

Abstract
Neuroinflammation contributes to Alzheimer’s disease (AD) progression. Secondary inflammatory insults trigger delirium and can accelerate cognitive decline. Individual cellular contributors to this vulnerability require elucidation. Using APP/PS1 mice and AD brain we studied secondary inflammatory insults to investigate hypersensitive responses in microglia, astrocytes, neurons and human brain tissue. The NLRP3 inflammasome was assembled surrounding β-amyloid and microglia were primed, facilitating exaggerated IL-1β responses to subsequent LPS stimulation. Astrocytes were primed to produce exaggerated chemokine responses to intrahippocampal IL-1β. Systemic LPS triggered microglial IL-1β, astrocytic chemokines, IL-6 and acute cognitive dysfunction, while IL-1β disrupted hippocampal gamma rhythm, all selectively in APP/PS1 mice. Brains from AD patients with infection showed elevated IL-1β and IL-6. Amyloid leaves the brain vulnerable to secondary inflammation at microglial, astrocytic, neuronal and cognitive levels, and infection amplifies neuroinflammatory cytokine synthesis. Exacerbation of neuroinflammation to produce deleterious outcomes like delirium and accelerated disease progression merits careful investigation in humans.
Keywords: Microglia, astrocyte, primed, priming, APP/PS1, delirium, dementia, vulnerability, neuroinflammation, chemokine, cytokine, IL-1β, CCL2, memory, gamma, network dysfunction
Part 1. Narrative
1.1. Contextual background
While patients with Alzheimer’s disease (AD) show cognitive decline over many years, decline in individuals is variable and non-linear1,2. Acute illness or injury, episodes of delirium and chronic co-morbidities can influence the rate and uniformity of decline3 and critical illness leads to hippocampal atrophy4 and long-term cognitive impairment5. Delirium, which frequently occurs during acute illness and correlates with baseline MMSE 6, is a significant predictor of these long-term outcomes. Delirium accelerates cognitive decline in AD patients7 and in unselected populations8 and increases risk for subsequent dementia 8-fold9. Systemic inflammation drives delirium in cases of acute trauma, surgery and infection and, even in the absence of delirium, systemic inflammation and elevated serum TNF-α are associated with increased rate of cognitive decline in AD patients10. Therefore, systemic inflammation can trigger delirium and significantly impact dementia trajectory, but the mechanisms by which it drives deleterious outcomes in patients with underlying dementia remains poorly understood. Addressing cellular levels at which the amyloid-laden brain shows disproportionate responses to secondary inflammatory insults is the central scientific question in the current study.
The existing neuroinflammatory state of the brain appears to influence subsequent responses to secondary inflammation.11 Microglia surrounding amyloid plaques in AD become activated and these cells can produce IL-1β, which may contribute to neuronal degeneration.12 More recently, the NLRP3 inflammasome, which cleaves immature pro-IL-1 to allow the release of mature IL-1β, has been shown to mediate key aspects of neuronal and cognitive dysfunction in the APP/PS1 model of AD.13 Notwithstanding these descriptions, IL-1β production is relatively muted in AD and in associated animal models. However IL-1β can be acutely induced in the brain following peripheral bacterial or viral infections14 and microglia have been shown to be ‘primed’ by evolving brain pathology15, facilitating exaggerated IL-1β production upon exposure to acute systemic inflammation induced by bacterial LPS.16 This is a potentially important mechanism for clinically relevant brain sequelae of acute systemic inflammation since it has been shown that microglia and IL-1β contribute to new Tau pathology, acute cognitive deficits and new brain injury after acute systemic inflammation in models of neurodegeneration, delirium and post-operative cognitive dysfunction.17–21 These brain responses to a single episode of acute systemic inflammation are distinct from studies of repeated dosing with LPS 22–25 which, cumulatively, affect amyloidosis and neuroinflammation (see review26). Although those prior studies sought to influence neuroinflammation from outside the brain, with largely detrimental consequences, chronic dosing regimens address fundamentally different questions to those posed by the interrogation of deleterious effects of a single acute systemic inflammatory episode on the vulnerable brain, leading to delirium and acute brain injury. The current study also does not seek to address the hypothesis that amyloidosis is insufficient for disease and that a hypothetical ‘first hit’ or ‘second hit’ infectious insult 27–29 is necessary for disease expression. The key motivation here is that, while several human and mouse model studies have contributed to the general idea that systemic inflammation contributes to acute exacerbations of cognitive function and worsening of AD, there are significant gaps in our knowledge about the early neuroinflammatory events in interactions of systemic inflammation with the AD brain during acute secondary inflammation.
There is, as discussed above, evidence to implicate microglia and IL-1β in these exacerbations but the direct consequences of IL-1β expression for other cell populations in the APP/PS1 brain and in AD brains are not clear. Both astrocytes and some neuronal populations can respond directly to IL-1β but whether these two populations show disproportionate responses to IL-1β in the amyloid-laden brain is largely unstudied.
Astrocytes are known to be activated in proximity to amyloid plaques, showing hypertrophy and up-regulation of GFAP.30 Astrocytes become ‘primed’ by neurodegeneration in prion disease, such that they produce exaggerated chemokine responses to subsequent acute IL-1β stimulation31 and astrocyte chemokine transcription was heightened in the aged brain upon LPS-induced sepsis.32 AD-like pathology may leave astrocytes hypersensitive to secondary inflammatory challenge and this represents a significant gap in our knowledge, with potential implications for delirium and exacerbation of dementia upon acute systemic inflammation.
In hippocampal neuronal networks of rodents and non-human primates, gamma oscillations support dynamic cognitive function, decision making and coding of novel stimuli.33–35 Prior studies in APP/PS1 slices revealed a mixture of persistent gamma and intermittent burst discharges, indicating a state of network hyperexcitability36 and suggesting mechanisms compensating for amyloid-related changes in function. IL-1β has been implicated in reduced power of hippocampal gamma oscillations in a multiple sclerosis model 37 and pyramidal neurons are hypersensitive to IL-1β in a chronic hippocampal neurodegeneration model.19 Therefore, the possibility that acutely elevated IL-1β might disrupt gamma rhythm, selectively in the APP/PS1 brain, could illustrate network vulnerability and might represent an important link between acute inflammatory challenge and acute alterations in cognitive function.
Though episodes of systemic inflammation induce delirium and exacerbate underlying dementia, we neither understand the cellular and molecular changes that underpin delirium nor understand how prior amyloidosis influences the wave of neuroinflammatory consequences that follows acute systemic inflammation. This represents a significant unmet clinical need because there are limited evidence-based strategies for neurologists or geriatric psychiatrists to actually manage dementia patients when they become acutely ill and/or delirious. This is largely because of the significant gaps in our knowledge about what happens to microglia, astrocytes and neurons during the period of acute inflammatory activation occurring during secondary inflammation. Here we hypothesised that microglia in the APP/PS1 brain would produce exaggerated levels of IL-1β upon secondary inflammatory challenge and, in turn, that both astrocytes and neuronal networks would respond in an exaggerated manner to locally applied, or locally produced, IL-1β. These questions were approached through intra-cerebral and intra-peritoneal challenges of APP/PS1 and wild-type animals with bacterial LPS or IL-1β and examination of appropriate cellular readouts in microglia, astrocytes and neuronal networks, as well as examination of acute cognitive dysfunction under these challenges. AD cases who died with infection versus those who did not were also examined to assess whether central cytokine tenets of that neuroinflammatory exacerbation hypothesis were preserved in humans.
1.2. Study conclusions and implications
The data herein indicate that both microglia and astrocytes are primed in the APP/PS1 model. Microglia show exaggerated IL-1β production upon LPS challenge and astrocytes show an exaggerated chemokine and Il6 response to acutely elevated IL-1β, suggesting an amplification loop that drives exaggerated inflammatory responses to acute stimulation in the amyloid-laden brain (Figure 1). In addition, IL-1β was sufficient to disrupt gamma rhythm selectively in brain slices from APP/PS1 mice and these mice were also more vulnerable to acute cognitive dysfunction triggered by bacterial LPS. Analysis of human AD brains showed that both IL-1β and IL-6 were elevated in AD patients who died with infection with respect to those without infection and that these two cytokines were correlated, supporting their sequential induction, as shown in the mouse experiments. Thus, vulnerability of the diseased brain to exacerbation of neuroinflammation upon acute systemic inflammation occurs in mice and in patients and this is mediated by multiple cellular populations.
Figure 1. Acute inflammatory events occurring in those with evolving amyloid pathology have disproportionate effects on neuroinflammation and cognitive and neurophysiological function compared to those in normal individuals.
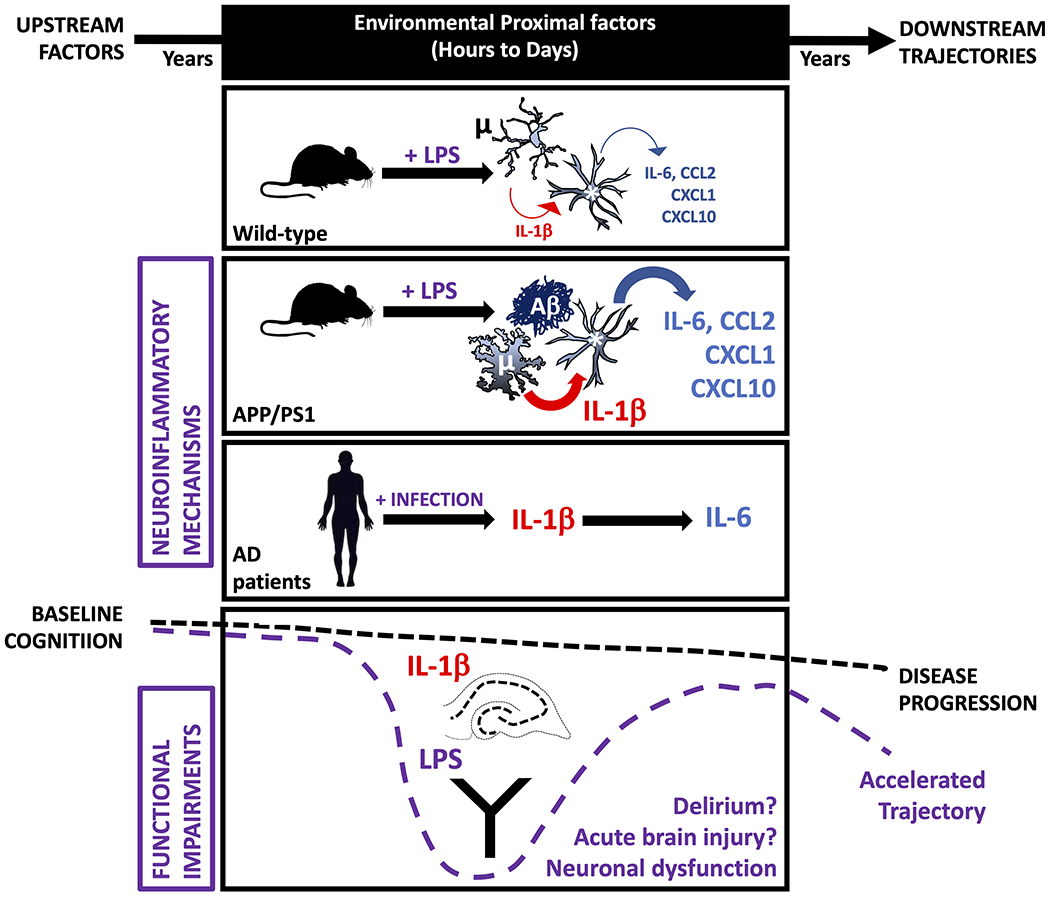
When APP/PS1 double transgenic mice and age-matched controls are exposed to equivalent acute LPS challenge, microglia (μ) surrounding amyloid plaques (Aβ) in APP/PS1 mice show exaggerated IL-1β responses, whether the LPS challenges were intracerebral (i.c.) or intraperitoneal (i.p.). In turn, astrocytes (*) from the APP/PS1 brain show exaggerated chemokine and IL-6 responses when exposed to acute IL-1β or LPS challenge. Acute peripheral LPS challenge was sufficient to produce acute cognitive impairment in a Y-maze task of cognitive flexibility and directly applied IL-1β was sufficient to disrupt gamma rhythm in ex vivo cortical-hippocampal networks. Both of these functional impairments occur selectively in APP/PS1 mice. Systemic infection also exacerbated brain inflammation in human Alzheimer’s disease (AD) cases: in patients who died with acute systemic infection, brain levels of IL-1β and IL-6 were higher than in those who did not experience infection and the levels of these 2 cytokines were directly correlated. Therefore the amyloid-laden brain is ‘primed’ at multiple cellular levels, causing heightened vulnerability to acute inflammatory events. Placing this within the context of the slowly evolving progression of Alzheimer’s Disease, one can propose that these cellular and molecular events, occurring within the ‘black box’ of proximal factors, are contributing to episodes of delirium and to the accelerated cognitive trajectory that has been observed in patients who experience delirium before or during their dementia.7,8
The study results emphasise that in brains with substantial amyloid deposition microglia, astrocytes and neuronal networks adopt new properties that leave them vulnerable to producing hypersensitive responses to subsequent secondary inflammation events. Upon secondary inflammation microglia produce exaggerated IL-1β and both astrocytes and neuronal networks show hypersensitive responses to acutely elevated IL-1β.
