

Abstract
Transmembrane 6 superfamily member 2 (TM6SF2) is located on chromosome 19 (19p12) and encodes for a protein of undetermined function. Genetic studies have reported the association between a nonsynonymous variant in TM6SF2 (E167K, rs58542926) with hepatic triglyceride content and its impact on the cardiovascular system. Clinical and epidemiological studies have confirmed the role of TM6SF2 in the development of nonalcoholic fatty liver disease (NAFLD). Recently, TM6SF2 was also shown to play an important role in promoting hepatic fibrosis and hepatocellular cancer in mouse models. This review aims to capture the physiological role of TM6SF2 in the regulation of lipid metabolism and its involvement in cardiometabolic diseases.
The nonsynonymous E167K variant in TM6SF2 is one of the strongest genetic risk factors in the development of NAFLD and hence understanding the molecular mechanism(s) governed by this variant can provide valuable insights into the disease pathogenesis, diagnostic and therapeutic potential.
Abbreviations
- ABCG
adenosine triphosphate‐binding cassette subfamily G member
- ALT
alanine aminotransferase
- APOB
apolipoprotein B
- ASCVD
atherosclerotic cardiovascular disease
- CVD
cardiovascular disease
- CVS
cardiovascular system
- DGAT
diacylglycerol O‐acyltransferase
- DHS
Dallas Heart Study
- EBP
emopamil binding protein
- ER
endoplasmic reticulum
- ERGIC
endoplasmic reticulum–Golgi intermediate compartment
- GWAS
genome‐wide association study
- HCC
hepatocellular carcinoma
- HDL‐C
high‐density lipoprotein cholesterol
- KO
knockout
- LDL‐C
low‐density lipoprotein cholesterol
- mRNA
messenger RNA
- MTTP
microsomal
triglyceride transfer protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PCs
phosphocholine
- PUFA
polyunsaturated fatty acid
- TC
total cholesterol
- TG
triglyceride
- TM6SF2
transmembrane 6 superfamily member 2
- VLDL
very low‐density lipoprotein
Nonalcoholic fatty liver disease (NAFLD) is a form of chronic liver disease characterized by abnormal accumulation of triglycerides (TGs) in the liver.( 1 , 2 ) Currently, NAFLD is the second most common etiology of liver transplantation in the United States and is projected to be the leading cause by the end of this decade.( 3 ) The earliest stage of NAFLD is hepatic steatosis, which is often self‐limiting and benign; this can progress to chronic inflammation, known as nonalcoholic steatohepatitis (NASH), cirrhosis, and eventually hepatocellular carcinoma (HCC), in a fraction of patients.( 2 , 4 ) Epidemiological studies have shown that a significant portion of patients with NAFLD (~20%) progress to NASH,( 5 ) but the risk determinants of disease progression remain unclear. Obesity and type 2 diabetes mellitus are the two major risk factors in the development of NAFLD.( 2 ) Disease susceptibility also varies among individuals and different ethnicities, underlying a genetic basis for the disease.
Evidence from twin studies document the role of heritability in hepatic fat accumulation and presence of NAFLD.( 6 , 7 ) A seminal study conducted on a multi‐ethnic cohort in the Dallas Heart Study (DHS) further confirmed the genetic basis for the development of NAFLD.( 8 ) In a first, a study showed variation of the degree of accumulation of TGs in the liver across races. Genome‐wide association study (GWAS) on the same cohort identified patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) and the I148M variant (rs738409) as the predominant genetic risk factor associated with development of NAFLD.( 9 , 10 ) A point mutation (rs58542926, c.499 C>T, P. Glu167Lys, E167K) in another gene, transmembrane 6 superfamily 2 (TM6SF2), was also independently associated with elevated liver TGs, higher circulating levels of alanine aminotransferase (ALT), and lower levels of low‐density lipoprotein cholesterol (LDL‐C).( 11 , 12 , 13 ) The association between the TM6SF2 E167K variant and NAFLD was also confirmed in subsequent studies.( 14 , 15 , 16 ) The minor (T) allele is more frequent in East Asians (approximately 34%) and Europeans (approximately 26%) and less common in Hispanics/Latinos (approximately 10%) and Africans (6%, Genome Aggregation Database).( 17 ) Existing literature also reports a greater proportion of patients who are lean with NAFLD carrying the TM6SF2 rs58542926 (T) allele than patients who are obese/overweight with NAFLD,( 18 ) further underscoring its independent role in the development of the disease.
The deleterious effect of the TM6SF2 E167K mutation in NAFLD is paradoxical in the context of the cardiovascular system (CVS), where it has been associated with a lower risk of cardiovascular disease (CVD).( 12 , 14 , 15 , 16 ) Mendelian randomization studies have also confirmed the cardioprotective effect of the TM6SF2 E167K variant.( 19 ) Burgeoning evidence has established an independent role of NAFLD in the risk of developing CVD,( 20 , 21 , 22 ) although mechanistic understanding remains obscure. The cardioprotective role of the TM6SF2 E167K variant implies a different disease pathogenesis with this genetic variant.( 23 ) Proper understanding of the physiological role of TM6SF2 in the body will aid us in providing insights to resolve the conundrum.
