

Abstract
Telomeres are nucleoprotein complexes located at the termini of eukaryotic chromosomes that prevent exonucleolytic degradation and end-to-end chromosomal fusions. Cancers often have critically shortened, dysfunctional telomeres contributing to genomic instability. Telomere shortening has been reported in a wide range of precancerous lesions and invasive carcinomas. However, the role of telomere alterations, including the presence of Alternative lengthening of telomeres (ALT), has not been studied in pituitary adenomas. Telomere length and the presence of ALT were assessed directly at the single cell level using a telomere-specific fluorescence in situ hybridization assay in tissue microarrays. Tumors were characterized as either ALT-positive or having short, normal, or long telomere lengths and then these categories were compared with clinicopathological characteristics. ATRX and DAXX expression was studied through immunohistochemistry. We characterized a discovery set of 106 pituitary adenomas including both functional and non-functional subsets (88 primary, 18 recurrent). Telomere lengths were estimated and we observed 64 (59.4%) cases with short, 39 (36.8%) cases with normal, and 0 (0%) cases with long telomeres. We did not observe significant differences in the clinicopathological characteristics of the group with abnormally shortened telomeres compared to the group with normal telomeres. However, three pituitary adenomas were identified as ALT-positive of which 2 were recurrent tumors. Two of these three ALT-positive cases had alterations in either of the chromatin remodeling proteins, ATRX and DAXX, which are routinely altered in other ALT-positive tumor subtypes. In a second cohort of 32 recurrent pituitary adenomas from 22 patients we found that the tumors from 36% of patients (n=8) were ALT-positive. This study demonstrates that short telomere lengths are prevalent in pituitary adenomas and that ALT-positive pituitary adenomas are enriched in recurrent disease.
INTRODUCTION
Pituitary adenomas are common primary intracranial tumors, and while most are benign, these tumors may still cause notable morbidity for patients (1, 2). As with other neuroendocrine tumors, pituitary adenomas are classified as either “functional” or “non-functional”. Functional tumors secrete excess levels of specific hormones, for example growth hormone (GH), adrenocorticotropic hormone (ACTH), prolactin (PRL), and follicle stimulating hormone/ luteinizing hormone (FSH/LH), or thyroid stimulating hormone (TSH). In some tumor subsets, continual over-production of any of these hormones can result in debilitating multisystem dysfunction and ultimately an increased risk of death (3). In contrast, non-functional adenomas do not secrete abnormally excessive hormones. However, these tumors often compress adjacent neurovascular structures, leading to mass effect and visual disturbances secondary to optic chiasm compression. On the genomic level, large cohorts of pituitary adenomas have recently been comprehensively profiled. These studies have revealed, in addition to limited somatic mutations, that a subset of pituitary adenomas are characterized by the presence of somatic copy-number alterations affecting whole chromosome arms that comprise a large fraction (~50%) of the genome (4–6).
In most human cancers, neoplastic cells achieve the ability to proliferate unlimitedly by maintaining their telomeres. Telomeres, the repetitive hexanucleotide sequences (TTAGGGn) located at the terminal ends of eukaryotic chromosomes, are pivotal for genome integrity. Critical telomere shortening is a common abnormality observed early in tumorigenesis, where it promotes malignant transformation and tumor progression via telomere destabilization and concomitant chromosomal instability (7). Consequently, alterations in cancer cell telomere lengths have been identified as prognostic factors in a variety of cancer subtypes (8, 9). Cancer cells require activation of a telomere maintenance mechanism, predominantly through activating the enzyme telomerase. In contrast, a subset of cancers, including a substantial proportion of pancreatic neuroendocrine tumors (PanNETs) and gliomas, utilize a telomerase-independent mechanism which is mediated by homology directed repair, termed Alternative Lengthening of Telomeres (ALT) (10).