The microglia of APP/PS1 mice increased in number, were altered in morphology, showed elevation of microglial priming and recently described DAM phenotype transcripts (Clec7a, Itgax, Tyrobp, Trem2)15,38 and suppression of the homeostatic gene Sall1.39 Functionally, microglia proximal to Aβ produced exaggerated IL-1β responses to acute stimulation with either LPS or IL-1β. Figure 2 Elegant single cell RNAseq studies show microglial heterogeneity within the AD transgenic brain,38,40 but the current studies, using classical immunohistochemistry are better able to show the precise spatial resolution of acute change in microglial phenotype. These microglia were ‘primed’ before the secondary challenge was applied and already showed evidence of inflammasome assembly, as has been previously described in APP transgenics and in AD patients.13 However, in the absence of secondary inflammation Il1b mRNA was limited and there was no visible nuclear localisation of NFκB p65 in plaque-associated microglia. There are prior reports of IL-1β expression in AD tissue and in animal models12,37 and we do not dispute those findings. However, our data support the idea that the inflammasome is already assembled in APP/PS1 brain (classically ‘signal II’), but Il1b transcription is restrained, so there is a weak supply of pro-IL-1β to cleave. However, later ‘pulses’ of pro-IL-1β arising from secondary inflammation (induced by LPS or IL-1β; Figure 3) provide much higher levels of IL-1β for processing. This inflammasome assembly, but with limited pro-IL-1β throughput, may represent a significant dimension of the primed microglial phenotype.
Figure 2. Microglial activation in APP/PS1.
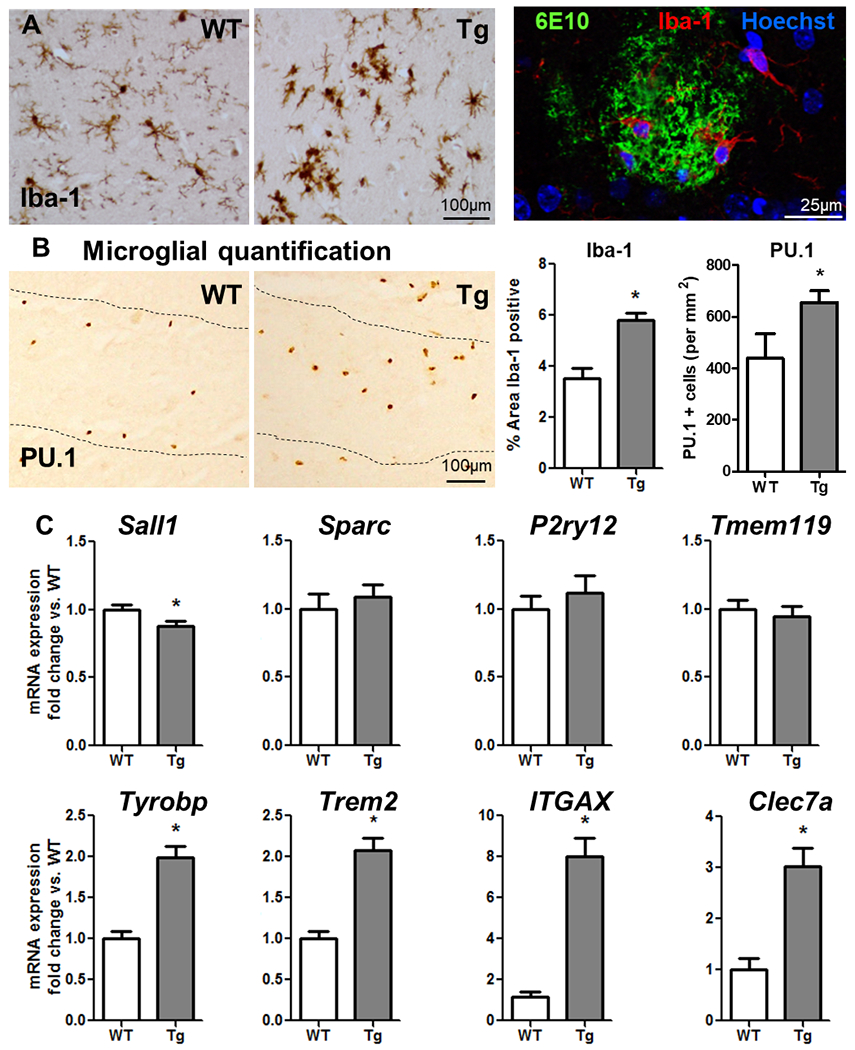
(A) Comparison of microglial activation in WT and Tg (APP/PS1, 19±3 months) by Iba-1 labelling, light microscopy and confocal image of Tg brain tissue showing (Aβ plaque labelled with 6E10 (green, 488nm) surrounded by Iba-1-positive microglia (red, 594nm). (B) Comparison of microglial numbers in WT and Tg (APP/PS1, 19±3 months) by PU.1 labelling, light microscopy. Microglial quantification in WT versus Tg and represented as the percentage of Iba-1-positive area and the number of PU.1 positive cells. (C) Hippocampal mRNA for specific microglial markers, expressed as fold change in Tg with respect to WT (age 19±3months). Mean±SEM (WT n=14; Tg n=19). t-test * vs. WT (p < 0.05).
Figure 3. Microglial priming in APP/PS1 mice.
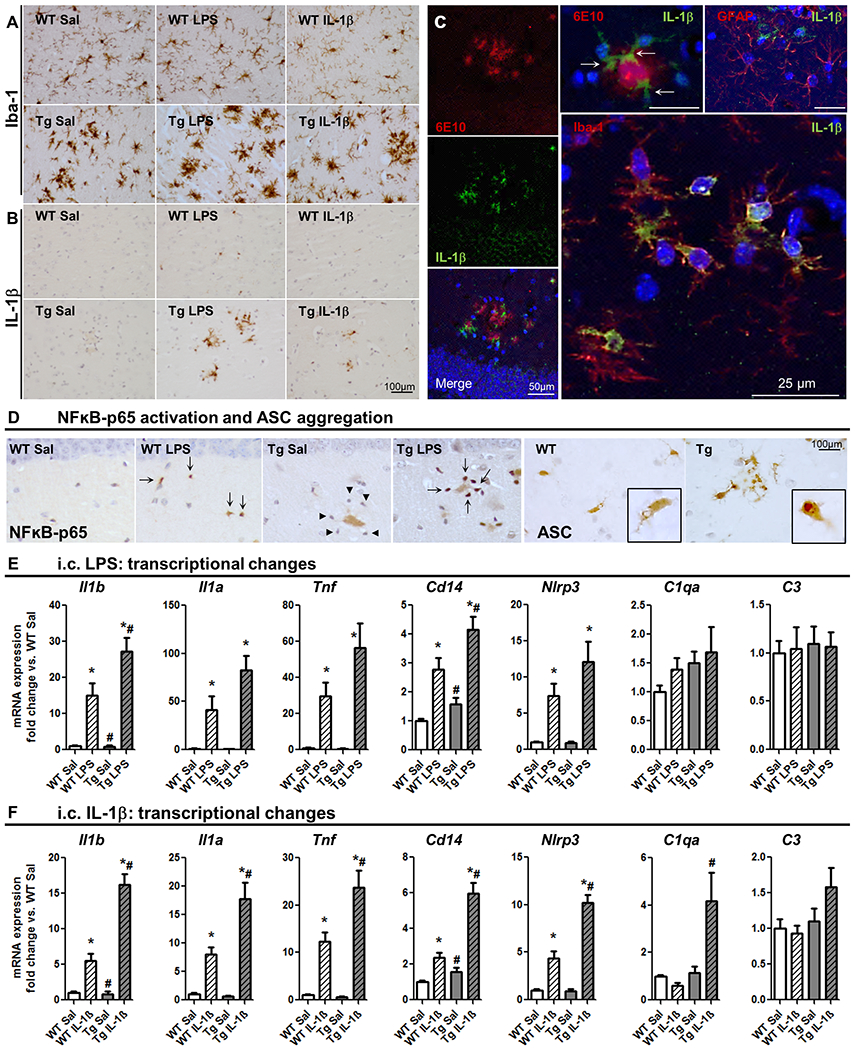
Acute challenges (LPS or IL-1β i.c., 2h) superimposed on primed microglia in APP/PS1 (19±3 months). (A) Iba-1 labelling in WT and Tg, treated with saline, 1 μg LPS or 10 ng IL-1β. (B) IL-1 β labelling in the same animals. (C, left stack) Confocal micrographs from Tg+IL1β showing IL-1β -positive cells (green, 488nm) around Aβ plaque, labelled with 6E10 (red, 630nm). Larger panel shows IL-1β positive cells (green, 488nm) co-localised with Iba-1-positive cells (red, 594nm). Upper right panel shows IL-1β positive cells (green, 488nm) that do not overlap with red GFAP-positive cells (594nm). (D) NFkB-p65 labelling (left panels) showing lack of activation in Tg+Sal nuclei (arrowheads) and activation in Tg+LPS (arrows). ASC aggregation (right panels) showing speck formation in Tg but not WT animals (zoom in the insets). (E) Hippocampal mRNA for specific microglial markers after i.c. LPS (fold change with respect to WT; 2h). (F) Hippocampal mRNA for selective microglial markers, after IL-1β i.c. (fold change; 2h). Mean±SEM (n=8-10). Kruskal Wallis Test followed by Mann-Whitney test. * effect of treatment; # effect of genotype (p< 0.05).
We also demonstrate, for the first time in any AD transgenic mouse model, that astrocytes become primed to produce exaggerated chemokine synthesis upon secondary inflammatory stimulation. Although both microglia and astrocytes can synthesise chemokines, astrocytes were the stronger producer of CCL2, CXCL1 and CXCL10 in response to IL-1β both in vitro (supplemental data) and in vivo (Figures 4, 5). These chemokines were not detectable in reactive astrocytes proximal to the plaques at baseline, but local application of IL-1β triggered clear chemokine expression in plaque-associated astrocytes. Exaggerated induction of these chemokines and the cytokine Il6 also occurred after systemic challenge with LPS, indicating that these hypersensitive responses can also be triggered from outside the brain. This phenotypic switching of astrocytes proximal to Aβ plaques is consistent with the idea of multiple and dynamic astrocyte phenotypes42,43 contrary to recently described astrocyte polarisation into just two phenotypes: A1 and A2.44,45 Data from other labs suggest that astrocytes from the APP/PS1 model do not conform closely to either A1 or A2 phenotypes30,46 and here we demonstrate that astrocytes proximal to Aβ plaques can be readily switched to adopt further phenotypes upon acute secondary inflammatory insults. Exaggerated responses to IL-1β included genes across putative A1 (Gbp2), A2 (Ptx3, Tgm1, Clcf1) and pan-reactive (Gfap, Cxcl10, S1pr3) phenotypes. This phenotypic switching replicates the recent description of primed astrocytes in the ME7 prion-diseased brain31 perhaps indicating that such priming of astrocytes may be a generic feature of astrocytes exposed to prior neurodegenerative stimuli. In our studies in prion disease 31, in APP/PS1 mice (this study), in aged mice32 and in earlier in vitro studies47 increased chemokine output was a key measure of exaggerated astrocyte responses. Elevated chemokine synthesis had consequences for neutrophil, monocyte and T-cell infiltration in the ME7 model31 and such leukocyte infiltration to the AD brain could have significant consequences for disease given reports that T cells48, monocytes49 and even neutrophils50 may influence AD progression. Consequences of acute episodes of heightened chemokine induction and immune cell infiltration, for the AD brain require further study.
Figure 4. Astrocytosis and differential expression of gene in isolated astrocytes and microglia in APP/PS1 and WT mice.
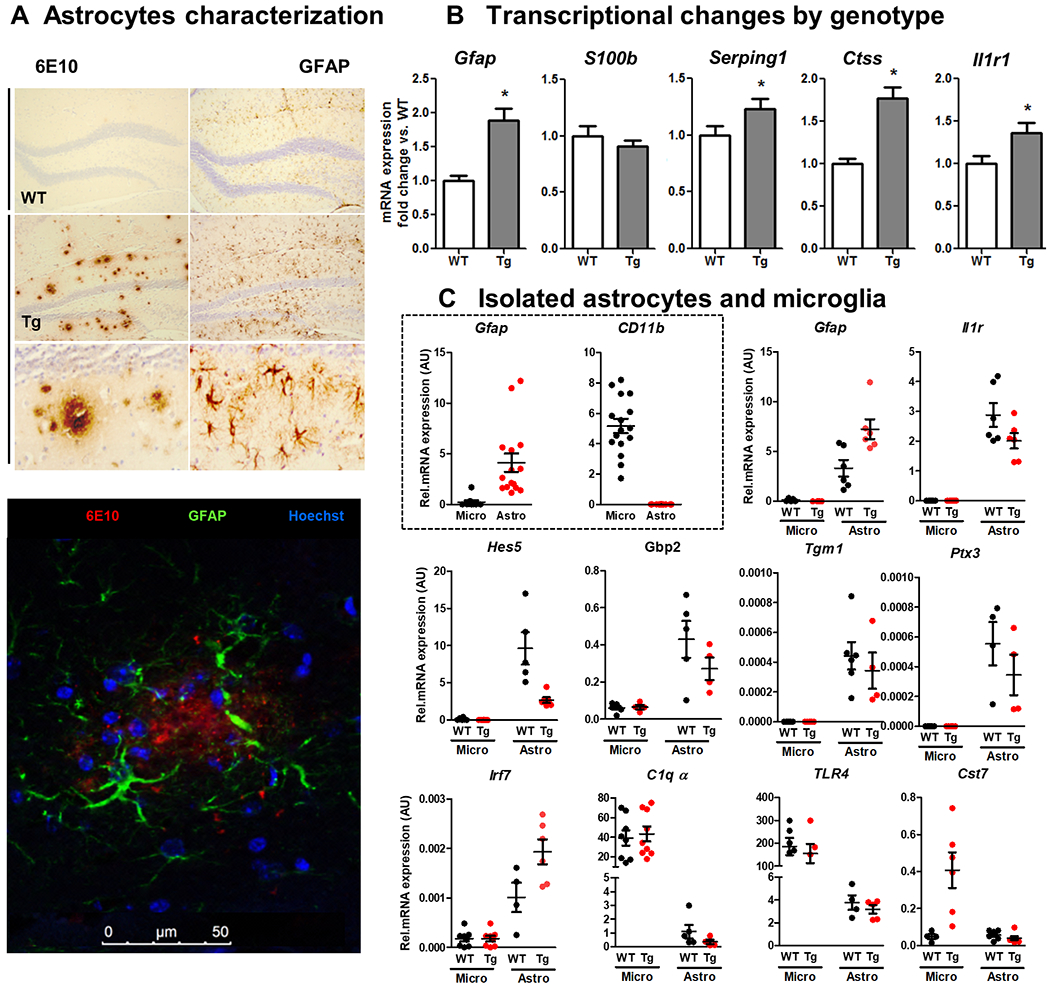
(A) Light microscopy labelling of 6E10 (left) and GFAP (right) of WT (first line) and Tg (second and third line). On the bottom, confocal image of Tg brain tissue labelled with 6E10 in red (630nm), green GFAP-positive astrocytes (488nm) surrounding the plaque. (B) B0 Transcriptional changes induced by genotype (WT vs. Tg) in hippocampal bulk mRNA. Aged 19±3 months. Mean±SEM (n=8-10) * vs. WT, t-test (p<0.05). (C) Transcriptional changes in isolated astrocytes and microglia from the hippocampus of WT and Tg mice. The first two graphs represent the purity of the isolated populations assessed by Gfap (for astrocytes) and CD11b (for microglia). The following panel are astrocytic-specific genes and the bottom row shows mostly microglia-specific genes (excepting Irf7, which is astrocytic and elevated in Tg).
