

Abstract
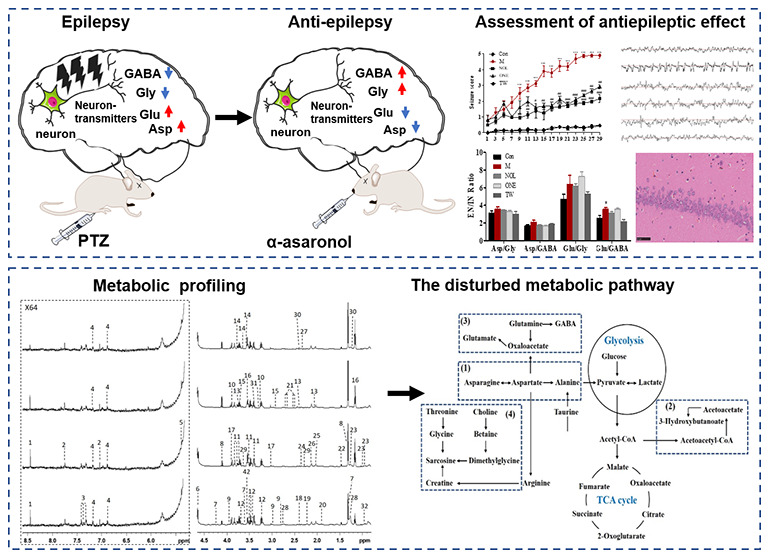
α-Asaronol from Acorus tatarinowii (known as “Shichangpu” in Traditional Chinese medicine) has been proved to possess more efficient antiepileptic activity and lower toxicity than α-asarone (namely “Xixinnaojiaonang” as an antiepileptic drug in China) in our previous study. However, the molecular mechanism of α-asaronol against epilepsy needs to be known if to become a novel antiepileptic medicine. Nuclear magnetic resonance (NMR)-based metabolomics was applied to investigate the metabolic patterns of plasma and the brain tissue extract from pentylenetetrazole (PTZ)-induced seizure rats when treated with α-asaronol or α-asarone. The results showed that α-asaronol can regulate the metabolomic level of epileptic rats to normal to some extent, and four metabolic pathways were associated with the antiepileptic effect of α-asaronol, including alanine, aspartate, and glutamate metabolism; synthesis and degradation of ketone bodies; glutamine and glutamate metabolism; and glycine, serine, and threonine metabolism. It was concluded that α-asaronol plays a vital role in enhancing energy metabolism, regulating the balance of excitatory and inhibitory neurotransmitters, and inhibiting cell membrane damage to prevent the occurrence of epilepsy. These findings are of great significance in developing α-asaronol into a promising antiepileptic drug derived from Traditional Chinese medicine.
1. Introduction
Epilepsy is a neurological disease characterized by recurrent spontaneous seizures due to hyperexcitability and hypersynchrony of brain neurons.1 Approximately 1% of the world population suffers from epilepsy, who have a bad life quality with the reduction of their consciousness and motor abilities. Unfortunately, even with optimal antiepileptic drug (AED) therapy, about one-third of patients have poor seizure control and become medically refractory.2 Even worse, adverse effects and drug resistance have become leading causes of treatment failure with current AEDs.3,4 Despite the improved effectiveness of surgical procedures, with more than half of operated patients achieving long-term freedom from seizures, epilepsy surgery is still suitable for a small subset of drug-resistant patients.5 The ketogenic diet, as an important adjunct to pharmacologic therapy, can improve seizure control and has the advantage of the relative absence of side effects.6 But recent reports suggested that gradual introduction of a high-fat diet causes brisk ketosis and a greater fraction of fat, as the polyunsaturated or monounsaturated species affords more intense ketosis.7,8 These therapeutic limitations have prompted a continuing search for new medicines, especially for drug-resistant epilepsy.
Previous research studies have demonstrated that α-asarone (E-1-propenyl-2,4,5-trimethoxy-benzene, see Figure 1), a major active component of Acorus tatarinowii (commonly known as “Shichangpu” in Traditional Chinese medicine),9,10 possesses neuroprotective effects, especially antiepileptic effects and/or anticonvulsant.11−13 In 1986, α-asarone (also known as “Xixinnaojiaonang”) was made into an antiepileptic drug (AED) in China to prevent seizures. Unfortunately, the long-term clinical use of α-asarone was limited due to its various side effects such as embryotoxicity and maternal toxicity, teratogenic, genotoxic, and hepato-carcinogenic properties, and ataxia or muscle incoordination.14−18 In our original studies, α-asaronol (E-3′-hydroxyasarone, see Figure 1) was discovered to be another component from A. tatarinowii and a metabolite in urine and blood when α-asarone was orally administrated to rats.19 The fact that α-asarone can be metabolized into α-asaronol was confirmed by liver microsomes of different species and human cytochrome P450 enzymes in other studies.20,21 Our recent study revealed that α-asaronol, against the maximal electroshock seizure (MES), subcutaneous injection pentylenetetrazole (PTZ)-induced seizure, and 3-mercaptopropionic acid (3-MP)-induced seizure, displayed a broad spectrum of antiepileptic activity, better neuroprotective effects, and lower acute toxicity than its metabolic parent compound α-asarone.22 α-Asaronol and its analogues, when compared with clinical drugs such as stiripentol, lacosamide, carbamazepine, and valproic acid, possessed excellent anticonvulsant activities as well as low neurotoxicity for developing into antiepileptic drugs, especially for the treatment of refractory epilepsies.23 These encouraging results prompted us to develop further α-asaronol into an efficient and safe antiepileptic drug as a substitute for α-asarone. Therefore, the molecular mechanism of α-asaronol with antiepileptic activity needs to be investigated in a chronic animal model of epilepsy.
Figure 1.

Chemical structures of α-asarone and α-asaronol.
The action mechanisms of antiepileptic drugs were complex and not fully understood. One of the well-known biochemical mechanisms is to modulate the imbalance between the inhibitory neurotransmitters and the excitatory neurotransmitters for sustaining the brain function.12,13,24−26 Both the increased activity of blood–brain barrier multidrug transporter proteins and alterations in drug targets rendering them drug-insensitive are two current hypotheses on the mechanisms of drug resistance in epilepsy.27 Inflammation processes,28 voltage-activated Na+ channels,29 and inhibition of lactate dehydrogenase (LDH)30,31 were also involved in the mechanism of antiepileptic drugs. Our recent research revealed that a peroxisome proliferator-activated receptor was related to the anticonvulsant activity of α-asaronol against PTZ-induced seizures.32 However, antiepileptic drugs that act on metabolic pathways are relatively unexplored.
Metabolomics is an important part of system biology along with genomics and proteomics. In such a system biology technique, metabolomics is a powerful tool for assessing the changes in global metabolites in a biological matrix, which can be directly associated with biological phenotypic responses to diseases as well as drug treatment or intervention.33−36 For instance, nuclear magnetic resonance (NMR)-based metabolomics was applied in characterizing drug-resistant epilepsy based on patient serum37−39 or rat brain,40 and gas chromatography coupled to mass spectrometry (GC-MS)-based metabolomics was used to investigate the metabolic alterations of epileptic patients and classify different types of seizures.41,42 In the present study, NMR-based metabolomics, combined with behavioral assessment, electroencephalography, the brain neurotransmitter measurement and histopathology, were applied to explore the antiepileptic activity of α-asaronol for deepening the understanding of biochemical mechanisms.
2. Results
2.1. Antiepileptic Activity of α-Asaronol on PTZ-Induced Seizure Rats
The seizure score was the behavior evaluation factor, which commonly assessed seizure intensity.43 It was classified according to the revised Racine’s scale44 as follows: stage 0 (no response), stage I (sudden behavioral arrest and/or motionless staring), stage II (head nodding, head clonus, and myoclonic jerk), stage III (unilateral forelimb clonus), stage IV (rearing with bilateral forelimb clonus), and stage V (generalized tonic–clonic seizures while lying on the side and/or wild jumping). Figure 2A shows time dependence on seizure scores for different groups. PTZ-induced animals (M) underwent from initial absence to visual tonic–clonic seizures, and their seizure scores were gradually increased from stage I to stage V, which was significantly increased to stage II at day 7, stage IV at day 15, and stage V at day 29 when compared to control (Con). In contrast, seizure scores were maximally increased to stage II in α-asaronol (NOL) and to stage III in α-asarone (ONE), and a significant decrease in seizure scores appeared at day 7 in NOL and ONE when compared with the M group. However, no seizure was observed in the drug vehicle group (TW) as control (Con), indicating that the cosolvent of TW-80 and saline has no interference on rats. The latency time, an interval between each injection of PTZ and the onset of myoclonic seizures in rats, was recorded to evaluate the latency of the attack (Figure 2B). During the experimental period, the latency time was about 100 s in the M group, while it was prolonged to 420 s in NOL and 300 s in ONE. It can be easily found that α-asaronol exhibited an anticonvulsant effect against PTZ-induced seizures by significantly increasing the latency for the onset of seizures.
