

Abstract
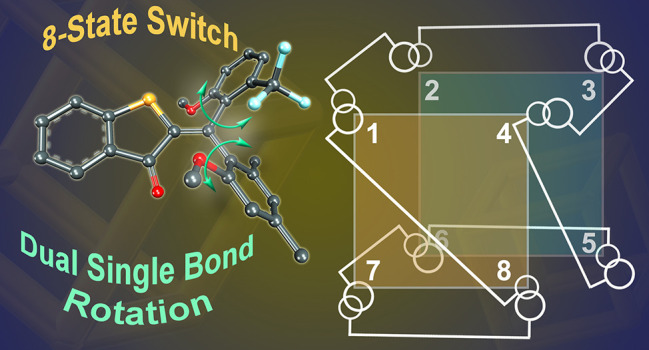
Typical photoswitches interconvert between two different states by simple isomerization reactions, which represents a fundamental limit for applications. To expand the switching capacity usually different photoswitches have to be linked together leading to strong increase in molecular weight, diminished switching function, and less precision and selectivity of switching events. Herein we present an approach for solving this essential problem with a different photoswitching concept. A basic molecular switch architecture provides precision photoswitching between eight different states via controlled rotations around three adjacent covalent bonds. All eight states can be populated one after another in an eight-step cycle by alternating between photochemical Hula-Twist isomerizations and thermal single-bond rotations. By simply changing solvent and temperature the same switch can also undergo a different cycle instead interconverting just five isomers in a selective sequence. This behavior is enabled through the discovery of an unprecedented photoreaction, a one-photon dual single-bond rotation.
Introduction
Molecular photoswitches are at the center of attention in smart nanotechnology research enabling high-precision control of events at the smallest scales and instilling bottom-up responsiveness and adaptability into materials, drugs, or catalytic reactions. Consequently, photoswitches are used for switching or gradual modulation of functions in myriads of applications nowadays and developments are ever steeply increasing.1−16 Roughly two different classes of photoswitches can be distinguished: bond isomerizing switches that undergo significant changes in molecular geometry and switches that provide substantial changes in their electronic structure.6,13,17,18 The overwhelming majority of light-induced bond isomerizations are achieved by simple one-bond rotations such as double-bond isomerization (DBI) reactions.8,12,19−21 Direct experimental proof for more complex coupled bond isomerizations, where more than one bond rotates concomitantly, are so far only available for bicycle pedal22 and Hula-Twist photoreactions23,24 (Figure 1a) although such simultaneous photochemical processes are proposed frequently in the literature.25−32
Figure 1.

Molecular photoswitching. (a) Bond rotations currently evidenced in photoswitching. (b) Multistate switching approaches. (c) Simple and compact eight-state photoswitch possessing eight different isomeric states and carrying out a hitherto unknown dual single-bond rotation (DSBR) photoreaction (top). Changing conditions allow to branch between Hula-Twist or DSBR photoreactions allowing for two different sequential switching cycles (bottom).
At the current state of the art most small molecular photoswitches can undergo reversible changes between two different states upon irradiation with light. To go beyond this binary behavior and open up more fundamental switching possibilities (Figure 1b), multiple photoresponsive units can be incorporated into a single larger molecule. This is achieved by linking either the same or different photochromic units together in a covalent fashion or through weaker interactions.16,33−38 Many well-known photoswitches like azobenzene,37,39−43 diarylethene,44−47 or spiropyranes48 have been interlinked in such a manner but the method can be extended to virtually any photochromic dye34,35,49 or dye-combination.50−52 An impressive escalation in the number of switching states is thus achievable, for example six different states are possible by connecting three light-responsive units covalently into one structure.51 Another possibility to enhance the switching-state number for a given photochromic molecule is provided by adding chemical signals like protonation and deprotonation.7,53−55 Alternatively, photon-driven molecular motors can be considered as compact multistate photoswitches,56−59 usually operating by alternating four different diastereomeric states.56,60−65 The advantage of the latter is their unequivocally more precise geometry control as opposed to the oftentimes rather loose changes obtained in multicomponent architectures. We recently contributed different types of visible-light-driven molecular motors to the field, featuring different rotation mechanisms and alternating between three to four different states.24,63,66 Up to five distinct states could be accessed in a macrocyclic integrated hemithioindigo (HTI) motor.67 Despite this steep progress in molecular photoswitches research, controlling more complex motions and using the full state-density capacity offered by small-molecular frameworks remain untackled challenges at present.
Herein we report on a simple and compact molecular setup 1 assuming eight different stable states, which interchange one after another by alternating visible light irradiation and heating steps. The photochemical steps encompass a variant of the Hula-Twist photoreaction as well as a hitherto unknown dual single-bond rotation (DSBR) photoreaction (Figure 1c). The thermal steps are sole single-bond rotations (SBRs). A change of the solvent polarity enables control of the sequence of isomer interconversions, and either eight or five states are interchanged under irradiation and heating in specific orders (Figure 1c, bottom). With this compact multistate photoswitch architecture, the next level in molecular addressability has been achieved providing exquisite control over precise molecular geometry changes via light signaling. At the same time, a so far unknown coupled photoisomerization reaction is discovered, which provides molecular scientists with a novel light-inducible molecular motion type.
Results and Discussion
Sequential switch 1 is derived from the parent HTI chromophore but contains two different aryl groups geminally connected to the photoisomerizable double bond. Both aryl groups are nonsymmetrically substituted thus establishing a chiral axis each, which coincides with the respective single bond connecting to the central double bond. Therefore, sequential switch 1 exists in four diastereomeric states and eight enantiomeric states. All states of 1, denoted as A (Z-(Sa)-(Ra) configuration), enantiomeric A′ (Z-(Ra)-(Sa) configuration), B (Z-(Ra)-(Ra) configuration), enantiomeric B′ (Z-(Sa)-(Sa) configuration), C (E-(Ra)-(Ra) configuration), enantiomeric C′ (E-(Sa)-(Sa) configuration), D (E-(Ra)-(Sa) configuration), and enantiomeric D′ (E-(Sa)-(Ra) configuration), are depicted in Figure 2a.