Figure 5. Astrocytes are primed to show exaggerated chemokine production in APP/PS1.
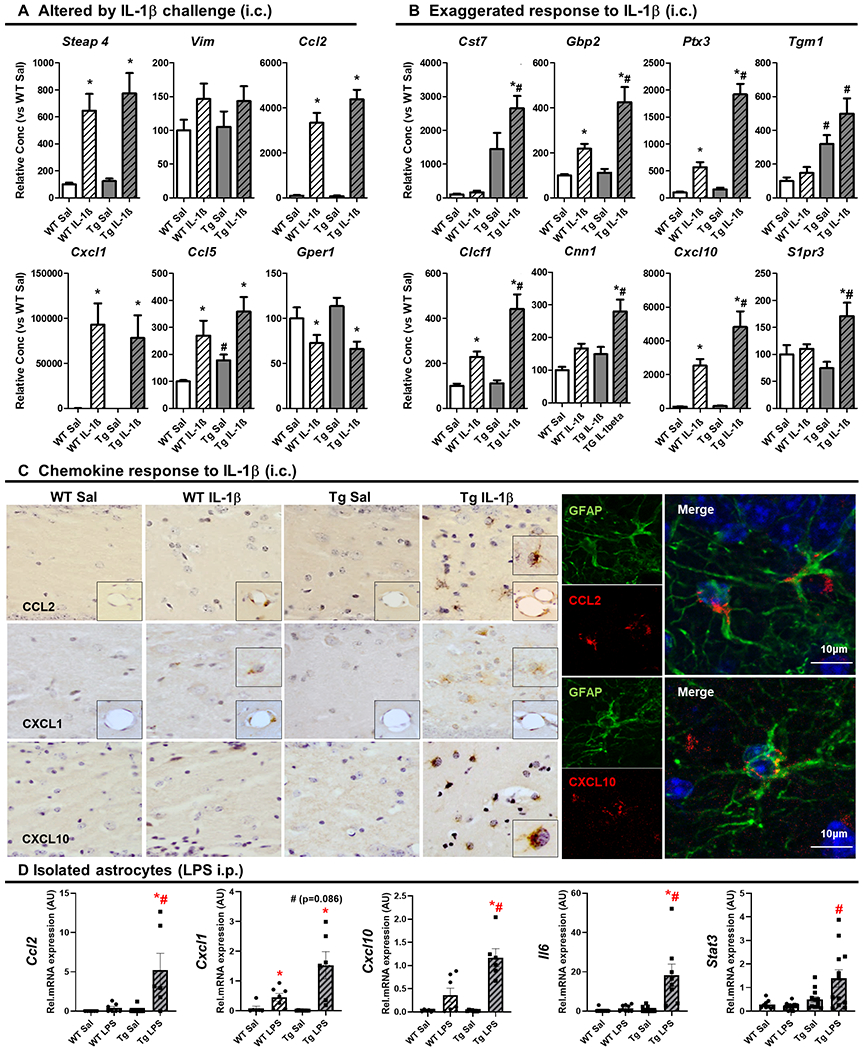
(A) Transcriptional changes induced by intracerebral (i.c.) IL-1β challenge (10ng) in hippocampal mRNA levels of selected astrocyte-associated genes, 2 h after challenge, expressed as fold change with respect to WT+Sal. Aged 19±3months. Mean ±SEM (n=8-10). (B) Exaggerated response to IL-1β challenge (10ng, i.c.) in hippocampal mRNA levels of selected astrocyte-associated genes, 2 h after challenge, expressed as fold change with respect to WT+Sal. Aged 19±3months. Mean ±SEM (n=8-10). (C) Chemokine response to IL-1β challenge (10ng, i.c.) Light microscopy labelling of CCL2, CXCL1 and CXCL10 in WT and Tg mice, 2 h after i.c. challenge with IL-1β (10 ng) or saline. Right panel shows the confocal imaging of Tg+IL-1β group for CCL2 (red; 594nm) and GFAP (green; 488nm) on the left and CXCL10 (red; 594nm) and GFAP (green; 488nm) on the right. (D) Transcriptional changes in isolated astrocytes of the selected chemokines together with Il6 and Stat3, Ccl2, Cxcl1 and Cxcl10, using a systemic inflammation model (3 h after LPS (250μg/kg, intraperitoneal, i.p.)). Aged 14±2months. Mean ±SEM (n=6-13). Kruskal Wallis Test followed by Mann-Whitney test, * effect of treatment; # effect of genotype (p < 0.05).
Thus, features of microglia and astrocytes, developing during disease, may place the AD brain at risk for more severe neuroinflammatory responses when systemic inflammatory events such as infection, surgery or injury occur in these populations and trigger these glial populations to adopt new phenotypic states.
The data discussed so far all come from the APP/PS1 mouse model and it is important to stress this significant caveat. While the APP/PS1 model does show considerable amyloidosis it lacks Tau pathology or significant neurodegeneration and these shortcomings may limit the generalisability of the current data. Likewise differences between immune responses in humans and mice raise further questions about whether these data will translate to humans. However we discuss a number of observations that mitigate the concerns noted above, detail some confirmatory human data collected in the current study and in the literature, and suggest a roadmap for validating, in humans, the phenomena described here.
Despite the limitations of the APP/PS1 model, it does provide robust amyloidosis. We used relatively old animals to interrogate priming of glial populations since amyloid transgenic mice are thought to better represent the mild cognitive impairment stage of disease51 and we reasoned that using older animals may therefore maximise their relevance to human disease and would be more appropriate to our aim: not to determine ‘first events’ in development of disease, but to elucidate the development of vulnerability to secondary stressors as disease progresses. Priming of both astrocytes and microglia by their proximity to human amyloid (formed from mutated human forms of APP and PSEN1), even in the absence of Tau, supports the idea that these may occur in the human AD brain, even though Tau and other neurodegenerative pathology may lead to additional glial phenotypes. There are also many differences between immune responses in humans and mice52 and significant differences between microglial profiles in AD57,103 and mouse models thereof38. However Habib et al. 104 describe activated astrocytes in the 5xFAD model of AD that overlap with aged astrocyte phenotypes in both mice and humans and Grubman et al.105 show enrichment, in human AD, of several transcripts involved in the regulation of cytokine production and in the response to cytokine stimulation. Those findings support the idea that astrocytes in human AD might indeed show hyper-responsiveness to cytokine stimulation. Moreover, despite their differences, human and mouse immune responses still show more convergence than divergence53: expression patterns of most orthologous inflammatory genes are conserved among humans and mice54 and many are shared between APP/PS1 mice and humans with AD.55 In particular, pathways of IL-1 expression, maturation and action are well conserved and are demonstrated to be up-regulated in both AD patients and mouse models.13 It is obviously essential to now address the extent to which these glial priming and phenotype switching phenomena, replicated across multiple mouse models,15 occur in AD brains.
Given the limitations of the APP/PS1 model we did address, in AD cases, some basic tenets of the neuroinflammatory ‘priming’ demonstrated in our mouse studies. AD patients who die with infection show altered microglial phenotype with respect to those without infection56 and here, using brain tissue homogenates from those same patients, we demonstrated elevated IL-1β in brains from AD patients with infections compared to those without infection (Figure 7), as predicted by the mouse data. We also show that IL-6 and IL-1β levels are correlated, but only in patients who died with infection, analogous to our observations, in mice, of secondary inflammation-induced IL-1β in microglia and IL-6 pathway induction in astrocytes. Though limited in scope, these initial human AD studies with and without infection tentatively support the mouse data and suggest that these glial priming phenomena could be relevant in AD.
Figure 7. IL-6 and IL-1β levels in brain homogenates from AD patients.
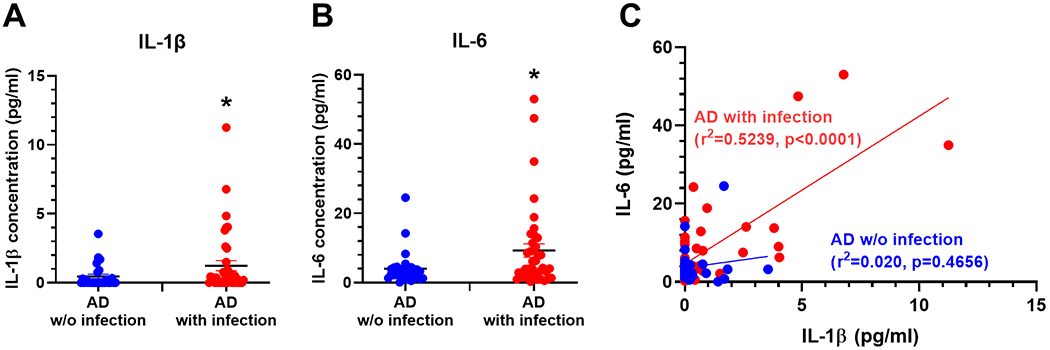
AD patients that concomitantly presented with infection at time of death (red dots, n=39) show significantly higher levels of IL-1β (A) and IL-6 (B) than AD patients without an infection (blue dots, n=28). Mann-Whitney test, one-tailed (a priori prediction of elevated IL-1β). Mean±SEM (AD without infection n=28; AD with infection n=39; * denotes p<0.05 by Mann-Whitney test). (C) Linear regression plot of IL-6 levels vs. IL-1β levels with separate equations of lines for infection and uninfected cases and Pearson correlations. Shows positive and significant correlation between IL-6 and IL-1β levels in AD patients with infection (y=3.780x+4.576; r2=0.5239, p<0.0001) but not in patients without infection (y=0.8298x+3.59; r2=0.020, p=0.4656).
The roadmap to validating these phenomena in humans requires localising these mediators to microglia and astrocytes in human tissue, upon secondary inflammatory insults. Further post-mortem studies should help to clarify whether these cell populations express heightened levels of specific mediators when acute inflammation is present at the time of death, while single cell RNAseq approaches offer opportunities to localise transcriptional signatures to specific cell types57. These approaches have not yet been used to study the impact of acute systemic inflammation on glial phenotypes on a background of AD. Moreover, those approaches are only applicable on post-mortem tissues. Identifying biomarkers that reliably inform on particular microglial and astrocyte phenotypes in living patients is more challenging. PET imaging of neuroinflammation can be achieved using 18kDa translocator protein (TSPO) imaging, and has been done post-LPS in humans58 but this has uncertain capacity to distinguish between microglial phenotypes or indeed even to distinguish between microglia and astrocytes.59 Better non-invasive markers are clearly required but those distinguishing different microglial and astrocyte phenotypes are beginning to emerge60,61. Among less invasive approaches, cerebrospinal fluid (CSF) offers an opportunity to sample brain levels of cytokines and other inflammatory markers that might inform on local glial phenotypes. To address inflammatory priming in AD, ideally one would be able to sample, during acute inflammatory episodes, the CSF of patients with and without established dementia. These types of studies are beginning to emerge from hip-fracture cohorts, facilitated by patients receiving spinal anaesthesia before hip replacement surgery. Although the cellular basis is far from clear at this point, there is evidence that IL-6, IL-8, CCL2 62–64 and soluble TREM2106 are increased in the CSF of hip-fracture patients suggesting acute changes in cytokine, chemokine and microglial markers in the brain upon acute systemic inflammatory insults.
