

Abstract
Objectives
Lipedema, a poorly understood chronic disease of adipose hyper-deposition, is often mistaken for obesity and causes significant impairment to mobility and quality-of-life. To identify molecular mechanisms underpinning lipedema, we employed comprehensive omics-based comparative analyses of whole tissue, adipocyte precursors (adipose-derived stem cells (ADSCs)), and adipocytes from patients with or without lipedema.
Methods
We compared whole-tissues, ADSCs, and adipocytes from body mass index–matched lipedema (n = 14) and unaffected (n = 10) patients using comprehensive global lipidomic and metabolomic analyses, transcriptional profiling, and functional assays.
Results
Transcriptional profiling revealed >4400 significant differences in lipedema tissue, with altered levels of mRNAs involved in critical signaling and cell function-regulating pathways (e.g., lipid metabolism and cell-cycle/proliferation). Functional assays showed accelerated ADSC proliferation and differentiation in lipedema. Profiling lipedema adipocytes revealed >900 changes in lipid composition and >600 differentially altered metabolites. Transcriptional profiling of lipedema ADSCs and non-lipedema ADSCs revealed significant differential expression of >3400 genes including some involved in extracellular matrix and cell-cycle/proliferation signaling pathways. One upregulated gene in lipedema ADSCs, Bub1, encodes a cell-cycle regulator, central to the kinetochore complex, which regulates several histone proteins involved in cell proliferation. Downstream signaling analysis of lipedema ADSCs demonstrated enhanced activation of histone H2A, a key cell proliferation driver and Bub1 target. Critically, hyperproliferation exhibited by lipedema ADSCs was inhibited by the small molecule Bub1 inhibitor 2OH-BNPP1 and by CRISPR/Cas9-mediated Bub1 gene depletion.
Conclusion
We found significant differences in gene expression, and lipid and metabolite profiles, in tissue, ADSCs, and adipocytes from lipedema patients compared to non-affected controls. Functional assays demonstrated that dysregulated Bub1 signaling drives increased proliferation of lipedema ADSCs, suggesting a potential mechanism for enhanced adipogenesis in lipedema. Importantly, our characterization of signaling networks driving lipedema identifies potential molecular targets, including Bub1, for novel lipedema therapeutics.
Subject terms: Obesity, Obesity
Introduction
Lipedema is a poorly understood, chronic disease characterized by bilaterally symmetrical deposition of abnormal adipose tissue, predominantly in the legs and variably in the arms [1, 2]. The classification of lipedema is based on the distribution of adipose tissue and on the severity of the disease (Stages I, II, III, and IV) [3] (Supplementary Fig. 1a). Most metabolic disorders relating to adipose tissue are associated with white adipose tissue (WAT), and dysfunction of its endocrine activities [4]. Excessive accumulation of WAT in abdominal organs (visceral adipose tissue) and subcutaneous regions disrupts metabolic homeostasis and likely drives metabolic disorders [4, 5]. In lipedema, patients accumulate excessive amounts of subcutaneous WAT, predominately in the legs [2].
Lipedema mostly affects women, typically starting during stages of hormonal change such as puberty, pregnancy, in vitro fertilization, or menopause. Symptoms include extreme leg sensitivity, hyperadiposity, swelling, pain, bruising, and joint complications (hyper-flexibility and later arthritis), resulting in impaired mobility and body image. Lipedema is often confused with obesity, lymphedema, lipodystrophies, and other fat disorders [1] and currently is diagnosed clinically. Palpation and intraoperative examination of lipedema fat reveals a nodular structure and consistency, in contrast to normal fat that is smooth and soft [6–8]. There is no cure for lipedema, and treatments are limited to invasive liposuction or excisional surgery to control symptoms and enhance mobility and esthetics. The etiology of lipedema is unknown, although clinical observations suggest genetic inheritance, hormonal influences, dilated blood vessels and lymphatics, and inflammation [3]. Despite a relatively high incidence of lipedema [6–8], molecular or genetic data to explain the pathogenesis of the disease are lacking [9–12]. To date, elucidation of the molecular mechanisms underlying lipedema has been hampered by the lack of lipedema animal models.
In adipose disorders, fat typically expands by increasing either the size or number of adipocytes [13]. While adipocytes are unable to divide, they can increase in size to accommodate greater amounts of lipid volume [13]. Adipocytes form via de novo differentiation from adipose-derived stem cells (ADSCs) upon adipogenic induction [13]. Increased ADSC numbers, therefore, can lead to increased differentiation into adipocytes and enhanced adipose deposition in pathologic fat disorders. It has been proposed that adipocytes increase in both size and number in lipedema; however, the molecular mechanisms by which ADSCs undergo proliferation or differentiation to adipocytes, resulting in excessive accumulation of lipedema adipose tissue, are poorly understood [13–16].
Here, we report on comprehensive comparative multi-omics analyses of adipose tissue, ADSCs, and adipocytes from lipedema and non-lipedema patients, which emphasize the profoundly distinct nature of lipedema and non-lipedema adipose tissue. Importantly, our omics analyses led us to identify signaling events central to enhanced ADSC proliferation in lipedema, from which we validate a gene candidate and derive a proof-of-principle therapeutic intervention. Our findings substantially improve understanding of the molecular mechanisms underpinning lipedema and will be useful for identifying disease biomarkers and targets for future therapeutics with which to combat this devastating disease.
Results
Lipedema adipose tissues show distinct gene signatures and ADSC densities to normal fat
Recently, it was demonstrated that lipedema adipose tissue has larger adipocytes, increased macrophage density and increased dermal blood and lymphatic vessels compared to normal fat [17]. Whether the large amounts of adipose tissue accumulated in lipedema (Supplementary Fig. 1a) differ from non-lipedema fat at the cellular and molecular levels, however, remains to be elucidated. To address this, adipose tissues surgically excised by the same surgeon (RS) from equivalent anatomical sites in lipedema patients and gender-, health-, age-, and BMI-matched (Supplementary Table 1) non-lipedema patients undergoing elective debulking surgery were compared. Macroscopic examination showed lipedema fat to be harder and more fibrotic and nodular than smooth, softer normal fat (Fig. 1a).
Fig. 1. Transcriptional profiling of adipose tissue from lipedema (LED) and non-lipedema (non-LED) patients.

a Lipedema and non-lipedema adipose tissue after surgery. b Volcano plot of significantly differentially expressed genes (adj. p < 0.05). c Heatmap of hierarchical clustering showing expression patterns of top 50 differentially expressed genes ranked by topTreat from limma. Z-score fold-change value: red indicates upregulation and blue indicates downregulation (lipedema n = 8; non-lipedema n = 4). d CD29, CD34, CD31, and perilipin staining of adipocytes from lipedema tissue. e Perilipin-stained adipocyte (purple) from lipedema adipose tissue with two small blood vessels (CD31+(green)), and ADSCs (CD34+(blue) CD29+(red)). Circles indicate CD29+/CD34+ and CD31– co-localized cells. White arrowheads indicate perilipin staining (purple) of adipocyte. f, g Quantification of CD29+/CD34+ cells by ImageJ from lipedema and non-lipedema adipose tissue (n = 3, 6 random fields were quantified).
