

Abstract
Background and Objectives
Recently accumulating evidence suggests the pivotal role of type 1 interferon (IFN-1) signature in the pathogenesis of neuromyelitis optica spectrum disorder (NMOSD). However, the mechanism of the initial trigger that augments IFN-1 pathway in the peripheral immune system of NMOSD has yet to be elucidated.
Methods
Clinical samples were obtained from 32 patients with aquaporin-4 antibody–positive NMOSD and 23 healthy subjects. IFN-1 induction in peripheral blood mononuclear cells (PBMCs) by serum-derived cell-free DNA (cfDNA) was assessed in combination with blockades of DNA sensors in vitro. CfDNA fraction was analyzed for DNA methylation profiles by bisulfite sequencing, elucidating the cellular origin of cfDNA. The induction of neutrophil extracellular trap related cell death (NETosis) was further analyzed in NMOSD and control groups, and the efficacy of pharmacologic intervention of NETosis was assessed.
Results
Enhanced IFN-1 induction by cfDNA derived from NMOSD was observed in PBMCs with cofactor of LL37 antimicrobial peptide. DNase treatment, cGAS inhibitor, and Toll-like receptor 9 antagonist efficiently inhibited IFN-1 production. DNA methylation pattern of cfDNA in patients with NMOSD demonstrated that the predominant cellular source of cfDNA was neutrophils. Whole blood transcriptome analysis also revealed neutrophil activation in NMOSD. In addition, enhanced NETosis induction was observed with NMOSD-derived sera, and efficient pharmacologic inhibition of NETosis with dipyridamole was observed.
Discussion
Our study highlights the previously unrevealed role of cfDNA predominantly released by neutrophil in the induction of IFN-1 signature in NMOSD and further indicate a novel pharmacologic target in NMOSD.
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune inflammatory disease of the CNS.1 Despite the major advancements that have been made on unraveling the pathogenic role of aquaporin-4 (AQP4)-Abs in NMOSD, the precise mechanism underlying the immune dysregulation in NMOSD has yet to be elucidated.
It has been reported that interferon (IFN)-β, a well-known treatment of multiple sclerosis (MS), worsens the disease course of NMOSD,2-5 and several studies have also demonstrated the correlation of elevated type 1 IFN (IFN-1) signature with severe disability in patients with NMOSD.6,7 Serologic coappearance of various antinuclear antibodies, the hallmark of prototypic IFN-1–mediated diseases such as systemic lupus erythematosus (SLE) and Sjögren syndrome, is frequently observed in patients with NMOSD. These clinical observations suggest the pivotal role of IFN-1 signature as an underlying mechanism involved in the pathogenesis of NMOSD.
IFN-1 activates the expression of genes involved in B-cell activation, such as B cell–activating factor,8,9 thus illustrating its involvement in B-cell proliferation, differentiation, and immunoglobulin class switching. Furthermore, IFN-α reportedly acts in concert with interleukin (IL)-6 to promote the differentiation of naive B cells into plasmablasts, thus resulting in the production of pathogenic autoantibodies in SLE.10
These previous observations highlight the critical role of IFN signature in dysregulated immune responses of NMOSD pathogenesis. However, the precise mechanism by which IFN signature is imprinted in patients afflicted with NMOSD has yet to be elucidated. Thus, the purpose of this study was to clarify the initial trigger that leads to the activation of IFN-1 signature in patients with NMOSD and further elucidate the precise mechanism and type of immune cells involved in the process.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The protocol of this study involving human participants was reviewed and approved by the Ethics Committee of Osaka University, Suita, Japan, and written informed consent was obtained from all subjects before their involvement in the study in accordance with the tenets set forth in the Declaration of Helsinki.
Clinical Sample
Clinical samples were obtained from patients with NMOSD positive for AQP4-Abs (n = 32) and healthy control subjects (n = 23). All the patients with NMOSD in this study fulfilled the 2015 NMOSD diagnostic criteria.11 Demographic data of patients with NMOSD and healthy control are described in eTable1. Peripheral blood mononuclear cells (PBMCs) were isolated with Cytiva Ficoll-Paque PLUS (GE Healthcare, Chicago, IL) in accordance with the manufacturer's instructions.
For the cell-free DNA (cfDNA) analysis, the patient's sera were assessed for the mRNA expression of IFN-α when added to healthy PBMCs, and samples were collected from those who showed high IFN-α inducibility of more than 25th percentile in the patients with NMOSD. All the experiments examining the stimulatory effects of either sera or cfDNA were performed on an aliquot of PBMC of allogenic individuals.
IFN-1 Induction by NMOSD Sera
PBMCs (3 × 105 cells per well) were stimulated with 20% NMOSD or healthy control sera and then collected after 8 hours.
