

SUMMARY
Mutations can be important biomarkers that influence the selection of specific cancer treatments. We recently combined mathematical modeling of RAS signaling network biochemistry with experimental cancer cell biology to determine why KRAS G13D is a biomarker for sensitivity to epidermal growth factor receptor (EGFR)-targeted therapies. The critical mechanistic difference between KRAS G13D and the other most common KRAS mutants is impaired binding to tumor suppressor Neurofibromin (NF1). Here, we hypothesize that impaired binding to NF1 is a ‘‘biophysical biomarker’’ that defines other RAS mutations that retain therapeutic sensitivity to EGFR inhibition. Both computational and experimental investigations support our hypothesis. By screening RAS mutations for this biophysical characteristic, we identify 10 additional RAS mutations that appear to be biomarkers for sensitivity to EGFR inhibition. Altogether, this work suggests that personalized medicine may benefit from migrating from gene-based and allele-based biomarker strategies to biomarkers based on biophysically defined subsets of mutations.
Graphical abstract
In brief
McFall and Stites investigate the relationship between RAS mutations and epidermal growth factor receptor (EGFR) inhibitors. They hypothesize that impaired binding of a RAS mutant to NF1, sensitivity to EGFR inhibition, and suppression of wild-type RAS-GTP by EGFR inhibition are interrelated. They establish these dependencies with systems biochemical approaches and identify 10 RAS mutant biomarkers.
INTRODUCTION
A major unmet problem in cancer medicine involves matching patients with effective treatments on the basis of their tumor genomes (Chin et al., 2011; Friedman et al., 2015). Many Food and Drug Association-approved drugs are initially approved for a well-defined and narrow set of biomarker mutations and are later found to be effective on a larger set of biomarker mutations. The identification of new subsets of patients who benefit from an existing agent on the basis of tumor genomic data is thus a proven approach for improving cancer outcomes.
RAS mutations were among the first actionable biomarkers, in which the presence of a KRAS mutation in a colorectal cancer (CRC) indicated resistance to treatments that target the epidermal growth factor receptor (EGFR) (Jonker et al., 2007). Initiation of EGFR signaling leads to the activation of the three RAS GTPases (KRAS, NRAS, and HRAS), which in turn promote the RAF/MEK/ERK mitogen activated protein kinase (MAPK) cascade that drives cellular proliferation (Figure S1A) (Yarden and Pines, 2012). Oncogenic RAS mutations, which are primarily found in KRAS and less commonly within NRAS (Figure 1A), are found in approximately 40% of patients with CRC (Cancer Genome Atlas and Cancer Genome Atlas Network, 2012; Prior et al., 2020). Many different mutant alleles have been observed (Figure 1B). These mutants are typically constitutively active in the absence of EGFR signals, and the mutants have the ability to initiate the ERK cascade (Moore et al., 2020).
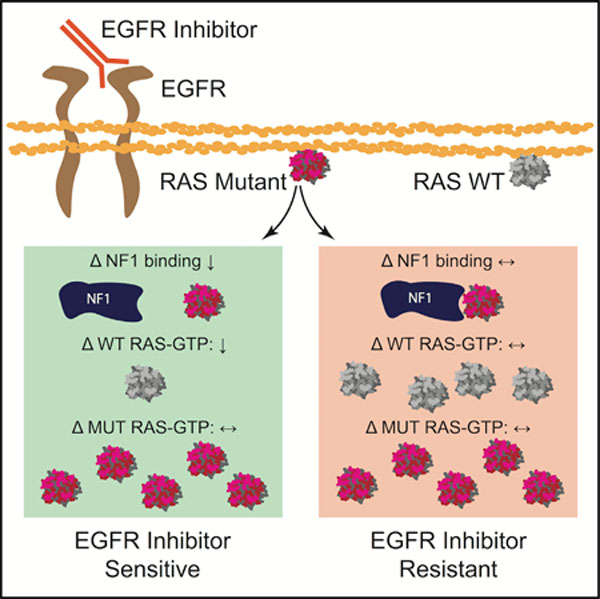
Figure 1. RAS-NF1 interactions are hypothesized to broadly determine whether a RAS mutant CRC is sensitive to EGFR inhibition.
(A) The distribution of RAS mutations between KRAS, NRAS, and HRAS within CRC.
(B) The different KRAS and NRAS mutations that have been observed in CRC.
(C) The establishment of KRAS G13D as an EGFR inhibitor-sensitive mutation necessitates the determination of whether patients with other KRAS and NRAS mutations may also benefit from treatment with EGFR inhibitors (EGFRi)
Therapeutic monoclonal antibodies that target EGFR, like cetuximab and panitumumab, are approved for the treatment of CRC (Normanno et al., 2009). Clinical trials have shown that EGFR inhibitors benefit patients without a RAS mutation, but not the subset of patients with a RAS mutation (Karapetis et al., 2008). Thus, patients with a constitutively active KRAS or NRAS mutation are recommended not to receive an EGFR inhibitor (Allegra et al., 2009; Jimeno et al., 2009). This relationship appears consistent with the general principles of EGFR/RAS signaling and is a paradigm in personalized medicine (Chin et al., 2011).
However, this relationship between RAS and EGFR inhibition appears overly simplistic. This first became evident after a retrospective analysis of clinical trial data revealed that patients with the KRAS G13D mutation (which involves a glycine [G] to aspartic acid [D] substitution at codon 13) benefited from EGFR inhibitor treatment (De Roock et al., 2010). Clinical guidelines did not change because it was unclear why KRAS G13D CRC should respond differently (Morelli and Kopetz, 2012). Recent studies, however, have provided a mechanistic basis that explains why KRAS G13D is sensitive to EGFR inhibition (McFall et al., 2019; Rabara et al., 2019).
It is possible that other RAS mutations may also indicate patients with CRC who could benefit from treatment with EGFR inhibitors (Figure 1C). In our previous work, we combined computational systems biology approaches that simulate protein biochemical activities with experimental cell biology to uncover the mechanism that explains KRAS G13D CRC sensitivity to EGFR inhibitors (McFall et al., 2019). Importantly, we found that a single biophysically measurable parameter, the affinity of the RAS mutant protein for Neurofibromin (NF1), was the critical property that determined sensitivity or resistance for cancer cells with one of the three most common KRAS mutants (G12D, G12V, and G13D). That work revealed that levels of GTP-bound, active, wild-type (WT) RAS decrease upon EGFR inhibitor treatment only within cells with the G13D mutant that binds NF1 poorly (Figures S1B and S1C).
Our work on KRAS G13D led us to speculate that a biophysical biomarker, the binding strength between a RAS mutant and NF1, may be able to classify whether other RAS mutations are likely to indicate sensitivity of a CRC to EGFR inhibition. To investigate, we first apply our computational model of RAS signaling to six NRAS mutants with available NF1 affinity data. Our model suggests, and we experimentally confirm, that two of the six should be sensitive to EGFR inhibition. We then empirically screen additional KRAS mutant isogenic cells and identify three more EGFR inhibitor-sensitive KRAS mutants. We find that each of these mutants has impaired NF1 binding, and we also find that cells with these mutants show reduced WT RAS-GTP upon EGFR inhibition. We use our computational model to show that reduced net interactions with NF1 are necessary for a RAS mutant cell to show sensitivity to EGFR inhibition. We then characterize the ability of an additional 12 KRAS mutant proteins to bind NF1, and we identify 5 more with impaired binding to NF1. Our assays also find that sensitivity and resistance correlate well with NF1 binding for these mutants. In total, our study identifies 10 additional RAS mutations that appear to be biomarkers for CRC cell sensitivity to EGFR inhibition. Overall, this study demonstrates the power of the mechanism-based systems biology approach as a tool to drive progress in personalized cancer medicine.
RESULTS
Modeling identifies additional RAS mutants that are sensitive to EGFR inhibition
Our previous work utilized a mathematical model of RAS signaling that includes guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), effector proteins that bind specifically to RAS-GTP, as well as WT and mutant RAS proteins (Figure 2A). The model includes GEF-driven (net) conversion of RAS-GDP to RAS-GTP, GAP-driven conversion of WT RAS-GTP to RAS-GDP, RAS-effector binding and unbinding, spontaneous GTP hydrolysis by RAS that occurs independently from GAPs, and spontaneous nucleotide dissociation and association that occurs independently from GEFs. General patterns of WT and mutant RAS signaling can be explained in terms of these processes, and the details of this model have been thoroughly described in multiple previous publications (McFall et al., 2019; Stites and Ravichandran, 2012; Stites and Shaw, 2018; Stites et al., 2007). The model has led to multiple prospective predictions about RAS biology that have been experimentally and reproducibly verified (McFall et al., 2019; Stites et al., 2007, 2015).
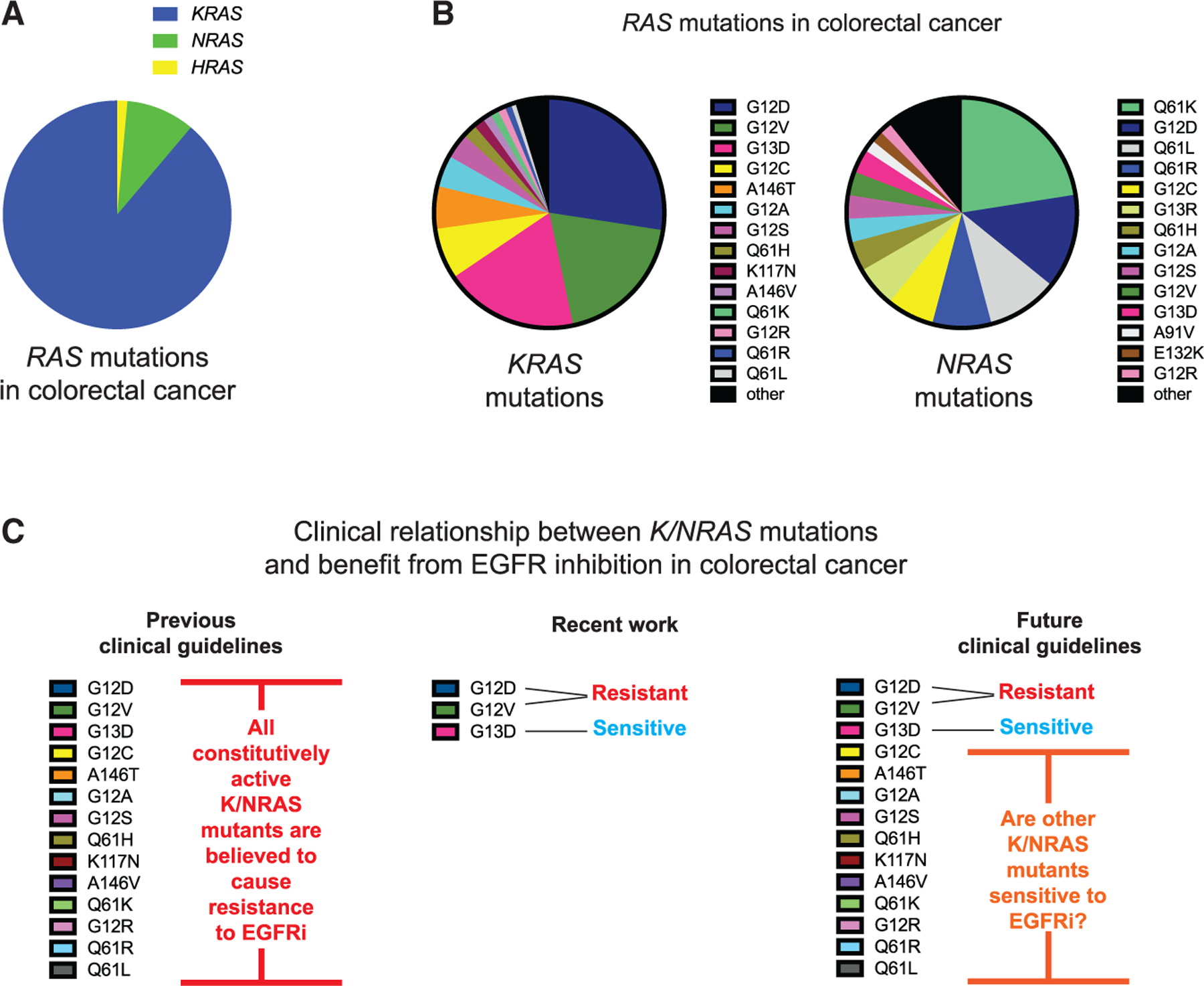
Figure 2. Computational predictions of the sensitivity of NRAS codon 61 mutations to EGFR inhibition and experimental confirmation of predicted sensitivities.
(A) Schematic of the RAS model.
(B) Reported relative change in the NF1 interaction Km for NRAS mutants, and RAS model prediction for EGFR inhibitor (EGFRi) sensitivity based on this Km value.
(C) Drug dose-response MTT assays for SW48 isogenic cells harboring the indicated RAS mutations. Data represent the mean ± SD (n = 8).
(D) Colony formation assays for SW48 isogenics treated (or not) with cetuximab.