To understand the molecular mechanisms underlying these macroscopic changes, we undertook transcriptome profiling to characterize and quantify the transcriptomes of lipedema and non-lipedema adipose tissue. Principle component analysis (PCA) indicated separation of lipedema from non-lipedema samples and showed clear differences between their transcriptomic profiles (Supplementary Fig. 1b), and differential gene expression analysis showed 4391 genes to be significantly differentially expressed (Fig. 1b and Supplementary Table 2). Hierarchical clustering of the top genes showed distinct subsets of lipedema and non-lipedema genes (Fig. 1c), with gene ontology (GO) annotation and pathway enrichment analysis highlighting cell proliferation, lipid metabolism, cell adhesion, inflammation, and immunity pathways (Supplementary Fig. 1c–e). Highly differential gene expression that biologically favored enhanced proliferation in lipedema involved the cell-cycle regulation- and proliferation-related genes ZIC1, UGT1A7, GREM1, TRIM67, Bub1, and HOTAIR. Overall, global gene expression data and pathway enrichment analyses showed significant alterations in the molecular signature of lipedema tissue, with a gene expression profile favoring adipose hyperproliferation, fibrosis, and inflammation, which are consistent with key clinical features of lipedema.
To evaluate potential cells within lipedema fat that may contribute to the hypertrophy detected by our tissue transcriptomics, we focused on ADSCs—critical effector cells in fat. ADSCs play diverse roles in immune-modulation, angiogenesis, apoptosis, and differentiation [18] and are precursors of adipocytes, the major cell constituent of adipose tissue. Of described ADSC markers [19], CD29 and CD34 cell surface markers were chosen to define ADSCs [20, 21]. To visualize the ADSCs within the perivascular niche, we performed multiplex immunostaining with the adipocyte marker perilipin, the endothelial cell marker CD31 and CD34 and CD29. We identified perilipin+ adipocytes in tissue colocalizing with CD31+ endothelial cells and CD29+/CD34+ ADSCs (Fig. 1d, e). Next, we quantified the numbers of CD29+/CD34+ cells in tissue specimens and found that lipedema adipose tissue showed greater numbers of CD29+/CD34+ ADSCs compared to control tissue (Fig. 1f, g), suggesting a greater propensity to generate fat when stimulated. Together, these results indicate that the adipose tissue microenvironment in lipedema differs significantly from that in non-lipedema patients, both in terms of its abundance of ADSCs and gene expression profiles.
ADSCs and adipocytes in lipedema tissue are functionally distinct from those in obese fat
Having demonstrated key differences in lipedema tissues, including higher numbers of CD29+/CD34+ ADSCs, we next focused on the role of these ADSCs, as stem cells play key roles in many diseases, and ADSCs are precursors to adipocytes that may underpin the pathogenesis of lipedema. We employed a method of ADSC enrichment from adipose tissues, enabling us to study these cells in cell-based assays (Fig. 2a). Single-cell suspensions were prepared from lipedema and control tissues and the ADSC-enriched stromal vascular fraction assessed by flow cytometry. We validated that ADSCs from both patient groups displayed characteristic spindle morphology and expression of ADSC markers [22] (Supplementary Fig. 2a–c).
Fig. 2. Isolation and characterization of lipedema and non-lipedema ADSCs from adipose tissue.

a Schematic showing isolation of ADSCs from adipose tissue. b, c ADSCs were isolated from lipedema and non-lipedema adipose tissue, grown in adipogenic differentiation media for 14 days and analyzed with Bodipy staining. Fat droplets stained green with Bodipy. d–f For adipogenic differentiation assays, lipedema ADSCs and non-lipedema ADSCs (40,000 cells/well in 48 well plate) were cultured for 24 h in growth media, followed by incubation in adipogenic differentiation media for 14 days. Adipogenic differentiation was then detected by Bodipy staining of neutral lipids in differentiated cells. Graphs represent quantification of fat droplets per well (d), fat droplets per cell (e), and percentage of fat-positive cells (f) (n = 3, mean ± SEM of three independent experiments, *p < 0.05, Student’s t test).
To assess the adipogenic properties of the ADSC populations, we performed differentiation studies of ADSCs to adipocytes. When stimulated to differentiate to adipocytes in vitro, ADSCs functionally resemble adipocytes in several key aspects, such as lipid accumulation [20, 21, 23], allowing differentiated ADSCs to be used as a surrogate in vitro adipogenesis model for physiological and functional investigations. Adipocyte differentiation of our enriched ADSCs was verified by staining for lipid droplets (consisting mainly of neutral lipids, triglyceride, and cholesterol esters [24]). BODIPY-lipid staining revealed that the number of fat droplets, percentage of fat-positive cells, and number of fat droplets/cell were significantly increased in lipedema adipocytes compared to controls (Fig. 2b–f). These findings suggest that lipedema ADSC populations differ from non-lipedema ADSCs in terms of growth and differentiation capacity, which may play a critical role in the pathogenesis of lipedema.
Global lipidomic analysis showed altered lipid composition in lipedema adipocytes
After showing that the lipid storage architecture within adipocytes differed in differentiated lipodema adipocytes, we next wanted to assess their lipid composition. Adipocyte lipid composition has been shown to contribute to several clinical disorders including obesity and cancer [25–27]. Therefore, to characterize the lipid profiles of adipocytes in lipedema, we used a non-targeted lipidomics approach, focusing on the profile of low molecular weight (m/z 300–3000) ionizable lipid molecules, and multivariate statistics to compare the lipid molecules in lipedema adipocytes against controls. Liquid chromatography mass spectrometry (LCMS) data acquisition identified 928 putative lipid species based on accurate mass [26, 27]. PCA analysis revealed distinct clusters for each of the adipocyte groups, suggesting a unique lipedema adipocyte lipidomic signature (Fig. 3a and Supplementary Fig. 3a). Next, the variable importance in projection (VIP) scores from a PLS-DA analysis were used to rank the most significant lipid differences. The leading significantly increased lipids in lipedema included glycerophospholipids (GPLs) LPE(24:1), PC(28:2), PC(26:0), PE(42:2), PE(42:1), LysoPC(24:1), and PC(42:3) (Supplementary Fig. 3b). To further characterize the lipid profiles of lipedema adipocytes, hierarchical clustering analysis was performed, which identified the altered lipids in lipedema (Fig. 3b). This analysis showed separation of the lipidomic signatures of lipedema from non-lipedema profiles, and after global characterization, we further filtered the 928 original lipid species detected to 112 significantly differing species between the groups. The lipid species of greatest differences between the groups included sphingolipids and GPLs and to a lesser extent, gangliosides, fatty acyls, and glycerolipids (Supplementary Fig. 3c and Supplementary Table 3). Utilizing a volcano plot to elucidate the lipid metabolites that are present at different levels in lipedema versus non-lipedema adipocytes showed a global effect (Fig. 3c). Lipids across all major lipid classes seem to be affected in lipedema adipocytes as can be seen from a heatmap of lipid classes (Supplementary Fig. 3d). These experiments demonstrated that, globally, lipids were significantly altered in adipocytes differentiated from the ADSCs of lipedema patients compared to non-lipedema patients.
Fig. 3. Comparison of lipid content in adipocytes from lipedema and non-lipedema patients.