Extraction and Measurement of Double-Stranded DNA in Human Sera
Extracellular double-stranded DNA in the sera was extracted by use of the DNA Extractor SP Kit (FujiFilm Wako Pure Chemical Corporation, Osaka, Japan) and quantified by the Quant-iT dsDNA Assay Kit (Invitrogen, Waltham, MA) with Absorbance/Fluorescence Microplate Reader SH9000lab (Corona Electric Co., Ibaraki, Japan) in accordance with the manufacturer's instructions.
Quantitative PCR
For the preparation of the quantitative PCR (qPCR) analysis, total RNA was isolated by the use of the RNeasy Micro Kit (Qiagen, Hilden, Germany). The total RNA was reverse transcribed with SuperScript IV VILO Master Mix (Thermo Fisher Scientific, Waltham, MA) in accordance with the manufacturer's instructions. qPCR analysis was performed with a SYBR Green qPCR Assay Kit (Bio-Rad Laboratories, Hercules, CA) against IFN-α (forward 5′-GACTCCATCTTGGCTGTGA-3′, reverse 5′-TGATTTCTGCTCTGACAACCT-3′), IFN-β (forward 5′-AAACTCATGAGCAGTCTGCA-3′, reverse 5′-AGGAGATCTTCAGTTTCGGAGG-3′) and GAPDH (forward 5′-AATCCCATCACCATCTTCCA-3′, reverse 5′-rTGGACTCCACGACGTACTCA-3′). The results were analyzed by use of the QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA).
Inhibitors of IFN-α Induction
DNaseI RNase-free (Thermo Fisher Scientific, Waltham, MA) was used at a final concentration of 25 units/mL. The sera were incubated with these enzymes for 100 minutes at 37°C. PBMCs (3 × 105 cells per well) were stimulated with 3 μM Toll-like receptor 9 (TLR9) antagonist ODN A151 (InvivoGen, San Diego, CA) for 6 hours or 20 μM cGAS inhibitor G140 (InvivoGen, San Diego, CA) for 3 hours before the PBMCs being stimulated with 20% NMOSD sera. The PBMCs were then collected at 8 hours after stimulation.
cfDNA Analysis
Peripheral blood samples were collected in tubes with ethylenediamine tetraacetic acid. The blood samples were first centrifuged at 1,600g for 10 minutes at 4°C. The plasma fraction was further centrifuged at 16,000g for 10 minutes at 4°C to pellet the residual cells. DNA was extracted from 5 mL of plasma using the MagMAX Cell-Free DNA Isolation Kit (Applied Biosystems, Waltham, MA) in accordance with the manufacturer's instructions. The concentrations of DNA were measured by the use of the Quant-iT dsDNA Assay Kit. For DNA sequencing, plasma DNA libraries were prepared using the Accel-NGS Methyl-Seq DNA library Kit and Methyl-Seq Set A Indexing Kit (Swift Biosciences, Ann Arbor, MI). The bisulfite-treated DNA libraries were sequenced on a NovaSeq 6000 Genome Sequencing System (Illumina, San Diego, CA). The KL divergence for each number of reads was calculated, and the variance was simulated. Based on the results, a sufficient number of reads above 20 ×106 reads were analyzed (eFigure 1A, links.lww.com/NXI/A698). The raw FASTQ sequencing files were downloaded and then trimmed using TrimGalore with the default parameters. Trimmed FASTQ sequencing files were aligned on the human genome reference, i.e., GENCODE version 34 using the Bismark software program with the default parameters. The deduplication and the extraction of methylation information were performed using deduplicate_bismark and bismark_methylation_extractor. Finally, the deconvolution of the cell of origin of cfDNA was performed using deconvolve.py12 with modification to apply the tool to BS-seq (github.com//yyoshiaki/methatlas).
LL37/HNP in Sera
The concentration of LL37 and HNP1-3 in the NMOSD sera and the healthy control sera was measured using a commercially available kit (Hycult Biotech, Uden, Netherlands).
LL37/HNP and cfDNA
The cfDNA was extracted from NMOSD or healthy control sera and quantified by the use of the Quant-iT dsDNA Assay Kit. PBMCs (3 × 105 cells per well) or dendritic cells (DCs) (2 × 104 cells per well) were incubated with cfDNA extracted from 3 mL of sera per well and LL37 (10 μM; AnaSpec, Fremont, CA) or HNP (10 μM; Hycult Biotechnology, Uden, Netherlands). After the cells were cultured overnight, the culture media was collected. IFN-α in the culture media was measured using the IFN alpha Human ELISA Kit (Invitrogen, Waltham, MA).
