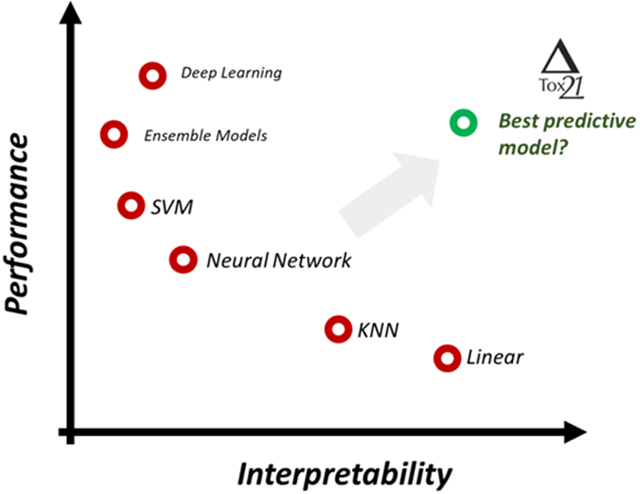
Selecting a model in predictive toxicology often involves a trade-off between prediction performance and explainability: should we sacrifice the model performance to gain explainability, or vice versa? Here we present a comprehensive study to assess algorithm and feature influences on model performance in chemical toxicity research. We conducted over 5000 models for a Tox21 bioassay dataset of 65 assays and ~7600 compounds. Seven molecular representations as features and twelve modeling approaches varying in complexity and explainability were employed to systematically investigate the impact of various factors on model performance and explainability. We demonstrated that endpoints dictated a model’s performance, regardless of the chosen modeling approach including deep learning and chemical features. Overall, more complex models such as (LS-)SVM and Random Forest performed marginally better than simpler models such as linear regression and KNN in the presented Tox21 data analysis. Since a simpler model with acceptable performance often also is easy to interpret for the Tox21 dataset, it clearly was the preferred choice due to its better explainability. Given that each dataset had its own error structure both for dependent and independent variables, we strongly recommend that it is important to conduct a systematic study with a broad range of model complexity and feature explainability to identify model balancing its predictivity and explainability.
Keywords: Tox21 bioassay, QSAR model, machine learning, predictive toxicology, explainable artificial intelligence (AI), explainability, interpretability
Graphical Abstract
Background
Artificial Intelligence (AI) has been playing an increasingly vital role in a broad range of scientific research and applications, including clinical diagnosis/prognosis, natural language processing, speech and face recognition, and machine translation. Recent development of neural networks, commonly known as Deep Learning (DL), have further speeded up development of AI by taking advantage of Big Data and increased computational power. Highlighted as one trigger event in 2012, the award-winning DL model (AlexNet) held a top-5 error rate of 15.3% in the ImageNet Large Scale Visual Recognition Challenge (ILSVRC), demonstrating a significant improvement over the second-best model’s top-5 error rate of 26.2%1. Since then, complex modeling algorithms such as DL have gained wide acceptance, leading to better model performance, especially in Big Data analysis.
In predictive toxicology, AI and Machine Learning (ML) also has been widely investigated for chemical risk assessment and drug safety evaluation. In the past decades, our group developed numerous predictive toxicology approaches and tools in this area, particularly for drug-induced liver injury (DILI)2–6 and toxicogenomics7–9. The combination of high throughput screening and ML has also become an important direction in predictive toxicology10–12. For instance, the Tox21 project has screened over 10000 chemical compounds via robotic automated high-throughput in vitro assays to measure corresponding bioactivities, an unprecedented achievement which provided millions of chemical bioactivity profiles and data points.10, 13
One of the key ML applications in predictive toxicology is to predict chemical bioactivities, including toxicity with molecule structure. Traditionally known as QSARs (quantitative structure activity relationships), this field has seen significant advancements with modern machine learning approaches, such as Support Vector Machine (SVM), Random Forest, and recently DL9, 14, 15. For instance, several DL approaches have been developed recently with QSAR studies16–19, most of which reported improved prediction accuracy for different tasks. Along with improved performance, another advantage of some DL approaches is their innate ability to work with molecular representation as SMILES, chemical graphs or images and thus bypassing the manual feature selection process.20, 21 While on one side this may remove a bias of a researcher to one or another type of descriptors it may also result in DL models which are more opaque since the reasoning for the model decisions is buried amid millions of neural network weights.
The problem of model interpretations is actively pursued in chemoinfomatics22 where it is going beyond the traditional QSAR and is highly relevant in other fields of science23. A prerequisite for a trustworthy model is that its performance can be explained. Explainability can be defined as an AI behavior that can be understood and accepted by humans, which involves many concepts and aspects such as transparency, interpretability, causality, transferability, accessibility, etc.24 This is a topic of active research in explainable Artificial Intelligence and some promising development in this area were recently reported elsewhere23, 25. Still some methods, such as a linear regression, k-nearest neighbors (kNN) or decision trees, which are used by researches since many years, are considered as more interpretable since, e.g., the weight of features in regression could be interpreted as its importance to the decision making. In predictive toxicology, the driving features are descriptors of a substance’s biological and chemical properties. Since human experts could explain influence of physiochemical descriptors, a predictive model developed with them generally could be more easily interpretable than the one using more complex features such as hashed fingerprints, graphical/geometric depictions and/or ML-derived molecular representations. With that said, the selection of chemical features and the complexity of modeling algorithm are currently the key factors to determine the explainability in predictive toxicological research.