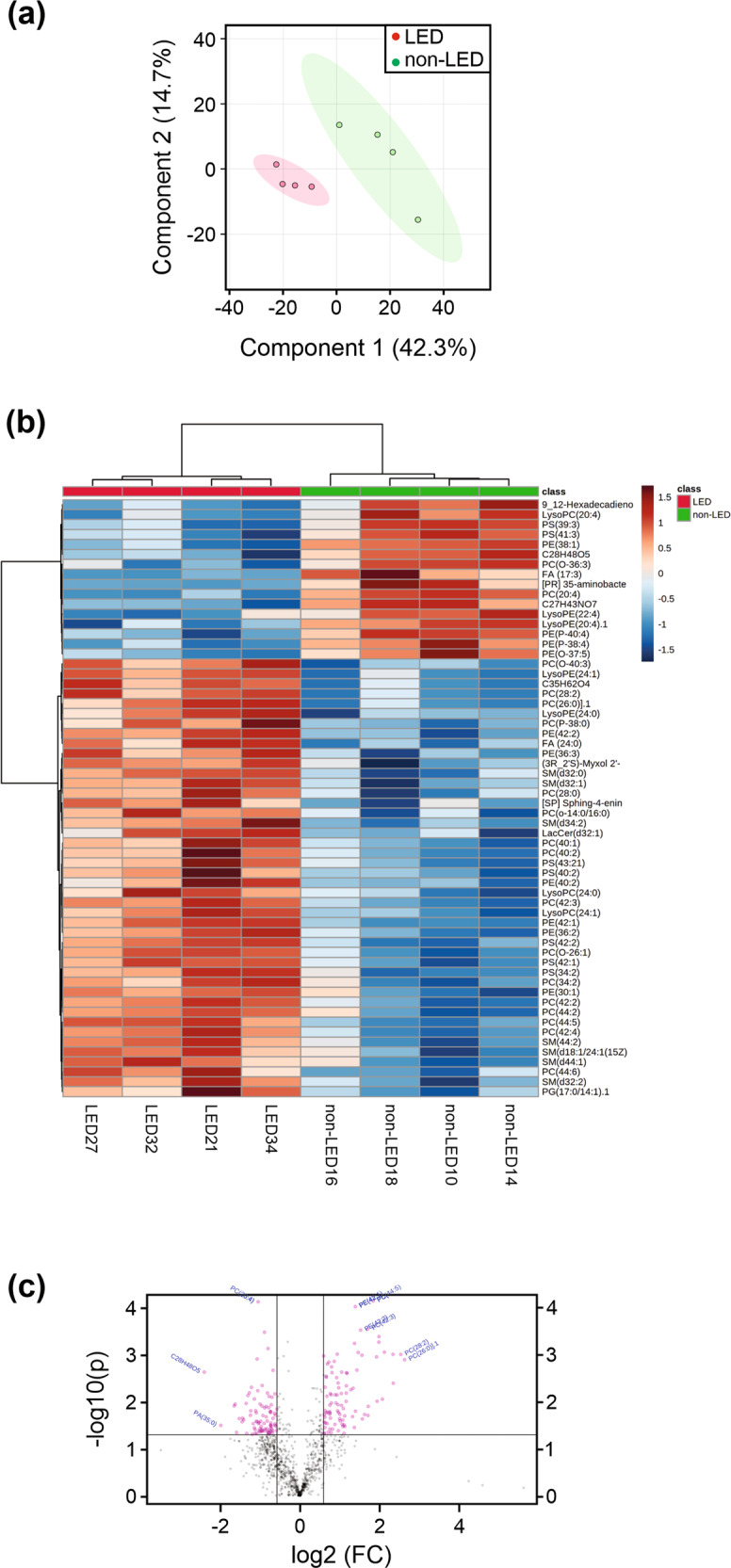
Lipedema (LED) ADSCs and non-lipedema (non-LED) ADSCs were treated with adipogenic differentiation medium for 14 days. Adipocytes were then collected and analyzed for differences in lipid content. a Supervised partial least squares discriminant analysis (PLS-DA) plot of four biological replicates from lipedema and non-lipedema adipocytes show clear separation of two different classes of metabolites. b Hierarchical clustering analysis of the 50 lipids with greatest differences in abundance between lipedema and non-lipedema adipocytes. c Volcano plot showing highly different lipid contents.
Global metabolic profiles are significantly altered in patients with lipedema
Previous studies showed that patients with adipose disorders but relatively normal metabolic status have lower mortality rates than patients with abnormal metabolic status [28]; however, the reason for this is unknown. Therefore, having demonstrated abnormalities in the lipidomic profile of lipedema adipocytes, we undertook a metabolomics analysis to profile the small endogenous molecules or metabolites in lipedema. A metabolic comparison of adipocytes was undertaken, and PCA showed separation between lipedema and non-lipedema groups (four biological replicates) (Supplementary Fig. 4a). Of the 640 distinct metabolites putatively identified in lipedema adipocytes, the most highly represented metabolite classes related to amino acid and carbohydrate metabolism (Supplementary Fig. 4b–d). VIP scores from a PLS-DA analysis were used to rank the most significant metabolite differences (Supplementary Fig. 4e). Pathway analysis suggested that perturbations in lipedema included pathways relating to amino acid metabolism (lysine biosynthesis and glutamate metabolism), peptides, and GLPs (Supplementary Table 4). It has been shown previously that metabolome imbalances result in adipocyte hypertrophy and/or hyperplasia that lead to fat disorders [29]. Taken together, our results indicate that the metabolites most deranged in lipedema may therefore critically contribute to disease pathogenesis.
ADSCs from lipedema adipose tissue display an enhanced proliferative state compared to those from BMI-matched patients
Having shown a gene expression profile in lipedema tissue that favors hyperproliferation, we evaluated the proliferative potential of lipedema ADSCs using proliferation assays to monitor their growth rates. Time-lapse microscopy showed that ADSCs from lipedema patients proliferated faster than control ADSCs (Fig. 4a). It is well documented that an accumulation of cells in S phase indicates cellular hyperproliferation and activation of the intra S phase checkpoint [30, 31]. In order to study cell division and elucidate the mechanisms by which lipedema ADSCs become hyperproliferative, cell-cycle analysis was performed. After synchronizing the ADSC groups by growth factor starvation to drive cells into the same phase (G0), complete medium was added to stimulate the cells to initiate proliferation. Results showed that 72 h after the addition of complete growth media, a higher percentage of lipedema ADSCs was in S phase (26.7%) compared to non-lipedema ADSCs (13.6%) (Fig. 4b, c). Finally, a colony formation unit (CFU) assay demonstrated an enhanced colony formation capacity of the hyperproliferative lipedema ADSCs (Fig. 4d, e). Together, these findings underscored the enhanced self-renewal and proliferative potential of lipedema ADSCs that may combine to drive adipose proliferation in lipedema.
Fig. 4. Lipedema ADSCs exhibit enhanced proliferative potential compared to non-lipedema ADSCs.

a Lipedema (LED) ADSCs and non-lipedema (non-LED) ADSCs were cultured in 96-well plates (3000 cells/well) for 72 h in growth media and cell growth was monitored by time-lapse microscopy (n = 6, mean ± SEM of two independent experiments, *p < 0.05, Student’s t test). b, c For cell-cycle assays, lipedema ADSCs (b) and non-lipedema ADSCs (c) were cultured in growth media for 48 h and stained with FXCycle Cell Cycle Assay Kit and analyzed by flow cytometry. d, e Clonogenicity potential of ADSCs as assessed by crystal violet staining of colony formation units (CFU). The stain was dissolved with 0.5% sodium dodecyl sulfate and absorbance was measured at 539 nm. Graph shows optical density for lipedema and non-lipedema (n = 3, mean ± SEM, *p < 0.05, Student’s t test).
Altered cell-cycle pathways drive adipogenesis and proliferation in lipedema ADSCs
We demonstrated transcriptomic differences in whole lipedema tissue and a hyperproliferative profile and functional behavior of lipedema ADSCs. To understand potential mechanism, we next explored the transcriptome of lipedema ADSCs themselves. RNA-seq followed by MDS and PCA analyses revealed clear separation, indicating different gene signatures, between lipedema and control ADSC transcriptomes (Supplementary Fig. 5a). In keeping with whole tissue profiles, differential expression analysis showed 3429 genes to be significantly differentially expressed between lipedema and control ADSCs (Fig. 5a, b and Supplementary Table 5). These data were further analyzed using the signaling pathway and GO terms cell-cycle, cell proliferation, cell division, and mitotic cell-cycle processes (Supplementary Fig. 5b). Cell-cycle mitotic pathways were among those most significantly differentially expressed (>50 genes) including cell-cycle genes that regulate and maintain the mitotic spindle checkpoint (Supplementary Fig. 5c, d). Significantly expressed genes were selected from the RNA-seq data set for validation by qRT-PCR. Among these genes, eight differentially expressed genes (Bub1, Bub1B, CDC20, CENPF, ASPM, BIRC5, KIF2C, and KIF14) were validated (Fig. 5c). These findings suggested that cell-cycle genes involved in regulating cell growth and proliferation are dysregulated in lipedema ADSCs and may contribute to the increased adipocyte number, and maldistribution and accumulation of dystrophic fat in lipedema.