Fluorescent-Activated Cell Sorting (FACS) Analysis
For FACS analysis, PBMCs from healthy individuals were stained using Pacific Blue-labeled anti-HLA-DR mAb (1:100; Becton Dickinson, Franklin Lakes, NJ), PE-Cy7-labeled anti-CD3, CD14, CD19, and CD56 mAb (1:100; Becton Dickinson, Franklin Lakes, NJ), APC-labeled anti-CD11c mAb (1:5; Becton Dickinson), and FITC-labeled anti-CD123 mAb (1:5; Becton Dickinson). DCs were isolated and quantified using a BD FACSAria II System (Becton Dickinson). HLA-DR (+) Lineage (−) CD11c (−) CD123 (+) was defined as plasmacytoid dendritic cells (pDCs), and HLA-DR (+) Lineage (−) CD11c (+) was defined as conventional dendritic cells (cDCs) as previously reported.13
Analysis of RNA-Seq Data
Read count data of whole blood RNA-seq in patients with NMOSD from the previous article6 were obtained and reanalyzed. RNA sequence data were analyzed with iDEP (an integrated web application for differential expression and pathway analysis of RNA-seq data).14 For the analysis, iDEP v0.92 was used, and the parameters were set at default. Principal component analysis, heatmap, and gene ontology (GO) analysis based on General Applicable Gene-set Enrichment (GAGE) was performed in the healthy control subjects and the patients with AQP4-Ab–positive NMOSD.
Human Neutrophil Purification
For neutrophil preparation, blood was collected in heparin tubes by standard phlebotomy techniques.15,16 The blood was fractionated by density-gradient centrifugation using Cytiva Ficoll-Paque PLUS. Neutrophils were further purified by dextran sedimentation of the red blood cell layer followed by lysis of residual erythrocytes with 0.2% sodium chloride. The purity of the neutrophils was confirmed by flow cytometry.
Detection and Quantitation of NET
Neutrophils were seeded on Iwaki chamber slides at a density of 106 cells/mL. Cells were incubated for 1 hour in a CO2 incubator at 37°C and then stimulated with 20% NMOSD, healthy control sera, phorbol myristate acetate (PMA) (100 nM), and phosphate-buffered saline (PBS). Dipyridamole (Sigma-Aldrich, St Louis, MO) was added to a final concentration of 40 μM. After stimulation for 4 hours, each well was washed with PBS, and then, SYTOX green stain (Invitrogen, Waltham, MA) was added to nonfixed live cells to ensure that only extracellular DNA was detected.17 Immunofluorescence confocal microscopy was performed by use of a BZ-X710 All-In-One Fluorescence Microscope (Keyence, Osaka, Japan), and the images were analyzed with ImageJ software (NIH, Bethesda, MD).
The human NET formation was quantitated by the use of the NETosis Assay Kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's protocol. Isolated neutrophils were primed with tumor necrosis factor (TNF)-α (2 ng/mL) for 15 minutes.
Statistical Analysis
p Values were calculated by the Mann-Whitney U test (2-tailed) for comparison between 2 groups and the Kruskal-Wallis H-test for comparison between multiple groups. The Wilcoxon signed-rank test was used to compare between 2 groups with correspondence. A p value of <0.05 was considered statistically significant.
Data Availability
Data that support the findings of this study are available from the corresponding author on reasonable request.
Results
Sera Derived From Patients With NMOSD Enhance IFN-1 Gene Expression in Peripheral Blood Mononuclear Cells
To clarify whether IFN-1 signature is enhanced in patients with NMOSD, we compared the gene expression of IFN-α and IFN-β by qPCR in PBMCs derived from either healthy control subjects or patients with NMOSD. In both groups, IFN-α gene expression was more prominent than IFN-β, and significantly enhanced gene expression of both IFN-α and IFN-β was observed in the patients with NMOSD compared with that in the healthy control subjects (Figure 1A).
Figure 1. Cell-Free DNA Derived From NMOSD Patient Sera Enhances IFN-1 Gene Expression in PBMCs.