Characterization of TM6SF2
Gene and Protein
TM6SF2, first reported in 2000,( 24 ) is located on chromosome 19p12 and encodes a protein containing 377 or 350 amino acids (two isoforms produced by alternative splicing). The protein is predicted to have seven to 10 transmembrane domains,( 11 ) while an additional expanded emopamil binding protein (EBP) superfamily (EXPERA) domain was found to be conserved, along with 3‐β‐hydroxysteroid‐8,7‐isomerase, through computational analysis.( 25 ) This can potentially indicate its role as an enzyme in cholesterol metabolism. The gene is moderately conserved across species, with the human protein showing 99.73%, 78.4%, and 78.57% homology with chimpanzee, mouse, and rat, respectively (https://www.ncbi.nlm.nih.gov/homologene/?term=Homo+sapiens +TM6SF2). A nonsynonymous point mutation in TM6SF2 (rs58542926, c.499 C>T) causes a glutamine to lysine substitution at residue 167 (p. Glu167Lys, E167K), resulting in a misfolded protein, accelerated protein degradation, and reduced protein levels in the body.( 11 )
Tissue Distribution and Regulation
TM6SF2 messenger RNA (mRNA) is abundantly expressed in liver, intestine, and kidney in both human and mouse.( 11 , 13 , 26 ) However, TM6SF2 protein in mouse is around 10‐fold higher in the small intestine than liver.( 26 ) Expression of full‐length TM6SF2 protein containing a C‐terminal green fluorescent protein tag in human hepatoma (Huh7) cells revealed perinuclear lattice‐like staining and considerable overlap with protein disulfide isomerase (endoplasmic reticulum [ER] marker) and ER‐Golgi intermediate compartment (ERGIC)‐53 (ERGIC marker).( 13 ) However, limited overlap was observed between TM6SF2 and Golgi marker Golgin subfamily B member 1 (GIANTIN),( 13 ) which led us to hypothesize that the subcellular localization of TM6SF2 is limited to the ER and ERGIC. However, subcellular fractionation of murine liver and biochemical analysis showed the presence of TM6SF2 in the ER and Golgi.( 26 ) The results were further validated in primary murine hepatocytes through confocal microscopy, where TM6SF2 colocalized with an ER marker (calnexin) and Golgi markers (receptor‐binding cancer antigen expressed on SiSo cells [RCAS1] and Giantin) but not with boron‐dipyrromethene staining.( 26 ) Given the technical challenges associated with the isolaton of ER and Golgi from mouse liver, contamination of ER with the Golgi compartment could not be completely ruled out. Unlike what was observed previously by immunocytochemistry, we recently found TM6SF2 mainly localized to the smooth ER but not to Golgi.( 27 ) In view of these conflicting lines of evidence, further research is required to confirm the subcellular localization of TM6SF2. Lei and colleagues( 28 ) reported carbohydrate‐responsive element‐binding protein (ChREBP)‐mediated transcriptional and translational up‐regulation of TM6SF2 protein.( 28 ) However, no change in levels of TM6SF2 protein was observed after fasting and refeeding the mice with a normal chow diet.( 26 ) An explanation for this discrepancy can be due to up‐regulation of hepatic ChREBP in response to high‐sucrose diet refeeding following fasting.( 29 ) Further experiments are required to validate the nutritional regulation of Tm6sf2.
Clinical Relevance of TM6SF2
Relationship Between TM6SF2 E167K Variant and NAFLD
Genetic evidence from multiple GWASs demonstrates the association of single nucleotide polymorphism (rs10401969) in the 19p12 locus with plasma levels of TG and total cholesterol (TC).( 30 , 31 , 32 , 33 , 34 , 35 ) In addition, GWASs established the association between the 19p12 locus and NALFD,( 36 , 37 ) coronary heart disease,( 34 , 38 ) and diabetes.( 39 , 40 ) However, no individual gene at this locus had been reported to regulate plasma lipids at that time. Mahdessian et al.( 13 ) performed gene expression analysis on 206 human liver tissues and reported the highest expression of TM6SF2 mRNA among genes in the 19p12 region. Subsequently, an expression quantitative trait locus analysis of rs10401969 was mapped to the expression of TM6SF2 mRNA (P = 0.0018), suggesting that TM6SF2 may play a dominant functional role in the NAFLD phenotype.
A growing body of evidence from population genetic studies implicates the TM6SF2 E167K variant as an independent risk factor for NAFLD (Table 1). A seminal study involving three independent populations (DHS, Dallas BioBank, and Copenhagen City Heart Study), confirmed the association between the TM6SF2 E167K variant and increased hepatic TG content and ALT.( 11 ) Subsequent studies in larger multi‐ethnic cohorts, except one,( 41 ) validated the association of the TM6SF2 E167K variant with NAFLD.( 15 , 41 , 42 , 43 , 44 , 45 ) A possible explanation of the null association between the TM6FS2 E167K variant and NAFLD in Hispanics could be due to small sample size and lack of statistical power. However, the relationship between the TM6SF2 E167K variant and NAFLD severity, fibrosis, cirrhosis, and HCC is less consistent.
TABLE 1.
Clinical Relevance of TM6SF2
| Reference | Sample Size | Ethnicity/Place of Study | Plasma Lipids | Liver Enzymes | HTGC/Method | NASH | Fibrosis | Cirrhosis | HCC | CVS |
|---|---|---|---|---|---|---|---|---|---|---|
| Kozlitina et al.( 11 ) | DHS (n = 2,736) | non‐Hispanic African Americans, non‐Hispanic, European Americans, Hispanics and other ancestry | LDL‐C↓ and TG↓ | ALT↑ | ↑/1H‐MRS | NM | NM | NM | NM | NM |
| Dallas BioBank (n = 8,585) | European‐Americans | LDL‐C↓ and TG↓ | ALT↑ | ↑/1H‐MRS | NM | NM | NM | NM | NM | |
| Copenhagen City Heart Study and Copenhagen General Population Study (n = 73,532) | Denmark | LDL‐C↓ and TG↓ | ALT↑ and AST↑ | ↑/1H‐MRS | NM | NM | NM | NM | NM | |
| Holmen et al.( 12 ) | N = 92,605 | Europeans | TC↓ and TG↓ | NM | NM | NM | NM | NM | NM | MI↓ |
| Liu et al.( 46 ) | Discovery cohort (n = 349) | European Caucasian | NM | NM | ↑/biopsy | ↑ | ↑ | Not clear | NM | NM |
| Validation cohort (n = 725) | European Caucasian | NM | NM | ↑/biopsy | NA | ↑ | Not clear | NM | NM | |
| NAFLD‐HCC cohort (n = 99) | European Caucasian | NM | NM | NM | NM | NM | NM | ↑ | NM | |
| Wong et al.( 47 ) | HK‐MRS (n = 922) | Chinese | TC↓, LDL‐C↓ and TG↓ | NM | ↑/1H‐MRS | NM | NA | NA | NM | NM |
| Sookoian et al.( 14 ) | N = 361 | Argentina | TC↓, LDL‐C: NA, and TG: NA | NA | ↑/biopsy | NM | NA | NM | NM | CVD risk↓ |
| Dongiovanni et al.( 16 ) | Liver biopsy cohort (n = 1,201) | Italy, Finland | TC↓, LDL‐C: NA and TG↓ | NA | ↑/biopsy | ↑ | ↑ | NM | NM | NM |
| carotid atherosclerosis cohort (427) | Italy | NM | NM | NM | NM | NM | NM | NM | Carotid plaques↓ | |
| SOS cohort (n = 1,819) | Swedish | ALT↑ and AST↑ | NM | NM | NM | NM | NM | NM | CVD events↓ | |
| Zhou et al.( 45 ) | N = 300 | Finnish | LDL‐C and TG: NA | ALT and AST: NA | ↑/1H‐MRS | NM | NM | NM | NM | NM |
| Akuta et al.( 48 ) | Patients with biopsy‐proven NAFLD (n = 211) | Japanese | NM | NM | NM | NA | NA | NM | NM | NM |
| Goffredo et al.( 41 ) | Children/adolescents with obesity (n = 402) | Caucasians | TC↓, and LDL‐C↓ | ↑ trend | ↑/MRI | NM | NM | NM | NM | NM |
| Children/adolescents with obesity (n = 266) | African Americans | NA | NA | ↑/MRI | NM | NM | NM | NM | NM | |