Fig. 5. Cell-cycle genes that are differentially expressed in lipedema enhance proliferative capacity of ADSCs.

a RNA-seq analyses of lipedema (LED) ADSCs from seven patients (LED21, 23, 25, 27, 28, 32, 34) and of non-lipedema (non-LED) ADSCs from three patients (non-LED10, 12, 14) indicate differences between lipedema ADSC and non-lipedema ADSC transcript signatures. Volcano plot analysis of significantly differentially expressed genes (adj. p < 0.05) showed that 1617 genes were downregulated and 1812 genes were upregulated in lipedema ADSCs compared to non-lipedema ADSCs. b Heatmap of hierarchical clustering showing representative gene expression patterns. Top 50 differentially expressed genes (ranked by topTreat from limma) in lipedema ADSCs compared to non-lipedema ADSCs. Z-score fold-change value: red indicates upregulation and blue indicates downregulation. c Differentially expressed genes involved in mitotic spindle checkpoint pathways of cell cycle were validated by quantitative qRT-PCR (n = 3, mean ± SEM of two independent experiments, *p < 0.05, Student’s t test). d Bub1 mRNA was quantified by qRT-PCR in lipedema ADSCs isolated from seven patients and non-lipedema ADSCs from three patients (*p < 0.05, Student’s t test). e Bub1 protein from lipedema ADSCs (LED21, 23, 27, 32, 34) and non-lipedema ADSCs (non-LED10, 12) was analyzed by western blotting. f Graph represents relative expression levels of Bub1 protein, as assessed by western blotting, normalized to GAPDH levels (mean ± SEM of two independent experiments, *p < 0.05, Student’s t test). g Levels of Histone H2A (H2A) (lower), a target of Bub1, and of phosphorylated H2A (upper), were analyzed in lipedema ADSCs (LED21, 27, 34) and non-lipedema ADSCs (non-LED10, 12, 14) by Western blotting.
Bub1 hyper-activation drives enhanced proliferation of ADSCs from lipedema patients
Of our validated candidates, Bub1 was selected for further study because: (1) Bub1 mRNA was significantly upregulated in both lipedema whole tissue and ADSCs; (2) Bub1 overexpression is associated with hyperproliferation and dysregulation of key processes in several cancers [32–37]; and (3) Bub1 is a mitotic checkpoint protein identified as a potential therapeutic target in cancer stem cells [32, 36, 38–43]. From a mechanistic perspective, Bub1 plays a role in transcriptional activation during G1/S transition by phosphorylating histone H2A [44], and knockdown of Bub1 by siRNA can arrest cells in the G1/S phase [45]. Conversely, overexpression of Bub1 results in increased cell proliferation and chromosomal instability via hyper-activation of histone H2A [44].
Our finding that Bub1 mRNA is overexpressed in lipedema ADSCs suggested to us that Bub1 may play a similar role in lipedema as it does in cancer (i.e., driving excessive cell proliferation), and could therefore be a potential therapeutic target. Consequently, we sought to analyze Bub1 transcript levels in ADSCs from lipedema and non-lipedema patients by qRT-PCR, which demonstrated that Bub1 mRNA was significantly upregulated in lipedema ADSC populations, compared to controls (Fig. 5d). Bub1 protein was also significantly upregulated in lipedema ADSCs as assessed by Western blotting (Fig. 5e, f). Having established upregulation of Bub1 gene expression by numerous methods, we analyzed phosphorylation of histone H2A, a downstream signaling target of Bub1 integral to Bub1-driven cell proliferation. Western blotting revealed a greater level of phosphorylated histone H2A in ADSCs from lipedema patients compared to controls (Fig. 5g), suggesting that ADSCs are “primed” to proliferate in lipedema.
To ascertain the effects of restricting Bub1 in lipedema ADSCs, we employed a CRISPR/Cas9-mediated lentiviral system to knockdown Bub1 and a small molecule Bub1 inhibitor. Using qRT-PCR analysis, we observed an approximately 80% reduction of Bub1 mRNA levels in Bub1-depleted ADSCs compared to negative controls (Fig. 6a). Furthermore, Western blot confirmed decreased Bub1 protein in Bub1-knockdown ADSCs (Fig. 6b). As expected, knockdown of Bub1 gene decreased proliferation of both lipedema and non-lipedema ADSCs; however, the reduction in proliferation was significantly more pronounced in lipedema ADSCs (Fig. 6c). To explore a pharmacological approach for inhibiting Bub1 in lipedema ADSCs, we next employed the small molecule 2OH-BNPP1, which specifically inhibits the Serine/Threonine kinase activity of Bub1 [46]. We found that 2OH-BNPP1 significantly and profoundly restricted proliferation of lipedema ADSCs by approximately 77%, after 3 days of treatment, whereas inhibition of non-lipedema ADSC proliferation was more modest at approximately 40% (Fig. 6d). We also tested the effect of 2OH-BNPP on the cell cycle in lipedema and non-lipedema ADSCs. As expected, cell-cycle analysis showed that ADSC treatment with 2OH-BNPP resulted in more cells arrested at G1/S phase for both lipedema and non-lipedema ADSCs, with lipedema ADSCs showing a more significantly pronounced effect (Supplementary Fig. 6a, b), suggesting that the hyperproliferative state of lipodema ADSCs makes them more sensitive to cell-cycle inhibitor agents than the non-lipedema controls.
Fig. 6. Bub1 drives enhanced proliferation of lipedema ADSCs.

a CRISPR/Cas9 lentiviral vectors that express gRNA for Bub1 gene (Bub1-KD), gRNA for the HPRT gene as positive control (pos-control), and scrambled gRNA as negative control (neg-control) were used in combination with lentiviral Cas9 to infect lipedema ADSCs and non-lipedema ADSCs with or without any treatment of viral vectors (untreated control; un-control). After 7 days of infection, ADSCs were used to extract RNA, and qRT-PCR analysis was performed to quantify Bub1 and HPRT transcripts. b Western blotting to quantify Bub1 protein in lipedema ADSCs and non-lipedema ADSCs after gene removal with CRISPR/Cas9 lentiviral vector expressing gRNA for the Bub1 gene. GAPDH was used as loading control. c Proliferation of ADSCs was measured following gene depletion of Bub1 (mean ± SEM of three independent experiments, *p < 0.05, Student’s t test). d Lipedema ADSCs and non-lipedema ADSCs were cultured in growth medium with or without Bub1 inhibitor (2OH-BNPP1, 25 μM) or DMSO. Cell proliferation was measured by DAPI staining after 3 days. Relative levels of proliferation for treated and control cells are shown (mean ± SEM of three independent experiments, *p < 0.05, Student’s t test). e Treatment of ADSCs with 2OH-BNPP1 inhibited phosphorylation of Histone H2A. Western blotting was performed to quantify phosphorylated H2A in lipedema ADSCs and non-lipedema ADSCs after treatment with or without 2OH-BNPP1. f Quantification of phosphorylated H2A normalized with levels of total H2A protein (mean ± SEM of two independent experiments, *p < 0.05, Student’s t test).