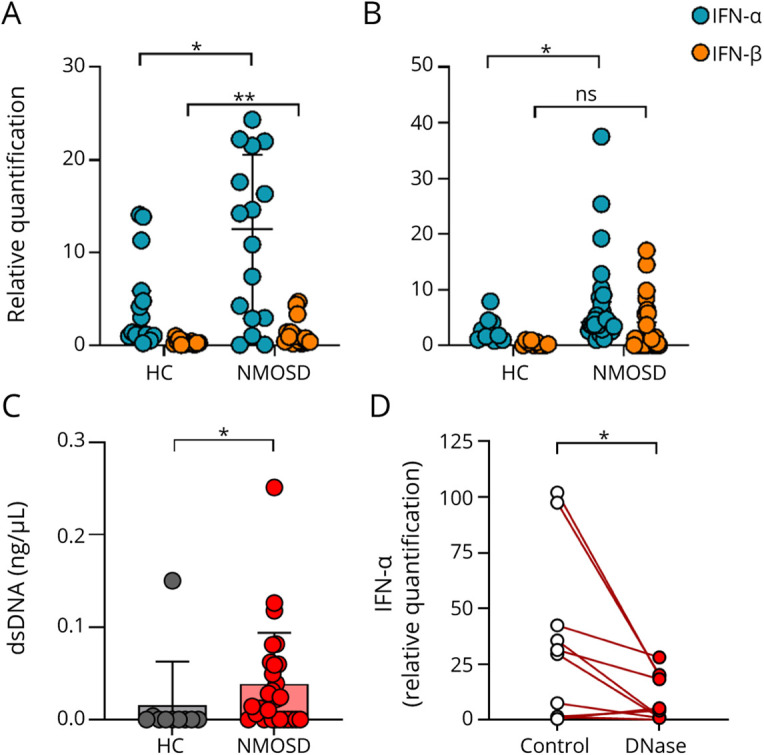
(A) IFN-α and IFN-β gene expression levels of PBMCs derived from patients with NMOSD (n = 16) and healthy control subjects (n = 15). The Mann-Whitney U test was used to determine statistical significance. (B) Gene expression levels of IFN-α and IFN-β of healthy PBMCs stimulated with either NMOSD sera (n = 30) or healthy control sera (n = 10). The Mann-Whitney U test was used to determine statistical significance. (C) Concentration of double-stranded DNA in sera. DNA was purified from NMOSD sera (n = 28) or healthy control sera (n = 10) and quantified. The Mann-Whitney U test was used to determine statistical significance. (D) The expression of IFN-α of healthy control PBMCs stimulated by NMOSD sera, which was pretreated with DNase (n = 10). The Wilcoxon signed-rank test was used to determine statistical significance. *p < 0.05, **p < 0.005. HC = healthy control; ns = not significant.
Next, we examined whether sera derived from patients with NMOSD possess higher capacity to induce IFN-1 gene expression in PBMCs. Sera of either the healthy control subjects or the patients with NMOSD were incubated with PBMCs derived from healthy controls and subsequently analyzed for the gene expression of IFN-α and IFN-β. Our findings showed that IFN-α was significantly induced exclusively in the PBMCs incubated with sera derived from the patients with NMOSD (Figure 1B). In contrast, the IFN-β gene expression level was comparable between both groups.
IFN-α Is Induced by Cell-Free DNA Derived From the Sera of Patients With NMOSD
Various factors have previously been reported as potent inducers of IFN-1 in autoimmune diseases. Recently, there is accumulating evidence that demonstrates the presence of extracellular DNA fraction, termed cfDNA, in biofluid. Moreover, it has been reported that in SLE, a prototypic IFN-1–mediated disease, serum cfDNA plays a pivotal role in the production of IFN-1.18,19
We revealed that DNA concentration was significantly higher in the sera derived from the patients with NMOSD compared with that of the control group (Figure 1C). To further clarify whether cfDNA fraction derived from patients with NMOSD has the capacity to induce IFN-1 production, PBMCs derived from healthy control subjects were incubated with NMOSD sera in both the presence and absence of DNase. DNase treatment significantly inhibited the gene expression of IFN-α in the PBMCs (Figure 1D).
cfDNA Is Predominantly Derived From Neutrophils in Patients With NMOSD
As serum cfDNA of NMOSD was found to be a potent inducer of IFN-1 production, we next aimed to investigate the primary source of cfDNA production. The cfDNA was collected from the plasma of both healthy control subjects and patients with NMOSD, and DNA methylation profiles were analyzed by bisulfite sequencing (Figure 2A). The cell of origin of the cfDNA was estimated by integrating DNA methylation rate references of 25 tissues and cell types as previously described.12 In both the healthy control subjects and the patients with NMOSD, the majority of cfDNA is produced by neutrophils and erythrocyte progenitors (Figure 2B).
Figure 2. Concentration of Neutrophil-Derived cfDNA Is Elevated in Patients With NMOSD.

(A) Schematic representation of the calculation of tissue contribution based on tissue-specific methylation profiles. (B) Heat map visualization of cfDNA concentration from different sources. (C) cfDNA concentration derived from the neutrophils in NMOSD plasma (n = 10) or healthy control plasma (n = 8). The Mann-Whitney U test was used to determine statistical significance. ***p < 0.002. HC = healthy control.
The total amount of cfDNA derived neutrophils was significantly elevated in NMOSD (Figure 2C), whereas no significant difference in the proportion of cfDNA derived from neutrophils was found between NMOSD and the control groups (eFigure 1B, links.lww.com/NXI/A698). In contrast, the total amount of cfDNA derived from other cell types, including erythroid progenitors, was not significantly increased. No significant correlation was found between the neutrophil-derived cfDNA and the total number of neutrophils in the peripheral blood or the dosage of prednisolone (eFigure 1C, links.lww.com/NXI/A698).