ALT-positive cancers often have lost function, through mutation or deletion, in the alpha thalassemia/mental retardation syndrome X-linked (ATRX) or death domain-associated protein (DAXX) genes. ATRX and DAXX are chromatin modifying proteins, and in concert, this ATRX/DAXX complex deposits histone variant H3.3 in heterochromatic regions of chromosomes containing highly repetitive sequence elements, such as retrotransposons, pericentromeric regions, and telomeres. These repetitive sequences are inherently unstable and are prone to increased replication errors and aberrant homologous recombination. As a result, disruption of ATRX/DAXX chromatin remodeling function at telomeres presumably allows for the development of ALT. In archived tissue specimens, ALT may be identified by the presence of unique ALT-associated features, including ultra-bright telomeric foci and dramatic cell-to-cell telomere heterogeneity. Using this approach, ALT and ATRX mutations have been associated with aggressive clinical behaviors in a variety of tumor types.
In the current study, we evaluated a discovery cohort of 106 pituitary adenomas (88 primary, 18 recurrent tumors), as well as an additional cohort of recurrent tumors (32 tumors from 22 patients). Using these cohorts, we evaluated the presence of telomere alterations, including the presence of Alternative lengthening of telomeres (ALT), as well as alterations in ATRX and DAXX protein expression and correlated these findings with clinicopathologic factors.
MATERIALS AND METHODS
Case selection and tissue microarray construction
This study was reviewed and approved by the human subjects Institutional Review Boards (IRBs) of the Dana-Farber/Brigham and Women’s Cancer Center (DF/BWCC), the Brigham and Women’s Hospital, Johns Hopkins Hospital, and the Broad Institute. Written informed consent or a waiver of consent was obtained for all participants. In the discovery cohort, histopathologic diagnosis based on WHO criteria, and tumor purity >80% was confirmed in all samples selected for study by two board-certified neuropathologists (S.S. and S.C.). Tumors were classified by their immunohistochemical expression of hormones (null, prolactin, ACTH, GH, FSH/LH, and TSH) as well as by the associated serum levels of the corresponding hormone. Corticotroph, somatotroph (including plurihormonal cases), and lactotroph adenomas were considered functional in our analyses, whereas gonadotroph and null-cell adenomas were considered nonfunctional. We classified tumors as nonfunctional in cases where there was immunohistologic staining for one or more hormones, but serum levels remained normal and there were no clinical signs of endocrinopathy. In the validation cohort of recurrent disease, we included cases of pituitary adenoma that required at least more than one surgery. Classification was performed similarly as above, based on immunohistochemical evaluation of at least GH, ACTH and prolactin, and serum levels of the corresponding hormone.
Telomere-specific FISH
Telomere-specific FISH was performed as previously outlined (11, 12). In brief, tissue slides were deparaffinized, hydrated, and steamed for 25 minutes in citrate buffer (Vector Laboratories). This was followed by dehydration and hybridization with a Cy3-labeled peptide nucleic acid (PNA) probe complementary to the mammalian telomere repeat sequence [(N-terminus to C-terminus) CCCTAACCCTAACCCTAA]. An Alexa Fluor-488–labeled PNA probe specific to human centromeric DNA repeats was also included as a control to assess the validity of the hybridization. Following post-hybridization washes, the slides were counterstained with DAPI following post-hybridization washes.
Microscopy
Slides were imaged with a Nikon 50i epifluorescence microscope equipped with X-Cite series 120 illuminator (EXFO Photonics Solutions, Ontario, CA) using a 40×/0.95 NA PlanApo lens with correction collar. For each color channel, separate grayscale images were captured using Nikon NIS-Elements software (NIS-Elements BR 3.2 64-bit) and an attached Photometrics CoolsnapEZ digital cooled charged coupled device camera and saved as 12-bit uncompressed TIFF files. Exposure times were set to avoid fluorescence signal saturation. For telomere length analysis, integration times were 500 milliseconds for Cy3 (telomere) and 100 milliseconds for the DAPI nuclear counterstain. In contrast, for ALT analysis, integration times were 500 milliseconds for Cy3 (telomere) and 100 milliseconds for the DAPI nuclear counterstain.