As it has been shown that Bub1 enhances cell proliferation by phosphorylating threonine 120 (T120) of histone H2A [33, 46–48], we finally tested the effect of 2OH-BNPP1 on the activation of histone H2A, which is downstream of Bub1. Treatment of both ADSC groups with 2OH-BNPP1 revealed marked reduction of histone H2A phosphorylation at T120 (Fig. 6e, f), a novel and important finding that suggested that the effect of Bub1 in lipedema is mediated by its capacity to phosphorylate downstream targets such as histone H2A.
Discussion
Despite increasing clinical recognition of lipedema, mechanisms underlying this problematic disease remain unknown. To explore these mechanisms, we undertook multi-faceted analyses of whole lipedema tissue, ADSCs, and adipocytes, using omics platforms. Our integrated approach uncovered deranged metabolic and lipid profiles, and dysregulated signaling pathways driving key biological processes, such as adipogenesis, cell cycle, lipid metabolism, and cell proliferation and differentiation. The global molecular profiles we characterized in lipedema uniquely distinguish lipedema from normal fat and define it as a distinct disease tissue. Importantly, the findings of our multimodal study were coherent across distinct biological platforms and provide new insights that will both help to address the deficit in lipoedema animal models and provide vital clues into potential novel therapeutic approaches.
Lipids play important roles in physiological processes such as energy homeostasis, bioenergetics, apoptosis, signal transduction, and cell recognition. Defective lipid catabolic pathways are involved in the pathogenesis of several metabolic diseases [49]. Neutral lipids, such as triglycerides, are less likely to promote metabolic disorders compared to other lipids classes such as sphingolipids and GPLs [50, 51], which have been documented to be involved in several metabolic diseases [52]. An example of the distinct nature of lipedema adipose tissue is illustrated by our discovery of metabolic differences in lipedema relating to levels of fatty acids and their conjugates that included GPLs (more highly abundant in lipedema adipocytes than non-lipedema adipocytes) that are metabolites known to regulate cell-cycle events in cell growth, and to enhance cell size and proliferation [53, 54]; notably, GPL inhibition can reduce cell proliferation [55, 56]. Sphingolipids were the second most different class of lipid metabolites, in terms of metabolite levels, between lipedema and non-lipedema adipose tissues. Sphingolipids are among the lipid classes implicated in lipotoxicity, and can modulate signaling pathways involved in fibrosis, apoptosis, triglyceride synthesis, and glucose metabolism [57–59]. Recent studies have shown a role for sphingolipids in the development of metabolic disorders such as atherosclerosis, hepatic-steosis, cardiomyopathy, diabetes, and insulin resistance [60, 61]. Our finding that the adipocyte lipidomics profile is significantly altered in lipedema, particularly in relation to sphingolipid metabolites, suggests that a disrupted lipid profile in adipocytes could contribute to metabolic dysfunction and pathogenesis in lipedema.
The gene expression profiles defined in lipedema whole tissue and ADSCs highlighted potential molecular drivers of lipedema. We identified over 7820 significantly differentially expressed genes in RNA-seq data from lipedema adipose tissue and ADSCs (4391 in adipose tissue and 3429 in ADSCs). The top-ranking signaling pathways and GO-molecular functions identified related to cell cycle and mitotic spindle checkpoints. Detailed pathway analysis further implicated the involvement of genes from mitotic spindle checkpoint pathways. Validation of the eight most promising genes involved in mitotic spindle checkpoint regulation by qRT-PCR identified Bub1 as a potential key mediator of mitotic spindle checkpoint pathways in lipedema tissue and ADSCs. Of the candidates, Bub1 was chosen for further study due to the role it plays in proliferation in other diseases, as aberrant Bub1 expression has been shown to promote cell proliferation in several cancers [33, 43, 62], and higher Bub1 expression in lipedema tissue and ADSCs could disrupt G1/S checkpoint inhibition resulting in increased cell proliferation. Furthermore, cell-cycle analysis identified differences between the cell-cycle phases of ADSCs in lipedema (high proportion in S phase) and normal ADSCs (low proportion in S phase). The higher Bub1 expression in lipedema tissue and ADSCs may act to disrupt G1/S checkpoint inhibition resulting in increased cell proliferation. Mitosis also plays a role in balancing stem cells between the states of self-renewal and differentiation from progenitor to mature cells by regulating symmetric and asymmetric division [43, 63]; hence, Bub1 upregulation in lipedema may contribute to abrogation of stem cell homeostasis. Importantly, the Bub1 downstream target histone H2A, phosphorylation of which is considered important for cell proliferation, was more highly phosphorylated in lipedema ADSCs.
We therefore conducted further investigation of the role of Bub1, studying the effects of Bub1 inhibition on ADSC proliferation using 2OH-BNPP1 [64]. Excitingly, either 2OH-BNPP1 treatment or CRISPR/Cas9-mediated Bub1 gene depletion can dramatically reduced lipedema ADSC proliferation. Treatment of lipedema ADSCs with 2OH-BNPP1 also profoundly inhibited histone H2A phosphorylation, further supporting a key role for Bub1 in enhanced ADSC proliferation and pathological adipogenesis in lipedema.
Taken together, our data—derived from multi-platform Omics interrogations across whole adipose tissue, adipocytes, and ADSCs derived from lipedema and control unaffected patients—have provided valuable insights into the mechanisms underlying lipedema. Using gene expression analysis and cell-based bioassays, we identified Bub1 as a novel therapeutic target in lipedema for which a small molecule inhibitor has been successfully tested in pre-clinical cancer models [65]. Our data indicate that Bub1 overexpression promotes ADSC hyperproliferation in lipedema and may therefore play a key role in lipedema disease onset and progression.
The findings of this study present, for the first time, a useful platform for pinpointing diagnostic biomarkers that may be predictive of lipedema disease progression. They may also help identify potential therapeutic targets for combating this chronic, incurable, and under-appreciated disease.
Materials and methods
Collection of human tissue and isolation of ADSCs
Adipose tissues were obtained by excision from the lower limbs of 24 non-obese female patients (14 with stage II–III lipedema (clinical criteria per Wold et al. [66]) and 10 non-lipedema healthy controls) who consented for inclusion in this study. Protocols were approved by the Human Research Ethics Committee, St Vincent’s Hospital, Melbourne (HREC-A 067/16 and HREC/16/SVHM/38) in accordance with the Declaration of Helsinki. ADSCs were isolated from adipose tissue from lipedema and non-lipedema control patients, using methods described by Zuk et al. [67] and as described in detail in Supplementary Methods.
RNA-seq analyses
Adipose tissue and ADSC samples from LED and non-LED patients were used for global gene expression analyses by RNA-sequencing. An average of 78 million 100-base long reads from each adipose tissue and ADSC sample were mapped to the human reference genome (b37 decoy), and raw gene counts were obtained for each sample using STAR (v2.7.2c) as described in detail in Supplementary Methods. Downstream differential gene expression and gene-set analyses were then conducted in R (v3.3.0) using packages mainly including edgeR (v3.16.5) [68] and limma (v3.30.12) [69].
Lipidomics and metabolomics analyses
ADSCs were cultured on 6-well culture dishes (n = 4) at 40,000 cells/cm2 in differentiation medium for 14 days. After 14 days, differentiated cells (adipocytes) were used for lipdomics and metabolomics analyses as described in detail in Supplementary Methods. Resulting lipidomic and metabolic profiling comparisons and PCA were undertaken using LCMS, and resulting untargeted data analyzed using IDEOM software [70]. Refer to Supplementary Methods for additional information.