(E) Colony formation assays for SW48 isogenic cells treated (or not) with EGF.
(F) EGFR inhibitor resistance assays. Data are means ± SD (n = 8). Significance was determined with *p < 0.01. Statistical significance was computed with one-way ANOVA followed by the post hoc Tukey test for multiple comparisons.
(G) RBD-IEF RAS activation assays for NRAS SW48 isogenics treated with cetuximab.
(H) Computational and experimental analyses both suggest that impaired binding to NF1 by a RAS mutant implies sensitivity of colon cancer cells with that mutant to EGFR inhibition, and also suggest that sensitivity follows from reductions of WT RAS-GTP upon EGFR inhibitor treatment.
(C) through (G) are each representative of three independent experiments.
Our previous simulations of G13D-, G12V-, and G12D-tailored versions of the model suggested that although mutant RAS-GTP levels were essentially unchanged when networks with any of these three mutants underwent EGFR inhibition, that WT RAS-GTP levels would fall more precipitously for G13D- than for G12D- and G12V-containing networks (McFall et al., 2019). This suggested WT RAS-GTP levels were the critical variable that could explain diverging response patterns, and we experimentally confirmed this prediction. We also found that impaired binding to NF1 was the critical parameter that determined whether the network with the RAS mutant would be sensitive to modeled EGFR inhibition. Here, we hypothesize that reduced binding to NF1 may be a feature of other RAS mutants and that mutants with reduced binding to NF1 may be sensitive to EGFR inhibition.
To investigate, we searched the literature for reports of RAS mutant proteins that bind poorly to NF1, the RAS GAP with a clear role in maintaining low levels of WT RAS-GTP and for which loss-of-function mutations have a clear driver role in cancer (Kiuru and Busam, 2017). Most biophysical studies that characterize the interaction between RAS mutants and RAS GAPs have focused on the p120 RAS GAP coded by RASA1 (Hunter et al., 2015; Wey et al., 2013). We found one study that considered six codon 61 NRAS mutations and their binding to NF1 and to p120 RAS GAP (Donovan et al., 2002). Interestingly, affinity levels for p120 RAS GAP do not correlate with those for NF1. Two of the six NRAS mutants, Q61K and Q61R, were reported to have significantly reduced binding to NF1 (~55× and ~40× weaker binding, respectively) (Figure 2B). This reduction was not as strong as KRAS G13D, for which a different group detected no binding to NF1 (Gremer et al., 2008), and which we approximated with a 100× reduction in the RAS-NF1 affinity (McFall et al., 2019). We extended our model to include these codon 61 mutations and simulated our model to determine whether the reported magnitude changes in NF1 binding may result in sensitivity to EGFR inhibition (Figure S2A). The model suggested that the two RAS mutants with the lowest affinity values toward NF1 (Q61K and Q61R) have a similar level of sensitivity to EGFR inhibition as the G13D mutant (McFall et al., 2019). In contrast, the other mutants showed a response to EGFR inhibition that was more like the resistant G12D and G12V RAS mutants. Additionally, the model suggested that levels of WT RAS-GTP would fall the most in Q61K- and Q61R-containing cancer cells and that there would be no change in mutant RAS-GTP for any of the mutants.
Experiments confirm NRAS Q61K and NRAS Q61R cells are sensitive to EGFR inhibition
We tested these predictions experimentally. We obtained NRAS mutant allele isogenic colon cancer cell lines derived from SW48 cells. These isogenic cells have been previously developed and utilized; they express the RAS proteins at similar levels between isogenic lines, express WT NF1, and are commercially available (De Roock et al., 2010; Hood et al., 2019; Mageean et al., 2015). There are four such NRAS mutant lines available, which are respectively heterozygous for NRAS Q61L, Q61H, Q61K, and Q61R. Thus, these cells allowed for us to test two mutants predicted to be sensitive and two predicted to be resistant. We performed cetuximab dose-response experiments on these cells along with KRAS G13D, G12V, and WT isogenics as controls. We observed NRAS Q61K and Q61R cells to show dose-dependent inhibition of proliferation at a level consistent with WT and KRAS G13D isogenic cells (Figure 2C). In contrast, NRAS Q61H and Q61L cells showed resistance to cetuximab at all doses, like KRAS G12V cells. We also observed reductions in phosphorylated ERK levels upon cetuximab treatment for the sensitive NRAS Q61K and Q61R cells, but not the Q61L and Q61H cells (Figure S2B).
To test long-term cetuximab sensitivity, we performed 4-week colony formation assays. We observed that NRAS Q61L, NRAS Q61H, and KRAS G12V isogenic cells formed colonies in the presence of cetuximab. In contrast, NRAS Q61K, NRAS Q61R, KRAS G13D, and WT isogenic cells produced a reduced number of colonies (Figure 2D). To confirm that the observed pattern of sensitivity followed from EGFR signaling, we also performed a colony formation assay in serum-depleted media supplemented with or without EGF. NRAS Q61L, NRAS Q61H, and KRAS G12V showed colony growth in the absence of EGF where NRAS Q61K, NRAS Q61R, KRAS G13D, and WT isogenics showed decreased colony formation in the absence of EGF (Figure 2E).
To confirm our findings with an orthogonal method, we evaluated whether the introduction of these mutants could cause parental (WT) SW48 cells to become resistant to cetuximab. For this resistance assay, parental SW48 cells were pretreated with cetuximab for 24 h and then transfected with an equal amount of construct for mutant RAS or WT RAS. We then evaluated proliferation after an additional 48 h of growth in the presence of cetuximab. We found that NRAS Q61L, NRAS Q61H, and KRAS G12V all significantly increased proliferation, while NRAS Q61K, NRAS Q61R, and KRAS G13D did not (Figures 2F and S2C).
NRAS Q61K and Q61R cells treated with EGFR inhibitors display reduced WT RAS-GTP
In our previous study of KRAS G13D, we demonstrated that sensitivity to cetuximab followed from reductions in WT RAS-GTP (HRAS and NRAS), but not in the mutant KRAS-GTP. Similarly, our computational model suggests that WT RAS-GTP levels should drop more in the Q61K and Q61R cells than in Q61L and Q61H cells (i.e., Figure S2A). To test this experimentally, we measured RAS activation in the presence or absence of cetuximab. We first performed RAS binding domain (RBD) pull-down to isolate GTP-bound RAS. We then separated RAS-GTP into KRAS, NRAS, and HRAS fractions with isoelectric focusing (IEF) using a protocol we developed and reported previously (RBD-IEF) (McFall et al., 2019). We detected reductions in WT RAS-GTP (KRAS and HRAS) within NRAS Q61K and Q61R isogenic cells, but we detected no significant changes in KRAS-GTP and HRAS-GTP within NRAS Q61L and Q61H cells (Figures 2G and S2D). There were no significant changes in NRAS-GTP after treatment for any of the NRAS mutant cells. Depletion of RAS-GTP by cetuximab in these experiments also resulted in decreased phospho-ERK, showing inhibition of the canonical MAPK pathway (Figure S2E).
The biophysical data we utilized in our model described a reduced affinity of NF1 for mutant NRAS Q61R and Q61K relative to NRAS WT, Q61L, and Q61H. These differences are critical for our predictions and our mechanism; for example, the substitution of the affinity of NRAS WT toward NF1 is sufficient to eliminate the computationally predicted sensitivity to EGFR inhibition (Figure S2F). Therefore, we desired to determine whether we could reproduce the previously described differences in NF1 affinity.
We first evaluated interaction strength by performing an NF1 co-immunoprecipitation (coIP) assay. We have previously shown G12V to bind NF1 well and G13D to be impaired in NF1 binding (McFall et al., 2019, 2020), and we used these mutants as positive and negative controls. We observed that NF1 strongly pulls down KRAS G12V, NRAS Q61H, and NRAS Q61L. Both NRAS Q61K and Q61R showed reduced binding to NF1 when compared to the other mutants, whereas KRAS G13D showed little binding (Figures S2G and S2H).
We also investigated NF1 binding within cells by bioluminescence resonance energy transfer (BRET). We observed decreased BRET signal for NRAS Q61K, NRAS Q61R, and KRAS G13D relative to NRAS Q61L, NRAS Q61H, KRAS G12D, and KRAS G12V (Figures S2I and S2J). We validated the specificity of our assay by utilizing Flag-tagged KRAS proteins, which do not produce a BRET signal (Figure S2K) (McFall et al., 2020), in experiments that evaluated competition for binding to NF1 (Figures S2L and S2M). Overall, our studies on codon 61 NRAS mutants validate our assertion that RAS mutants with impaired binding to NF1 are less effective at promoting resistance to EGFR inhibitors (Figure 2H).
Orthogonal methods confirm mutant-specific sensitivity to EGFR inhibition
We next investigated whether other EGFR inhibitors show a similar pattern of sensitivity and resistance for the NRAS genotypes. We find Q61K and Q61R cells to be sensitive to the EGFR inhibitors erlotinib and panitumumab, and Q61L and Q61H cells to be resistant (Figure S3A). We then treated three different NRAS codon 61 isogenic cells with EGFR small interfering RNA (siRNA) and/or erlotinib. We observed reduced EGFR phosphorylation for both EGFR inhibition and EGFR knockdown in all three NRAS genotypes tested (Figure S3B). We observed both perturbations to cause reduced proliferation of Q61K and Q61R cells and to cause no change for Q61L cells (Figure S3C). We also observed reduced ERK phosphorylation, KRAS-GTP, and HRAS-GTP in the Q61K and Q61R cells, but not in the Q61L cells (Figures S3B and S3C). Altogether, these experiments suggest that RAS mutant-specific sensitivity is dependent on EGFR signaling, regardless of the class of EGFR inhibitor. This is consistent with previous observations about KRAS G13D and EGFR inhibition (McFall et al., 2019, 2020; Rabara et al., 2019).
Orthogonal methods confirm that mutant-specific sensitivity to EGFR inhibition depends on NF1 activity and on WT RAS-GTP suppression
We investigated the effect of NF1 knockdown on the response to cetuximab (Figures S3D and S3E). We observed that reduced NF1 expression resulted in a loss of sensitivity to cetuximab for the NRAS Q61K and Q61R SW48 cells as measured by proliferation, RAS-GTP levels, and ERK phosphorylation. This suggests that NF1 activity is required for sensitivity to cetuximab. This is consistent with our previous studies of KRAS G13D CRC (McFall et al., 2019, 2020) and with similar, independent, contemporaneous results from the National Cancer Institute (NCI) RAS Initiative’s investigation into the sensitivity of KRAS G13D CRC to EGFR inhibition (Rabara et al., 2019). Additionally, we found that siRNA-mediated knockdown of NF1 in cetuximab-resistant SW48 NRAS Q61L cells caused no observable change in RAS-GTP levels, ERK phosphorylation, or proliferation.