| Children/adolescents with obesity (n = 289) | Hispanics | TC↓, and LDL‐C↓ | ALT↑ and AST↑ | ↑/MRI | NM | NM | NM | NM | NM | |
| Grandone et al.( 57 ) | Children/adolescents with obesity (n = 1,010) | Italy | TC↓, LDL‐C↓ and TG↓ | ALT↑ and AST: NA | ↑/ultrasound | NM | NM | NM | NM | NM |
| Mancina et al.( 56 ) | Children/adolescents with obesity (n = 423) | Italy | NM | NA | ↑/ultrasound | NM | NM | NM | NM | NM |
| O'Hare et al. ( 60 ) | GHS bariatric surgery cohort (n = 983) | NA | NM | ↑/biopsy | ↑ | ↑ | NA | NM | NM | |
| ACDRP cohorts (n = 3,556) | Caucasian | TC↓, LDL‐C↓ and TG↓ | NA | NM | NM | NM | NM | NM | NM | |
| Krawczyk et al.( 49 ) | NAFLD Clinical Study Group project (n = 515) | German | NM | ALT↑ and AST↑ | ↑/MRI | NM | NA | NM | NM | NM |
| Liu et al.( 15 ) | N > 300,000 | European, African, South Asian, Hispanic or other ancestry | TC↓ and TG↓ | NM | ↑/CT | NM | NM | NM | NM | CAD↓ |
| Basyte‐Bacevice et al.( 50 ) | N = 1,012 | Lithuania | NM | NM | NM | NM | NA | NA | NM | NM |
| Anstee et al.( 42 ) | Histologically characterized cohort (1,483 biopsied NAFLD cases and 17,781 controls) | Europeans | NM | NM | ↑/biopsy | ↑ | NM | NM | NM | NM |
| Parisinos et al.( 43 ) | UK BioBank Cohort (n = 14,440) | Europeans | LDL‐C↓ and TG↓ | ALT↑ and AST↑ | ↑/MRI | NM | NM | ↑ | NM | CAD↓ |
| Chen et al.( 44 ) | UK BioBank (1,088 cases vs. 407,873 controls) and Michigan Genomics Initiative (875 cases of cirrhosis vs. 30,346 controls) | UK, US | LDL‐C↓ and TG↓ | ALT↑ | NM | NM | NM | ↑ | NM | NM |
Abbreviations: ↑, increase or positive association; ↓, decrease or negative association; ACDRP, Amish Complex Disease Research Program; AST, aspartate transaminase; CAD, coronary artery disease; CT, computed tomography; GHS, Geisinger Health System; HK‐MRS, Hong Kong, magnetic resonance spectroscopy; 1H‐MRS, proton nuclear magnetic resonance spectroscopy; HTGC, hepatic triglyceride content; MI, myocardial infarction; MRI, magnetic resonance imaging; NA, no association; NM, not mentioned; SOS, Obese Subjects Study; UK, United Kingdom; US, United States.
An initial study by Liu et al.( 46 ) demonstrated an association between the TM6SF2 E167K variant with hepatic steatosis and histologic stage of fibrosis but no correlation with histopathological evidence of steatohepatitis and HCC. Similar results were reported by other groups, linking the TM6SF2 E167K variant with hepatic steatosis but not with fibrosis or histopathological steatohepatitis.( 14 , 47 , 48 , 49 , 50 )
However, in a recent large cross‐sectional study in a European cohort consisting of 1,201 patients, Dongiovanni and colleagues( 16 ) showed a positive association between the TM6SF2 E167K variant and hepatic steatosis, steatohepatitis, and fibrosis. A meta‐analysis of four studies and 4,325 patients also confirmed the increased risk of steatosis, fibrosis, and cirrhosis in carriers of the variant.( 51 )
The conflicting nature of the evidence can be attributed to the cumulative risk due to the presence of confounding risk factors, like alcoholic liver disease and hepatitis C infection. The lower allele frequency and relatively small size of cohorts could explain conflicting results. A GWAS identified the TM6SF2 E167K variant as a risk locus for alcohol‐related cirrhosis in individuals of European descent, with subsequent validation in two independent European cohorts.( 52 ) Moreover, the same variant was strongly associated with alcohol‐related and non‐hepatitis B HCC.( 53 , 54 ) Future prospective studies with proper phenotyping for NAFLD and genotyping, apart from comparison between groups that are well matched for risk factors for chronic liver disease, need to be conducted to ascertain the risk of disease severity of NASH and of fibrosis with the TM6SF2 variant. Finally, a Mendelian randomization study using genetic risk variants for NAFLD also confirmed a robust causal relationship between hepatic fat content and fibrosis, which further points toward the role of TM6SF2 in accelerating liver fibrosis.( 55 )
Apart from adults, the TM6SF2 E167K variant has also been associated with NAFLD in children and adolescents.( 56 , 57 ) Using imaging modalities (ultrasonography and magnetic resonance imaging) and liver biopsies, independent groups have demonstrated the association between the TM6SF2 E167K variant and liver fat content( 11 , 16 , 45 , 46 ) and severity of liver disease.( 14 , 41 , 56 )
TM6SF2 E167K Variant and CVD
The American College of Cardiology/American Heart Association Atherosclerotic Cardiovascular Disease (ASCVD) Risk Estimator identifies dyslipidemia, including elevated plasma TC, LDL‐C, and TG levels and decreased high‐density lipoprotein cholesterol (HDL‐C) levels, as independent risk factors for CVD.( 58 ) In a seminal GWAS that included 5,643 Norwegians, a strong association between the TM6SF2 E167K variant and reduction of serum TC and TG and concomitant reduced risk of myocardial infarction was described (Table 1).( 12 ) The results have since been replicated in subsequent studies involving independent cohorts, highlighting the paradoxical effect of the TM6SF2 variant in heart and liver.( 11 , 15 , 16 ) The cardioprotective effect of the TM6SF2 variant is primarily due to its effect on a more favorable plasma lipid profile.( 16 ) A meta‐analysis that included 101,326 subjects showed that carriers of the KK genotype of the TM6SF2 E167K variant had lower levels of TC and LDL‐C than those homozygous for the TM6SF2 EE genotype.( 59 ) This has been supported by data from the Amish Complex Disease Research Program cohort, which reported an increase in plasma level of HDL‐C in addition to a decrease in TC, LDL‐C, and TG in patients with the TM6SF2 E167K variant.( 60 ) The relation between dyslipidemia and subclinical atherosclerosis has been well documented.( 61 ) Carotid plaque measurements, which are a surrogate marker of subclinical atherosclerosis, were used by Dongiovanni and colleagues( 16 ) in patients with NASH. While the overall risk of cardiovascular events increased in patients with NASH, TM6SF2‐mediated NASH was noted to have lower subclinical atherosclerosis as well as a lower incidence of cardiovascular events. However, the cardioprotective effect of the TM6SF2 E167K variant was lost after adjusting for plasma TC levels,( 16 ) implying a TM6SF2‐induced favorable plasma lipid profile as the major contributor. Inflammation plays a significant role in subclinical ASCVD.( 62 , 63 ) C‐reactive protein, a well‐validated biomarker of subclinical ASCVD, was markedly reduced in patients with NAFLD carrying the TM6SF2 E167K variant when compared to controls.( 14 ) The effect of the TM6SF2 NAFLD risk variant over CVS requires more attention from both clinicians and researchers. Aggressive management of hepatic steatosis in patients with NAFLD may negate its positive effect on the heart. Replication of the cardioprotective effects of TM6SF2 in longitudinal studies and better mechanistic understanding of its role in hepatic steatosis will aid in therapeutic manipulation of downstream effects of TM6SF2 without compromising its beneficial effect on the CVS.