Figure 2.

Structures of all eight isomers of sequential HTI switch 1 and corresponding ECD-spectra of enantiomerically pure samples. (a) Schematic representation of the molecular structures of 1 with stereo labels. (b) Structures of isomers A′ (from racemic crystals), B′ (from racemic crystals), C′ and C in the crystalline state and corresponding ECD-spectra in MeCN at 20 °C of A, A′, B, B′, C/D, and C′/D′. Experimental spectra (solid lines) were compared with calculated spectra (B3LYP/6-311G(d,p) level of theory using a PCM solvent model for MeCN, broken lines).
Synthesis of 1 followed an established protocol for the generation of 4-fold double-bond substituted HTIs (see the Supporting Information).68 Because of the increased steric hindrance of the two aryl substituents, all atropisomers show increased thermal stability facilitating isolation and analysis. Isomers rac-A and rac-B are stable enough at 22 °C to enable separation by chromatography methods. Isomers rac-C and rac-D interconvert rapidly at 22 °C, making separation impossible. However, since isomer rac-C is significantly more stable, it is mainly populated at ambient temperatures as confirmed by NMR spectroscopy. Consequently, it is also the only isomer, which crystallizes from a solution of rac-C and rac-D isomers and can thus be obtained in highly enriched form. These results are in good agreement with the theoretical description predicting isomers rac-D to be 0.63 kcal/mol higher in energy as compared to rac-C (see Figure 3 and the Supporting Information).
Figure 3.

Thermal properties of sequential HTI switch 1. (a) Kinetics of the thermal atropisomerization from A′ to B′ in MeCN at 60 °C followed by chiral HPLC. Experimental data were fitted using a Markov matrix kinetic model (see Supporting Information for details). (b) Kinetics of the thermal atropisomerization from rac-D to rac-C in MeCN-d3 at −20 °C followed by 1H NMR spectroscopy (400 MHz, −20 °C). (c) Ground-state energy profile of HTI 1 experimentally determined in MeCN-d3 solution (red) and calculated at the B3LP/6-311G(d,p) of level of theory with a PCM solvent model for MeCN (blue).
The relative configurations of the different diastereomers were elucidated by 1D and 2D NMR methods in combination with crystal structure analysis by X-ray diffraction (see Figure 2b and the Supporting Information). The absolute configurations could be revealed after separation of the enantiomers by chiral HPLC and comparison of experimentally obtained ECD-spectra with calculated ECD spectra (B3LYP/6-311G(d,p) level of theory using a PCM solvent model for MeCN). The structural assignments were confirmed by X-ray crystal structure analysis of racemic A/A′, B/B′, and C/C′ and enantiopure C′ and C isomers (see Figure 2b,c and the Supporting Information).
Thermal isomerization reactions and the ground-state energy profile of HTI 1 and its different isomers were determined in a combined theoretical (B3LYP/6-311G(d,p) level of theory using a PCM solvent model for MeCN) and experimental approach as summarized in Figure 3. The relative Gibbs free energy differences ΔG of the stable isomers were determined experimentally by either heating a sample of rac-A to 62 °C over prolonged time in MeCN-d3 solution in the dark to establish thermal equilibrium or measuring the equilibrium ratio between rac-C and rac-D directly in the dark at 20 °C. From the relative abundance of each enantiomeric pairs rac-A and rac-B or rac-C and rac-D, the corresponding ΔG values could be obtained (see Table S1). The Gibbs energy of activation for thermal atropisomerization of rac-A to rac-B was determined to be ΔG⧧ = 26.8 kcal mol–1 in MeCN-d3 solution by following thermal decay of rac-A at 62 °C with 1H NMR spectroscopy. The theoretical description predicts isomer interconversion by selective rotation of the trifluormethylanisol substituent whereby the smaller methoxy substituent faces the side of the sulfur atom (TS2, Figure 3c). The selective rotation of one aromatic ring was confirmed by following the atropisomerization of enantiomers by chiral HPLC at 60 °C (see Figure 3a and Supporting Information for details). Isomer A is thermally converted exclusively into isomer B and A′ is thermally converted exclusively into B′. This selectivity is only possible by sole rotation around the trifluormethylanisol substituent. It should be mentioned at this point that a significant energy difference between the two possible transition states TS1 and TS2 is found in the theoretical description. Therefore, theory predicts an unidirectional rotation for this atropisomerization, which can however not be proven in a straightforward manner via experiment. A strong indication is given by the good agreement between the absolute ΔG⧧ values obtained from theory and experiment.
The corresponding Gibbs energy of activation for thermal rac-C/rac-D interconversion was found to be ΔG⧧ = 18.8 kcal mol–1 in MeCN-d3 solution by following the decay kinetics of a rac-D enriched sample with 1H NMR spectroscopy at −20 °C in the dark (see Figure 3b and the Supporting Information). The theoretical description predicts that this atropisomerization takes place by sole rotation of the dimethylanisol substituent whereby again the smaller methoxy substituent faces the side of the sulfur atom (i.e., via transition state TS5, Figure 3c). The selective rotation could be confirmed experimentally (see the Supporting Information). After chiral HPLC separation of enantiomer pairs C/D from pairs C′/D′ at 22 °C thermal interconversion of the enantiopure samples was monitored using chiral HPLC as analytical method. Only the thermal conversion of D to C as well as that of D′ to C′ is observed. Rotation of the trifluormethylanisol substituent would lead to interconversions from D′ to C and from D to C′, respectively, which was not detected in the experiments. Again, an unidirectional rotation of the single bond is predicted from theory for the atropisomerization between rac-D and rac-C. The thermal isomerization reactions of HTI 1 were thus found to be highly selective, and only the aromatic residue with the Z relation to the sulfur atom rotates in this process. No thermal double bond isomerization was observed even after prolonged heating to 140 °C in tetrachloroethane-d2 solution and thus the relative stabilities of the Z and E configured isomers could not be determined experimentally.