Empirical screening of isogenic cells identifies additional EGFR inhibitor-sensitive KRAS mutants that are also poor NF1 binders
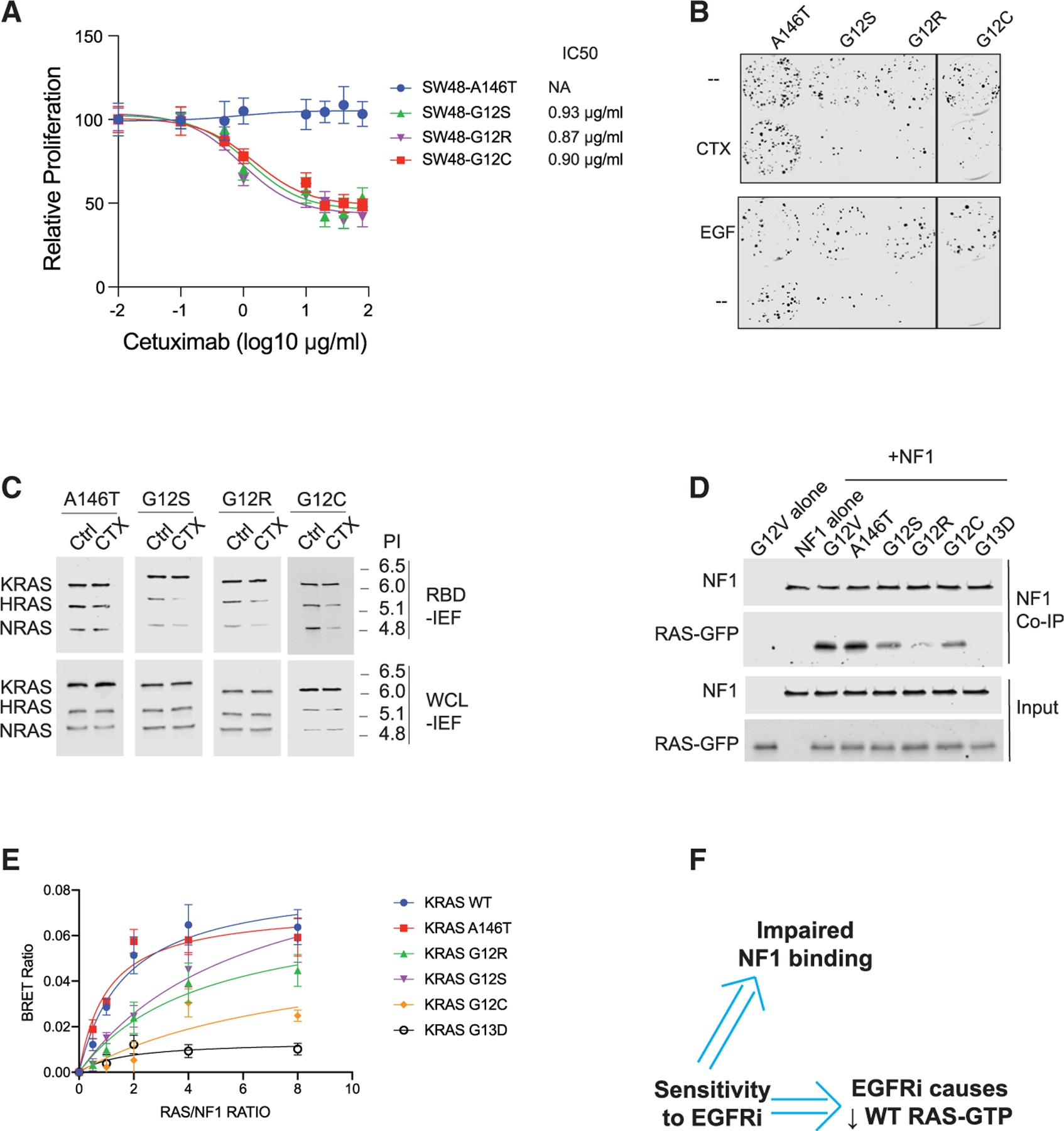
We speculated that there are more RAS mutants that are sensitive to EGFR inhibition by this same mechanism. We investigated this hypothesis empirically by obtaining the then remaining commercially available members of the KRAS mutant SW48 isogenic panel: KRAS G12C, G12R, G12S, and A146T. We evaluated each for sensitivity to cetuximab. We observed a dose-dependent reduction in proliferation in G12C, G12R, and G12S cells, while cells with the fast cycling A146T mutant (Poulin et al., 2019) showed complete resistance to EGFR inhibition by cetuximab (Figure 3A). We also observed reduced colony formation for G12S, G12R, and G12C cells, but not for A146T cells, when grown in cetuximab and when grown in the absence of EGF (Figure 3B).
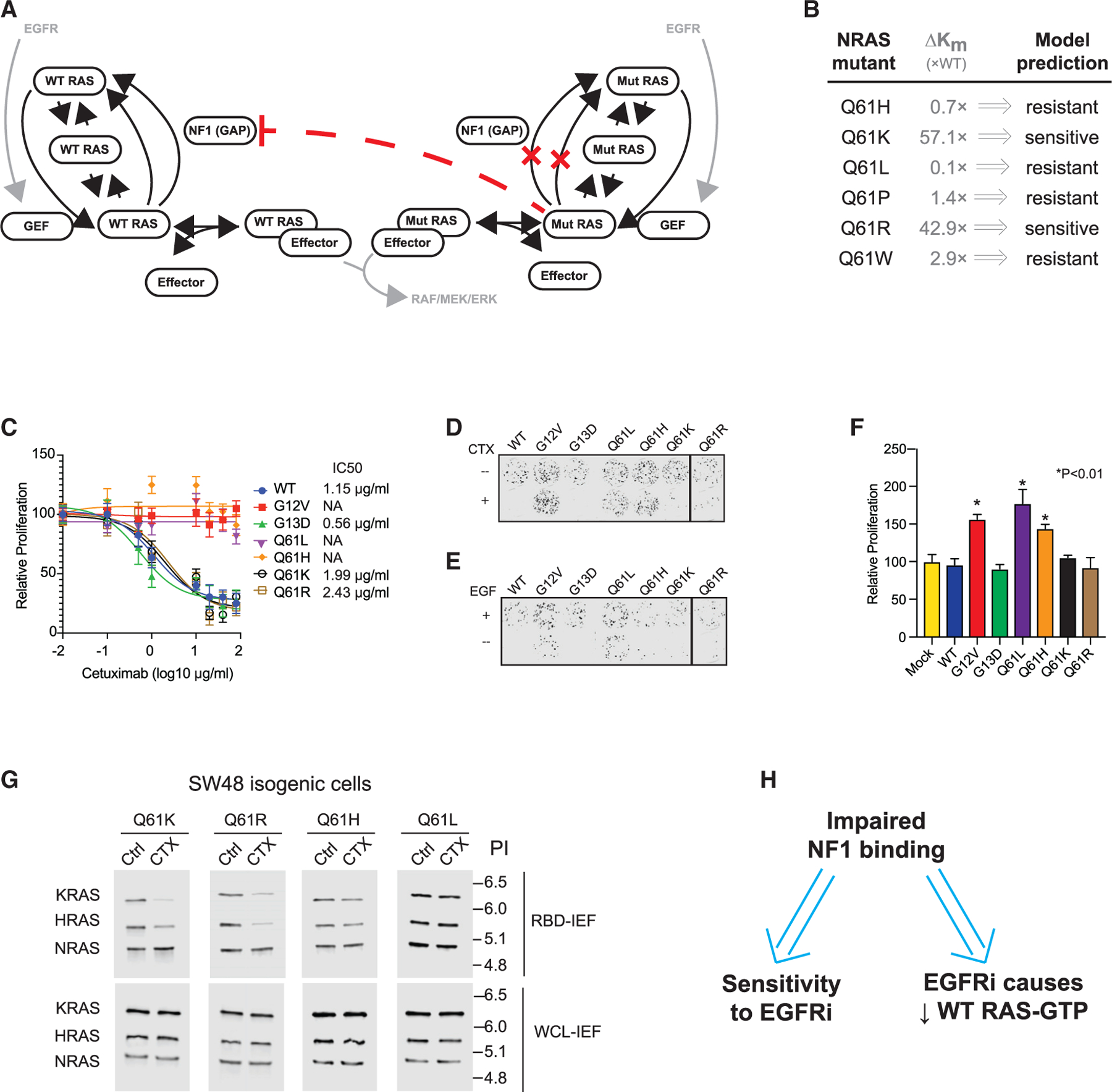
Figure 3. Isogenic cell line-based empiric screening finds three KRAS mutants that indicate sensitivity to EGFR inhibition, each of which supports the reduced NF1-binding and WT RAS-GTP level mechanism.
(A) Drug dose-response assays for KRAS A146T, G12C, G12R, and G12S SW48 isogenic cells. Data represent the mean ± SD (n = 8).
(B) Colony formation assays for SW48 isogenics treated (or not) with cetuximab (top) and treated (or not) with EGF (bottom).
(C) RBD-IEF RAS activation assays for SW48 isogenics treated with cetuximab.
(D) NF1 co-immunoprecipitation (coIP) assay for NF1 with KRAS G12V, A146T, G12S, G12R, G12C, and KRAS G13D.
(E) BRET measurements of interactions between NF1 and RAS. Data represent the BRET ratio ± SD from eight biological replicates (n = 8).
(F) Empirical screens to find EGFR inhibitor (EG-FRi)-sensitive RAS mutant genotypes suggest EGFR inhibitor sensitivity implies impaired binding of that RAS mutant to NF1, and implies that the sensitive cell displays reductions in WT RAS-GTP upon EGFR inhibition.
(B) through (E) are each representative of three independent experiments. CTX, cetuximab; NA, not applicable; Ctrl, control; WCL, whole cell lysate; PI, isoelectric point.
To determine whether the sensitivity of these RAS mutant cells to EGFR inhibition follows from reductions in WT RAS-GTP, we treated the cells with cetuximab and performed RBD-IEF (Figures 3C, S3F, and S3G). We observed reductions in HRAS-GTP, NRAS-GTP, and total RAS-GTP in G12C, G12S, and G12R isogenics, but these reductions were statistically significant only in the G12C and G12S cells. We also observed reduced ERK phosphorylation in the G12C, G12S, and G12R cells treated with cetuximab, but not the A146T cells (Figures S3H and S3I).
We investigated how well each of these KRAS mutants bound to NF1. We observed that KRAS G12R, G12S, and G12C all had reduced affinity to NF1 when compared to KRAS G12V and A146T when assessed by NF1 coIP (Figures 3D and S3J) and when assessed by BRET (Figure 3E). We note that G12R appeared more impaired by coIP than by BRET, but it displayed reduced binding to NF1 by both approaches. We also investigated the effect of NF1 knockdown on EGFR inhibitor-sensitive KRAS G12R and G12S cells (Figures S3K and S3L). We observed that reduced NF1 expression resulted in a loss of sensitivity to cetuximab as well as a loss of reductions in RAS-GTP and phosphorylated ERK levels. This is consistent with our observations on EGFR inhibitor-sensitive NRAS mutant cells. Overall, the RAS mutants found in these EGFR inhibitor-sensitive cells had weakened binding to NF1 and showed reduced WT RAS-GTP on EGFR inhibition, consistent with our motivating hypothesis (Figure 3F).
Modeling suggests that WT RAS-GTP reductions from EGFR inhibition imply reduced RAS mutant binding to RAS GAPs
The sensitivity of KRAS G13D CRC to EGFR inhibition perplexed clinicians and cancer biologists for nearly a decade. Two recent studies confirmed the sensitivity but explained it with slightly different mechanisms (Figure 4A). The mechanism that we advanced and build on here (mechanism 1) requires a consideration of both mutant and WT RAS (McFall et al., 2019). We demonstrated that KRAS G13D, unlike the two most common KRAS mutants (G12D and G12V), is impaired at binding to the RAS GAP NF1. As G12D and G12V are NF1 insensitive (no kcat for the conversion of mutant RAS-GTP to mutant RAS-GDP by NF1), their interaction with NF1 does not alter mutant RAS-GTP levels. However, their interaction with NF1 does sequester NF1 away from WT RAS-GTP, which shifts the dynamic equilibrium between WT RAS-GTP and WT RAS-GDP toward increased WT RAS-GTP. As G13D binds poorly to NF1 (i.e., the interaction is characterized by a high Km), NF1 is not sequestered into nonproductive complexes with the G13D mutant, and NF1 can therefore maintain low levels of WT RAS-GTP when EGFR is not driving RAS-GTP production. In contrast, mechanism 2 proposes that KRAS G13D retains sensitivity to NF1-mediated GTP hydrolysis (i.e., retains a high kcat on the same order of magnitude as WT RAS-GTP) (Rabara et al., 2019). The authors of that study propose that KRAS G13D is therefore EGFR dependent for GTP loading and that EGFR inhibition results in reduced KRAS G13D-GTP. The mechanism 2 study did not address or investigate WT RAS-GTP signaling.
Figure 4. Computational modeling suggests a reduction in WT RAS-GTP upon EGFR inhibition implies impaired NF1 binding and EGFR inhibitor sensitivity.
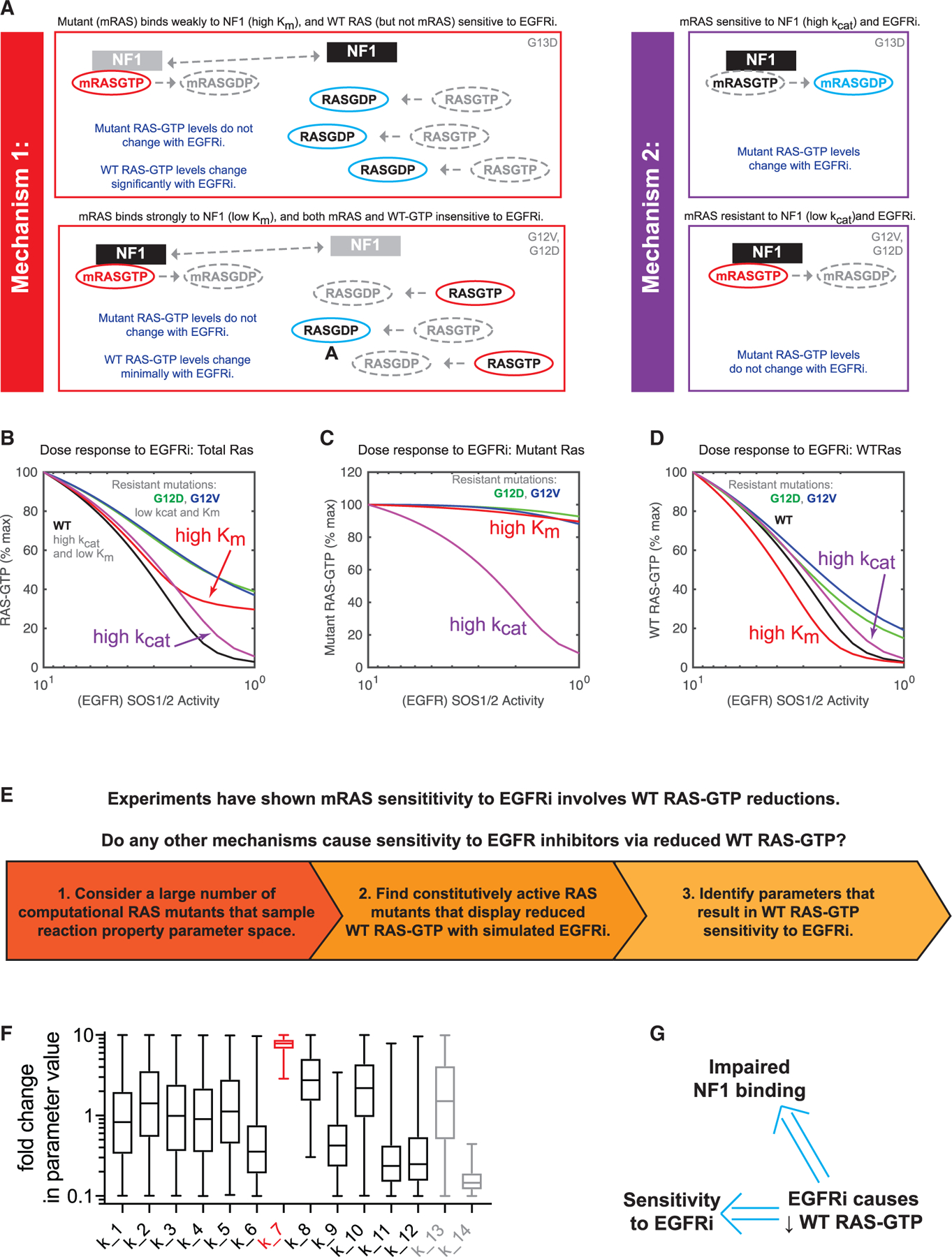
(A) Schematics presenting two different proposed mechanisms to explain how KRAS G13D CRCs respond to EGFR inhibitors (EGFRi).