Though many of our findings use intracerebral challenges (in order to address specific hypotheses about glial hypersensitivity), LPS gains access to the brain parenchyma to a very limited extent65–67 so it is important that we have also shown that both primed astrocytes and microglia can switch phenotype upon systemic inflammation. Brain effects of systemic infection and sepsis often clinically manifest as septic encephalopathy or delirium68,69 but pathophysiological understanding of inflammation-induced delirium remains limited. The demonstration here that moderate dose LPS produces acute cognitive deficits, selectively in APP/PS1 animals resonates with recent mouse data revealing acute attentional deficits following surgery (tibial fracture)70. Even after LPS, animals retained a well-trained reference memory but APP/PS1 animals treated with LPS, were less able to shift attention to the new exit when it’s location changed and abandoning their prior strategy was required (despite sickness behaviour responses being equivalent). This delay to shift attention to the new location is typical of human delirium, in which patients show impairments in cognitive tasks involving attention and processing of novel, trial specific, information despite preservation of previously acquired long-term memories.71 CSF levels of IL-1β62 and IL-672 are associated with delirium in trauma/surgery patients and here we show acute IL-1β and IL-6 synthesis in APP/PS1 mice after LPS, which is mirrored in the brains of the AD cases with infection. We propose that the acute cytokine changes described here in mice and humans are relevant to human neuro-psychiatric outcomes of acute systemic inflammation in patients with underlying dementia.
Pursuing ways in which these acute elevations of cytokines might contribute to neuropsychiatric changes, we show that acute application of IL-1β to hippocampal brain slices also revealed a selective vulnerability, triggering a disruption of gamma network activity in CA3 of slices from APP/PS1 mice that does not occur in WT animals (Figure 6). Although one cannot conclude that this neurophysiological disruption is that which underpins the acute cognitive deficits, disruption of gamma rhythm does interfere with dynamic cognitive functions like working memory and executive function.33,34 How the selectivity for disruption in APP/PS1 slices arises requires further work. Gamma oscillations, in vitro, depend upon AMPA receptor-mediated excitatory drive onto parvalbumin-containing GABA interneurons and the recruitment of pyramidal neurons via GABAA receptor-mediated inhibition.73–76 Prior studies in APP/PS1 slices reveal a mixture of persistent gamma and intermittent burst discharges, indicating a state of network hyperexcitability36 and suggesting compensatory remodelling of circuits affected by Aβ. A remodelled form of gamma activity that is particularly sensitive to disruption by IL-1β, might include changes in GABAA77,78 and AMPA79 receptor function since both can be altered by IL-1β. Increased sensitivity may also arise via increased neuronal expression of IL-1 receptors. We found elevated hippocampal mRNA for IL-1R1 in APP/PS1 mice, which was not explained by astrocytic or microglial expression. IL-1-induced changes in gamma require further investigation and may help clarify how secondary inflammation acutely impairs network and cognitive function, putatively informing our understanding of delirium during dementia.
Figure 6. Systemic LPS, IL-1β, acute cognitive dysfunction & gamma rhythm disruption in APP/PS1 mice.
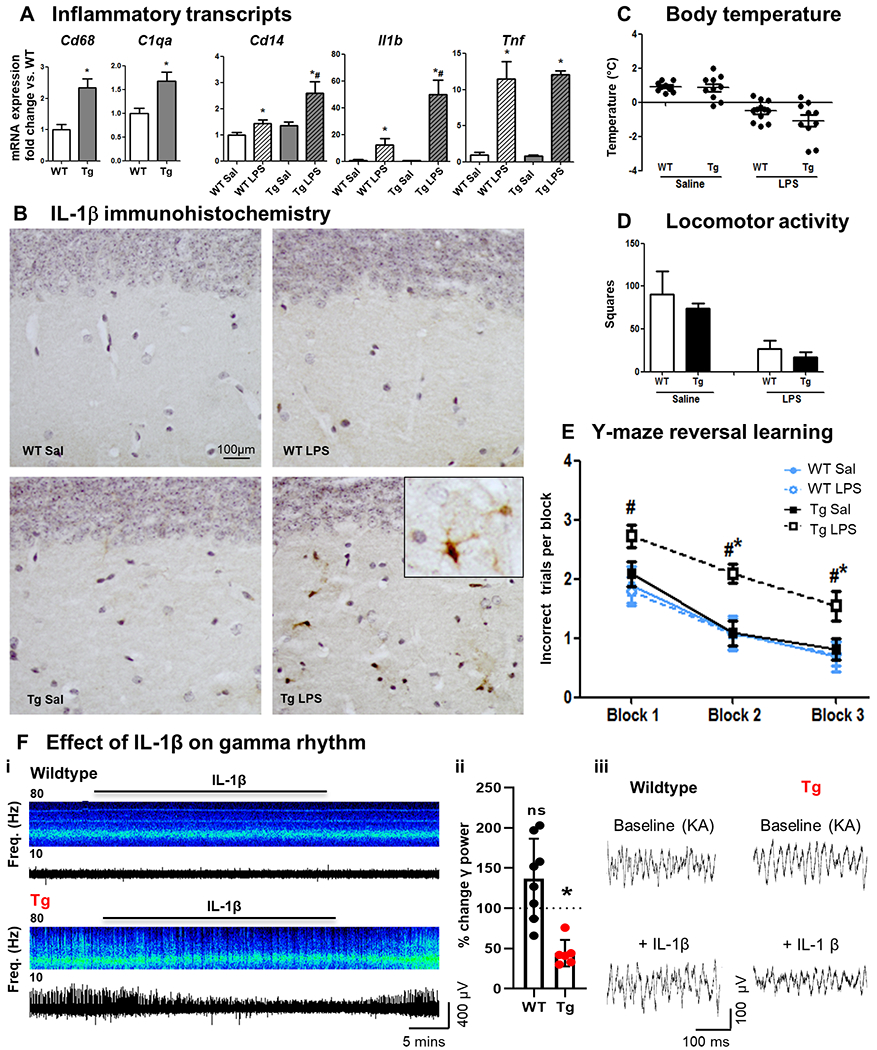
(A) Hippocampal mRNA (fold change) in selected microglial markers in APP/PS1 and WT mice (16±1 month) 2 h after LPS (100 μg/kg i.p.) challenge (Mean±SEM, n=5-7). (B) IL-1β staining in animals under the same conditions. (C) Deviation from baseline body temperature. (D) Locomotor activity in 3 minutes in the open field. (E) Reversal learning in Y maze. Mean±SEM (n=10-15). Kruskal Wallis Test. * effect of treatment; # effect of genotype (p < 0.05). (F) Effect of IL-1β (10 ng/ml) on gamma rhythm in WT and Tg brain slices. (i) Example long time-course local field potential traces of kainate-evoked (50-100 nM) persistent gamma frequency oscillations, treated acutely with IL-1β and recovering on washout. (ii) % Reduction in the power of gamma oscillations induced by acute IL-1β in WT/Tg mice. Mean±SEM (WT: n=9; Tg: n=22). * WT vs. Tg (p <0.05). (iii) Example short time course local field potential traces showing kainate-evoked gamma oscillations challenged acutely with IL-1β.
Prior studies have shown that serial/chronic LPS challenge schedules drive increased APP processing, Aβ plaque area and Tau hyperphosphorylation 17,22–25,80. Disease progression was not pursued here, and we also make no claim for acute systemic inflammation as a crucial first or second hit in precipitating AD. However, prior animal data suggest that transient exacerbation of neuroinflammation can alter degenerative trajectory81,82 and, via IL-1β, can produce new brain injury.17,19 Systemic inflammation and circulating TNF-α are associated with accelerated cognitive decline in AD patients10, emphasising its relevance in humans. The increased risk for subsequent dementia and long-term cognitive decline imposed by delirium9,83 does not appear to be accounted for by increased amyloid or Tau8, suggesting that some de novo pathology occurs during these episodes. Therefore dual approaches, using model systems to probe interactions of systemic inflammation with amyloid pathology and human CSF analysis that demonstrates inflammatory mediator associations with brain injury markers, such as neurofilament light and synaptic proteins84,85 are clearly necessary.
Although falling short of defining the signalling processes that cause the delirium-like deficits observed here, the hypersensitivity of the three cell populations examined here and the central involvement of IL-1β at all three levels, as well as increased vulnerability at the brain endothelium70 identifies these hypersensitivities as likely contributors to neuroinflammatory exacerbation. This is also consistent with dual roles for IL-1β in acute cognitive dysfunction and in acute brain injury already shown in an alternative model for delirium superimposed on dementia.19 The cellular and molecular vulnerabilities demonstrated in the current study are summarised in Figure 1 and seated within our overarching hypothesis for how systemic inflammation triggers delirium and contributes to long term cognitive trajectories in AD. Targeting this interaction between underlying disease and secondary inflammation, to limit the impacts of secondary inflammation on exacerbation of disease should, therefore, become an area of significant therapeutic interest86 and may also synergise with the current focus on disease modification using new anti-inflammatory medications.
Part 2. Consolidated results and study design
2.1. Design
The overall aim of the study was to elucidate the multiple cellular levels at which the amyloid-laden brain becomes hypersensitive to secondary acute inflammation. APP/PS1 double transgenic mice (mixed sex, 16±1 months and 19±3 months, in separate experiments) were exposed to secondary inflammation in order to directly measure the in vivo responses of microglia, astrocytes and neurons to acute pro-inflammatory stimulation. We used immunohistochemistry and quantitative PCR to measure key descriptors of the responses of these cells and then sought to verify the effects of acute systemic inflammation on cognitive function in mice. We also validated, in brain tissue from human AD cases, key brain inflammatory outcomes of acute systemic inflammation.
Microglial priming:
Bacterial endotoxin (LPS, 1 μg intracerebral; i.c.) was administered to APP/PS1 and WT mice to examine the response of microglia surrounding amyloid plaques, examining inflammasome activation, NFκB activation, IL-1β production and expression of transcriptional markers to assess acute phenotypic changes.
Astrocytes:
Acute in vivo IL-1β challenge (10 ng i.c.) was used to examine astrocyte responses, through PCR assays of chemokines (Ccl2, Cxcl1, Cxcl10), cytokines (Il6) and other astrocyte phenotypic markers arising from recent descriptions of A1/A2 astrocyte polarisation. Chemokine expression was further pursued by immunohistochemistry and confocal microscopy in separate animals.
Neuronal networks:
Neuronal network responses to secondary inflammation were measured: examining the impact of acute IL-1β application on hippocampal gamma oscillations in ex vivo brain slices from 16 month old APP/PS1 and WT animals.
Systemic inflammation:
Impacts of secondary systemic inflammation on microglia, astrocytes and dynamic cognitive function were verified via peripheral challenge with LPS (100μg/kg i.p.), measuring microglial IL-1β expression, astrocyte Ccl2, Cxcl1, Cxcl10, Il6 and Stat3 expression and cognitive flexibility assessment (Y-maze reversal learning) in the hours after LPS challenge.
Human validation:
Key outcomes of acute systemic infection in human AD cases were then examined: brain tissue expression of IL-1β and the downstream mediator IL-6.
2.2. Findings
Microglial activation was verified around plaques in the APP/PS1 brain at 19±3 months of age, with increased numbers (by Pu.1 labelling ), condensed morphology (by Iba1 labelling) and altered transcriptional phenotype (elevated levels of Tyrobp, Trem2, Itgax and Clec7a) compared to age-matched controls (Figure 2). This previously described set of core genes 15,38 suggested that these microglia were ‘primed’ to show exaggerated responses to subsequent stimulation and, therefore, LPS i.c. was administered to examine this hypothesis. LPS induced limited IL-1β in microglia in WT brains but exaggerated levels in the APP/PS1 brain. The IL-1β was clearly expressed in microglia, and not in astrocytes, specifically around amyloid plaques (Figure 3A–C). Activation of the IL-1-maturing enzyme complex the NLRP3 inflammasome was demonstrated in plaque-associated microglia in APP/PS1 mice in the absence of secondary inflammation, as previously described13, but p65 labelling showed that the NFκB transcription factor was not activated (i.e. localised to the nucleus) until secondary inflammation was induced. Consistent with this muted NFκB activation, several NFκB-dependent genes were minimally expressed in APP/PS1 mice per se, but Il1b, Il1a, Cd14 and Nlrp3 all showed exaggerated induction 2 h after acute LPS or IL-1β challenge in APP/PS1 mice (Figure 3D–F). Therefore, secondary inflammation switches the microglial phenotype towards prominent IL-1β expression and processing, selectively in APP/PS1 mice.
Astrocytes also showed morphological evidence of activation around plaques (Figure 4A) and qPCR on hippocampal homogenates revealed elevated expression of astrocyte markers Gfap, Serping1 and Ctss (Figure 4B). We used astrocytes and microglia, freshly isolated from 16 month old APP/PS1 and WT mice, to validate a panel of specific astrocyte transcripts: Ptx3, Gbp2, Tgm1, Gfap (Figure 4C) and others identified in transcriptomic studies of isolated astrocytes 30,45. Thereafter, these were pursued in transcriptional analysis of astrocyte phenotype after acute challenge with IL-1β (astrocytes respond directly to microglial IL-1β in vivo, but not to LPS itself). Two hours after acute challenge with IL-1β, several genes were induced in both WT and APP/PS1 mice but a substantial panel showed exaggerated induction in APP/PS1 mice, including A1 and A2 astrocyte transcripts and the chemokine Cxcl10 (Figure 5A,B). Based on our prior demonstration of astrocyte ‘priming’ in a prion model of neurodegeneration31 we used immunohistochemistry to show exaggerated production of the chemokines CCL2, CXCL1 and CXCL10 and double-labelling with confocal microscopy to show that CCL2 and CXCL10 were expressed in astrocytes, specifically proximal to amyloid plaques (Fig. 5C). This showed that astrocytes were ‘primed’ to produce exaggerated chemokine responses with respect to astrocytes from non-diseased animals.