Figure 2.

Antiepileptic activity of α-asaronol and α-asarone on PTZ-induced seizure rats was assessed by the seizure score (A), latency time of seizure (B), electroencephalography (C), the ratio of the brain neurotransmitter level (D), and histology in the hippocampal CA1 region (E) at different groups, including control (Con), PTZ-induced group (M), α-asaronol-treated group (NOL), α-asarone-treated group (ONE), and drug vehicle-treated group (TW). Data were presented as mean ± S.E. and were statistically compared using Student’s t-test. Differences were considered significantly with p < 0.05 (#), p < 0.01 (##), or p < 0.001 (###) between NOL/ONE and M and p < 0.05 (*) between M and Con.
It has been reported that the essence of epilepsy is in the abnormal activity of the intracranial neural network caused by the excessively synchronized discharge of neurons.45 Hence, we further evaluated the antiepileptic effect of NOL by examining electroencephalograms (EEG) and the measurement of neurotransmitters as well as pathological examination. EEG was monitored for different groups of rats at the waking states (Figure 2C). In comparison with the fundamental wave in control, EEG in the M group was characterized by typical spontaneous epileptiform discharges with frequent sharp waves, spike waves, and spike–slow synthesis waves. Nevertheless, the amplitude and frequency of EEG in both NOL and ONE, with scattered spikes and spike waves, were lower than those in the M group. Figure 2D shows the ratio of excitatory neurotransmitters (Asp and Glu) to inhibitory ones (Gly and GABA) in rat brain tissue . The ratio of Glu to GABA was significantly increased in the M group compared to control, indicating that PTZ disrupts the balance between excitatory neurotransmitters and inhibitory ones. However, no significant difference in the ratio values of Glu/Gly, Asp/Gly, and Asp/GABA was observed among the other different groups. Figure 2E shows the histology of the rat hippocampal CA1 region in different groups of rats. Severe degeneration of neurons in the M group, which emerged more cytoplasmic eosinophilia, nuclear irregularities, and mild edema, can be distinguished clearly from normal round-shaped cells in control. However, acidophilic neurons were rarely observed in the hippocampus regions of both NOL and NOE. Moreover, histopathological analysis of the NOL group did not show any marked morphological alterations associated with neurodegeneration in the hippocampal regions. It was clearly perceived from the level of neurotransmitters, electroencephalography, and histopathology that α-asaronol has antiepileptic activity on rats, which seems superior to α-asarone.
2.2. Assignment of Metabolites with NMR Spectra
Figure 3 shows the typical 1H NMR spectra of plasma (P) and the brain tissue extract (B) obtained from control (Con), PTZ-induced group (M), α-asaronol-treated group (NOL), and α-asarone-treated group (ONE). NMR resonances were assigned to specific metabolites based on the reported studies,46−48 Chenomx NMR Suite 6.0 (Chenomx, Edmonton, Canada), and publicly accessible metabolomics databases such as HMDB (http://www.hmdb.ca) and KEGG (http://www.kegg.jp). A total of 43 metabolites were identified (Table 1), which were further confirmed individually with 2D NMR data from J-resolved, COSY, TOCSY, HSQC, and HMBC spectra. It was found that plasma and the brain tissue extract mainly contained glucose, lactate, choline, GABA, lipids, TCA intermediate metabolites, and a series of amino acids. To obtain more detailed metabolomic changes, we performed multivariate statistical analysis of these NMR data.
Figure 3.

Typical 600 MHz 1H NMR spectra of plasma (P) and the brain tissue extract (B) from four different groups of rats including control (Con), PTZ-induced group (M), α-asaronol-treated group (NOL), and α-asarone-treated group (ONE). Dot box regions (left) were expanded 64 times and 16 times in comparison with no box regions (right). Metabolite keys are shown in Table 1.
Table 1. 1H NMR Data for Metabolites in Rat Plasma (P) and Brain Tissue Extract (B)a.
| key | metabolite | moieties | δ1H (multiplicity) | sample |
|---|---|---|---|---|
| 1 | formate | CH | 8.46 (s) | P |
| 2 | histidine | 2-CH | 7.05 (s) | P, B |
| 4-CH | 7.57 (s) | |||
| 3 | phenylalanine | 4-CH | 7.38 (m) | P |
| 2,6-CH | 7.33 (m) | |||
| 3,5-CH | 7.43 (m) | |||
| 4 | tyrosine | 2,6-CH | 7.20 (d) | P |
| 3,5-CH | 6.91 (d) | |||
| 5 | α-glucose | 1-CH | 5.24 (d) | P |
| 5-CH | 3.84 (m) | |||
| 6-CH | 3.78 (m) | |||
| 3-CH | 3.71 (dd) | |||
| 2-CH | 3.54 (dd) | |||
| 4-CH | 3.42 (dd) | |||
| 6 | β-glucose | 6-CH′ | 3.90 (dd) | P |
| 6-CH | 3.71 (dd) | |||
| 5-CH | 3.47 (m) | |||
| 4-CH | 3.41(dd) | |||
| 2-CH | 3.26 (dd) | |||
| 3-CH | 3.50 (t) | |||
| 1-CH | 4.66 (d) | |||
| 7 | threonine | βCH | 4.24 (m) | P, B |
| αCH | 3.58 (d) | |||
| γCH3 | 1.32 (d) | |||
| 8 | lactate | αCH | 4.12 (q) | P, B |
| βCH3 | 1.33 (d) | |||
| 9 | asparagine | αCH | 3.95 (m) | P |
| βCH2 | 2.84 (dd) | |||
| β′CH2 | 2.94 (dd) | |||
| 10 | betaine | CH2 | 3.91 (s) | P |
| CH3 | 3.27 (s) | |||
| 11 | choline | βCH2 | 3.53 (t) | B |
| N-CH3 | 3.21 (s) | |||
| αCH2 | 4.07 (t) | |||
| 12 | glutamine | αCH | 3.78 (m) | B |
| βCH2 | 2.17 (m) | |||
| γCH2 | 2.46 (m) | |||
| 13 | glutamate | αCH | 3.75 (m) | P, B |
| γCH2 | 2.35 (m) | |||
| βCH2 | 2.07 (m) | |||
| 14 | glycerol | CH | 3.77 (m) | P, B |
| CH2′ | 3.65 (dd) | |||
| CH2 | 3.56 (dd) | |||
| 15 | dimethylglycine | CH2 | 3.71 (s) | P |
| CH3 | 2.92 (s) | |||
| 16 | ether | CH2 | 3.55 (q) | P |
| CH3 | 1.18 (t) | |||
| 17 | creatine | CH2 | 3.93 (s) | P |
| CH3 | 3.04 (s) | |||
| 18 | succinate | CH2COOH | 2.41 (s) | P, B |
| 19 | acetone | CH3 | 2.23 (s) | P |
| 20 | acetate | CH3 | 1.93 (s) | P, B |
| 21 | citrate | CH2′ | 2.71 (d) | P |
| CH2 | 2.69 (d) | |||
| 22 | alanine | αCH | 3.77 (q) | P, B |
| βCH3 | 1.48 (d) | |||
| 23 | isoleucine | βCH | 1.01 (d) | P |
| β′CH | 1.99 (m) | |||
| γCH2 | 1.26 (m) | |||
| γ′CH2 | 1.47 (m) | |||
| δCH3 | 0.94 (t) | |||
| 24 | pyruvate | CH3 | 2.37 (s) | P |
| 25 | N-acetyl-glycoprotein | CH3 | 2.04 (s) | P |
| 26 | O-acetyl-glycoprotein | CH3 | 2.13 (s) | P |
| 27 | acetoacetate | CH3 | 2.28 (s) | P |
| 28 | lipid | (CH2)n | 1.27 (m) | P |
| CH2C=C | 2.03 (m) | |||
| =C–CH2–C= | 2.77 (m) | |||
| CH=CH | 5.30 (m) | |||
| 29 | valine | αCH | 3.62 (d) | P, B |
| βCH | 2.28 (m) | |||
| γ′CH3 | 1.05 (d) | |||
| γCH3 | 1.00 (d) | |||
| 30 | 3-hydroxybutyrate | αCH2′ | 2.42 (dd) | P |
| γCH3 | 1.20 (d) | |||
| 31 | taurine | CH2S | 3.29 (t) | P, B |
| CH2N | 3.43 (t) | |||
| 32 | leucine | δCH3 | 0.96 (d) | P, B |
| δ′CH3 | 0.97 (d) | |||
| βCH2 | 1.71 (m) | |||
| γCH | 1.72 (m) | |||
| 33 | NAA | CH3 | 2.03 (s) | B |
| 34 | GABA | CH3 | 2.30 (t) | B |
| CH2 | 1.93 (s) | |||
| 35 | aspartate | αCH | 3.91 (m) | B |
| βCH2 | 2.68 (dd) | |||
| β′CH2 | 2.82 (dd) | |||
| 36 | phosphorylcholine | CH3 | 3.23 (s) | B |
| βCH2 | 3.60 (t) | |||
| αCH2 | 4.17 (t) | |||
| 37 | myo-inositol | CH | 3.63 (t) | B |
| 2-CH | 3.53 (dd) | |||
| 2-CH | 3.28 (t) | |||
| CH | 4.06 (t) | |||
| 38 | xanthosine | 2-CH | 7.89 (s) | B |
| 2′-CH | 5.86 (d) | |||
| 3′-CH | 4.73 (m) | |||
| 4′-CH | 4.41 (m) | |||
| 39 | inosine | 2-CH | 8.36 (s) | B |
| 7-CH | 8.25 (s) | |||
| 2′-CH | 6.10 (d) | |||
| 3′-CH | 4.78 (t) | |||
| 4′-CH | 4.48 (t) | |||
| 5′-CH | 4.29 (m) | |||
| CH2 | 3.85 (m) | |||
| 40 | fumarate | CH | 6.52 (s) | B |
| 41 | hypoxanthine | 2-CH | 8.22 (s) | B |
| 7-CH | 8.20 (s) | |||
| 42 | glycine | CH2 | 3.58 (s) | P, B |
| 43 | lysine | CH | 3.77 (m) | B |
| αCH | 3.76 (t) | |||
| βCH2 | 1.92 (m) | |||
| γCH2 | 1.49 (m) | |||
| εCH2 | 3.03 (t) |
s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and dd, doublet of doublets.