ALT and telomere length assessment
ALT status was interpreted using previously published criteria and was characterized by the presence of distinct large telomeric FISH DNA signals (10, 13, 14). In the ALT-negative cases, telomere lengths were qualitatively scored by direct visual assessment of the stained slides, comparing the intensity of telomere signals from cancer cells to the intensity of telomere signals of entrapped non-neoplastic cells. Thus, telomere lengths were evaluated as being either normal, or abnormally short or long.
Immunohistochemistry
Immunohistochemical studies were systematically performed using an ATRX antibody (rabbit polyclonal, 1:200 dilution, catalog# HPA001906 Sigma-Aldrich). Immunostaining was performed on automated instruments (BenchMark, Ventana Medical Systems, Tucson, AZ, USA). The immunohistochemical protocol included deparaffinization, hydration, antigen retrieval, primary antibody incubation, and detection and visualization as per manufacturer’s instructions. Immunohistochemistry for DAXX (rabbit polyclonal, 1:100 dilution, catalog# HPA008736, Atlas Antibodies) was performed manually. Sections were incubated with primary antibody for 2 hours at room temperature followed by secondary antibody (Leica Microsystems) for 30 min and detected with 3,30-diaminobenzidine (Sigma-Aldrich) after 10 min.
Next Generation Sequencing
To identify possible somatic genetic alterations associated with ALT, next generation sequencing was performed on the 3 ALT-positive cases. While one of the cases was previously assessed by whole exome sequencing (4), the other cases were assessed using Oncopanel, an established hybrid-capture and massively parallel sequencing assay (15).
Statistical Analysis
Categories of telomere length (short, normal, long) were correlated with the clinicopathological characteristics. Variables were described using proportions, ranges, means, medians, and standard deviations as appropriate. Proportions were compared using Chi-Square or Fisher’s exact tests as appropriate. Statistical analyses were performed using GraphPad Prism version 8.0 (San Diego, CA).
RESULTS
Clinicopathological characteristics
The clinicopathological characteristics for the entire study population of the discovery set (N=106) are shown in Table 1 and some details have been previously published (4). Briefly, the mean age of patients was 53.4 years (range, 16–86) and 61.3% were male. 88 tumors were newly diagnosed and 18 were recurrent tumors. The majority of the tumors evaluated were non-functional (78.3%); while 21.7% were functional, including 15 HGH expressing tumors (somatotroph adenomas) associated with acromegaly, 2 HGH and prolactin expressing tumors (somatomammotroph adenomas) associated with acromegaly, 3 prolactin expressing tumors (prolactinomas) associated with amenorrhea, galactorrhea, or low testosterone, 1 ACTH expressing tumor (corticotroph adenoma) associated with Cushing’s syndrome, and 1 adenoma with no immunohistochemically detectable hormone expression associated with Cushing’s syndrome. Finally, while most tumors did not display the previously defined disrupted genotype (61.3%), 21.7% did present with this disrupted genotype.
Table 1.
Clinicopathologic and molecular features of pituitary adenomas
| Pituitary Adenomas (N=106) | |
|---|---|
|
| |
| Mean age (range) | 53.4 (16–86) |
| Gender (%) | |
| Male | 61.3 |
| Female | 38.7 |
| Clinical endocrinologic status (%) | |
| Non-functional | 76.5 |
| Functional | 21.7 |
| Missing | 1.8 |
| Recurrent (%) | |
| No | 83.0 |
| Yes | 17.0 |
| Disrupted genotype (%) | |
| No | 61.3 |
| Yes | 21.7 |
| Missing | 17.0 |
| Telomere status (%) | |
| Normal | 36.8 |
| Short | 59.4 |
| ALT | 2.8 |
In situ telomere analysis in pituitary adenomas
Telomere lengths were qualitatively scored through direct visual assessment by comparing the intensity of telomere signals in the cancer cells to the intensity of telomere signals of entrapped non-neoplastic cells within the same case. Based on the cancer cell telomere length, the cases were grouped into either short (59.4% cases; n = 64), normal (36.8% cases; n = 39), or long (0% cases; n = 0) categories. In Figure 1, representative pituitary adenomas demonstrating cancer cells with short telomeres and normal telomeres are shown. The cases with short telomeres did not significantly differ from the cases with normal telomeres in any of the clinicopathological characteristics, including gender, clinical endocrinologic status, recurrent status, or displaying the disrupted genotype.