Importantly, further reference to various clinical data revealed a correlation between neutrophil-derived cfDNA concentration and the length of spinal-cord lesions in patients with NMOSD (eFigure 1D, links.lww.com/NXI/A698). However, no correlation was found between the neutrophil-derived cfDNA concentration and other clinical data such as serum AQP4-Abs titers and annualized relapse rate (data not shown). These results together suggest that neutrophils are an essential source of cfDNA and that the amount of the neutrophil-derived cfDNA is more prominently present in patients with NMOSD.
cfDNA of Patients With NMOSD Induces IFN-1 Production in the Presence of LL37
It is known that nuclear-derived self-DNA is not taken up by DCs without the presence of cofactors such as antimicrobial peptides.20,21 To further clarify the mechanism by which cfDNA induces the production of IFN-1, we examined the amount of LL37 and HNP, antimicrobial peptides that reportedly elicit host-derived DNA by DCs.20,21 Although some patients with NMOSD tended to show higher concentration of both LL37 and HNP in serum compared with that in the control group, no statistically significant difference in the levels was found between the 2 groups (Figure 3A.a). To examine whether cfDNA alone can induce the production of IFN-1 in patients with NMOSD, PBMCs derived from healthy control subjects were incubated with both healthy control and NMOSD patient cfDNA fraction samples. When PBMCs were cultured with cfDNA in the presence of LL37, but not HNP, IFN-α in the culture media significantly increased in both the NMOSD patient and healthy control groups (Figure 3A.b and A.c). In addition, a significant increase in IFN-α was found in the culture media of the NMOSD group patients in comparison to that in the healthy control group subjects (Figure 3A.b).
Figure 3. cfDNA Derived From Patients With NMOSD Induces the Production of IFN-α in the Presence of LL37.

(A.a) LL37 (ng/mL) and HNP (ng/mL) in NMOSD sera (n = 23) or healthy control sera (n = 16). The Mann-Whitney U test was used to determine statistical significance. (A.b) (A.c) IFN-α (pg/mL) in culture media. After stimulation of PBMCs with cfDNA extracted from NMOSD sera (n = 10) or healthy control sera (n = 10) in the presence or absence of LL37 (10 μM) (A.b) or HNP (10 μM) (A.c), culture media were collected and assayed by ELISA. The Kruskal-Wallis H-test (Bonferroni adjusted) was used to determine statistical significance. (B) The expression of IFN-α of healthy control PBMCs stimulated by NMOSD sera incubated with cGAS inhibitor (G140) or TLR9 antagonist (ODN A151) (n = 10). The Wilcoxon signed-rank test was used to determine statistical significance. (C) Gating strategy for cell sorting. HLA-DR (+) Lineage (−) CD11c (−) CD123 (+) was defined as pDCs. HLA-DR (+) Lineage (−) CD11c (+) was defined as cDCs. (D) IFN-α (pg/mL) in culture media. After treatment of pDCs (D.a) or cDCs (D.b) with cfDNA extracted from NMOSD sera (n = 11) or healthy control sera (n = 11) in the presence of LL37 (10 μM), culture media were collected and assayed by ELISA. The Kruskal-Wallis H-test (Bonferroni adjusted) was used to determine statistical significance. *p < 0.05, **p < 0.005. HC = healthy control; ns = not significant.
cfDNA Derived From NMOSD Induces IFN-1 Production via the cGAS-STING and Toll-like Receptor 9 Pathways
Recognition of nucleic acid derived from host cells is mediated by several innate receptors, leading to IFN-1 production. Thus, we first investigated whether IFN-α gene expression induced by NMOSD sera is inhibited by G140, a cGAS-specific inhibitor. When PBMCs collected from healthy donors were cultured with NMOSD sera, either with or without G140, there was a reduction of IFN-α gene expression in the G140-treated group (Figure 3B). In addition to the cGAS-STING pathway, TLR9 is a well-known innate receptor involved in nucleic acid sensing, which is present in endosomal compartments. Thus, we further investigated the inhibitory effect of ODN A151, a TLR9 antagonist, on IFN-α gene induction by NMOSD sera. When PBMCs collected from healthy donor subjects were cultured with NMOSD sera, either with or without ODN A151, ODN A151 also showed a significantly suppressed IFN-α gene expression pattern (Figure 3B).
To clarify the major type of cells involved in IFN-α production induced by NMOSD-derived cfDNA, we next separately isolated populations of pDCs and cDCs from PBMCs of healthy control subjects (Figure 3C). cfDNA extracted from either NMOSD or healthy control sera was added to pDCs and cDCs, respectively, in the presence of LL37, and IFN-α secreted in the culture media of each group was then compared. The pDCs incubated with NMOSD-derived cfDNA alone showed significant production of IFN-α (Figure 3D.a). In contrast, no statistically significant difference was observed among the populations of all groups of cDCs (Figure 3D.b).