To verify that these glial populations could be triggered to switch phenotype even during systemic inflammation, we challenged animals with LPS (100 μg/kg) i.p. and showed that microglia made exaggerated levels of IL-1β (Figure 6A) and isolated astrocytes made exaggerated levels of all of the chemokines above and also the cytokine Il6 (Figure 5D). Associated with these exaggerated microglial and astrocyte responses, APP/PS1 animals, selectively, showed acute deficits in cognitive flexibility/executive function in the hours after LPS challenge (Figure 6E), demonstrating an increased vulnerability to acute cognitive dysfunction upon acute systemic inflammation. Consistent with this, we showed that a key electrophysiological parameter underlying dynamic cognitive processes, gamma oscillations measured in hippocampal slices, was also significantly disrupted by acute treatment with IL-1β, selectively in APP/PS1 mice (Figure 6F). Therefore, microglia and astrocytes are primed to show heightened cytokine and chemokine responses and neuronal networks and cognitive function are hypersensitive to acute disruption by acute inflammation, selectively in APP/PS1 mice. This selective vulnerability to disrupted function upon systemic inflammation, in mice, resonates with the occurrence of delirium in patients with dementia6,87,88 and with the exacerbation of disease progression10 upon systemic inflammation.
Caveats with transgenic models prompted us to validate some key findings in human AD cases. In grey matter tissue from AD patients who died with systemic infection, IL-1β was significantly elevated compared to those who died without systemic infection. Moreover, IL-6, which is expressed in astrocytes in mice, also showed elevated expression in patients with infection and was strongly correlated with IL-1β expression.
These data confirm that acute systemic inflammation, in mice and in AD patients, switched the brain inflammatory phenotype to one producing elevated IL-1β and IL-6, and these hypersensitive inflammatory responses, at least in APP/PS1 mice, are sufficient to drive neuronal and cognitive dysfunction.
Part 3. DETAILED METHODS AND RESULTS
3.1. Methods
3.1.1. Animals and pro-inflammatory stimuli
APPSwe/PS1dE9 mice (Jax strain #005864, +/0, hereafter referred to as Tg) of 19±3 months were housed at 21°C with a 12 h light/dark cycle. We used relatively old animals since amyloid transgenic mice are thought to better represent the mild cognitive impairment stage of disease51 and we reasoned that using older animals may therefore maximise their relevance to human disease. Although some animals in the colony did die at earlier ages, likely due to reported seizure activity89, none died as result of the secondary inflammatory challenges made here. Food and water access was ad libitum. For surgery, mice were anesthetized i.p. with Avertin (50% w/v in tertiary amyl alcohol, diluted 1:40 in H2O; 20 ml/kg, i.p.; Sigma) and positioned in a stereotaxic frame (Kopf Instruments). Holes were drilled at 2.0 mm (anteroposterior) and 1.7 mm (right side of the midline) from Bregma, and 1 μl of LPS (1μg/μl) or IL-1β (10 ng/μl) was injected into the right hippocampus (depth 1.6 mm) using a glass microcapillary (Sigma). Control animals were administered 1 μl of saline 0.9%. After surgery, mice were placed in a recovery box at 25°C and were closely monitored. A different cohort of animals (16±1 months) was challenged i.p. with LPS (100μg/kg) before examination of acute sickness or assessment of cognitive function during acute systemic inflammation and to examine hippocampal expression of inflammatory transcripts. All animal experimentation was performed under licenses granted by the Minister for Health and Children and from the Health Products Regulatory Authority, Ireland, with approval from the local ethical committee and in compliance with the Cruelty to Animals Act, 1876 and the European Community Directive, 86/609/EEC. Every effort was made to minimize stress to the animals.
3.1.2. Acute sickness measurements
Temperature was measured at 0 h (immediately before LPS challenge) and 2 h after LPS challenge by a rectal probe (Thermalert TH5, Physitemp, Clifton, New Jersey), placed approximately 1.5 cm into the rectum of the mouse. Temperature deviation from baseline was calculated by subtracting the measurement at 0 h from the measurement at 2 h. To investigate locomotor and rearing activity the open field task was used. Mice were placed in a box (58×33×19 cm) and the number of times the mouse reared and crossed the squares in the box (distance travelled) was recorded for 3 min, since animals move very little during acute LPS-induced sickness, even when monitored for longer periods. This activity was measured 2 h after LPS or saline challenge and immediately before taking their temperature at this time point.
3.1.3. Reference Memory and cognitive flexibility
To investigate reference memory and cognitive flexibility, the “paddling” Y-maze visuospatial task was used as described in 81. This task was designed specifically to examine acute changes in cognitive function, relevant to delirium in the hours post-challenge with LPS, rather than to examine any change in the trajectory of memory dysfunction associated with the model per se. The task specifically interrogates a failure of cognitive flexibility or inability to shift attention, manifest by increased perseveration at a previously learned location, upon the moving of the exit to a new location. A clear perspex Y-maze consisting of three arms with dimensions 30 x 8 x 13 cm was mounted on a white plastic base. The distal end of each arm contained a hole, 4 cm in diameter and two arms could be blocked by insertion of a closed black plastic tube thus preventing mice from exiting. The third hole had an open black tube, 2 cm above the floor, where mice could exit the maze and enter a black burrowing tube to be returned to their home cage. Each of the three plastic tubes had a burrowing tube over them on the outside of the maze so that from the centre of the maze all arms looked identical. The maze was filled with 2 cm of water at 20 to 22°C, sufficient to motivate mice to leave the maze by paddling to an exit tube, from where they are returned to their home cage. Mice were placed in one of two possible start arms in a pseudorandomised sequence for 10 trials and the groups were counter-balanced with respect to the location of the exit and start arm. For any individual mouse the exit arm was fixed (to test reference memory). Wild type (WT) and transgenic APPSwe/PS1dE9 (Tg) mice (16±1 month) were trained for 6 days (10 trials per day). An arm entry was defined as entry of the whole body, excluding the tail and a correct trial was defined as entry to the exit tube without entering other arms. On day 7 mice were injected i.p with LPS (100μg/kg or vehicle) and at 2 h post-challenge were tested on retention of memory in the Y-maze for 1 trial to confirm retention of prior learning (based on pilot data indicating that retention was preserved under LPS challenge). The exit for each mouse was then switched to a different arm and mice had to learn the location of the new exit over 12 trials. The 12 trials were divided into 3 blocks of 4 and incorrect trials per block were recorded.
3.1.4. Tissue preparation
Animals for analyses of cytokine-induced transcriptional changes were terminally anesthetized with sodium pentobarbital at 2 h post challenge (Euthatal; Merial Animal Health) and transcardially perfused with heparinized saline before dissection of hippocampus and snap freezing in liquid nitrogen and storage at −80°C until use. The 2 h time point was chosen based on several years of experience with acute challenges to the brain. The acute cytokine response to LPS (IL-1, TNF etc) and the acute cytokine/chemokine response to IL-1 (CCL2, CXCL1, CXCL10, IL-6 etc) all peak at approximately 2 h post-challenge.31,90
Animals for immunohistochemical examination were terminally anesthetized with the previously described method and perfused with heparinized saline followed by 10% neutral buffered formalin (Sigma). Brains were postfixed in formalin and then embedded in paraffin wax. Coronal sections (10 μm) were cut on a Leica RM2235 Rotary Microtome (Leica Microsystems) at the level of the hippocampus and floated onto electrostatically charged slides (Menzel-Glaser) and dried at 37°C overnight.
3.1.5. RNA extraction, cDNA synthesis and quantitative PCR
Total RNA was isolated using the RNeasy Plus Mini method (Qiagen, Limburg, Netherlands) following the manufacturer’s instructions. To ensure complete DNA elimination from the column bound RNA, an on-column DNase step was performed. The RNA yield and quality of each sample were quantified based on Optical Density (OD) using the ‘NanoDrop’ ND-1000 UV–vis spectrophotometer (Thermo Fisher Scientific). cDNA synthesis was carried out using a High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, Warrington, UK). Primer and probe sets were designed using Applied Biosystems Primer Express software and amplified a single sequence of the correct amplicon size, as verified by SDS-PAGE. Where no probe sequence is shown, the DNA binding dye SYBR green was used in its place. Pre-made primers were also used and run following the manufacturer’s instructions (Thermo Fisher Scientific). Primer pair/probe sequences are shown in Table 1. Samples for RT-PCR were run in duplicate using FAM/SYBR (Roche) in a StepOne Real-Time PCR system (Applied Biosystems, Warrington, UK) under the cycling conditions: 95°C for 10 min followed by 95°C for 10 secs and 60°C for 30 secs for 40-45 cycles. Quantification was achieved by exploiting the relative quantitation method, using cDNA from LPS-injected mouse brain as a standard expressing all genes of interest and serial 1 in 4 dilutions of this cDNA to construct a linear standard curve relating cycle threshold (CT) values to relative concentrations, as previously described 16. Gene expression data were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and expressed relative to WT saline-treated values.
Table 1. PCR Primer sequences.
Where no probe sequence is shown, SYBR green dye was used. For pre-made primers, assay ID is provided. Forward (F), reverse (R), probe (P). Primer sets for Clcf1, Gbp2, Cst7, Myl9, S1pr3, Serping1 and Tgm1 were predesigned TAQMAN gene expression assays.
| Gene | Access. num. | BP | Sequence |
|---|---|---|---|
| C1qa | NM_007572.2 | 268 | F: 5’GCCGAGCACCCAACGGGAAGG3’ R: 5’GGCCGGGGCTGGTCCCTGATA3’ |
| C3 | NM_009778.2 | 115 | F: 5’AAAGCCCAACACCAGCTACA3’ R: 5’GAATGCCCCAAGTTCTTCGC3’ |
| Ctss | NM_021281 | 80 | F: 5’GCCACTAAAGGGCCTGTCTCT3’ R: 5’TCGTCATAGACACCGCTTTTGT3’ |
| Ccl2 | NM_011333 | 81 | F: 5’GTTGGCTCAGCCAGATGCA3’ R: 5’AGCCTACTCATTGGGATCATCTTG3’ P: 5’TAACGCCCCACTCACCTGCTGCTACT3’ |
| CD14 | NM_009841.4 | 128 | F: 5’GCCAAATTGGTCGAACAAGC3’ R: 5’CCATGGTCGGTAGATTCTGAAAGT3’ |
| Cxcl1 | NM_008176 | 82 | F: 5’CACCCAAACCGAAGTCATAGC3’ R:5’AATTTTCTGAACCAAGGGAGCTT3’ |
| Cxcl10 | M33266.1 | 127 | F: 5’GCCGTCATTTTCTGCCTCAT3’ R: 5’GCTTCCCTATGGCCCTCATT3’ P: 5’TCTCGCAAGGACGGTCCGCTG3’ |
| Gbp2 | NM_010260.1 | 94 | F: 5’CAGCTGCACTATGTGACGGA3’ R: 5’AGCCCACAAAGTTAGCGGAA3’ |
| Gfap | K01347.1 | 90 | F: 5’CTCCAACCTCCAGATCCGAG3’ R: 5’TCCACAGTCTTTACCAGATGT3’ |
| Gper1 | NM_029771.3 | 115 | F: 5’TGCTGCCATCCAGATTCAAG3’ R: 5’GGGAACGTAGGCTATGGAAAGAA3’ |
| Il1b | M15131 | 69 | F: 5’GCACACCCACCCTGCA3’ R: 5’ACCGCTTTTCCATCTTCTTCTT3’ P: 5’TGGAGAGTCTGGATCCCAAGCAATACCC3’ |
| Il1r1 | NM_001123382.1 | 117 | F: 5’GCAATATCCGGTCACACGAGTA3’ R: 5’ATCATTGATCCTGGGTCAGCTT3’ P: 5’TCCTGAGCCCTCGGAATGAGACGATC3’ |
| Il1a | NM_010554 | 150 | F: 5’TGTTGCTGAAGGAGTTGCCAG3’ R: 5’CCCGACTTTGTTCTTTGGTGG3’ |
| Nlpr3 | AY355340.1 | 115 | F: 5’GAGCCTACAGTTGGGTGAAATGT3’ R: 5’CCACGCCTACCAGGAAATCTC3’ |
| Ptx3 | X83601 | 62 | F: 5’ACAACGAAATAGACAATGGACTTCAT3’ R: 5’CTGGCGGCAGTCGCA3’ P: 5’CCACCGAGGACCCCACGCC3’ |
| Ccl5 | NM_013653 | 75 | F: 5’GCAGTCGTGTTTGTCACTCGAA3’ R: 5’GATGTATTCTTGAACCCACTTCTTCTC3’ |
| S100a10 | NM_009112.2 | 71 | F: 5’CTAGCCTCATCGTGGTGTGC3’ R: 5’GAGTCTGTCGAAACCTGGGC3’ |
| Steap4 | NM_054098.3 | 93 | F: 5’TGCAAGCCGGCAGGTGTTTGT3’ R: 5’TCCAGTGGGGTGAGCCCAAGA3’ |
| Tnf | M11731 | 149 | F: 5’CTCCAGGCGGTGCCTATG3’ R: 5’GGGCCATAGAACTGATGAGAGG3’ P: 5’TCAGCCTCTTCTCATTCCTGCTTGTGG3’ |
| Vim | NM_011701.4 | 100 | F: 5’GCTGCAGGCCCAGATTCA3’ R: 5’TTCATACTGCTGGCGCACAT3’ |
3.1.6. Immunohistochemistry
Animals were labelled with GFAP (1:2000; Dako Z0334), 6E10 (1:1000; Biolegend 803001), Iba-1 (1:2000; Abcam ab5076), PU.1 (1:400; Cell Signaling 2266S), CCL2 (1:200; part 840288 of DY479 (R&D Systems), IL-1β (1:50; Peprotech 500-P51), CXCL1 (1:50; R&D AF-453-NA), CXCL10 (1:10000; Peprotech P129), NFκB (1:100; Santa Cruz sc-8008) and ASC (1:1000; AdipoGen AL177 006-C100). Briefly, all sections were quenched for 20 min with 1% H2O2/methanol and incubated overnight at 4°C with the primary antibody and the appropriate serum. The next day, sections were incubated for 1 h with biotinylated secondary antibody (1:100, Vector). After several washes in phosphate buffer saline (PBS), the ABC method was used (Vectastain Kit, PK6100 Vector) and the reaction product was revealed using 3,3’ diaminobenzidine as chromogen (Sigma Aldrich) and H2O2 as substrate. Slides were counterstained using Haemotoxylin (VWR International Ltd, Dublin, Ireland), dehydrated, coverslipped, examined and photographed using an Olympus DP25 camera (Mason) mounted on a Leica DM3000 microscope (Laboratory Instruments and Supplies, Ashbourne), captured using CellA™ software (Olympus, Mason). For Iba-1 quantification, the percentage area covered by Iba-1 positive cells in thresholded images was calculated using ImageJ software (ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA), using two sections per animal (WT n=9; Tg n=7). Pu.1 quantification was performed in the hilus of the hippocampal region (ispsilateral to the i.c. challenge), three frames per section, four sections per animal (WT n=4; Tg n=8). Experimentor was blinded to identity of samples.