2.3. NMR-Based Metabolomic Analysis for Antiepileptic Activity of α-Asaronol
Multivariate statistical analysis and the heatmap were derived from 1H NMR spectra of plasma and the brain tissue extract at the different groups, including control (Con), PTZ-induced group (M), α-asaronol-treated group (NOL), and α-asarone-treated group (ONE, Figure 4). The 3D principal component analysis (PCA) score plots of NMR data, where each point represents an individual sample, indicate similar metabolic profiling when points are clustered together but differential metabolic profiling when dispersed. The 3D PCA plots obtained from plasma (Figure 4A) and the brain tissue extract (Figure 4B) demonstrated where NOL and ONE groups had distinct separations from the M group, but some overlapping with the Con group. These results suggested that PTZ-induced seizures can cause the metabolic alteration of normal rats but the metabolic status seemed to be a recovery when treated with α-asaronol and α-asarone. To determine the metabolites contributing to the antiepilepsy effects of α-asaronol and α-asarone, OPLS-DA models were conducted for plasma and brain tissue extract samples, in which there were excellent separations between M and Con (Figure 4A1,B1), between NOL and M (Figure 4A2,B2), and between ONE and M (Figure 4A3,B3). The parameters of the permutation test were as follows: R2X = 0.62, R2Y = 0.95, and Q2 = 0.71 for plasma and R2X = 0.76, R2Y = 0.94, and Q2 = 0.53 for the brain tissue extract. OPLS-DA models were ensured to be valid with CV-ANOVA (p < 0.05). The significantly changed metabolites for distinguishing their differences were identified (Table S1) based on the correlation coefficient with a cutoff of |p(corr)| ≥ 0.50 and the variable importance of the projection greater than 1.5 (VIP > 1.5).
Figure 4.

Multivariate statistical analysis and the heatmaps derived from 1H NMR spectra of plasma and the brain tissue extract at four different groups, including control (Con), PTZ-induced group (M), α-asaronol-treated group (NOL), and α-asarone-treated group (ONE). 3D PCA score plots for plasma (A) and the brain tissue extract (B), and their OPLS-DA score plots between M and Con (A1, B1), between NOL and M (A2, B2), and between ONE and M (A3, B3). The significantly changed metabolites for plasma (C) and brain tissue extract (D) were shown in the heatmaps, where the red cell stands for the signal intensity of a metabolite higher than its mean signal intensity within one group and the blue cell stands for the signal intensity of a metabolite lower than its mean signal intensity within one group.
The differential metabolites for plasma and the brain tissue extract from the four groups were chosen and visualized in the heatmaps. As shown in Figure 4C, six metabolites in plasma including dimethylglycine, β-glucose, α-glucose, betaine, threonine, and O-acetyl-glycoprotein displayed higher levels in the M group than those in control. However, 12 metabolites including lipid, glycerol, 3-hydroxybutyrate, glycine, N-acetyl-glycoprotein, leucine, valine, lactate, taurine, glutamate, alanine, and succinate were shown to be at lower levels in the M group than those in control. As a result, most of these altered metabolites showed a reversal trend in NOL and ONE and tend to be normal. In the heatmap for the brain tissue extract (Figure 4D), compared with control, the PTZ-induced group (M) was characterized by the elevated concentrations of 10 metabolites including valine, glycine, choline, lactate, acetate, alanine, aspartate, inosine, taurine, and glutamate and the reduced concentrations of six metabolites including myo-inositol, GABA, glycerol, phosphorylcholine, threonine, and xanthosine. Nevertheless, the levels of the above-mentioned metabolites appear to be reversed to a certain extent in NOL and ONE.
Based on NMR metabolomic analysis, 18 metabolites in plasma and 16 metabolites in the brain tissue extract were altered when PTZ-induced rats were treated with α-asaronol. These altered metabolites were imported to perform pathway impact analysis to discover the most relevant biochemical processes and visualize them. Accordingly, based on the Impact Values greater than 0.50, the four metabolic pathways (Figure 5A) are in the following order: alanine, aspartate, and glutamate metabolism (1), synthesis and degradation of ketone bodies (2), glutamine and glutamate metabolism (3), and glycine, serine, and threonine metabolism (4). The disrupted metabolites in these metabolic pathways were identified as to be associated with the treatment of α-asaronol and are visualized in Figure 5B.
Figure 5.

Pathway impact analysis was associated with the treatment of α-asaronol (A) and four disrupted metabolic pathways (B) were visualized based on the impact values larger than 0.50, including alanine, aspartate, and glutamate metabolism (1); synthesis and degradation of ketone bodies (2); glutamine and glutamate metabolism (3); and glycine, serine, and threonine metabolism (4).
3. Discussion
Pentyleneterazole (PTZ), a selective GABA-A receptor chloride channel blocker, is used widely to develop chemical kindling in both rats and mice at subconvulsive doses within 30 days.50 Such a kindling model is commonly used to establish tonic–clonic seizure and screen antiseizure drugs in animals.49 We found that the treatment with α-asaronol or α-asarone can reduce seizure scores from stage V (generalized tonic–clonic seizures while lying on the side and/or wild jumping) to stage III (unilateral forelimb clonus), extend the latency time of seizure from 100 to 400 s, normalize EEG, and protect neurons from damage for PTZ-induced seizure rats. These observations were in line with the previous report that α-asaronol can increase the latency to seizure onset and decrease the mortality rates in the PTZ-induced seizure.22
3.1. α-Asaronol Can Improve Energy Metabolism by Enhancing Glucose Utilization and Lipid Consumption
Our NMR metabolomic analysis showed an elevation in the amount of glucose (α-glucose and β-glucose), a decline of lactate, and a reduction in succinate (one of the TCA cycle intermediates) in plasma from PTZ-induced rats compared with control. The alterations of these metabolites suggested the compromised energy metabolism related to poor glucose utilization due to PTZ stimulation. PTZ-induced mice in the previous study were also characterized by a significant increase in blood glucose.51 It was conceived that blood glucose even in physiologic concentration may increase neuronal excitability,52 and hyperglycemia lowers seizure threshold in adult rats.53 Epilepsy-related glucose hypometabolism was associated with mitochondrial dysfunction,54−56 presumably as a consequence of deficient glycolysis and thus the elevation of glucose. The persistent convulsion of the entire body during seizures would lead to a temporary oxygen shortage and an inhibition of the tricarboxylic acid cycle, resulting in the reduced formation of succinate. Conversely, the diminished glucose concentration as well as the increased concentrations of lactate and succinate were observed in α-asaronol-treated rats and especially in α-asarone-treated rats. A diminution of blood glucose, which resembles occurrence with a low-carbohydrate diet control for epilepsy rats,57,58 might lower neuronal excitability and thereby attenuate an epileptic diathesis.59 Elevated levels of lactate and succinate may hint at an increase in glucose utilization. It was conferred that α-asaronol and α-asarone may facilitate glucose consumption for lowering neuronal excitability and exert antiepileptic effect via enhancing glycolysis to improve energy metabolism.