Whole Blood Transcriptome Reveals Neutrophil Activation in Patients With NMOSD
To clarify the unbiased immune landscape of the peripheral immune dysregulation of patients with NMOSD, whole blood transcriptome of patients with NMOSD was analyzed referencing to the previously reported RNA sequencing data set.6 The data sets were used to perform a principal component analysis to compare the patients with NMOSD and healthy control subjects (Figure 4A).
Figure 4. Whole Blood Transcriptome Reveals Neutrophil Activation in Patients With NMOSD.

(A) Principal component analysis indicating transcriptional clustering in whole blood of healthy control subjects (n = 18) and patients with AQP4 antibody–positive NMOSD (n = 26). (B) Visualization of networks between the gene ontology (GO) biological process in AQP4 antibody–positive NMOSD whole blood RNA-seq. GO analysis based on GAGE was performed in healthy controls (n = 18) and patients with AQP4 antibody–positive NMOSD (n = 26). (C) The top 10 GO terms associated with biological process in AQP4 antibody–positive NMOSD whole blood RNA-seq. HC = healthy control.
When GO analysis was performed between the patients with NMOSD and healthy control subjects using the GAGE method, various pathways related to neutrophil-mediated responses were found to be activated in the NMOSD group (Figure 4, B and C). The identified GO pathways encompassed neutrophil activation, neutrophil degranulation, and neutrophil-mediated immunity.
NMOSD Patient–Derived Sera Are a Potent Inducer of NETosis
Based on the observation suggesting that neutrophils are constitutively activated, as well as the finding that the majority of cfDNA is neutrophil-derived in NMOSD, we next aimed to investigate whether NETosis, a form of neutrophil cell death implicated in aberrant IFN-1 production, is efficiently induced by NMOSD sera.
Neutrophils isolated from healthy donors were stimulated with sera derived from either patients with NMOSD or healthy control subjects for 4 hours and subsequently stained with SYTOX green, a cell-impermeable DNA-binding dye. The number of SYTOX green–positive cells was significantly increased in NMOSD sera–treated group compared with that in the controls (Figure 5A). In addition, neutrophil elastase (a well-known marker that represents the degree of NETosis) was quantified to exclude the possibility that DNA is released independently of NETosis. As a result, in the NMOSD sera–treated group, significantly enhanced detection of elastase was observed compared with that in the control group (Figure 5B).
Figure 5. Sera Derived From Patients With NMOSD Induce NETosis.

(A.a) Immunofluorescence analysis with SYTOX green nucleic acid stain was performed on healthy control neutrophils stimulated by PMA (100 nM), PBS, HC sera, or NMOSD sera. Scale bar: 200 μm. (A.b) Quantification of NET formation in healthy control neutrophils, which stimulated by NMOSD sera (n = 8) or healthy control sera (n = 3). Data are expressed as total SYTOX green–positive extracellular DNA area. The Mann-Whitney U test was used to determine statistical significance. (B) NET-associated elastase in healthy control neutrophils stimulated by NMOSD sera (n = 8) or healthy control sera (n = 4) was measured by ELISA. The Mann-Whitney U test was used to determine statistical significance. (C.a) SYTOX green immunofluorescence analysis was performed on healthy control neutrophils stimulated by NMOSD sera with or without dipyridamole (40 μM). Scale bar: 200 μm (C.b) Quantification of NET formation in healthy control neutrophils stimulated by NMOSD sera (n = 8) with or without dipyridamole (40 μM). Data are expressed as total SYTOX green–positive extracellular DNA area. The Wilcoxon signed-rank test was used to determine statistical significance. (D) NET-associated elastase in healthy control neutrophils stimulated by NMOSD sera (n = 11) with or without dipyridamole (40 μM) was measured by ELISA. The Wilcoxon signed-rank test was used to determine statistical significance. *p < 0.05, **p < 0.005. HC = healthy control.
Because these results demonstrated that NMOSD sera are a potent inducer of NETosis, we next examined whether AQP4-Abs alone are capable of eliciting NETosis. Neutrophils exposed to patient-derived recombinant AQP4-Abs did not induce NETosis when assessed by SYTOX green (eFigure 2, links.lww.com/NXI/A698), indicating that certain unidentified factors other than AQP4-Abs present in NMOSD sera possess the capacity to induce NETosis.