After establishing the thermal behavior of 1 its photochemistry was studied in detail (Figure 4a). Isomers A, A′, B, and B′ could be isolated in pure form and their photochemical reactions were elucidated individually. Isomers C/D and C′/D′ could only be isolated as enantiomerically pure mixtures of diastereomers at ambient temperatures. Their photochemical properties were thus determined in thermodynamic equilibrium at 22 °C or separately at low temperatures after selective isomer enrichment. Irradiation of pure rac-A in MeCN did not lead to significant population of other isomers and therefore productive photochemistry of these enantiomers is strongly subdued. In contrast irradiation of isomers rac-B in MeCN at −40 °C with 450 nm leads to exclusive formation of rac-D and a ratio of rac-B/rac-D = 52:48 is established in the photostationary state (PSS, see Figure 4b and the Supporting Information). Because of this PSS composition it was also established that rac-D only undergoes photoisomerization back to rac-B. When warming the solution above −30 °C, the thermal rac-D to rac-C atropisomerization was observed as discussed above. If irradiation was continued at the higher temperature, the photochemical rac-C to rac-A isomerization occurred. In a similar experiment at 22 °C, illumination of an equilibrium rac-D/rac-C mixture with 405 nm light resulted in strong accumulation of rac-A in the PSS with a ratio of rac-A:rac-B:rac-C:rac-D = 95:3:2:0 (see Figure 4c and the Supporting Information). It is thus possible to enrich rac-A almost quantitatively under irradiation with blue light. Quantum yields for the two photoreactions of rac-B and rac-C were obtained using photon counting in conjunction with 1H NMR or UV/vis spectroscopy and chiral HPLC analysis and are discussed further below (also see the Supporting Information for more details).
Figure 4.

Photoreactions of HTI 1 during 450 nm irradiation. (a) Photoreactions of 1 and associated quantum yields measured in MeCN solution. iQuantum yield determined taking into account the thermal equilibrium of C and D at 20 °C. iiDSBR = double single-bond rotation, quantum yield indirectly determined by multiplying the ratio of the photoproducts determined by chiral HPLC analysis with the quantum yield for the Hula-Twist reaction from rac-B to rac-D measured at −40 °C. iiiQuantum yield determined at −40 °C. (b) Photoconversion of isomer rac-B in MeCN-d3 solution at −40 °C followed by 1H NMR-spectroscopy (400 MHz). Starting with pure rac-B (top spectrum) only population of isomer rac-D is observed (top to bottom spectra recorded after the indicated irradiation times). (c) Photoconversion of isomer mixture rac-C/rac-D in MeCN-d3 solution at 22 °C followed by 1H NMR-spectroscopy (400 MHz). Starting with a mixture of rac-C/rac-D (top spectrum) almost exclusive population of isomer rac-A is observed (top to bottom spectra recorded after the indicated irradiation times). (d) Photoconversion of isomers B in MeCN solution at −40 °C followed by chiral HPLC. Starting with enantiomerically pure B (chromatogram 1) HPLC runs were conducted after different times of continuous irradiation at −40 °C monitoring the photoisomerization process (chromatograms 2–10). * denotes an impurity.
Both photoreactions rac-B to rac-D and rac-C to rac-A are Hula-Twist reactions, a photoreaction in which the central double bond and one adjacent single bond rotate in the same step.23,69 However, the molecular setup of HTI 1 allows for two different Hula-Twist photoreactions for each rac-B to rac-D and rac-C to rac-A conversion, respectively as such coupled bond rotation can involve either of the two aryl substituents. To elucidate, which single bond is rotated in the Hula-Twist reaction enantiopure samples of B, B′, C/D, and C′/D′ were illuminated and the photoreaction kinetics were followed by quantitative 1H NMR or chiral HPLC (see Figure 4b–d and the Supporting Information). Irradiation of B with 450 nm light at −40 °C led to formation of D in MeCN with a quantum yield of 1.4% while no notable reaction into D′ was observable (see Figure 4b,d). Thus, the Hula-Twist photoreaction proceeds exclusively by rotation around the central double bond and the single bond connecting to the dimethylanisol substituent. However, another photoreaction was also observed in this experiment leading from B to B′ with a significant quantum yield of 0.9% (Figure 4d). The associated motion is a DSBR in which two atropisomerization photoreactions occur at the same time, while the central double bond is not isomerized. To the best of our knowledge, this reaction is not described in the literature (a theoretically possible two-photon process can be excluded especially in apolar solvents since all other isomers do not undergo photoreactions that populate isomers B or B′ significantly). Interestingly, a sole single-bond rotation (SBR) from B to A or to A′ was not observed photochemically; if one single bond rotates, then the other single bond also undergoes rotation. As a result of this dual atropisomerization, isomer B is converted into its enantiomer B′. Isomers B and B′ therefore represent a branching point for the photochemistry of 1. Full racemization of the sample via DSBR is however prevented since a significant amount of B is converted to D in the competing Hula-Twist photoreaction. As expected, it was found that enantiomeric isomer B′ behaves in a mirror-symmetric manner and photoisomerizes into D′ via a Hula-Twist reaction and into B via DSBR. Isomer C predominantly underwent photoisomerization into A′ with a quantum yield of 5.2% at 450 nm irradiation. This corresponds to a Hula-Twist photoreaction involving the central double bond and the adjacent single bond to the dimethylanisol fragment. In this case, no DSBR photoreaction or other photoreactions were observed. However, the thermal equilibrium between C and D accounts for a small population of isomer D in the experiment; therefore, photoisomerization of the latter via Hula-Twist to isomer B was also detected to some degree. Irradiation of enantiomeric mixture C′/D′ showed the expected mirror-symmetric behavior and led to population of A and small amounts of B′, respectively. Finally, the photochemistry of both enantiomers A and A′ was scrutinized and (as observed already for the racemic mixture) showed strongly diminished efficiency by at least a factor of 10 compared to photochemical processes that produce A and A′ as products. Some minimal DSBR converting A into A′ and vice versa were found together with similarly inefficient conversion to isomers C/C′ (for details, see the Supporting Information).