Here, another characteristic metabolic alteration was the elevated lactate in the brain tissue extract in spite of the diminished lactate in plasma, when PTZ-induced rats were in comparison with control. A higher level of lactate was also found in the cerebellum and hippocampus of PTZ-kindled animals40 and cortex and hippocampus or cerebrospinal fluid of epileptic patients.60−62 Unfortunately, the current data cannot permit a precise localization of the lactate formation because the extract of the whole brain tissue, rather than the separated brain compartment, was analyzed using NMR. Anyway, it is universally known that lactate plays a crucial role in several biochemical processes, especially in anaerobic glycolysis. The elevation of lactate in the brain tissue extract indicated an enhancement of anaerobic glycolysis and reflected an imbalance of oxygen supplement and demand during seizures. Differently, the blood lactate concentration was reported to be either elevated or diminished during seizures for epilepsy patients.37,39,41 Lactate is an alternated fuel for brain cells as glucose, but lactate in the blood cannot sustain normal brain function. The epileptic brain will take up and metabolize exogenous lactate serving as an energy source like the brain during recovery from maximal exercise.63 Hence, the mechanism responsible for the elevation of lactate in the brain tissue extract may be (i) an increase in lactate production in the epileptic focus and CSF, as mentioned above and (ii) uptake from peripheral blood, thereby reflecting the low level of lactate in plasma. However, the level of lactate in the brain tissue extract was downregulated to normal in NOL and even lower than normal in ONE. A previous study suggested that stiripentol, a clinically used antiepileptic drug, can inhibit lactate dehydrogenase (LDH) enzymes and strongly suppress seizures in vivo, mainly by decreasing the level of lactate in the brain.30 Our previous study showed that α-asaronol has better LDH inhibition than stiripentol.23 It was inferred that α-asaronol, especially α-asarone, can inhibit anaerobic glycolysis and thereby modulate nervous system energy metabolism, to prevent the development of epilepsy.
If energy metabolism is compromised due to disease, the body becomes reliant on fatty acids as energy substrates; thus, the consumption of lipid increases. Converse to the elevated level of glucose in the current study, the decreased levels of lipid, glycerol, and 3-hydrobulyrate (ketone body) were observed in the plasma of PTZ-induced rats. Glycerol is the skeleton component of triglyceride molecules and is transformed from dihydroxyacetone phosphate formed by glycolysis. Energy metabolism is compromised and the body consumes glycerol ester, which in turn supplies energy through lipid metabolism. However, in comparison with the PTZ-induced seizure group, the levels of lipid, glycerol, and 3-hydrobulyrate were increased to a comparative normal level in ONE, whereas no significant change was in NOL. It was concluded that α-asaronol exerted antiepileptic effects on enhancing energy metabolism by lipid consumption coupled with glucose utilization, whereas the improved energy metabolism for α-asarone mainly relied on glycolysis.
3.2. α-Asaronol Can Regulate the Equilibrium of Amino Acid Neurotransmitters
The present study showed that a noticed metabolic feature with a simultaneous increase in glutamate and a decrease in GABA was characterized in the PTZ-induced rat’s brain tissue extract. There was much evidence of abnormally elevated glutamate levels in human or animal epilepsy during the seizure/interictal stage.60,64,65 Glutamate is the most abundant fast excitatory neurotransmitter in the nervous system, and its level in the epileptogenic brain is correlated to the severity of seizures.66 The excessive glutamate in the synaptic cleft induces excitotoxicity by activating the N-methyl-d-aspartate (NMDA) receptor, which promotes calcium influx that leads to neuronal death.67 GABA is a key inhibitory neurotransmitter in the brain, and the deficiency of GABA would induce neuronal hyperexcitability, which contributed to the occurrence of seizures. GABA acts on the GABA-A receptor after being released from the presynaptic vesicles, which promotes the opening of Cl– channels and causes hyperpolarization of the postsynaptic cell.68 Our results agreed with a common opinion that seizures are correlated to the disruption of the balance between excitatory and inhibitory neurotransmitters.69,70 Indeed, repeated PTZ-induced seizures alter the GABA-mediated inhibition and glutamate-mediated excitation, which may contribute to the increased seizure susceptibility.49 However, a decreased level of glutamate and an increased level of GABA were observed in NOL-treated and ONE-treated animals. Inhibition of glutamate by grafting GABA-ergic progenitors into the epileptic regions of animals can reduce seizures. The elevation of GABA inhibited the regulation of neuronal overdischarge and postsynaptic facilitation, producing inhibitory postsynaptic potentials. It was documented that the promotion of GABA release to enhance the GABA-mediated inhibitory action has become an important target of antiepileptic drugs.13,24,71 That is to say, the antiepileptic effect of α-asaronol may be accounted for reducing neuronal hyperexcitability by correcting the neuronal excitation–inhibition imbalance.
It was noted that the increased levels of alanine, taurine, aspartate, glycine, leucine, and valine were accompanied by the elevation of glutamate in the brain tissue extract from PTZ-kindled rats. Branched-chain amino acids (BCAAs) play an important role in regulating the levels of the major excitatory neurotransmitter glutamate in the central nervous system.72 The baseline concentrations of alanine, taurine, and glutamate were synchronously elevated in the hippocampus of PTZ fully kindled rats.73 Alanine, an important NMDA receptor coagonist, is a precursor of glutamate and is highly expressed during recovery from ischemia and hypoxia.74 Accordingly, it was found that alanine production increases to replenish glutamate stores when glutamate stores are depleted.75 A massive increase in the release of excitatory amino acids during epileptic seizures can activate glutamate receptors such as NMDA, and the activation of NMDA receptors stimulates taurine release in many brain structures including the hippocampus.76 The onset of PTZ-induced convulsive seizures seemed mainly related to a marked increase of glutamate, aspartate, taurine, and glycine, while the maintenance and frequency of seizures seemed related to a marked increase of glycine, in combination with an increase of glutamate.77 Concentrations of aspartate, glycine, and glutamate are simultaneously increased in the epileptogenic cerebral cortex since the activities of the enzymes, glutamate dehydrogenase and aspartate aminotransferase involved in glutamate and aspartate metabolism, are increased.78 Moreover, a striking change in PTZ-induced animals in our study was the simultaneous accumulation of leucine and the formation of glutamate and aspartate. The brain rapidly transaminates leucine, which is the source of as much as one-third of all brain amino nitrogen. Accumulation of brain leucine, therefore, should increase the levels of glutamate and aspartate.79 However, our current data showed decreased levels of glutamine and threonine. This posit partially occurs via the exchange of leucine for brain glutamine since the increased leucine levels in the brain are accompanied by decreased glutamine levels. In the epileptic brain, when the neuronal release of glutamate is high and flux through glutamine synthetase probably is greater, the system releases glutamine in exchange for leucine.80 These observations underscored the role of other amino acids in setting the equilibrium between excitatory and inhibitory processes in the brain.
Converse to the higher concentrations of glutamate, alanine, taurine, aspartate, glycine, leucine, and valine in the brain tissue extract, PTZ-kindled rats had significantly lower levels of most amino acids (glutamate, alanine, glycine, leucine, isoleucine, and valine) in plasma when compared with control. This disturbance of amino acid metabolism was also shown in patients with juvenile myoclonic epilepsy and patients with refractory epilepsy.81 Because of the bidirectional flow of amino acids between blood and the brain, the reverse amounts of these amino acids between plasma and the brain suggested that PTZ can enhance amino acid uptake from blood to the brain. It is interesting that when PTZ-kindled rats were treated with α-asaronol and α-asarone, amino acids such as glutamate, alanine, and leucine were decreased in the brain but were observed to increase in plasma. It was suggested that α-asaronol and α-asarone may also facilitate the mechanism by which the brain exports to blood compounds, in the process favoring the removal of glutamate carbon and nitrogen.
3.3. α-Asaronol Can Exert the Neuroprotective Effect by Suppressing Cell Membrane Damage
The current NMR data showed that a simultaneously increased level of choline and a decreased level of phosphorylcholine was found in the brain tissue extract from PTZ-induced seizure rats, which gave one possibility of the enhanced hydrolysis of cell membrane phosphatidylcholine (a precursor of membrane formation). The increase of choline concentration in the brain can be attributed to abnormal phospholipid metabolism in a number of pathophysiological conditions, such as neural trauma and chronic degenerative disorders including seizure82 and ischemia.83 It is well known that choline plays an important role in the structural integrity of cell membranes and lipoprotein composition, and therefore, the increased choline may reflect myelin breakdown, increased cell density, or gliosis. As a result, excessive concentrations of choline may cause seizure maintenance and neuronal damage and lethality associated with status epilepticus.84 PTZ-kindling resulted in neuronal death and neurostructural changes in the hippocampus, the amygdala, and its neighboring cortex, leading to the development of generalized tonic–clonic seizures.85,86 Therefore, it can be conferred that PTZ is most likely to produce cell membrane damage, which is reflected with increased choline and decreased phosphorylcholine, due to the enhanced hydrolysis of phosphatidylcholine. However, the levels of choline and phosphorylcholine detected in PTZ-induced rats were reversed when treated with NOL and ONE, which was thought to reflect altered membrane turnover, indicating that α-asaronol can exert the neuroprotective effect on suppressing cell membrane damage.