Time-lapse microscopy
Live imaging was performed over 72 h at 37 °C with 5% CO2 on an Operetta high-content screening system (PerkinElmer) using a 10× objective. Detail has been described in Supplementary Methods.
Colony forming unit (CFU) assay
The CFU assay was used to assess ADSC self-renewal capacity. Individual colonies of >50 cells were counted using a microscope, dissolved with 0.5% SDS and absorbance measured at 539 nm (POLARstar Optima plate reader (BMG Labtek)). Detail has been described in Supplementary Methods.
Cell-cycle assay
ADSC cell-cycle phase changes were monitored with FxCycle Cell Cycle Assay Kits Cat#F10348 (Invitrogen) as described in detail in Supplementary Methods. After 48–96 h incubation, cells were fixed with 4% PFA and stained with FxCycle Far Red stain (30 min). Samples were analyzed by flow cytometry (BD FACS ARIA Flow cytometer) using FlowJo software (Tree Star, Oregon, USA).
Protein analysis by immunoblotting
ADSCs were cultured overnight (density 5000–8000 cells/cm2), lysed in RIPA buffer (Thermo Fisher Scientific, USA) and total cellular protein quantified (Pierce BCA Protein Assay kit (Thermo Fisher Scientific, USA)). Membranes were blocked (Nupage Blocking buffer, cat#927–40000, Li-COR, USA) and incubated with primary antibodies (Supplementary Table 6) as described in detail in Supplementary Methods.
qPCR and gene expression analysis
Total RNA was extracted from ADSCs (RNeasy mini kit (Qiagen)), and 1 μg was transcribed to cDNA (gDNA Clear cDNA Synthesis Kit (Biorad)) and gene-specific primers (Supplementary Table 7) were used to perform PCR using the QuantStudio 6 (Applied Biosystem, Life Technologies). Detail has been described in Supplementary Methods.
Flow cytometry
ADSCs were trypsinized, resuspended, and washed with FACS wash buffer. Cells were stained using antibodies against CD44 (1:20), CD105 (1:20), CD73 (1:20), or CD90 (1:20), or appropriate isotype controls (all antibodies from BD Pharmingen, Supplementary Table 6) according to the protocol as described in detail in Supplementary Methods. FACS analysis was performed on BD FACS ARIA Flow cytometer and FlowJo software (Tree Star, Oregon, USA).
Cell proliferation assay
ADSCs were seeded in 96-well plates (3000 cells/well) with fresh culture media supplied 48 hourly. After 3 days in culture, cells were fixed (4% PFA), DAPI-nuclear stained and cell quantification performed using the Operetta high-content screening system (PerkinElmer, objectives ×10 magnification) as described in Supplementary Methods.
Adipogenic differentiation assay and lipid droplet quantification
ADSCs were cultured on 96-, 48-, or 6-well culture dishes at 40,000 cells/cm2 in differentiation medium for 14 days. After 14 days, differentiated cells were fixed with 4% PFA (1 h), washed (PBS), and stained for lipid droplets with Bodipy (2 μg/mL in 150 mM NaCl) [71], before quantification was performed (Operetta High-Content Screening System, PerkinElmer, ×10 objective) as described in detail in Supplementary Methods.
CRISPR/Cas9 to generate Bub1-knockdown ADSCs
We used lentiviral CRISPR/Cas9 particles (Invitrogen LentiArray CRISPR, Thermo Fisher Scientific, USA) to knock down Bub1 in ADSCs as described in detail in Supplementary Methods. Lentivector permanently expressing cas9 with a blasticidin-resistance gene, and lentivector expressing gRNA for the Bub1 gene with a puromycin resistance gene, were used to infect ADSCs. Bub1 gene knockdown was confirmed by qRT-PCR and western blotting.
Immunohistochemistry of human tissue
Human tissue samples were fixed in neutral-buffered formalin and processed to paraffin wax. Five-millimeter sections were cut and mounted on Polysine slides and allowed to dry. Opal multiplex staining was performed according to the manufacturer’s (PerkinElmer) instructions, with minor modifications as described in detail in Supplementary Methods.
Statistical analyses
Assays were performed in triplicate, each with ADSCs from at least three lipedema and non-lipedema patients. Data were reported as mean ± SEM. Statistical comparisons used unpaired two-tailed Student’s t test. Analyses were performed using GraphPad Prism 7.1 software. Comparison with p values < 0.05 were considered significantly different.
Supplementary information
Acknowledgements
We thank Wayne Morrison, Matt Ritchie, Xueyi Dong, Xiaoliang Han, Jason Palmer, Geraldine Mitchell, Anne Kong, Yiwen Gerand, Janna Taylor, and members of O’Brien Institute Department at St Vincent’s Institute of Medical Research for discussions and technical assistance.
Author contributions
MI designed the study, processed tissue samples, conducted all the experiments, analyzed data, interpreted results, and wrote the first draft of the manuscript. NB assisted in the flow cytometry experiments. SM, AMM, RL, and DM conducted RNA-seq data analysis. CN contributed in the imaging data analysis. DA and DJC assisted in lipidomics and metabolomics experiments. MGA provided feedback on the manuscript. RS and TK supervised the recruitment of study participants, supervised MI with the study design, and assisted in the final draft of the manuscript.
Funding
This work was supported by generous grants from the McMullin Family Trust, Stafford Fox Foundation, Lipoedema Australia, Wicking Foundation, and the O’Brien Foundation Hummingbirds.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Musarat Ishaq, Email: ishaqmusarat@gmail.com.
Tara Karnezis, Email: tkarnezis@svi.edu.au.
Supplementary information
The online version contains supplementary material available at 10.1038/s41366-021-01002-1.