Taking the thermal interconversions and the photoreactions together a repetitive isomerization cycle of racemic HTI 1 can be established in four distinct steps using the same solvent acetonitrile (Figure 5a). Starting with isomer rac-B the rac-D isomer can be obtained with 48% in the PSS (note that up to 80% of rac-D can be formed by irradiation of rac-B at −80 °C in MeOH solution as shown in the Supporting Information). By thermal SBR rac-D is converted into the rac-C isomer with 86%, as the thermal equilibrium ratio between rac-C and rac-D is 86:14. By further irradiation at 20 °C isomer rac-A can be enriched up to 95% (rac-A:rac-B:rac-C:rac-D = 95:3:2:0). In the next thermal SBR rac-A is converted to rac-B with 78% (rac-A:rac-B:rac-C:rac-D = 20:78:2:0). Therefore, a complete cycle populating one of the four diastereomers selectively after another is carried out by HTI 1. This cycle can be repeated without isolation of one of the diastereomers by “refocusing” the mixture in the isomer rac-A enriched state. This is achieved by irradiation of the mixture with 405 nm light at 20 °C, which reestablishes reliably the rac-A:rac-B:rac-C:rac-D = 95:3:2:0 ratio in solution. Consequently, the following heating and photoisomerization steps produce the same isomer ratios as in the first cycle. The fact that this cycle is reproducible when starting from an arbitrary mixture of isomers of HTI 1 was demonstrated by executing the cycle experiment 3 times in a row with the same NMR-sample (see the Supporting Information). Also, after 10 cycles, corresponding to 20 photochemical and 20 thermal steps, only a minimal decrease of the performance by about 5% was observed (Figure 5b).
Figure 5.

Isomerization properties of the sequential photoswitch 1. (a) Comprehensive scheme showing the selective and sequential switching behavior of HTI 1. The nature of individual transformations are assigned. (b) Repetitive reversible switching cycles of racemic HTI 1. The depicted steps represent a complete rac-B → rac-D → rac-C → rac-A cycle but without monitoring the photoreaction from rac-B to rac-D (up to 48% formed in the bulk transformation in MeCN-d3 and up to 80% formed in MeOH) followed by the thermal reaction to rac-C individually (up to 33% formed in the bulk transformation in MeCN-d3). Minimal loss of performance is observed after 10 full cycles. Refocusing occurs by the same enrichment of isomer rac-A. (c) Propensity for eight- (green trace) or five-step (red trace) sequential switching cycles depending on the ratio of Hula-Twist (x-axis) versus DSBR (black trace) photoreactions of isomers B/B′. (d) Schematic representation of a molecular motor mechanism as opposed to the herein presented sequential switch mechanism.
When also taking the enantiomers into account, a selective cycle of isomer interconversions A → B → D → C → A′ → B′ → D′ → C′ → A is constituted from the main photoreactions and thermal reactions. This is possible because each individual isomer transformation is highly selective under well-controlled conditions. The A → B and A′ → B′ as well as D → C and D′ → C′ interconversions proceed thermally and without racemization as sole SBRs, and all photochemical steps proceed as Hula-Twist reactions again with high selectivities. After a total of eight steps, the starting isomer is obtained again. This eight-step cycle can however be bypassed by the one-photon-DSBR converting isomer B into B′. In this case, HTI 1 carries out the two enantiomeric cycles A → B → B′ → D′ → C′ → A and A′ → B′ → B → D → C → A′.
By changing the environmental conditions during photoirradiation steps switching between the five- and eight-step cycles is possible (Figure 5c). If isomers B and B′ carry out a Hula-Twist photoreaction, then the eight-step cycle is followed, and if the one photon DSBR occurs, then one of the five-step cycles is pursued. Obviously, the probability of the eight step-cycle is 100% if both B isomers exclusively carry out the Hula-Twist reaction. This situation is approached in MeOH solution at −80 °C with a preference for the Hula-Twist reaction of 85% corresponding to 72% eight-step cycle overall (see Figure 5c and the Supporting Information for calculation details). Optimizing the conditions for the five-step cycle is not so obvious, as both the Hula-Twist and DSBR reaction of B and B′ are required for this cycle. Maximum efficiency for the five-step cycles is therefore reached in a situation where 50% Hula-Twist and 50% DSBR are present. This situation can nearly be reached in MeCN solution at 20 °C where 61% DSBR and 39% Hula-Twist leads to 48% propensity for the five-step cycle (Figure 5c). At the same time, 15% of the eight-step cycle is present under these conditions. Similarly irradiation of B/B′ at −40 °C in MeCN also leads to 48% propensity for the five-step cycle, owed to 61% Hula-Twist and 39% DSBR photoreactions taking place under these conditions. However, now 35% of the eight-step cycle is also present, which makes this situation less selective. When further increasing the overall DSBR probability by strongly reducing solvent polarity and conducting experiments in toluene solution, the efficiency for the five-step cycles decreases again slightly, as more “unproductive” switching back and forth between B and B′ is taking place. However, the decrease of the Hula-Twist reaction is much more severe in this case, and a comfortable situation can be reached in which the latter is almost completely suppressed. Thus, in toluene solution at 20 °C the eight-step cycle is only present to 8%, and the five-step cycle is much more likely with 40%. In this way, the preferences of HTI 1 to undergo a specific isomer interconversion cycle can be switched upside down by simply changing solvent and temperatures. Related strong effects on the photochemistry of HTI photochromes have been described by our group70−72 especially for the competing Hula-Twist and sole double bond photoisomerization.23,24 Thus, the eight-step cycle is preferred 5-fold over the five-step cycle in MeOH solvent at −80 °C (i.e., 72% total likelihood for eight-step versus 15% total likelihood for the five-step cycle). The five-step cycle is preferred over the eight-step cycle also by a factor of 5 in toluene solution at 20 °C (8% total likelihood for eight-step versus 40% total likelihood for the five-step cycle).