(B) Computational simulations of EGFR inhibition for both mechanism 1 (red) and mechanism 2 (purple). Simulated dose responses of RAS G12V (green), G12D (blue), and WT (black) are provided for comparison. The proportion of total RAS (WT and mutant) that is bound to GTP is the model output.
(C) Computational simulations, as in (B), but with the model output limited to the proportion of mutant RAS that is bound to GTP.
(D) Computational simulations, as in (B), but with the model output limited to the proportion of WT RAS that is bound to GTP.
(E) Schematic displaying the computational approach to search for alternative mechanisms of RAS mutant network sensitivity to EGFR inhibition that involve WT RAS-GTP reduction at levels approximately the same as found when G13D parameters are utilized.
(F) Values from all of the parameter sets that resulted in EGFR sensitivity through WT RAS-GTP reduction without mutant RAS-GTP reduction. Parameters are presented normalized to the value of the same parameter in WT RAS-GTP. Whiskers span from minimum to maximum values. The Km of the interaction between NF1 and a modeled RAS mutant (k_7) is indicated in red. Parameter definitions are provided in Figure S4A.
(G) Computational screens suggest that a reduction in WT RAS-GTP upon EGFR inhibition implies both an overall sensitivity to EGFR inhibition and that any such RAS mutant has a reduced Km for NF1
We utilized our computational model of the RAS signaling dynamic equilibrium to evaluate both hypotheses. We modeled mechanism 2 with the reported NF1:G13D kcat from that study, and we utilized the same parameters we used previously to model KRAS G13D to model mechanism 1. Simulations find that both mechanisms would result in reduced total RAS-GTP upon EGFR inhibition (Figure 4B). One notable difference is that mechanism 1 (high Km) has the G13D mutant being constitutively active, while mechanism 2 (high kcat) would not be sufficient to cause the G13D mutant to be constitutively active.
Additional differences between the two mechanisms become evident when mutant RAS-GTP levels are considered. Modeling mechanism 1 finds essentially no change in the quantity of mutant RAS-GTP with EGFR inhibition, whereas modeling mechanism 2 finds a large change in mutant RAS-GTP levels upon EGFR inhibition (Figure 4C). Thus, the measurement of change in mutant RAS-GTP levels can distinguish between the two mechanisms. With regard to WT RAS-GTP levels, both mechanism 1 and mechanism 2 suggest reductions in wild-type RAS-GTP levels that are larger than observed for KRAS G12V and KRAS G12D (Figure 4D). Our previous experimental work on KRAS G13D CRC observed reductions in WT RAS-GTP but not mutant RAS-GTP (McFall et al., 2019, 2020), providing strong evidence for mechanism 1.
We asked whether there may be other mechanisms that could also cause reductions in WT RAS-GTP in a constitutively active RAS mutant cell treated with an EGFR inhibitor. To investigate, we returned to our computational model (Figure 4E). To search for mutants with other possible mechanisms of sensitivity, we created 3 million different computational RAS mutants. Each RAS mutant can be specified by how each of the 12 independent RAS reaction parameters differs ratiometrically from WT RAS (Stites et al., 2015) (Figure S4A). These mutants sample the space of potential RAS mutant biochemistries. Although we cannot map individual computational mutants to a specific RAS mutation, this approach allows us to determine what behaviors could potentially be displayed by RAS mutations. This may be thought of as somewhat analogous, and complementary, to experimental methods that sample different single amino acid substitution mutants to explore the space of potential RAS mutations (Bandaru et al., 2017).
We considered the behavior of RAS mutants from three different sets of 1 million computational random RAS mutants, each set sampling an additional order-of-magnitude range of both upward and downward fold-change variation for all of the model’s RAS reaction free parameters. Within each set, we considered computational random RAS mutants that were constitutively active at a level similar to KRAS G13D, G12D, and G12V under conditions with and without EGFR stimulation, and with a similar net change in total RAS-GTP between modeled stimulated and unstimulated conditions. We then simulated full EGFR inhibitor dose responses for all of these mutants and identified the dose responses that displayed a reduction in WT RAS-GTP that was approximately the same as computationally observed for KRAS G13D. We then investigated the parameters of all of these mutants to determine whether mechanisms other than an elevated NF1:RAS Km could cause this pattern of RAS activation.
When we evaluated the first set of 1 million mutants, where each parameter was within an order of magnitude increase or decrease from WT RAS, we found that all mutants that showed WT RAS-GTP reductions at a level comparable to G13D upon EGFR inhibition had an elevated Km (Figure 4F). The other parameters had a much wider range of values, suggesting that Km was the parameter that needed to be within a certain range for EGFR inhibitor sensitivity. Additionally, all of these mutants that resulted in WT RAS-GTP sensitivity to EGFR inhibition also resulted in overall sensitivity to EGFR inhibition, as evaluated by total (mutant and wild-type) RAS-GTP (Figure S4B). When we evaluated the mutants with larger magnitude changes in parameter values, we again observed that an elevated Km caused sensitivity to EGFR inhibition through WT RAS-GTP reductions (Figure S4C). As the parameters grew, we encountered some mutants that did not show an elevated Km but did show reduced WT RAS-GTP upon EGFR inhibition. On further inspection, we observed that all of these mutants had large increases in the affinity of the RAS mutant for its effectors (Figure S4D). Although not initially anticipated, this finding is consistent with reduced NF1 binding; a RAS mutant that binds more tightly to effectors is less able to bind and competitively inhibit NF1. Thus, our simulations with computational random RAS mutants that sampled increasingly broad portions of parameter space suggest that reduction of WT RAS-GTP upon EGFR inhibition implies that the RAS mutant has impaired NF1 binding, and this also implies that a cell with such a RAS mutant is sensitive to EGFR inhibition (Figure 4G).
BRET screens identify additional RAS mutants that are poor NF1 binders
Our studies suggest that a CRC cell with a RAS mutant protein that has impaired binding to NF1 will be sensitive to EGFR inhibition (Figure 5A). Thus, it should be possible to screen RAS mutants for how strongly they bind to NF1 as an approach to infer which indicate sensitivity to EGFR inhibition. We utilized our NF1-RAS BRET protocol to screen 12 of the most common mutants we had not yet studied (A59E, D33E, G13R, G12F, G12A, K117N, G13C, T35S, R68S, G12P, G12Y, and A146V) (Cancer Genome Atlas and Cancer Genome Atlas Network, 2012; Cerami et al., 2012; Sondka et al., 2018), and we identified 5 additional mutants that showed reduced NF1-RAS binding: G13R, G12F, G12A, G12P, and G12Y (Figures 5B, S5A, and S5B). CoIP assays with the same 12 mutants found that the same 5 mutants do not bind well to NF1 (Figures 5C and S5C).
Figure 5. NF1 interaction strength screening identifies an additional five KRAS mutants that are sensitive to EGFR inhibition.
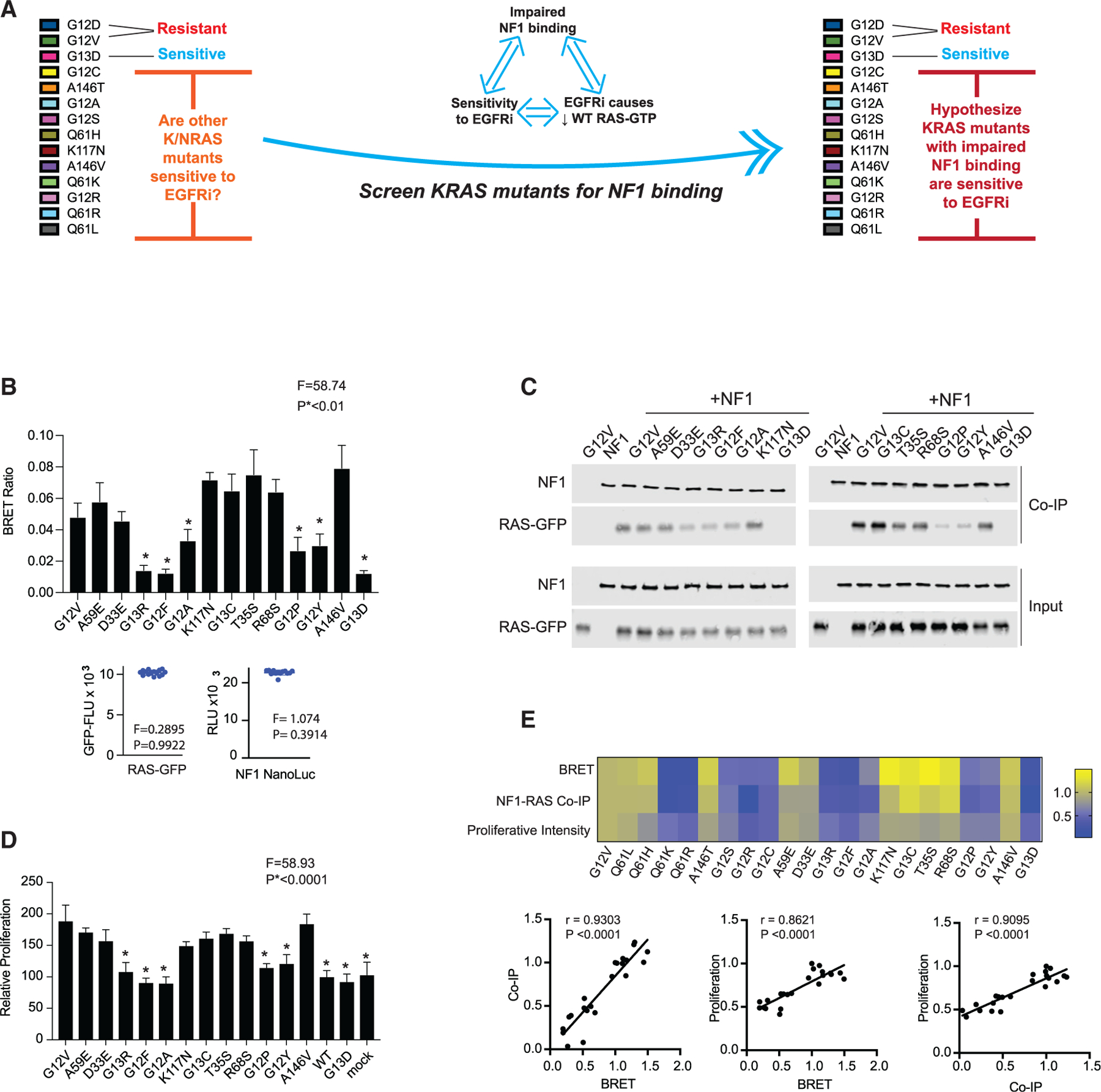
(A) Schematic to summarize that our analysis up to this point suggests that sensitivity to EGFR inhibition, impaired binding of a RAS mutant to NF1, and the reduction of WT RAS-GTP upon treatment of a mutant RAS CRC cell with an EGFR inhibitor (EGFRi) are associated. We thereby propose that the measurement of the relative strength of binding between mutant RAS and NF1 can serve as a method to infer sensitivity to EGFR inhibition.