The choice of modeling algorithm frequently involves a perceived trade-off between predictivity and explainability. In other words, increasing predictivity sometimes could lead to lower explainability; the reverse also could be true. The challenge is how we can balance predictivity and explainability to achieve a trustworthy model. To achieve that, we first need to understand how much the selection of model algorithms, as well as chemical features, would impact predictivity. In this study, we report a case study using the Tox21 bioassay activity dataset.26, 27 We mostly focus on the transparency and interpretability versus prediction performance of analyzed methods for Tox21 endpoints, which is attributed to the types of the modeling algorithms and the descriptors. We provide a broad view of ML applications in predictive toxicology by systematically investigating the influence of assay endpoints (68 assays), modeling algorithms (twelve ML and deep learning algorithms), and features (seven chemical representations).
Methods and Materials
Tox21 Dataset
Tox21 bioassay activity data (the Tox21 Dataset13) were collected for 68 bioassay endpoints27 and 8948 compounds, of which each was tested in at least one assay. Chemical bioactivity data was preprocessed and categorized into four major classes: active agonist, active antagonist, inactive and inconclusive28. In this study, both active agonists and active antagonists were considered positive compounds, and inactives were considered negative. Only one active category was considered in each assay; i.e., if one assay contained more active agonists than active antagonists, we used the active agonists as positive in the analysis. Inconclusive compounds were excluded from all assays/endpoints. Chemicals were further deduplicated based on their InChI keys. The final number of positive and negative samples in each assay was summarized in Supplementary Table S1 and all processed data are available as Supplementary Table S2.
In addition, the chemical similarity of a Tox21 assay endpoint was calculated using a within-group chemical similarity (S) score. S represented the chemical diversity in accordance with the endpoint, which was measured according to formula (1). In the formula, m and n represent the number of compounds in positive and negative classes, respectively. Jaccardij indicates the Jaccard similarity coefficient (index) between compounds i and j, which was calculated based on RDkit fingerprints. As formulated, higher S value corresponds to endpoint with compounds that are more similar within the class of actives.
| (1) |
Predictive modeling algorithms
In total, 12 modeling algorithms were included in this study; These modeling algorithms can be categorized into four classes: (1) neural networks; (2) decision trees ensemble methods; (3) SVMs; and (4) simple algorithms. For neural networks, we deployed a 3-Layer neural-network (MLP-3), 7-Layer neural-network (DNN19), Associative Neural Network (ASNN-MTL)29, and a Multi-task Learning version of the 7-Layer Neural-network (DNN-MTL)19. Algorithm ending with MTL were performed with multi-task learning framework. Four decision trees ensemble methods, XGBoost30, AdaBoost, GradientBoosting, and Random Forest, were used. For SVMs, we applied rbf SVM (SVM)31, 32 and Least-Squares SVM (LS-SVM)33 optimized for GPU-based computing. Lastly, Linear Regression and KNN were used as for simple algorithms32. These modeling algorithms provided broad coverage of modeling complexity while representing popular modeling algorithms in the field. Linear regression and KNN are simple methods that are easier to understand, while SVMs and neural networks are relatively more complicated and difficult to interpret. The use of ensembles for decision trees despite it added more complexity was also shown to significantly improve their performances30, 34. Deep neural-networks are one of the recent popular modeling algorithms and often have good predictivity compared to other algorithms especially in studies dealing with large data14, 21, 35.
Among the 12 studied algorithms, XGBOOST, LS-SVM, DNN, ASNN-MTL and DNN-MTL were performed in OCHEM platform (http://ochem.eu), which has LS-SVM and DNN(-MTL) implemented using GPU.36 The other seven modeling algorithms were performed on local computing cluster at FDA. As designed, the training and testing compounds for two experiment sites were exactly the same, but we applied slightly different data-preprocessing strategy on each site. For example, correlation filters (>0.95) was used in OCHEM platform for feature reduction during data pre-processing. We checked the influence of the preprocessing on the performance of the Random Forest, which was calculated with the same setting of hyperparameters in OCHEM and FDA sites but did not observe any systematic bias. The parameters used in these modeling algorithms, if not specified, were set to defaults, which were optimized in multiple studies performed by their authors. The details of modeling algorithms used in this study are summarized in Table 1.
Table 1.