Although carrying out sequential switching cycles HTI 1 is not a molecular motor, obviously both five-step cycles are mirror-symmetric to each other; therefore, any partial directional motions cancel each other out. The eight-step cycle possesses no enantiomeric counterpart, but it does have a mirror plane that mirrors one-half of the cycle with the other one. This situation is comparable with a meso-compound, which is achiral despite the presence of stereo information. While one-half of the rotations within the eight-step cycle of HTI 1 is forward, the other half must be backward. Both motions cancel each other out (see Figure 5d). However, HTI 1 is also not a simple switch as the forward and backward motions are not random but follow a specific sequence and are isomer selective. Therefore, we refer to HTI 1 as a sequential switch. Interestingly, under observation with achiral techniques like NMR or UV–vis spectroscopy the five- and eight-step cycles are indistinguishable, and both cycles lead to the same apparent conversion in the sequence rac-A → rac-B → rac-C → rac-D.
In conclusion, we herein present an unprecedented type of a sequential molecular switch allowing the interconversion of eight different isomers selectively and in defined sequence. This interconversion can be carried out in an eight-step cycle featuring sequential photoactivated Hula-Twist reactions and thermal single-bond rotations or by two enantiomeric five-step cycles involving an unprecedented one-photon DSBR photoreaction. By simply changing the solvent and temperature, each cycle can be preferred over the other by a factor of 5. Furthermore, enrichment of one racemic isomer up to 95% in the PSS is possible enabling a continuous and identical repeatable sequence of isomer interconversions, even when starting with an arbitrary mixture of isomers. This new type of compact molecular tool providing complex sequential movements and eight different accessible states will add new fundamental opportunities for photoswitch applications and future bottom-up building of nanomaterials. In addition, the here discovered one-photon DSBR photoreaction enables a new type of light-induced motion at the molecular scale, which should be of great interest for photoswitching applications and especially for future advanced molecular machine building.
Acknowledgments
H.D. thanks the Deutsche Forschungsgemeinschaft (DFG) for an Emmy Noether fellowship (DU 1414/1-2). We further thank the European Research Council under the European Union’s Horizon 2020 research and innovation program (PHOTOMECH, grant agreement no. 101001794), the Deutsche Forschungsgemeinschaft (SFB 749, A12), and the Cluster of Excellence “Center for Integrated Protein Science Munich” (CIPSM) for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c11183.
Details of synthesis, isomer separation and identification, photochemical, photophysical and thermal behavior, quantum yield measurements, NMR spectra, theoretical description, and crystal structural data (PDF)
Accession Codes
CCDC 2100907–2100911 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Lubbe A. S.; Szymanski W.; Feringa B. L. Recent developments in reversible photoregulation of oligonucleotide structure and function. Chem. Soc. Rev. 2017, 46 (4), 1052–1079. 10.1039/C6CS00461J. [DOI] [PubMed] [Google Scholar]
- Lerch M. M.; Hansen M. J.; van Dam G. M.; Szymanski W.; Feringa B. L. Emerging Targets in Photopharmacology. Angew. Chem., Int. Ed. 2016, 55 (37), 10978–10999. 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: beyond proof of principle. J. Am. Chem. Soc. 2014, 136 (6), 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Morstein J.; Trauner D. New players in phototherapy: photopharmacology and bio-integrated optoelectronics. Curr. Opin. Chem. Biol. 2019, 50, 145–151. 10.1016/j.cbpa.2019.03.013. [DOI] [PubMed] [Google Scholar]
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Light-controlled tools. Angew. Chem., Int. Ed. 2012, 51 (34), 8446–8476. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- Irie M.; Fukaminato T.; Matsuda K.; Kobatake S. Photochromism of diarylethene molecules and crystals: memories, switches, and actuators. Chem. Rev. 2014, 114 (24), 12174–12277. 10.1021/cr500249p. [DOI] [PubMed] [Google Scholar]
- Andreasson J.; Pischel U. Molecules with a sense of logic: a progress report. Chem. Soc. Rev. 2015, 44 (5), 1053–1069. 10.1039/C4CS00342J. [DOI] [PubMed] [Google Scholar]
- Shao B.; Aprahamian I. Hydrazones as New Molecular Tools. Chem. 2020, 6 (9), 2162–2173. 10.1016/j.chempr.2020.08.007. [DOI] [Google Scholar]
- Boelke J.; Hecht S. Designing Molecular Photoswitches for Soft Materials Applications. Adv. Optical Mater. 2019, 7 (16), 1900404. 10.1002/adom.201900404. [DOI] [Google Scholar]