References
- 1.Child AH, Gordon KD, Sharpe P, Brice G, Ostergaard P, Jeffery S, et al. Lipedema: an inherited condition. Am J Med Genet A. 2010;152A:970–6. doi: 10.1002/ajmg.a.33313. [DOI] [PubMed] [Google Scholar]
- 2.Buck DW, II, Herbst KL. Lipedema: a relatively common disease with extremely common misconceptions. Plast Reconstr Surg Glob Open. 2016;4:e1043. doi: 10.1097/GOX.0000000000001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paolacci S, Precone V, Acquaviva F, Chiurazzi P, Fulcheri E, Pinelli M, et al. Genetics of lipedema: new perspectives on genetic research and molecular diagnoses. Eur Rev Med Pharmacol Sci. 2019;23:5581–94. doi: 10.26355/eurrev_201907_18292. [DOI] [PubMed] [Google Scholar]
- 4.Tchernof A, Despres JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93:359–404. doi: 10.1152/physrev.00033.2011. [DOI] [PubMed] [Google Scholar]
- 5.Porter SA, Massaro JM, Hoffmann U, Vasan RS, O’Donnel CJ, Fox CS. Abdominal subcutaneous adipose tissue: a protective fat depot? Diabetes Care. 2009;32:1068–75. doi: 10.2337/dc08-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canning C, Bartholomew JR. Lipedema. Vasc Med. 2018;23:88–90. doi: 10.1177/1358863X17739698. [DOI] [PubMed] [Google Scholar]
- 7.Caruana M. Lipedema: a commonly misdiagnosed fat disorder. Plast Surg Nurs. 2018;38:149–52. doi: 10.1097/PSN.0000000000000245. [DOI] [PubMed] [Google Scholar]
- 8.Torre YS, Wadeea R, Rosas V, Herbst KL. Lipedema: friend and foe. Horm Mol Biol Clin Investig. 2018;33. [DOI] [PMC free article] [PubMed]
- 9.Al-Ghadban S, Diaz ZT, Singer HJ, Mert KB, Bunnell BA. Increase in leptin and PPAR-gamma gene expression in lipedema adipocytes differentiated in vitro from adipose-derived stem cells. Cells. 2020;9:430. [DOI] [PMC free article] [PubMed]
- 10.Al-Ghadban S, Pursell IA, Diaz ZT, Herbst KL, Bunnell BA. 3D spheroids derived from human lipedema ASCs demonstrated similar adipogenic differentiation potential and ECM remodeling to non-lipedema ASCs in vitro. Int J Mol Sci. 2020;21:8350. [DOI] [PMC free article] [PubMed]
- 11.Pereira de Godoy LM, Pereira de Godoy HJ, Pereira de Godoy Capeletto P, Guerreiro Godoy MF, Pereira de Godoy JM. Lipedema and the evolution to lymphedema with the progression of obesity. Cureus. 2020;12:e11854. doi: 10.7759/cureus.11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Priglinger E, Strohmeier K, Weigl M, Lindner C, Auer D, Gimona M, et al. SVF-derived extracellular vesicles carry characteristic miRNAs in lipedema. Sci Rep. 2020;10:7211. doi: 10.1038/s41598-020-64215-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryden M, Andersson DP, Bergstrom IB, Arner P. Adipose tissue and metabolic alterations: regional differences in fat cell size and number matter, but differently: a cross-sectional study. J Clin Endocrinol Metab. 2014;99:E1870–6. doi: 10.1210/jc.2014-1526. [DOI] [PubMed] [Google Scholar]
- 14.Hoffstedt J, Arner E, Wahrenberg H, Andersson DP, Qvisth V, Lofgren P, et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia. 2010;53:2496–503. doi: 10.1007/s00125-010-1889-3. [DOI] [PubMed] [Google Scholar]
- 15.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SM, Lun M, Wang M, Senyo SE, Guillermier C, Patwari P, et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014;20:1049–58. doi: 10.1016/j.cmet.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Ghadban S, Cromer W, Allen M, Ussery C, Badowski M, Harris D, et al. Dilated blood and lymphatic microvessels, angiogenesis, increased macrophages, and adipocyte hypertrophy in lipedema thigh skin and fat tissue. J Obes. 2019;2019:8747461. doi: 10.1155/2019/8747461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattar P, Bieback K. Comparing the immunomodulatory properties of bone marrow, adipose tissue, and birth-associated tissue mesenchymal stromal cells. Front Immunol. 2015;6:560. doi: 10.3389/fimmu.2015.00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourin P, Bunnell BA, Casteilla L, Dominici M, Katz AJ, March KL, et al. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: a joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT) Cytotherapy. 2013;15:641–8. doi: 10.1016/j.jcyt.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez AM, Elabd C, Delteil F, Astier J, Vernochet C, Saint-Marc P, et al. Adipocyte differentiation of multipotent cells established from human adipose tissue. Biochem Biophys Res Commun. 2004;315:255–63. doi: 10.1016/j.bbrc.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 21.Dicker A, Le Blanc K, Astrom G, van Harmelen V, Gotherstrom C, Blomqvist L, et al. Functional studies of mesenchymal stem cells derived from adult human adipose tissue. Exp Cell Res. 2005;308:283–90. doi: 10.1016/j.yexcr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 22.Nepali S, Park M, Lew H, Kim O. Comparative analysis of human adipose-derived mesenchymal stem cells from orbital and abdominal fat. Stem Cells Int. 2018;2018:3932615. doi: 10.1155/2018/3932615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perrini S, Laviola L, Cignarelli A, Melchiorre M, De Stefano F, Caccioppoli C, et al. Fat depot-related differences in gene expression, adiponectin secretion, and insulin action and signalling in human adipocytes differentiated in vitro from precursor stromal cells. Diabetologia. 2008;51:155–64. doi: 10.1007/s00125-007-0841-7. [DOI] [PubMed] [Google Scholar]
- 24.Fei W, Du X, Yang H. Seipin, adipogenesis and lipid droplets. Trends Endocrinol Metab. 2011;22:204–10. doi: 10.1016/j.tem.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Shukla L, Yuan Y, Shayan R, Greening DW, Karnezis T. Fat therapeutics: the clinical capacity of adipose-derived stem cells and exosomes for human disease and tissue regeneration. Front Pharmacol. 2020;11:158. doi: 10.3389/fphar.2020.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Leon H, Boue S, Szostak J, Peitsch MC, Hoeng J. Systems biology research into cardiovascular disease: contributions of lipidomics-based approaches to biomarker discovery. Curr Drug Discov Technol. 2015;12:129–54. doi: 10.2174/1570163812666150702123319. [DOI] [PubMed] [Google Scholar]
- 27.Lappas M, Mundra PA, Wong G, Huynh K, Jinks D, Georgiou HM, et al. The prediction of type 2 diabetes in women with previous gestational diabetes mellitus using lipidomics. Diabetologia. 2015;58:1436–42. doi: 10.1007/s00125-015-3587-7. [DOI] [PubMed] [Google Scholar]
- 28.Hamer M, Stamatakis E. Metabolically healthy obesity and risk of all-cause and cardiovascular disease mortality. J Clin Endocrinol Metab. 2012;97:2482–8. doi: 10.1210/jc.2011-3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berger JP. Role of PPARgamma, transcriptional cofactors, and adiponectin in the regulation of nutrient metabolism, adipogenesis and insulin action: view from the chair. Int J Obes (Lond) 2005;29(Suppl 1):S3–4. doi: 10.1038/sj.ijo.0802904. [DOI] [PubMed] [Google Scholar]
- 30.Lavrov AV, Smirnichina SA. [Nuclear heterogeneity and proliferative capacity of human adipose derived MSC-like cells] Tsitologiia. 2010;52:616–20. [PubMed] [Google Scholar]
- 31.Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, et al. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8:14728. doi: 10.1038/ncomms14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabsch H, Takeno S, Parsons WJ, Pomjanski N, Boecking A, Gabbert HE, et al. Overexpression of the mitotic checkpoint genes BUB1, BUBR1, and BUB3 in gastric cancer-association with tumour cell proliferation. J Pathol. 2003;200:16–22. doi: 10.1002/path.1324. [DOI] [PubMed] [Google Scholar]
- 33.Zhu LJ, Pan Y, Chen XY, Hou PF. BUB1 promotes proliferation of liver cancer cells by activating SMAD2 phosphorylation. Oncol Lett. 2020;19:3506–12. doi: 10.3892/ol.2020.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asghar A, Lajeunesse A, Dulla K, Combes G, Thebault P, Nigg EA, et al. Bub1 autophosphorylation feeds back to regulate kinetochore docking and promote localized substrate phosphorylation. Nat Commun. 2015;6:8364. doi: 10.1038/ncomms9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moustakas A. The mitotic checkpoint protein kinase BUB1 is an engine in the TGF-beta signaling apparatus. Sci Signal. 2015;8:fs1. doi: 10.1126/scisignal.aaa4636. [DOI] [PubMed] [Google Scholar]