Brief summary of modeling algorithms used in this study.
| SYNONYM | MODELING ALGORITHM | CATEGORY | PARAMETERS |
|---|---|---|---|
|
| |||
| LINEAR | Linear regression | Simple Model | |
| KNN | K-Nearest Neighbors | Simple Model | K=5 (default) |
| RF | Random Forest | Ensemble Model | n=100 (default) |
| ABOOST | Adaptive Boosting | Ensemble Model | n=50 (default) |
| GBOOST | Gradient Boosting | Ensemble Model | n=100 (default) |
| XGBOOST | Extreme Gradient Boosting | Ensemble Model | |
| SVM | Support Vector Machine | SVMs | Kernel=rbf; C=100; gamma=’scale’ |
| LS-SVM | Least-Squares SVM | SVMs | |
| MLP3 | 3 layers Multi-layer Perceptron | Neural Networks | nodes=[32, 64, 32]; |
| DNN | 7 layers Neuro Networks | Neural Networks | nodes=[512:256:128:64:32:16] |
| DNN-MTL | Multi-Task Learning of DNN | Neural Networks | Same as above |
| ASNN-MTL | Associative neural network | Neural Networks | Ensemble of 64 models with one hidden layer containing 3 neurons |
Chemical Features
We evaluated seven different types of chemical features to represent the chemical structures, five chemical fingerprints (i.e., RDKit37, ECFP4, FCFP4, Extended Functional Groups (EFG)38 and ToxPrint39) and two QSAR descriptors (i.e., MordRed40 and Mold241). RDKit fingerprint was developed by RDKit37 and was calculated with default parameters (nbits=2048). Extended-Connectivity Fingerprints (ECFP4 and FCFP4) are atom-based and feature-based chemical fingerprints, both of which were calculated by using Morgan fingerprints generated by the RDKit, with radius=2 and bit length 1024. ToxPrints are based on the publicly available ToxPrint chemotypes (v2.0_r711, https://toxprint.org/) generated within the associated ChemoTyper application (https://chemotyper.org/). EFG is an extension of a functional group set previously implemented by the CheckMol42 that also covers heterocyclic compound classes and periodic table groups38. ToxPrint chemotypes consist of 729 uniquely defined chemical features coded in XML based Chemical Subgraphs and Reactions Markup Language (CSRML). The numbers of features generated for each type of descriptors are listed in Table 2.
Table 2.
Brief summary of feature generation tools used in this study.
| SYNONYM | #FEATURES | CATEGORY | REFERENCE |
|---|---|---|---|
|
| |||
| RDKIT | 2048 | Chemical fingerprints | https://www.rdkit.org |
| ECFP4 | 1024 | Chemical fingerprints | https://www.rdkit.org |
| FCFP4 | 1024 | Chemical fingerprints | https://www.rdkit.org |
| EFG | 583 | Chemical fingerprints | Citation38 |
| TOXPRINT | 729 | Chemical fingerprints | https://toxprint.org, 39 |
| MORDRED | 1825 | QSAR descriptors | Citation40 |
| MOLD2 | 777 | QSAR descriptors | Citation41 |
Results
Study workflow
The overall study design is depicted in Figure 1a. We used all publicly available assays from the Tox21 dataset13 to take advantage of the diversity in assayed endpoints (i.e., 68 bioactivity endpoints). In total, 8948 compounds were profiled by at least one assay. After data pre-processing to remove chemical duplicates, 7660 compounds were retained for the analyses. These compounds were split on training and test set comprising 7000 and 660 compounds, respectively (see Supplementary Table S2). We conducted binary classification and for each assay only active and inactive compounds were considered. The active compounds could either be active agonists or active antagonists, depending on the majority group of the assay (see Materials and Methods for details). For duplicated compounds, only compound with consistent bioactivities in the same assay were considered: others were labeled as inconclusive and discarded. Finally, we removed three assays that did not have enough active compounds (<=20), therefore 65 Tox21 assays remained for modeling analysis. The final processed-ready dataset is available for download from http://ochem.eu and is also included as Supplementary Table S2.
Figure 1.

The overview of the workflow used to analyze the data. (a) Overall study design. (b) Construct and evaluate predictive model with selected predictor, modeling algorithm and endpoint.
For each endpoint, we developed 84 models with exhaustive combination of seven types of molecular features in conjunction with 12 modeling algorithms. Specifically, we used RDkit, ECFP4, FCFP4, EFG, ToxPrint, Mold2 and MordRed to measure/represent different types of compound fingerprints or QSAR descriptors. Four major categories of 12 modeling algorithms were used to represent the varying degrees of complexity in modeling algorithms.
Figure 1b shows the general modeling pipeline with a single experiment (i.e., one feature set and one modeling algorithm applied to data from a single assay), in which a model pairing a modeling method with chemical features was evaluated by 5-fold cross-validation. During the cross-validation, we split the training dataset into 5 folds, where 4 folds were used for training and the other fold was used for validation. Next, the models were tested on the hold-out samples from the test set, which contains 660 unseen compounds. Final model performance thereafter was measured by the average AUC of the training set as well as the AUC of testing set.
Model predictive performance
Overall results on the testing data are shown in Figure 2a. As shown, each cell in this hierarchical clustering map (HCA) is the average testing AUC from the nested cross validation results in one model. The x-axis contains 65 Tox21 assays. The HCA map contains 84 rows, which represented all combinations of seven feature sets and 12 modeling algorithms.