- Göstl R.; Senf A.; Hecht S. Remote-controlling chemical reactions by light: Towards chemistry with high spatio-temporal resolution. Chem. Soc. Rev. 2014, 43 (6), 1982–1996. 10.1039/c3cs60383k. [DOI] [PubMed] [Google Scholar]
- Feringa B. L., Browne W. R., Eds. Molecular Switches, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA, 2011. [Google Scholar]
- Petermayer C.; Dube H. Indigoid Photoswitches: Visible Light Responsive Molecular Tools. Acc. Chem. Res. 2018, 51 (5), 1153–1163. 10.1021/acs.accounts.7b00638. [DOI] [PubMed] [Google Scholar]
- Kortekaas L.; Browne W. R. The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome. Chem. Soc. Rev. 2019, 48 (12), 3406–3424. 10.1039/C9CS00203K. [DOI] [PubMed] [Google Scholar]
- Hull K.; Morstein J.; Trauner D. In Vivo Photopharmacology. Chem. Rev. 2018, 118 (21), 10710–10747. 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- Klajn R. Spiropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43 (1), 148–184. 10.1039/C3CS60181A. [DOI] [PubMed] [Google Scholar]
- Andreasson J.; Pischel U. Molecules for security measures: from keypad locks to advanced communication protocols. Chem. Soc. Rev. 2018, 47 (7), 2266–2279. 10.1039/C7CS00287D. [DOI] [PubMed] [Google Scholar]
- Fischer E.; Hirshberg Y. Formation of coloured forms of spirans by low-temperature irradiation. J. Chem. Soc. 1952, (Nov), 4522–4524. [Google Scholar]
- Irie M.; Mohri M. Thermally Irreversible Photochromic Systems. Reversible Photocyclization of Diarylethene Derivatives. J. Org. Chem. 1988, 53 (4), 803–808. 10.1021/jo00239a022. [DOI] [Google Scholar]
- Waldeck D. H. Photoisomerization Dynamics of Stilbenes. Chem. Rev. 1991, 91 (3), 415–436. 10.1021/cr00003a007. [DOI] [Google Scholar]
- Bandara H. M.; Burdette S. C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41 (5), 1809–1825. 10.1039/C1CS15179G. [DOI] [PubMed] [Google Scholar]
- Weinstein J.; Wyman G. M. Spectroscopic Studies on Dyes. II. The Structure of N,N’-Dimethylindigo1. J. Am. Chem. Soc. 1956, 78 (16), 4007–4010. 10.1021/ja01597a038. [DOI] [Google Scholar]
- Saltiel J.; Papadimitriou D.; Krishna T. S.; Huang Z. N.; Krishnamoorthy G.; Laohhasurayotin S.; Clark R. J. Photoisomerization of all-cis-1,6-diphenyl-1,3,5-hexatriene in the solid state and in solution: a simultaneous three-bond twist process. Angew. Chem., Int. Ed. 2009, 48 (43), 8082–8085. 10.1002/anie.200902724. [DOI] [PubMed] [Google Scholar]
- Gerwien A.; Schildhauer M.; Thumser S.; Mayer P.; Dube H. Direct evidence for hula twist and single-bond rotation photoproducts. Nat. Commun. 2018, 9 (1), 2510. 10.1038/s41467-018-04928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwien A.; Mayer P.; Dube H. Photon-Only Molecular Motor with Reverse Temperature-Dependent Efficiency. J. Am. Chem. Soc. 2018, 140 (48), 16442–16445. 10.1021/jacs.8b10660. [DOI] [PubMed] [Google Scholar]
- Liu R. S.; Asato A. E. The primary process of vision and the structure of bathorhodopsin: a mechanism for photoisomerization of polyenes. Proc. Natl. Acad. Sci. U. S. A. 1985, 82 (2), 259–263. 10.1073/pnas.82.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuß W.; Kosmidis C.; Schmid W. E.; Trushin S. A. The photochemical cis-trans isomerization of free stilbene molecules follows a hula-twist pathway. Angew. Chem., Int. Ed. 2004, 43 (32), 4178–4182. 10.1002/anie.200454221. [DOI] [PubMed] [Google Scholar]
- Warshel A. Bicycle-pedal model for the first step in the vision process. Nature 1976, 260 (5553), 679–683. 10.1038/260679a0. [DOI] [PubMed] [Google Scholar]
- Schapiro I.; Weingart O.; Buss V. Bicycle-Pedal Isomerization in a Rhodopsin Chromophore Model. J. Am. Chem. Soc. 2009, 131 (1), 16–17. 10.1021/ja805586z. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Chen X.; Cui G.; Fang W.-H.; Thiel W. Concerted Asynchronous Hula-Twist Photoisomerization in the S65T/H148D Mutant of Green Fluorescent Protein. Angew. Chem., Int. Ed. 2014, 53 (33), 8649–8653. 10.1002/anie.201405303. [DOI] [PubMed] [Google Scholar]
- Jung Y. O.; Lee J. H.; Kim J.; Schmidt M.; Moffat K.; Srajer V.; Ihee H. Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nat. Chem. 2013, 5 (3), 212–220. 10.1038/nchem.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila V. R. I.; Schotte F.; Cho H. S.; Hummer G.; Anfinrud P. A. Contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein. Nat. Chem. 2014, 6 (4), 258–259. 10.1038/nchem.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y. O.; Lee J. H.; Kim J.; Schmidt M.; Moffat K.; Šrajer V.; Ihee H. Reply to ’contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein’. Nat. Chem. 2014, 6 (4), 259–260. 10.1038/nchem.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust D.; Andreasson J.; Pischel U.; Moore T. A.; Moore A. L. Data and signal processing using photochromic molecules. Chem. Commun. 2012, 48 (14), 1947–1957. 10.1039/C1CC15329C. [DOI] [PubMed] [Google Scholar]