4. Conclusions
In this study, NMR-based metabolomics was applied to reveal the antiepileptic effect of α-asaronol for the first time. The varied metabolome, including 18 metabolites in plasma and 16 metabolites in the brain from PTZ-induced seizure rats, was modulated to the normal level to some extent when treated with α-asaronol and α-asarone. Four metabolic pathways were associated with the antiepileptic effect of α-asaronol, including alanine, aspartate, and glutamate metabolism; the synthesis and degradation of ketone bodies; glutamine and glutamate metabolism; and glycine, serine, and threonine Metabolism. α-Asaronol can exert the antiepileptic effect on PTZ-induced seizure rats by improving energy metabolism, regulating the equilibrium of amino acid neurotransmitters, and suppressing cell membrane damage, which demonstrated a great promising future for the development of novel antiepileptic medicine.
5. Materials and Methods
5.1. Chemicals
Analytical grade sodium azide (NaN3), K2HPO4·3H2O, and NaH2PO4·2H2O were obtained from Guoyao Chemical Reagent Co., Ltd. (Shanghai, China) and used without further treatment. Sodium-3-trimethylsilyl [2,2,3,3]-d4 propionate (TSP) and deuterium oxide (D2O) were purchased from Cambridge Isotope Laboratories, Inc. (Cambridge, MA). Pentylenetetrazol (PTZ) and amino acid standards including glutamate, glycine, GABA, and aspartate were procured from Sigma-Aldrich (St. Louis). α-Asarone was purchased from Macklin Biochemical Technology Co., Ltd. (Shanghai, China), and α-asaronol was synthesized and purified by our laboratory (chemical purity >99.5%). Physiological saline (0.9%) was purchased from Cisen Pharmaceutical Co., Ltd. (Jining, China) and 0.5% Tween-80 was bought from Dengfeng Chemical Reagent Co., Ltd. (Tianjin, China). Ether was obtained from Chron Chemical Co., Ltd. (Chengdu, China), and acetonitrile was obtained from Fisher Scientific. Chloral hydrate was purchased Shanpu Chemical Co., Ltd. (Shanghai, China).
5.2. Animal and Experimental Design
All of the experiments were in accordance with the National Institute of Health’s Guidelines regarding the principles of animal care (PR China, 2004) and approved by the Animal Care and Use Committee of Northwest University in China. Adult male Sprague–Dawley rats (6–8 weeks old, weighing 220–250 g, SCXK-Shaan-2017-003) were purchased from the Experimental Animal Centre of Xi’an Jiaotong University. They were housed in individual cages (5 per cage) under controlled conditions of temperature (23 ± 2 °C), relative air humidity (60 ± 5%), and 12 h light/dark cycles, with free access to food and tap water.
Rat chronic epilepsy was developed with the pentylenetetrazol (PTZ) kindling model according to the previously reported method.49 After a week of acclimatization, 65 rats were randomly divided into five groups (n = 13), namely, control group (Con), drug vehicle-treated group (TW), PTZ-induced group (M), α-asaronol-treated group (NOL), and α-asarone-treated group (ONE). Prior to administration, the PTZ solution was prepared with physiological saline (0.9%) while α-asaronol and α-asarone suspensions were dispersed, respectively, in 0.5% Tween-80 with the titration of physiological saline (0.9%) until they became transparent. Animals of the M group were intraperitoneally only treated with PTZ (35 mg/kg body weight) once every other day within 30 days. NOL and ONE groups were orally administrated with α-asaronol and α-asarone at a dosage of 50 mg/kg body weight, respectively, on 1 day after the injection of PTZ at the same dosage with the M group. TW group rats were orally administrated with only drug vehicle (0.5% Tween-80 containing saline).
5.3. Neurobehavioral Score, Latency Time, and Electroencephalogram (EEG) Monitoring
After each PTZ injection, rats were gently placed in isolated transparent plexiglass cages and their behavior was observed carefully within 60 min to assign appropriate seizure scores according to the revised Racine’s scale.44 The latency time was recorded to evaluate the latency of the attack. To prevent the results affected by observer preference, the participants did not know which compound they administrated to rats. Treatment groups were identified with letters like A, B, C, etc., and the investigator was blinded as to which letter denoted which of the treatment groups. All data are presented as mean ± S.E. The results were analyzed by a one-way ANOVA, followed by Student’s t-test (p < 0.05, p < 0.01, or p < 0.001).
Rats were anesthetized by 7% chloral hydrate (i.p.) and placed in a stereotactic frame, and a 2 cm incision in the middle of the rat cranial crest was cut and the periosteum was peeled. Following the holes punched with a dental drill, two needle electrodes with a diameter of 0.5 mm were inserted in the cortex (2.0 mm anterior, 2.0 mm lateral to bregma, and 0.5 mm subdural) and in the hippocampus (3.8 mm posterior, 2.0 mm lateral to bregma, and 2.6 mm subdural), while one needle electrode with a diameter of 0.5 mm was inserted in the tip of the nose as a reference. All of the electrodes were fixed with dental acrylic cement. The electroencephalograms (EEGs) for rats were monitored over 60 min for 5 days using the BL-420S biological function test system.
5.4. Sample Collection
Blood (1.5 mL) was collected from the ophthalmic vein into an Eppendorf tube with sodium heparin on the last day (day 30) and centrifuged (5000 rpm, 10 min) to get plasma. After the collection of blood, two rats for each group were sacrificed under anesthesia and their brains were removed for histopathological assessment. The brain tissues of the left animals were harvested and then divided into two parts, one for neurotransmitter measurement and the other one for NMR measurement. These biosamples were immediately snap-frozen in liquid nitrogen and stored at −80 °C until analyzed.
5.5. Histopathological Assessment
After the brain was immersed in a 4% paraformaldehyde solution for 24 h at 4°C, the coronal sections of 10 μm passing through the hippocampus were sliced, mounted, and stained by hematoxylin and eosin (H&E). Histopathological assessments were performed under microscopes by a qualified pathologist.
5.6. Brain Neurotransmitter Measurement
The levels of amino acid neurotransmitters, including Glu, Gly, Asp, and GABA in rat brain tissue, were determined with an external standard method by LC-MS/MS. The brain tissue (0.5 g) was homogenized and extracted ultrasonically with 1 mL of acetonitrile/water (v/v = 1/1) for 30 min, and the supernatant was collected with centrifugation (13 200 rpm, 10 min) and diluted with 10 times water. Then, 1 μL of the extract sample was injected into a QTRAP-LC-MS/MS (Shimadzu-AB SCIEX). The separation was performed on a BEH Amide column (1.7 μm, 100 mm × 2.1 mm) at 40 °C. The mobile phase was composed of water (0.1% formic acid) and acetonitrile (5 mM ammonium acetate, 0.1% formic acid) at a flow rate of 0.3 mL/min. The gradient elution was set at 95% B (0–1.0 min), 95% B to 50% B (1.1–10.0 min), and 95% B (10.1–12.0 min). All samples were detected by multiple reactions monitoring (MRM) scan mode in positive electrospray ionization (ESI+). The turbo spray temperature (TEM) was set at 550 °C and ion spray voltage (IS) was 5000 V. The flow rates of ion source gas (GS1), ion source gas (GS2), and curtain gas (CUR) were 50, 60, and 40 L/min, respectively.
5.7. NMR Measurement and Data Processing
Each plasma sample (200 μL) was mixed with 400 μL of saline solution (containing 75% D2O, 0.9% NaCl, 0.04% NaN3) and was centrifuged (5000 rpm, 10 min), and then 550 μL of the supernatant was transferred into 5 mm NMR tubes for analysis. Brain tissue (200 mg) was cut with surgical scissors and placed in a 4 mL tube and was then added with 800 μL of phosphate buffer (0.1 M, K2HPO4/NaH2PO4, pH 7.4, 50% D2O, and 0.01% NaN3) containing 0.025% TSP, homogenized at 4 °C, and centrifuged at 12 000 rpm for 10 min. Each supernatant (550 μL) was transferred into a 5 mm NMR tube for analysis.
1H NMR spectra were acquired at 298 K on a Varian VNMRS 600 MHz NMR spectrometer (599.904 MHz for proton frequency). The water-suppressed Carr-Purcell-Meiboom-Gill sequence (RD-90°-[τ-180°-τ]n-acquisition; τ = 267.7 μs and n = 100) was acquired for plasma samples. The NOESYPR pulse sequence was acquired (RD-90°-t1-90°-tm-90°-acquisition; t1 = 4 μs and tm = 100 ms) for brain tissue extract samples. For all experiments, 128 transients were collected with 64k data points and a spectral width of 12 ppm with a relaxation delay of 2.0 s and an acquisition time of 2.38 s. The free induction decays were weighed by an exponential function with a line-broadening factor of 0.5 Hz prior to Fourier transformation.
The 1H NMR spectra were processed using MestReNova software (Mestrelab Research, Santiago de Compostella, Spain) and were phase-corrected and baseline-corrected manually. The plasma spectra were referenced to the methyl proton signal of lactate (δ 1.33), and the brain tissue extract spectra were referenced to TSP (δ 0.00). All of these spectra were then integrated into regions with a bucket width of 0.004 ppm using MestReNova software. To avoid the interference of exogenous substances with endogenous metabolites, some signal regions were carefully excluded, including the regions of δ 1.14–1.22 and 3.54–3.60 (for ether) and δ 4.45–5.22 (for water) in the plasma spectra, as well as the region of δ 4.40–5.80 (for water) in the brain spectra. The data were then normalized to the total sum of the spectra prior to analysis.