Figure 2.

(a) Heatmap of all Tox21 assays across different modeling algorithms and features. (b) Overall, collapsed performance for all Tox21 assays. The validation and testing results are consistent across all endpoints.
First, we found that these 65 Tox21 assay endpoints showed very different performance patterns. Some endpoints always had a high AUC regardless of the type of feature or modeling algorithm used. Contrary to that some endpoints showed a consistently low AUC across all feature-algorithm combinations. With respect of their performance similarity models using the same chemical features tended to cluster together.
The overall predicting performance for each Tox21 assay was summarized in the box-plot representation (Figure 2b). Each bar represented a collapsed result of one particular modeling algorithm, by combining all features and endpoints. Training (i.e., Cross-validation) and testing (i.e., hold-out) results were presented in blue and orange boxes, respectively. As shown, there was no significant performance gap between Training and Testing results, indicating that the developed models were robust. Detailed model performances (AUC) for each Tox21 assay were summarized in Table S1.
Overall performance of model algorithms and features
Further analysis was applied to inspect the overall influence of chemical features and modeling algorithms, by averaging the results across all 65 Tox21 endpoints. The 5-fold CV and the hold-out testing results were summarized in Table 3.
Table 3.
Averaged performance of seven feature types and twelve modeling algorithms.
| Model/Feature | RDKIT | ECFP4 | FCFP4 | EFG | TOXPRINT | MOLD2 | MORDRED | Average | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| 5-fold CV | Test | 5-fold CV | Test | 5-fold CV | Test | 5-fold CV | Test | 5-fold CV | Test | 5-fold CV | Test | 5-fold CV | Test | 5-fold CV | Test | |
|
| ||||||||||||||||
| DNN-MTL | 0.79 | 0.83 | 0.79 | 0.80 | 0.79 | 0.76 | 0.86 | 0.81 | 0.72 | 0.77 | 0.82 | 0.84 | 0.81 | 0.84 | 0.8 | 0.81 |
| DNN | 0.78 | 0.81 | 0.77 | 0.79 | 0.78 | 0.79 | 0.82 | 0.77 | 0.71 | 0.70 | 0.78 | 0.81 | 0.77 | 0.81 | 0.77 | 0.78 |
| ASNN-MTL | 0.77 | 0.76 | 0.80 | 0.84 | 0.79 | 0.84 | 0.84 | 0.81 | 0.73 | 0.79 | 0.80 | 0.81 | 0.66 | 0.67 | 0.77 | 0.79 |
| MLP3 | 0.76 | 0.80 | 0.75 | 0.78 | 0.75 | 0.77 | 0.77 | 0.75 | 0.76 | 0.80 | 0.83 | 0.83 | 0.83 | 0.85 | 0.78 | 0.8 |
| XGBOOST | 0.79 | 0.81 | 0.76 | 0.77 | 0.75 | 0.77 | 0.80 | 0.78 | 0.67 | 0.69 | 0.81 | 0.85 | 0.81 | 0.85 | 0.77 | 0.79 |
| GBOOST | 0.83 | 0.85 | 0.81 | 0.83 | 0.80 | 0.81 | 0.83 | 0.83 | 0.81 | 0.83 | 0.85 | 0.87 | 0.84 | 0.87 | 0.83 | 0.84 |
| ABOOST | 0.77 | 0.79 | 0.76 | 0.78 | 0.77 | 0.78 | 0.80 | 0.79 | 0.78 | 0.81 | 0.82 | 0.83 | 0.89 | 0.84 | 0.79 | 0.8 |
| RF | 0.84 | 0.84 | 0.83 | 0.83 | 0.82 | 0.81 | 0.86 | 0.83 | 0.82 | 0.81 | 0.86 | 0.88 | 0.86 | 0.88 | 0.84* | 0.84* |
| LS-SVM | 0.84 | 0.86 | 0.82 | 0.83 | 0.81 | 0.84 | 0.84 | 0.82 | 0.73 | 0.74 | 0.86 | 0.88 | 0.87* | 0.88* | 0.83 | 0.83 |
| SVM | 0.84 | 0.85 | 0.83 | 0.84 | 0.81 | 0.82 | 0.81 | 0.80 | 0.81 | 0.81 | 0.85 | 0.87 | 0.85 | 0.88 | 0.83 | 0.84 |
| KNN | 0.73 | 0.77 | 0.73 | 0.74 | 0.72 | 0.73 | 0.71 | 0.70 | 0.72 | 0.75 | 0.76 | 0.79 | 0.77 | 0.80 | 0.73 | 0.75 |
| Linear | 0.79 | 0.81 | 0.81 | 0.82 | 0.81 | 0.82 | 0.81 | 0.78 | 0.81 | 0.83 | 0.82 | 0.83 | 0.82 | 0.85 | 0.81 | 0.82 |
|
| ||||||||||||||||
| Average | 0.79 | 0.81 | 0.79 | 0.80 | 0.78 | 0.80 | 0.81 | 0.79 | 0.76 | 0.78 | 0.82* | 0.84* | 0.81 | 0.84 | ||
Stars indicate the best performing descriptor sets and algorithms.