Figure 1. Telomere length analysis by FISH in pituitary adenomas.
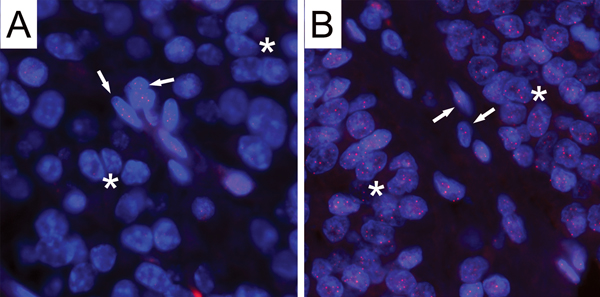
Two representative examples of cases showing either short or normal telomere lengths in tumor cells are shown. (A) This case shows strikingly diminished telomere signals in tumor cells (asterisks) as compared to the entrapped non-neoplastic cells (arrows). (B) This case displays comparable telomere intensities in tumor cells (asterisks) with those observed in the entrapped non-neoplastic cells (arrows). In both images, the DNA is stained with DAPI (blue) and telomere DNA is stained with the Cy3-labeled telomere-specific peptide nucleic acid probe (red). It is noteworthy that the centromere DNA, stained with the FITC-labeled centromere-specific peptide nucleic acid probe, has been omitted from the image to emphasize the differences in telomere lengths. Original magnification, × 400.
Characterization of ALT-positive pituitary adenomas
ALT was identified in 3 of 106 (2.8%) pituitary tumors (cases 15, 95, and 111). Two were male and 1 was female, with ages ranging from 39–73. Two of these cases were non-functional, whereas 1 case was HGH expressing (densely or sparsely granulated, associated with acromegaly). Two of the 3 cases (66%) were recurrent, as compared to only 16 of 103 (15.5%) of the ALT-negative pituitary adenomas (chi-square test; p=0.01). As shown in Figure 2, ultra-bright telomeric foci indicative of ALT were present in all three cases. ATRX loss was observed in case 95 and DAXX loss observed in case 15. In contrast, case 111 retained nuclear protein positivity for both ATRX and DAXX. As expected, ATRX and DAXX protein expression was retained in all of the ALT-negative pituitary adenomas, although 4 cases did display partial loss of ATRX and 1 case displayed partial loss of DAXX. Finally, we evaluated the somatic mutational spectrum in these 3 ALT-positive cases. Case 15 was previously assessed by whole exome sequencing (4) and 117 mutations were identified, including in ARID1B, ATP2B2, CHD4, PI3KR1, and RUNX1. The other two ALT-positive cases were assessed using an established hybrid-capture and massively parallel sequencing assay (OncoPanel) (15). Case 95 had mutations in AR, CHEK2, CYLD, FGFR3, KAT6A, and TRAF7; whereas, case 111 only had one somatic mutation (EP300). Interestingly, mutations in ATRX or DAXX were not identified in any of the 3 cases.
Figure 2. ALT-positive pituitary adenomas and ATRX and DAXX immunostaining.