(B) BRET measurements of interactions between NF1 and RAS to evaluate 12 additional KRAS mutants. Data represent the BRET ratio ± SD. Statistical difference was determined by one-way ANOVA followed by the post hoc Tukey test for multiple comparisons. Equal amounts of BRET donor and acceptor (NF1-NanoLuc/ mutant RAS-GFP) were expressed as seen in distribution plots (bottom), where data points are representative of mean relative luciferase units (NF1-NanoLuc) and mean GFP fluorescence units (RAS mutant) from the same samples in the histogram. There was no statistical difference among NF1-NanoLuc or RAS-GFP signals. *p < 0.01.
(C) NF1 co-immunoprecipitation (coIP) assays for the 12 additional KRAS mutants.
(D) EGFR inhibitor resistance assays for the 12 additional KRAS mutants. Histograms and error bars represent the mean ± SD (n = 8). Statistical difference was determined by one-way ANOVA followed by the post hoc Tukey test for multiple comparisons.*p < 0.0001.
(E) Comparisons of BRET assays, NF1 coIP assays, and proliferation assays. Data are normalized to KRAS G12V within each group of assays. Pearson correlation coefficients and p values are provided for each set of comparisons.
(B) through (D) are each representative of three independent experiments.
We next evaluated each of these mutants with our cetuximab resistance assay. We observed that G13R, G12F, G12A, G12P, and G12Y showed no difference in proliferation when compared to WT KRAS, like KRAS G13D and mock transfected conditions. Each of the other mutants showed a significant gain in proliferation (Figures 5D and S5D). These data reflect the pattern of resistance and sensitivity suggested by our NF1 interaction data. Comparison of the normalized mean effect size in BRET signal, the NF1 coIP intensity, and the cetuximab-rescue proliferation index for all 12 mutants showed strong, statistically significant, correlations between all three measures (Figure 5E). This suggests that the impairments we measure in binding to NF1 are not strongly dependent on the specific assay or specific conditions of the test.
Validation of EGFR inhibitor-sensitive RAS mutants in additional model systems
Our prior experimental data utilized SW48 heterozygous RAS mutant isogenic cells. To evaluate generalizability, we obtained four colon cancer cell lines that harbored a RAS mutation we found to be a biomarker for sensitivity to EGFR inhibition and two colon cancer cell lines with a RAS mutation we found to be a biomarker for resistance (Figure 6A). Of note, all six cell lines are NF1 WT (Barretina et al., 2012). We observed cetuximab to cause a strong reduction in growth in LS123 (KRAS G12S), SW1116 (KRAS G12A), and SW837 (KRAS G12C) cells, consistent with our observations in SW48 cells (Figure 6B). Also in agreement with our work on SW48 cells, LS1034 (KRAS A146T) and SW948 (KRAS Q61L) did not respond to cetuximab at any dose. Of note, SW1463 (KRAS G12C) cells were resistant to cetuximab, in contrast to the SW837 (KRAS G12C) and SW48 KRAS G12C isogenic cells. We believe this is due to the fact these cells are homozygous for KRAS G12C.
Figure 6. Validation of identified EGFR inhibitor-sensitive RAS mutants in additional model systems.
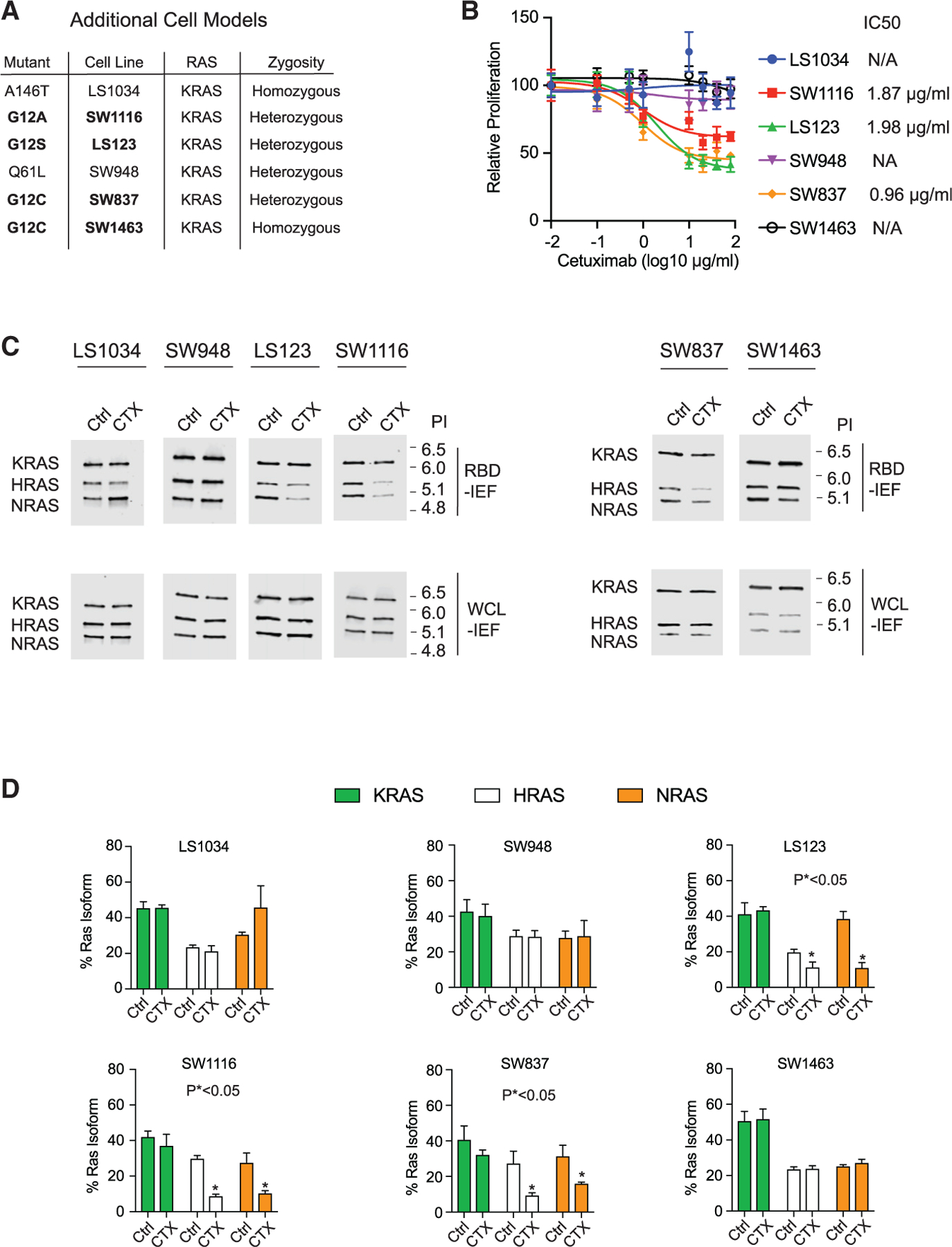
(A) Table of additional RAS mutant CRC cell lines that were investigated for EGFR inhibitor sensitivity.
(B) Drug dose-response assays for the additional cell lines. Data points and error bars represent the mean ± SD (n = 8).
(C) RBD-IEF after cetuximab treatment for theadditional cell lines.
(D) Quantification of RAS-GTP levels from RBD-IEF. Data points and error bars represent the mean ± SD from three separate experiments. Statistical significance was determined by unpaired two-tailed t est between untreated and treated conditions. *p < 0.05.
(B) and (C) are each representative of three independent experiments. CTX, cetuximab; NA, not applicable; Ctrl, control; WCL, whole cell lysate, PI, isoelectric point.
We next performed colony formation assays for all of these cells (Figure 6A). We found that LS123, SW1116, and SW837 were sensitive to cetuximab, but LS1034, SW948, and SW1463 were not. We also tested the long-term effects of EGF depletion on these cells, and we observed that LS123, SW1116, and SW837 relied on EGF for colony formation but LS1034, SW948, and SW1463 did not.
We next tested to see whether cetuximab treatment resulted in reduced WT RAS-GTP within these cell lines. RBD-IEF revealed statistically significant reductions in WT RAS-GTP (HRAS and NRAS) and total RAS-GTP in the LS123, SW116, and SW837 cells (Figures 6C, 6D, and S6B). In contrast, the cell lines that showed normal growth under cetuximab treatment showed no reductions in WT RAS-GTP. Importantly, the G12C homozygous SW1463 cells that were resistant to cetuximab also showed no change in WT RAS-GTP levels upon cetuximab treatment. We hypothesize that the increased dosage of KRAS G12C may overcome the reduced affinity and allow for NF1 competitive inhibition that in turn results in an elevated level of total WT RAS-GTP.
Mutations are observed to behave similarly whether in KRAS, NRAS, or HRAS
KRAS, NRAS, and HRAS are highly homologous and share the same hotspots at codons 12, 13, and 61 (Stephen et al., 2014). Additionally, the biochemical rate constants for the reactions that regulate RAS signaling are similar between the three (Ahmadian et al., 1997; Lenzen et al., 1998). We evaluated whether RAS mutations interact similarly with NF1 when they occur in KRAS, NRAS, or HRAS. We performed BRET saturation using Q61L, Q61H, Q61K, Q61R, G12V, and G12D mutations within KRAS, NRAS, and HRAS expression constructs. We found no noticeable difference between the NF1 interaction with each RAS mutation across the three RAS proteins (Figure S6C).
We have previously reported that KRAS G12V and G12D bind to NF1 with strong affinity and render cells resistant to EGFR inhibition (McFall et al., 2019). We obtained heterozygous HRAS G12V and heterozygous NRAS G12D isogenic SW48 cells to evaluate whether these mutant cells behave similarly to KRAS G12V and KRAS G12D cells. We found that both isogenic lines were resistant to cetuximab (Figure S6D) and had no change in WT RAS-GTP upon treatment with cetuximab (Figure S6E), similar to what we observed for KRAS G12V and KRAS G12D isogenics (McFall et al., 2019). These observations suggest that the specific mutation may be more important than whether the mutated gene is KRAS, NRAS, or HRAS when evaluating possible sensitivity to cetuximab.
DISCUSSION
Our findings suggest that impaired binding to NF1 may be a biophysical biomarker that can classify RAS mutants into EGFR inhibitor-sensitive and EGFR inhibitor-resistant classes. We used this insight to find 10 more RAS mutations that appear to indicate sensitivity to EGFR inhibitors. We anticipate that additional RAS mutants with impaired binding to NF1 can be identified in further studies and that CRC cells with these mutations will often have sensitivity to EGFR inhibition.
The value of ‘‘biophysical biomarkers’’ that can subclassify mutant forms of a protein becomes evident when one considers that KRAS G13D CRC was originally discovered to be sensitive to EGFR inhibitors as a statistically significant survival signal in clinical trial data (De Roock et al., 2010). However, in the absence of a supporting mechanism, this was originally considered a possible statistical anomaly and expert opinion stated that a prospective, randomized controlled trial was needed to prove that patients with KRAS G13D CRC benefit from EGFR inhibition (De Roock et al., 2010; Morelli and Kopetz, 2012). However, there are many challenges and costs to pursuing a clinical trial, and a controlled trial of cetuximab for patients with KRAS G13D CRC has not yet occurred.
We will refer to the process of evaluating clinical trial data for responsive subsets of patients, the same process that originally identified KRAS G13D as a biomarker for sensitivity to EGFR inhibitors (De Roock et al., 2010), as the ‘‘discovery approach.’’ The discovery approach offers slow progress for personalized medicine because it requires treating a large number of patients, with the goal of identifying rare subsets of patients that appear to benefit, and then searching for the relevant biomarker(s). Importantly, the desire to search for less common mutant biomarkers and the ability to justify the ongoing cost would both likely decrease once the most common biomarkers are evaluated. (It is important to note that KRAS G13D is the third most common RAS mutation in CRC, and that there is a rapid drop off in frequency for the increasingly less common RAS mutations.) Additionally, the identification of true responders and true biomarkers must be balanced by the likelihood of identifying chance outliers because multiple hypothesis testing is involved. The path to personalized medicine by the discovery approach could also require each mutant biomarker to be studied alone in a prospective, randomized controlled trial. The inclusion of less common to rare mutants in discovery clinical trial cohorts as well as the enrollment of validation randomized controlled trials that focus on patients with rare mutants both pose significant challenges to the advancement of personalized cancer medicine through the discovery approach.
In contrast, we here advocate a ‘‘biophysical biomarker approach.’’ This approach focuses on the identification of the measurable biophysical properties of mutant proteins that are the critical variables that determine sensitivity and resistance to treatment. The identification of biophysical biomarkers could potentially be done without a large ‘‘discovery’’ clinical trial, thus reducing the cost and time required to identify candidate mutation biomarkers. Additionally, prospective clinical trial validation could focus on a cohort that includes patients with any of the different mutant forms of a protein that share the same biophysical biomarker. This would facilitate clinical trials by reducing the number of trials needed (one per biophysical biomarker, rather than one per mutation), and it would also be easier to accrue patients to a clinical trial that tests classes of mutations believed to behave similarly.