The processed NMR data were then imported to the SIMCA-P 14.1 software package (Umetric, Sweden) and visualized by multivariate statistical analysis. Principal component analysis (PCA) was performed using the mean-centered NMR data to differentiate each group. Orthogonal partial least squares discriminate analysis (OPLS-DA) was utilized to reveal the difference between two groups. To avoid overinterpreting, only two principal components were calculated for the models with the obtained R2 and Q2 values as initial indicators of model quality. The validity of the models against overfitting was assessed by the parameter R2, and the predictive ability was described by Q2. All of the OPLS-DA models were cross-validated using a 200-time permutation and evaluated by 7-fold cross-validation and further ensured with analysis of variance coefficient of variation (CV-ANOVA) with p < 0.05 considered as valid. The selection of significant metabolites was based on the correlation-scaled predictive loading vector with a cutoff of |p(corr)| = 0.5 and the variable importance of the projection greater than 1.5 (VIP > 1.5).
The heatmap and pathway impact analysis were made by the online MetaboAnalyst 4.0 software package (www.metaboanalyst.ca). The heatmap provides an intuitive visualization of a data table, where each cell was color-coded with the mean value of metabolite NMR signal intensities within one group to reveal a significant change in metabolite concentration at different groups. Pathway impact analysis was determined using the hypergeometric test and relative betweenness centrality to analyze the concentration with high sensitivity for identifying subtle metabolic changes involved in the same biological pathway.
Acknowledgments
This research was funded by Shaanxi Key Research and Development Project (2018KWZ-05) and the Project Shaanxi Provincial Key Research and Development Program (2019SF-056).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c06922.
OPLS-DA correlation coefficients for metabolites (Table S1) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Devinsky O.; Annamaria V.; Souhel N.; Nihal C. D. L.; Michael A. R. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Kwan P.; Brodie M. J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- McCorry D.; Chadwick D.; Marson A. Current drug treatment of epilepsy in adults. Lancet Neurol. 2004, 3, 729–735. 10.1016/S1474-4422(04)00935-4. [DOI] [PubMed] [Google Scholar]
- Schmidt D.; Schachter S. C. Drug treatment of epilepsy in adults. Br. Med. J. 2014, 348, g254 10.1136/bmj.g254. [DOI] [PubMed] [Google Scholar]
- Moshé S. L.; Perucca E.; Ryvlin P.; Tomson T. Epilepsy: new advances. Lancet 2015, 385, 884–898. 10.1016/S0140-6736(14)60456-6. [DOI] [PubMed] [Google Scholar]
- Stafstrom C. E.; Rho J. M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3, 59 10.3389/fphar.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuehrlein B. S.; Rutenberg M. S.; Silver J. N.; Warren M. W.; Theriaque D. W.; Duncan G. E.; Stacpoole P. W.; Brantly M. L. Differential metabolic effects of saturated versus polyunsaturated fats in ketogenic diets. J. Clin. Endocrinol. Metab. 2004, 89, 1641–1645. 10.1210/jc.2003-031796. [DOI] [PubMed] [Google Scholar]
- Bergqvist A. G. C.; Schall J. I.; Gallagher P. R.; Cnaan A.; Stallings V. A. Fasting versus gradual initiation of the ketogenic diet: A prospective, randomized clinical trial of efficacy. Epilepsia 2005, 46, 1810–1819. 10.1111/j.1528-1167.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- Bai X. M.; Liu T.; Liu D. L.; Wei Y. J. Simultaneous determination of alpha-asarone and beta-asarone in Acorus tatarinowii using excitation-emission matrix fluorescence coupled with chemometrics methods. Spectrochim. Acta, Part A 2018, 191, 195–202. 10.1016/j.saa.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Feng X. L.; Yu Y.; Qin D. P.; Gao H.; Yao X. S. Acorus Linnaeus: a review of traditional uses, phytochemistry and neuropharmacology. RSC Adv. 2015, 5, 5173–5182. 10.1039/C4RA12049C. [DOI] [Google Scholar]
- Miao J. K.; Chen Q. X.; Wu X. M.; Li C.; Zhang X. P. Antiepileptic properties of alpha-asarone from Acori Graminei Rhizoma in mice and rats seizure models. Int. J. Pharmacol. 2012, 8, 567–571. 10.3923/ijp.2012.567.571. [DOI] [Google Scholar]
- Miao J. K.; Chen Q. X.; Li C.; Li X. W.; Zhang X. P.; et al. Modulation effects of alpha-asarone on the GABA homeostasis in the Lithium-Pilocarpine model of Temporal Lobe Epilepsy. Int. J. Pharmacol. 2012, 9, 24–32. 10.3923/ijp.2013.24.32. [DOI] [Google Scholar]
- Huang C.; Li W. G.; Zhang X. B.; Wang Li.; Xu T. L.; Wu D. Z.; Li Y. Alpha-asarone from Acorus gramineus alleviates epilepsy by modulating A-Type GABA receptors. Neuropharmacology 2013, 65, 1–11. 10.1016/j.neuropharm.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Salazar M.; Salazar S.; Ulloa V.; Mendoza T.; Pages N.; Chamoro G. Teratogenic action of alpha-asarone in the mouse. J. Toxicol. Clin. Exp. 1992, 12, 149–154. [PubMed] [Google Scholar]
- Karwicka E.; Marczewska J.; Anuszewska E.; Łozowicka B.; Chilmonczyk Z. Genotoxicity of alpha-asarone analogues. Bioorg. Med. Chem. 2008, 16, 6069–6074. 10.1016/j.bmc.2008.04.049. [DOI] [PubMed] [Google Scholar]
- Marczewska J.; Drozd E.; Anuszewska E.; Chilmonczyk Z.; Łozowicka B. Assessment of the genotoxic activity of alpha-asarone and its derivatives in the comet assay. Acta Pol. Pharm. 2013, 70, 349–354. [PubMed] [Google Scholar]
- Cai Q.; Li Y. Y.; Mao J. X.; Pei G. Neurogenesis-promoting natural product alpha-asarone modulates morphological dynamics of activated microglia. Front. Cell. Neurosci. 2016, 10, 280 10.3389/fncel.2016.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebel T.; Hermes L.; Haupenthal S.; Müller L.; Esselen M. alpha-Asarone, beta-asarone, and gamma-asarone: Current status of toxicological evaluation. J. Appl. Toxicol. 2021, 41, 1166–1179. 10.1002/jat.4112. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Bai Y. J.; Zeng M.; Chen X. F.; Xie J.; Li B.; He X. R.; Bai Y. J.; Jia P.; Meng X.; Liang J.; Wang S. X.; Fan T. P.; Wu B.; Zheng X. H. Pharmacokinetics and tissue distribution evaluation of alpha-asaronol and its main metabolite in rats by HPLC method. J. Pharm. Biomed. Anal. 2019, 172, 349–356. 10.1016/j.jpba.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Cartus A. T.; Schrenk D. Metabolism of carcinogenic alpha-asarone by human cytochrome P450 enzymes. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 213–223. 10.1007/s00210-019-01724-0. [DOI] [PubMed] [Google Scholar]
- Cartus A. T.; Schrenk D. Metabolism of the carcinogen alpha-asarone in liver microsomes. Food Chem. Toxicol. 2016, 87, 103–112. 10.1016/j.fct.2015.11.021. [DOI] [PubMed] [Google Scholar]
- He X.; Yajun Bai Y. J.; Zeng M.; Zhao Z. F.; Zhang Q.; Xu N.; Qin F. G.; Wei X. Y.; Zhao M. M.; Wu N.; Li Z. H.; Zhang Y. J.; Fan T. P.; Zheng X. H. Anticonvulsant activities of alpha-asaronol ((E)-3 ’-hydroxyasarone), an active constituent derived from alpha-asarone. Pharmacol. Rep. 2018, 70, 69–74. 10.1016/j.pharep.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Bai Y. J.; He X. R.; Bai Y. J.; Sun Y.; Zhao Z. F.; Chen X. F.; Li B.; Xie J.; Li Y.; Jia P.; Meng X.; Zhao Y.; Ding Y. R.; Xiao C. N.; Wang S. X.; Yu J.; Liao S.; Zhang Y. J.; Zhu Z. L.; Zhang Q.; Zhao Y. H.; Qin F. G.; Zhang Y.; Wei X. Y.; Zeng M.; Liang J.; Cuan Y.; Shan J. Z.; Fan T. P.; Wu B.; Zheng X. H. Polygala tenuifolia-Acori tatarinowii herbal pair as an inspiration for substituted cinnamic alpha-asaronol esters: Design, synthesis, anticonvulsant activity, and inhibition of lactate dehydrogenase study. Eur. J. Med. Chem. 2019, 183, 111650 10.1016/j.ejmech.2019.111650. [DOI] [PubMed] [Google Scholar]