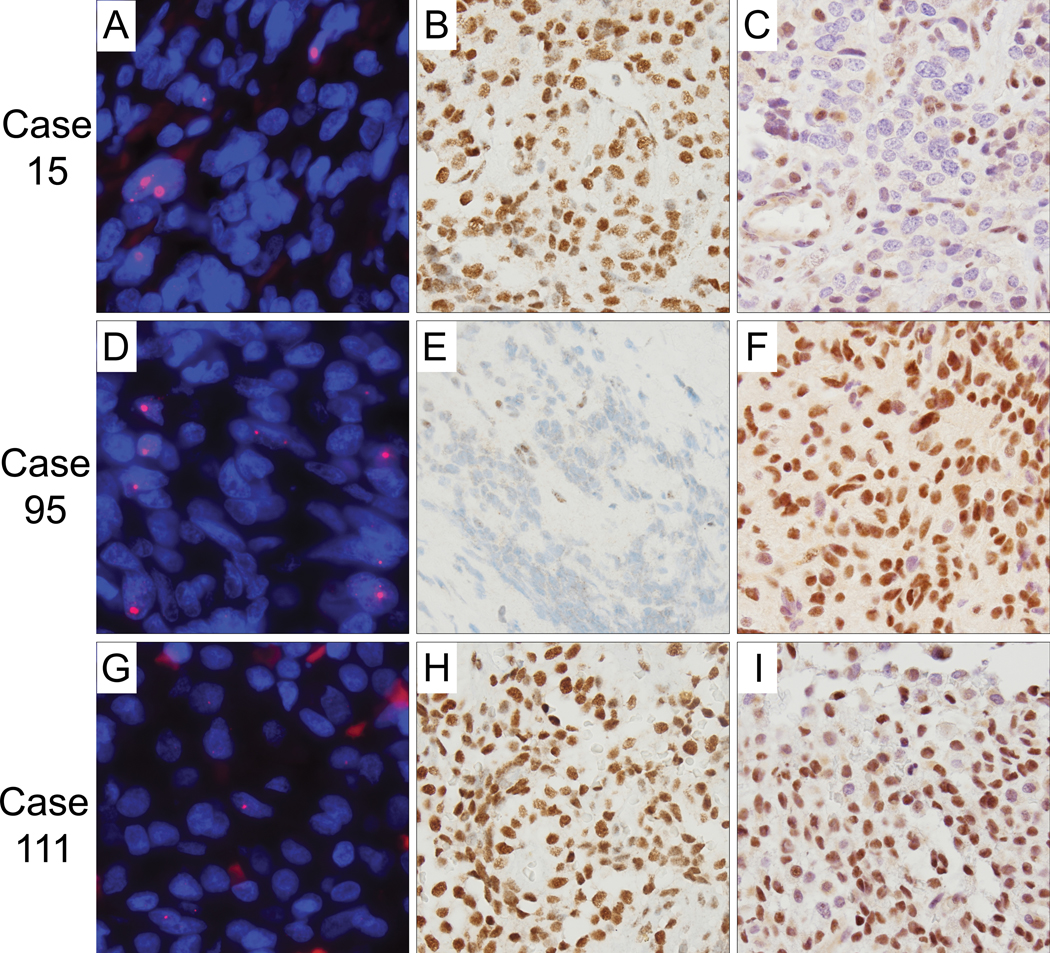
Representative regions of the three cases (15, 95, 111) are shown. (A,D,G) Representative images of ALT-positive cases displaying ultrabright telomeric FISH signals as indicative of ALT. The DNA is stained with DAPI (blue) and telomere DNA is stained with the Cy3-labeled telomere-specific peptide nucleic acid probe (red). (B,E,H) Case 95 shows tumor cell-specific loss of nuclear ATRX expression, whereas cases 15 and 111 display intact ATRX nuclear expression. (Original magnification ×400). (C,F,I) Case 15 shows tumor cell-specific loss of nuclear DAXX expression, whereas cases 95 and 111 display intact DAXX nuclear expression. For all images presented - original magnification, × 400.
Assessment of additional recurrent pituitary adenomas
Because we identified a small number of ALT-positive pituitary adenomas that appeared to be enriched in recurrent disease, we next assessed tumors from a different cohort of patients that had all developed recurrent disease. In total, we assessed 32 recurrent pituitary adenomas from 22 patients. Of the 22 patients, ALT was identified in 8 (36%) patients using the telomere-specific FISH assay. Of the 10 patients for whom tumor tissue was available to be studied from two different surgeries, we found that both tumors were ALT-negative in seven patients and both tumors were ALT-positive in two patients. We did identify one case in which the original pituitary adenoma was ALT-negative, but 2 years later, the subsequent recurrent adenoma was ALT-positive with partial ATRX loss, thereby suggesting that ALT activation can occur upon progression.