Dipyridamole Inhibits NETosis Induced by Sera Derived From Patients With NMOSD
Reportedly, surface adenosine receptors trigger cyclic AMP (cAMP) formation in neutrophils and subsequently regulate excess NETosis induction. Moreover, it has been reported that dipyridamole, a phosphodiesterase inhibitor, increases extracellular concentrations of adenosine, thereby interfering with the breakdown of cAMP.22 To examine whether NETosis induction by NMOSD patient sera can be pharmacologically suppressed by dipyridamole, neutrophils stimulated with NMOSD sera were cultured in the presence and absence of dipyridamole. We revealed that treatment with dipyridamole resulted in a significant reduction of SYTOX green–positive cells (Figure 5C). Moreover, neutrophil elastase was consistently significantly inhibited via the addition of dipyridamole (Figure 5D). Altogether, our data lend support to a model in which NETosis is involved in the pathogenesis of patients with NMOSD via IFN-1 signature, and dipyridamole reduces the aberrant induction of NETosis in patients with NMOSD (eFigure 3, links.lww.com/NXI/A698).
Discussion
The cfDNA is released on cell death and is known to contain fragments of nuclear DNA. There are several approaches for identifying the origin of cfDNA based on tissue-specific epigenetics. Using that cfDNA preserves tissue- or cell type–specific DNA methylation patterns of the original tissues,23 methods have been developed to predict the origin of cfDNA by integrating the predefined reference of tissue- or cell type–specific DNA methylation patterns. In a recent study, the development of a highly specific and sensitive method for estimating the origin of cfDNA by deconvolution of plasma methylation is reported.12 Thus, in this study, we used those methods to estimate the origin of cfDNA in NMOSD. To the best of our knowledge, there have been few reports on the qualitative evaluation of cfDNA in autoimmune diseases,24 and evaluation of the origin of cfDNA based on the DNA methylation patterns by bisulfite sequencing in autoimmune diseases, including NMOSD, remains to be clarified. In this study, we found neutrophils to be the predominant source of cfDNA. Thus, it is possible that neutrophil-derived cfDNA forms a complex with LL37, which then stimulates cGAS or TLR9 to produce IFN-1. It has been shown that human DNA binds to LL37 and forms a complex that allows it to be readily incorporated into pDCs.20,21 Reportedly, this LL37-DNA complex is retained in the early endocytotic compartments and stimulates TLR9.21 Similarly, in monocytes, the LL37-DNA complex has been shown to efficiently escape from endosomal vesicles, enter the cytosolic compartments, and stimulate the cGAS-STING pathway to induce IFN production.25
It is generally accepted that various stimuli can induce NETosis, including pathogens inflammatory cytokines, and activated platelets.26,27 For example, it has been suggested that in rheumatoid arthritis, not only antibodies such as anticitrullinated vimentin antibodies but also cytokines such as IL17A and TNF-α induce NETosis.28 It has also been reported that TNF-α and IL-1β, which are known to be elevated in NMOSD,29 induce NETosis in vitro.27,30 In our study, we found that NETosis was upstream of IFN signature. There are also reports that recombinant human IFN-β with complement induced NETosis in vitro.31 Thus, it is possible that NETosis is not a simple 1-way relationship but induces a positive feedback loop of IFN-1 production.
Although recently approved monoclonal antibodies efficiently prevent clinical relapses by inhibiting the production or pathogenicity of AQP4-Abs, a therapeutic approach that effectively intervenes with the dysregulated upstream immune signature of NMOSD has yet to be developed. On the other hand, in patients with SLE, anifrolumab, a monoclonal antibody against IFN-1 receptor subunit 1, is currently undergoing clinical trials and has shown efficacy against the placebo.32 Our results suggest that the regulation of IFN-1 and, potentially, NETosis and cfDNA may be applicable to the treatment of patients with NMOSD.
In this study, we reanalyzed the read count data of AQP4-Ab–positive NMOSD and healthy control from the previous article.6 One of the limitations in our study is that we could not consider covariates in the analysis of the whole blood transcriptome, as the demographic data on healthy control were not publicly available.
In conclusion, the findings in this study highlight the previously unrevealed roles of the neutrophil pathway in the NMOSD immune signature and provide a novel avenue for the development of a pharmacologic agent.
Acknowledgment
The authors thank Yoko Goto and Akiko Mikami for their technical assistance.
Glossary
- AQP4
aquaporin-4
- cAMP
cyclic AMP
- cDC
conventional dendritic cell
- cfDNA
cell-free DNA
- DC
dendritic cell
- FACS
fluorescent-activated cell sorting
- GAGE
General Applicable Gene-set Enrichment
- GO
gene ontology
- IFN
interferon
- IL
interleukin
- MS
multiple sclerosis
- NMOSD
neuromyelitis optica spectrum disorder
- PBMC
peripheral blood mononuclear cell
- pDC
plasmacytoid dendritic cell
- PMA
phorbol myristate acetate
- PBS
phosphate-buffered saline
- qPCR
quantitative PCR
- SLE
systemic lupus erythematosus
- TLR9
Toll-like receptor 9
- TNF
tumor necrosis factor
Appendix. Authors
Contributor Information
Hisashi Murata, Email: hisa310v_v@yahoo.co.jp.
Yoshiaki Yasumizu, Email: yyasumizu@ifrec.osaka-u.ac.jp.