Preclinical Studies Involving TM6SF2
Murine Models
To explore the mechanistic role of TM6SF2 in NAFLD pathogenesis and blood lipid regulation, murine models of Tm6sf2 knockdown or knockout (KO) were established. Kozlitina et al.( 11 ) knocked down TM6SF2 by injecting adenovirus‐associated virus (AAV) into the tail vein of C57BL/6J mice to achieve significant reduction of protein translation. Knockdown of TM6SF2 resulted in significant lowering of plasma TC and TG levels in the knockdown group compared to control, while their liver TG levels increased significantly. The difference in liver TG deposition was more conspicuous when fed with a high‐sucrose diet.( 11 ) Although ALT levels (marker of hepatic dysfunction) were significantly increased in Tm6sf2 KO mice, there was no evidence of inflammation, differential expression of fibrosis‐associated genes, and histopathological characteristics of liver injury.( 26 ) Another group used adenovirus with short hairpin RNAs to target the Tm6sf2 coding region in C57BL/6J mice; they reported a significant decrease in circulating TC levels with no concurrent changes in liver TG and hepatic functions.( 12 ) A possible explanation of the discrepancy in results could be due to either a different mode of interfering RNA delivery, extent of the mRNA knockdown, or variable hepatic TM6SF2 protein levels. To circumvent these limitations, permanent gene deletion of established Tm6sf2 employing the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system was undertaken to create Tm6sf2 KO mice.( 64 ) Under a normal diet, serum TC and HDL‐C were significantly reduced compared to their wild‐type littermates, with no cholesterol or TG deposition in the liver,( 64 ) which is inconsistent with previous reports.( 26 ) We found inactivation of TM6SF2 resulted in hepatic steatosis in rat under a normal diet.( 27 ) More recently, two other independent groups reported that whole‐body or hepatic ablation of Tm6sf2 in mice leads to increase hepatic TG accumulation.( 65 , 66 ) Liver‐specific inactivation of TM6SF2 was reported to promote hepatic steatosis and fibrosis and accelerate development of HCC.( 66 ) Whether TM6SF2 plays a role in development of hepatic fibrosis and HCC needs further investigation.
Ehrhardt et al.( 67 ) achieved transient genetic overexpression of Tm6sf2 in mice by injecting AAV into wild‐type C57BL/6J mice; they noted significant reduction of plasma TG, TC, and HDL. Moreover, Tm6sf2 overexpression resulted in accumulation of TG in the liver but not cholesterol.( 67 ) However, stable hepatic overexpression of human TM6SF2 in mice resulted in increased circulating plasma lipids (TC and HDL‐C) and hepatic TG and cholesterol.( 64 ) Of note, both the KO and overexpression of Tm6sf2 in murine models result in hepatic steatosis, implying a more complex association between mutant genotype and phenotype, which could not be explained through merely gain or loss of function alone. Because TM6SF2 is localized in ER and binding to apolipoprotein B (ApoB), there is a high possibility that overexpression of Tm6sf2 may sequester ApoB‐containing lipoproteins in ER. Ehrhardt et al.( 67 ) showed Tm6sf2 overexpression reduced ApoB secretion and resulted in its accumulation within ER, which is consistent with this hypothesis.
Data regarding the cardiovascular effects of murine models of Tm6sf2 KO remain lacking, which necessitates further research.
Zebrafish Model
The expression of tm6sf2 is confined to liver and intestine in zebrafish. Using the CRISPR/Cas9 system to inactivate tm6sf2 expression in zebrafish, O’Hare et al.( 60 ) reported significant TG accumulation in the liver and intestine.
Mechanism of Action of TM6SF2 in Lipid Metabolism
TG in TM6SF2 and Hepatic Lipid Metabolism
Liver and intestine are the two major sources of lipids in the body. The intestine secretes lipid in the form of chylomicrons, while liver can synthesize and package lipids into very low‐density lipoprotein (VLDL) and secrete them into circulation.( 68 , 69 ) TM6SF2 is predominantly expressed in the small intestine and liver, raising the possibility that TM6SF2 may regulate lipid metabolism in these organs. Liver lipids are mainly derived from de novo synthesis of fatty acids or absorption of remnant lipoprotein particles and free fatty acids from the circulation. A fraction of hepatic lipids is mainly used for hepatic energy production, and another part is secreted into circulation in the form of VLDLs. Hepatic steatosis due to loss of TM6SF2 can be either due to abnormality in TG synthesis or secretion of VLDL‐TG or a combination of both. Inactivation of Tm6sf2 in mice did not affect sterol responsive element binding protein (SREBP‐1 and SREBP‐2), the master transcriptional regulator of fatty acid and cholesterol biosynthesis,( 26 ) ruling out the possibility of excess synthesis causing hepatic steatosis in Tm6sf2 KO mice. Increased hepatic fat and decreased circulating lipids in Tm6sf2 KO mice are consistent with a defect in VLDL‐TG secretion, conferred by loss of functional protein. The defect of VLDL‐TG secretion can be explained through the following two potential mechanisms: first, reduction in the number of secreted VLDL particles from the liver as in the microsomal triglyceride transfer protein (MTTP) KO model( 70 , 71 ); and second, through the reduction in the TG content of VLDL particles while the number of VLDL particles remains constant.( 72 ) APOB concentration is a surrogate marker of the number of VLDL particles, and TG content is a reflection of the particle size. Smagris et al.( 26 ) found that VLDL particles of Tm6sf2 KO mice were smaller in size than wild‐type mice and secretion of VLDL‐TG was significantly reduced without any reduction in plasma APOB levels, indicating that defective VLDL‐TG but not APOB secretion may be predominantly involved in eliciting the phenotype. The role of TM6SF2 on APOB secretion was also explored in vitro. TM6SF2 knockdown in human cell lines (Huh7 and HepG2) results in a remarkable decrease in TG secretion with a modest reduction in ApoB levels.( 13 ) However, in a pulse‐chase experiment to monitor APOB secretion, the investigators observed that the level of newly synthesized and secreted APOB100 in plasma of Tm6sf2 KO mice was similar to that of the wild type while a mild increase in APOB48 was observed in KO mice,( 26 ) which was also validated in primary hepatocytes.( 66 ) No changes in hepatic APOB levels were reported in Tm6sf2 KO mice.( 26 ) These findings indicate that loss of TM6SF2 function results in decreased VLDL lipidation without alteration of APOB synthesis or secretion.