Every year there are nearly 150,000 new CRC diagnoses within the United States (Siegel et al., 2020a, 2020b). Although the 10 mutations we identify here are individually less common than KRAS G13D (which is found in 7% of patients with CRC), the 10 mutations are collectively found in nearly 9% of CRC cases. We estimate that 13,000 patients with CRC per year will have one of these potentially targetable mutations in the United States, with many more worldwide (Cerami et al., 2012). We propose that a prospective clinical trial should evaluate the value of EGFR inhibitor treatment for patients with CRC whose tumor harbors one of the 10 mutations identified here (in either KRAS, NRAS, or HRAS). Additional preclinical studies using animal models (i.e., xenografts, genetically engineered mice) and/or additional in vitro approaches (i.e., spheroids and organoids) may be required to motivate such a clinical trial. Of note, previous studies have found similar responses to RAS pathway inhibition for two-dimensional (2D) cell culture and three-dimensional (3D) cell culture, with the 3D system actually demonstrating greater sensitivity to RAS inhibition (Canon et al., 2019; Hallin et al., 2020; Janes et al., 2018; Patricelli et al., 2016; Santana-Codina et al., 2020). Additionally, elegant organoid studies that dissect the phenotypes of different KRAS mutants find patterns of mutant-specific drug sensitivity that are similar to those observed in 2D and 3D culture (Zafra et al., 2020).
Although EGFR inhibitors like cetuximab and panitumumab are sometimes used as single agents in WT RAS CRC, they are more commonly utilized in combination with chemotherapy (Van Cutsem et al., 2009; Xie et al., 2020). Similarly, we would anticipate that the benefit of EGFR inhibitors for patients with CRC who also have one of the identified RAS mutants would be higher if EGFR inhibitors were used as part of a treatment combination. Relatedly, EGFR inhibitors have reproducibly been observed to synergize with KRAS G12C inhibitors, including studies that use the same SW837 and SW1463 cell lines (Canon et al., 2019; Hallin et al., 2020; Ryan et al., 2020; Xue et al., 2020). Of note, several of these studies that investigated combinations of EGFR and KRAS G12C inhibitors include conditions with only EGFR inhibitor treatment; these data reveal partial sensitivity of the KRAS G12C mutant cells to EGFR inhibition, consistent with our findings (Canon et al., 2019; Hallin et al., 2020; Ryan et al., 2020; Xue et al., 2020).
Cancer genomes display a great deal of diversity, and for that reason we do not expect every patient with CRC with one of these mutations to be clinically responsive to EGFR inhibition. For example, although KRAS G12C cells were generally EGFR inhibitor sensitive, we found KRAS G12C homozygous mutant SW1463 cells to be EGFR inhibitor resistant. This suggests that patients with a homozygous mutation will be more resistant than those with a heterozygous mutation. Similarly, we would hypothesize that patients with both a mutation and an amplification of that same mutation are likely to be resistant. Co-occurring mutations can also promote resistance; in earlier work, both we and a separate group identified NF1 loss-of-function/loss-of-expression mutations as a mechanism of resistance for KRAS G13D mutant cancers (McFall et al., 2019; Rabara et al., 2019). Our experiments also find that loss of NF1 function causes resistance in cell lines with an EGFR inhibitor-sensitive RAS mutation. We would anticipate that NF1 mutations will cause resistance to EGFR inhibition for the 10 additional RAS mutations identified here.
In future studies, it would be interesting to uncover the structural basis for impaired binding between RAS mutants and NF1. Codons 12 and 13 are both glycine in WT KRAS, and glycine is the simplest amino acid. It seems reasonable to speculate that some side chains could create a steric obstruction to binding with NF1. Additionally, many of the side chains in RAS mutants observed to be impaired at NF1 binding are electrostatically charged, suggesting the additional possibility of electrostatic repulsion as a factor that impairs NF1 binding. These possibilities could be investigated with a variety of methods (Hunter et al., 2015; Lu et al., 2016; Rabara et al., 2019).
The idea that mutant forms of a gene can be subdivided into classes that influence sensitivity or resistance to targeted therapy has precedent. For example, KIT mutations in gastrointestinal stromal gumor (GIST) commonly occur within exon 9 and exon 11, but those at exon 11 are generally more responsive to treatment with imatinib. More recently, BRAF mutations have been subdivided into three classes, with each class having different sensitivities to targeted therapies that follow from their distinct biochemical properties (Yao et al., 2017). Overall, the identification of biophysically defined subsets of specific oncoproteins offers an efficient path forward for personalized cancer medicine. Biochemical mechanism-based, computational systems biology dynamical system models should further be able to facilitate the identification of bio-physical subclassifications for common oncoproteins.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Ed Stites (estites@salk.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed material transfer agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
All original code for the computational analysis of RAS mutations is publicly available at github (https://github.com/StitesLab/RAS_EGFRi). A doi to the version of record is provided in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line models and culture
LS1034, SW837, SW1463, SW48 cells and SW48 isogenic counterparts were cultured in RPMI 1640 medium supplemented with fetal bovine serum (FBS) (10%), penicillin (100 U/ml), streptomycin (100 µg/ml), and l-glutamine (2 mM). SW1116 and SW948 cells were cultured in Lebovitz L-15 medium supplemented with fetal bovine serum (FBS) (10%), penicillin (100 U/ml), streptomycin (100 µg/ml), and l-glutamine (2 mM). LS123 cells were cultured in minimum essential medium (EMEM) supplemented with fetal bovine serum (FBS) (10%), penicillin (100 U/ml), streptomycin (100 µg/ml), and l-glutamine (2 mM). All cells were grown in the indicated medium and incubated at 37°C in 5% CO2 unless indicated otherwise in experimental methods. SW48 cells were obtained from Horizon Discovery. SW948, LS123, SW1116, LS1034, SW837 and SW1463 cells were obtained from the American Type Culture Collection (ATCC). All cell lines tested negative for mycoplasma. Clinical grade cetuximab and panitumumab were generous gifts from Dr. Shumei Kato at the University of California San Diego. Erlotinib was purchased from Selleck Chemicals (S7786).
METHOD DETAILS
Mathematical model of RAS signaling
The key details of the RAS model for this manuscript are summarized here. Additional discussions of the RAS model, including code, equations, and parameters, are available in previous publications (McFall et al., 2019; Stites and Ravichandran, 2012; Stites and Shaw, 2018; Stites et al., 2007, 2015). The model is used to identify steady-state behavior of the RAS signaling module. Mass action kinetics to describe intrinsic GTPase activity, nucleotide binding and unbinding, and effector binding and unbinding. The competitive, irreversible, Michaelis-Menten equation was used to model NF1 activity on RAS because the experimental literature provides values for kcat and Km values, and also because our emphasis on steady-state solutions eliminates concerns about initial transients being inaccurate. The competitive, reversible, Michaelis-Menten equation was used to model RAS GEF activity on RAS. The same equations are used for WT and mutant RAS proteins, but the parameter values may differ for WT and mutant proteins.
Parameters values for WT RAS, G12V RAS, G12D RAS, and G13D RAS have been previously provided and utilized (McFall et al., 2019; Stites et al., 2007, 2015), those same values are used here. Parameter values for codon 61 RAS mutants were obtained from published literature (Donovan et al., 2002). Data include assessments of the NF1:RAS Km, the intrinsic GTPase rate, and the spontaneous (non-GEF mediated) dissociation of GDP and GTP. For all of these parameters, the ratio of the mutant parameter to the wild-type parameter from the same study was used to scale the value of the corresponding WT RAS parameter used in the original RAS model.
Computational random RAS mutants are specified by how each of their independent parameters varies from the value of WT RAS. We generated three distinct sets of computational random RAS mutants where parameters were randomly generated with a log normal distribution. Each set had one million random mutants; for one set all independent and dependent parameters were limited to those where each parameter was no more than one order of magnitude different from wild-type, one with a maximal two order of magnitude difference from wild-type, and one with a maximal three order of magnitude difference from wild-type. For each set of one million mutants, we focused on those that were constitutively active at a level that was within 1% total RAS-GTP and total Effector-RAS-GTP complex from the levels obtained for modeled G12D, G12V, and G13D RAS for both modeled unstimulated (1 × basal GEF) and stimulated (10 × basal GEF) conditions, and that also spanned a total change of RAS-GTP and Effector-RAS-GTP between unstimulated and stimulated conditions that was within 1% total RAS-GTP and total Effector-RAS-GTP of the net change for G12D, G12V, and G13D. Those mutants that met these metrics were considered constitutively active like the other known RAS mutants. Each of these computational constitutively active random RAS mutants was then simulated for a full EGFR inhibitor dose response by evaluating different levels of SOS/GEF activation of RAS, from 10 × to 1 × in 0.25× increments. Dose responses were then normalized to span from 1 (at 10× ) to 0 (at 1× ), and the integral of the dose response was approximated by summing the normalized values. A value of 10% of the difference between the integral of the G13D dose response and the minimum of the integral dose response for G12D and G12V, and a value of 10% of the difference between G13D and the minimum of G12D and G12V at 2× basal GEF were both used to define mutants that were used as a cutoff to define EGFR inhibitor sensitivity. Both Q61K and Q61R met this cutoff for EGFR inhibitor sensitivity, and the other four codon 61 mutants studied did not.
Western blot analysis
Cell lysates were generated using lysis buffer (Thermo Fisher Scientific, 1862301) containing protease inhibitor cocktail (Cell Signaling Technology) and incubated on ice for 1 hour, with brief vortexing every 5 minutes. The total protein concentration was determined with the Pierce Protein assay (Thermo Fisher Scientific). Protein samples were resolved by electrophoresis on either 12% SDS–polyacrylamide gels or 4%–18% gradient gels and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corporation) for 30 min at 25 V with the Trans-Blot Turbo (Bio-Rad Laboratories). The blots were probed with the appropriate primary antibody and the appropriate fluorophore-conjugated secondary antibody. The protein bands were visualized using the Li-Cor CLx Odyssey imaging station (Li-Cor Biosystems). Comparative changes were measured with Li-Cor Image Studio Lite software from three independent experiments (N = 3). Comparisons were made by normalizing to indicated loading control for an internal reference, and the control lane for an external reference.
Proliferation assay
Cells (5000 per well) were seeded in 96-well plates in complete media. Treatments were initiated after the cells were attached (24h). At the appropriate time points, cell viability was determined by MTT assay; 5 mg/ml in phosphate-buffered saline was added to each well followed by incubation at 37°C for 2 hours. The formazan crystal sediments were dissolved in 100 µL of dimethyl sulfoxide and absorbance was measured at 590 nm using a Tecan Infinite 200 PRO plate reader. Each treatment was performed in eight replicate wells (n = 8) and repeated three different times (N = 3). For proliferation assays involving transfected SW48 cells, cells were plated in a 96-well plate at 5000 cells per well in antibiotic-free medium. Twenty-four hours later, cells were transfected with expression plasmids with duplex containing 0.2 µg of DNA and 0.25 µL of Lipofectamine 2000 per well. Cell proliferation was assayed at 48 hours.
Active RAS pull-down assay
Isolation of active RAS-GTP was performed using the Active Ras Pull-Down and Detection Kit (Thermo Fisher Scientific), following the manufacturer’s protocol. RAS abundance was measured by western blot or by RBD-IEF. Analysis of RBD pull-down lysates was performed with mouse anti-KRAS antibody (WH0003845M1, Sigma), rabbit anti-NRAS (ab167136, Abcam), rabbit anti-HRAS (ab32417, Abcam), mouse anti–pan-RAS antibody (1862335, Thermo Fisher Scientific). Input lysates were analyzed with mouse anti-pERK (675502, Biolegend), rat anti-ERK (686902, Biolegend), mouse anti-GAPDH (sc-4772, Santa Cruz Biotechnology), mouse anti-NF1 (sc-376886, Santa Cruz Biotechnology) and mouse anti-EGFR (sc-373746, Santa Cruz Biotechnology).
IEF of active RAS isoforms and total endogenous RAS
Cells were cultured in T-75 adherent culture flasks. Cells were grown in growth medium alone or growth medium plus cetuximab (20 µg/ml) for 48 hours. Medium was removed, and cells were washed with ice-cold tris-buffered saline. Cells were scraped in 1 mL of lysis wash buffer [25 mM tris-HCl (pH 7.2), 150 mM NaCl, 5 mM MgCl2, 1% NP-40, and 5% glycerol]. Cells were lysed on ice and vortexed every 10 s. Cell lysates were subjugated to RBD co-immunoprecipitation as previously described above. RBD co-immunoprecipitation product was resolved by SDS–polyacrylamide gel electrophoresis in a 12% polyacrylamide gel. Bands were excised from the 21-kDa region of the gel. Gel products were liquified at 95°C for 5 min. Protein was extracted and purified using the ReadyPrep 2-D Cleanup Kit (Bio-Rad Laboratories) following the manufacturer’s protocol. Protein samples were added to 50% glycerol loading buffer and incubated at room temperature for 20 min. Samples and IEF ladder were resolved on a Criterion Bio-Lyte IEF Gel with a 3 to 10 pH range (Bio-Rad Laboratories). Gels were run at the following power conditions with constant voltage: 100 V for 60 min, 250 V for 60 min in a stepwise fashion with a total run time of 120 min. The IEF gel was then soaked in 5% SDS buffer for 24 hours with gentle rocking at 4°C. Protein was electrophoretically transferred to PVDF membranes (Millipore Corporation) for 1 hour at a constant 25 V with Trans-Blot Turbo transfer station (Bio-Rad Laboratories). The PVDF blots were probed with the anti–pan-RAS primary antibody from the Active Ras Pull-Down and Detection Kit (Thermo Fisher Scientific) and the anti-mouse DyLight 800 fluorophore-conjugated secondary antibody (Invitrogen). The protein bands were visualized using the Li-Cor CLx Odyssey imaging station (Li-Cor Biosystems). Comparative changes were measured with Li-Cor Image Studio Lite software. Quantification was performed using Li-Cor Odyssey Image Studio Lite. Input sample was used for internal reference and drug treatment was compared against control sample.