Regarding to the feature types, the best feature type was Mold2 (AUC-CV=0.82; AUC-test=0.84). The weakest feature type was ToxPrint (AUC-CV=0.76; AUC-test=0.78). In general, QSAR descriptors (i.e., Mold2 and MordRed) performed better than chemical fingerprints. We also observed that EFG outperformed ToxPrint, both of which were kinds of structural alerts or functional groups.
On the other hand of modeling algorithms, we found GBoost, RF and SVMs showed better predicting performance than other categories of algorithms, and the best modeling algorithms among 12 we tested was RF (AUC-CV=0.84; AUC-test=0.84); the relatively weakest modeling algorithm was KNN (AUC-CV=0.73; AUC-test=0.75). In addition, we did not observe better performance of more complicated models such as the neural networks, compared to simple models of Linear Regression, which provided similar results on average. In addition, XGBOOST did not improve the model predictivity compared to GBoost. We also observed that Multi-task learning framework may improve the modeling predictivity since DNN-MTL outperformed DNN thus confirming results of other studies.
For the single pair of model-feature combination, LS-SVM with MordRed held the best performance in average 65 Tox21 endpoints prediction (AUC-CV=0.87; AUC-test=0.88). Note that the predictivity differences among top combinations were marginal; such as LS-SVM-Mold2, LS-SVM-MordRed, RF-Mold2 and RF-MordRed, all of which held AUC around 0.88 in hold-out testing result.
Parameter influence on predictivity of SVM and RF
Based on the overall prediction results, SVM and Random Forest (RF) showed a good predictivity among all feature types and endpoints we tested. Both RF and SVM have hyper-parameters, which may have significant impacts on the model performance. In aims to understand how much the hyper-parameter tuning could affect the predictivity of both SVM and RF, we performed a grid search analysis to fine-tune “cost parameter” (C) and gamma (g) for SVM (with default RBF kernel) and “number of trees” (n) and “minimal number of samples in leaf node” (L) for RF, respectively.
As shown in Figure 3a, the hyper-parameters showed a large impact on SVM models. As a “proper” set of hyper-parameters (e.g., C=10 and g = “scale”)43 would achieve over 0.8 AUC across all types of features and endpoints where an “improper” set of hyper-parameters (e.g., C=0.1 and g=1) would fail completely (Averaged AUC<0.5). On the other hand, we found that the influence of hyper-parameters on RF models was much smaller in comparison to SVM models (Figure 3b), as the “worst” set of hyper-parameters (e.g., n=10; L=20) still could get an averaged AUC around 0.75. Taken together, our findings demonstrated that hyper-parameter selection can have a larger impact on some modeling algorithms such as SVM, while less affects other modeling algorithms such as RF.
Figure 3.

Hyper-Parameter tuning result for (a) SVM and (b) Random Forest.
Influence of sample size and chemical similarity on endpoint predictability
Figure 4 shows an assessment of the other two important innate properties of a dataset that affect model performance, sample size and chemical similarity. Due to data availability, sample size varied from endpoint to endpoint, and the positive sample size (i.e., number of active agonist or antagonist in the dataset) was usually much smaller than negative sample size. A smaller positive sample leads to more imbalanced dataset, which could affect the model performance. We therefore analyzed the correlation between model performance and positive sample ratio for the analyzed endpoint (Figure 4, light gray bars). As shown, we found that the positive sample size had little effect on the averaged model performance. However the sample size influenced the variation of the performance and we observed wider boxes and quantiles in assays with smaller positive sample sizes. In addition, lower performed assays also tended to have larger variations of performance, which indicated the modeling algorithm and feature selection could more affect these low-performed and less sample size endpoints.
Figure 4.

Influences of positive sample ratio (light-gray bars) and sample within-group similarity (green/red/black bars) on endpoint performance (blue box plot). Green and red bars are the top ten highest performed assays with positive and negative S score, respectively.
With respect to the chemical similarity of endpoints, the S scores were negative for most of the Tox21 assays, indicating that for most assays, compounds within the respective positive/active class shared low structural similarity (Figure 4, Red/Green/Black bars). Meanwhile, we found that the S scores for eight of ten endpoints with high predictability were positive, implying that higher structural similarity within the positive/active compound class may contribute to the higher predictive performance of the models for these assays. However, endpoints with negative S score did not necessarily result in poor performance; for example, although assays of tox21-ahr-p1 and tox21-pr-bla-antagonist (green bars in Figure 4) had negative S score −0.018 and −0.037 respectively, they still had high AUC (>0.85) in both training and testing results.