Daisuke Motooka, Email: daisukem@gen-info.osaka-u.ac.jp.
Shohei Beppu, Email: makku_syo@hotmail.com.
Naoyuki Shiraishi, Email: pinan23145@gmail.com.
Yasuko Sugiyama, Email: mirulala0929@yahoo.co.jp.
Keigo Kihara, Email: kinoko1658@yahoo.co.jp.
Satoru Tada, Email: route135@gmail.com.
Toru Koda, Email: toru.koda@dmi.med.osaka-u.ac.jp.
Hachiro Konaka, Email: h-konaka@imed3.med.osaka-u.ac.jp.
Hyota Takamatsu, Email: thyota@imed3.med.osaka-u.ac.jp.
Atsushi Kumanogoh, Email: kumanogo@imed3.med.osaka-u.ac.jp.
Tatsusada Okuno, Email: okuno@neurol.med.osaka-u.ac.jp.
Hideki Mochizuki, Email: hmochizuki@neurol.med.osaka-u.ac.jp.
Study Funding
This study was supported by a JSPS KAKENHI Grant Number JP20K07759.
Disclosure
The authors report no disclosures related to the manuscript. Go to Neurology.org/NN for full disclosures.
Publication History
Received by Neurology: Neuroimmunology & Neuroinflammation October 27, 2021. Accepted in final form January 12, 2022. Submitted and externally peer reviewed. The handling editor was Scott S. Zamvil, MD, PhD, FAAN.
References
- 1.Liu Y, Zhang H, Zhang TX, et al. Effects of tocilizumab therapy on circulating B cells and T helper cells in patients with neuromyelitis optica spectrum disorder. Front Immunol. 2021;12:703931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler. 2012;18(10):1480-1483. [DOI] [PubMed] [Google Scholar]
- 3.Palace J, Leite MI, Nairne A, Vincent A. Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010;67(8):1016-1017. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu J, Hatanaka Y, Hasegawa M, et al. IFNβ-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010;75(16):1423-1427. [DOI] [PubMed] [Google Scholar]
- 5.Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol. 2010;17(5):672-676. [DOI] [PubMed] [Google Scholar]
- 6.Agasing AM, Wu Q, Khatri B, et al. Transcriptomics and proteomics reveal a cooperation between interferon and T-helper 17 cells in neuromyelitis optica. Nat Commun. 2020;11(1):2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asgari N, Voss A, Steenstrup T, Kyvik KO, Stenager E, Lillevang ST. Interferon alpha association with neuromyelitis optica. Clin Dev Immunol. 2013;2013:713519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A. Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol. 2012;90(5):498-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sjöstrand M, Johansson A, Aqrawi L, Olsson T, Wahren-Herlenius M, Espinosa A. The expression of BAFF is controlled by IRF transcription factors. J Immunol. 2016;196(1):91-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menon M, Blair PA, Isenberg DA, Mauri C. A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity. 2016;44(3):683-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9(1):5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villani AC, Satija R, Reynolds G, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356(6335):eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. 2018;19(1):534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhns DB, Priel DAL, Chu J, Zarember KA. Isolation and functional analysis of human neutrophils. Curr Protoc Immunol. 2015;111:7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishide M, Nojima S, Ito D, et al. Semaphorin 4D inhibits neutrophil activation and is involved in the pathogenesis of neutrophil-mediated autoimmune vasculitis. Ann Rheum Dis. 2017;76(8):1440-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato Y, Park J, Takamatsu H, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. 2018;77(10):1507-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duvvuri B, Lood C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. Front Immunol. 2019;10:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3(73):73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564-569. [DOI] [PubMed] [Google Scholar]
- 22.Ali RA, Gandhi AA, Meng H, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. 2019;10(1):1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kundaje A, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan RW, Jiang P, Peng X, et al. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci U S A. 2014;111(49):E5302-E5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chamilos G, Gregorio J, Meller S, et al. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood. 2012;120(18):3699-3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661-2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keshari RS, Jyoti A, Dubey M, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One. 2012;7(10):e48111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5(178):178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang T, Wang S, Zheng Q, et al. Increased plasma levels of epithelial neutrophil-activating peptide 78/CXCL5 during the remission of Neuromyelitis optica. BMC Neurol. 2016;16:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta AK, Joshi MB, Philippova M, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584(14):3193-3197. [DOI] [PubMed] [Google Scholar]
- 31.Herges K, de Jong BA, Kolkowitz I, et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult Scler. 2012;18(4):398-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka Y, Tummala R. Anifrolumab, a monoclonal antibody to the type I interferon receptor subunit 1, for the treatment of systemic lupus erythematosus: an overview from clinical trials. Mod Rheumatol. 2021;31(1):1-12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data that support the findings of this study are available from the corresponding author on reasonable request.