- Mutoh K.; Kobayashi Y.; Yamane T.; Ikezawa T.; Abe J. Rate-Tunable Stepwise Two-Photon-Gated Photoresponsive Systems Employing a Synergetic Interaction between Transient Biradical Units. J. Am. Chem. Soc. 2017, 139 (12), 4452–4461. 10.1021/jacs.6b13322. [DOI] [PubMed] [Google Scholar]
- Moran M. J.; Magrini M.; Walba D. M.; Aprahamian I. Driving a Liquid Crystal Phase Transition Using a Photochromic Hydrazone. J. Am. Chem. Soc. 2018, 140 (42), 13623–13627. 10.1021/jacs.8b09622. [DOI] [PubMed] [Google Scholar]
- Fihey A.; Perrier A.; Browne W. R.; Jacquemin D. Multiphotochromic molecular systems. Chem. Soc. Rev. 2015, 44 (11), 3719–3759. 10.1039/C5CS00137D. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Weidner S.; Risse T.; Hecht S. The role of statistics and microenvironment for the photoresponse in multi-switch architectures: The case of photoswitchable oligoazobenzene foldamers. Chem. Sci. 2013, 4 (11), 4156–4167. 10.1039/c3sc51664d. [DOI] [Google Scholar]
- Zhang J.; Zou Q.; Tian H. Photochromic materials: more than meets the eye. Adv. Mater. 2013, 25 (3), 378–99. 10.1002/adma.201201521. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Grubert L.; Hecht S.; Bleger D. Orthogonal switching in four-state azobenzene mixed-dimers. Chem. Commun. 2017, 53 (23), 3323–3326. 10.1039/C7CC00504K. [DOI] [PubMed] [Google Scholar]
- Bléger D.; Liebig T.; Thiermann R.; Maskos M.; Rabe J. P.; Hecht S. Light-Orchestrated Macromolecular “Accordions”: Reversible Photoinduced Shrinking of Rigid-Rod. Polymers. Angew. Chem. Int. Ed. 2011, 50 (52), 12559–12563. 10.1002/anie.201106879. [DOI] [PubMed] [Google Scholar]
- Heindl A. H.; Becker J.; Wegner H. A. Selective switching of multiple azobenzenes. Chem. Sci. 2019, 10 (31), 7418–7425. 10.1039/C9SC02347J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Slavov C.; Wegner H. A.; Wachtveitl J.; Dreuw A. Computational design of a molecular triple photoswitch for wavelength-selective control. Chem. Sci. 2018, 9 (46), 8665–8672. 10.1039/C8SC03379J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisnetti F.; Ballardini R.; Credi A.; Gandolfi M. T.; Masiero S.; Negri F.; Pieraccini S.; Spada G. P. Photochemical and electronic properties of conjugated bis(azo) compounds: an experimental and computational study. Chem. - Eur. J. 2004, 10 (8), 2011–2021. 10.1002/chem.200305590. [DOI] [PubMed] [Google Scholar]
- Peters A.; Branda N. R. Limited photochromism in covalently linked double 1,2-dithienylethenes. Adv. Mater. Opt. Electron. 2000, 10 (6), 245–249. . [DOI] [Google Scholar]
- Yagi K.; Irie M. Photochromic and Fluorescent Properties of a Diarylethene Dimer. Chem. Lett. 2003, 32 (9), 848–849. 10.1246/cl.2003.848. [DOI] [Google Scholar]
- Han M.; Luo Y.; Damaschke B.; Gomez L.; Ribas X.; Jose A.; Peretzki P.; Seibt M.; Clever G. H. Light-Controlled Interconversion between a Self-Assembled Triangle and a Rhombicuboctahedral Sphere. Angew. Chem., Int. Ed. 2016, 55 (1), 445–449. 10.1002/anie.201508307. [DOI] [PubMed] [Google Scholar]
- Han M.; Michel R.; He B.; Chen Y. S.; Stalke D.; John M.; Clever G. H. Light-triggered guest uptake and release by a photochromic coordination cage. Angew. Chem., Int. Ed. 2013, 52 (4), 1319–1323. 10.1002/anie.201207373. [DOI] [PubMed] [Google Scholar]
- Kortekaas L.; Ivashenko O.; van Herpt J. T.; Browne W. R. A Remarkable Multitasking Double Spiropyran: Bidirectional Visible-Light Switching of Polymer-Coated Surfaces with Dual Redox and Proton Gating. J. Am. Chem. Soc. 2016, 138 (4), 1301–1312. 10.1021/jacs.5b11604. [DOI] [PubMed] [Google Scholar]
- Hoffmann K.; Guentner M.; Mayer P.; Dube H. Symmetric and nonsymmetric bis-hemithioindigos – precise visible light controlled shape-shifters. Org. Chem. Front. 2019, 6 (8), 1244–1252. 10.1039/C9QO00202B. [DOI] [Google Scholar]
- Vlasceanu A.; Koerstz M.; Skov A. B.; Mikkelsen K. V.; Nielsen M. B. Multistate Photoswitches: Macrocyclic Dihydroazulene/Azobenzene Conjugates. Angew. Chem., Int. Ed. 2018, 57 (21), 6069–6072. 10.1002/anie.201712942. [DOI] [PubMed] [Google Scholar]
- Andreasson J.; Pischel U.; Straight S. D.; Moore T. A.; Moore A. L.; Gust D. All-photonic multifunctional molecular logic device. J. Am. Chem. Soc. 2011, 133 (30), 11641–11648. 10.1021/ja203456h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balter M.; Li S.; Nilsson J. R.; Andreasson J.; Pischel U. An all-photonic molecule-based parity generator/checker for error detection in data transmission. J. Am. Chem. Soc. 2013, 135 (28), 10230–10233. 10.1021/ja403828z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrin C.; Aidibi Y.; Sanguinet L.; Leriche P.; Aloise S.; Orio M.; Delbaere S. When Light and Acid Play Tic-Tac-Toe with a Nine-State Molecular Switch. J. Am. Chem. Soc. 2019, 141 (48), 19151–19160. 10.1021/jacs.9b11048. [DOI] [PubMed] [Google Scholar]