Discussion
We conducted a systematic investigation on the choice of modeling approaches (12 ML/DL algorithms used) and chemical features (seven molecular representations) in predictive toxicology, with a specific focus on comparative analysis of model performance and explainability and the trade-off between them. Results demonstrated that the assay or endpoint itself was the largest determining factor for model performance; a good performance was reached for a predictable endpoint (high predictability) regardless of the choice of modeling algorithm or feature type used44. As implied in Figure 4, assays with lower performance tended to have larger variations. The influence of modeling algorithm and features was more pronounced for such endpoints as well as for those with more imbalanced dataset (i.e., smaller positive sample sizes). For such endpoints it could be important to conduct a systematic analysis by using a broad range of approaches of various model complexity and feature explainability. For the Tox21 datasets studied here, we found that using simple modeling approaches such as Linear Regression in a number of cases provided models with similar performance to those of more complex approaches, such as neural networks. Such models could be more preferable in the context of the computational toxicology due to better balancing of their predictivity and interpretability. Of course, the reported in this study results could be influenced by type of the data and used descriptors, but the use of simple baseline models should not be ignored.
Not all datasets have the same complexity, which further emphasizes the need for evaluating a broad range of modeling approaches and molecular representations. Three innate data properties are of special importance: endpoint predictability, data imbalance, and size. With respect to endpoint predictability, we only examined the chemical structure similarity within and across class labels without considering the quality of the endpoints measurements themselves. The results implied that a high chemical structure-driven predictability likely resulted in a good performance, but the reverse was not entirely apparent.
An algorithm with a simple architecture, such as linear regression may not be the most powerful, but it will be easily explainable when using interpretable descriptors. Algorithms with more complicated architectures, such as (LS-)SVM or (deep) neural networks, may have better statistical performance but can be more difficult to interpret. In this study, we did observe that some more complex modeling algorithms had overall better prediction accuracy as compared to simple algorithms such as linear regression and KNN. However, the influence of a modeling algorithm was not as significant as the nature of the endpoint itself and in a number of cases simpler and easier to interpret models with similar performances were obtained. Selecting a complex but less explainable model could hinder its use due to possible concerns with interpretation of its predictions for new data.
Therefore, when considering both predictivity and explainability in the context of chemical feature based Tox21 data analysis, we recommend do not overlook using simple models: they provide higher explainability while still can have similar performance as some more complicated approaches. Another strategy is to increase the explainability of complex models via model-agnostic approaches45. With more such approaches being developed46–48 and with their wider availability, complex models such as DL networks may hold great potential for improved explainability. In their absence an interpreting a complex model is much more difficult than interpreting a simple model such as linear regression. Given that each dataset has its own error structure both for dependent and independent variables, we strongly recommend that it is important to conduct a systematic study with a broad range of method complexity and feature explainability to select a model, which balance its predictivity and explainability
Note that in this study we only considered chemical feature-based models, while recently we observed many deep learning models now directly analyse chemical structure such as SMILE string, InChI key, 3D image as the model input49, 50. Directly using chemical structure instead of features may be a game changer to the predictive toxicology, just like the current image analysis nowadays will directly use the raw image rather than human-engineered features. The investigation and comparison between feature-based and feature-free models also need to be comprehensively performed. In addition, consensus modeling could also be an effective way to improve model predictivity51. Evaluation the explainability of consensus model is also a challenge and the objective of future studies. While we plan to perform such studies ourselves we also encourage the other researchers to analyze the data of this study, which contain nearly 440k measurements for 65 properties (Supplementary table S2) in order to propose and benchmark approaches balancing predictivity and explainability of models.
Future directions can also include evaluating metrics to qualitatively or quantitatively measure the interpretability on demand, in order to investigate how much interpretability could be gained by using different approaches and whether they contribute the same interpretations. In addition, multi-task learning was proved to be an efficient way to improve the model performance by sharing the modeling architectures among different predictive endpoints14, 17, 19. We also observed the same tendency in this study but a more comprehensive investigation on the effects of multi-task learning compared to single-task learning would be critical to better elucidate impact of these methods on the predictive toxicology.
Supplementary Material
Table S1. Averaged training and testing AUC for 65 Tox21 assays
Table S2. Processed dataset of the Chemicals and Tox21 Endpoints used in this study
Acknowledgements
This research was supported in part by the Intramural/Extramural research program of the NCATS, NIH and by the China Scholarship Council (CSC) for ZX (201706880010). The authors thank Joanne Berger, FDA Library, for manuscript editing assistance.
Footnotes
Disclaimer: The views presented in this article do not necessarily reflect those of the U.S. Food and Drug Administration or the National Institutes of Health. Any mention of commercial products is for clarification and is not intended as an endorsement.