DISCUSSION
Pituitary adenomas represent a heterogeneous group of neoplasms that derive and/or share immunophenotypic and functional properties with endocrine cells of the adenohypophysis. The WHO Classification of the Tumours of the Endocrine Organs recognizes seven major categories of pituitary adenomas based on endocrine cell identity and several rare subtypes (16). Recent genomic studies have emerged in the past years characterizing alterations associated with several adenoma subtypes. For example, activating mutations involving GNAS leading to cAMP/protein kinase A pathway activation is a recurrent event in somatotroph adenomas (17, 18), and somatic mutations in the deubiquitinase gene USP8, resulting in increased EGF pathway signaling are present in a subset (~40–60%) of functioning corticotrophic adenomas (19, 20). In this study, we identified that the majority of pituitary adenomas have shortened telomere lengths. Additionally, we identify a subset of pituitary adenomas that display the ALT phenotype and are enriched in recurrent disease.
While 10–15% of all cancers are ALT-positive, the prevalence of ALT is enriched in multiple tumor types, including pancreatic neuroendocrine tumors (PanNETs), gliomas, neuroblastomas, and sarcomas (10, 13). The vast majority of ALT-positive cell lines and cancers have lost functional ATRX (21). However, DAXX alterations have been identified in cell lines (22, 23) and some cancer types (24), and DAXX alterations are twice as prevalent as ATRX alterations in ALT-positive PanNETs (11, 25, 26). Across multiple studies of primary well-differentiated PanNETs, loss of function of the ATRX/DAXX chromatin remodeling complex, mainly through somatic mutations, and acquisition of ALT are associated with decreased recurrence-free survival (11, 25–29) and a molecular subtype that resembles islet α-cells at the epigenetic and transcriptomic levels (30, 31). Additional neuroendocrine tumor types that have ATRX mutations in a subset of cases are pheochromocytomas and paragangliomas (32–34). Similar to the PanNETs, pheochromocytomas and paragangliomas with somatic ATRX mutations are strongly associated with ALT and an aggressive clinical behavior. However, in the ALT-positive cases identified in this current study in which sequencing was performed, mutations in ATRX or DAXX were not found. Although, two cases displayed ATRX or DAXX protein loss, suggesting either homozygous loss of the gene or the presence of another silencing mechanism (e.g. promoter methylation). In contrast, case 111 was wild-type for ATRX and DAXX at the gene and protein levels and only one somatic mutation in EP300, a histone acetyltransferase that regulates transcription via chromatin remodeling, was found (35). While EP300 has not been previously linked to telomere maintenance, these results suggest the possibility of an additional driver of ALT, similar to recent studies that have identified SMARCAL1 mutations in a subset of ALT-positive glioblastomas (36) and SLX4IP mutations in a subset of ALT-positive osteosarcomas (37).
In studies of pituitary adenomas, a published cohort of 42 pituitary adenomas from pediatric and adolescent patients revealed that 3 cases displayed loss of nuclear ATRX protein expression, although the vast majority of adenomas retained ATRX (38). Similarly, ATRX and DAXX protein expression were retained in a large series of adenohypophyseal endocrine tumors of different hormonal and clinical types from 246 patients; however 1 (or 2) corticotroph carcinomas examined displayed ATRX loss (39). Although only in a single pituitary carcinoma, a rare tumor that is pathologically indistinguishable from adenoma but strictly defined by the presence of leptomeningeal or extracranial metastases, Guo and colleagues documented the presence of a somatic mutation in ATRX, along with mutations in PTEN and TP53 (40).
There are a number of strengths of our current study. The discovery cohort consisted of a large number of pituitary adenomas with well-annotated clinical and pathological data. In addition, analysis of a second cohort that we collected to characterize recurrent pituitary adenomas in more detail confirmed the presence of ALT in a substantial fraction of recurrent disease. Also, we assessed two different cancer-specific telomere alterations, telomere shortening and presence of ALT, using a robust telomere-specific FISH assay. However, despite these strengths, there are also limitations to our study. This is a retrospective study with imperfect clinical follow-up. In addition, there is a relatively small sample size with limited number of ALT-positive cases and a limited number of cases with available tumor from primary and recurrent disease resections. The clinical significance of the subset of ALT-positive pituitary adenomas remains to be determined.