Existing data suggest that lipidation of VLDL is a two‐step process.( 73 ) The first step involves lipidation associated with APOB synthesis taking place in the rough ER, which requires the presence of the MTTP protein.( 71 ) The second step in the lipidation process involves further lipidation of nascent VLDL. In the event of any defect in the first lipidation step, the nonlipidated APOB protein will get degraded and will lead to a significant reduction in plasma APOB level.( 71 ) Data from transmission electron microscopy of liver showed a decrease in the size of the nascent VLDL particles in Golgi of liver‐specific Tm6sf2 KO mice and undetectable levels of VLDL particles in the ER or Golgi of liver‐specific Mttp KO mice, indicating TM6SF2 may be involved in the second step of lipidation. In vitro data from TM6SF2 knockdown in HuH7 and HCC (HepG2) cells showed a mild reduction in secretion of APOB.( 13 ) However, as mentioned above, APOB levels in liver and serum of Tm6sf2 KO mice did not show any decrease in comparison to wild‐type mice,( 26 , 66 ) suggesting that Tm6sf2 may not be involved in the early lipidation of VLDL. Reduced APOB secretion was also reported in carriers of the TM6SF2 E167K genetic variant in human hepatic three‐dimesional spheroids.( 74 ) Possible explanations for the difference in APOB levels could be functional change in cellular metabolism and interspecies difference in the presence of APOB subtypes between humans and mouse. More recently, it was reported that TM6SF2 interacts and stabilizes APOB.( 65 ) Knockout of Tm6sf2 leads to decreased hepatic APOB protein level in mice according to the time of day the mice were fasted.( 65 ) However, under both conditions, plasma TC and TG were increased in the liver and decreased in serum of Tm6sf2‐deficient mice,( 65 ) ruling out any dependence of hepatic steatosis phenotype in Tm6sf2 KO mice on “time‐of‐day” fasting.
Phospholipids and polyunsaturated fatty acids (PUFAs) are critical players in the second stage of lipidation of VLDL.( 72 ) Luukkonen and colleagues( 75 ) reported reduction in the level of PUFAs in liver and serum TGs and liver phosphocholine (PCs) in patients carrying the TM6SF2 E167K variant, but hepatic free fatty acids were relatively enriched in PUFA. Concomitantly, decreased incorporation of PUFAs into TGs and PCs in TM6SF2 knockdown hepatocytes was reported.( 76 ) Another study also reported a significant reduction of polyunsaturated lipid species and an increase in saturated and monounsaturated species in HuH7 cells following TM6SF2 knockdown.( 76 ) They reported relative and absolute arachidonic acid (AA, 20:4n‐6) depletion in the PCs.( 76 ) These findings have not been confirmed in animal studies. Although marginal differences of hepatic and plasma fatty acids were observed in wild‐type and Tm6sf2 KO mice, the AA content of the PC fraction was similar between the two groups.( 26 )
Knockdown of TM6SF2 in Huh7 and HepG2 cell lines also reduces transcription of diacylglycerol O‐acyltransferase 1 (DGAT1) and DGAT2,( 13 ) which are key enzymes in TG synthesis.( 77 ) However, there is no evidence to confirm that transcriptional perturbation of DGAT1 and DGAT2 leads to a decrease in translational efficiency. Taken together, the effect of Tm6sf2 KO appears to be mediated through decreased lipidation of VLDL (Fig. 1).
FIG. 1.
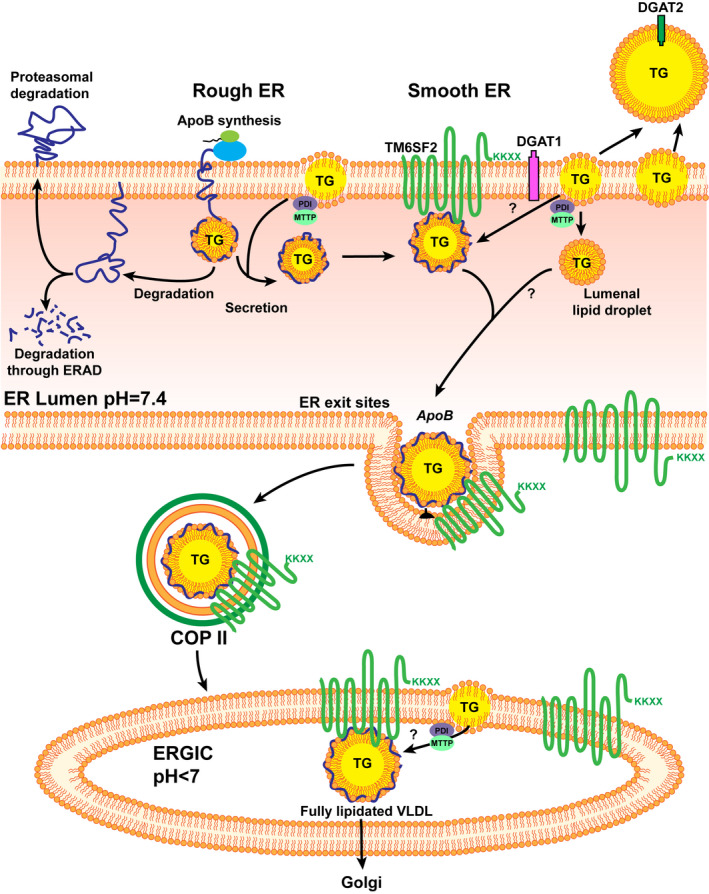
The potential mechanisms of TM6SF2 in hepatic lipid metabolism. TM6SF2 localized to ER and ERGIC. (Adapted from Mahdessian et al., Proc Natl Acad Sci U S A 2014;111:8913‐8918.). TM6SF2 interacts with APOB and is involved in the second lipidation of VLDLs. Abbreviations: COP II, coat protein complex II; ERAD, endoplasmic‐reticulum‐associated protein degradation.