For confocal microscopy, the brain sections were incubated using 6E10, Iba-1, IL-1β, GFAP, CCL2 and CXCL10 at the same dilution described in the previous paragraph and the appropriated serum overnight at 4 °C. The day after, they were incubated with the corresponding secondary antibody for 1h at room temperature at (1:800) (Invitrogen. Alexa Fluor 488 anti-mouse; Alexa Fluor 488 anti-rabbit; Alexa Fluor 594 anti-goat; Alexa Fluor 594 anti-rabbit; Alexa Fluor 633 anti-mouse) and counterstained with Hoechst (1:2000; Sigma Aldrich 33258) for 10 min at RT. Slides were then coverslipped with ProLong™ Gold Antifade Mountant medium (Thermo Fisher Scientific) and examined with a Leica SP8 Scanning confocal microscope (Leica Microsystems). Abbreviations: GFAP (Glial Fibrillary Acidic Protein); 6E10 (antibody against Aβ); Iba-1(ionized calcium-binding adapter molecule 1); CCL2 (CC chemokine ligand 2); IL-1β (interleukin-1β); CXCL1/10 (CXC chemokine ligand 1/10); NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells); ASC (Apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain).
3.1.7. Fluorescence-activated cell sorting (FACS)
Further cohorts of animals were used to isolate microglia and astrocytes from the brains of WT and Tg mice. The hippocampus of WT and Tg animals (14±2 months old) with or without LPS i.p. (250μg/kg) was punched out and kept in ice cold 1ml HBSS and single cell suspension protocol was followed as previously described.91 Sorted cells were collected in 1.5 ml Lobind RNAse/DNAse free tubes containing 350μl of sorting buffer (HBSS without Phenol Red supplemented with 7.5 mM HEPES and 0.6% glucose). 40-50,000 CD45lowCD11b+ microglia and 80-100,000 GLAST+CD45-astrocytes were sorted. Purity of sorted astrocytes and microglia was determined by qPCR and subsequently RNA was isolated from sorted populations to run quantitative PCR.
3.1.8. Ex vivo brain slice electrophysiology
Coronal slices (400μm) containing the hippocampus and entorhinal cortex were prepared from WT and Tg mice (21±2 months old). The experimentalist was blind to the genotype of the animal. Animals were anaesthetised with inhaled isoflurane and immediately euthanized by cervical dislocation. The brain was quickly removed and brain slices were taken at low temperatures (4-5°C) in a modified cerebrospinal fluid (CSF) solution containing: Sucrose (252 mM), KCl (3 mM), NaH2PO4 (1.25 mM), MgSO4 (2 mM), CaCl2.2H2O (2 mM), Glucose (10 mM). All salts were obtained from Sigma Aldrich. Slices were transferred to a holding chamber and maintained at room temperature at the interface between wetted carbogen (95% O2, 5% CO2) and artificial cerebrospinal fluid (aCSF) containing NaCl (126 mM), KCl (3 mM), NaH2PO4 (1.25 mM), MgSO4 (1mM), CaCl2.2H2O (1.2 mM), Glucose (10mM), and NaHCO3 (24mM). Slices were allowed to equilibrate for at least 30 min and then were transferred to a recording chamber and maintained at 34°C at the interface between warm, wet carbogen (95% O2, 5% CO2) and aCSF. Slices were allowed to equilibrate for 15 min before recordings began. Extracellular recordings from the CA3 region were made using borosilicate glass micropipettes (from Harvard Apparatus) filled with aCSF (resistance 2-5MΩ) connected to an extracellular amplifier (EXT-10-2F, NPI Electronics GmbH, Tamm, Germany). Signals were analog filtered at 1-300 Hz and digitised at 5kHz. Bath perfusion of kainate (50nM-100nM) in aCSF was used to induce gamma frequency (20-80 Hz) oscillations. Following the emergence of stable gamma activity, IL-1β (10 ng/ml) was added to the circulating perfusion medium. After 30 min, the IL-1β was removed by switching the perfusion medium to aCSF with warm, wet carbogen (95% O2, 5% CO2). Recordings were analysed using Spike2. A power spectrum (FFT size 8192, Hanning window) was generated for the final 60 seconds before the IL-1β was added to the slice as well as for the final 60 seconds of the IL-1β application. These power spectra were analysed by measuring the area under the curve in the gamma frequency band (20-80 Hz). The peak frequency was also recorded in this band. The change induced by IL-1β was normalised relative to the baseline values recorded in the absence of IL-1β. The number and average amplitude of the interictal events in these periods were also quantified.
3.1.9. Human tissue collection
Autopsy-acquired brain tissue from donors was sourced from the South West Dementia Brain Bank (University of Bristol) and BRAIN UK (Queen Elizabeth University Hospital, Glasgow). Clinical history and cause of the death as included in post-mortem reports was used to subdivide cases into 2 subgroups: AD patients, who died without systemic infection (AD w/o infection, n=28) and those who died with systemic infection (AD with infection, n=39). Alzheimer’s cases had a clinical diagnosis of AD made during life and satisfied post-mortem neuropathological consensus criteria for AD.92 The current data represent new analyses of an existing dataset. For details and extended information see56.
3.1.10. MesoScale Discovery multiplex analyses of brain tissue
Fresh frozen tissue available for the AD groups with and without systemic infection was used for MesoScale Discovery as (MSD) multiplex assays. Inflammatory proteins were measured on the V-Plex MSD electrochemiluminescence multi-spot assay platform (MesoScale Diagnostics, Rockville USA).100 mg of fresh frozen grey matter from AD cases (n = 67) was homogenised at a tissue concentration of 20% w/v in RIPA lysis buffer (Thermo Fisher Scientific) by use of a handheld homogeniser (Thermo Fisher Scientific); the buffer was supplemented with protease inhibitors (Complete Mini, Sigma Aldrich) and phosphatase inhibitors (Thermo Fisher Scientific). Total protein concentration in the supernatant was measured by BCA Protein Assay Kit (Thermo Fisher Scientific). 12.5μl of brain homogenate (1:4 dilution) was used for each assay according to the manufacturer’s protocol. The cytokine panel 1 V-PLEX human biomarker 40-PLEX kit was used and imaged on the Meso QuickplexSQ120 (MesoScale Discovery) according to manufacturers’ instructions to obtain absolute protein levels (pg/ml). Frozen blocks from 4 controls and 2 multiple sclerosis brains containing chronic inactive, acute and chronic active lesions were used as negative and positive controls, respectively. To address a specific hypothesis arising from mouse experiments in the current study, we analysed expression of IL-1β and IL-6, proposing that these cytokines would be elevated by infection and would be correlated with each other.
3.1.11. Statistical analyses
For multiple comparisons two-way analysis of variance (ANOVA) was performed, with factors being genotype (Wild Type or Transgenic) and treatment (LPS, Saline or IL-1β). Data were not always normally distributed, and, in these cases, nonparametric tests were used (Kruskal–Wallis and post hoc pair-wise comparisons with Mann–Whitney U-test). Post hoc comparisons were performed with a level of significance set at p ≤ 0.05. For data that were normally distributed and homoscedastic, we used a standard parametric post-hoc test (Bonferroni’s test) and for those that were normally distributed, but non-homoscedastic, we performed non-parametric post-hoc comparisons (Games–Howell’s test). For two-group comparisons, data were analysed using Student-t test when they were normally distributed, and the Mann-Whitney test was run if data did not pass the assumptions for parametric analyses. Data are presented as mean ± standard error of the mean (SEM). Symbols in the graphs denote post-hoc tests. For correlation analyses, data from AD patients with and without infection were plotted separately and Pearson linear regression tests were run. Statistical analyses were carried out with the SPSS 22.0 software package (SPSS, Inc., Chicago, IL, USA).
3.2. RESULTS
Microglia.
Microglia are known to increase in number and reactivity around Aβ plaques93–95. Here we show, by immunohistochemistry, increased microglial activation in APP/PS1 (Tg) mice quantified by the percentage area that is Iba-1 positive (Fig.2A) and the number of Pu.1 positive cells per mm2 (Fig.2B). RT-PCR was used to assess a set of microglial-specific genes that are part of the core gene signature for microglial priming reported to change in AD models.15 We observed a significant decrease in Sall1 (p=0.048) but no significant change in Sparc, P2ry12 and Tmem119, all of which have a role in the homeostatic maintenance of the resident microglia phenotype 38,39. Since the numbers of microglia are significantly increased, the unchanged tissue levels on P2ry12 and Tmem119, actually indicate decreased expression of these markers on a ‘per cell’ basis, as described elsewhere38. However, there was a marked increase in Tyrobp (p<0.001), which reliably correlates with increased microglial numbers15,96,97, and significant increases in Trem2, Itgax and Clec7a (p<0.001; Fig.2C). These data suggest that these microglia have lost regulatory control and have become primed.
Microglial priming.
Therefore we examined whether superimposed acute challenges (LPS or IL-1β i.c.), would produce exaggerated IL-1β responses in these primed microglia at 2 h post-challenges (an optimal time for post-LPS cytokine induction31). Despite Tg mice showing a greater number of Iba-1 positive cells and more activated morphology than WT mice (Fig.3A), IL-1β expression was not evident in Tg animals per se (3B, left). Acute stimulation with LPS induced exaggerated production of IL-1β in clusters of cells surrounding amyloid plaques in Tg animals (3B, centre) compared to isolated single IL-1β-positive cells in WT animals. This was also apparent in Tg mice challenged with IL-1β (Fig.3B, right). This IL-1β is produced by microglial cells, specifically in those microglia adjacent to 6E10-positive Aβ plaques (Fig.3C), all IL-1β positive cells co-localised with Iba-1 and none co-localised with astrocytes (Fig.3C, top right), as shown by double-labelling with antibodies against IL-1β and Iba-1 or GFAP. The intense IL-1β labelling is due to resident microglia since they were measured 2 h after LPS challenge, too soon to be explained by infiltrating monocytes and also cannot be explained by microglial phagocytosis of other IL-1-expressing cells since these microglia contained only one nucleus (prior studies have shown two nuclei inside microglial cells during phagocytosis of apoptotic cells).98
To better understand this phenotypic switch in plaque-associated microglia we assessed two key steps in the synthesis and maturation of IL-1β: the nuclear translocation of the p65 subunit of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and the assembling of ASC (Apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain) mediated by the NLPR3 inflammasome pathway.99,100 Microglia adjacent to Aβ plaques in the hippocampus of Tg mice challenged with saline do not show nuclear localisation of NFkB-p65, with microglial nuclei (identified by their small, darker, slightly oblong nuclei), surrounding plaques showing only blue haematoxylin counterstain (arrowheads in Fig. 3D) while the Tg+LPS group show clear nuclear translocation of NFkB-p65 in those same small oblong nuclei surrounding plaques (arrows Fig. 3D). Minimal expression of either Il1b or Nlrp3 in Tg+Sal animals but distinct and indeed exaggerated induction of these genes in Tg+LPS (Fig.3E) supports the conclusion that NFκB is minimally activated in APP/PS1 per se.
However, in untreated Tg animals we observed intense ASC labelling with a non-uniformity of distribution within the cell consistent with ASC “speck” formation in plaque-associated cells of microglial morphology (Fig.3D, right), compared to modest and uniform distribution in WT animals. Therefore, plaque-associated microglia in Tg animals show inflammasome assembly as a result of amyloidosis per se 13 (classically termed signal II) 99,100. Therefore, they are primed to transcribe, translate and efficiently mature IL-1β upon secondary stimulation, but because they have not translocated NFκB p65 to the nucleus and because Il1b mRNA was not significantly elevated in Tg animals these animals do not produce significant IL-1β until they have received the secondary inflammatory stimulus.
RT-PCR analysis 2 h after an acute LPS challenge (i.c.) showed that LPS-treated animals of both genotypes showed an increase in the cytokines Il1b, Il1a, Tnf, CD14 and inflammasome component Nlrp3 mRNA levels, with no changes in the complement genes C1qa or C3.