In summary, short telomeres are prevalent in pituitary adenomas. Additionally, a substantial subset of pituitary adenomas, enriched in recurrent tumors, are ALT-positive. Future studies are necessary to validate and extend these findings.
ACKNOWLEDGEMENTS
This study was supported by a Basic/Translational Science Investigator Award from the North American Neuroendocrine Tumor Society supported by the Neuroendocrine Tumor Research Foundation (CMH), Department of Defense grant W81XWH-18-1-0496 (FJR), NIH grant P30 CA006973 to the Sidney Kimmel Comprehensive Cancer Center (PI: W. Nelson), NIH grant T32 GM007748 (SC), and an award from the Brain Science Foundation (SS).
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
All authors have no conflicts of interest to declare.
REFERENCES
- 1.Gittleman H, Ostrom QT, Farah PD, Ondracek A, Chen Y, Wolinsky Y, et al. Descriptive epidemiology of pituitary tumors in the United States, 2004–2009. J Neurosurg. 2014;121:527–35. [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;17 Suppl 4:iv1-iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jane JA Jr., Laws ER Jr. The surgical management of pituitary adenomas in a series of 3,093 patients. J Am Coll Surg. 2001;193:651–9. [DOI] [PubMed] [Google Scholar]
- 4.Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, et al. Landscape of Genomic Alterations in Pituitary Adenomas. Clin Cancer Res. 2017;23:1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hage M, Viengchareun S, Brunet E, Villa C, Pineau D, Bouligand J, et al. Genomic Alterations and Complex Subclonal Architecture in Sporadic GH-Secreting Pituitary Adenomas. J Clin Endocrinol Metab. 2018;103:1929–39. [DOI] [PubMed] [Google Scholar]
- 6.Salomon MP, Wang X, Marzese DM, Hsu SC, Nelson N, Zhang X, et al. The Epigenomic Landscape of Pituitary Adenomas Reveals Specific Alterations and Differentiates Among Acromegaly, Cushing’s Disease and Endocrine-Inactive Subtypes. Clin Cancer Res. 2018;24:4126–36. [DOI] [PubMed] [Google Scholar]
- 7.Meeker AK, Hicks JL, Iacobuzio-Donahue CA, Montgomery EA, Westra WH, Chan TY, et al. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res. 2004;10:3317–26. [DOI] [PubMed] [Google Scholar]
- 8.Simpson K, Jones RE, Grimstead JW, Hills R, Pepper C, Baird DM. Telomere fusion threshold identifies a poor prognostic subset of breast cancer patients. Mol Oncol. 2015;9:1186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heaphy CM, Yoon GS, Peskoe SB, Joshu CE, Lee TK, Giovannucci E, et al. Prostate cancer cell telomere length variability and stromal cell telomere length as prognostic markers for metastasis and death. Cancer Discov. 2013;3:1130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JY, Brosnan-Cashman JA, An S, Kim SJ, Song KB, Kim MS, et al. Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin Cancer Res. 2017;23:1598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cesare AJ, Heaphy CM, O’Sullivan RJ. Visualization of Telomere Integrity and Function In Vitro and In Vivo Using Immunofluorescence Techniques. Curr Protoc Cytom. 2015;73:12.40.1–12.40.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez FJ, Vizcaino MA, Blakeley J, Heaphy CM. Frequent alternative lengthening of telomeres and ATRX loss in adult NF1-associated diffuse and high-grade astrocytomas. Acta Neuropathol. 2016;132:761–3. [DOI] [PubMed] [Google Scholar]
- 15.Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lloyd RV, Osamura RY, Kloppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs. Fourth ed. Lyon: International Agency for Research on Cancer; 2017. [Google Scholar]
- 17.Ronchi CL, Peverelli E, Herterich S, Weigand I, Mantovani G, Schwarzmayr T, et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur J Endocrinol. 2016;174:363–72. [DOI] [PubMed] [Google Scholar]
- 18.Valimaki N, Demir H, Pitkanen E, Kaasinen E, Karppinen A, Kivipelto L, et al. Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J Clin Endocrinol Metab. 2015;100:3918–27. [DOI] [PubMed] [Google Scholar]