Cholesterol in TM6SF2 and Hepatic Lipid Metabolism
The role of Tm6sf2 in cholesterol metabolism has also been explored. The 3‐β‐hydroxysteroid‐8,7‐isomerase coded by EBP is a key enzyme in the cholesterol biosynthetic pathway.( 78 ) TM6SF2 protein was identified as an EBP homologue and shares a functional domain with EBP.( 25 ) In HuH7 cells, overexpression of TM6SF2 increases cholesterol biosynthesis, while inhibition of downstream enzyme Δ7‐dehydrocholesterol reductase (an enzyme converting 7‐dehydrocholesterol to cholesterol at the final step of cholesterol biosynthesis) abolishes this effect.( 64 ) Compared with wild‐type mice, the expression of cholesterol metabolism‐related genes (such as LDL receptor; scavenger receptor, class B type 1 [SR‐BI]; and proprotein convertase subtilisin/kexin type 9 serine protease [PCSK9]) in Tm6sf2 KO mice did not show any difference( 26 ) except for 7‐dehydrocholesterol reductase, which had a higher expression in KO mice.( 64 ) Adenosine triphosphate‐binding cassette subfamily G member (ABCG) 5 and ABCG8 can promote the removal of liver cholesterol through hepatobiliary secretion.( 79 ) The levels of ABCG5 and ABCG8 were elevated in the livers of Tm6sf2 KO mice in comparison to their wild‐type littermates.( 64 ) However, the underlying mechanism is unclear. These findings indicate that TM6SF2 may also promote liver cholesterol biosynthesis.
TM6SF2 and Intestinal Lipid Absorption
O'Hare et al.( 60 ) reported that carriers of the TM6SF2 167K allele had significantly lower postprandial serum TG level than the carriers of TM6SF2 167E allele after high‐fat stimulation in the Amish Complex Disease Research Program. This indicated that the E167K variant is associated with lipid absorption, which was further confirmed in another independent study.( 80 ) Smagris et al.( 26 ) reported that Tm6sf2 KO mice fed a high‐fat diet for 12 weeks had a large accumulation of neutral lipids in jejunum but no accumulation was observed in wild‐type mice. They further conducted a lipid tolerance test by providing mice corn oil (10 µL/g) through oral gavage and then measured plasma TG levels. The results showed that absorption of lipids in wild‐type mice and Tm6sf2 KO mice was similar, although the absorption peak of Tm6sf2 KO mice was shifted compared to that of wild‐type mice, suggesting impairment in the rate of intestinal lipid absorption.( 26 ) Consequently, TM6SF2 deficiency might be responsible for the reduced secretion of chylomicrons from intestinal cells. Furthermore, inactivation of tm6sf2 in zebrafish can induce TG accumulation in cytoplasmic lipid droplets of intestinal epithelial cells.( 60 ) Because the absorption of dietary lipid involves multiple steps (starting from storage of absorbed lipids to assembly of chylomicrons), the precise role of TM6SF2 activity in the process cannot be delineated.( 60 )
Challenges and Future Directions
NAFLD, a burgeoning etiology of chronic liver disease in the developed world, is a complex metabolic disease leading to systemic manifestations that can also affect the cardiovascular system. Currently, therapies targeting disease progression in NAFLD are lacking. Leveraging human genetics to identify risk determinants in NAFLD has unearthed a strong association between the TM6SF2 E167K variant and hepatic steatosis. Genetic risk variants, like TM6SF2 E167K, have preventive and therapeutic importance in the context of NAFLD. The insidious nature of NAFLD necessitates early detection and intervention to prevent late sequelae of the disease. Genetic risk scores based on genetic variants alone or taken together may allow clinicians to screen for hepatic steatosis to identify at‐risk individuals and intervene through modifiable risk factors, like dietary restriction and weight loss.( 81 ) Incorporating genetic risk variants, such as TM6SF2, may aid in improving noninvasive diagnostic accuracy of NAFLD as well.( 55 ) As shown by Paternostro et al.,( 82 ) adding TM6SF2 to risk stratification models improved the diagnostic accuracy of the model for the prediction of advanced fibrosis. From a therapeutic standpoint, patients carrying the TM6SF2 E167K variant can be targeted for early intervention, despite not having histologic evidence of NASH at a time point. Secondly, this genetic risk variant can be beneficial in enrolling these at‐risk patients in clinical trials for the development of novel therapies for NAFLD/NASH, thereby increasing the power of these studies and addressing drug efficacy against genetic risk‐mediated NAFLD.( 83 ) Finally, therapies targeting the relevant biological pathways affected by genetic risk variants, like TM6SF2, may aid in reducing the increased risk of developing NAFLD in this group of patients. Primary prevention strategies, including close clinical monitoring of individuals carrying this variant, may be beneficial in reducing the disease burden in the population. An evolving body of clinical and preclinical literature implicates the role of this risk variant in reducing VLDL‐TG secretion from the liver, resulting in accumulation of hepatic TGs leading to NAFLD. However, mechanistic insights about the exact role of this variant in causing NAFLD remains unknown and represents an area of active interest and research. Better understanding of the disease pathogenesis due to this risk variant may aid in developing targeted therapies to treat NAFLD. Of note, carriers of the TM6SF2 variant also have favorable cardiovascular outcomes. Hence, identification of downstream pathways altered by the TM6SF2 E167K variant may aid in the disentanglement of its deleterious role in NAFLD development. It will also help in developing therapeutic strategies to retain its cardioprotective effect without offsetting the benefit of targeting the variant itself for the treatment of NAFLD.