NF1-RAS co-immunoprecipitation
HEK293T cells were individually transfected with the expression plasmid for NF1-Flag, or the indicated RAS-GFP. Cells were harvested in IP Lysis/Wash Buffer (0.025 M tris-HCl, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, and 5% glycerol; pH 7.4 and 1 × protease inhibitor) 24 hours after transfection. Whole-cell lysates (500 µg) were precleared for 0.5 hours using Control Agarose Resin slurry (Thermo Fisher Scientific). Immunoprecipitation was performed by first incubating 800 µL of HEK293T NF1-Flag precleared lysate with 200 µL of the indicated mutant RAS-GFP precleared cell lysate. Each cell lysate mixture had EDTA (pH 8.0) added to make a final concentration of 10 mM. GTP-γ-S was added to the solution to a final concentration of 100 nM. This solution was incubated at room temperature for 20 min with gentle rocking. The reaction was terminated by adding MgCl2 to the solution at a final concentration of 50 mM. The final steps of the co-immunoprecipitation were performed using the Pierce Immunoprecipitation Kit (Thermo Fisher Scientific) with immobilized anti-NF1 antibody (Santa Cruz Biotechnology, CA). A total of 500 µg of the cell lysate was added and incubated at room temperature under rotary agitation for 2 hours. At the end of the incubation, the complexes were washed five times with lysis buffer. The western blot was probed with mouse monoclonal NF1 antibody (Santa Cruz Biotechnology, CA, sc-20017) and with rabbit anti-GFP antibody (Cell Signaling Technology, MA, 2956S). Quantification was performed using Li-Cor Odyssey Image Studio Lite. All samples were normalized to input for internal reference, and compared against the positive control KRAS G12V for an external reference.
Bioluminescence resonance energy transfer (BRET) assay
Human embryonic kidney HEK293T cells were grown in DMEM/10% FBS without antibiotic treatment. Cells were seeded at 5000 cells per well in a 96-well white opaque Perkin Elmer microplate. Twenty-four hours after seeding, cells were co-transfected with a constant concentration of 0.1 µg of NF1-NanoLuc pcDNA expression plasmid with increasing concentrations of GFP-tagged KRAS (WT or Mutant) with 0.25 µL of Lipofectamine 2000 per well following the manufacturer’s protocol (Thermo Fisher Scientific). Twenty-four hours later, the culture plate was read with GFP fluorescence setting on the Tecan Infinite M200 PRO to measure GFP expression. Next, medium was aspirated from each well and 25 µL of Nano-Glo Live Cell Reagent was added to each well per the manufacturer’s protocol (Promega). Plates were placed on an orbital shaker for 1 min at 300 rpm. After incubation, the plate was read on the Tecan Infinite M200 PRO with LumiColor Dual Setting with an integration time of 1000 ms. BRET ratio was calculated from the dual emission readings. For each BRET saturation curve, a NF1-NanoLuc transfection was included for background reading. Background was subtracted and BRET ratio was plotted as a function of the RAS-GFP/NF1-NanoLuc plasmid ratio. BRET assays were repeated three times (N = 3), each with eight biological replicates (n = 8).
Competitive bioluminescence resonance energy transfer (BRET) assay
Human embryonic kidney HEK293 T cells were grown in DMEM/10% FBS without antibiotic. Cells were seeded at 5 × 103 cells per well in a 96-well white opaque Perkin Elmer microplate. Twenty-four hours after seeding, cells were co-transfected with a constant concentration of 0.1 µg of NF1-NanoLuc pcDNA expression plasmid and increasing concentrations of the indicated RAS-EGFP pcDNA or RAS-Flag pcDNA expression plasmid with 0.25 ml of Lipofectamine 2000 per well following the manufacturer’s protocol (ThermoFisher). Twenty-four hours later, the culture plate was read with GFP fluorescence setting on the Tecan Infinite M200 PRO to measure GFP expression. Next, medium was aspirated from each well and 25 µl of NanoGlo Live Cell Reagent was added to each well per the manufacturer’s protocol (Promega). Plates were placed on orbital shaker for 1 min at 300 rpm. After incubation, the plate was read on the Tecan Infinite M200 PRO with LumiColor Dual Setting with an integration time of 1000 ms. BRET ratio was calculated from the dual emission readings. For the saturation curve, BRET ratio was plotted as a function of the RAS-GFP/NF1-NanoLuc plasmid ratio. Competitive BRET was performed by transfecting equal (0.2 µg) amounts of donor (RAS-EGFP pcDNA) and competing (RAS-Flag pcDNA) plasmids (indicated in the figure) with 0.1 µg of NF1-NanoLuc pcDNA plasmid. Assays were performed as described above in the saturation curve, raw values for GFP fluorescence and luciferase are reported, showing equal transfection efficiency across tests. Each competitive BRET was performed three times with eight biological replicates.
Colony formation assay
Cells were trypsinized, and 100 cells per well were plated in triplicate six-well (60mm) plates in either complete media (10%FBS) or serum reduced media (1.5% FBS). Colonies were either left untreated or supplemented with 20 µg/ml of cetuximab or 20ng/ml of EGF. Colonies were formed after 4 weeks. The cells were fixed with ice-cold methanol and stained with crystal violet. Images were obtained using the Li-Cor CLx Odyssey imaging station (Li-Cor Biosystems). A total of three experimental replicates were performed.
Expression constructs
Ras expression constructs from the NCI Ras Initiative clone collection were Gateway-cloned into enhanced green fluorescent protein (EGFP) expression vector pEZYegfp (Addgene #18671) or pEZYflag (Addgene #18700). NF1 expression construct (Addgene #70424) was Gateway-cloned into pcDNA3.1-ccdB-NanoLuc (Addgene #87067).The RAS Clone Collection was a gift from D. Esposito at FNL (Addgene kit #1000000070 and kit #1000000089). pEZYegfp and pEZYflag were gifts from Y.-Z. Zhang at Illinois Institute of Technology (Addgene plasmid #18671; http://n2t.net/; RRID:Addgene_18671 and Addgene plasmid #18700; http://n2t.net/addgene:18700; RRID:Addgene_18700). pcDNA3.1-ccdB-NanoLuc were gifts from M. Taipale at University of Toronto (Addgene plasmid #87075; http://n2t.net/addgene:87075; RRID:Addgene_87075 and Addgene plasmid #87067; http://n2t.net/addgene:87067; RRID:Addgene_87067).
siRNA knockdown
Cells were plated in adherent culture plates containing siRNA and Lipofectamine RNAi Max (13778150, ThermoFisher) with a concentration of 10pmol siRNA per 10,000 cells in optiMEM reduced medium (per manufacturers instruction). siRNAs used were EGFR siRNA (S565, ThermoFisher), Control siRNA (AM4611,ThermoFisher) and NF1 siRNA (S57341, ThermoFisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of biological replicates is denoted by ‘‘n’’ and the number of experimental replicates is specified in the figure legend. All data are reported as mean values ± standard deviation and were analyzed using Prism 8 software (GraphPad Software, Incorporated, La Jolla, CA, USA). Statistical significance testing between two conditions was done using unpaired two-tailed t-test, assuming equal variances, and from at least three independent experiments, unless stated otherwise. Statistical significance for three or more conditions was calculated via one-way ANOVA followed by post hoc Tukey’s test for multiple comparisons, unless stated otherwise. Significances were reported as P* < 0.05, 0.01, 0.001, 0.0001 or exact values and are reported in each figure, and the statistical test(s) used is included in each figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
Antibodies
| ||
| mouse anti-KRAS | Sigma | cat#WH0003845M1 |
| rabbit anti-NRAS | Abcam | cat#ab167136 |
| mouse anti–pan-RAS | Thermo Fisher | cat#1862335 |
| mouse anti-pERK | Biolegend | cat#675502 |
| rat anti-ERK | Biolegend | cat#686902 |
| mouse anti-GAPDH | Santa Cruz Biotechnology | cat#sc-4772 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, DyLight 800 | Thermo Fisher | cat#SA5–10176 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, DyLight 680 | Thermo Fisher | cat#35569 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher | cat#A-21096 |
| mouse anti-NF1 | Santa Cruz Biotechnology | cat#sc-376886 |
| mouse anti-EGFR | Santa Cruz Biotechnology | cat#sc-373746 |
| rabbit anti-HRAS | Abcam | cat#ab32417 |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Nano-Glo® Live Cell Assay System | Promega | cat#N2011 |
| Pierce 660nm Protein Assay Reagent | Thermo Fisher | cat#22660 |
| Erlotinib | Selleck Chemicals | cat#S7786 |
| Cetuximab | Eli Lilly and Co | CAS: 205923–56-4 |
| Panitumumab | Amgen | CAS: 339177–26-3 |
|
Critical Commercial Assays | ||
| Active Ras Pull-Down and Detection Kit | Thermo Fisher | cat#16117 |
| Pierce Co-Immunoprecipitation Kit | Thermo Fisher | cat#26149 |
|
Experimental Models: Cell Lines | ||
| SW48 Parental | ATCC | cat#CCL-231 |
| KRAS (G12A/+)SW48 | Horizon Discovery | cat#HD 103–009 |
| KRAS (G12V/+)SW48 | Horizon Discovery | cat#HD 103–007 |
| KRAS (G12R/+)SW48 | Horizon Discovery | cat#HD 103–010 |
| KRAS (G13D/+)SW48 | Horizon Discovery | cat#HD 103–002 |
| KRAS (G12S/+)SW48 | Horizon Discovery | cat#HD 103–013 |
| KRAS (A146T/+)SW48 | Horizon Discovery | cat#HD 103–036 |
| KRAS (G12D/+)SW48 | Horizon Discovery | cat#HD 103–011 |
| KRAS (G12C/+)SW48 | Horizon Discovery | cat#HD 103–006 |
| NRAS (Q61L/+)SW48 | Horizon Discovery | cat#HD 103–038 |
| NRAS (Q61R/+)SW48 | Horizon Discovery | cat#HD 103–022 |
| NRAS (Q61H/+)SW48 | Horizon Discovery | cat#HD 103–035 |
| NRAS (Q61K/+)SW48 | Horizon Discovery | cat#HD 103–017 |
| NRAS (G12D/+)SW48 | Horizon Discovery | cat#HD 103–062 |
| LS1034 | ATCC | cat#CRL-2158 |
| SW1116 | ATCC | cat#CCL-233 |
| LS123 | ATCC | cat#CCL-255 |
| SW948 | ATCC | cat#CCL-237 |
| SW837 | ATCC | cat#CCL-235 |
| SW1463 | ATCC | cat#CCL-234 |
| HRAS (G12V/+) SW48 | Horizon Discovery | cat#HD 103–034 |
|
Oligonucleotides | ||
| EGFR siRNA | Thermo Fisher | Assay ID S565 |
| Control siRNA | Thermo Fisher | Assay ID AM4611 |
| NF1 siRNA | Thermo Fisher | Assay ID S57341 |
|
Recombinant DNA | ||
| Hs.HRAS G12D | Gift from Dominic Esposito at Frederick National Laboratory (FNL) | Addgene #83183 |
| Hs.NRAS Q61K | Gift from Dominic Esposito at FNL | Addgene #83180 |
| Hs.NRAS Q61R | Gift from Dominic Esposito at FNL | Addgene #83179 |
| Hs.NRAS Q61L | Gift from Dominic Esposito at FNL | Addgene #83178 |
| Hs.NRAS G12V | Gift from Dominic Esposito at FNL | Addgene #83174 |
| Hs.KRAS4b T35S | Gift from Dominic Esposito at FNL | Addgene #83157 |
| Hs.KRAS4b A146T | Gift from Dominic Esposito at FNL | Addgene #83150 |
| Hs.KRAS4b G12F | Gift from Dominic Esposito at FNL | Addgene #83149 |
| Hs.KRAS4b R68S | Gift from Dominic Esposito at FNL | Addgene #83148 |
| Hs.KRAS4b A146V | Gift from Dominic Esposito at FNL | Addgene #83147 |
| Hs.KRAS4b K117N | Gift from Dominic Esposito at FNL | Addgene #83146 |
| Hs.KRAS4b G12S | Gift from Dominic Esposito at FNL | Addgene #83144 |
| Hs.KRAS4b G12R | Gift from Dominic Esposito at FNL | Addgene #83143 |
| Hs.KRAS4b G12A | Gift from Dominic Esposito at FNL | Addgene #83142 |
| Hs.KRAS4b Q61H | Gift from Dominic Esposito at FNL | Addgene #83140 |
| Hs.KRAS4b Q61R | Gift from Dominic Esposito at FNL | Addgene #83135 |
| Hs.KRAS4b Q61L | Gift from Dominic Esposito at FNL | Addgene #83134 |
| Hs.KRAS4b G13D | Gift from Dominic Esposito at FNL | Addgene #83133 |
| Hs.KRAS4b G12V | Gift from Dominic Esposito at FNL | Addgene #83132 |
| Hs.KRAS4b G12D | Gift from Dominic Esposito at FNL | Addgene #83131 |
| Hs.KRAS4b G12C | Gift from Dominic Esposito at FNL | Addgene #83130 |
| Hs.KRAS4b | Gift from Dominic Esposito at FNL | Addgene #83129 |
| Hs.HRAS G12V | Gift from Dominic Esposito at FNL | Addgene #83184 |
| Hs.KRAS4b G13C | Gift from Dominic Esposito at FNL | Addgene #83145 |
| Hs.KRAS4b G12Y | Gift from Dominic Esposito at FNL | Addgene #82799 |
| Hs.KRAS4b D33E | Gift from Dominic Esposito at FNL | Addgene #81659 |
| Hs.KRAS4b G13R | This work | N/A |
| Hs.KRAS4b G12P | This work | N/A |