References
- (1).Krizhevsky A, Sutskever I, and Hinton GE (2012) Imagenet classification with deep convolutional neural networks, In Advances in neural information processing systems pp 1097–1105. [Google Scholar]
- (2).Chen M, Borlak J, and Tong W (2013) High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 58, 388–396. [DOI] [PubMed] [Google Scholar]
- (3).Chen M, Borlak J, and Tong W (2016) A Model to predict severity of drug-induced liver injury in humans. Hepatology 64, 931–940. [DOI] [PubMed] [Google Scholar]
- (4).Wu L, Liu Z, Auerbach S, Huang R, Chen M, McEuen K, Xu J, Fang H, and Tong W (2017) Integrating Drug’s Mode of Action into Quantitative Structure–Activity Relationships for Improved Prediction of Drug-Induced Liver Injury. Journal of chemical information and modeling 57, 1000–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hong H, Thakkar S, Chen M, and Tong W (2017) Development of decision forest models for prediction of drug-induced liver injury in humans using a large set of FDA-approved drugs. Scientific reports 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Khadka KK, Chen M, Liu Z, Tong W, and Wang D (2020) Integrating adverse outcome pathways (AOPs) and high throughput in vitro assays for better risk evaluations, a study with drug-induced liver injury (DILI). ALTEX-Alternatives to animal experimentation 37, 187–196. [DOI] [PubMed] [Google Scholar]
- (7).Tong W, Cao X, Harris S, Sun H, Fang H, Fuscoe J, Harris A, Hong H, Xie Q, and Perkins R (2003) ArrayTrack--supporting toxicogenomic research at the US Food and Drug Administration National Center for Toxicological Research. Environmental health perspectives 111, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bushel PR, and Tong W (2018) Integrative Toxicogenomics: Analytical Strategies to Amalgamate Exposure Effects With Genomic Sciences. Frontiers in genetics 9, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu Z, Huang R, Roberts R, and Tong W (2019) Toxicogenomics: a 2020 vision. Trends in Pharmacological Sciences 40, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huang R, Xia M, Sakamuru S, Zhao J, Shahane SA, Attene-Ramos M, Zhao T, Austin CP, and Simeonov A (2016) Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nature communications 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Luechtefeld T, Marsh D, Rowlands C, and Hartung T (2018) Machine learning of toxicological big data enables read-across structure activity relationships (RASAR) outperforming animal test reproducibility. Toxicological Sciences 165, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Thakkar S, Li T, Liu Z, Wu L, Roberts R, and Tong W (2020) Drug-induced liver injury severity and toxicity (DILIst): Binary classification of 1279 drugs by human hepatotoxicity. Drug discovery today 25, 201–208. [DOI] [PubMed] [Google Scholar]
- (13).Richard AM, Huang R, Waidyanatha S, Shinn P, Collins BJ, Thillainadarajah I, Grulke CM, Williams AJ, Lougee RR, and Judson RS (2020) The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chemical Research in Toxicology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mayr A, Klambauer G, Unterthiner T, and Hochreiter S (2016) DeepTox: toxicity prediction using deep learning. Frontiers in Environmental Science 3, 80. [Google Scholar]
- (15).Ghasemi F, Mehridehnavi A, Perez-Garrido A, and Perez-Sanchez H (2018) Neural network and deep-learning algorithms used in QSAR studies: merits and drawbacks. Drug Discov. Today 23, 1784–1790. [DOI] [PubMed] [Google Scholar]
- (16).Ghasemi F, Mehridehnavi A, Fassihi A, and Pérez-Sánchez H (2018) Deep neural network in QSAR studies using deep belief network. Applied Soft Computing 62, 251–258. [Google Scholar]
- (17).Zakharov AV, Zhao T, Nguyen D-T, Peryea T, Sheils T, Yasgar A, Huang R, Southall N, and Simeonov A (2019) Novel consensus architecture to improve performance of large-scale multitask deep learning QSAR models. Journal of Chemical Information and Modeling 59, 4613–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chakravarti SK, and Alla SRM (2019) Descriptor free QSAR modeling using deep learning with long short-term memory neural networks. Frontiers in Artificial Intelligence 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sosnin S, Karlov D, Tetko IV, and Fedorov MV (2018) Comparative study of multitask toxicity modeling on a broad chemical space. Journal of chemical information and modeling 59, 1062–1072. [DOI] [PubMed] [Google Scholar]
- (20).LeCun Y, Bengio Y, and Hinton G (2015) Deep learning. nature 521, 436–444. [DOI] [PubMed] [Google Scholar]
- (21).Chen H, Engkvist O, Wang Y, Olivecrona M, and Blaschke T (2018) The rise of deep learning in drug discovery. Drug discovery today 23, 1241–1250. [DOI] [PubMed] [Google Scholar]
- (22).Tetko IV, and Engkvist O (2020) From Big Data to Artificial Intelligence: chemoinformatics meets new challenges. Journal of Cheminformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Samek W, Montavon G, Vedaldi A, Hansen LK, and Müller K-R (2019) Explainable AI: interpreting, explaining and visualizing deep learning. Vol. 11700, Springer Nature. [Google Scholar]