Supported by the Chinese Scholarship Council (grant 201806370096 to F.L.), Hunan Provincial Natural Science Foundation of China (grant no. 2021JJ40852 to F.L.), and National Natural Science Foundation of China (grant 82100495 to F.L.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547‐555. [DOI] [PubMed] [Google Scholar]
- 4. Hobbs HH. Science, serendipity, and the single degree. J Clin Invest 2018;128:4218‐4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Argo CK, Caldwell SH. Epidemiology and natural history of non‐alcoholic steatohepatitis. Clin Liver Dis 2009;13:511‐531. [DOI] [PubMed] [Google Scholar]
- 6. Makkonen J, Pietiläinen KH, Rissanen A, Kaprio J, Yki‐Järvinen H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: a study in monozygotic and dizygotic twins. J Hepatol 2009;50:1035‐1042. [DOI] [PubMed] [Google Scholar]
- 7. Loomba R, Schork N, Chen C‐H, Bettencourt R, Bhatt A, Ang B, et al.; Genetics of NAFLD in Twins Consortium . Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015;149:1784‐1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387‐1395. [DOI] [PubMed] [Google Scholar]
- 9. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan X, Waterworth D, Perry JRB, Lim N, Song K, Chambers JC, et al. Population‐based genome‐wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet 2008;83:520‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg‐Hansen A, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holmen OL, Zhang HE, Fan Y, Hovelson DH, Schmidt EM, Zhou W, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco‐Cereceda A, Hamsten A, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 2014;111:8913‐8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sookoian S, Castaño GO, Scian R, Mallardi P, Fernández Gianotti T, Burgueño AL, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015;61:515‐525. [DOI] [PubMed] [Google Scholar]
- 15. Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, et al. Exome‐wide association study of plasma lipids in >300,000 individuals. Nat Genet 2017;49:1758‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506‐514. [DOI] [PubMed] [Google Scholar]
- 17. Kozlitina J. Genetic risk factors and disease modifiers of nonalcoholic steatohepatitis. Gastroenterol Clin North Am 2020;49:25‐44. [DOI] [PubMed] [Google Scholar]
- 18. Chen F, Esmaili S, Rogers GB, Bugianesi E, Petta S, Marchesini G, et al. Lean NAFLD: a distinct entity shaped by differential metabolic adaptation. Hepatology 2020;71:1213‐1227. [DOI] [PubMed] [Google Scholar]
- 19. Lauridsen BK, Stender S, Kristensen TS, Kofoed KF, Køber L, Nordestgaard BG, et al. Liver fat content, non‐alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta‐analysis of 279 013 individuals. Eur Heart J 2018;39:385‐393. [DOI] [PubMed] [Google Scholar]
- 20. Söderberg C, Stål P, Askling J, Glaumann H, Lindberg G, Marmur J, et al. Decreased survival of subjects with elevated liver function tests during a 28‐year follow‐up. Hepatology 2010;51:595‐602. [DOI] [PubMed] [Google Scholar]
- 21. Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver‐related mortality in non‐alcoholic fatty liver disease. J Hepatol 2008;49:608‐612. [DOI] [PubMed] [Google Scholar]
- 22. Kim D, Choi S‐Y, Park EH, Lee W, Kang JH, Kim W, et al. Nonalcoholic fatty liver disease is associated with coronary artery calcification. Hepatology 2012;56:605‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sliz E, Sebert S, Würtz P, Kangas AJ, Soininen P, Lehtimäki T, et al. NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum Mol Genet 2018;27:2214‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carim‐Todd L, Escarceller M, Estivill X, Sumoy L. Cloning of the novel gene TM6SF1 reveals conservation of clusters of paralogous genes between human chromosomes 15q24–>q26 and 19p13.3–>p12. Cytogenet Cell Genet 2000;90:255‐260. [DOI] [PubMed] [Google Scholar]
- 25. Sanchez‐Pulido L, Ponting CP. TM6SF2 and MAC30, new enzyme homologs in sterol metabolism and common metabolic disease. Front Genet 2014;5:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem 2016;291:10659‐10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo F, Hobbs HH, Cohen JC. Abstract 13164: Tm6sf2 is a regulator of liver fat metabolism in smooth ER influencing VLDL lipidation [Abstract]. Circulation 2020;142:A13164. [Google Scholar]
- 28. Lei Y, Hoogerland JA, Bloks VW, Bos T, Bleeker A, Wolters H, et al. Hepatic carbohydrate response element binding protein activation limits nonalcoholic fatty liver disease development in a mouse model for glycogen storage disease type 1a. Hepatology 2020;72:1638‐1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Linden AG, Li S, Choi HY, Fang F, Fukasawa M, Uyeda K, et al. Interplay between ChREBP and SREBP‐1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J Lipid Res 2018;59:475‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008;40:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, et al. Six new loci associated with blood low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol or triglycerides in humans. Nat Genet 2008;40:189‐197. Erratum in: Nat Genet 2008;40:1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, et al.; ENGAGE Consortium . Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet 2009;41:47‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 2009;41:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al.; Global Lipids Genetics Consortium . Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gorden A, Yang R, Yerges‐Armstrong LM, Ryan KA, Speliotes E, Borecki IB, et al.; GOLD Consortium . Genetic variation at NCAN locus is associated with inflammation and fibrosis in non‐alcoholic fatty liver disease in morbid obesity. Hum Hered 2013;75:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Speliotes EK, Yerges‐Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al.; NASH CRN; GIANT Consortium; MAGIC Investigators; GOLD Consortium . Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 2013;45:1345‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, et al.; Wellcome Trust Case Control Consortium; Meta‐Analyses of Glucose and Insulin‐related traits Consortium (MAGIC) Investigators; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN‐T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; DIAbetes Genetics Replication And Meta‐analysis (DIAGRAM) Consortium . Large‐scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saxena R, Elbers C, Guo Y, Peter I, Gaunt T, Mega J, et al. Large‐scale gene‐centric meta‐analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet 2012;90:410‐425. Erratum in: Am J Hum Genet 2012;90:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goffredo M, Caprio S, Feldstein AE, D'Adamo E, Shaw MM, Pierpont B, et al. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: a multiethnic study. Hepatology 2016;63:117‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al.; EPoS Consortium Investigators . Genome‐wide association study of non‐alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J Hepatol 2020;73:505‐515. Erratum in: J Hepatol 2021;74:1274‐1275. [DOI] [PubMed] [Google Scholar]
- 43. Parisinos CA, Wilman HR, Thomas EL, Kelly M, Nicholls RC, McGonigle J, et al. Genome‐wide and Mendelian randomisation studies of liver MRI yield insights into the pathogenesis of steatohepatitis. J Hepatol 2020;73:241‐251. Erratum in: J Hepatol 2020;73:1594‐1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen VL, Chen Y, Du X, Handelman SK, Speliotes EK. Genetic variants that associate with cirrhosis have pleiotropic effects on human traits. Liver Int 2020;40:405‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou Y, Llauradó G, Orešič M, Hyötyläinen T, Orho‐Melander M, Yki‐Järvinen H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J Hepatol 2015;62:657‐663. [DOI] [PubMed] [Google Scholar]