| HS.KRAS4b A59E | This work | N/A |
| KRAS4b WT GFP | This work | N/A |
| KRAS4b G12V GFP | This work | N/A |
| KRAS4b G12D GFP | This work | N/A |
| KRAS4b G12S GFP | This work | N/A |
| KRAS4b G21R GFP | This work | N/A |
| KRAS4b G12P GFP | This work | N/A |
| KRAS4b G12Y GFP | This work | N/A |
| KRAS4b G12A GFP | This work | N/A |
| KRAS4b G12C GFP | This work | N/A |
| NRAS G12V GFP | This work | N/A |
| NRAS G12D GFP | This work | N/A |
| HRAS G12V GFP | This work | N/A |
| HRAS G12D GFP | This work | N/A |
| HRAS WT | This work | N/A |
| NRAS Q61R GFP | This work | N/A |
| HRAS Q61R GFP | This work | N/A |
| KRAS Q61R GFP | This work | N/A |
| NRAS Q61K GFP | This work | N/A |
| HRAS Q61K GFP | This work | N/A |
| KRAS Q61K GFP | This work | N/A |
| NRAS Q61L GFP | This work | N/A |
| NRAS Q61L GFP | This work | N/A |
| KRAS4b A146T GFP | This work | N/A |
| KRAS4b A59E GFP | This work | N/A |
| KRAS4b G13D GFP | This work | N/A |
| KRAS4b G13C GFP | This work | N/A |
| KRAS4b G13R GFP | This work | N/A |
| KRAS4b D33E GFP | This work | N/A |
| KRAS4b K117N GFP | This work | N/A |
| KRAS4b T35S GFP | This work | N/A |
| KRAS4b R68S GFP | This work | N/A |
| KRAS4b A146V GFP | This work | N/A |
| KRAS4b A146T GFP | This work | N/A |
| The RAS Clone Collection | Gift from Dominic Esposito at FNL | Addgene kit #1000000070 and Addgene kit #1000000089 |
| pEZYegfp | Guo et al. (2008) | Addgene #18671 |
| NF1 | Gift from Dominic Esposito at FNL | Addgene #70423 |
| pcDNA3.1-ccdB-NanoLuc | Gift from Mikko Taipale at University of Toronto | Addgene #87067 |
| pEZYflag | Guo et al. (2008) | Addgene #18700 |
|
Software and algorithms | ||
| MATLAB | Mathworks | RRID:SCR_001622 |
| GraphPad Prism | GraphPad Software | RRID:SCR_002798 |
| Adobe Illustrator | Adobe | RRID:SCR_010279 |
| Image Studio Lite | Li-Cor | RRID:SCR_013715 |
| RAS mutation analyses | This manuscript | https://doi.org/10.5281/zenodo.5699432 |
Highlights.
Mutant RAS cancer sensitivity to EGFR inhibition involves suppression of wild-type RAS-GTP
Sensitivity to EGFR inhibition follows from impaired binding of a RAS mutant to NF1
Ten RAS mutant biomarkers for sensitivity to EGFR inhibition are identified
ACKNOWLEDGMENTS
We thank Tony Hunter, the Hunter laboratory, the Stites laboratory, and members of the Salk Institute Cancer Center for helpful conversations and feedback. We thank Shumei Kato for providing cetuximab and panitumumab and for helpful conversations on personalized medicine. Support for this work was provided by NIH grants K22CA216318, DP2AT011327, T32CA009370, and P30CA014195 and Department of Defense (DOD) grant W81XWH-20–1-0538.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110096.
DECLARATION OF INTERESTS
A patent application has been submitted for the identified RAS mutant biomarker alleles.
REFERENCES
- Ahmadian MR, Hoffmann U, Goody RS, and Wittinghofer A (1997). Individual rate constants for the interaction of Ras proteins with GTPase-activating proteins determined by fluorescence spectroscopy. Biochemistry 36, 4535–4541. 10.1021/bi962556y. [DOI] [PubMed] [Google Scholar]
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, and Schilsky RL (2009). American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol 27, 2091–2096. 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- Bandaru P, Shah NH, Bhattacharyya M, Barton JP, Kondo Y, Cofsky JC, Gee CL, Chakraborty AK, Kortemme T, Ranganathan R, and Kuriyan J (2017). Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife 6, e27810. 10.7554/eLife.27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N.; Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. 10.1158/21598290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Andersen JN, and Futreal PA (2011). Cancer genomics: from discovery science to personalized medicine. Nat. Med 17, 297–303. 10.1038/nm.2323. [DOI] [PubMed] [Google Scholar]
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, et al. (2010). Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304, 1812–1820. 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- Donovan S, Shannon KM, and Bollag G (2002). GTPase activating proteins: critical regulators of intracellular signaling. Biochim. Biophys. Acta 1602, 23–45. [DOI] [PubMed] [Google Scholar]
- Friedman AA, Letai A, Fisher DE, and Flaherty KT (2015). Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer 15, 747–756. 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L, Gilsbach B, Ahmadian MR, and Wittinghofer A (2008). Fluoride complexes of oncogenic Ras mutants to study the Ras-RasGap interaction. Biol. Chem 389, 1163–1171. 10.1515/BC.2008.132. [DOI] [PubMed] [Google Scholar]
- Guo F, Chiang MY, Wang Y, and Zhang YZ (2008). An in vitro recombination method to convert restriction- and ligation-independent expression vectors. Biotechnol. J 3, 370–377. 10.1002/biot.200700170. [DOI] [PubMed] [Google Scholar]
- Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 10, 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood FE, Klinger B, Newlaczyl AU, Sieber A, Dorel M, Oliver SP, Coulson JM, Blüthgen N, and Prior IA (2019). Isoform-specific Ras signaling is growth factor dependent. Mol. Biol. Cell 30, 1108–1117. 10.1091/mbc.E18-10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res 13, 1325–1335. 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589.e17. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, and Eckhardt SG (2009). KRAS mutations and sensitivity to epidermal growth factor receptor inhibitors in colorectal cancer: practical application of patient selection. J. Clin. Oncol 27, 1130–1136. 10.1200/JCO.2008.19.8168. [DOI] [PubMed] [Google Scholar]
- Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, et al. (2007). Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med 357, 2040–2048. 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. (2008). K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med 359, 1757–1765. 10.1056/NEJ-Moa0804385. [DOI] [PubMed] [Google Scholar]
- Kiuru M, and Busam KJ (2017). The NF1 gene in tumor syndromes and melanoma. Lab. Invest 97, 146–157. 10.1038/labinvest.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen C, Cool RH, Prinz H, Kuhlmann J, and Wittinghofer A (1998). Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry 37, 7420–7430. 10.1021/bi972621j. [DOI] [PubMed] [Google Scholar]
- Lu S, Jang H, Nussinov R, and Zhang J (2016). The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep 6, 21949. 10.1038/srep21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mageean CJ, Griffiths JR, Smith DL, Clague MJ, and Prior IA (2015). Absolute Quantification of Endogenous Ras Isoform Abundance. PLoS ONE 10, e0142674. 10.1371/journal.pone.0142674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall T, Diedrich JK, Mengistu M, Littlechild SL, Paskvan KV, Sisk-Hackworth L, Moresco JJ, Shaw AS, and Stites EC (2019). A systems mechanism for KRAS mutant allele-specific responses to targeted therapy. Sci. Signal 12, eaaw8288. 10.1126/scisignal.aaw8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall T, Schomburg NK, Rossman KL, and Stites EC (2020). Discernment between candidate mechanisms for KRAS G13D colorectal cancer sensitivity to EGFR inhibitors. Cell Commun. Signal 18, 179. 10.1186/s12964-020-00645-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Rosenberg SC, McCormick F, and Malek S (2020). RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov 19, 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli MP, and Kopetz S (2012). Hurdles and complexities of codon 13 KRAS mutations. J. Clin. Oncol 30, 3565–3567. 10.1200/JCO.2012.43.6535. [DOI] [PubMed] [Google Scholar]
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, and Ciardiello F (2009). Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol 6, 519–527. 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al. (2016). Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 6, 316–329. 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al. (2019). Tissue-Specific Oncogenic Activity of KRASA146T. Cancer Discov 9, 738–755. 10.1158/2159-8290.CD-18-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Hood FE, and Hartley JL (2020). The Frequency of Ras Mutations in Cancer. Cancer Res 80, 2969–2974. 10.1158/0008-5472.CAN-19-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabara D, Tran TH, Dharmaiah S, Stephens RM, McCormick F, Simanshu DK, and Holderfield M (2019). KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 116, 22122–22131. 10.1073/pnas.1908353116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, Hong CB, and Corcoran RB (2020). Vertical Pathway Inhibition Over-comes Adaptive Feedback Resistance to KRASG12C Inhibition. Clin. Cancer Res 26, 1633–1643. 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana-Codina N, Chandhoke AS, Yu Q, Małachowska B, Kuljanin M, Gikandi A, Stańczak M, Gableske S, Jedrychowski MP, Scott DA, et al. (2020). Defining and Targeting Adaptations to Oncogenic KRASG12C Inhibition Using Quantitative Temporal Proteomics. Cell Rep 30, 4584–4599.e4. 10.1016/j.celrep.2020.03.021. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, and Jemal A (2020a). Colorectal cancer statistics, 2020. CA Cancer J. Clin 70, 145–164. 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2020b). Cancer statistics, 2020. CA Cancer J. Clin 70, 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, and McCormick F (2014). Dragging ras back in the ring. Cancer Cell 25, 272–281. 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Stites EC, and Ravichandran KS (2012). Mathematical investigation of how oncogenic ras mutants promote ras signaling. Methods Mol. Biol 880, 69–85. 10.1007/978-1-61779-833-7_5. [DOI] [PubMed] [Google Scholar]
- Stites EC, and Shaw AS (2018). Quantitative Systems Pharmacology Analysis of KRAS G12C Covalent Inhibitors. CPT Pharmacometrics Syst. Pharmacol 7, 342–351. 10.1002/psp4.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stites EC, Trampont PC, Haney LB, Walk SF, and Ravichandran KS (2015). Cooperation between Noncanonical Ras Network Mutations. Cell Rep 10, 307–316. 10.1016/j.celrep.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stites EC, Trampont PC, Ma Z, and Ravichandran KS (2007). Network analysis of oncogenic Ras activation in cancer. Science 318, 463–467. 10.1126/science.1144642. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, et al. (2009). Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med 360, 1408–1417. 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- Wey M, Lee J, Jeong SS, Kim J, and Heo J (2013). Kinetic mechanisms of mutation-dependent Harvey Ras activation and their relevance for the development of Costello syndrome. Biochemistry 52, 8465–8479. 10.1021/bi400679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie YH, Chen YX, and Fang JY (2020). Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther 5, 22. 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, Kim D, Li C, de Stanchina E, Mazutis L, et al. (2020). Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425. 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, Drosten M, Zhao H, Cecchi F, Hembrough T, et al. (2017). Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238. 10.1038/nature23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, and Pines G (2012). The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 12, 553–563. 10.1038/nrc3309. [DOI] [PubMed] [Google Scholar]
- Zafra MP, Parsons MJ, Kim J, Alonso-Curbelo D, Goswami S, Schatoff EM, Han T, Katti A, Fernandez MTC, Wilkinson JE, et al. (2020). An In Vivo Kras Allelic Series Reveals Distinct Phenotypes of Common Oncogenic Variants. Cancer Discov 10, 1654–1671. 10.1158/2159-8290.CD-20-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
All original code for the computational analysis of RAS mutations is publicly available at github (https://github.com/StitesLab/RAS_EGFRi). A doi to the version of record is provided in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.