- (24).Arrieta AB, Díaz-Rodríguez N, Del Ser J, Bennetot A, Tabik S, Barbado A, García S, Gil-López S, Molina D, and Benjamins R (2020) Explainable Artificial Intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Information Fusion 58, 82–115. [Google Scholar]
- (25).Jiménez-Luna J, Grisoni F, and Schneider G (2020) Drug discovery with explainable artificial intelligence. Nature Machine Intelligence 2, 573–584. [Google Scholar]
- (26).Huang R, Xia M, Sakamuru S, Zhao J, Shahane SA, Attene-Ramos M, Zhao T, Austin CP, and Simeonov A (2016) Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun 7, 10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tox21. (2017) Tox21 assays. [Google Scholar]
- (28).Huang R (2016) A Quantitative High-Throughput Screening Data Analysis Pipeline for Activity Profiling, In High-Throughput Screening Assays in Toxicology (Zhu H, and Xia M, Eds.), Humana Press. [Google Scholar]
- (29).Tetko IV (2008) Associative neural network, In Artificial Neural Networks pp 180–197, Springer. [Google Scholar]
- (30).Chen T, and Guestrin C (2016) Xgboost: A scalable tree boosting system, In Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining pp 785–794. [Google Scholar]
- (31).Chang C-C, and Lin C-J (2011) LIBSVM: A library for support vector machines. ACM transactions on intelligent systems and technology (TIST) 2, 1–27. [Google Scholar]
- (32).Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, and Dubourg V (2011) Scikit-learn: Machine learning in Python. the Journal of machine Learning research 12, 2825–2830. [Google Scholar]
- (33).Suykens JA, and Vandewalle J (1999) Least squares support vector machine classifiers. Neural processing letters 9, 293–300. [Google Scholar]
- (34).Breiman L (1996) Bagging predictors. Machine learning 24, 123–140. [Google Scholar]
- (35).Baskin II, Winkler D, and Tetko IV (2016) A renaissance of neural networks in drug discovery. Expert opinion on drug discovery 11, 785–795. [DOI] [PubMed] [Google Scholar]
- (36).Sushko I, Novotarskyi S, Körner R, Pandey AK, Rupp M, Teetz W, Brandmaier S, Abdelaziz A, Prokopenko VV, and Tanchuk VY (2011) Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. Journal of computer-aided molecular design 25, 533–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).(2020) The RDKit Book (https://www.rdkit.org/docs/RDKit_Book.html, access on 11/22/2020).
- (38).Salmina ES, Haider N, and Tetko IV (2016) Extended functional groups (EFG): an efficient set for chemical characterization and structure-activity relationship studies of chemical compounds. Molecules 21, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yang C, Tarkhov A, Marusczyk J. r., Bienfait B, Gasteiger J, Kleinoeder T, Magdziarz T, Sacher O, Schwab CH, and Schwoebel J (2015) New publicly available chemical query language, CSRML, to support chemotype representations for application to data mining and modeling. Journal of Chemical Information and Modeling 55, 510–528. [DOI] [PubMed] [Google Scholar]
- (40).Moriwaki H, Tian Y-S, Kawashita N, and Takagi T (2018) Mordred: a molecular descriptor calculator. Journal of cheminformatics 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hong H, Xie Q, Ge W, Qian F, Fang H, Shi L, Su Z, Perkins R, and Tong W (2008) Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. Journal of chemical information and modeling 48, 1337–1344. [DOI] [PubMed] [Google Scholar]
- (42).Haider N (2010) Functionality pattern matching as an efficient complementary structure/reaction search tool: an open-source approach. Molecules 15, 5079–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Scikit-Learn. (2020) SVC (https://scikit-learn.org/stable/modules/generated/sklearn.svm.SVC.html, access on 11/22/2020). [Google Scholar]
- (44).Consortium, M. (2010) The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nature biotechnology 28, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ribeiro MT, Singh S, and Guestrin C (2016) Model-agnostic interpretability of machine learning. arXiv preprint arXiv:1606.05386. [Google Scholar]
- (46).Ribeiro MT, Singh S, and Guestrin C (2016) “ Why should i trust you?” Explaining the predictions of any classifier, In Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining pp 1135–1144. [Google Scholar]
- (47).Zafar MR, and Khan NM (2019) DLIME: A deterministic local interpretable model-agnostic explanations approach for computer-aided diagnosis systems. arXiv preprint arXiv:1906.10263. [Google Scholar]
- (48).Finn C, Abbeel P, and Levine S (2017) Model-agnostic meta-learning for fast adaptation of deep networks. arXiv preprint arXiv:1703.03400. [Google Scholar]
- (49).Karpov P, Godin G, and Tetko IV (2019) Transformer-CNN: Fast and Reliable tool for QSAR. arXiv preprint arXiv:1911.06603. [Google Scholar]
- (50).Sosnin S, Vashurina M, Withnall M, Karpov P, Fedorov M, and Tetko IV (2019) A survey of multi-task learning methods in chemoinformatics. Molecular informatics 38, 1800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Novotarskyi S, Abdelaziz A, Sushko Y, Körner R, Vogt J, and Tetko IV (2016) ToxCast EPA in vitro to in vivo challenge: insight into the Rank-I model. Chemical research in toxicology 29, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Averaged training and testing AUC for 65 Tox21 assays
Table S2. Processed dataset of the Chemicals and Tox21 Endpoints used in this study