- Kammerer M.; Rassner M. P.; Freiman T. M.; Feuerstein T. J. Effects of antiepileptic drugs on GABA release from rat and human neocortical synaptosomes. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 384, 47–57. 10.1007/s00210-011-0636-8. [DOI] [PubMed] [Google Scholar]
- Naseer M. I.; Li S. P.; Kim M. O. Maternal epileptic seizure induced by Pentylenetetrazol: Apoptotic neurodegeneration and decreased GABA(B1) receptor expression in prenatal rat brain. Mol. Brain 2009, 2, 11 10.1186/1756-6606-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel A.; Mikkelsen M.; Edden R. A. E.; Robertson C. E. Regional balance between glutamate plus glutamine and GABA plus in the resting human brain. Neuroimage 2020, 220, 117112 10.1016/j.neuroimage.2020.117112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margineanu D. G.; Klitgaard H. Mechanisms of drug resistance in epilepsy: relevance for antiepileptic drug discovery. Expert Opin. Drug Discovery 2009, 4, 23–32. 10.1517/17460440802611729. [DOI] [PubMed] [Google Scholar]
- de Vries E. E.; Munckhof B. V. D.; Braun K. P. J.; Kerkhof A. V. R.; Jager W. D.; Jansen F. E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. 10.1016/j.neubiorev.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Wang Z.-J.; Levinson S. R.; Sun L. Q.; Heinbockel T. H. Identification of both GABA(A) receptors and voltage-activated Na+ channels as molecular targets of anticonvulsant alpha-asarone. Front. Pharmacol. 2014, 11, 40 10.3389/fphar.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sada N.; Lee S.; Katsu T.; Otsuki T.; Inoue T. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015, 347, 1362–1367. 10.1126/science.aaa1299. [DOI] [PubMed] [Google Scholar]
- Cho C. H. Commentary: Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Front. Cell. Neurosci. 2015, 9, 264 10.3389/fncel.2015.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Zhang B. Y.; Sun Y.; Zhang S. S.; Li X.; Sik A.; Bai Y. J.; Zheng X. H.; Liu K. C. Involvement of peroxisome proliferator-activated receptor gamma in anticonvulsant activity of alpha-asaronol against pentylenetetrazole-induced seizures in zebrafish. Neuropharmacology 2020, 162, 107760 10.1016/j.neuropharm.2019.107760. [DOI] [PubMed] [Google Scholar]
- Kaddurah-Daouk R.; Krishnan K. R. R. Metabolomics: A global biochemical approach to the study of central nervous system diseases. Neuropsychopharmacology 2009, 34, 173–186. 10.1038/npp.2008.174. [DOI] [PubMed] [Google Scholar]
- Kaddurah-Daouk R.; Kristal B. S.; Weinshilboum R. M. Metabolomics: A global biochemical approach to drug response and disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- McGarrah R. W.; Crown S. B.; Zhang G. F.; Shah S. H.; Christopher B. Newgard cardiovascular metabolomics. Circ. Res. 2018, 122, 1238–1258. 10.1161/CIRCRESAHA.117.311002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. L.; Atherton H.; Shockcor J.; Atzori L. Metabolomics as a tool for cardiac research. Nat. Rev. Cardiol. 2011, 8, 630–643. 10.1038/nrcardio.2011.138. [DOI] [PubMed] [Google Scholar]
- Boguszewicz Ł.; Jamroz E.; Ciszek M.; Widera E. E.; Kijonka M.; Banasik T.; Skorupa A.; Sokół M. NMR-based metabolomics in pediatric drug resistant epilepsy - preliminary results. Sci. Rep. 2019, 9, 15 10.1038/s41598-019-51337-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detour J.; Bund C.; Behr C.; Cebula H.; Cicek E. A.; Hirsch M. P. V.; Lannes B.; Lhermitte B.; Nehlig A.; Kehrli P.; Proust F.; Hirsch E.; Namer I. J. Metabolomic characterization of human hippocampus from drug-resistant epilepsy with mesial temporal seizure. Epilepsia 2018, 59, 607–616. 10.1111/epi.14000. [DOI] [PubMed] [Google Scholar]
- Murgia F.; Muroni A.; Puligheddu M.; Polizzi L.; Barberini L.; Orofino G.; Solla P.; Poddighe S.; Carratore F. D.; Griffin J. L.; Atzori L.; Marrosu F. Metabolomics as a tool for the characterization of drug-resistant Epilepsy. Front. Neurol. 2017, 8, 459 10.3389/fneur.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody S.; Brennan L. Effects of pentylenetetrazole-induced seizures on metabolomic profiles of rat brain. Neurochem. Int. 2010, 56, 340–344. 10.1016/j.neuint.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Wang D.; Wang X. X.; Kong J.; Wu J. Y.; Lai M. C. GC-MS-Based metabolomics discovers a shared serum metabolic characteristic among three types of epileptic seizures. Epilepsy Res. 2016, 126, 83–89. 10.1016/j.eplepsyres.2016.07.003. [DOI] [PubMed] [Google Scholar]
- Wei C. M.; Li Y.; Yao H.; Liu H. J.; Zhang X. M.; Guo R. C. A metabonomics study of epilepsy in patients using gas chromatography coupled with mass spectrometry. Mol. BioSyst. 2012, 8, 2197–2204. 10.1039/c2mb25105a. [DOI] [PubMed] [Google Scholar]
- Pavlova T.; Stepanichev M.; Gulyaeva N. Pentylenetetrazole kindling induces neuronal cyclin B1 expression in rat hippocampus. Neurosci. Lett. 2006, 392, 154–158. 10.1016/j.neulet.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Lüttjohann A.; Fabene P. F.; Luijtelaar G. A revised Racine’s scale for PTZ-induced seizures in rats. Physiol. Behav. 2009, 98, 579–586. 10.1016/j.physbeh.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Liu G. L.; Wang K. Y.; Zhao C. Z.; Jun X. D.; Zuo C. T. Functional imaging and pathology in brain of interictal cats kindled by pentylenetetrazol. Neurol. Asia 2013, 18, 9–15. [Google Scholar]
- Xiao C. N.; Jia P.; Wu M.; Zhang Y. J.; Wang S. X.; Zhao X. F.; Zheng X. H. Cold water forced swimming stress induced metabolic alterations in rats. Anal. Methods 2014, 6, 4144–4151. 10.1039/c4ay00374h. [DOI] [Google Scholar]
- Shi X. H.; Xiao C. N.; Wang Y. L.; Tang H. R. Gallic Acid Intake Induces Alterations to Systems Metabolism in Rats. J. Proteome Res. 2013, 12, 991–1006. 10.1021/pr301041k. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Foxall P. J.; Spraul M.; Farrant R. D.; Lindon J. C. 750 MHz 1H and 1H-13C NMR Spectroscopy of Human Blood Plasma. Anal. Chem. 1995, 67, 793–811. 10.1021/ac00101a004. [DOI] [PubMed] [Google Scholar]
- Samokhina E.; Samokhin A. Neuropathological profile of the pentylenetetrazol (PTZ) kindling model. Int. J. Neurosci. 2018, 128, 1086–1096. 10.1080/00207454.2018.1481064. [DOI] [PubMed] [Google Scholar]
- Huang R. Q.; Bell-Horner C. L.; Dibas M. I.; Covey D. F.; Drewe J. A.; Dillon G. H. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: Mechanism and site of action. J. Pharmacol. Exp. Ther. 2001, 298, 986–995. [PubMed] [Google Scholar]
- Yudkoff M.; Daikhin Y.; Nissim I.; Horyn O.; Lazarow A.; Nissim I. Metabolism of brain amino acids following pentylenetetrazole treatment. Epilepsy Res. 2003, 53, 151–162. 10.1016/S0920-1211(02)00260-7. [DOI] [PubMed] [Google Scholar]
- Burdakov D.; Gerasimenko O.; Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J. Neurosci. 2005, 25, 2429–2433. 10.1523/JNEUROSCI.4925-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechter E. M.; Veliskova J.; Velisek L. Correlation between extracellular glucose and seizure susceptibility in adult rats. Ann. Neurol. 2003, 53, 91–101. 10.1002/ana.10415. [DOI] [PubMed] [Google Scholar]
- Kudin A. P.; Zsurka G.; Elger C. E.; Kunz W. S. Mitochondrial involvement in temporal lobe epilepsy. Exp. Neurol. 2009, 218, 326–332. 10.1016/j.expneurol.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Rowley S.; Patel M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radical Biol. Med. 2013, 62, 121–131. 10.1016/j.freeradbiomed.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S. J.; Yu B. C. Mitochondrial matters of the brain: mitochondrial dysfunction and oxidative status in epilepsy. J. Bioenerg. Biomembr. 2010, 42, 457–459. 10.1007/s10863-010-9317-4. [DOI] [PubMed] [Google Scholar]