- 19.Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47:31–8. [DOI] [PubMed] [Google Scholar]
- 20.Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25:306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012;8:e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mason-Osann E, Dai A, Floro J, Lock YJ, Reiss M, Gali H, et al. Identification of a novel gene fusion in ALT positive osteosarcoma. Oncotarget. 2018;9:32868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yost KE, Clatterbuck Soper SF, Walker RL, Pineda MA, Zhu YJ, Ester CD, et al. Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci Rep. 2019;9:4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gopal RK, Kubler K, Calvo SE, Polak P, Livitz D, Rosebrock D, et al. Widespread Chromosomal Losses and Mitochondrial DNA Alterations as Genetic Drivers in Hurthle Cell Carcinoma. Cancer Cell. 2018;34:242–55 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146:453–60 e5. [DOI] [PubMed] [Google Scholar]
- 26.Singhi AD, Liu TC, Roncaioli JL, Cao D, Zeh HJ, Zureikat AH, et al. Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin Cancer Res. 2017;23:600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. [DOI] [PubMed] [Google Scholar]
- 28.Pipinikas CP, Dibra H, Karpathakis A, Feber A, Novelli M, Oukrif D, et al. Epigenetic dysregulation and poorer prognosis in DAXX-deficient pancreatic neuroendocrine tumours. Endocr Relat Cancer. 2015;22:L13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pea A, Yu J, Marchionni L, Noe M, Luchini C, Pulvirenti A, et al. Genetic Analysis of Small Well-differentiated Pancreatic Neuroendocrine Tumors Identifies Subgroups With Differing Risks of Liver Metastases. Ann Surg. 2020;271:566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan CS, Laddha SV, Lewis PW, Koletsky MS, Robzyk K, Da Silva E, et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun. 2018;9:4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cejas P, Drier Y, Dreijerink KMA, Brosens LAA, Deshpande V, Epstein CB, et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat Med. 2019;25:1260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Job S, Draskovic I, Burnichon N, Buffet A, Cros J, Lepine C, et al. Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res. 2019;25:760–70. [DOI] [PubMed] [Google Scholar]
- 34.Comino-Mendez I, Tejera AM, Curras-Freixes M, Remacha L, Gonzalvo P, Tonda R, et al. ATRX driver mutation in a composite malignant pheochromocytoma. Cancer Genet. 2016;209:272–7. [DOI] [PubMed] [Google Scholar]
- 35.Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, et al. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–3. [DOI] [PubMed] [Google Scholar]
- 36.Diplas BH, He X, Brosnan-Cashman JA, Liu H, Chen LH, Wang Z, et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat Commun. 2018;9:2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panier S, Maric M, Hewitt G, Mason-Osann E, Gali H, Dai A, et al. SLX4IP Antagonizes Promiscuous BLM Activity during ALT Maintenance. Mol Cell. 2019;76(1):27–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Schmidt RE, Dahiya S. Pituitary Adenoma in Pediatric and Adolescent Populations. J Neuropathol Exp Neurol. 2019;78:626–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casar-Borota O, Botling J, Granberg D, Stigare J, Wikstrom J, Boldt HB, et al. Serotonin, ATRX, and DAXX Expression in Pituitary Adenomas: Markers in the Differential Diagnosis of Neuroendocrine Tumors of the Sellar Region. Am J Surg Pathol. 2017;41:1238–46. [DOI] [PubMed] [Google Scholar]
- 40.Guo F, Wang G, Wang F, Xu D, Liu X. Identification of Novel Genes Involved in the Pathogenesis of an ACTH-Secreting Pituitary Carcinoma: A Case Report and Literature Review. Front Oncol. 2018;8:510. [DOI] [PMC free article] [PubMed] [Google Scholar]