The effects of LPS (*) and genotype (#) on expression were assessed using Kruskal Wallis test, followed by Mann-Whitney analyses. Within this set of genes, Il1b and CD14 presented an exaggerated response in Tg+LPS in comparison with WT+LPS (Fig.3E; all p values <0.043).
Since administering LPS directly to the brain is relatively unphysiological experiment, which we used to address a specific hypothesis predicting exaggerated microglial response to LPS when the stimulus was applied proximally, we also assessed whether microglia would show exaggerated responses to IL-1β itself, since this cytokine frequently arises in the brain following traumatic or infectious episodes. We directly injected IL-1β i.c. and showed that only microglia in the Tg brain presented detectable de novo IL-1β protein expression at 2 h (Fig.3B). Examining transcriptional changes 2 h after IL-1β challenge (Fig.3F), we showed that IL-1β elevated Il1b, Il1a, Tnf, CD14 and Nlrp3 mRNA levels in both genotypes but produced exaggerated increases in Il1b, Il1a, Tnf, CD14, Nlrp3 and C1qa mRNA expression in Tg+IL-1β animals (all p values < 0.041, effect of IL-1β (*) or genotype (#)).
Astrocytes.
Using immunohistochemical techniques we show that astrocytes show a more activated phenotype and strong GFAP staining in Tg astrocytes encircling 6E10-positive Aβ plaques (Fig.4A). We analysed a small panel of astrocyte genes in brain homogenates (Fig.4B) and showed that Tg mice have significantly higher levels of Gfap, Ctss, Serping1 and Il1r1 than WT animals (all p values <0.036). In order to validate further astrocyte transcripts identified by previous investigators as being elevated in models of stroke, sepsis and AD 30,44,101 while also attending to recently described A1, A2 and pan reactive signatures45 we isolated astrocytes and microglia from WT and Tg animals (14±2 months old). (Fig.4B). Sorted microglia and astrocytes showed high purity by qPCR (boxed panel, Fig.4C) and further transcriptional analysis validated that Gfap, Il1r, Ptx3, Tgm1, Hes5, Irf7 and Gbp2 are highly enriched in astrocytes with respect to microglia, while C1qα, Tlr4 and Cst7 are predominantly microglial transcripts. These analyses validate the subsequent examination of a selection of these genes as reliable astrocytic markers in bulk RNA together with other genes of interest, including chemokines, Il6 and the IL-6-dependent transcription factor STAT3.
Astrocyte priming.
Having demonstrated exaggerated microglial production of IL-1β we then asked whether astrocytes would, downstream, show heightened responses to IL-1β. Using the genes validated above, we investigated the hypothesis that astrocytic ‘priming’ also occurs in the APP/PS1 model of AD by examining those astrocytic transcripts 2 h after an acute challenge with IL-1β (10ng, i.c.) in WT and Tg mice (19±3 months). For several genes IL-1β had significant effects that were equivalent in both strains: mRNA for Steap4, Ccl2, Cxcl1, Ccl5 was increased and that for Gper1 decreased in both genotypes in an equivalent manner (Fig.5A; all p values < 0.018), while Vim (vimentin) did not change. Moreover a number of genes were robustly increased in disease per se (Ccl5, Tgm1 and Cst7 all p values < 0.042) and a number were increased by IL-1β in WT mice (Clcf1, Cxcl10, Ptx3 and Gbp2; all p values <0.030). However, a large group of transcripts (Cst7, Gbp2, Ptx3, Tgm1, Clcf1, Cnn1, Cxcl10, S1pr3) showed an exaggerated response to IL-1β challenge in Tg compared to the same challenge in WT (# on the Tg+IL-1β in Fig.5B). Most data were non-parametric and using Kruskal-Wallis test followed by Mann-Whitney analyses, we found an effect of genotype and treatment exclusively in Tg+IL-1β group (interaction between treatment and genotype for all transcripts in Fig.5B; p < 0.035). Several of those, including Ptx3, Tgm1 and Gbp2, were shown to be astrocytic using isolated cells (Fig.4C). These data support the idea that the astrocyte population is primed to show phenotypic switching in response to acute IL-1 stimulation. To confirm this hypothesis, we focussed on chemokine expression (CXCL1, CCL2 and CXCL10) in light of their implication in our prior demonstration of astrocyte priming in chronic neurodegeneration.31 This was assessed by light microscopy (Fig.5C upper panel) and confocal imaging (Fig.5C bottom) in animals (19±3 months) challenged with IL-1β (10ng, i.c.). These chemokines are induced at the brain endothelium by IL-1β, serving as a positive control for the immunolabelling (insets in figure 5C). CCL2 and CXCL1 labelling were clear in the vessels of both WT and Tg animals challenged with IL-1β but only Tg+IL-1β animals showed labelling of parenchymal cells. CCL2- and CXCL1-positive cells in the dentate gyrus had large circular nuclei typical of astrocytes and clearly distinguishable from the smaller, darker microglial nuclei (Fig.5C first and second rows). CCL2-positive cells we confirmed as astrocytes by confocal imaging (Fig.5C bottom left), showing perinuclear punctate CCL2 labelling (red) immediately proximal to the green labelling of the GFAP intermediate filaments of astrocytes.
IL-1β induced astrocyte CXCL10 in cells of astrocytic morphology (inset) selectively in APP/PS1 mice (Fig.5C third row). Confocal imaging (Fig.5C, bottom right), showing vesicle-like, perinuclear CXCL10 labelling (red) immediately proximal to GFAP-positive intermediate filament (green) confirmed astrocyte expression. CXCL10 was occasionally also observed in non-astrocytic cells but no CXCL10 was evident in astrocytes of WT animals, even when challenged with IL-1β. Highly pure primary astrocytes were also shown to be more robust than primary microglia in their synthesis of these chemokines upon acute IL-1β treatment in vitro (2.5 ng/ml for 6 h; supplementary figure 1). Thus, astrocytes proximal to Aβ-plaques in Tg mice are primed to show exaggerated chemokine responses to acute IL-1β stimulation.
Since these acute challenges were intracerebral, it was important to show that key tenets of glial priming were demonstrable even when acute inflammatory challenges were made systemically, using LPS (250μg/kg i.p.) in a different cohort of animals (14±2 months). We focussed on the transcription of these three chemokines together with Il6 and Stat3 (a transcript directly downstream of IL-6 signalling), as measured by qPCR on FACS sorted astrocytes (Fig.5D). Data were not normally distributed and were analysed by non-parametric Kruskal Wallis test. Although systemic LPS challenge did induce modest increases in WT astrocyte mRNA levels of Ccl2 (p=0.082, ns), Cxcl1 (p=0.01), Cxcl10 (p=0.063, ns), and no changes in Il6 or Stat3 (p=0.99), it produced exaggerated chemokine and IL-6 pathway responses in astrocytes from APP/PS1 mice (Ccl2, p=0.005; Cxcl1 p=0.086; Cxcl10, p=0.007; Il6, p=0.020; Stat3 p=0.025). These results in astrocytes isolated from mice i.p challenged with LPS, confirm that astrocytes are primed in the APP/PS1 model of AD, resulting in exaggerated chemokine and IL-6 pathway responses to either central or systemic acute inflammatory challenges.
Exaggerated microglial responses could also be triggered by systemic LPS, using CD68 and C1qa mRNA levels (Fig.6A) as reliable measures of plaque-associated microglia activation102 and Il1b, Cd14 and Tnfa as indicators of acute microglial activation. APP/PS1 animals showed typical complement and phagocytic activation and we found exaggerated Il1b and CD14 responses in the Tg+LPS group (all p values < 0.047). These data were supported by light microscopy imaging (Fig.6B) showing that only Tg mice challenged with i.p. LPS (100 μg/kg) showed clear IL-1β positive microglia-like cells (see insert) in the hippocampal dentate gyrus.
Regarding functional consequences of systemic inflammation with LPS (100 μg/kg, i.p.), we show that challenged animals from both strains presented equivalent LPS-induced sickness behaviour, showing a decrease in body temperature (Fig.6C) and in distance travelled in the open field (Fig.6D). With respect to maintenance of cognitive function in the face of acute inflammation: when animals were tested on retention of previously learned visuospatially-led exit from the Y-maze, LPS did not impair this function (data not shown). However, when the location of the exit was moved to an alternate arm, thus requiring cognitive flexibility and the adoption of a new strategy for maze escape, we observed a significant impairment, exclusively in Tg+LPS animals (Fig.6E). Kruskal-Wallis Test revealed a significant effect of genotype (#) and i.p. LPS (*) and significantly worse cognitive impairment in Tg+LPS (all p values < 0.032).
Given that primed microglia synthesise increased IL-1β upon secondary challenge with LPS, whether by central or peripheral challenge, we examined the ability of IL-1β (10 ng/mL) to disrupt neuronal network activity in brain slices from Tg and WT mice (21±3 months; Fig.6F). Persistent gamma frequency oscillations were elicited using kainate (50 – 100 nM) in the CA3 region of the hippocampus. In Tg mice, the power of the oscillation in the gamma band (20 - 80 Hz) was significantly reduced relative to that in the absence of IL-1β (baseline 4437 ± 1854 μV2 versus IL-1β 1782 ± 818.5 μV2, 6 slices from 5 animals, p<0.03). This represents an IL-1-induced, 44% reduction, in gamma oscillation power. This reduction in power was not observed in brain slices from WT mice, similarly challenged with IL-1β (baseline 1768 ± 1043 μV2 versus IL-1β 1920 ± 929.5 μV2, p<0.25, 8 slices from 3 animals). Acute challenge with IL-1β slightly reduced the mean peak frequency of the oscillations in the gamma band (4.3± 3.29 Hz in WT vs. 3.2±3.93 in Tg), but this effect does not represent a band shift in the type of oscillatory activity present and was not strain-dependent (two-way ANOVA main effect of IL-1β: p<0.0071, no effect of genotype p < 0.34; WT: 8 slices from 3 animals, Tg: 6 slices from 5 animals). The data suggest that hippocampal gamma frequency oscillations in APP/PS1 brain are more susceptible to disruption by IL-1β than is the case in age-matched control brain.
The data from our mouse experiments (Figs. 3, 6) led to the a priori hypothesis that IL-1β would be elevated in AD patients with infection. Fresh frozen tissue from AD patients with and without infection at the time of death was used for IL-1β and IL-6 analysis using Mesoscale assays. IL-1β in brain homogenates from AD patients with infection (n=39) was significantly higher than in AD patients without infection (n=28, p=0.0484; Fig.7A). IL-6 was also elevated in patients with infection (Fig.7B; p=0.0236). Consistent with the hypothesis of IL-1β-dependent IL-6 synthesis, the levels of these two cytokines were significantly positively correlated only in AD patients with infection (r2=0.5239, p<0.0001) while no correlation was found in AD patients without infection (r2=0.020, p=0.4656) (Fig.7C).
In summary, we present evidence that upon secondary inflammatory challenge, microglia in APP/PS1 mice, produce acutely elevated IL-1β. In turn, IL-1β is sufficient to trigger exaggerated levels of chemokines and IL-6 in astrocytes. These acutely elevated IL-1β and IL-6 changes are replicated in AD patients who died with systemic infection. LPS, in mice, acutely disrupted cognitive flexibility and IL-1β was sufficient to disrupt hippocampal gamma rhythm, selectively in the APP/PS1 mouse brain.
Supplementary Material
Acknowledgements
This study was supported by a Wellcome Trust Senior Research Fellowship to Colm Cunningham (SRF090907) and by the NIH (R01AG050626). Hugh Delaney is generously supported by a John Scott Studentship (Schools of Medicine and Biochemistry & Immunology) and Edel Hennessy and Dáire Healy were supported by College Awards from the Trinity Foundation. The technical assistance of Gavin MacManus in the Biomedical Sciences Imaging suite is gratefully acknowledged. We would like to thank Caroline Herron, University College Dublin for assistance with pilot experiments and Sonja Rakic, University of Southampton, for assistance with human samples, supported by Alzheimer’s Research UK (grant ARUK-PG2012–8).
Footnotes
Conflicts of interest. The authors have no competing interests.