- Gobbi L.; Seiler P.; Diederich F. A Novel Three-Way Chromophoric Molecular Switch: pH and Light Controllable Switching Cycles. Angew. Chem., Int. Ed. 1999, 38 (5), 674–678. . [DOI] [PubMed] [Google Scholar]
- Kink F.; Collado M. P.; Wiedbrauk S.; Mayer P.; Dube H. Bistable Photoswitching of Hemithioindigo with Green and Red Light: Entry Point to Advanced Molecular Digital Information Processing. Chem. - Eur. J. 2017, 23 (26), 6237–6243. 10.1002/chem.201700826. [DOI] [PubMed] [Google Scholar]
- Koumura N.; Zijlstra R. W. J.; van Delden R. A.; Harada N.; Feringa B. L. Light-driven monodirectional molecular rotor. Nature 1999, 401 (6749), 152–155. 10.1038/43646. [DOI] [PubMed] [Google Scholar]
- Kassem S.; van Leeuwen T.; Lubbe A. S.; Wilson M. R.; Feringa B. L.; Leigh D. A. Artificial molecular motors. Chem. Soc. Rev. 2017, 46 (9), 2592–2621. 10.1039/C7CS00245A. [DOI] [PubMed] [Google Scholar]
- Roke D.; Wezenberg S. J.; Feringa B. L. Molecular rotary motors: Unidirectional motion around double bonds. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (38), 9423–9431. 10.1073/pnas.1712784115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistemaker H. A.; Stacko P.; Visser J.; Feringa B. L. Unidirectional rotary motion in achiral molecular motors. Nat. Chem. 2015, 7 (11), 890–896. 10.1038/nchem.2362. [DOI] [PubMed] [Google Scholar]
- Klok M.; Boyle N.; Pryce M. T.; Meetsma A.; Browne W. R.; Feringa B. L. MHz Unidirectional Rotation of Molecular Rotary Motors. J. Am. Chem. Soc. 2008, 130 (32), 10484–10485. 10.1021/ja8037245. [DOI] [PubMed] [Google Scholar]
- Štacko P.; Kistemaker J. C. M.; van Leeuwen T.; Chang M.-C.; Otten E.; Feringa B. L. Locked synchronous rotor motion in a molecular motor. Science 2017, 356 (6341), 964–968. 10.1126/science.aam8808. [DOI] [PubMed] [Google Scholar]
- Greb L.; Lehn J. M. Light-driven molecular motors: imines as four-step or two-step unidirectional rotors. J. Am. Chem. Soc. 2014, 136 (38), 13114–13117. 10.1021/ja506034n. [DOI] [PubMed] [Google Scholar]
- Guentner M.; Schildhauer M.; Thumser S.; Mayer P.; Stephenson D.; Mayer P. J.; Dube H. Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor. Nat. Commun. 2015, 6 (1), 8406. 10.1038/ncomms9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber L. A.; Hoffmann K.; Thumser S.; Böcher N.; Mayer P.; Dube H. Direct Observation of Hemithioindigo-Motor Unidirectionality. Angew. Chem., Int. Ed. 2017, 56 (46), 14536–14539. 10.1002/anie.201708178. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Schildhauer M.; Rott F.; Huber L. A.; Guentner M.; Thumser S.; Hoffmann K.; Oesterling S.; de Vivie-Riedle R.; Riedle E.; Dube H. Complete Mechanism of Hemithioindigo Motor Rotation. J. Am. Chem. Soc. 2018, 140 (15), 5311–5318. 10.1021/jacs.8b02349. [DOI] [PubMed] [Google Scholar]
- Gerwien A.; Mayer P.; Dube H. Green light powered molecular state motor enabling eight-shaped unidirectional rotation. Nat. Commun. 2019, 10 (1), 4449. 10.1038/s41467-019-12463-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl E.; Mayer P.; Dube H. Active and Unidirectional Acceleration of Biaryl Rotation by a Molecular Motor. Angew. Chem., Int. Ed. 2020, 59 (14), 5730–5737. 10.1002/anie.201913798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwien A.; Reinhardt T.; Mayer P.; Dube H. Synthesis of Double-Bond Substituted Hemithioindigo Photoswitches. Org. Lett. 2018, 20 (1), 232–235. 10.1021/acs.orglett.7b03574. [DOI] [PubMed] [Google Scholar]
- Liu R. S. H. Photoisomerization by Hula-Twist: A Fundamental Supramolecular Photochemical Reaction. Acc. Chem. Res. 2001, 34 (7), 555–562. 10.1021/ar000165c. [DOI] [PubMed] [Google Scholar]
- Wiedbrauk S.; Maerz B.; Samoylova E.; Reiner A.; Trommer F.; Mayer P.; Zinth W.; Dube H. Twisted Hemithioindigo Photoswitches: Solvent Polarity Determines the Type of Light-Induced Rotations. J. Am. Chem. Soc. 2016, 138 (37), 12219–12227. 10.1021/jacs.6b05981. [DOI] [PubMed] [Google Scholar]
- Wiedbrauk S.; Maerz B.; Samoylova E.; Mayer P.; Zinth W.; Dube H. Ingredients to TICT Formation in Donor Substituted Hemithioindigo. J. Phys. Chem. Lett. 2017, 8 (7), 1585–1592. 10.1021/acs.jpclett.7b00371. [DOI] [PubMed] [Google Scholar]
- Stallhofer K.; Nuber M.; Schüppel F.; Thumser S.; Iglev H.; de Vivie-Riedle R.; Zinth W.; Dube H. Electronic and Geometric Characterization of TICT Formation in Hemithioindigo Photoswitches by Picosecond Infrared Spectroscopy. J. Phys. Chem. A 2021, 125 (20), 4390–4400. 10.1021/acs.jpca.1c02646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.