- 36.Paganelli L, Caillaud MC, Quentin M, Damiani I, Govetto B, Lecomte P, et al. Three BUB1 and BUBR1/MAD3-related spindle assembly checkpoint proteins are required for accurate mitosis in Arabidopsis. New Phytol. 2015;205:202–15. doi: 10.1111/nph.13073. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Katsaros D, Shen Y, Fu Y, Canuto EM, Benedetto C, et al. Biological and clinical significance of MAD2L1 and BUB1, genes frequently appearing in expression signatures for breast cancer prognosis. PLoS One. 2015;10:e0136246. doi: 10.1371/journal.pone.0136246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myrie KA, Percy MJ, Azim JN, Neeley CK, Petty EM. Mutation and expression analysis of human BUB1 and BUB1B in aneuploid breast cancer cell lines. Cancer Lett. 2000;152:193–9. doi: 10.1016/s0304-3835(00)00340-2. [DOI] [PubMed] [Google Scholar]
- 39.Ouyang B, Knauf JA, Ain K, Nacev B, Fagin JA. Mechanisms of aneuploidy in thyroid cancer cell lines and tissues: evidence for mitotic checkpoint dysfunction without mutations in BUB1 and BUBR1. Clin Endocrinol (Oxf) 2002;56:341–50. doi: 10.1046/j.1365-2265.2002.01475.x. [DOI] [PubMed] [Google Scholar]
- 40.Grabsch HI, Askham JM, Morrison EE, Pomjanski N, Lickvers K, Parsons WJ, et al. Expression of BUB1 protein in gastric cancer correlates with the histological subtype, but not with DNA ploidy or microsatellite instability. J Pathol. 2004;202:208–14. doi: 10.1002/path.1499. [DOI] [PubMed] [Google Scholar]
- 41.Grigorova M, Staines JM, Ozdag H, Caldas C, Edwards PA. Possible causes of chromosome instability: comparison of chromosomal abnormalities in cancer cell lines with mutations in BRCA1, BRCA2, CHK2 and BUB1. Cytogenet Genome Res. 2004;104:333–40. doi: 10.1159/000077512. [DOI] [PubMed] [Google Scholar]
- 42.Breit C, Bange T, Petrovic A, Weir JR, Muller F, Vogt D, et al. Role of intrinsic and extrinsic factors in the regulation of the mitotic checkpoint kinase Bub1. PLoS One. 2015;10:e0144673. doi: 10.1371/journal.pone.0144673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han JY, Han YK, Park GY, Kim SD, Lee CG. Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Sci Rep. 2015;5:15993. doi: 10.1038/srep15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol. 2011;193:1049–64. doi: 10.1083/jcb.201012035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Z, Sun Y, Harley SE, Zou H, Yu H. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc Natl Acad Sci U S A. 2004;101:18012–7. doi: 10.1073/pnas.0408600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nyati S, Schinske-Sebolt K, Pitchiaya S, Chekhovskiy K, Chator A, Chaudhry N, et al. The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-beta signaling. Sci Signal. 2015;8:ra1. doi: 10.1126/scisignal.2005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brimacombe CA, Burke JE, Parsa JY, Catania S, O’Meara TR, Witchley JN, et al. A natural histone H2A variant lacking the Bub1 phosphorylation site and regulated depletion of centromeric histone CENP-A foster evolvability in Candida albicans. PLoS Biol. 2019;17:e3000331. doi: 10.1371/journal.pbio.3000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi Y, Kawashima SA. Bub1 kinase- and H2A phosphorylation-independent regulation of Shugoshin proteins under glucose-restricted conditions. Sci Rep. 2019;9:2826. doi: 10.1038/s41598-019-39479-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Chaurasia B, Summers SA. Ceramides – lipotoxic inducers of metabolic disorders. Trends Endocrinol Metab. 2015;26:538–50. doi: 10.1016/j.tem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 52.Lamari F, Mochel F, Sedel F, Saudubray JM. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J Inherit Metab Dis. 2013;36:411–25. doi: 10.1007/s10545-012-9509-7. [DOI] [PubMed] [Google Scholar]
- 53.Anel A, Naval J, Gonzalez B, Torres JM, Mishal Z, Uriel J, et al. Fatty acid metabolism in human lymphocytes. I. Time-course changes in fatty acid composition and membrane fluidity during blastic transformation of peripheral blood lymphocytes. Biochim Biophys Acta. 1990;1044:323–31. doi: 10.1016/0005-2760(90)90076-a. [DOI] [PubMed] [Google Scholar]
- 54.Calder PC, Yaqoob P, Harvey DJ, Watts A, Newsholme EA. Incorporation of fatty acids by concanavalin A-stimulated lymphocytes and the effect on fatty acid composition and membrane fluidity. Biochem J. 1994;300:509–18. doi: 10.1042/bj3000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scaglia N, Caviglia JM, Igal RA. High stearoyl-CoA desaturase protein and activity levels in simian virus 40 transformed-human lung fibroblasts. Biochim Biophys Acta. 2005;1687:141–51. doi: 10.1016/j.bbalip.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 56.Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer. 2005;41:1316–22. doi: 10.1016/j.ejca.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 57.Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017;38:649–65. doi: 10.1016/j.tips.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. 2019;365:386–92. doi: 10.1126/science.aav3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dekker MJ, Baker C, Naples M, Samsoondar J, Zhang R, Qiu W, et al. Inhibition of sphingolipid synthesis improves dyslipidemia in the diet-induced hamster model of insulin resistance: evidence for the role of sphingosine and sphinganine in hepatic VLDL-apoB100 overproduction. Atherosclerosis. 2013;228:98–109. doi: 10.1016/j.atherosclerosis.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 60.Glaros EN, Kim WS, Wu BJ, Suarna C, Quinn CM, Rye KA, et al. Inhibition of atherosclerosis by the serine palmitoyl transferase inhibitor myriocin is associated with reduced plasma glycosphingolipid concentration. Biochem Pharmacol. 2007;73:1340–6. doi: 10.1016/j.bcp.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 61.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 62.Gayyed MF, El-Maqsoud NM, Tawfiek ER, El Gelany SA, Rahman MF. A comprehensive analysis of CDC20 overexpression in common malignant tumors from multiple organs: its correlation with tumor grade and stage. Tumour Biol. 2016;37:749–62. doi: 10.1007/s13277-015-3808-1. [DOI] [PubMed] [Google Scholar]
- 63.White J, Dalton S. Cell cycle control of embryonic stem cells. Stem Cell Rev. 2005;1:131–8. doi: 10.1385/SCR:1:2:131. [DOI] [PubMed] [Google Scholar]
- 64.Jia L, Li B, Yu H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20 phosphorylation. Nat Commun. 2016;7:10818. doi: 10.1038/ncomms10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siemeister G, Mengel A, Fernandez-Montalvan AE, Bone W, Schroder J, Zitzmann-Kolbe S, et al. Inhibition of BUB1 kinase by BAY 1816032 sensitizes tumor cells toward taxanes, ATR, and PARP inhibitors in vitro and in vivo. Clin Cancer Res. 2019;25:1404–14. doi: 10.1158/1078-0432.CCR-18-0628. [DOI] [PubMed] [Google Scholar]
- 66.Wold LE, Hines EA, Jr., Allen EV. Lipedema of the legs; a syndrome characterized by fat legs and edema. Ann Intern Med. 1951;34:1243–50. doi: 10.7326/0003-4819-34-5-1243. [DOI] [PubMed] [Google Scholar]
- 67.Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–28. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 68.Lun AT, Chen Y, Smyth GK. It’s DE-licious: a recipe for differential expression analyses of RNA-seq experiments using quasi-likelihood methods in edgeR. Methods Mol Biol. 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
- 69.Su S, Law CW, Ah-Cann C, Asselin-Labat ML, Blewitt ME, Ritchie ME. Glimma: interactive graphics for gene expression analysis. Bioinformatics. 2017;33:2050–2. doi: 10.1093/bioinformatics/btx094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Creek DJ, Jankevics A, Burgess KE, Breitling R, Barrett MP. IDEOM: an Excel interface for analysis of LC-MS-based metabolomics data. Bioinformatics. 2012;28:1048–9. doi: 10.1093/bioinformatics/bts069. [DOI] [PubMed] [Google Scholar]
- 71.Gocze PM, Freeman DA. Factors underlying the variability of lipid droplet fluorescence in MA-10 Leydig tumor cells. Cytometry. 1994;17:151–8. doi: 10.1002/cyto.990170207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.