References
- 1.Clark CM, Sheppard L, Fillenbaum GG, et al. Variability in annual Mini-Mental State Examination score in patients with probable Alzheimer disease: a clinical perspective of data from the Consortium to Establish a Registry for Alzheimer’s Disease. Arch Neurol. 1999;56(7):857–862. [DOI] [PubMed] [Google Scholar]
- 2.Holmes C, Lovestone S. Long-term cognitive and functional decline in late onset Alzheimer’s disease: therapeutic implications. Age Ageing. 2003;32(2):200–204. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham C, Hennessy E. Co-Morbidity and Systemic Inflammation as Drivers of Cognitive Decline: New Experimental Models Adopting a Broader Paradigm in Dementia Research. Vol 7. BioMed Central; 2015:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semmler A, Widmann CN, Okulla T, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84(1):62–69. [DOI] [PubMed] [Google Scholar]
- 5.Pandharipande PP, Girard TD, Jackson JC, et al. Long-Term Cognitive Impairment after Critical Illness. N Engl J Med. 2013;369(14):1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis DHJ, Skelly DT, Murray C, et al. Worsening Cognitive Impairment and Neurodegenerative Pathology Progressively Increase Risk for Delirium. Am J Geriatr Psychiatry 2015;23(4):403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology. 2009;72(18):1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis DHJ, Muniz Terrera G, Keage H, et al. Delirium is a strong risk factor for dementia in the oldest-old: a population-based cohort study. Brain. 2012;135(9):2809–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis DHJ, Muniz-Terrera G, Keage HAD, et al. Association of Delirium With Cognitive Decline in Late Life. JAMA Psychiatry. 2017;74(3):244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes C, Cunningham C, Zotova E, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perry VH, Nicoll JAR, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. [DOI] [PubMed] [Google Scholar]
- 12.Griffin WST, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J Leukoc Biol. 2002;72(2):233–238. [PMC free article] [PubMed] [Google Scholar]
- 13.Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2012;493(7434):674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyor WR, Glass JD, Griffin JW, et al. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31(4):349–360. [DOI] [PubMed] [Google Scholar]
- 15.Holtman IR, Raj DD, Miller JA, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 2015;3(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25(40):9275–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cibelli M, Fidalgo AR, Terrando N, et al. Role of interleukin-1β in postoperative cognitive dysfunction. Ann Neurol. 2010;68(3):360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skelly DT, Griffin ÉW, Murray CL, et al. Acute transient cognitive dysfunction and acute brain injury induced by systemic inflammation occur by dissociable IL-1-dependent mechanisms. Mol Psychiatry. 2019;24(10):1533–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: A role for the cytokine IL-1β. Neurosci Biobehav Rev. 2001;25(1):29–41. [DOI] [PubMed] [Google Scholar]
- 21.Feng X, Valdearcos M, Uchida Y, Lutrin D, Maze M, Koliwad SK. Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI insight. 2017;2(7):e91229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, Lee YK, Yuk DY, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAlpine FE, Lee JK, Harms AS, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34(1):163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25(39):8843–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14(1):133–145. [DOI] [PubMed] [Google Scholar]
- 26.Neher JJ, Cunningham C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019;40(4):358–374. [DOI] [PubMed] [Google Scholar]
- 27.Readhead B, Haure-Mirande JV, Funk CC, et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron. 2018;99(1):64–82.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itzhaki RF. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front Aging Neurosci. 2014;6(AUG). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018;99(1):56–63.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orre M, Kamphuis W, Osborn LM, et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging. 2014;35(12):2746–2760. [DOI] [PubMed] [Google Scholar]
- 31.Hennessy E, Griffin ÉW, Cunningham C. Astrocytes Are Primed by Chronic Neurodegeneration to Produce Exaggerated Chemokine and Cell Infiltration Responses to Acute Stimulation with the Cytokines IL-1β and TNF-α. J Neurosci. 2015;35(22):8411–8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci. February 2018:201800165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson A, Redish AD. Neural ensembles in CA3 transiently encode paths forward of the animal at a decision point. J Neurosci. 2007;27(45):12176–12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montgomery SM, Buzsáki G. Gamma oscillations dynamically couple hippocampal CA3 and CA1 regions during memory task performance. Proc Natl Acad Sci U S A. 2007;104(36):14495–14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jutras MJ, Fries P, Buffalo EA. Gamma-band synchronization in the macaque hippocampus and memory formation. J Neurosci. 2009;29(40):12521–12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palop JJ, Chin J, Roberson ED, et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer’s Disease. Neuron. 2007;55(5):697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nisticò R, Mango D, Mandolesi G, et al. Inflammation Subverts Hippocampal Synaptic Plasticity in Experimental Multiple Sclerosis. PLoS One. 2013;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keren-Shaul H, Spinrad A, Weiner A, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 39.Buttgereit A, Lelios I, Yu X, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. 2016;17(12):1397–1406. [DOI] [PubMed] [Google Scholar]
- 40.Mrdjen D, Pavlovic A, Hartmann FJ, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity. 2018;48(2):380–395.e6. [DOI] [PubMed] [Google Scholar]
- 41.Benzing WC, Wujek JR, Ward EK, et al. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20(6):581–589. [DOI] [PubMed] [Google Scholar]
- 42.Cunningham C, Dunne A, Lopez-Rodriguez AB. Astrocytes: Heterogeneous and Dynamic Phenotypes in Neurodegeneration and Innate Immunity. Neuroscientist. 2019;25(5):455–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Escartin C, Guillemaud O, Carrillo-de Sauvage MA. Questions and (some) answers on reactive astrocytes. Glia. 2019;67(12):2221–2247. [DOI] [PubMed] [Google Scholar]
- 44.Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceyzériat K, Ben Haim L, Denizot A, et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henn A, Kirner S, Leist M. TLR2 Hypersensitivity of Astrocytes as Functional Consequence of Previous Inflammatory Episodes. J Immunol. 2011;186(5):3237–3247. [DOI] [PubMed] [Google Scholar]
- 48.Gate D, Saligrama N, Leventhal O, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hohsfield LA, Humpel C. Migration of blood cells to β-amyloid plaques in Alzheimer’s disease. Exp Gerontol. 2015;65:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zenaro E, Pietronigro E, Bianca V Della, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21(8):880–886. [DOI] [PubMed] [Google Scholar]
- 51.Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66(5):631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Junhee Seok H Shaw Warren, Alex GC, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franco Bocanegra DK, Nicoll JAR, Boche D. Innate immunity in Alzheimer’s disease: the relevance of animal models? J Neural Transm. 2018;125(5):827–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shay T, Jojic V, Zuk O, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci U S A. 2013;110(8):2946–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.López-González I, Schlüter A, Aso E, et al. Neuroinflammatory signals in alzheimer disease and APP/PS1 transgenic mice: Correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. 2015;74(4):319–344. [DOI] [PubMed] [Google Scholar]
- 56.Rakic S, Hung YMA, Smith M, et al. Systemic infection modifies the neuroinflammatory response in late stage Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570(7761):332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hannestad J, Gallezot JD, Schafbauer T, et al. Endotoxin-induced systemic inflammation activates microglia: [11C]PBR28 positron emission tomography in nonhuman primates. Neuroimage. 2012;63(1):232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Notter T, Coughlin JM, Gschwind T, et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol Psychiatry. 2018;23(2):323–334. [DOI] [PubMed] [Google Scholar]
- 60.Boche D, Gerhard A, Rodriguez-Vieitez E. Prospects and challenges of imaging neuroinflammation beyond TSPO in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2019;46(13):2831–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Narayanaswami V, Dahl K, Bernard-Gauthier V, Josephson L, Cumming P, Vasdev N. Emerging PET Radiotracers and Targets for Imaging of Neuroinflammation in Neurodegenerative Diseases: Outlook Beyond TSPO. Mol Imaging. 2018;17:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cape E, Hall RJ, van Munster BC, et al. Cerebrospinal fluid markers of neuroinflammation in delirium: A role for interleukin-1β in delirium after hip fracture. J Psychosom Res. 2014;77(3):219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacLullich AMJ, Edelshain BT, Hall RJ, et al. Cerebrospinal fluid interleukin-8 levels are higher in people with hip fracture with perioperative delirium than in controls. J Am Geriatr Soc. 2011;59(6):1151–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skrede K, Wyller TB, Watne LO, Seljeflot I, Juliebø V. Is there a role for monocyte chemoattractant protein-1 in delirium? Novel observations in elderly hip fracture patients. BMC Res Notes. 2015;8(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain Behav Immun. 2010;24(1):102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology. 2004;201(1-3):197–207. [DOI] [PubMed] [Google Scholar]
- 67.Chakravarty S, Herkenham M. Toll-Like Receptor 4 on Nonhematopoietic Cells Sustains CNS Inflammation during Endotoxemia, Independent of Systemic Cytokines. J Neurosci. 2005;25(7):1788–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ebersoldt M, Sharshar T, Annane D. Sepsis-associated delirium. Intensive Care Med. 2007;33(6):941–950. [DOI] [PubMed] [Google Scholar]
- 69.Cunningham C, MacLullich AMJ. At the extreme end of the psychoneuroimmunological spectrum: Delirium as a maladaptive sickness behaviour response. Brain Behav Immun. 2013;28:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang P, Velagapudi R, Kong C, et al. Neurovascular and immune mechanisms that regulate postoperative delirium superimposed on dementia. Alzheimer’s Dement. 2020;16(5):734–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown LJE, Ferner HS, Robertson J, et al. Differential effects of delirium on fluid and crystallized cognitive abilities. Arch Gerontol Geriatr. 2011;52(2):153–158. [DOI] [PubMed] [Google Scholar]
- 72.Vasunilashorn SM, Ngo L, Inouye SK, et al. Cytokines and Postoperative Delirium in Older Patients Undergoing Major Elective Surgery. Journals Gerontol - Ser A Biol Sci Med Sci. 2014;70(10):1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378(6552):75–78. [DOI] [PubMed] [Google Scholar]
- 74.Fuchs EC, Zivkovic AR, Cunningham MO, et al. Recruitment of Parvalbumin-Positive Interneurons Determines Hippocampal Function and Associated Behavior. Neuron. 2007;53(4):591–604. [DOI] [PubMed] [Google Scholar]
- 75.Tamás G, Buhl EH, Lörincz A, Somogyi P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nat Neurosci. 2000;3(4):366–371. [DOI] [PubMed] [Google Scholar]
- 76.Whittington MA, Traub RD, Jefferys JGR. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature. 1995;373(6515):612–615. [DOI] [PubMed] [Google Scholar]
- 77.Yu B, Shinnick-Gallagher P. Interleukin-1?? inhibits synaptic transmission and induces membrane hyperpolarization in amygdala neurons. J Pharmacol Exp Ther. 1994;271(2):590–600. [PubMed] [Google Scholar]
- 78.Wang S, Cheng Q, Malik S, Yang J. Interleukin-1β inhibits γ-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther. 2000;292(2):497–504. [PubMed] [Google Scholar]
- 79.Lai AY, Swayze RD, El-Husseini A, Song C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. 2006;175(1–2):97–106. [DOI] [PubMed] [Google Scholar]
- 80.Wendeln A-C, Degenhardt K, Kaurani L, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556(7701):332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cunningham C, Campion S, Lunnon K, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65(4):304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Torvell M, Hampton DW, Connick P, MacLullich AMJ, Cunningham C, Chandran S. A single systemic inflammatory insult causes acute motor deficits and accelerates disease progression in a mouse model of human tauopathy. Alzheimer’s Dement Transl Res Clin Interv. 2019;5:579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Witlox J, Eurelings LSM, de Jonghe JFM, Kalisvaart KJ, Eikelenboom P, van Gool WA. Delirium in Elderly Patients and the Risk of Postdischarge Mortality, Institutionalization, and Dementia. JAMA. 2010;304(4):443. [DOI] [PubMed] [Google Scholar]
- 84.Casey CP, Lindroth H, Mohanty R, et al. Postoperative delirium is associated with increased plasma neurofilament light. Brain. 2020;143(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leavey N, Hammond SP, Shepstone L, et al. Study protocol: ASCRIBED: the impact of Acute SystematiC inflammation upon cerebRospinal fluId and blood BiomarkErs of brain inflammation and injury in dementia: A study in acute hip fracture patients. BMC Neurol. 2019;19(1):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khachaturian AS, Hayden KM, Devlin JW, et al. International drive to illuminate delirium: A developing public health blueprint for action. Alzheimer’s Dement. 2020;16(5):711–725. [DOI] [PubMed] [Google Scholar]
- 87.Lee HB, Mears SC, Rosenberg PB, Leoutsakos JMS, Gottschalk A, Sieber FE. Predisposing factors for postoperative delirium after hip fracture repair in individuals with and without dementia. J Am Geriatr Soc. 2011;59(12):2306–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahmed S, Leurent B, Sampson EL. Risk factors for incident delirium among older people in acute hospital medical units: A systematic review and meta-analysis. Age Ageing. 2014;43(3):326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Minkeviciene R, Rheims S, Dobszay MB, et al. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29(11):3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Skelly DT, Hennessy E, Dansereau M-A, Cunningham C. A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1β, [corrected] TNF-α and IL-6 challenges in C57BL/6 mice. Block ML, ed. PLoS One. 2013;8(7):e69123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nazmi A, Field RH, Griffin EW, et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia. 2019;67(7):1254–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012;8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2(1):a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wes PD, Holtman IR, Boddeke EWGM, Möller T, Eggen BJL. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia. 2016;64(2):197–213. [DOI] [PubMed] [Google Scholar]
- 97.Wes PD, Sayed FA, Bard F, Gan L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia. 2016;64(10):1710–1732. [DOI] [PubMed] [Google Scholar]
- 98.Hughes MM, Field RH, Perry VH, Murray CL, Cunningham C. Microglia in the degenerating brain are capable of phagocytosis of beads asncd of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia. 2010;58(16):2017–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.White CS, Lawrence CB, Brough D, Rivers-Auty J. Inflammasomes as therapeutic targets for Alzheimer’s disease. Brain Pathol. 2017;27(2):223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liddelow SA, Barres BA. Not everything is scary about a glial scar. Nature. 2016;532(7598):182–183. [DOI] [PubMed] [Google Scholar]
- 102.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Srinivasan K, Friedman BA, Etxeberria A,Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, and Hansen DV. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Reports 2020. 31: 107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A and Schwartz M. Disease-associated astrocytes in Alzheimer’s disease and aging. Nature Neuroscience 2020. 23:701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, Pflueger J, Lister R, Rackham OJL, Petretto E and Jose M. Polo JM A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation Nature Neuroscience. 2019. 22:2087–2097 [DOI] [PubMed] [Google Scholar]
- 106.Henjum K, Quist-Paulsen E, Zetterberg H, Blennow Kaj, Nilsson LNG, Watne LO. CSF sTREM2 in delirium—relation to Alzheimer’s disease CSF biomarkers Aβ42, t-tau and p-tau J. Neuroinflammation 15:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.