- Bough K. J.; Schwartzkroin P. A.; Rho J. M. Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia 2003, 44, 752–760. 10.1046/j.1528-1157.2003.55502.x. [DOI] [PubMed] [Google Scholar]
- Eagles D. A.; Boyd S. J.; Kotak A.; Allan F. Calorie restriction of a high-carbohydrate diet elevates the threshold of PTZ-induced seizures to values equal to those seen with a ketogenic diet. Epilepsy Res. 2003, 54, 41–52. 10.1016/S0920-1211(03)00041-X. [DOI] [PubMed] [Google Scholar]
- Yudkoff M.; Daikhin Y.; Melø T. M.; Nissim I.; Sonnewald U.; Nissim I. The ketogenic diet and brain metabolism of amino acids: Relationship to the anticonvulsant effect. Annu. Rev. Nutr. 2007, 27, 415–430. 10.1146/annurev.nutr.27.061406.093722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I.; Kasoff W. S.; Cassaday M. P.; Jacob R.; Gueorguieva R.; Sherwin R. S.; Krystal J. H.; Spencer D. D.; Abi-Saab W. M. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol. 2005, 57, 226–235. 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Tumani H.; Jobs C.; Brettschneider J.; Hoppner A. C.; Kerling F.; Fauser S. Effect of epileptic seizures on the cerebrospinal fluid - A systematic retrospective analysis. Epilepsy Res. 2015, 114, 23–31. 10.1016/j.eplepsyres.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Chatzikonstantinou A.; Ebert A. D.; Hennerici M. G. Cerebrospinal fluid findings after epileptic seizures. Epileptic Disord. 2015, 17, 453–459. 10.1684/epd.2015.0779. [DOI] [PubMed] [Google Scholar]
- Ide K.; Schmalbruch I. K.; Quistorff B.; Horn A.; Secher N. H. Lactate, glucose and O-2 uptake in human brain during recovery from maximal exercise. J. Physiol. 2000, 522, 159–164. 10.1111/j.1469-7793.2000.t01-2-00159.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. L.; Maidment N. T.; Shomer M. H.; Behnke E. J.; Ackerson L.; Fried I.; Engel J. Comparison of seizure related amino acid release in human epileptic hippocampus versus a chronic, kainate rat model of hippocampal epilepsy. Epilepsy Res. 1996, 26, 245–254. 10.1016/S0920-1211(96)00057-5. [DOI] [PubMed] [Google Scholar]
- Haglid K. G.; Wang S.; Qiner Y.; Hamberger A. Excitotoxicity. Experimental correlates to human epilepsy. Mol. Neurobiol. 1994, 9, 259–263. 10.1007/BF02816125. [DOI] [PubMed] [Google Scholar]
- Thomas P. M.; Phillips J. P.; O’Connor W. T. Hippocampal microdialysis during spontaneous intraoperative epileptiform activity. Acta Neurochir. 2004, 146, 143–151. 10.1007/s00701-003-0189-9. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Qin Z. H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- Guerriero R. M.; Giza C. C.; Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27 10.1007/s11910-015-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C.; Cossart R.; Hirsch J. C.; Esclapez M.; Ben-Ari Y. What is GABAergic inhibition? How is it modified in epilepsy?. Epilepsia 2000, 41, S90–S95. 10.1111/j.1528-1157.2000.tb01564.x. [DOI] [PubMed] [Google Scholar]
- Mishra C. B.; Kumari S.; Angeli A.; Monti S. M.; Buonanno M.; Tiwari M.; Supuran C. T. Discovery of benzenesulfonamides with potent human carbonic anhydrase inhibitory and effective anticonvulsant action: design, synthesis, and pharmacological assessment. J. Med. Chem. 2017, 60, 2456–2469. 10.1021/acs.jmedchem.6b01804. [DOI] [PubMed] [Google Scholar]
- Koo B. S.; Park K. S.; Ha J. H.; Park J. H.; Lim J. C.; Lee D. U. Inhibitory effects of the fragrance inhalation of essential oil from Acorus gramineus on central nervous system. Biol. Pharm. Bull. 2003, 26, 978–982. 10.1248/bpb.26.978. [DOI] [PubMed] [Google Scholar]
- Hutson S. M.; Lieth E.; LaNoue K. F. Function of leucine in excitatory neurotransmitter metabolism in the central nervous system. J. Nutr. 2001, 131, 846S–850S. 10.1093/jn/131.3.846S. [DOI] [PubMed] [Google Scholar]
- Maciejak P.; Szyndler J.; Turzyńska D.; Sobolewska A.; Taracha E.; Skórzewska A.; Lehner M.; Bidziński A.; Hamed A.; Wisłowska-Stanek A.; Płaźnik A. The effects of group III mGluR ligands on pentylenetetrazol-induced kindling of seizures and hippocampal amino acids concentration. Brain Res. 2009, 1282, 20–27. 10.1016/j.brainres.2009.05.049. [DOI] [PubMed] [Google Scholar]
- Armano S.; Coco S.; Bacci A.; Pravettoni E.; Schenk U.; Verderio C.; Varoqui H.; Erickson J. D.; Matteoli M. Localization and functional relevance of system A neutral amino acid transporters in cultured hippocampal neurons. J. Biol. Chem. 2002, 277, 10467–10473. 10.1074/jbc.M110942200. [DOI] [PubMed] [Google Scholar]
- Griffin J. L.; Rae C.; Dixon R. M.; Radda G. K.; Matthews P. M. Excitatory amino acid synthesis in hypoxic brain slices: does alanine act as a substrate for glutamate production in hypoxia?. J. Neurochem. 1998, 71, 2477–2486. 10.1046/j.1471-4159.1998.71062477.x. [DOI] [PubMed] [Google Scholar]
- Saransaari P.; Oja S. S. Characterization of N-methyl-D-aspartate-evoked taurine release in the developing and adult mouse hippocampus. Amino Acids 2003, 24, 213–221. 10.1007/s00726-002-0310-z. [DOI] [PubMed] [Google Scholar]
- Sechi G.; Rosati G.; Deiana G. A.; Petruzzi V.; Deriu F.; Correddu P.; Riu P. L. D. Co-variation of free amino acids in brain interstitial fluid during pentylenetetrazole-induced convulsive status epilepticus. Brain Res. 1997, 764, 230–236. 10.1016/S0006-8993(97)00487-3. [DOI] [PubMed] [Google Scholar]
- Sherwin A. L. Neuroactive amino acids in focally epileptic human brain: a review. Neurochem. Res. 1999, 24, 1387–1395. 10.1023/A:1022580506443. [DOI] [PubMed] [Google Scholar]
- Kanamori K.; Ross B. D.; Kondrat R. W. Rate of glutamate synthesis from leucine in rat brain measured in vivo by 15N NMR. J. Neurochem. 1998, 70, 1304–1315. 10.1046/j.1471-4159.1998.70031304.x. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Zielke H. R.; Tildon J. T.; Zielke C. L.; Baab P. J. Elevation of amino acids in the interstitial space of the rat brain following infusion of large neutral amino and ketocids by microdialysis:leucine infusion. Dev. Neurosci. 1996, 18, 415–419. 10.1159/000111435. [DOI] [PubMed] [Google Scholar]
- Rainesalo S.; Keränen T.; Palmio J.; Peltola J.; Oja S. S.; Saransaari P. Plasma and cerebrospinal fluid amino acids in epileptic patients. Neurochem. Res. 2004, 29, 319–324. 10.1023/B:NERE.0000010461.34920.0c. [DOI] [PubMed] [Google Scholar]
- Flynn C. J.; Wecker L. Concomitant increases in the levels of choline and free fatty acids in rat brain: evidence supporting the seizure-induced hydrolysis of phosphatidylcholine. J. Neurochem. 1987, 48, 1178–1184. 10.1111/j.1471-4159.1987.tb05644.x. [DOI] [PubMed] [Google Scholar]
- Scremin O. U.; Jenden D. J. Time-dependent changes in cerebral choline and acetylcholine induced by transient global ischemia in rats. Stroke 1991, 22, 643–647. 10.1161/01.STR.22.5.643. [DOI] [PubMed] [Google Scholar]
- Jope R. S.; Gu X. Seizures increase acetylcholine and choline concentrations in rat brain regions. Neurochem. Res. 1991, 16, 1219–1226. 10.1007/BF00966699. [DOI] [PubMed] [Google Scholar]
- Park J. H.; Cho H.; Kim H.; Kim K. Repeated brief epileptic seizures by pentylenetetrazole cause neurodegeneration and promote neurogenesis in discrete brain regions of freely moving adult rats. Neuroscience 2006, 140, 673–684. 10.1016/j.neuroscience.2006.02.076. [DOI] [PubMed] [Google Scholar]
- Pavlova T.; Stepanichev M.; Gulyaeva N. Pentylenetetrazole kindling induces neuronal cyclin B1 expression in rat hippocampus. Neurosci. Lett. 2006, 392, 154–158. 10.1016/j.neulet.2005.09.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.