- 46. Liu Y‐L, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JBS, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non‐alcoholic fatty liver disease. Nat Commun 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wong VWS, Wong GLH, Tse CH, Chan HLY. Prevalence of the TM6SF2 variant and non‐alcoholic fatty liver disease in Chinese. J Hepatol 2014;61:708‐709. [DOI] [PubMed] [Google Scholar]
- 48. Akuta N, Kawamura Y, Arase Y, Suzuki F, Sezaki H, Hosaka T, et al. Relationships between genetic variations of PNPLA3, TM6SF2 and histological features of nonalcoholic fatty liver disease in Japan. Gut Liver 2016;10:437‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krawczyk M, Rau M, Schattenberg JM, Bantel H, Pathil A, Demir M, et al.; NAFLD Clinical Study Group . Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy‐based study. J Lipid Res 2017;58:247‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Basyte‐Bacevice V, Skieceviciene J, Valantiene I, Sumskiene J, Petrenkiene V, Kondrackiene J, et al. TM6SF2 and MBOAT7 gene variants in liver fibrosis and cirrhosis. Int J Mol Sci 2019;20:1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Z, Que S, Zhou L, Zheng S, Romeo S, Mardinoglu A, et al. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: a meta‐analysis. Sci Rep 2017;7:9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD, et al. A genome‐wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol‐related cirrhosis. Nat Genet 2015;47:1443‐1448. [DOI] [PubMed] [Google Scholar]
- 53. Falleti E, Cussigh A, Cmet S, Fabris C, Toniutto P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig Liver Dis 2016;48:69‐75. [DOI] [PubMed] [Google Scholar]
- 54. Yang J, Trépo E, Nahon P, Cao Q, Moreno C, Letouzé E, et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int J Cancer 2019;144:533‐544. [DOI] [PubMed] [Google Scholar]
- 55. Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283:356‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mancina RM, Sentinelli F, Incani M, Bertoccini L, Russo C, Romeo S, et al. Transmembrane‐6 superfamily member 2 (TM6SF2) E167K variant increases susceptibility to hepatic steatosis in obese children. Dig Liver Dis 2016;48:100‐101. [DOI] [PubMed] [Google Scholar]
- 57. Grandone A, Cozzolino D, Marzuillo P, Cirillo G, Di Sessa A, Ruggiero L, et al. TM6SF2 Glu167Lys polymorphism is associated with low levels of LDL‐cholesterol and increased liver injury in obese children. Pediatr Obes 2016;11:115‐119. [DOI] [PubMed] [Google Scholar]
- 58. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation 2019;139:e1082‐e1143. Erratum in: Circulation 2019;139:e1182‐e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2‐rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: a meta‐analysis. Hepatology 2015;62:1742‐1756. [DOI] [PubMed] [Google Scholar]
- 60. O'Hare EA, Yang R, Yerges‐Armstrong LM, Sreenivasan U, McFarland R, Leitch CC, et al. TM6SF2 rs58542926 impacts lipid processing in liver and small intestine. Hepatology 2017;65:1526‐1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martin SS, Blaha MJ, Blankstein R, Agatston A, Rivera JJ, Virani SS, et al. Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: implications for statin therapy from the multi‐ethnic study of atherosclerosis. Circulation 2014;129:77‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fernández‐Friera L, Fuster V, López‐Melgar B, Oliva B, Sánchez‐González J, Macías A, et al. Vascular inflammation in subclinical atherosclerosis detected by hybrid PET/MRI. J Am Coll Cardiol 2019;73:1371‐1382. [DOI] [PubMed] [Google Scholar]
- 63. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 2012;32:2045‐2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fan Y, Lu H, Guo Y, Zhu T, Garcia‐Barrio MT, Jiang Z, et al. Hepatic transmembrane 6 superfamily member 2 regulates cholesterol metabolism in mice. Gastroenterology 2016;150:1208‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li B‐T, Sun M, Li Y‐F, Wang J‐Q, Zhou Z‐M, Song B‐L, et al. Disruption of the ERLIN‐TM6SF2‐APOB complex destabilizes APOB and contributes to non‐alcoholic fatty liver disease. PLoS Genet 2020;16:e1008955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Newberry EP, Hall Z, Xie Y, Molitor EA, Bayguinov PO, Strout GW, et al. Liver‐specific deletion of mouse Tm6sf2 promotes steatosis, fibrosis, and hepatocellular cancer. Hepatology 2021. 10.1002/hep.31771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB‐trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet 2017;26:2719‐2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hussain MM. Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidol 2014;25:200‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Y, Liu L, Zhang H, Fan J, Zhang F, Yu M, et al. Mea6 controls VLDL transport through the coordinated regulation of COPII assembly. Cell Res 2016;26:787‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Raabe M, Véniant MM, Sullivan MA, Zlot CH, Björkegren J, Nielsen LB, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue‐specific knockout mice. J Clin Invest 1999;103:1287‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Raabe M, Flynn LM, Zlot CH, Wong JS, Veniant MM, Hamilton RL, et al. Knockout of the abetalipoproteinemia gene in mice: reduced lipoprotein secretion in heterozygotes and embryonic lethality in homozygotes. Proc Natl Acad Sci U S A 1998;95:8686‐8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rong X, Wang BO, Dunham MM, Hedde PN, Wong JS, Gratton E, et al. Lpcat3‐dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. Elife 2015;4:e06557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Olofsson SO, Stillemark‐Billton P, Asp L. Intracellular assembly of VLDL: two major steps in separate cell compartments. Trends Cardiovasc Med 2000;10:338‐345. [DOI] [PubMed] [Google Scholar]
- 74. Prill S, Caddeo A, Baselli G, Jamialahmadi O, Dongiovanni P, Rametta R, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep 2019;9:11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Luukkonen PK, Zhou Y, Nidhina Haridas PA, Dwivedi OP, Hyötyläinen T, Ali A, et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J Hepatol 2017;67:128‐136. [DOI] [PubMed] [Google Scholar]
- 76. Ruhanen H, Nidhina Haridas PA, Eskelinen E‐L, Eriksson O, Olkkonen VM, Käkelä R. Depletion of TM6SF2 disturbs membrane lipid composition and dynamics in HuH7 hepatoma cells. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:676‐685. [DOI] [PubMed] [Google Scholar]
- 77. Gluchowski NL, Gabriel KR, Chitraju C, Bronson RT, Mejhert M, Boland S, et al. Hepatocyte deletion of triglyceride‐synthesis anzyme acyl CoA: diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology 2019;70:1972‐1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Long T, Hassan A, Thompson BM, McDonald JG, Wang J, Li X, et al. Structural basis for human sterol isomerase in cholesterol biosynthesis and multidrug recognition. Nat Commun 2019;10:2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee JY, Kinch LN, Borek DM, Wang J, Wang J, Urbatsch IL, et al. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 2016;533:561‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Musso G, Cipolla U, Cassader M, Pinach S, Saba F, De Michieli F, et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J Lipid Res 2017;58:1221‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pelusi S, Baselli G, Pietrelli A, Dongiovanni P, Donati B, McCain MV, et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci Rep 2019;9:3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Paternostro R, Staufer K, Traussnigg S, Stättermayer A‐F, Halilbasic E, Keritam O, et al. Combined effects of PNPLA3, TM6SF2 and HSD17B13 variants on severity of biopsy‐proven non‐alcoholic fatty liver disease. Hepatol Int 2021;15:922‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Romeo S, Sanyal A, Valenti L. Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab 2020;31:35‐45. [DOI] [PubMed] [Google Scholar]